

Abstract
Lung cancer is the leading cause of cancer-related deaths worldwide. The paralogous transcriptional cofactors Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ, also called WWTR1), the main downstream effectors of the Hippo signal transduction pathway, are emerging as pivotal determinants of malignancy in lung cancer. Traditionally, studies have tended to consider YAP and TAZ as functionally redundant transcriptional cofactors with similar biological impact. However, there is growing evidence that each of them also possesses distinct attributes. Here we sought to systematically characterize the division of labor between YAP and TAZ in non–small cell lung cancer (NSCLC), the most common histological subtype of lung cancer. Representative NSCLC cell lines as well as patient-derived data showed that the two paralogs orchestrated nonoverlapping transcriptional programs in this cancer type. YAP preferentially regulated gene sets associated with cell division and cell-cycle progression, whereas TAZ preferentially regulated genes associated with extracellular matrix organization. Depletion of YAP resulted in growth arrest, whereas its overexpression promoted cell proliferation. Likewise, depletion of TAZ compromised cell migration, whereas its overexpression enhanced migration. The differential effects of YAP and TAZ on key cellular processes were also associated with differential response to anticancer therapies. Uncovering the different activities and downstream effects of YAP and TAZ may thus facilitate better stratification of patients with lung cancer for anticancer therapies.
Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide. Despite significant progress in understanding the causes of lung cancer, the 5-year survival is still below 15% (1). Therefore, there is an urgent need for better understanding of the molecular mechanisms that drive lung cancer to devise more effective treatments.
The paralogous transcriptional cofactors Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ, also called WWTR1), the main downstream effectors of the Hippo signaling pathway, are emerging as pivotal determinants of malignancy in lung cancer (2, 3). However, Hippo pathway genes, including YAP/TAZ, are rarely mutated in tumors, with only a few exceptions (4, 5), and thus YAP/TAZ dysregulation in cancer is likely to be driven by other mechanisms.
One open question in the YAP/TAZ field is to which extent these paralogs are functionally redundant. Although YAP and TAZ possess only nearly 57% amino acid sequence similarity (Supplementary Fig. S1), they are often regarded as functionally redundant, with similar biological impact (3, 6–9). Indeed, both participate in diverse biological processes, including tissue homeostasis, development, organ growth, regeneration, stem cell regulation, and mechanotransduction (10). Yet, a variety of features suggest that they may differ in their regulation and downstream activities (11–13). For example, YAP and TAZ display distinct knockout phenotypes during nephron development (14). Notably, evidence for their different functions in lung cancer is also emerging (15–17). For example, YAP, but not TAZ, inhibits squamous trans-differentiation of lung adenocarcinoma cells (15). Furthermore, TAZ protein levels are higher in lung cancer cell lines relative to normal lung epithelial cells, whereas YAP levels are more or less comparable (17). Moreover, lung cancer-derived cells depleted of TAZ, but not YAP, are more sensitive to bromodomain and extraterminal (BET) domain protein inhibitors (16). Nevertheless, the extent of non-redundancy between YAP and TAZ in lung cancer cells, as well as the functional impact of such non-redundancy, remains to be comprehensively determined.
Here we sought to explore the division of labor between YAP and TAZ in non–small cell lung cancer (NSCLC), the most common histologic subtype of lung cancer (1). Employing representative NSCLC cell lines, we show that the two paralogs orchestrate non-identical transcriptional programs, giving rise to distinct biological phenotypes. Specifically, YAP preferentially regulates cell division and cell-cycle progression, whereas TAZ preferentially regulates extracellular matrix (ECM), cell adhesion, and cell migration. These findings imply that YAP and TAZ have distinct, yet complementary, roles in lung cancer cells. Identification of differential activities and downstream effects of YAP and TAZ may enable better stratification of lung cancer tumors, and uncover cancer vulnerabilities that might be exploited therapeutically.
Materials and Methods
Cell lines and treatments
Cells were maintained at 37°C with 5% CO2. H1299 and H460 cells were cultured in RPMI1640 (Gibco) supplemented with 10% FBS (Biological Industries, BI) and 1% penicillin–streptomycin (BI). A549 cells were grown in DMEM (BI) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone) and 1% penicillin–streptomycin (BI). mCherry-labeled H1299 cells (18) were a kind gift of Uri Alon (Weizmann Institute of Science). H460 and A549 cells were obtained from ATCC; H460 cells were labeled with mCherry (19). Cell lines were authenticated by STR profiling and tested negative for Mycoplasma, and did not exceed 12 in vitro passages. Taxol [paclitaxel; Sigma (T7402)] was used at a final concentration of 1 μmol/L for 42 hours.
Western blot analysis
Western blot analysis was performed as described (20), using the following antibodies: GAPDH (Millipore, Catalog No. MAB374, RRID:AB_2107445), Flag (Sigma-Aldrich, Catalog No. F3165, RRID:AB_259529), Vinculin (Sigma-Aldrich, Catalog No. V9131, RRID:AB_477629), YAP/TAZ (Cell Signaling Technology, Catalog No. 8418, RRID:AB_10950494), and YAP (Santa Cruz Biotechnology, Catalog No. sc-376830, RRID:AB_2750899). Conjugated anti-mouse or anti-rabbit secondary antibodies were from Jackson ImmunoResearch. Imaging and quantification were performed using ChemiDoc MP Imager with Image Lab 4.1 software (Bio-Rad).
siRNA and plasmid transfections
For siRNA-mediated knockdown, SMARTpools or single oligonucleotides (Dharmacon; Supplementary Table S1) were used with the Dharmafect#1 transfection reagent (Dharmacon), at a final concentration of 30 nmol/L. For siPLK1 knockdown, we used 10 nmol/L PLK1 siRNA and 20 nmol/L siControl. The medium containing oligonucleotides and reagents was replaced after 6 hours. Plasmid transfection was done using jetPEI DNA transfection reagent (Polyplus Transfection). The final DNA amount was 10 μg per 10 cm dish, and the transfection medium was replaced after five hours. pcDNA3-Flag-YAP, with residues 90, 91, 94, 95, 96 replaced with Alanine (YAPmTEAD) and pcDNA3-Flag-TAZ were a generous gift of Yosef Shaul (Weizmann Institute of Science). TAZ-GFP and TAZ S51A (TAZmTEAD; ref. 21) were a generous gift of Kunxin Luo (University of California, Berkeley, CA).
RNA extraction, reverse transcription, and RT-qPCR
Total RNA was isolated using a NucleoSpin RNA Kit (Macherey Nagel). Reverse transcription and qPCR were performed as described (22). Values were normalized to either HPRT or GAPDH. Primers are listed in Supplementary Table S1.
RNA sequencing
Cells were transfected with siControl, siYAP, or siTAZ SMARTpool oligonucleotides for 48 hours and serum-starved for the last 18 hours. RNA was extracted and the polyA fraction was purified from 500 ng of total RNA, followed by fragmentation and generation of double-stranded cDNA, Agencourt Ampure XP beads cleanup (Beckman Coulter), end repair, A base addition, adapter ligation and PCR amplification. Libraries were quantified by Qubit (Thermo Fisher Scientific) and TapeStation (Agilent) and sequenced on HiSeq 2500 (Illumina; single read sequencing).
Transcriptomic analysis
RNA-sequencing (RNA-seq) analysis was done using the UTAP transcriptome analysis pipeline (23). Reads were trimmed to remove adapters and low-quality bases using cutadapt [-a “A(10)” -a “T(10)” –times 2 -q 20 -m 25; ref. 24] and mapped to genome GRCh38 (Gencode, UCSC) using STAR v2.4.2a (–alignEndsType EndToEnd, –outFilterMismatchNoverLmax 0.05, –twopassMode Basic; ref. 25). Reads were counted using STAR, and genes having minimum five reads in at least one sample were considered. Counts normalization and differential expression detection were analyzed with DESeq2 (betaPrior, cooksCutoff and independent filtering parameters set to False; ref. 26). Differentially expressed genes were selected with |log2Fold change|≥1 and adjusted P-value ≤0.05 (Benjamini and Hochberg). For A549 RNA-seq, batch correction was done using sva (3.26.0) R package. Gene expression heatmaps were generated with Partek Genomics Suite 7.0 (Partek Inc.), using log normalized values (rld), with row standardization. Volcano plots were generated with Matlab. Gene Ontology (GO) enrichment analyses were performed with Metascape (27) and Gene Set Enrichment Analysis (GSEA; ref. 28) preranked tool. The data are accessible through GEO Series accession number GSE151201.
Cell-cycle profiling
Cells were grown in 6 cm plates and transfected with the indicated siRNAs or plasmids for 48 hours. Following serum starvation (0% serum) for 18 hours, cells were analyzed with Phase-Flow BrdU Cell Proliferation Kit (BioLegend). Briefly, cells were incubated with BrdU for 75 minutes and labeled with Alexa Fluor-647 conjugated anti-BrdU antibody. Total DNA was stained with DAPI. Then, 50,000 cells were collected and analyzed by multispectral imaging flow cytometry. The percentage of cells in each cell-cycle phase was manually determined on the basis of BrdU intensity and total DNA content, using FlowJo (Becton, Dickinson and Company).
Scratch assays
Cells were transfected with the indicated siRNAs or plasmids for a total of 48 hours, reaching 90% to 100% confluence. A scratch was introduced with a 200 μL pipette tip; detached cells were washed off, and medium was replaced with serum-free medium. Gap closure was imaged at 0 and 24 hours with a Nikon eclipse Ti-E microscope at ×4 magnification, capturing at least four fields for each condition. The migration distance was assessed manually using ImageJ (NIH). Gap closure was calculated as follows: (0 hour gap width – 24 hours gap width)/0 hour gap width.
