

Abstract
Purpose
Molecular similarities have been reported between basal-like breast cancer (BLBC) and high-grade serous ovarian cancer (HGSOC). To date, there have been no prognostic biomarkers that can provide risk stratification and inform treatment decisions for both BLBC and HGSOC. In this study, we developed a molecular signature for risk stratification in BLBC and further validated this signature in HGSOC.
Methods
RNA-seq data was downloaded from The Cancer Genome Atlas (TCGA) project for 190 BLBC and 314 HGSOC patients. Analyses of differentially expressed genes between recurrent vs. non-recurrent cases were performed using different bioinformatics methods. Gene Signature was established using weighted linear combination of gene expression levels. Their prognostic performance was evaluated using survival analysis based on progression-free interval (PFI) and disease-free interval (DFI).
Results
63 genes were differentially expressed between 18 recurrent and 40 non-recurrent BLBC patients by two different methods. The recurrence index (RI) calculated from this 63-gene signature significantly stratified BLBC patients into two risk groups with 38 and 152 patients in the low-risk (RI-Low) and high-risk (RI-High) groups, respectively (p = 0.0004 and 0.0023 for PFI and DFI, respectively). Similar performance was obtained in the HGSOC cohort (p = 0.0131 and 0.004 for PFI and DFI, respectively). Multivariate Cox regression adjusting for age, grade, and stage showed that the 63-gene signature remained statistically significant in stratifying HGSOC patients (p = 0.0005).
Conclusion
A gene signature was identified to predict recurrence in BLBC and HGSOC patients. With further validation, this signature may provide an additional prognostic tool for clinicians to better manage BLBC, many of which are triple-negative and HGSOC patients who are currently difficult to treat.
Keywords: Basal-like breast cancer, High-grade serous ovarian cancer, Recurrence, Gene signature, Prognosis
Introduction
Breast cancer is the most common cancer in women around the world. While early detection by mammography has greatly reduced the mortality of breast cancer, patients continue to develop recurrences many years after diagnosis. Basal-like breast cancer (BLBC) is one of the intrinsic subtypes of breast cancer and is often negative for estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2), the so-called triple-negative breast cancer (TNBC) [1]. This subtype, only accounting for 10–20% of breast cancer, is more aggressive than hormone receptor-positive breast cancer (ER+ and/or PR+) in that it grows and spreads to lymph nodes or other distant organs more quickly and more frequently [2, 3]. While anti-estrogen and anti-HER2 therapies do not work for BLBC, BLBC does respond better to platinum-based chemotherapy regimens than hormone receptor-positive breast cancer, yet it does have a higher risk of developing recurrence within 5 years of treatment [3, 4]. Newer targeted therapies (PARP inhibitors and PD-L1 immunotherapy) are emerging treatment paradigms for BLBC/TNBC [5, 6].
On the other hand, ovarian cancer is diagnosed in fewer women than breast cancer, but it ranks 5th in cancer deaths among women and causes more deaths than any other female gynecological cancer [7]. High-grade serous ovarian cancer (HGSOC) accounts for 70% of all ovarian cancer cases and is the most malignant form of ovarian cancer [8]. A substantial proportion of HGSOC has inherited mutations in the BRCA1/2 genes, and by the time of becoming symptomatic, they are usually at an advanced stage with poor outcome [9]. Similar to BLBC, HGSOC responds to platinum-based chemotherapy, and for those with BRCA1/2 mutations, targeted therapy with a PARP inhibitor is recommended [10]. Nevertheless, effective treatment of BLBC/TNBC and HGSOC remains a challenge.
Development of gene expression-based signatures for assessment of the risk of recurrence has been of great interest across many cancer types. Particularly in hormone receptor-positive breast cancer, several multigene expression signatures have been commercialized to provide prognostic information on individual risk of recurrence and predictive information on the likelihood of benefit from chemotherapy and extended endocrine therapy [11–15]. To date, there have been some reports of developing gene signatures for TNBC recurrence or progression, but there have been no gene signatures able to predict recurrence in both BLBC/TNBC and HGSOC [16–18]. In this paper, we report the development of a prognostic gene expression signature, based on The Cancer Genome Atlas (TCGA) RNA-seq data, specifically for BLBC and the evaluation of its prognostic performance in HGSOC.
Methods
Study patients and data
The TCGA program (https://cancergenome.nih.gov/) generated and characterized genomic, epigenomic, transcriptomic, and proteomic profiles for over 11,000 primary cancers and matched normal samples across 33 cancer types. Genome-wide mRNA-seq raw count data for breast cancer and HGSOC were downloaded from TCGA harmonized database via the GDC data portal using the TCGAbiolinks package using Bioconductor. Data derived from recurrent tumors and matched normal samples were excluded from this study. Clinicopathologic data were obtained from the TCGA Pan-Cancer Clinical Data Resource (TCGA-CDR) [19]. PAM50 intrinsic subtype classification was performed following the method by Parker et al. [20].
Data processing
To reduce noise from no/low expression genes, genes that had 10 or less raw counts in more than 90% of the samples were excluded from further analyses. Raw count data were normalized using the method of trimmed mean of M-values (TMM) prior to the calculation of recurrence index based on developed gene expression signatures [21]. Gene annotation was done via BioMart databases (https://www.biomart.org) using Bioconductor package biomaRt [22].
Differential gene expression analysis
Three RNA-seq analysis methods were used to identify gene features that were differentially expressed between those patients that showed progression within 2 years versus those having no progression events for at least 5 years. DESeq2 and edgeR were based on negative binomial generalized linear models for count-based expression data. DESeq2 uses gene-specific dispersion parameters, while edgeR includes both common and gene-specific dispersion parameters moderated by empirical Bayes to borrow information across genes [23, 24]. The voom/limma method does not assume negative binomial distribution for the RNA-seq data [25, 26]. Instead, it estimates a mean–variance relationship of the log-counts, generating a precision weight for each normalized observation. As such, the normalized log-counts and the associated precision weights can be used with any statistical methods that are precision weight aware. Therefore, voom/limma methods open the analysis of RNA-seq data to a wide variety of statistical techniques that were previously developed for microarray data analysis. The multiple testing adjustment was done by Benjamini & Hochberg False Discovery Rate (FDR) [27]. Thresholds for determining statistical significance are provided in the Results section. Common differentially expressed genes were identified among the three analysis methods as the basis for developing prognostic gene signatures.
