

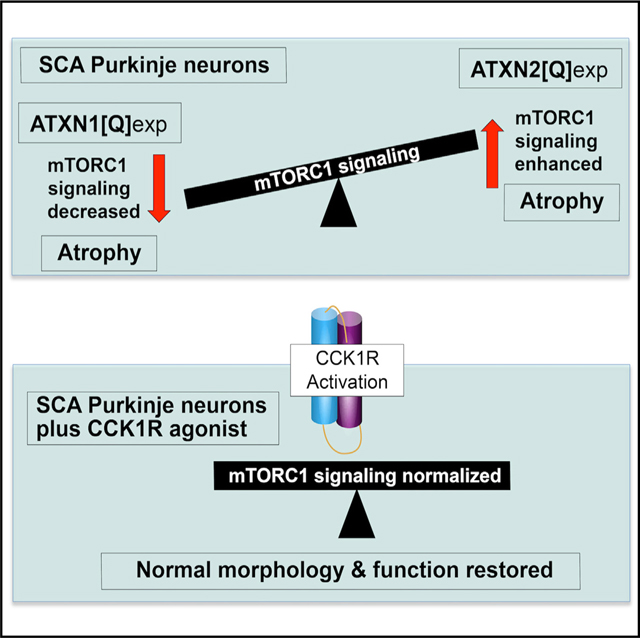
SUMMARY
Spinocerebellar ataxias (SCAs) are a group of genetic diseases characterized by progressive ataxia and neurodegeneration, often in cerebellar Purkinje neurons. A SCA1 mouse model, Pcp2-ATXN1[30Q]D776, has severe ataxia in absence of progressive Purkinje neuron degeneration and death. Previous RNA-seq analyses identify cerebellar upregulation of the peptide hormone cholecystokinin (Cck) in Pcp2-ATXN1[30Q]D776 mice. Importantly, absence of Cck1 receptor (Cck1R) in Pcp2-ATXN1[30Q]D776 mice confers a progressive disease with Purkinje neuron death. Administration of a Cck1R agonist, A71623, to Pcp2-ATXN1[30Q] D776;Cck−/− and Pcp2-AXTN1[82Q] mice dampens Purkinje neuron pathology and associated deficits in motor performance. In addition, A71623 administration improves motor performance of Pcp2-ATXN2[127Q] SCA2 mice. Moreover, the Cck1R agonist A71623 corrects mTORC1 signaling and improves expression of calbindin in cerebella of AXTN1[82Q] and ATXN2[127Q] mice. These results indicate that manipulation of the Cck-Cck1R pathway is a potential therapeutic target for treatment of diseases involving Purkinje neuron degeneration.
In brief
Wozniak et al. show that administration of the Cck1R agonist A71623 to AXTN1[82Q] SCA1 and ATXN2[127Q] SCA2 mice dampens their deficits in motor performance, corrects mTORC1 signaling, and improves cerebellar expression of calbindin, a Purkinje neuron marker. These results indicate that the Cck-Cck1R pathway is a therapeutic target for SCAs.
Graphical Abstract
INTRODUCTION
Among inherited neurodegenerative diseases are the severe forms of the dominantly inherited spinocerebellar ataxia (SCA) that are due to an in-frame CAG trinucleotide expansion encoding a polyglutamine (polyQ) tract (Durr, 2010; Klockgether, 2011). These SCAs (SCA1–SCA3, SCA6, SCA7, and SCA17) are clinically characterized by a progressive ataxia with cerebellar degeneration that in most of these SCAs consists of severe degeneration and loss of Purkinje cells, the major integrative neuron of the cerebellar cortex (Koeppen, 2005; Seidel et al., 2012).
To gain insight into the molecular function of ATXN1 as well as cellular pathways critical for SCA1-like disease in Purkinje cells, a series of SCA1 mouse models were developed and characterized that manifest SCA1-like disease phenotypes, including cerebellar ataxia and loss of Purkinje cells (Burright et al., 1995; Watase et al., 2002; Duvick et al., 2010). One outcome of this work in relation to SCA1 Purkinje cell disease is the concept that a critical ATXN1 function is its role in regulating transcription and RNA processing (Paulson et al., 2017). Two ATXN1 transgenic mouse lines formed the basis of a study that utilized RNA sequencing (RNA-seq) to reveal cerebellar gene expression changes associated with disease progression and protection (Ingram et al., 2016). In these mice, expression of ATXN1 is specifically directed to Purkinje cells. One line included mice expressing ATXN1 with an expanded polyQ, ATXN1[82Q], that manifest a progressive disease culminating in Purkinje cell death (Burright et al., 1995; Clark et al., 1997). The second line included mice expressing ATXN1 with a wild-type (WT) polyQ stretch but with an Asp at position 776 that mimics some aspects of S776 phosphorylation. This amino acid substitution transforms human WT ATXN1 [30Q] into a pathogenic protein, with ATXN1[30Q]-D776 mice manifesting ataxia that is as severe as that manifested by ATXN1[82Q] animals. Yet, in contrast to ATXN1[82Q] mice, cerebellar disease in ATXN1[30Q]-D776 mice does not manifest with a progressive cerebellar pathology that cumulates with Purkinje neuron death (Duvick et al., 2010). Analysis of the ATXN1 transgenic RNA-seq data demonstrated a substantial, unique, and Purkinje-cell-specific upregulation of cholecystokinin (Cck) RNA in Pcp2-ATXN1[30Q]D776 mice (Ingram et al., 2016). Upon crossing ATXN1[30Q]D776 mice to Cck−/− and Cck1R−/− mice, it was found that absence of either Cck or Cck1 receptor (Cck1R) in Pcp2-ATXN1[30Q]D776 mice resulted in a Purkinje cell disease in which pathology progressed to cell death. These results strongly supported a hypothesis where elevated Cck and its subsequent processing to a peptide that binds to Purkinje cell Cck1R in an autocrine manner causes the lack of progressive Purkinje cell pathology in ATXN1[30Q]D776 mice.
Here, we show that administration of the Cck1R agonist A71623 was protective against progressive ataxia and Purkinje cell pathology in ATXN1[30Q]D776/Cck−/− mice. Moreover, RNA-seq analysis of ATXN1[30Q]D776/Cck−/− cerebella disclosed a weighted gene coexpression network analysis (WGCNA) gene module significantly overlaps with a gene network recently associated with Purkinje cell disease progression in ATXN1[82Q] mice (Ingram et al., 2016), indicating that the progressive disease in ATXN1[30Q]D776/Cck−/− and ATXN1[82Q] mice have molecular features in common. Correspondingly, administration of the Cck1R agonist A71623 corrected mTORC1 signaling and was protective against progressive ataxia in ATXN1[82Q] mice. Lastly, administration of A71623 also improved motor performance in ATXN2[127Q] mice and restored normal cerebellar mTORC1 signaling. These results provide proof of principle for Cck1R activation as a therapeutic option across the SCAs characterized by Purkinje cell dysfunction/degeneration.