Transwell migration assays
Cells were seeded on a transwell insert (8 μm pore size; Costar) with medium containing 1% serum. Medium containing 10% serum was added to the bottom well. After 16 hours, inserts were fixed with ice cold methanol (100%) for 5 minutes, and migrated cells were stained with crystal violet for 30 minutes; nonmigrated cells were removed with cotton plugs. Fixed and stained cells were imaged with a Nikon eclipse Ti-E microscope at ×10 magnification, capturing at least four fields for each condition, and crystal violet stained areas were quantified with an ImageJ macro. Coverage by migrating cells was calculated as percentage of stained area relative to total area.
Cell counting
Cells were seeded in a 96-well plate, transfected with the indicated siRNAs or plasmids in medium containing 10% serum for either 24, 48, or 72 hours, and counted with a LUNA automated cell counter (Logos Biosystems).
Cell death assays
Cell death was assessed using the CellTox Green Cytotoxicity Assay (Promega). Cells were seeded in a 96-well plate and transfected with indicated siRNAs or plasmids for a total 48 hours. Following incubation with either 1 μmol/L Taxol or DMSO for 42 hours, including serum starvation for the last 18 hours, CellTox reagent was added and the GFP fluorescence of each well was measured using an Infinite M200 microplate reader (Tecan). Three technical replicates were done for each condition. Cell death was calculated by subtracting the background signal and normalizing to DMSO-treated cells (control).
Statistical analysis
Independent biological replicates were performed and group comparisons were done as detailed in the figure legends. P-values below 0.05 were considered significant. Statistical significance between two experimental groups is indicated by asterisks.
Synthetic lethality
We employed a recently published synthetic lethality (SL) pipeline, ISLE (29) in a four-step process, to compute SL scores for all paralogous gene pairs (n = 6353, derived from ref. 30), from expression profiles of 433 lung adenocarcinoma (LUAD) human tumors from The Cancer Genome Atlas (TCGA; ref. 31) and 87 LUAD cancer cell lines (32). To compute the SL score for a given paralogous pair, we first investigated the gene essentiality upon inactivation of its partner in LUAD cell lines. By definition, it is expected that gene A will be essential only when its SL partner gene B is inactive in a given cancer cell line (29). Using a set of genome-wide shRNA/sgRNA screens (as in ref. 29), we tested whether knockdown/knockout of one paralog is significantly more lethal when the other paralog is lowly expressed (bottom third quantile) versus highly expressed (top third quantile) across LUAD cell lines, via Wilcoxon rank-sum test (FDR corrected P < 0.1). Underrepresented SL pairs were next identified by quantifying the significance of simultaneous inactivation of both paralogs (under-expressed, via a hypergeometric test, FDR corrected P < 0.1) in LUAD TCGA cohort. Then, we investigated the clinical relevance score, by performing a Cox multivariate regression testing whether the patients with an active SL interaction (both paralogs are inactive) have better survival than the rest of the patients, while controlling for cancer types, age, gender, race, tumor purity, genomic instability, and the effect of individual gene activation (FDR corrected P < 0.1). In the last step, we calculated phylogenetic similarity measure for each paralogous pair using a nonnegative matrix factorization, which measures the Euclidean distance while taking into account their phylogenetic profile across 86 species. For a final SL score, we averaged the scores from all steps, and scaled the scores from 0 to 1 (1 indicates high SL interaction). Paralogous pairs were ranked according to their final SL scores.
YAP-differential and TAZ-differential drug sensitivity analysis
To identify YAP or TAZ distinct associations with drug response, we utilized drug sensitivity profiles (PRISM) of 4,518 drugs tested across 47 LUAD cancer cell lines (33). We identified drugs that showed differential response between cell lines with high expression of YAP (top 33%) compared with those with low YAP expression (bottom 33%), and likewise for TAZ. P-value of the comparison was determined by Wilcoxon rank-sum test. Then, by using Drugbank (34) annotation, the targets for each of the YAP-differential or TAZ-differential drugs were defined. YAP-differential and TAZ-differential drugs, their targets and P-values are listed in Supplementary Table S2.
Taxol sensitivity analysis
PRISM dataset
By utilizing single-dose Taxol cell viability data across cancer cell lines (n = 578; ref. 33), we tested whether lines with high expression of YAP (top 33%, n = 192) are differentially more sensitive to Taxol compared with those with low YAP (bottom 33%, n = 192), and likewise for TAZ. P-value of the comparison was determined by Wilcoxon rank-sum test.
GDSC dataset
Taxol sensitivity data was obtained from ref. 35; sensitivity was measured by area under the dose–response curve in 136 lung cancer cell lines. The Spearman correlation coefficient of Taxol sensitivity with either YAP or TAZ log2-transformed protein levels (determined by RPPA in Cancer Cell Line Encyclopedia; ref. 36) was calculated across these cell lines.
TCGA YAP and TAZ correlation analysis
We queried the LUAD expression dataset of TCGA, comprising 20,167 expressed genes and 515 tumor samples (after cleaning and filtering the data). LUAD TCGA mRNA-seq RSEM normalized data were downloaded from http://gdac.broadinstitute.org/. Expression data (X) was transformed to X_transformed = log2(X + 1). The Pearson correlation coefficient with either YAP or TAZ mRNA levels was calculated for each gene. Genes were sorted according to absolute R, and the top 8% (1,600 genes) were selected for further analysis. Nonoverlapping genes between the YAP-correlated and TAZ-correlated lists (1,276 genes) were subjected for GO annotation analysis.
Chromatin immunoprecipitation sequencing
Cells were serum starved for 18 hours. Cross-linking with 1% formaldehyde and chromatin immunoprecipitation (ChIP) were performed, as in ref. 37, using either YAP1 (Thermo Fisher Scientific, Catalog No. PA1–46189, RRID:AB_2219137) or TAZ antibody (Proteintech, Catalog No. 23306–1-AP, RRID:AB_2721185), and Pierce ChIP-grade Protein G magnetic beads (Thermo Fisher Scientific). Reverse crosslinking and DNA purification were with ChIP DNA Clean & Concentrator Kit (Zymo Research). 4 to 20 ng ChIP DNA was processed as described (38). Libraries were prepared and multiplexed at the Crown Institute for Genomics (G-INCPM, Weizmann Institute of Science), using NextSeq 75 cycles high output kit (Illumina; single read sequencing). Reads were preprocessed with cutadapt to remove adapters and low-quality bases (parameters: –times 2 -q 20 -m 25). Reads were mapped to human genome (hg19) using bowtie2 (local mode). Broad peaks were called using MACS2, relative to background input samples that had been cross-linked and sonicated, but not immunoprecipitated (parameters: –bw 300 -B -f –SPMR -keep-dup auto -q 0.01). Peaks were annotated using Homer software. Peaks with at least 30% overlap between replicates were considered. For differential peaks analysis, DiffBind package was used (http://bioconductor.org/packages/release/bioc/html/DiffBind.html). Significant peaks were defined as FDR ≤ 0.05 and |log2Fold change|≥1. Peaks were visualized on genomic features with IGV (39). The data are accessible through GEO Series accession number GSE151201.
Results
YAP and TAZ are associated with distinct transcriptional programs
YAP and TAZ act primarily as transcriptional cofactors (6, 9, 10). To compare YAP and TAZ impact on the transcriptome of lung cancer-derived cells, we performed RNA-seq analysis following siRNA-mediated transient knockdown of either YAP (siYAP) or TAZ (siTAZ) in NSCLC-derived H1299 cell line (knockdown validation in Fig. 1A and Supplementary Fig. S2). As expected, a subset of 82 common genes were impacted similarly by partial depletion of either YAP or TAZ (Fig. 1B; Supplementary Fig. S3). However, expression of a larger number of genes was distinctly affected in a paralog-specific manner. Thus, 204 genes were significantly downregulated or upregulated at least two-fold upon YAP depletion, but were only mildly affected by TAZ depletion (Fig. 1B and C). Conversely, expression of 324 other genes was significantly affected by TAZ depletion, but less so by YAP depletion (Fig. 1B and D). For simplicity, the sets of genes preferentially regulated by YAP or TAZ will hereafter be referred to as YAP- or TAZ-regulated genes, respectively.
Figure 1.