GO enrichment analysis
Gene ontology (GO) enrichment analysis was performed using The Gene Ontology Resource (https://geneontology.org/). Gene IDs from the prognostic gene signature were entered into the search engine for GO Enrichment Analysis on the GO homepage using the GO aspect of biological process. Analysis performed by the PANTHER Classification System provided a list of GO terms that were over- and under-represented from the submitted gene list using the Fisher’s exact test to determine significance (p < 0.05) [28, 29].
Statistical analysis
Prognostic risk models were built based on the commonly identified differentially expressed genes, and a raw recurrence index was calculated based on log2 transformed TMM normalized count data by weighted linear combination using Wald statistic for each gene from DESeq2 analysis as the weight [30]. The calculated raw recurrence index was then linearly scaled to between 0 and 10 to derive the final recurrence index (RI). For BLBC, the threshold to classify patients into RI-Low (less than the threshold) vs. RI-High (at or above the threshold) groups was determined to ensure there were minimal recurrent cases within the stratified RI-Low group, because the clinical utility for the generally considered high-risk BLBC was to identify those with relatively low risk of recurrence to avoid aggressive treatments. On the other hand, for HGSOC that is highly aggressive, the selection of the threshold was to identify a relatively small, yet reasonable proportion of patients (e.g., 20%) as clinically super-high risk of recurrence to warrant new regimens of even more aggressive therapies.
The primary study endpoint was progression-free interval (PFI) with events including local or distant recurrences, new primary tumor in the breast, or death from disease; the secondary endpoint was disease-free interval (DFI) which had the same endpoint as PFI but required that the patient first achieve disease-free status after receiving the first course of treatment. Kaplan–Meier analysis with log-rank test was used to assess the equality of the survival curves of the prognostic risk groups [31]. Cox proportional hazard regression was used to derive hazard ratios (HRs) for comparing the risks of differential risk groups [32]. All analyses were conducted using R statistical package (version 3.5.2; https://www.r-project.org).
Results
RNA-seq raw counts were downloaded from TCGA for 1102 breast cancer samples and 314 high-grade serous stage I-III ovarian cancer samples with clinical outcome data available for 1090 breast cancer and all ovarian cancer samples. The median number of total reads per library was 58 million, ranging from 13 to 114 million. The breast cancer data included 563 luminal A, 215 luminal B, 82 Her2-enriched, 190 basal-like and 40 normal-like breast cancer patients. Among the 190 BLBC patients that were the focus of this study, 62% of patients were postmenopausal, 86% had T1 or T2 tumors, 62% had lymph node negative disease, 87% had only primary tumors, while 2% had metastatic disease, and 85%, 89% and 95% were ER, PR, and HER2 negative, respectively (Table 1). For the HGSOC dataset, the median age was 59 years old, 93% were stage III, and 87% had grade 3 tumors (Table 2).
Table 1.
Patient characteristics of basal-like breast cancer cohort
| Factors | RNA-seq (N = 190) |
|---|---|
| N (%) | |
| Age | |
| Median (min, max) | 54 (29, 90) |
| Menopausal status Pre | 38 (20%) |
| Peri | 10 (5%) |
| Post | 117 (62%) |
| Indeterminate | 11 (6%) |
| Unknown | 14 (7%) |
| Race White | 111 (58%) |
| Black | 64 (34%) |
| Asian | 7 (4%) |
| Unknown | 8 (4%) |
| T stage | |
| T1 | 37 (19%) |
| T2 | 127 (67%) |
| T3 | 19 (10%) |
| T4 | 6 (3%) |
| Unknown | 1 (1%) |
| N stage | |
| N0 | 118 (62%) |
| N1 | 51 (27%) |
| N2 | 15 (8%) |
| N3 | 6 (3%) |
| M stage | |
| M0 | 165 (87%) |
| M1 | 4 (2%) |
| Unknown | 21 (11%) |
| ER | |
| Positive | 21 (11%) |
| Negative | 162 (85%) |
| Unknown | 7 (4%) |
| PR | |
| Positive | 12 (6%) |
| Negative | 169 (89%) |
| Unknown | 9 (5%) |
| HER2 | |
| Positive | 6 (3%) |
| Negative | 180 (95%) |
| Unknown | 4 (2%) |
| DFI | |
| Event | 22 (12%) |
| Event-free | 148 (78%) |
| Unknown | 20 (10%) |
| PFI | |
| Event | 29 (15%) |
| Event-free | 161 (85%) |
Table 2.
Patient characteristics of high-grade serous ovarian cancer cohort
| Factors | RNA-seq (N = 314) |
|---|---|
| N (%) | |
| Age | 59 (30, 87) |
| Median (min, max) | |
| Race | 269 (86%) |
| White | |
| Black | 23 (7%) |
| Asian | 9 (3%) |
| Others or unknown | 13 (4%) |
| Clinical stage | |
| I | 1 (0%) |
| II | 21 (7%) |
| III | 292 (93%) |
| Grade | |
| G2 | 35 (11%) |
| G3 | 273 (87%) |
| GX | 6 (2%) |
| DFI | |
| Event | 126 (40%) |
| Event-free | 50 (16%) |
| Unknown | 138 (44%) |
| PFI | |
| Event | 227 (72%) |
| Event-free | 87 (28%) |
The RNA-seq datasets examined 56,963 annotated genomic features (terminology “genes” used in the following for simplicity). To remove low expressing genes, 31,375 genes (55% of all genes) that had ≤ 10 counts in 90% of the samples were excluded, leaving 25,588 genes for the analyses. To identify those genes that were differentially expressed between recurrent and non-recurrent patients with BLBC, 18 patients who had progression events within 2 years were compared to 40 patients who had no progression events for at least 5 years using three RNA-seq specific analysis methods: DESeq2, edgeR and voom/limma.
DESeq2 identified 3295 (13%) genes as differentially expressed with a p value < 0.05. Using Benjamini & Hochberg FDR adjustment, 307 genes remained significant with an adjusted p value < 0.05. edgeR analysis identified 3296 (13%) differentially expressed genes (p < 0.05), and 343 genes remained significant (adjusted p < 0.01) after Benjamini & Hochberg FDR adjustment. The voom/limma method appears to be the most conservative in this analysis, identifying 1152 and 228 genes as differentially expressed with p < 0.05 and p < 0.01, respectively; no genes were statistically significant (adjusted p < 0.05) after Benjamini & Hochberg FDR adjustment. To avoid the bias of one method over the other when building the prognostic signatures, an approximately similar number of differentially expressed genes for each method were obtained as guided by differing thresholds for p-value or adjusted p-value. Gene sets from the three analysis methods (n = 307, 343 and 228 from DESeq2, edgeR and voom/limma, respectively) were intersected to derive common differentially expressed genes for building prognostic gene signatures. 63, 58 and 21 genes were commonly differentially expressed by DESeq2/edgeR, DESeq2/limma and edgeR/limma analysis, respectively (Fig. 1). Due to the fewer number of common genes between edgeR and limma analyses and no differentially expressed genes by voom/limma after FDR adjustment, only the 63-gene set was examined further for its prognostic ability in BLBC and HGSOC. Annotation information and differential expression analysis results for the 63 genes are available in Supplemental Table S1.