RESULTS
Administration of a Cck1R agonist, A71623, reduces disease in Pcp2-ATXN1[30Q]D776/Cck−/− mice
Pcp2-ATXN1[30Q]D776 animals have ataxia in absence of Purkinje cell progressive pathology (Duvick et al., 2010). Previously, we found that Pcp2-ATXN1[30Q]D776 mice have an increase in cerebellar expression of Cck and that deletion of Cck or Cck1R genes from ATXN1[30Q]D776 animals converted disease from being nonprogressive to one in which Purkinje cell pathology is progressive and readily detected by 36 weeks of age (Ingram et al., 2016; Figure S1). In addition, Cck RNA levels are decreased in Pcp2-ATXN1[82Q] (Ingram et al., 2016) and Pcp2-ATXN2[127Q] cerebella (Pflieger et al., 2017). This led to the suggestion that a Cck/Cck1R pathway protects Pcp2-ATXN1[30Q]D776 Purkinje cells from a progressive pathology (Ingram et al., 2016). We reasoned that a test of this idea would assess whether activation of Purkinje cell Cck1Rs in ATXN1[30Q] D776/Cck−/− mice dampened disease progression.
A71623, a Cck tetrapeptide analog, is a highly selective agonist for Cck1R. In rodents, peripheral administration is able to elicit CNS-mediated behavioral effects (Asin et al., 1992a, 1992b). To assess the ability of A71623 administered peripherally to activate cerebellar Cck1Rs, phosphorylation of cerebellar Erk1 and Erk2, downstream targets of Cck1R activation (Dufresne et al., 2006), was assessed in mice following intraperitoneal (IP) injections of A71623. Doses selected were 0.026 mg (30 nmol)/kg, a dose previously shown to have central effects in mice (Asin et al., 1992a), and 0.132 mg/kg, the maximum dose given solubility of A71623. As shown in Figures S2A and S2B, IP injections of A71623 at these doses resulted in a dose-dependent increase in cerebellar Erk1 and Erk2 phosphorylation.
Figure 1A depicts the A71623 treatment strategy utilized to assess whether this Cck1R agonist impacts Purkinje cell disease progression in ATXN1[30Q]D776/Cck−/− mice. Since the extent of recovery from ATXN1-induced neurodegeneration decreases with age in SCA1 transgenic mice (Zu et al., 2004; Friedrich et al., 2018), we selected to initiate Cck1R agonist treatment early in disease progression. Briefly, baseline motor assessments were performed at 5 and 6 weeks using balance beam and rotarod, respectively. Upon completion of baseline rotarod assessments, IP osmotic pumps were implanted, and the Cck1R agonist A71623 was administered at a rate of 0.026 mg (30 nmol)/kg/day. Balance beam and rotarod assessments were performed, and pumps were replaced at times indicated. Mice were sacrificed after the final rotarod evaluation at 36 weeks of age and extent of cerebellar pathology determined. By both balance beam (Figure 1B) and rotarod (Figure 1C) assessments, two cohorts of ATXN1[30Q]D776/Cck−/− mice were equally compromised neurologically prior to treatment. ATXN1[30Q]D776/Cck−/− animals that received the A72613 agonist performed significantly better than ATXN1[30Q]D776/Cck−/− mice that received vehicle alone (Figures 1B and 1C). Purkinje cell pathology in the ATXN1[30Q]D776/Cck−/− animals receiving the Cck1R A72613 agonist, as assessed by extent of molecular layer atrophy (Figures 1D, S4C, and S4D) and loss of Purkinje cells (Figure 1D), was significantly less than ATXN1[30Q]D776/Cck−/− mice that received vehicle alone. Of note, while Cck has been shown to reduce food intake by activating Cck1R (reviewed in Miller and Desai, 2016), we found no difference in body weight between A71623- and vehicle-treated ATXN1 [30Q]D776/Cck−/− animals at 36 weeks of age (Figure S2C).
Figure 1. Cck1R agonist A71623 treatment dampens Purkinje neuron pathology in ATXN1[30Q]D776;Cck−/− mice.

(A) Scheme of assessment and treatment course. Mice were tested for motor performance using two tests. Then, either 0.02 mg/kg/day A71623 or vehicle (20 mM PBS) was administered until 36 weeks of age, at which time mice were sacrificed for pathology. Osmotic minipumps were replaced every 6 weeks.
(B) Time to cross the 10-mm round balance beam.
(C) Latency to fall on the rotarod.
(D) Molecular layer thickness in 36-week-old untreated and treated mice.
(E) Number of Purkinje neurons per 250 μm in cerebellar primary fissure in 36-week-old untreated and treated mice.
Error bars represent SEM. *p < 0.05, **p < 0.01, and ***p < 0.001, two-way ANOVA with Tukey post hoc test. N’s (B–E) are indicated within each bar on the graphs.
Overlap between ATXN1[30Q]D776/Cck−/− and ATXN1 [82Q] disease-associated gene coexpression modules
Previously, we showed that a WGCNA of cerebellar RNA-seq data revealed a module reflecting Purkinje-cell-disease-correlated signature of gene expression alterations in ATXN1[82Q] animals (Ingram et al., 2016; Table S1). To gain an understanding of the disease-correlated gene expression alterations in ATXN1[30Q]/Cck−/− mice, RNA-seq was performed on ATXN1 [30Q]/Cck−/− and Cck−/− cerebella at 5, 12, and 28 weeks followed by WGCNA (Langfelder and Horvath, 2008). Two WGCNAs were performed; the first included the ATXN1[30Q]/Cck−/− RNA-seq data plus that obtained previously from WT, ATXN1[30Q]D776, and ATXN1[82Q] cerebella, and in the second one, the ATXN1[82Q] RNA-seq data were not included in the WGCNA.
Of the 36 WGCNA modules obtained on all RNA-seq data excluding that from ATXN1[82Q] cerebella, the 374-gene Pink module (Table S2) was found to significantly correlate with disease (p = 7e-13), as determined by thickness of the cerebellar molecular layer. The eigengene, a single representative expression profile (i.e., the first principal component of the gene expression for genes in a WGCNA module and a summary of the standardized module expression) was used to evaluate the coexpression pattern of the cerebellar WGCNA Pink module. The Pink module eigengene in both ATXN1[82Q] and ATXN1[30Q] D776/Cck−/− and not in ATXN1[30Q]D776 mice decreased with age (Figure S3A) in a manner similar to the Magenta eigengene (Ingram et al., 2016). Consistent with the time course of molecular layer thinning in ATXN1[82Q] and ATXN1[30Q]D776/Cck−/− mice (Figure S1), the Pink eigengene decreased most pronouncedly between 5 and 12 weeks in ATXN1[82Q] and not until 28 weeks in ATXN1[30Q]D776/Cck−/− cerebella (Figure S3A). Interestingly, the Pink WGCNA module obtained using ATXN1[30Q]D776/Cck−/− RNA-seq data overlapped extensively with the previous disease-associated Magenta WGCNA module (Ingram et al., 2016) (Figure S3B). To rule out that this overlap was driven by ATXN1[82Q] RNA-seq data, a second WGCNA was performed excluding the ATXN1[82Q] data (Table S3). Once again a Pink module was found to significantly correlate with molecular layer thickness that also overlapped highly significantly with the previous Magenta WGCNA module (Figure 2B). Thus, we conclude that the RNA-seq/WGCNA of ATXN1[82Q] and ATXN1[30Q]D776/Cck−/− supports the conclusion that the Purkinje cell disease in these two ATXN1 transgenic lines shares an underlying molecular basis.