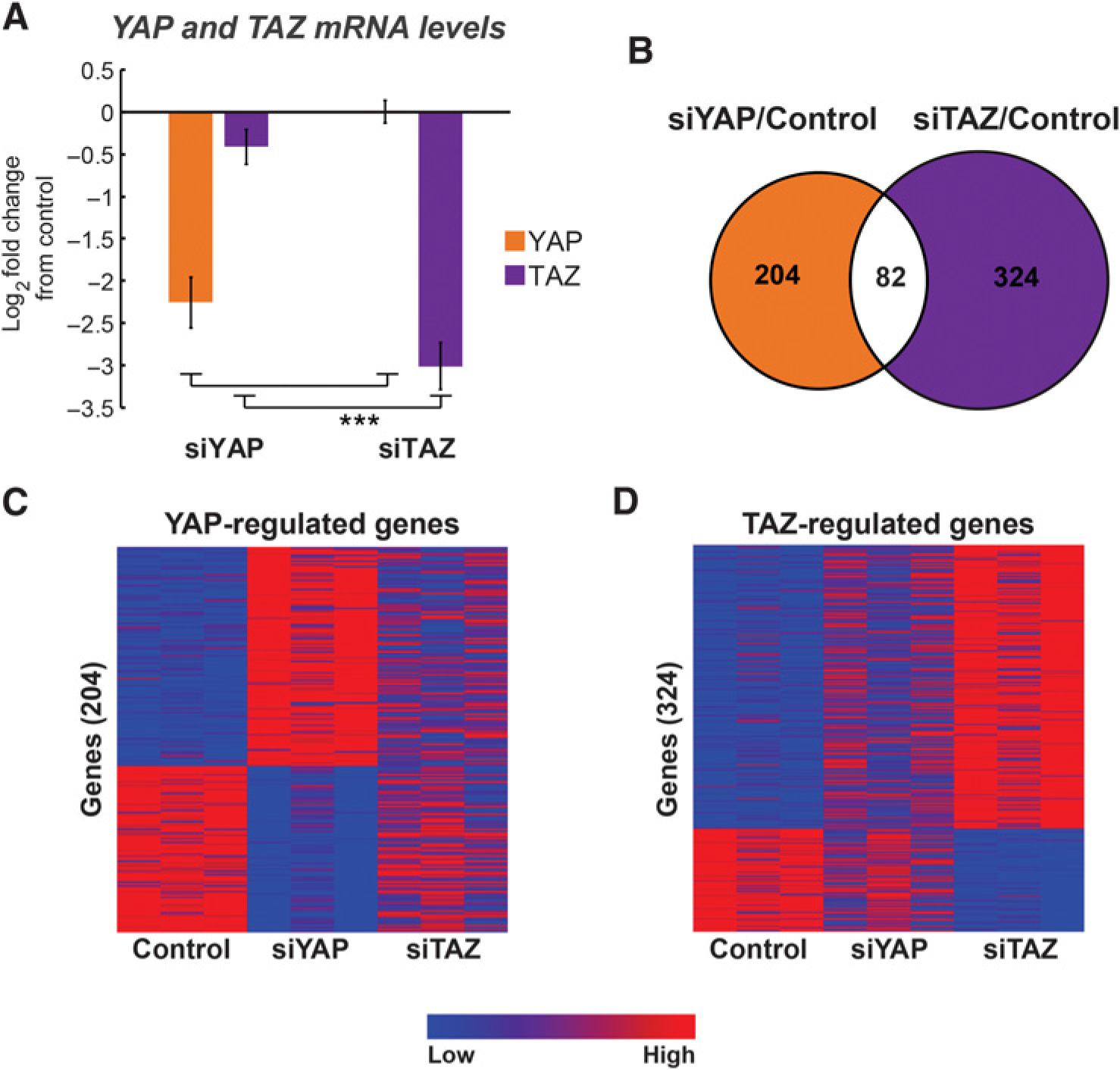
YAP and TAZ are associated with distinct transcriptional programs in H1299 cells. Human NSCLC-derived H1299 cells were transiently transfected with siRNA SMARTpools targeting either YAP (siYAP) or TAZ (siTAZ) or with control siRNA. RNA was extracted 48 hours later and subjected to RNA-seq analysis. A, RT-qPCR analysis of YAP and TAZ mRNA levels to validate knockdown efficiency. Data represent log2 mRNA expression (mean ± SEM) normalized to GAPDH and control transfected cells from three independent biological repeats. ***, P < 0.001 one-way ANOVA and Tukey’s post hoc test of the indicated comparisons. B, Venn diagram of the overlap between significantly differentially expressed genes upon transfection with either siYAP or siTAZ compared with control, from three biological repeats. Orange and purple indicate genes whose expression was selectively altered (either positively or negatively) by siYAP or siTAZ, respectively. Absolute fold change ≥2, adjusted P-value ≤ 0.05. C and D, Heatmaps of gene-expression levels of the nonoverlapping 204 siYAP significantly differentially expressed genes (C) and the 324 siTAZ significantly differentially expressed genes (D) depicted in B.
YAP preferentially regulates cell-cycle progression, whereas TAZ preferentially regulates cell migration
To identify biological processes regulated by either YAP or TAZ in H1299 cells, we performed GO analysis. Interestingly, genes preferentially regulated by YAP were strongly enriched for cell division- and mitosis-related-terms (Fig. 2A; Supplementary Fig. S4A; Supplementary Table S3), whereas the two top enriched terms for TAZ-regulated genes were related to ECM organization (Fig. 2B; Supplementary Fig. S4B; Supplementary Table S3). Likewise, GSEA on all informative genes, ranked by their expression fold change (relative to control) upon either siYAP or siTAZ, showed that the top downregulated genes upon YAP silencing were strongly enriched for cell-proliferation-related genes (Fig. 2C; ref. 40), whereas genes upregulated upon TAZ silencing were enriched for adhesion- and ECM-related genes (Fig. 2D; ref. 41). To assess the generality of our observations, we performed RNA-seq analysis also with NSCLC-derived A549 cells. Importantly, GSEA revealed again that YAP-regulated genes were enriched for cell-proliferation-related genes whereas TAZ-regulated genes were enriched for adhesion- and ECM-related terms (Fig. 2E and F).
Figure 2.
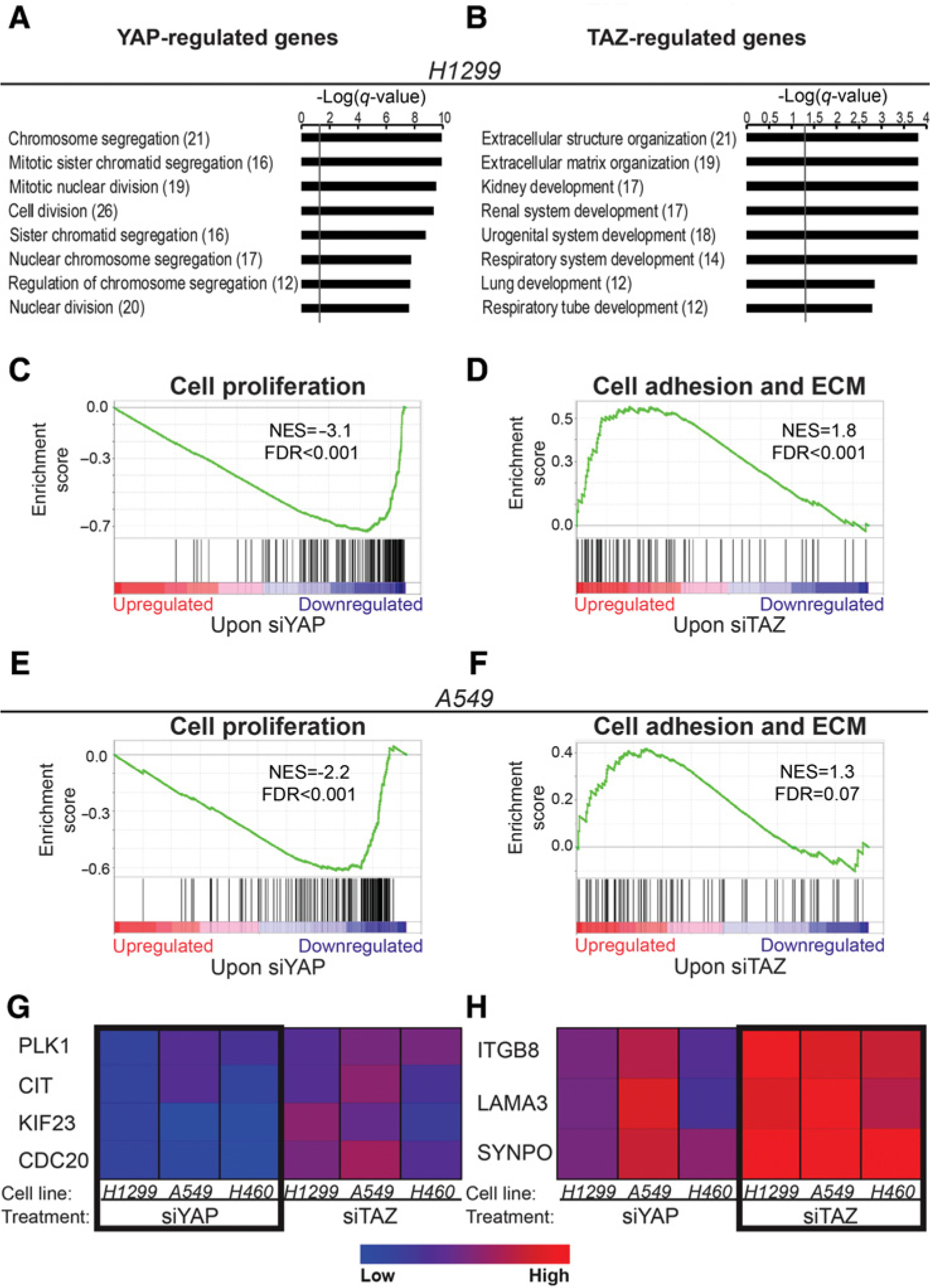
YAP-regulated genes are enriched for cell division terms, whereas TAZ-regulated genes are enriched for adhesion- and ECM-related-terms. A and B, Eight top enriched GO Biological Processes terms of the YAP-regulated (A) and TAZ-regulated (B) genes in H1299 cells, as determined by Metascape. Q-value is −log10 transformed; gray line, q-value of 0.05. In brackets is the number of enriched genes in each term. C–F, GSEA enrichment plots for H1299 RNA-seq (C and D) or A549 RNA-seq (E and F) genes, ranked by fold change upon either siYAP or siTAZ relative to control, using the following gene sets: cell proliferation (40) and cell adhesion and ECM (41). G and H, Heatmaps of RT-qPCR analysis of representative YAP-regulated cell-proliferation-related (G) and TAZ-regulated ECM-related (H) genes in three NSCLC-derived cell lines. Data represent mean log2 mRNA expression normalized to either HPRT (for H1299 and H460) or GAPDH (for A549) and control transfected cells from three independent biological repeats in each cell line. NES, normalized enrichment score.
Selected genes from each subset were also validated by RT-qPCR analysis in several NSCLC-derived cell lines. Although the extent of preferential regulation varied between cell lines, canonical cell-cycle regulators (PLK1, CIT, KIF23, CDC20) were indeed preferentially downregulated by siYAP (Fig. 2G), whereas genes affecting cell adhesion and migration [integrin beta8 (ITGB8), laminin subunit alpha-3 (LAMA3), Synaptopodin (SYNPO)] were preferentially upregulated by siTAZ (Fig. 2H).
As expected, expression of common YAP/TAZ-regulated genes was affected more strongly by simultaneous knockdown of YAP and TAZ, as compared with knockdown of each paralog alone (Supplementary Figs. S5A and S5B). In contrast, YAP-regulated genes were affected to a similar extent by YAP single knockdown and YAP/TAZ double knockdown (Supplementary Fig. S5C), and likewise for TAZ-regulated genes (Supplementary Fig. S5D), further confirming their paralog-specific regulation.