Fig. 1.
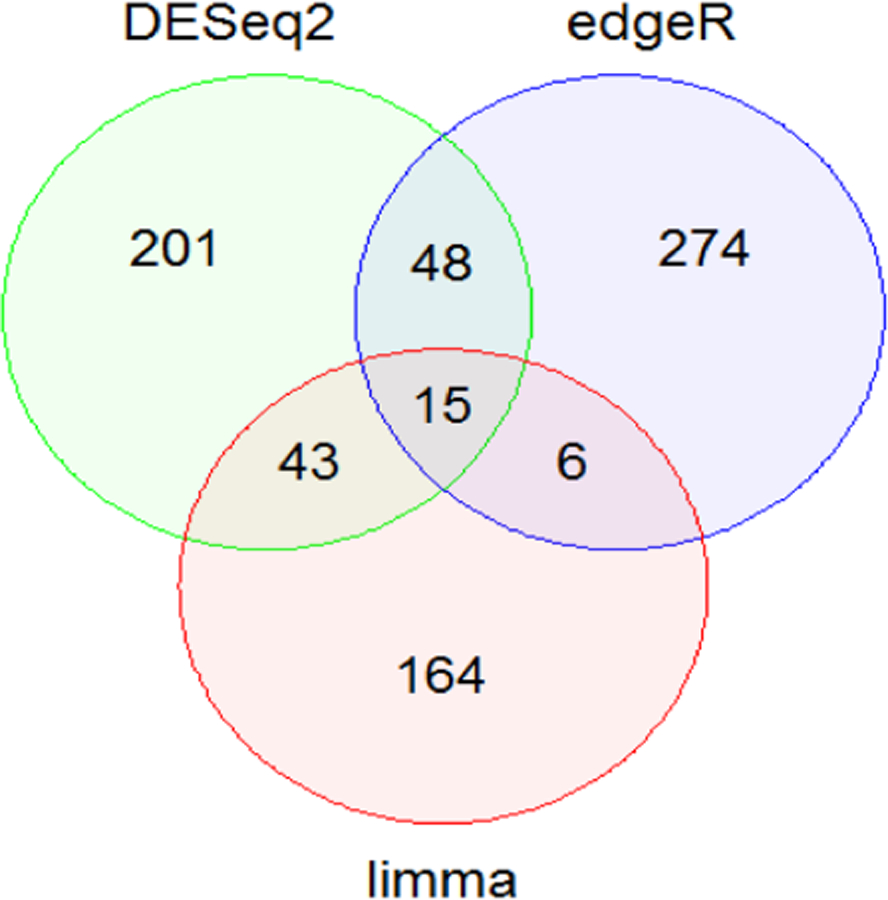
Common differentially expressed genes among the three RNA-seq analytic methods
A prognostic gene signature was developed based on the 63 genes and the RI was calculated by linear combination weighted by the Wald statistic for each gene derived from DESeq2 analysis. The cut-point for BLBC was chosen to stratify 20% of patients as the RI-Low group to ensure a minimal number of recurrent cases in this low-risk group. In the cohort of 190 BLBC patients, those classified as RI-High (n = 152) by the 63-gene signature had a statistically significantly higher risk of PFI events than those classified as RI-Low (n = 38) with no PFI events in the low-risk group (p = 0.004) (Fig. 2A). The RI remained to be statistically significant (p < 0.0001) after adjusting for age, T stage and N stage. Even in the subset of BLBC patients that excluded those used in the training and model building, the 63-gene signature remained to be prognostic with clear separation of the survival curves of the two risk groups (Fig. 2B), even though the p-value was only close to marginally significant (p = 0.057), apparently due to a limited number of events in this subset. Using the secondary endpoint of DFI, the 63-gene signature also significantly stratified all BLBC patients into different prognostic risk groups (Fig. 2C, p = 0.0023). Similarly, in the subset excluding those used in the training, the signature showed a clear trend in stratifying patients into different prognostic risk groups, even though the p-value is not statistically significant (Fig. 2D, p = 0.08). As a continuous risk score, the 63-gene signature derived RI showed increased risk of recurrence as its value increased (Fig. 3).
Fig. 2.
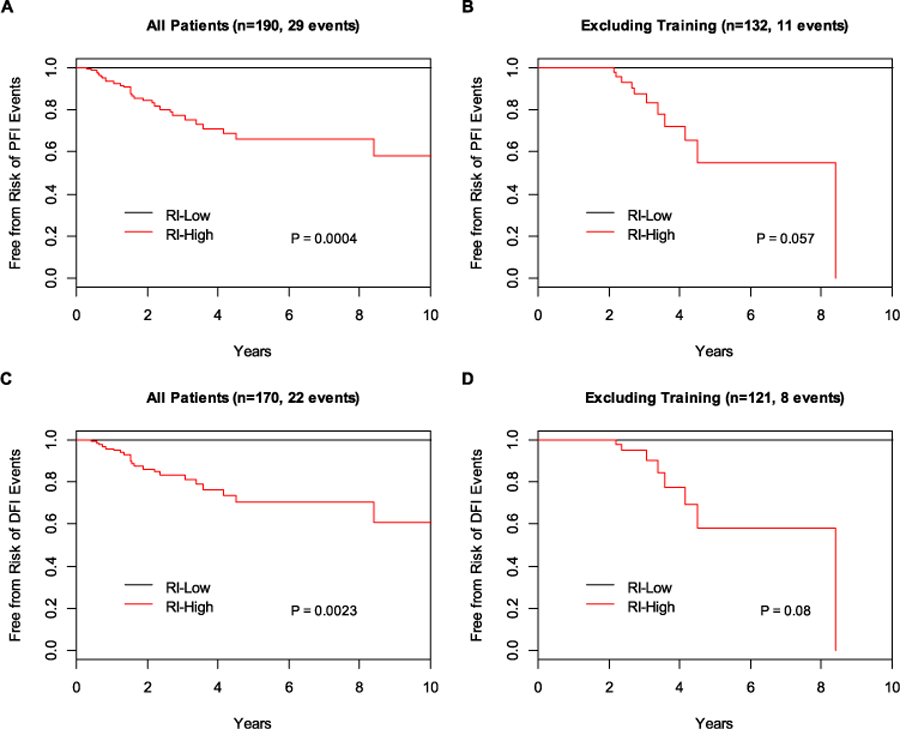
Prognostic performance of the 63-gene signature in basal-like breast cancer. A PFI of all patients; B PFI excluding patients in training set; C DFI of all patients; D DFI excluding patients in training set. RI-High high recurrence index (RI), RI-Low low recurrence index (RI)
Fig. 3.