Figure 2. Cck1R agonist A71623 treatment dampens Purkinje neuron pathology ATXN1[82Q] mice and improves motor performance in ATXN2[127Q] mice.

(A) Scheme of the assessment and treatment course. Mice were tested for motor performance using two tests. Then either 0.02 mg/kg/day A71623 or vehicle (20 mM PBS) was given until 12 weeks of age, at which time mice were sacrificed for pathology.
(B) Number of footslips on the 10-mm round balance beam.
(C) Time to cross the 10-mm round balance beam.
(D) Latency to fall on the rotarod.
(E) Molecular thickness of 6-week-old untreated mice compared to 12-week-old treated mice.
(F) Schematic depiction of the assessment and treatment course of ATXN2[127Q] mice. Mice were tested for motor performance at baseline and 3 and 11 weeks of age using the 10-mm round balance beam. At 4 weeks of age, mice were given daily IP injections of 0.02 mg/kg/day A71623 or vehicle (20 mM PBS). At 6 weeks of age, osmotic pumps were implanted intraperitoneally, and the Cck1R agonist or vehicle was administered.
(G) Number of footslips on the 10-mm round balance beam for ATXN2[127Q].
(H) Time to cross the 10-mm round balance for ATXN2[127Q].
N’s are indicated inside each bar for each genotype/test. Error bars represent SEM. *p < 0.05, **p < 0.01, and ***p < 0.001, two-way ANOVA with Tukey post hoc test.
Cck1R agonist A71623 alleviates disease in ATXN1 [82Q] mice
With the significant similarity in age-related gene coexpression changes in ATXN1[82Q] and ATXN1[30Q]D776/Cck−/− cerebella, along with a decrease in Cck expression in Purkinje cells of ATXN1[82Q] mice (Ingram et al., 2016), we reasoned that the Cck1R agonist A71623 would likely dampen disease in ATXN1[82Q] mice. Thus, a treatment scheme based on the one used to show the ability of the Cck1R agonist A71623 (dosed at 0.026 mg/kg/day) to impact Purkinje cell disease progression in ATXN1[30Q]D776/Cck−/− mice was applied to ATXN1[82Q] mice (Figure 2A). Due to the more progressive nature of disease in ATXN1[82Q] animals, the strategy utilized included two modifications from that used with ATXN1[30Q]D776/Cck−/− mice. First, agonist treatment was initiated earlier, at 4 weeks of age. Therefore, until animals reached a size such that the IP pumps could be implanted, they were given IP injections of A71623 for 2 weeks. The second modification was that the initial trial was run for 12 weeks, to the age when ATXN1[82Q] mice show the most dramatic decrease in the thickness of the molecular layer (Figure S1). Following a final motor assessment at 11 and 12 weeks of age, A71623- and vehicle-treated animals were sacrificed and analyzed using molecular and morphological approaches.
By beam walk assessments (Figures 2B and 2C) assessing footslips and time to cross a 10-mm round beam, respectively, the cohorts that were subsequently treated with A71623 or vehicle were equally impaired at 3 weeks of age. However, at 11 weeks of age, following 7 weeks of treatment, ATXN1[82Q] mice treated with the Cck1R agonist A71623 performed significantly better on the beam than those administered vehicle alone. Likewise, rotarod evaluation indicated that both cohorts performed equally well at 4 weeks of age. Yet, by 12 weeks of age, performance on the rotarod of vehicle-treated ATXN1 [82Q] mice deteriorated more significantly than that of ATXN1 [82Q] animals that received the agonist A71623 (Figure 2D). Similarly, molecular layer thinning at 12 weeks of age was significantly worse in vehicle-treated compared to A71623-treated ATXN1[82Q] mice (Figures 2E, S4A, and S4B). Importantly, the mechanism by which A71623 treatment alleviated disease in the ATXN1[82Q] mice did not involve a reduction in levels of the ATXN1[82Q] protein (Figure S5).
Cck1R agonist A71623 improves motor performance of ATXN2[127Q] mice
To assess whether the Cck1R agonist A71623 has a protective effect on another SCA mouse model, we utilized a mouse model of SCA2 expressing ATXN2[127Q] under the Purkinje-cell-specific regulatory element from the Pcp2 gene (Hansen et al., 2013). The treatment scheme and dosage for the Cck1R agonist A71623 was identical to that used for Pcp2-ATXN1[82Q] mice (Figure 2F). At 11 weeks of age, Pcp2-ATXN2[127Q] mice had a significant deficit in performance on the balance beam by both number of footslips and time to cross (Figure 2G and 2H). Importantly, at 11 weeks of age and following 7 weeks of treatment, Pcp2-ATXN2[127Q] mice treated with the Cck1R agonist A71623 performed significantly better on the balance beam than those administered vehicle alone (Figure 2G).
Proper mTORC1 signaling is restored by the Cck1R agonist A71623 in ATXN1[82Q] and ATXN2[127Q] cerebella
Ruegsegger et al. demonstrated that reduced mTORC1 signaling contributes to Purkinje cell disease in the cerebellum of the Atxn1154Q/2Q knockin mouse model of SCA1 (Ruegsegger et al., 2016). Importantly, mTORC1 is a downstream signaling pathway activated via Cck1R stimulation (reviewed in Cawston and Miller, 2010). Thus, we assessed the effect of the Cck1R agonist A71623 on mTORC1 signaling status in Pcp2-ATXN1[82Q] and Pcp2-ATXN2[127Q] cerebella by measuring levels of phosphorylated ribosomal protein S6 (pS6) and the translational repressor 4e-bp1 (p4e-bp1), two well-established targets of mTORC1 (Wullschleger et al., 2006). Figures 3A and 3B show that mTORC1 signaling was significantly decreased in 11-week-old ATXN1[82Q] cerebella as evaluated by measuring pS6 and p4e-bp1 levels. Next, we examined whether administration of the Cck1R agonist A71623 restored mTORC1 signaling in ATXN1[82Q] cerebella. 11-week-old ATXN1[82Q] mice were given an IP injection of A71623 (0.264 mg/kg). 24 h later, cerebella were harvested and mTORC1 signaling was found to be restored to a WT level, as evaluated by pS6 and p4e-bp1 levels (Figures 3A and 3B). Consistent with the ability of A71623 to decrease pathology in ATXN1[82Q] mice (Figure 3), a small but significant increase in the amount of calbindin protein was detected in cerebellar extracts from A71623-treated mice (Figures 3A and 3B). To validate that the action of A71623 on mTORC1 signaling was via the Cck1R, ATXN1[82Q] mice were crossed to Cck1R−/− mice (Kopin et al., 1999). Figures 3C and 3D show that in 8-week-old ATXN1[82Q] mice lacking Cck1R, A71623 was no longer able to restore cerebellar mTORC1 signaling.