Next, to determine whether the differential transcriptomic effects of YAP and TAZ drive different functional outcomes, we examined the effects of depletion of each paralog on the proliferation and migration of NSCLC-derived cells. Notably, in all three cell lines tested, YAP silencing (knockdown validation in Supplementary Figs. S6A–S6C) attenuated markedly the increase in cell number over time (Fig. 3A). In contrast, TAZ knockdown had only a mild (H1299, A549) or marginal (H460) impact. Concordantly, transient overexpression of YAP, but not TAZ (overexpression validation in Supplementary Figs. S7A–S7C), augmented the proliferation of H1299 and A549 cells (Fig. 3B).
Figure 3.
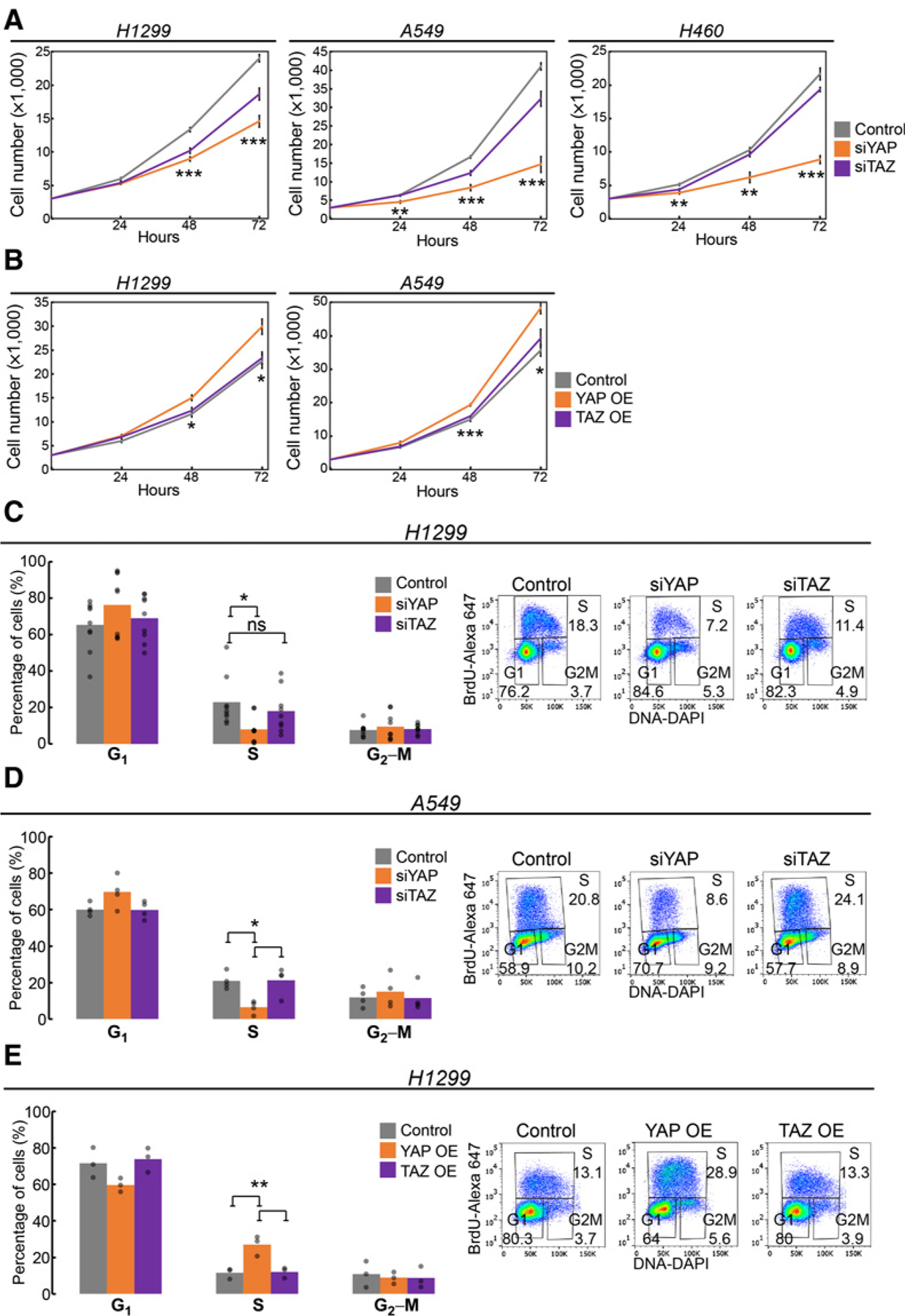
YAP preferentially promotes cell-cycle progression in NSCLC-derived cell lines. A, The indicated cell lines were transfected transiently with siRNA SMARTpools targeting either YAP (siYAP) or TAZ (siTAZ) or control siRNA, and counted at the indicated times posttransfection. B, The indicated cell lines were transfected with plasmids encoding either YAP-flag (YAP OE) or TAZ-flag (TAZ OE), or control plasmid, and counted as in A. y-axis, average cell number from three independent biological repeats. Error bars, SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001, one-way ANOVA. C and D, Cell-cycle profiling, by BrdU + DAPI analysis, of H1299 (n = 9) and A549 (n = 4) cell cultures transfected with the indicated siRNAs. E, BrdU + DAPI cell-cycle profiling of H1299 cell cultures (n = 3) transfected transiently with the indicated plasmids. Left, average percentages of cells in each cell-cycle phase; each dot represents an independent biological repeat. Right, representative FACS analysis images. ns, not significant; *, P < 0.05; **, P < 0.01 determined by one-way ANOVA and Tukey’s post hoc test of the indicated comparisons.
Depletion of either PLK1 or CIT, both preferentially regulated by YAP (Fig. 2G), attenuates cell-cycle progression in multiple cancer cell types (42–44), leading to depletion of the S-phase subpopulation and accumulation of cells in G1-phase. Indeed, as seen in Fig. 3C and D, silencing of YAP, but not TAZ, resulted in a prominent S-phase depletion in both H1299 and A549 cells (siYAP/Control ratio = 0.4 and 0.3, respectively) and elicited a mild increase in G1-phase, relative to control. Similarly, PLK1 silencing mimicked the cell-cycle effects of siYAP (Supplementary Figs. S8A and S8B). Conversely, overexpression of YAP, but not TAZ, increased the S-phase fraction (Fig. 3E). Thus, YAP preferentially acts as a positive regulator of cell-cycle progression in these lung cancer cells.
Excessive ECM production can impede cell migration, owing to aberrant cell adhesion (45). Hence, we assessed the impact of YAP and TAZ on cell migration. Indeed, in a gap closure (“scratch”) assay, TAZ knockdown, which augmented the expression of adhesion- and ECM-related genes (Fig. 2D, F, and H), strongly attenuated cell migration in H1299, A549, and H460 cells (Fig. 4A and B; Supplementary Fig. 9A), whereas silencing of YAP had almost no effect. Concordantly, transient overexpression of TAZ, but not YAP, led to a mild increase in cell migration in both H1299 and A549 cells (Fig. 4C; Supplementary Fig. S9B). These observations were further validated in a transwell migration assay (Supplementary Figs. S9C and S9D). Silencing of YAP or TAZ by single siRNA oligonucleotides phenocopied the effects of the corresponding siRNA SMARTpools (Supplementary Figs. S10A–S10D). Altogether, our observations imply that YAP and TAZ have nonoverlapping roles in these lung cancer cells, whereas YAP preferentially promotes cell-cycle progression, TAZ preferentially promotes cell migration.
Figure 4.