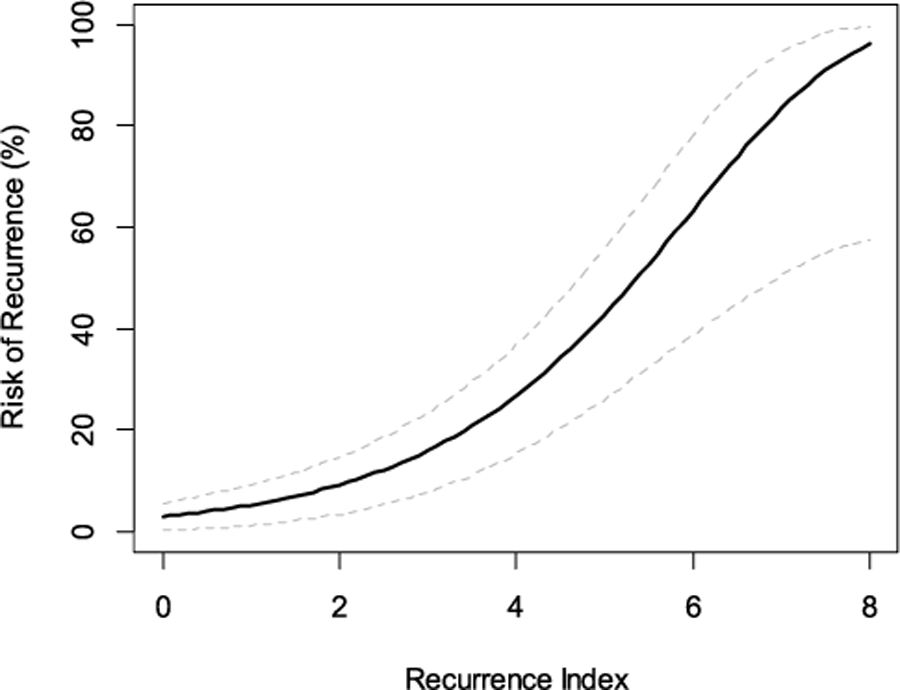
Risk of recurrence as a function of continuous risk score by the 63-gene signature in basal-like breast cancer
In the HGSOC cohort, the cut-point was chosen to classify approximately 20% of patients as clinically super-high risk. Those classified as RI-High (n = 57) by the 63-gene signature had a statistically significant higher risk of PFI events than those classified as RI-Low (n = 255) (Fig. 4A; HR: 1.49, 95% CI 1.09–2.06; p = 0.0131) and remained to be statistically significant after adjusting for age, clinical stage and grade (p = 0.0259). Using the secondary endpoint of DFI, the clinical outcome difference between the two risk groups was even larger, with those classified as RI-High having double the risk of recurrence than those classified as RI-Low (Fig. 4B; HR: 2.16, 95% CI 1.40–3.34; p = 0.0004). Even after adjusting for age, tumor grade and clinical stage, the RI remained a statistically significant prognostic factor (HR: 2.18, 95% CI 1.41–3.37; p = 0.0005) in HGSOC. RI, derived from the 63-gene signature, as a continuous risk score, showed increased risk of recurrence as its value increased (Fig. 5).
Fig. 4.
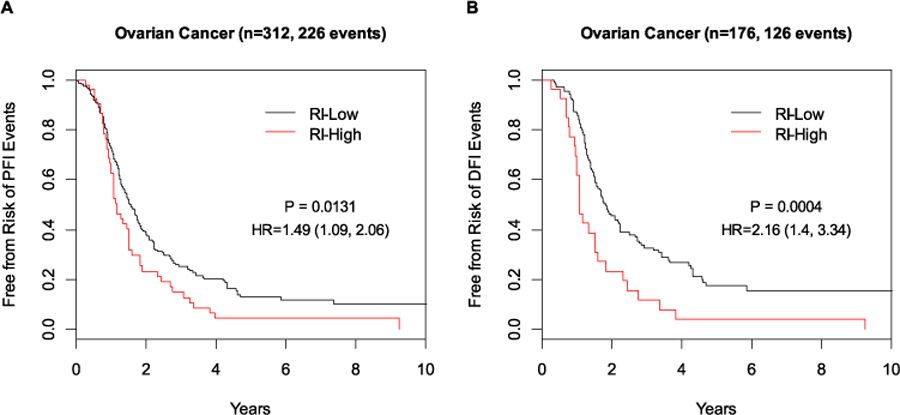
Prognostic performance of the 63-gene signature in high-grade serous ovarian cancer. A PFI; B DFI. RI-High high recurrence index (RI), RI-Low low recurrence index (RI)
Fig. 5.