Figure 3. mTORC1 signaling activity and cerebellar marker Calb1 are reduced in ATXN1[82Q] mice and restored by Cck1R activation.

(A) Phosphorylation of the ribosomal protein S6 (pS6) and eukaryotic translation initiation factor 4E binding protein 1 (p-4e-bp1) are significantly reduced in the cerebellum of ATXN1[82Q] mice (11 weeks of age) compared to WT/FVB control mice. Administration of the Cck1R-selective agonist A71623 to ATXN1[82Q] mice for 24 h improves cerebellar phosphorylation of S6 and 4e-bp1 and levels of Calb1. Each lane represents extract from an individual mouse. Actb is used as a loading control, and the blots are from replicate experiments.
(B) Quantification of pS6, p4e-bp1, and Calb1 expression levels in cerebella of ATXN1[82Q] mice.
(C) Absence of the Cck1R in 8-week-old ATXN1 [82Q] mice prevents A71623 restoration of cerebellar S6 phosphorylation.
Data are mean ± SD. ns, p > 0.05; *p < 0.05; **p < 0.01; and ***p < 0.001; one-way ANOVA followed by Bonferroni’s multiple comparisons test.
Figure 4 shows that the Cck1R agonist A71623 also normalized cerebellar mTORC1 signaling in ATXN2[127Q] mice. In contrast to ATXN1[82Q], where mTORC1 signaling is decreased, ATXN2[127Q] mice show hyperactivation of mTOR signaling resulting in autophagy aberration (Paul et al., 2021). However, both mouse models share similar cerebellar marker gene dysregulations, including Calb1. To investigate if Cck1R agonist (A71623) has an effect on mTORC1 signaling, we performed intraperitoneal (IP) bolus injection of A71623 (0.0264, 0.264, and 1.0 mg/kg) in ATXN2[127Q] mice at 13 weeks of age for 24 h. On western blot analyses, ATXN2[127Q] mice showed increased phospho-S6 and phospho-4e-bp 1 levels as a function of mTORC1 activation (Figures 4A and 4B). CcK1R agonist treatment successfully reduced pS6 and p-4e-bp 1 levels (Figures 4A and 4B). Complementary to reduced pS6 levels, we also observed significant restoration of a cerebellar gene, Calb1, in ATXN2[127Q] mice by Cck1R agonist treatment (Figures 4A and 4B).
Figure 4. Cck1R activation normalizes mTORC1 signaling activity and restores cerebellar marker Calb1 in ATXN2[Q127] mice.

(A) Phosphorylation of the ribosomal protein S6 (pS6) and eukaryotic translation initiation factor 4E binding protein 1 (p4e-bp1) is increased and the cerebellar marker Calb1 is decreased in the cerebellum of ATXN2[127] mice (13 weeks of age) compared to WT mice. Administration of the Cck1R-selective agonist A71623 to ATXN2[127Q] mice for 24 h improves cerebellar phosphorylation of S6 and 4e-bp1 and increases levels of Calb1. Each lane represents extract from an individual mouse. Actb is used as a loading control, and blots are from replicate experiments.
(B) Quantification of pS6, p4e-bp1, and Calb1 is shown.
Data are mean ± SD. ns, p > 0.05; *p < 0.05; **p < 0.01; and ***p < 0.001; one-way ANOVA followed by Bonferroni’s multiple comparisons test.
DISCUSSION
In many SCAs, cerebellar Purkinje neurons manifest pathology and dysfunction (Robinson et al., 2020). Previous RNA-seq analyses of mouse models of SCA1 revealed an enhanced expression of Cck in cerebellar RNA from ATXN1[30Q]D776 mice that manifest severe ataxia in absence of progressive Purkinje cell pathology (Ingram et al., 2016). Development of ataxia with progressive Purkinje cell pathology upon crossing ATXN1[30Q] D776 mice with Cck−/− and Cck1R−/− mice suggested that the elevated Cck in ATXN1[30Q]D776 mice and its activation of Cck1R brought about the lack of Purkinje cell pathology in these mice. Here, we show that a Cck1R-selective agonist, A71623, was able to mitigate the ataxia and progressive Purkinje cell pathology in ATXN1[30Q]D776/Cck−/−, ATXN1[82Q], and ATXN2 [127Q] mice, providing direct evidence that activation of Cck1Rs is neuroprotective in SCA cerebellar disease.
CCK was first identified as a gastrointestinal (GI) tract hormone regulating GI motility, pancreatic enzyme secretion, gastric emptying, and gastric acid secretion. Subsequently CCK was found to be a widely expressed and abundant neuropeptide in the CNS where it is reported to regulate output of neuronal circuits, notably satiation (Lee and Soltesz, 2011). Colocalization and interaction of CCK with other key neurotransmitters in CNS areas suggests that CCK has an extensive role regulating neuronal activity. CCK interacts with two G-protein-coupled receptors, CCK1R and CCK2R, to mediate its biological actions (Dufresne et al., 2006). In the cerebellum, a prominent site of Cck and Cck1R expression are Purkinje neurons (Allen Brain Atlas; https://portal.brain-map.org/).
Typically, the mTORC1 signaling pathway is viewed as being inhibited by various stressors (reviewed in Su and Dai, 2017). Based on the results presented in this study, along with our previous work (Ingram et al., 2016), in the case of ATXN1 [82Q], we propose a speculative model whereby a CCK/CCK1R/mTORc1 pathway has a role in Purkinje cells adjusting to stress and that this pathway is negatively impacted by ATXN1 with an expanded polyQ tract. In ATXN1[82Q] Purkinje cells, cumulative stress induced by mutant ATXN1 promotes the cleavage of CCK to the octapeptide CCK-8, the natural ligand with the highest affinity for the Cck1R (Dufresne et al., 2006) that upon secretion binds to and activates CCK1R on Purkinje cells. Expression of ATXN1[82Q] enhances Purkinje cell stress. The fact that ATXN1 with an expanded polyQ also reduces CCK expression dampens the ability of Purkinje cells to respond to stress, thus promoting ATXN1 pathogenesis. Activation of Purkinje cell CCK1R stimulates mTORC1, which we speculate is a critical component by which CCK1R activation dampens the pathogenic effects of ATXN1 with an expanded PolyQ. Inactivation of mTORC1 induces a progressive loss of Purkinje cells by apoptosis (Angliker et al., 2015). mTORC1 signaling, in addition to responding to many stresses, is impaired in ATXN1154Q knockin mice, and absence of mTORC1 in Purkinje cells of ATXN1154Q mice worsens disease (Ruegsegger et al., 2016).