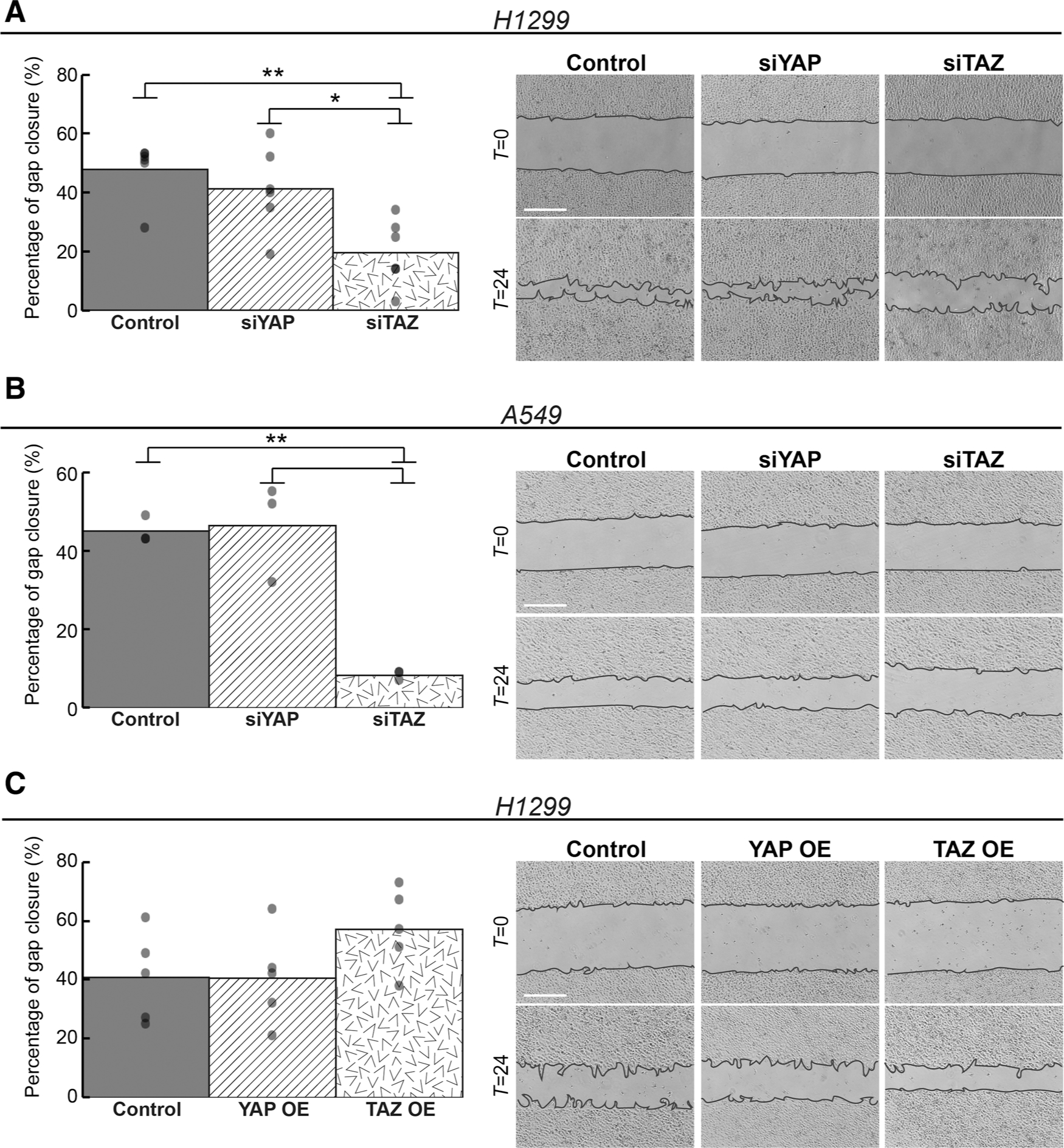
TAZ, but not YAP, preferentially promotes cell migration. A and B, Gap closure (“Scratch”) assays of H1299 (n = 6) and A549 (n = 3) cell cultures transfected with the indicated siRNAs. C, Gap closure assay of H1299 cell cultures (n = 5) transfected transiently with plasmids encoding either YAP-flag (YAP OE) or TAZ-flag (TAZ OE), or control plasmid. Left, average percentage of gap closure calculated from all biological repeats; each dot represents an independent biological repeat. Right, representative images of gap closure at T = 0 and T = 24 hours. *, P < 0.05; **, P < 0.01 determined by one-way ANOVA and Tukey’s post hoc test of the indicated comparisons. Scale bar, 500 μm.
Partial nonredundancy of YAP and TAZ and association with distinct phenotypes in lung tumors
SL is a broadly accepted indication of functional backup and redundancy between genes (46). SL implies a genetic interaction between two genes, whereby individual inactivation of either gene retains cell fitness, while their combined inactivation reduces fitness (and in extreme cases, is lethal; ref. 29). Redundant paralogous pairs often display strong SL, such that cells expressing intrinsically low levels of one paralog are highly dependent on retention of the other paralog (30). Hence, the extent of SL informs on the extent of functional redundancy of a paralogous pair. The partially nonredundant transcriptional and functional effects of YAP and TAZ in lung cancer cells suggested that these paralogs might display only partial SL. We therefore utilized a SL identification computational pipeline (29) to compute SL scores of human paralogs, as previously defined (n = 6,353; ref. 30), in lung adenocarcinoma (LUAD) cells and tumors. Indeed, we found that YAP-TAZ have a fairly low degree of SL when compared with the ranking of all paralogous pairs (Fig. 5A; SL score = 0.37, Rank = 5643). In contrast, ARID1A and ARID1B, known to be functionally redundant (47), were ranked much higher (SL score = 0.95, Rank = 8, Fig. 5A, “+”). As negative controls, we paired ARID1A-ARID1B with either YAP or TAZ (YAP-ARID1A, YAP-ARID1B, TAZ-ARID1A, TAZ-ARID1B); as expected, these pairings yielded very low SL scores (mean SL score = 0.12, Fig. 5A, “−”). Thus, the SL analysis further confirms that YAP and TAZ are only partially redundant in lung cancer cells and tumors.
Figure 5.
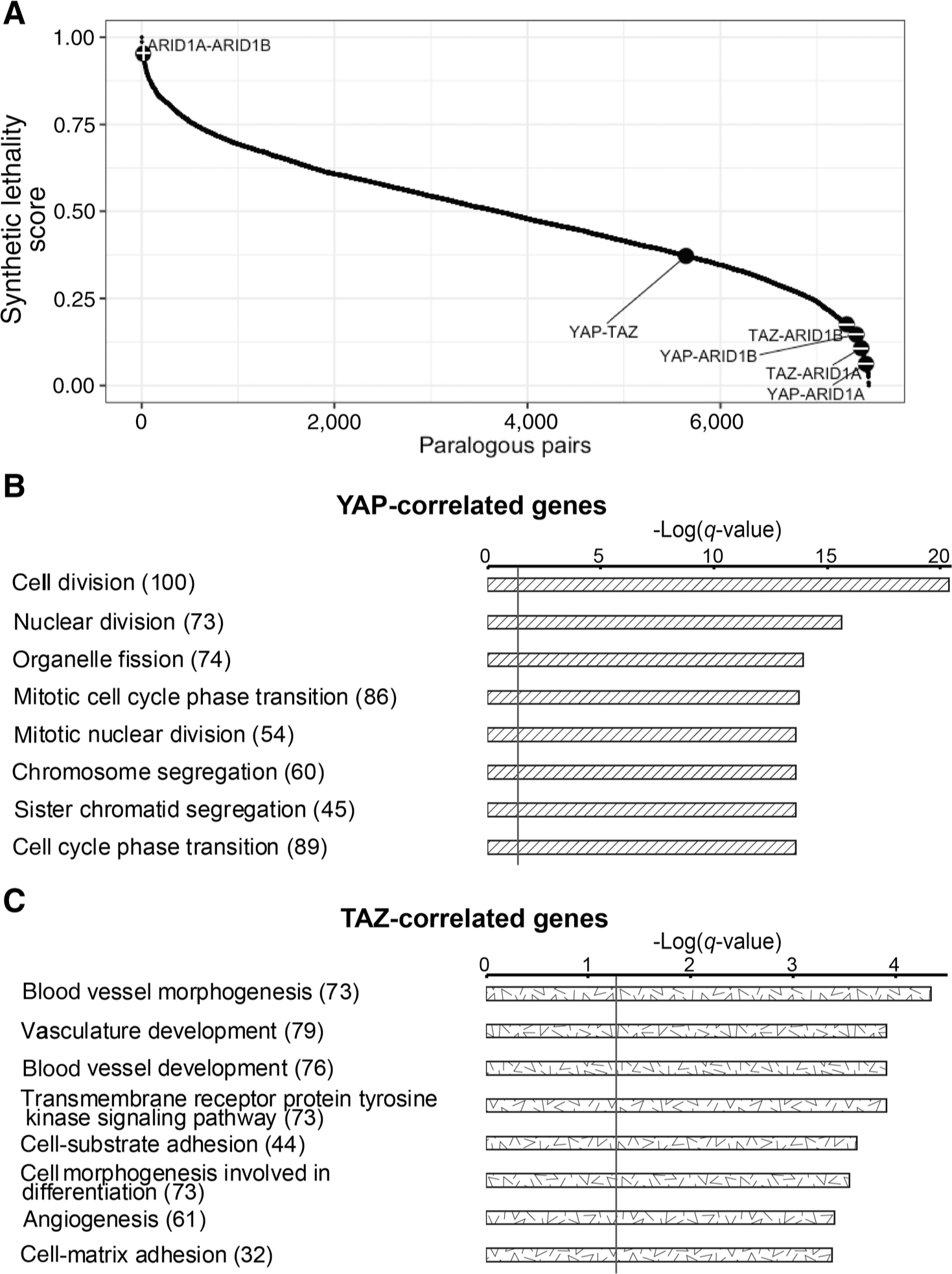
YAP and TAZ display partial non-redundancy and their expression is correlated with distinct biological processes in lung adenocarcinoma cell lines and tumors. A, Ranked synthetic lethality scores of all paralogous pairs, computed via the ISLE pipeline (29), in LUAD cell lines and tumors (see Materials and Methods). Higher score indicates higher synthetic lethality. Representative positive control and negative control pairs are shown with “+” and “−,” respectively. B and C, GO analysis (by Metascape) of the nonoverlapping YAP-correlated and TAZ-correlated genes, regardless of directionality (absolute R) in lung adenocarcinoma tumors (TCGA LUAD). The top eight enriched GO Biological Processes terms are shown. Gray line, q-value of 0.05. In brackets is the number of enriched genes in each term.
Next, we asked whether expression levels of YAP and TAZ in lung tumors are associated with cell-cycle- or ECM-related terms. Toward this aim, we queried the LUAD expression dataset of TCGA. Utilizing an unbiased correlation-based method, the correlation coefficient with either YAP or TAZ mRNA was calculated for each gene in the dataset. Interestingly, YAP and TAZ mRNA levels are only partially correlated (Supplementary Fig. S11). The top 1,600 genes displaying the highest correlation coefficients, regardless of directionality, were further analyzed. Remarkably, only 324 of the 1,600 genes overlapped between YAP-correlated and TAZ-correlated gene sets. Importantly, GO analysis of the nonoverlapping genes (n = 1,276) showed that YAP-correlated genes were enriched in cell division- and cell-cycle-related terms (Fig. 5B; Supplementary Table S4), whereas TAZ-correlated genes were enriched in morphogenesis-, development-, and adhesion-related terms (Fig. 5C; Supplementary Table S4); of note, morphogenesis is closely linked with adhesion (48). Hence, our in vitro observations, implying distinct roles for YAP and TAZ in lung cancer cell biology, also appear to hold for actual human tumors.