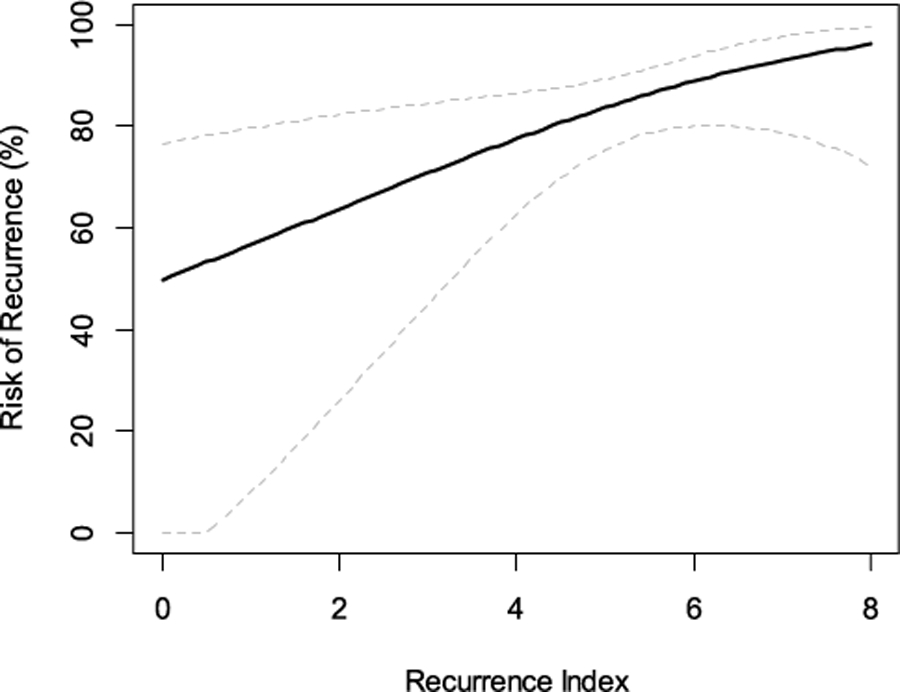
Risk of recurrence as a function of continuous risk score by the 63-gene signature in high-grade serous ovarian cancer
To further explore the biological processes underlying the prognostic gene signature, GO enrichment analysis was performed on the 63-gene signature. Forty-one out of 63 genes in the gene signature (63 total genes including 10 long non-coding RNA (lncRNA) genes and 11 unmapped genes) mapped to the human whole-genome reference database (20,996 IDs). One hundred fifty-six GO terms were found to be significantly over-represented (p < 0.05) in the prognostic gene signature with 18 of these GO terms having a p-value less than 0.01. Among the most significant GO terms are vascular endothelial growth factor signaling pathway, cell–cell signaling, and peptide hormone processing (Supplemental Table S2). The hierarchical clustering of BLBC and HGSOC datasets based on 63 genes are shown in Supplemental Figs. S1 and S2.
Discussion
In this study, we were able to develop a gene expression-based prognostic signature, based on 63 genes, to quantify the likelihood of recurrence events in BLBC. Out of the 63 genes, there were 39 protein coding genes, 10 lncRNA genes involved in bone development, DNA repair, cell adhesion, proliferation, signal transduction etc. Among the genes were known biomarkers for breast (PGF, KLK12) and ovarian cancers (PAX1, OLFM4) [33–36]. PTHLH is a marker for bone metastasis in breast cancer patients whose overexpression upregulates P2RX6 which is specific for the calcium signaling pathway [37]. Several known epigenetic markers like KLHDC7B, SCGB3A1 and PRMT8 that catalyzes the transfer of methyl groups on proteins were also present in our signature [38–40]. Some of the 63 genes in our signature have been reported to be related to both breast cancer and ovarian cancer tumors. For example, SLC5A5 was upregulated in breast cancer and was also reported as a poor prognostic factor in ovarian cancer [41, 42]. HSPB1 is involved in breast cancer causing drug resistance, and it was also reported to be associated with aggressive ovarian cancer with inherent resistance to chemotherapy [43, 44]. SCGB3A1 is a methylation marker for both breast and ovarian carcinoma [39, 45]. Recent studies show that lncRNAs play a significant role in cancer progression and may serve as an independent predictor for patient outcomes [46]. Similarly, numerous studies have reported that non-coding genes can also act as cancer driver genes by affecting gene expression [47].
The signature was able to classify 20% of BLBC patients, clinically considered high risk, as genomically low risk who experienced minimal recurrence clinically. More interestingly, this signature was also able to stratify HGSOC patients into risk groups with significantly different risks of recurrence. To our knowledge, this is the first report of a gene expression signature that showed prognostic activity in both BLBC and HGSOC. The fact that this gene panel not only can predict a lower chance of recurrence when the recurrence rate is not very high (for BLBC, c.f. PFI events in Supplemental Figure S1), but also can discern a more risky group for recurrence when the recurrence rate is high (for HGSOC, c.f. PFI events in Supplemental Figure S2), demonstrates its potential robustness in predicting different risks of recurrence depending on clinical needs.
A few of the 63 genes in our signature overlap with other reported gene signatures for BLBC or HGSOC. Finkernagel et al. reported CLEC11A in the protein signature for HGSOC recurrence [48]. PAX1 was present in the DNA methylation signature associated with serous ovarian cancer progression [35]. Another interesting observation in this study is that the majority of the recurrences in the BLBC dataset occurred prior to 3 years after diagnosis, which is consistent with a previous study on TNBC showing that the risk of recurrence peaked at 3 years and declined rapidly afterwards, and that TNBC had increased likelihood of recurrence than hormone receptor-positive breast cancer within 5 years but not thereafter [2].
Considerable effort has been invested to seek appropriate public gene expression datasets to validate the signature in BLBC and HGSOC. Unfortunately, the vast majority of the available gene expression profiling studies were based on microarray platforms; no other RNA-seq datasets with adequate clinical follow-up were found available at this time. For example, we had access to the microarray dataset (Illumina HT-12 v3) from the METABRIC project [49]. However, due to the platform difference, only 34 out of the 63 genes can be successfully mapped to the METABRIC dataset. Even with the significantly reduced number of genes, there was a clear trend of prognostic stratification of the 297 TNBC patients from the METABRIC project by the partial signature (data not shown), although statistical significance was not reached (P = 0.14).
Currently, the main treatment modality for TNBC is cytotoxic chemotherapy. While there has been some progress with new targeted therapies such as PARP inhibitors and immune check point inhibitors in TNBC, due to high disease heterogeneity, BLBC/TNBC has not seen the same level of success with targeted therapies as other cancer types [5, 6, 50]. Molecular subtyping of BLBC/TNBC and biomarkers predictive of therapeutic response are critically needed [51]. Many of the prognostic/predictive gene expression signatures developed for breast cancer have been mainly for hormone receptor-positive breast cancer; much less work has been done on BLBC or TNBC [11, 12, 20, 52, 53].