In contrast to ATXN1[82Q]-expressing Purkinje cells, where mTORC1 signaling is decreased (Figure 3), ATXN2[127Q] Purkinje cells have enhanced mTORC1 signaling (Figure 4). Previously, Angliker et al. (2015) noted a striking similarity in the effect between Purkinje cells with decreased mTORC1 signaling compared to those in which mTORC1 signaling was enhanced. In mice where mTORC1 signaling was decreased due to a loss of mTORC1 or in Purkinje cells in which mTORC1 signaling was enhanced due to the absence of the mTORC1 inhibitor TSC1, the function and survival of Purkinje cells were adversely affected. The data presented here on mTORC1-signaling levels in cerebella of ATNX1[82Q] and ATXN2[127Q] transgenic mice indicate that SCAs might also be categorized according to mTORC1-signaling activity, with SCA1 having reduced Purkinje cell mTORC1 signaling and SCA2 being a SCA with enhanced Purkinje cell mTORC1 signaling. Intriguingly, activation of the Cck1R in both instances restored mTORC1 signaling to a normal level. Understanding the mechanism and cellular pathways by which Cck1R activation in ATNX1[82Q] and ATXN2[127Q] transgenic Purkinje cells restores mTORc1 signaling to a normal level is important when considering the potential of Cck1R activation as a therapeutic target in SCAs. Moreover, aberrant mTROC1 signaling is seen in the striatum of a Huntington’s disease (HD) mouse model (Lee et al., 2015), while restoring proper mTORC1 signaling reduced disease severity in this HD model. It is intriguing to speculate that the ability of the CCK/CCK1R pathway to restore mTORC1 signaling has therapeutic potential for several neurodegenerative diseases.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Harry T. Orr (orrxx002@umn.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All sequencing datasets generated as part of this study are publicly available in NCBI GEO under accession # GSE180969.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The Institutional Animal Care and Use Committee approved all animal use protocols. ATXN1[82Q] (Burright et al., 1995), ATXN1 [30Q]-D776, ATXN1[82Q]-D776 (Duvick et al., 2010), ATXN2[127Q] (Hansen et al., 2013), and WT/FVB/NJ mice were housed and managed by Research Animal Resources under SPF conditions in an AAALAC-approved facility. In all experiments involving the use of mice, equal numbers of male and females were used. Ages of mice used in this study were from 5 to 36 weeks in Figures 1 and 3 to 12 weeks in Figure 2. In Figure 3 the mice were 11 weeks old and in Figure 4 they were 13 weeks of age.
METHOD DETAILS
RNA isolation and sequencing
Total RNA was isolated from dissected cerebella using TRIzol Reagent (Life Technologies, Carlsbad, California) following the manufacturer’s protocols. Cerebella were homogenized using an RNase-Free Disposable Pellet Pestles in a motorized chuck. For RNA-sequencing, RNA was further purified to remove any organic carryover using the RNeasy Mini Kit (QIAGEN, Venlo, Netherlands) following the manufacturer’s RNA Cleanup protocol.
Cerebellar RNA from three biological replicates for each genotype was isolated. Purified RNA was sent to the University of Minnesota Genomics Center for quality control, including quantification using fluorimetry (RiboGreen assay, Life Technologies) and RNA integrity assessed with capillary electrophoresis (Agilent BioAnalyzer 2100, Agilent Technologies, Inc.) generating an RNA integrity number (RIN). All submitted samples had greater than 1ug total mass and RINs 7.9 or greater. Library creation was completed using oligo-dT purification of polyadenylated RNA, which was reverse transcribed to create cDNA. cDNA was fragmented, blunt-ended, and ligated to barcoded adaptors. Library was size selected to 320bp ± 5% to produce average inserts of approximately 200bp, and size distribution validated using capillary electrophoresis and quantified using fluorimetry (PicoGreen, Life Technologies) and Q-PCR. Libraries were then normalized, pooled and sequenced. 12 and 28 week ATXN1[82Q], ATXN1[30Q]-D776, and WT/FVB samples were sequenced on an Illumina GAIIX using a 76nt paired-end read strategy, while 5 week samples from these genotypes were sequenced on an Illumina HiSeq 2000 using a 100nt paired-end read strategy. Data were analyzed on University of Minnesota Supercomputing Institute’s (MSI) high-performance computing cluster.
Agonist treatments
Osmotic pumps
The Cck1 receptor (Cck1R) agonist A71623 (Tocris Biosciences) was suspended in 20mM PBS according to the manufacturer’s directions. For the experiment with ATXN1[30Q]D776;Cck−/− mice, osmotic minipumps (Alzet, 1004) containing either A71623 (0.02mg/kg/day) or Vehicle (20mM PBS) were implanted intraperitoneally (IP) in 6 week old mice. Briefly, 5 week old mice were deeply anesthetized by intramuscular injection of a ketamine/xylazine cocktail (100mg/kg ketamine and 10mg/kg xylazine). Fur on IP implantation site (1cm below ribcage) was shaved. Incision sites were scrubbed with povidone-iodine (Betadine) scrub. A 1cm-long incision was made under the ribcage. The peritoneal wall was gently incised beneath the cutaneous incision and the pump cannula was placed into the peritoneal cavity. The musculoperitoneal layer was closed with 4.0 absorbable suture and the skin wound was closed using surgical staples (Alzet). Ten days after surgery, the staples were removed and wounds examined for healing. For the duration of the experiment, pumps were removed and replaced every 7 weeks. Behavioral data was collected at the time points indicated in Figure 4B. Because of the size of the osmotic minipumps, the mice have to be ~20 g or larger for safe implantation into the IP space. In ATXN1[82Q] mice, single bolus injections of A71623 (0.02mg/kg) or Vehicle were administered daily beginning at week 5 and continuing until the mice were ~20 g (for approximately 7 days, or until the mice were 6 weeks old). At this time the pumps were implanted for the remainder of the experimental timeline.
IP injections
ATXN2[127Q] mice (13 weeks of age) were given intraperitoneally (IP) bolus injection of Cck1R agonist (A71623: 0.0264, 0.264 and 1.0 mg/kg) or vehicle (20 mM PBS) for 24 hr. Following A71623 treatment, cerebella were harvested and stored at −80°C until used.