Differential association of YAP versus TAZ with response to anticancer drugs
Direct pharmacologic inhibition of YAP/TAZ activity still remains a clinical challenge (49). The distinct functions of YAP and TAZ suggest that the two paralogs may affect differently the response to particular anticancer therapies. Indeed, using drug sensitivity data across 47 LUAD cell lines (PRISM dataset; ref. 33), we identified 142 drugs whose response profile differs preferentially between high versus low YAP mRNA levels, and 38 drugs whose response profile differs preferentially between high versus low TAZ mRNA (Supplementary Table S2).
Taxol (paclitaxel), a microtubule poison, is used to treat NSCLC (50). Interestingly, inspection of a drug sensitivity dataset (33) revealed that cell viability upon Taxol treatment was negatively correlated with levels of YAP, but not TAZ mRNA (Fig. 6A). Further analysis of the largest publicly available cancer cell line drug sensitivity collection (GDSC dataset; ref. 35) confirmed that Taxol sensitivity was more significantly correlated with YAP protein levels (R = 0.22, P-value <0.02; Supplementary Fig. S12) than with TAZ protein levels (R = 0.10, P-value = 0.23) in lung cancer cell lines.
Figure 6.
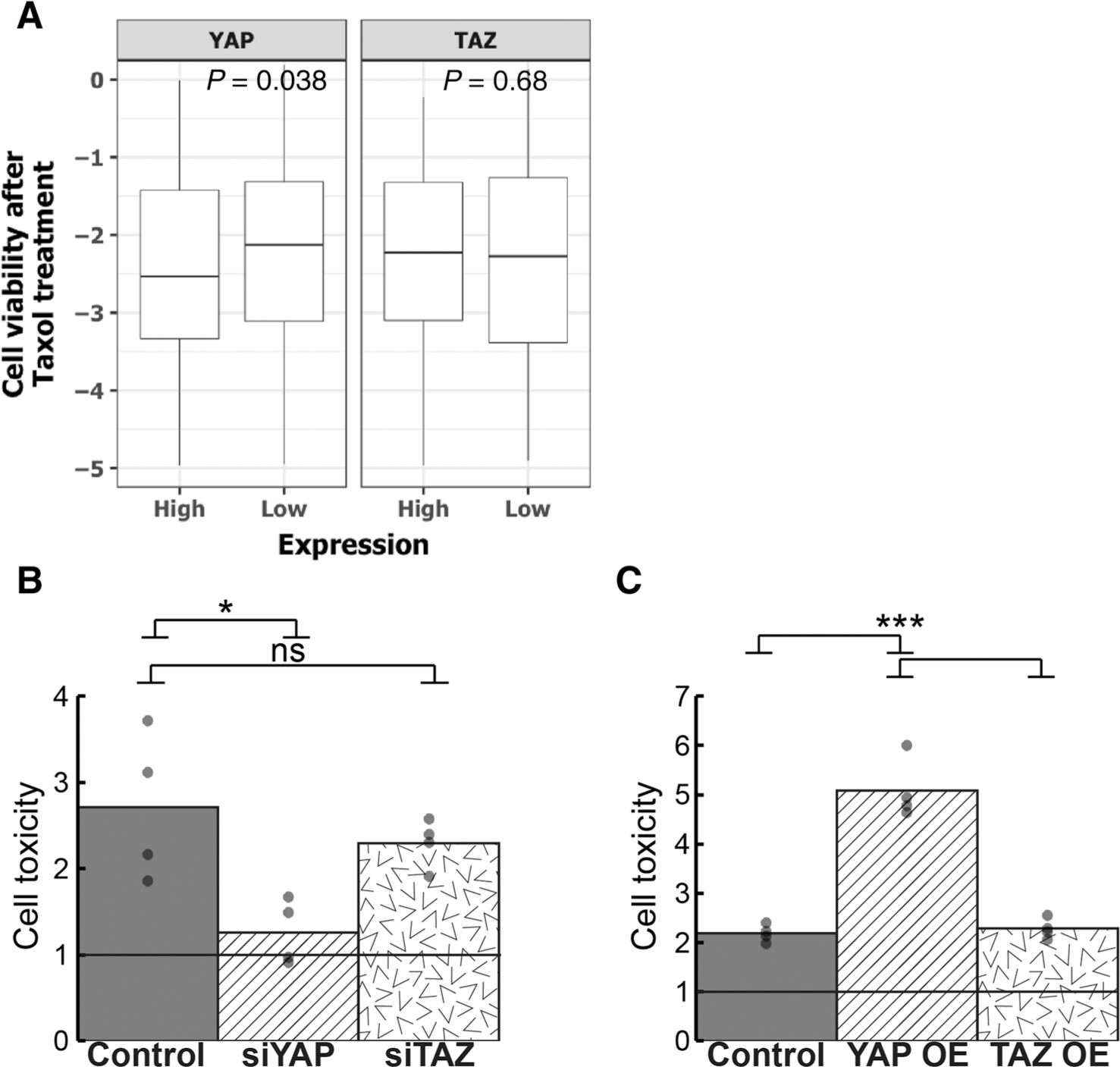
Taxol sensitivity is preferentially dependent on YAP. A, Box plot of Taxol cell viability scores across cancer cell lines (n = 578) derived from PRISM (33), binned by YAP or TAZ expression; lower cell viability implies greater sensitivity to Taxol. P-value, Wilcoxon rank-sum test. B and C, H1299 cells were transfected with the indicated siRNA SMARTpools (B) or expression plasmids (C), and 6 hours later were treated with 1 μmol/L Taxol for an additional 42 hours, including serum starvation for the last 18 hours. The percentage of dead cells was determined by the CellTox Green Cytotoxicity Assay (see Materials and Methods) and normalized to DMSO-treated cells (control). Shown is the average from four biological repeats; each dot represents an independent repeat. ns, not significant; *, P < 0.05; ***, P < 0.001, determined by one-way ANOVA and Tukey’s post hoc test of the indicated comparisons.
To compare directly the impact of YAP versus TAZ on Taxol sensitivity, we treated H1299 cells with Taxol after knockdown of either YAP or TAZ. As seen in Fig. 6B, YAP silencing indeed greatly compromised the cytotoxicity of Taxol, whereas TAZ silencing had only a marginal effect. Conversely, transient overexpression of YAP, but not TAZ, increased Taxol sensitivity (Fig. 6C). Thus, YAP and TAZ also affect differentially the sensitivity to specific anticancer agents.
Involvement of TEAD in expression of YAP- and TAZ-regulated genes
YAP and TAZ do not bind directly to DNA, and much of their transcriptional activity is dependent on “piggybacking” to TEA domain proteins 1–4 (TEAD1–4) DNA binding proteins (10). To assess whether YAP/TAZ distinct transcriptional effects depend on TEAD, we overexpressed mutant forms of either YAP or TAZ defective in TEAD binding, and monitored their impact on relevant genes (overexpression validation in Supplementary Fig. S13A). As seen in Supplementary Figs. S13B and S13C, loss of TEAD binding attenuated the induction of common YAP/TAZ-regulated genes and of YAP-regulated genes. Interestingly, while the upregulation of CLDN5 by TAZ was compromised by loss of TEAD binding, this was not so for three TAZ-repressed genes. Hence, the role of TEAD in the differential transcriptional effects of YAP and TAZ is gene and mechanism dependent.
TAZ binds preferentially to chromatin regions encompassing TAZ-regulated migration-related genes
To further explore the mechanisms underlying YAP/TAZ differential transcriptional programs, we performed ChIP sequencing (ChIP-seq) analysis of H1299 cells with antibodies against either YAP or TAZ. As expected, there was approximately 50% overlap between YAP and TAZ binding sites (Supplementary Fig. S14A). Furthermore, consistent with previously published data (6), most peaks were within 10 to 100 kb from the nearest transcription start site (TSS; Supplementary Fig. S14B).
We then explored the peaks (approximately 1,200) displaying a significant differential binding of YAP or TAZ (|fold change| ≥ 2, FDR ≤ 0.05; Fig. 7A), hereafter referred to as YAP-differential or TAZ-differential peaks. Remarkably, whereas 61% of TAZ-differential peaks were located less than 10 kb from the nearest TSS (yellow and green in Fig. 7B), the majority of YAP-differential peaks were located over 100 kb away from any annotated gene (red in Fig. 7B), suggesting that differential YAP/TAZ binding sites might control gene regulation via distinct regulatory mechanisms.
Figure 7.