Notably, Lehmann et al. identified 6 TNBC subtypes (2 basal-like [BL1 and BL2], immunomodulatory [IM], mesenchymal [M], mesenchymal stem like [MSL], and luminal androgen receptor [LAR]) using 587 TNBC tumors from public microarray datasets [54]. These subtypes appeared to have differential responses to various therapies based on cell line models. However, validation studies of these molecular subtypes in clinical samples are still lacking. Rody et al. showed that a signature associated with the high B-cell metagene and low IL-8 metagene demonstrated prognostic ability in TNBC [55]. Similarly, Iglesia et al. found that increased metastasis-free survival is correlated with B-cell gene expression signatures, which was mainly limited to BLBC and immunoreactive ovarian cancer. Another immune-related signature of 4 genes was identified by Criscitiello et al. that showed significant association with distant recurrence-free survival in a cohort of 115 patients [56]. Al-Ejeh et al. established an 8-gene signature based on the Oncomine database that was shown to be prognostic in TNBC [57]. Hallett et al. identified a 14-gene signature from a training set of 85 BLBC patients and validated it in a small cohort of 49 patients to identify those who recurred within 5 years versus those who showed excellent long-term outcome [58]. Yau et al. developed a 5-gene predictor termed the Integrated Cytokine Score (ICS) for TNBC based on 2 previously identified signatures and confirmed its prognostic value in two public microarray datasets of 95 TNBC patients [17]. More recently, a comprehensive whole-genome sequencing study based on 254 TNBC tumors was completed. This study showed that a previously developed mutational-signature-based algorithm HRDetect for homologous recombination repair deficiency (HRD) had prognostic value in 144 TNBC patients who had received adjuvant chemotherapy [59]. In another study based on a signature of 36 genes measuring MHC class II (MHCII) pathway expression, Stewart et al. showed that in an independent cohort of 56 TNBC patients, the signature was significantly associated with longer disease-free survival [16]. Compared to these studies, the signature identified in this study is unique in that this signature classified low-risk patients with very low risk of recurrence so that they can be safely and sufficiently treated with standard chemotherapy, while those classified as high risk may be considered as candidates for new targeted treatments. We would like to note that although our signature does contain a few immune response genes (OLFM4, IGHV1–3, VSIG8), no immune signaling-related biological processes were found enriched within our signature.
A substantial proportion of ovarian cancer patients achieve complete response to initial platinum and paclitaxel-based chemotherapy; however, most of those with advanced disease will develop recurrence within 18 months [60]. Traditional prognostic factors, such as age, performance status, FIGO stage, tumor grade and initial surgery results, are insufficient to predict therapeutic response and survival. Currently, there are no biomarkers that can predict which patients will benefit from systemic first-line platinum and taxane-based chemotherapy. Thus, nearly all women are given the same regimen although they will not display the same response and have the same outcome. Like TNBC, epithelial ovarian cancer is a heterogeneous disease with each subtype harboring different genetic mutations that can be molecularly targeted for improved treatment. HGSOC is characterized by mutations in p53, BRCA1, BRCA2, NF1, CDK12, as well as abnormalities in NOTCH and FOXM1 signaling pathways. Of note, nearly all ovarian cancers that harbor deleterious mutations in BRCA1 and BRCA2 are HGSOC [61]. While PARP inhibitors have been approved for ovarian cancer patients who carry mutations in BRCA1 and BRCA2, no other predictive biomarkers have been validated for routine clinical use. Large-scale gene expression profiling studies have been performed to identify biomarker/gene signatures for responses to specific chemotherapy regimens and prognosis [62–69]. However, no multigene genomic signatures are currently commercially available either as predictive or prognostic tests. The gene expression signature discovered in this study, although preliminary in validation, has the potential to provide prognostic utility in these difficult-to-treat HGSOC patients.
Importantly, integrated analyses from TCGA Research Network, based on genomic DNA copy number, DNA methylation, exome sequencing and mRNA expression, demonstrated that BLBC/TNBC tumors and HGSOC tumors shared many molecular commonalities (TP53 mutations, RB1 and BRCA1 loss, MYC amplification, genomic instability and common copy number gains, etc.), indicating a related etiology and that common therapeutic approaches should be considered. This is further supported by the activity of platinum analogs and taxanes in both BLBC/TNBC and HGSOC [70]. Our study supported this commonality between these two cancers by, for the first time, identifying a common prognostic signature for both cancers.
Supplementary Material
Acknowledgements
We thank patients for participating in the study. Portions of this work were supported by funds from the US Department of Defense for the Breast Cancer Center of Excellence (BC-COE)/Clinical Breast Care Project (CBCP) through Uniformed Services University of the Health Sciences (HU0001–16-2–0004 Subawards 3406 and 3425). The contents of this publication are the sole responsibility of the author(s) and do not necessarily reflect the views, opinions or policies of Uniformed Services University of the Health Sciences (USUHS), The Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., the Department of Defense (DoD), the Departments of the Army, Navy, or Air Force. Mention of trade names, commercial products, or organizations does not imply endorsement by the U.S. Government.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s10549-020-05884-z) contains supplementary material, which is available to authorized users.
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. N Engl J Med 363(20):1938–1948. 10.1056/NEJMra1001389 [DOI] [PubMed] [Google Scholar]
- 2.Dent R, Trudeau M, Pritchard KI et al. (2007) Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 13(15):4429–4434. 10.1158/1078-0432.ccr-06-3045 [DOI] [PubMed] [Google Scholar]
- 3.Reddy SM, Barcenas CH, Sinha AK et al. (2018) Long-term survival outcomes of triple-receptor negative breast cancer survivors who are disease free at 5 years and relationship with low hormone receptor positivity. Br J Cancer 118(1):17–23. 10.1038/bjc.2017.379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.National Comprehensive Cancer Network Breast Cancer (Version 3.2019). https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf.