Western blots
Mouse cerebellar extracts were prepared by homogenization of tissues in extraction buffer [25 mM Tris-HCl pH 7.6, 250 mM NaCl, 0.5% Nonidet P-40, 2 mM EDTA, 2 mM MgCl2, 0.5 M urea and protease inhibitors (Sigma-Aldrich, P-8340)] followed by centrifugation at 4°C for 20 min at 14,000 RPM (Paul et al., 2021). Only supernatants were used for western blotting. Protein extracts were resolved by SDS-PAGE and transferred to Hybond P membranes (Amersham Bioscience, USA), and processed for western blotting. Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore, Cat# WBKLSO500) was used to visualize the signals, which were detected on the ChemiDoc MP imager (Bio-Rad) and the band intensities were quantified by ImageJ software analyses after inversion of the images. Relative protein abundances were expressed as ratios to Actb or Gapdh. Antibodies used for western blotting and their dilutions were as follows: Phospho-S6 Ribosomal Protein (Ser235/236) (D57.2.2E) XP® Rabbit mAb [(1:4000), Cell Signaling, Cat#4858], S6 Ribosomal Protein (5G10) Rabbit mAb [(1:4000), Cell Signaling, Cat# 2217], Phospho-4E-BP1 (Thr37/46) (236B4) Rabbit mAb [(1:3000), Cell Signaling, Cat#2855], 4E-BP1 Antibody [(1:4000), Cell Signaling, Cat#9452], monoclonal anti-Calbindin-D-28K antibody [(1:5,000), Sigma-Aldrich, C9848], monoclonal anti-β-Actin–peroxidase (clone AC-15) [(1:30,000), Sigma-Aldrich, A3854], and GAPDH (14C10) Rabbit mAb [(1:5000), Cell Signaling, Cat#2118]. Secondary antibodies: Peroxidase-conjugated AffiniPure goat anti-rabbit IgG (H + L) [(1:5000), Jackson ImmunoResearch Laboratories, Cat# 111–035-144] and Horse anti-mouse IgG Antibody (Peroxidase) [(1:5000), Vector laboratories: Cat# PI-2000].
Behavioral analyses
Rotarod
Mice were tested on the rotarod apparatus (Ugo Basile, Comerio, Italy) using an accelerating protocol; 5 to 50 rpm, 5 min ramp duration, 5 min max trial length. The test consisted of a total of 4 trials per day for 4 consecutive days. Mice were habituated to the testing room 15 minutes prior to the start of testing on each day. Animals were segregated by sex during testing and run in consistent groups (up to 5 at a time). To ensure enough recovery time between trials, animals were given 10–15 minutes between the end of a trial and the following trial, which included the time to test the other groups in the trial. Trials ended whenever an animal failed to stay on the rotarod or if they made 2 consecutive rotations clinging to the rod and not ambulating. Animals were returned to their home cages after trial completion. The apparatus was cleaned between each animal group within a trial with a 70% ethanol.
Beam walk
The beam walk protocol consisted of 3 consecutive training days followed by one test day. The beam walk apparatus was built in-house by the Mouse Behavior Core using wood and plastic components, consisting of beams (of varying sizes and shapes) that extend to a goal box. The apparatus is elevated ~50 cm off a table surface. Testing was performed in a dark room with only a single light directed at the start position of the beam to encourage crossing to the goal box. Males and females were segregated during testing. Animals were run at approximately the same time every day and habituated to the behavior room for about 15 minutes prior to the start of training. At the start of the first trial of each day, an animal was placed into the goal box for ~15–30 s to become familiar before beginning the first trial. After familiarization with the goal box, the mouse was brought to the start end of the beam and placed with the nose right behind the ‘start’ line. Beam walk training (using the 15 mm square beam) consisted of 4 trials per day, with a maximum of 60 s allowed for an animal to cross. Animals were tested in smaller groups of 4–7 to allow for ~5 min intertrial interval. Hindpaw foot slips and time (sec) to cross the beam were carefully recorded by an investigator blind to treatment and genotype. A foot slip was defined as a hind leg and paw coming completely off the beam in a fast downward sweeping motion. Animals that successfully crossed the beam were left in the goal box for ~15 s while the beam was wiped down with a 70% ethanol solution. The animal was then returned to their home cage until the next trial. The goal platform was cleaned with the ethanol solution after each trial and between animals on the first trial of each day.
QUANTIFICATION AND STATISTICAL ANALYSIS
Gene expression analyses
Gene expression analyses were performed with the Tuxedo pipeline (Kim et al., 2013; Trapnell et al., 2010). Initial read quality was assessed using FastQC v.0.10.1 (Andrews - Babraham Bioinformatics, FastQC A quality control tool for high throughput sequence data) and reads were trimmed to remove low quality 30 ends and adaptor contamination using Trimmomatic (Bolger et al., 2014).
Reads were aligned to the mouse reference genome (mm10) with Tophat2 v2.0.11 by using mostly default parameters with two exceptions: mate inner distance and standard deviation were adjusted to the data and a gene annotation model only looking for supplied junctions (mm10 gtf file from iGenomes). Gene abundances were estimated with Cufflinks v.2.2.1, and differential gene expression was determined with Cuffdiff using default parameters (Trapnell et al., 2012). Genes with a q ≤ 0.05 were considered significant.
WGCNA
FPKM abundance estimates for all 45 samples were produced by CuffNorm (Cufflinks v.2.2.1, Trapnell et al., 2012) using the mm10 iGenomes gtf excluding miRNAs and snoRNAs, which were too short to be captured by the RNA library preparation. Genes were retained for analysis if they had at least 10FPKM for at least 3 samples, and data were log2 transformed (log2(FPKM+1)) and quantile normalized for WGCNA analysis (Langfelder and Horvath 2012). Hierarchical clustering revealed one outlier sample (one wt/FVB week 28 sample), which was removed from the dataset prior to network construction. The WGCNA R package (WGCNA v. 1.46, R v.3.1.1) was used to construct an unsigned gene coexpression network with a soft threshold power [beta] of 10. Twenty-two modules were detected, including ten that were significantly associated with ML thickness. Of the 10, one 374 gene module, the pink module, had an extremely high correlation (0.91, p = 9e-18), compared to a correlation of 0.55 for the next-most-highly correlated module. To determine whether this module was stable even without the presence of the ATXN1[82Q] samples, the WGCNA was repeated for the dataset with the AGXN1[82Q] samples removed. Two outliers (The same wt/FVB week 28 sample, and one D30 week 12 sample) were removed from the dataset for this analysis. Using a soft threshold power of 10, an unsigned gene coexpression network of 36 modules was constructed. The 36 modules create by this WGCNA were then compared via pairwise Fisher’s exact test to the modules created by the WGCNA with the ATXN1[82Q] samples included, to identify significantly overlapping modules between the two analyses.