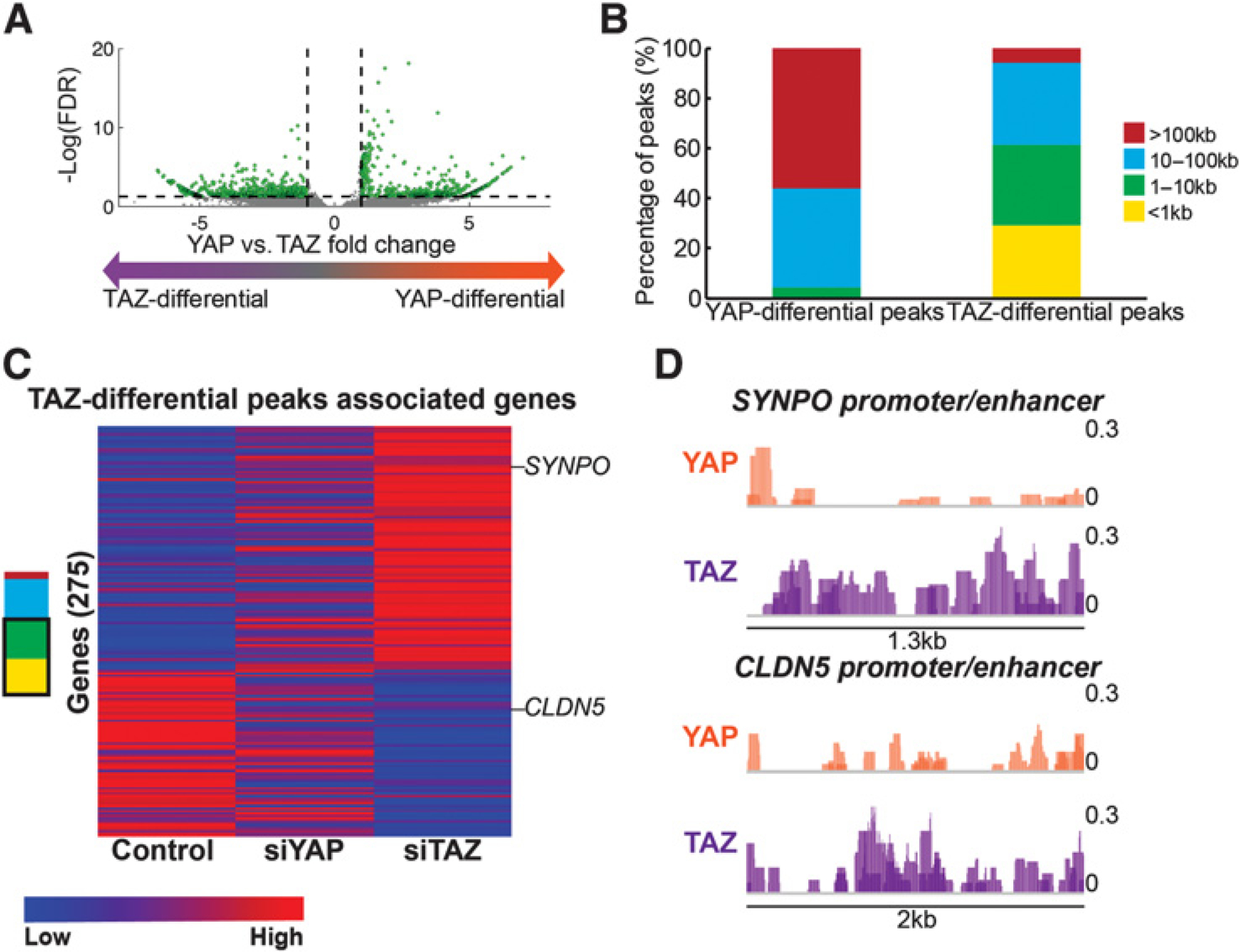
TAZ-differential ChIP peaks are associated with TAZ-regulated and migration-related genes. H1299 cells were subjected to ChIP-seq analysis with either anti-YAP or anti-TAZ antibodies, in two biological repeats. A, Volcano plot of YAP- and TAZ-differentially bound peaks (see Materials and Methods). Green dots represent peaks whose binding was significantly differential between YAP and TAZ (absolute fold change ≥2, FDR ≤0.05). x-axis, log2 anti-YAP peak score −log2 anti-TAZ peak score. Dashed lines, FDR of 0.05 (bottom), and log2-fold change of 2 (right) and 0.5 (left). B, Absolute distance of YAP-differential (n = 610) and TAZ-differential peaks (n = 604) from the nearest TSS, as determined by HOMER. C, Heatmap of mean expression levels (log2) of 275 genes (from the H1299 RNA-seq in Fig. 1) associated with TAZ-differential ChIP-seq peaks. Only peaks located less than 10 kb from the nearest TSS (yellow and green in B) were included. D, YAP and TAZ binding profiles at two representative promoter/enhancer regions of TAZ preferentially regulated genes (CLDN5: chr22:195,064,59–195,090,20; SYNPO: chr5:149,993,718–149,995,070). ChIP-seq coverage is shown for two biological repeats (overlaid color) and scaled to 1,000,000/(totalReadCount). Black lines and number indicate length of the presented genomic area.
We next assigned TAZ-differential peaks to the nearest gene, considering only peaks residing within 10 kb from the nearest TSS [owing to the negligible number of YAP-differential peaks meeting this condition (yellow and green in Fig. 7B), these were not further analyzed]. As expected, genes assigned to TAZ-differential peaks were differentially expressed upon siTAZ, but not siYAP, compared with control (Fig. 7C). Interestingly, several of these genes are known to regulate cancer cell migration (51, 52). For instance, CLDN5 promotes cell migration, whereas SYNPO inhibits migration (51, 52). As shown in Fig. 7D, TAZ bound more than YAP to regulatory regions of CLDN5 and SYNPO. Hence, the preferential effects of TAZ on migration-related genes may be partly due to preferential binding to their regulatory regions.
Discussion
In this study, we compared the functional impact of YAP versus TAZ in NSCLC-derived cells. We found that although both paralogs modulated to a similar extent a common subset of genes, distinctly larger subsets of genes were preferentially affected in a paralog-specific manner. A pronounced difference in their biological impact was revealed: whereas YAP preferentially affected cell-cycle progression, TAZ preferentially modulated cell migration. This finding was apparent not only in cell lines, but also in data from human lung tumors. Thus, there exists a division of labor between the two paralogs in lung cancer cells and tumors. Moreover, this division of labor is associated with differential responses to anticancer drugs, such as Taxol.
Of note, many YAP-regulated genes were also mildly affected by TAZ depletion, and vice versa (Fig. 1C and D), suggesting that the relative contribution of YAP versus TAZ to the expression of such genes may depend on the relative abundance of each paralog in a given cell. A similar idea was recently proposed for protein–protein interactions: two paralogs may share potential binding partners, but the actual interaction profile of each paralog will be determined by its relative abundance and relative affinities for the different interaction partners (53). Indeed, TAZ silencing caused a mild decrease in cell proliferation in some but not all cell lines (Fig. 3A): likewise, YAP slightly affected cell migration in some experiments (Supplementary Figs. S9A, S9C, and S9D), in agreement with earlier reports (17, 54, 55). Therefore, the division of labor between YAP and TAZ is preferential rather than absolute, and may differ between different types of cancer and perhaps even between individual cases of the same cancer type. Interestingly, Sun and colleagues (56) have recently shown that although YAP and TAZ similarly promote the proliferation of non-differentiated myoblasts, they exert opposing effects when such cells are induced to undergo myogenic differentiation. Furthermore, Plouffe and colleagues (57) found that inactivation of YAP in human embryonic kidney cells had greater effects than TAZ inactivation, supporting the notion that one paralog may dominate over the other, in a context-dependent manner, in regulating specific biological functions.
Many gene duplication events play major roles in shaping the genomes of different species. Paralogs originating from a gene duplication event can diverge and acquire changes that contribute to organismal diversity. Invertebrates have only a single YAP/TAZ orthologue; in Drosophila this orthologue, Yorkie (Yki), is required for both cell proliferation and cell migration in the course of multiple biological processes (58–62). In vertebrates, YAP and TAZ regulate both proliferation and migration, but with different efficiencies, enabling refined regulation of complex biological processes. We propose that the gene duplication and the consequent division of labor between the two paralogs might explain their retention throughout vertebrate evolution.
Our analysis demonstrates that YAP and TAZ display both shared and differential genome-wide chromatin binding patterns. In the case of TAZ, this may explain in part its ability to preferentially regulate a subset of migration-related genes. However, a comparable mechanistic explanation for YAP-regulated genes is still lacking. Thus, more work is needed to fully explain the molecular basis for the differential transcriptional effects of the two paralogs. Plausibly, YAP and TAZ may vary in their relative affinities for particular interacting proteins, which may lead to different transcriptional preferences. This might be subject to further regulation, for example, by posttranslational modifications, providing an additional layer of context-dependent diversity. Interestingly, it was recently demonstrated (21) that TAZ, but not YAP, readily forms nuclear condensates via liquid–liquid phase separation (LLPS), whereas YAP was shown to form LLPS condensates under other conditions (63). Notably, we revealed hundreds of genomic sites located far away from any annotated gene, which were bound selectively by YAP and not TAZ. This raises the intriguing possibility that YAP may have an additional function, not shared with TAZ, perhaps associated with long-range chromatin interactions or high-order genome organization.
In sum, our findings suggest that tumorigenesis may take advantage of the division of labor between YAP and TAZ to orchestrate complementary oncogenic functions. These findings may enable a better stratification of lung cancer tumors, and perhaps reveal cancer vulnerabilities that might be exploited toward more effective therapies.
Supplementary Material
Significance:
Thease findings show that oncogenic paralogs YAP and TAZ have distinct roles in NSCLC and are associated with differential response to anticancer drugs, knowledge that may assist lung cancer therapy decisions.