- 5.Litton JK, Rugo HS, Ettl J et al. (2018) Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med 379(8):753–763. 10.1056/NEJMoa1802905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmid P, Adams S, Rugo HS et al. (2018) Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med 379(22):2108–2121. 10.1056/NEJMoa1809615 [DOI] [PubMed] [Google Scholar]
- 7.The Gene Ontology’s Reference Genome Project: a unified framework for functional annotation across species (2009). PLoS Comput Biol 5(7):e1000431. 10.1371/journal.pcbi.1000431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Botesteanu D-A, Lee J-M, Levy D (2016) Modeling the dynamics of high-grade serous ovarian cancer progression for transvaginal ultrasound-based screening and early detection. PLoS ONE 11(6):e0156661. 10.1371/journal.pone.0156661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alsop K, Fereday S, Meldrum C et al. (2012) BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol 30(21):2654–2663. 10.1200/jco.2011.39.8545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowtell DD, Böhm S, Ahmed AA et al. (2015) Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer 15:668. 10.1038/nrc4019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paik S, Shak S, Tang G et al. (2004) A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 351(27):2817–2826. 10.1056/NEJMoa041588 [DOI] [PubMed] [Google Scholar]
- 12.van ‘t Veer LJ, Dai H, van de Vijver MJ et al. (2002) Gene expression profiling predicts clinical outcome of breast cancer. Nature 415(6871):530–536. 10.1038/415530a [DOI] [PubMed] [Google Scholar]
- 13.Sgroi DC, Sestak I, Cuzick J et al. (2013) Prediction of late distant recurrence in patients with oestrogen-receptor-positive breast cancer: a prospective comparison of the breast-cancer index (BCI) assay, 21-gene recurrence score, and IHC4 in the TransATAC study population. Lancet Oncol 14(11):1067–1076. 10.1016/S1470-2045(13)70387-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paik S, Tang G, Shak S et al. (2006) Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol 24(23):3726–3734. 10.1200/jco.2005.04.7985 [DOI] [PubMed] [Google Scholar]
- 15.Goss PE, Ingle JN, Martino S et al. (2005) Randomized trial of letrozole following tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: updated findings from NCIC CTG M.A17. JNCI 97(17):1262–1271. 10.1093/jnci/dji250 [DOI] [PubMed] [Google Scholar]
- 16.Stewart RL, Updike KL, Factor RE et al. (2019) A multigene assay determines risk of recurrence in patients with triple-negative breast cancer. Can Res 79(13):3466–3478. 10.1158/0008-5472.can-18-3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yau C, Sninsky J, Kwok S et al. (2013) An optimized five-gene multi-platform predictor of hormone receptor negative and triple negative breast cancer metastatic risk. Breast Cancer Res 15(5):R103. 10.1186/bcr3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iglesia MD, Vincent BG, Parker JS et al. (2014) Prognostic B-cell signatures using mRNA-Seq in patients with subtype-specific breast and ovarian cancer. Clin Cancer Res 20(14):3818–3829. 10.1158/1078-0432.ccr-13-3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Lichtenberg T, Hoadley KA et al. (2018) An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173(2):400–416e411. 10.1016/j.cell.2018.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parker JS, Mullins M, Cheang MCU et al. (2009) Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 27(8):1160–1167. 10.1200/jco.2008.18.1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11(3):R25. 10.1186/gb-2010-11-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durinck S, Spellman PT, Birney E et al. (2009) Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4(8):1184–1191. 10.1038/nprot.2009.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCarthy DJ, Chen Y, Smyth GK (2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40(10):4288–4297. 10.1093/nar/gks042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Law CW, Chen Y, Shi W et al. (2014) voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15(2):R29. 10.1186/gb-2014-15-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Symth GK (2005) limma: linear models for microarray data. Bioinformatics and computational biology solutions using R and bioconductor. Springer, New York. 10.1007/0-387-29362-0_23 [DOI] [Google Scholar]
- 27.Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc 57(1):289–300 [Google Scholar]
- 28.Thomas PD, Kejariwal A, Campbell MJ et al. (2003) PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res 31(1):334–341. 10.1093/nar/gkg115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fisher RA (1992) Statistical methods for research workers. In: Kotz S, Johnson NL (eds) Breakthroughs in statistics springer. Series in statistics (perspectives in statistics). Springer, New York. 10.1007/978-1-4612-4380-9_6 [DOI] [Google Scholar]
- 30.Wald A (1945) Sequential tests of statistical hypotheses. Ann Math Statist 16(2):117–186. 10.1214/aoms/1177731118 [DOI] [Google Scholar]
- 31.Kaplan EL, Meier P (1958) Nonparametric estimation from incomplete observations. J Am Statist Assoc 53(282):457–481. 10.1080/01621459.1958.10501452 [DOI] [Google Scholar]
- 32.Cox DR (1972) Regression models and life-tables. J Roy Stat Soc 34(2):187–220 [Google Scholar]
- 33.Yousef GM, Magklara A, Diamandis EP (2000) KLK12 Is a novel serine protease and a new member of the human kallikrein gene family—differential expression in breast cancer. Genomics 69(3):331–341. 10.1006/geno.2000.6346 [DOI] [PubMed] [Google Scholar]
- 34.Maae E, Olsen DA, Steffensen KD et al. (2012) Prognostic impact of placenta growth factor and vascular endothelial growth factor A in patients with breast cancer. Breast Cancer Res Treat 133(1):257–265. 10.1007/s10549-012-1957-0 [DOI] [PubMed] [Google Scholar]
- 35.Keita M, Wang Z-Q, Pelletier J-F et al. (2013) Global methylation profiling in serous ovarian cancer is indicative for distinct aberrant DNA methylation signatures associated with tumor aggressiveness and disease progression. Gynecol Oncol 128(2):356–363. 10.1016/j.ygyno.2012.11.036 [DOI] [PubMed] [Google Scholar]
- 36.Ma H, Tian T, Liang S et al. (2016) Estrogen receptor-mediated miR-486–5p regulation of OLFM4 expression in ovarian cancer. Oncotarget 7(9):10594–10605. 10.18632/oncotarget.7236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson RW, Sun Y, Ho PWM et al. (2018) Parathyroid hormone-related protein negatively regulates tumor cell dormancy genes in a PTHR1/cyclic AMP-independent manner. Front Endocrinol 1:1. 10.3389/fendo.2018.00241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeong G, Bae H, Jeong D et al. (2018) A Kelch domain-containing KLHDC7B and a long non-coding RNA ST8SIA6-AS1 act oppositely on breast cancer cell proliferation via the interferon signaling pathway. Sci Rep 8(1):12922. 10.1038/s41598-018-31306-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Q, Lothe RA, Ahlquist T et al. (2007) DNA methylation profiling of ovarian carcinomas and their in vitro models identifies HOXA9, HOXB5, SCGB3A1, and CRABP1 as novel targets. Mol Cancer 6(1):45. 10.1186/1476-4598-6-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Y, Bedford MT (2013) Protein arginine methyltransferases and cancer. Nat Rev Cancer 13(1):37–50. 10.1038/nrc3409 [DOI] [PubMed] [Google Scholar]