STATISTICAL ANALYSIS
Statistical tests used for behavioral data were Two-Way ANOVA with Tukey post hoc test and for western blot data the One-way ANOVA followed by Bonferroni’s multiple comparisons test. The details of these analyses are provided in Figure legends. To assess overlap of genes between WGCNA modules, we followed the procedure of Oldham et al. (2008) to conduct a hypergeometric test for significant overlap. These statistical details are presented in the Gene Expression method sections. The exact n values ranged from 4 to 13 with n representing number of animals tested or number of animal samples evaluated. Exact n’s are indicated in each Figure legend. For analyses means and SEMs are depicted except for main Figures 3 and 4 where SDs are shown. *p < 0.05, **p < 0.01, ***p < 0.001 for all analyses and are depicted in the legend of each Figure.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit polyclonal anti-ATXN1 | In house (Servadio et al., 1995) | 11750; RRID: AB_2721278 https://pubmed.ncbi.nlm.nih.gov/7647801/ |
| Rabbit polyclonal anti-ATXN1 | In house (Servadio et al., 1995) | 11NQ; RRID: AB_2721279 https://pubmed.ncbi.nlm.nih.gov/7647801/ |
| Rabbit polyclonal anti-P-ERK1/2 (p44/42 MAPK) T202/Y204 | Cell Signaling Technology | 9101; RRID: AB_2721280 |
| Rabbit polyclonal anti-ERK1/2 (p44/42 MAPK) | Cell Signaling Technology | 9102; RRID: AB_2721281 |
| Rabbit monoclonal anti-P-S6 S235/236 (D57.2.2E) | Cell Signaling Technology | 4858; RRID: AB_916156 |
| Rabbit polyclonal anti-S6 ribosomal protein | Cell Signaling Technology | 2217; RRID: AB_331355 |
| Rabbit monoclonal anti-Phospho-4E-BP1 (Thr37/46) (236B4) | Cell Signaling Technology | 2855; RRID:AB_560835 |
| Rabbit Polyclonal anti-4E-BP1 Antibody | Cell Signaling Technology | 9452; RRID:AB_331692 |
| Rabbit monoclonal anti-GAPDH (14C10) | Cell Signaling Technology | 2118; RRID:AB_561053 |
| monoclonal anti-β-Actin–peroxidase (clone AC-15) | Sigma | A3854; RRID:AB_262011 |
| Mouse monoclonal anti-Calbindin (D-28K) | Sigma | C9848; RRID: AB_2314067 |
| Mouse monoclonal anti-α-Tubulin | Sigma | T5168; RRID: AB_477579 |
| Alexa Fluor 488-AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson Immunoresearch | 711-545-152; RRID: AB_2313584 |
| Cy3-AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immunoresearch | 715-165-150; RRID: AB_2340813 |
| Peroxidase-conjugated AffiniPure goat anti-rabbit IgG (H + L) | Jackson Immunoresearch | 111-035-144; RRID:AB_2307391 |
| HRP Horse anti-mouse IgG Antibody | Vector Laboratories | PI-2000; RRID:AB_2336177 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| TRIzol reagent | ThermoFisher Scientific | 15596-026 |
| Phosphatase Inhibitor Cocktail 2 | Sigma | P5726 |
| Phosphatase Inhibitor Cocktail 3 | Sigma | P0044 |
| cOmplete Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 11836170001 |
| Protease Inhibitor Cocktail | Sigma | P8340 |
|
| ||
| Critical commercial assays | ||
|
| ||
| iScript Reverse Transcription Supermix for RT-qPCR | Bio-Rad | 1708840 |
| Light Cycler 480 Probes Master kit | Roche | 04-707-494-001 |
| Quant-iT RiboGreen assay | Life Technologies | R11490 |
| RNeasy Mini Kit | QIAGEN | 74104 |
| PicoGreen | ThermoFisher Scientific | P11496 |
| Agilent BioAnalyzer 2100 | Agilent Technologies, Inc. | SCR_018043 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq data from wt/FVB, ATXN1[30Q]D776, ATXN1[82Q] | Ingram et al., 2016 | GSE75778 |
| RNA-seq data from Cck−/−, ATXN1[30Q]D776/Cck−/− | This paper | GSE180969 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6 | The Jackson Laboratory | Stock No: 005304; RRID: IMSR_JAX:005304 |
| Mouse: FVB | The Jackson Laboratory | Stock No: 001800; RRID:IMSR_JAX:001800 |
| Mouse: Cck1R−/− | The Jackson Laboratory | Stock No: 006367; RRID:IMSR_JAX:006367 |
| Mouse: Cck−/− | The Jackson Laboratory | Stock No: 017710; RRID:IMSR_JAX:017710 |
| Mouse: PCP2-ATXN1[82Q] | In house (Rousseaux et al., 2018) | N/A; MGI:6113813 https://pubmed.ncbi.nlm.nih.gov/29526553/ |
| Mouse: PCP2-ATXN1[82Q];Cck1R−/− | This paper | N/A |
| Mouse: PCP2-ATXN1[30Q]D776 | In house (Duvick et al., 2010) | https://pubmed.ncbi.nlm.nih.gov/20869591/ |
| Mouse: PCP2-ATXN1[30Q]D776;Cck−/− | This paper | |
| Mouse: PCP2-ATXN2[127Q] | In house (Hansen et al., 2013) | https://pubmed.ncbi.nlm.nih.gov/23087021/ |
|
| ||
| Software and algorithms | ||
|
| ||
| FastQC v.0.10.1 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trimmomatic v.0.33 | Bolger et al., 2014 | https://pubmed.ncbi.nlm.nih.gov/24695404/ |
| Tophat2 v.2. | Kim et al., 2013 | https://pubmed.ncbi.nlm.nih.gov/23618408/ |
| Cufflinks 2.2.1 | Trapnell et al., 2010; Roberts et al., 2011; Trapnell et al., 2012 | https://pubmed.ncbi.nlm.nih.gov/20436464/ https://pubmed.ncbi.nlm.nih.gov/21410973/ https://pubmed.ncbi.nlm.nih.gov/22383036/ |
| R v.3.1.1 | R foundation for statistical computing | http://www.r/project.og/ |
| WGCNA v1.4.6 | Langfelder and Horvath, 2008, 2012 | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2631488/ https://pubmed.ncbi.nlm.nih.gov/23050260/ |
|
| ||
| Other | ||
|
| ||
| Micro-osmotic pump Model 1004 | ALZET | N/A; Cat. # 0009922 |
Highlights.
In murine Purkinje neurons expressing ATXN1[82Q], mTORC1 signaling is depressed
In murine Purkinje neurons expressing ATXN2[127Q], mTORC1 signaling is enhanced
CCK1R activation restores Purkinje cell function/morphology and mTORC1 signaling
ACKNOWLEDGMENTS
This study was supported by NIH/NINDS grant RO1NS 045667 (H.T.O.) and NIH/NINDS grants R37NS033123, R21NSNS103009, and UO1NS103883 (S.M.P.). The authors thank the Biomedical Genomics Center, Mouse Phenotyping Core at the University of Minnesota and Orion Rainwater for propagating and maintaining the mouse colony.
Footnotes
DECLARATION OF INTERESTS
E.A.L.W. and H.T.O. declare patent US 10,973,812 B2 (issued April 13, 2021) as relevant to the work in this paper.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109831.