Acknowledgments
We thank all members of the Oren lab for fruitful discussions, Yosef Shaul and Kunxin Luo for their generous gift of plasmids, Yarden Nuriel for technical assistance, Itay Tirosh and Benny Geiger for scientific advice, Uri Alon and Tamar Danon for access to their research facilities, and scientists of the Weizmann Institute Department of Life Sciences core facilities for help and guidance. This work was supported in part by grants from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation and the DKFZ-MOST Cooperation in Cancer Research, a grant from Anat and Amnon Shashua, and the Moross Integrated Cancer Center. M. Oren is incumbent of the Andre Lwoff chair in molecular biology.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Herbst SR, Heymach JV, Lippman SM. Lung cancer. N Engl J Med 2008;359:1367–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lo Sardo F, Strano S, Blandino G. YAP and TAZ in lung cancer: oncogenic role and clinical targeting. Cancers 2018;10:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lau AN, Curtis SJ, Fillmore CM, Rowbotham SP, Mohseni M, Wagner DE, et al. Tumor-propagating cells and Yap/Taz activity contribute to lung tumor progression and metastasis. EMBO J 2014;33:468–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Xu X, Maglic D, Dill MT, Mojumdar K, Ng PK, et al. Comprehensive molecular characterization of the Hippo signaling pathway in cancer. Cell Rep 2018;25:1304–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen HY, Yu SL, Ho BC, Su KY, Hsu YC, Chang CS, et al. R331W missense mutation of oncogene YAP1 is a germline risk allele for lung adenocarcinoma with medical actionability. J Clin Oncol 2015;33:2303–10. [DOI] [PubMed] [Google Scholar]
- 6.Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol 2015;17:1218–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beyer TA, Weiss A, Khomchuk Y, Huang K, Ogunjimi AA, Varelas X, et al. Switch enhancers interpret TGF-beta and Hippo signaling to control cell fate in human embryonic stem cells. Cell Rep 2013;5:1611–24. [DOI] [PubMed] [Google Scholar]
- 8.Hiemer SE, Szymaniak AD, Varelas X. The transcriptional regulators TAZ and YAP direct transforming growth factor beta-induced tumorigenic phenotypes in breast cancer cells. J Biol Chem 2014;289:13461–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galli GG, Carrara M, Yuan WC, Valdes-Quezada C, Gurung B, Pepe-Mooney B, et al. YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol Cell 2015;60:328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piccolo S, Dupont S, Cordenonsi M. The biology of YAP-TAZ: Hippo signaling and beyond. Physiol Rev 2014;94:1287–312. [DOI] [PubMed] [Google Scholar]
- 11.Yuen HF, McCrudden CM, Huang YH, Tham JM, Zhang X, Zeng Q, et al. TAZ expression as a prognostic indicator in colorectal cancer. PLoS One 2013;8:e54211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grijalva JL, Huizenga M, Mueller K, Rodriguez S, Brazzo J, Camargo F, et al. Dynamic alterations in Hippo signaling pathway and YAP activation during liver regeneration. Am J Physiol Gastrointest Liver Physiol 2014;307:G196–204. [DOI] [PubMed] [Google Scholar]
- 13.Liu CY, Chan SW, Guo F, Toloczko A, Cui L, Hong W. MRTF/SRF dependent transcriptional regulation of TAZ in breast cancer cells. Oncotarget 2016;7:13706–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reginensi A, Scott RP, Gregorieff A, Bagherie-Lachidan M, Chung C, Lim DS, et al. Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet 2013;9:e1003380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao Y, Zhang W, Han X, Li F, Wang X, Wang R, et al. YAP inhibits squamous transdifferentiation of Lkb1-deficient lung adenocarcinoma through ZEB2-dependent DNp63 repression. Nat Commun 2014;5:4629. [DOI] [PubMed] [Google Scholar]
- 16.Gobbi G, Donati B, Do Valle IF, Reggiani F, Torricelli F, Remondini D, et al. The Hippo pathway modulates resistance to BET proteins inhibitors in lung cancer cells. Oncogene 2019;38:6801–17. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Z, Hao Y, Liu N, Raptis L, Tsao MS, Yang X. TAZ is a novel oncogene in non-small cell lung cancer. Oncogene 2011;30:2181–6. [DOI] [PubMed] [Google Scholar]
- 18.Cohen AA, Geva-Zatorsky N, Eden E, Frenkel-Morgenstern M, Issaeva I, Sigal A, et al. Dynamic proteomics of individual cancer cells in response to a drug. Science 2008;322:1511–6. [DOI] [PubMed] [Google Scholar]
- 19.Arandkar S, Furth N, Elisha Y, Nataraj NB, van der Kuip H, Yarden Y, et al. Altered p53 functionality in cancer-associated fibroblasts contributes to their cancer-supporting features. Proc Natl Acad Sci U S A 2018;115:6410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spolverini A, Fuchs G, Bublik DR, Oren M. let-7b and let-7c microRNAs promote histone H2B ubiquitylation and inhibit cell migration by targeting multiple components of the H2B deubiquitylation machinery. Oncogene 2017;36:5819–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Y, Wu T, Gutman O, Lu H, Zhou Q, Henis YI, et al. Phase separation of TAZ compartmentalizes the transcription machinery to promote gene expression. Nat Cell Biol 2020;22:453–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furth N, Pateras IS, Rotkopf R, Vlachou V, Rivkin I, Schmitt I, et al. LATS1 and LATS2 suppress breast cancer progression by maintaining cell identity and metabolic state. Life Sci Alliance 2018;1:e201800171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohen R, Barlev J, Hornung G, Stelzer G, Feldmesser E, Kogan K, et al. UTAP: user-friendly transcriptome analysis pipeline. BMC Bioinformatics 2019;20:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011;17:10–12. [Google Scholar]
- 25.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 2019;10:1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JS, Das A, Jerby-Arnon L, Arafeh R, Auslander N, Davidson M, et al. Harnessing synthetic lethality to predict the response to cancer treatment. Nat Commun 2018;9:2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Viswanathan SR, Nogueira MF, Buss CG, Krill-Burger JM, Wawer MJ, Malolepsza E, et al. Genome-scale analysis identifies paralog lethality as a vulnerability of chromosome 1p loss in cancer. Nat Genet 2018;50:937–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 2013;45:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corsello SM, Nagari RT, Spangler RD, Rossen J, Kocak M, Bryan JG, et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat Cancer 2020;1:235–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018;46:D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 2013;41:D955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569:503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Agostino S, Sorrentino G, Ingallina E, Valenti F, Ferraiuolo M, Bicciato S, et al. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep 2016;17:188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blecher-Gonen R, Barnett-Itzhaki Z, Jaitin D, Amann-Zalcenstein D, Lara-Astiaso D, Amit I. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat Protoc 2013;8:539–54. [DOI] [PubMed] [Google Scholar]
- 39.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013;14:178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 2008;40:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nardone G, Oliver-De La Cruz J, Vrbsky J, Martini C, Pribyl J, Skladal P, et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat Commun 2017;8:15321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giraldez S, Galindo-Moreno M, Limon-Mortes MC, Rivas AC, Herrero-Ruiz J, Mora-Santos M, et al. G1/S phase progression is regulated by PLK1 degradation through the CDK1/betaTrCP axis. FASEB J 2017;31:2925–36. [DOI] [PubMed] [Google Scholar]
- 43.Medina-Aguilar R, Marchat LA, Arechaga Ocampo E, Gariglio P, Garcia Mena J, Villegas Sepulveda N, et al. Resveratrol inhibits cell cycle progression by targeting Aurora kinase A and Polo-like kinase 1 in breast cancer cells. Oncol Rep 2016;35:3696–704. [DOI] [PubMed] [Google Scholar]
- 44.Wu Z, Zhu X, Xu W, Zhang Y, Chen L, Qiu F, et al. Up-regulation of CIT promotes the growth of colon cancer cells. Oncotarget 2017;8:71954–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gupton SL, Waterman-Storer CM. Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration. Cell 2006;125:1361–74. [DOI] [PubMed] [Google Scholar]
- 46.Cereda M, Mourikis TP, Ciccarelli FD. Genetic redundancy, functional compensation, and cancer vulnerability. Trends Cancer 2016;2:160–2. [DOI] [PubMed] [Google Scholar]
- 47.Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat Med 2014;20:251–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Horton ER, Byron A, Askari JA, Ng DHJ, Millon-Fremillon A, Robertson J, et al. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat Cell Biol 2015;17:1577–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shreberk-Shaked M, Oren M. New insights into YAP/TAZ nucleo-cytoplasmic shuttling: new cancer therapeutic opportunities? Mol Oncol 2019;13:1335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramalingam S, Belani CP. Paclitaxel for non-small cell lung cancer. Expert Opin Pharmacother 2004;5:1771–80. [DOI] [PubMed] [Google Scholar]
- 51.Escudero-Esparza A, Jiang WG, Martin TA. Claudin-5 is involved in breast cancer cell motility through the N-WASP and ROCK signalling pathways. J Exp Clin Cancer Res 2012;31:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wong JS, Iorns E, Rheault MN, Ward TM, Rashmi P, Weber U, et al. Rescue of tropomyosin deficiency in Drosophila and human cancer cells by synaptopodin reveals a role of tropomyosin alpha in RhoA stabilization. EMBO J 2012;31:1028–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dandage R, Landry CR. Paralog dependency indirectly affects the robustness of human cells. Mol Syst Biol 2019;15:e8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Noguchi S, Saito A, Horie M, Mikami Y, Suzuki HI, Morishita Y, et al. An integrative analysis of the tumorigenic role of TAZ in human non-small cell lung cancer. Clin Cancer Res 2014;20:4660–72. [DOI] [PubMed] [Google Scholar]
- 55.Zheng CH, Chen XM, Zhang FB, Zhao C, Tu SS. Inhibition of CXCR4 regulates epithelial mesenchymal transition of NSCLC via the Hippo-YAP signaling pathway. Cell Biol Int 2018;42:1386–94. [DOI] [PubMed] [Google Scholar]
- 56.Sun C, De Mello V, Mohamed A, Ortuste Quiroga HP, Garcia-Munoz A, Al Bloshi A, et al. Common and distinctive functions of the Hippo effectors Taz and Yap in skeletal muscle stem cell function. Stem Cells 2017;35:1958–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Plouffe SW, Lin KC, Moore JL 3rd, Tan FE, Ma S, Ye Z, et al. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. J Biol Chem 2018;293:11230–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsai CR, Anderson AE, Burra S, Jo J, Galko MJ. Yorkie regulates epidermal wound healing in Drosophila larvae independently of cell proliferation and apoptosis. Dev Biol 2017;427:61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin TH, Yeh TH, Wang TW, Yu JY. The Hippo pathway controls border cell migration through distinct mechanisms in outer border cells and polar cells of the Drosophila ovary. Genetics 2014;198:1087–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ren F, Wang B, Yue T, Yun EY, Ip YT, Jiang J. Hippo signaling regulates Drosophila intestine stem cell proliferation through multiple pathways. Proc Natl Acad Sci U S A 2010;107:21064–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005;122:421–34. [DOI] [PubMed] [Google Scholar]
- 62.Thompson BJ, Cohen SM. The Hippo pathway regulates the bantam micro-RNA to control cell proliferation and apoptosis in Drosophila. Cell 2006;126:767–74. [DOI] [PubMed] [Google Scholar]
- 63.Cai D, Feliciano D, Dong P, Flores E, Gruebele M, Porat-Shliom N, et al. Phase separation of YAP reorganizes genome topology for long-term YAP target gene expression. Nat Cell Biol 2019;21:1578–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.