- 41.Tazebay UH, Wapnir IL, Levy O et al. (2000) The mammary gland iodide transporter is expressed during lactation and in breast cancer. Nat Med 6(8):871–878. 10.1038/78630 [DOI] [PubMed] [Google Scholar]
- 42.Riesco-Eizaguirre G, Leoni SG, Mendiola M et al. (2014) NIS mediates iodide uptake in the female reproductive tract and is a poor prognostic factor in ovarian cancer. J Clin Endocrinol Metab 99(7):E1199–E1208. 10.1210/jc.2013-4249 [DOI] [PubMed] [Google Scholar]
- 43.Oesterreich S, Weng C-N, Qiu M et al. (1993) The small heat shock protein hsp27 is correlated with growth and drug resistance in human breast cancer cell lines. Can Res 53(19):4443–4448 [PubMed] [Google Scholar]
- 44.Langdon SP, Rabiasz GJ, Hirst GL et al. (1995) Expression of the heat shock protein HSP27 in human ovarian cancer. Clin Cancer Res 1(12):1603–1609 [PubMed] [Google Scholar]
- 45.Verschuur-Maes AHJ, de Bruin PC, van Diest PJ (2012) Epigenetic progression of columnar cell lesions of the breast to invasive breast cancer. Breast Cancer Res Treat 136(3):705–715. 10.1007/s10549-012-2301-4 [DOI] [PubMed] [Google Scholar]
- 46.Gupta RA, Shah N, Wang KC et al. (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464(7291):1071–1076. 10.1038/nature08975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taft RJ, Pang KC, Mercer TR et al. (2010) Non-coding RNAs: regulators of disease. J Pathol 220(2):126–139. 10.1002/path.2638 [DOI] [PubMed] [Google Scholar]
- 48.Finkernagel F, Reinartz S, Schuldner M et al. (2019) Dual-platform affinity proteomics identifies links between the recurrence of ovarian carcinoma and proteins released into the tumor microenvironment. Theranostics 9(22):6601–6617. 10.7150/thno.37549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Curtis C, Shah SP, Chin SF et al. (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486(7403):346–352. 10.1038/nature10983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robson M, Im S-A, Senkus E et al. (2017) Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med 377(6):523–533. 10.1056/NEJMoa1706450 [DOI] [PubMed] [Google Scholar]
- 51.Hassan S, Esch A, Liby T et al. (2017) Pathway-enriched gene signature associated with 53BP1 response to PARP inhibition in triple-negative breast cancer. Mol Cancer Ther 16(12):2892–2901. 10.1158/1535-7163.mct-17-0170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jerevall PL, Ma XJ, Li H et al. (2011) Prognostic utility of HOXB13: IL17BR and molecular grade index in early-stage breast cancer patients from the Stockholm trial. Br J Cancer 104(11):1762–1769. 10.1038/bjc.2011.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Filipits M, Rudas M, Jakesz R et al. (2011) A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin Cancer Res 17(18):6012–6020. 10.1158/1078-0432.ccr-11-0926 [DOI] [PubMed] [Google Scholar]
- 54.Lehmann BD, Bauer JA, Chen X et al. (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Investig 121(7):2750–2767. 10.1172/jci45014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rody A, Karn T, Liedtke C et al. (2011) A clinically relevant gene signature in triple negative and basal-like breast cancer. Breast Cancer Res 13(5):R97–R97. 10.1186/bcr3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Criscitiello C, Bayar MA, Curigliano G et al. (2017) A gene signature to predict high tumor-infiltrating lymphocytes after neoadjuvant chemotherapy and outcome in patients with triple-negative breast cancer. Ann Oncol 29(1):162–169. 10.1093/annonc/mdx691 [DOI] [PubMed] [Google Scholar]
- 57.Al-Ejeh F, Simpson PT, Sanus JM et al. (2014) Meta-analysis of the global gene expression profile of triple-negative breast cancer identifies genes for the prognostication and treatment of aggressive breast cancer. Oncogenesis 3(4):e100–e100. 10.1038/oncsis.2014.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hallett RM, Dvorkin-Gheva A, Bane A et al. (2012) A gene signature for predicting outcome in patients with basal-like breast cancer. Sci Rep 2:227–227. 10.1038/srep00227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Staaf J, Glodzik D, Bosch A et al. (2019) Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study. Nat Med 25(10):1526–1533. 10.1038/s41591-019-0582-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jayson GC, Kohn EC, Kitchener HC et al. (2014) Ovarian cancer. The Lancet 384(9951):1376–1388. 10.1016/s0140-6736(13)62146-7 [DOI] [PubMed] [Google Scholar]
- 61.Bell D, Berchuck A, Birrer M et al. (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474(7353):609–615. 10.1038/nature10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spentzos D, Levine DA, Kolia S et al. (2005) Unique gene expression profile based on pathologic response in epithelial ovarian cancer. J Clin Oncol 23(31):7911–7918. 10.1200/jco.2005.02.9363 [DOI] [PubMed] [Google Scholar]
- 63.Bild AH, Yao G, Chang JT et al. (2006) Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 439(7074):353–357. 10.1038/nature04296 [DOI] [PubMed] [Google Scholar]
- 64.Lage H, Denkert C (2007) In: Dietel M (ed) Resistance to chemotherapy in ovarian carcinoma. 10.1007/978-3-540-46091-6_6 [DOI] [PubMed] [Google Scholar]
- 65.Jazaeri AA, Awtrey CS, Chandramouli GVR et al. (2005) Gene expression profiles associated with response to chemotherapy in epithelial ovarian cancers. Clin Cancer Res 11(17):6300–6310. 10.1158/1078-0432.ccr-04-2682 [DOI] [PubMed] [Google Scholar]
- 66.Hinchcliff E, Paquette C, Roszik J et al. (2019) Lymphocyte-specific kinase expression is a prognostic indicator in ovarian cancer and correlates with a prominent B cell transcriptional signature. Cancer Immunol Immunother 68(9):1515–1526. 10.1007/s00262-019-02385-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hartmann LC, Lu KH, Linette GP et al. (2005) Gene sexpression profiles predict early relapse in ovarian cancer after platinum-paclitaxel chemotherapy. Clin Cancer Res 11(6):2149–2155. 10.1158/1078-0432.ccr-04-1673 [DOI] [PubMed] [Google Scholar]
- 68.Sabatier R, Finetti P, Bonensea J et al. (2011) A seven-gene prognostic model for platinum-treated ovarian carcinomas. Br J Cancer 105:304. https://doi.org/10.1038/bjc.2011.219. https://www.nature.com/articles/bjc2011219#supplementary-information [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Le Page C, Ouellet V, Quinn MCJ et al. (2008) BTF4/BTNA3.2 and GCS as candidate mRNA prognostic markers in epithelial ovarian cancer. Cancer Epidemiol Biomark Prev 17(4):913–920. 10.1158/1055-9965.epi-07-0692 [DOI] [PubMed] [Google Scholar]
- 70.Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490(7418):61–70. 10.1038/nature11412 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.