REFERENCES
- Angliker N, Burri M, Zaichuk M, Fritschy J-M, and Rüegg MA (2015). mTORC1 and mTORC2 have largely distinct functions in Purkinje cells. Eur. J. Neurosci. 42, 2595–2612. [DOI] [PubMed] [Google Scholar]
- Asin KE, Bednarz L, Nikkel AL, Gore PA Jr., Montana WE, Cullen MJ, Shiosaki K, Craig R, and Nadzan AM (1992a). Behavioral effects of A71623, a highly selective CCK-A agonist tetrapeptide. Am. J. Physiol. 263, R125–R135. [DOI] [PubMed] [Google Scholar]
- Asin KE, Bednarz L, Nikkel AL, Gore PA Jr., and Nadzan AM (1992b). A-71623, a selective CCK-A receptor agonist, suppresses food intake in the mouse, dog, and monkey. Pharmacol. Biochem. Behav. 42, 699–704. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burright EN, Clark HB, Servadio A, Matilla T, Feddersen RM, Yunis WS, Duvick LA, Zoghbi HY, and Orr HT (1995). SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 82, 937–948. [DOI] [PubMed] [Google Scholar]
- Cawston EE, and Miller LJ (2010). Therapeutic potential for novel drugs targeting the type 1 cholecystokinin receptor. Br. J. Pharmacol. 159, 1009–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, Matilla A, Zoghbi HY, and Orr HT (1997). Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J. Neurosci. 17, 7385–7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufresne M, Seva C, and Fourmy D (2006). Cholecystokinin and gastrin receptors. Physiol. Rev. 86, 805–847. [DOI] [PubMed] [Google Scholar]
- Durr A (2010). Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 9, 885–894. [DOI] [PubMed] [Google Scholar]
- Duvick L, Barnes J, Ebner B, Agrawal S, Andresen M, Lim J, Giesler GJ, Zoghbi HY, and Orr HT (2010). SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron 67, 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich J, Kordasiewicz HB, O’Callaghan B, Handler H, Wagener C, Duvick L, Swayze EE, Rainwater O, Hofstra B, Benneyworth M, et al. (2018). Antisense oligonucleotide-mediated Ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI Insight 3, 123193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen ST, Meera P, Otis TS, and Pulst SM (2013). Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum. Mol. Genet. 22, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram M, Wozniak EAL, Duvick L, Yang R, Bergmann P, Carson R, O’Callaghan B, Zoghbi HY, Henzler C, and Orr HT (2016). Cerebellar transcriptome profiles of ATXN1 transgenic mice reveal SCA1 disease progression and protection pathways. Neuron 89, 1194–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klockgether T (2011). Update on degenerative ataxias. Curr. Opin. Neurol. 24, 339–345. [DOI] [PubMed] [Google Scholar]
- Koeppen AH (2005). The pathogenesis of spinocerebellar ataxia. Cerebellum 4, 62–73. [DOI] [PubMed] [Google Scholar]
- Kopin AS, Mathes WF, McBride EW, Nguyen M, Al-Haider W, Schmitz F, Bonner-Weir S, Kanarek R, and Beinborn M (1999). The cholecystokinin-A receptor mediates inhibition of food intake yet is not essential for the maintenance of body weight. J. Clin. Invest. 103, 383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2012). Fast R functions for robust correlations and hierarchical clustering. J. Stat. Softw. 46, i11. [PMC free article] [PubMed] [Google Scholar]
- Lee SY, and Soltesz I (2011). Cholecystokinin: a multi-functional molecular switch of neuronal circuits. Dev. Neurobiol. 71, 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Tecedor L, Chen YH, Monteys AM, Sowada MJ, Thompson LM, and Davidson BL (2015). Reinstating aberrant mTORC1 activity in Huntington’s disease mice improves disease phenotypes. Neuron 85, 303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LJ, and Desai AJ (2016). Metabolic actions of the type 1 cholecystokinin receptor: Its potential as a therapeutic target. Trends Endocrinol. Metab. 27, 609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, and Geschwind DH (2008). Functional organization of the transcriptome in human brain. Nat. Neurosci. 11, 1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Dansithong W, Figueroa KP, Gandelman M, Scoles DR, and Pulst SM (2021). Staufen1 in Human Neurodegeneration. Ann. Neurol. 89, 1114–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson HL, Shakkottai VG, Clark HB, and Orr HT (2017). Polyglutamine spinocerebellar ataxias - from genes to potential treatments. Nat. Rev. Neurosci. 18, 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflieger LT, Dansithong W, Paul S, Scoles DR, Figueroa KP, Meera P, Otis TS, Facelli JC, and Pulst SM (2017). Gene co-expression network analysis for identifying modules and functionally enriched pathways in SCA2. Hum. Mol. Genet. 26, 3069–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, Trapnell C, Donaghey J, Rinn JL, and Pachter L (2011). Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12, R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KJ, Watchon M, and Laird AS (2020). Aberrant cerebellar circuitry in the spinocerebellar ataxias. Front. Neurosci. 14, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseaux MWC, Tschumperlin T, Lu H-C, Lackey EP, Bondar VV, Wan Y-W, Tan Q, Adamski CJ, Friedrich J, Twaroski K, et al. (2018). ATXN1-CIC complex is the primary driver of cerebellar pathology in Spinocerebellar ataxia type 1 through a gain-of-function mechanism. Neuron 97, 1235–1243.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegsegger C, Stucki DM, Steiner S, Angliker N, Radecke J, Keller E, Zuber B, Rüegg MA, and Saxena S (2016). Impaired mTORC1-dependent expression of Homer 3 influences SCA1 pathophysiology. Neuron 89, 129–146. [DOI] [PubMed] [Google Scholar]
- Seidel K, Siswanto S, Brunt ERP, den Dunnen W, Korf HW, and Rüb U (2012). Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 124, 1–21. [DOI] [PubMed] [Google Scholar]
- Servadio A, Koshy B, Armstrong D, Antalffy B, Orr HT, and Zoghbi HY (1995). Expression analysis of the ataxin-1 protein in tissues from normal and spinocerebellar ataxia type 1 individuals. Nat. Genet. 10, 94–98. [DOI] [PubMed] [Google Scholar]
- Su K-H, and Dai C (2017). mTORC1 senses stresses: coupling stress to proteostasis. Bioassays 39, 1600268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watase K, Weeber EJ, Xu B, Antalffy B, Yuva-Paylor L, Hashimoto K, Kano M, Atkinson R, Sun Y, Armstrong DL, et al. (2002). A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 34, 905–919. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, and Hall MN (2006). TOR signaling in growth and metabolism. Cell 124, 471–484. [DOI] [PubMed] [Google Scholar]
- Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB, and Orr HT (2004). Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J. Neurosci. 24, 8853–8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing datasets generated as part of this study are publicly available in NCBI GEO under accession # GSE180969.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request