

Abstract
Quantification of alternative splicing to detect the abundance of differentially spliced isoforms of a gene in total RNA can be accomplished via RT-PCR using both quantitative real-time and semi-quantitative PCR methods. These methods require careful PCR primer design to ensure specific detection of particular splice isoforms. We will also describe analysis of alternative splicing using a splicing “minigene” in mammalian cell tissue culture to facilitate investigation of the regulation of alternative splicing of a particular exon of interest.
Keywords: Alternative splicing, RNA, RT-PCR, variable exon, minigene, splicing factors, splicing regulation
1. Introduction
Alternative splicing is a key cellular process whereby particular combinations of exons in a nascently transcribed pre-mRNA are either included or excluded to generate different mature mRNA splice isoform transcripts, resulting in multiple protein isoforms encoded by a single gene. It is estimated that nearly all protein-coding genes in the human genome are alternatively spliced, providing an essential source of protein diversity[1, 2]. Differentially spliced isoforms may exert distinct biological functions, therefore accurate quantification of the relative amounts of splice isoforms of a gene is essential to explore the role of alternative splicing in biological processes as well as disease[3]. The pre-mRNA of an alternative spliced gene contains both constitutive exons and variable exons interspaced by introns that are ultimately spliced out and excluded from the final mRNA transcript. In this chapter, we will describe the characterization of exon-skipping, the most common form of alternative splicing, where one or more variable exons are either included or skipped from the final transcript to make up the array of spliced isoforms of a gene[4].
Reverse Transcription-Polymerase Chain Reaction (RT-PCR) is one of the most convenient methods for studying RNA transcripts and can generally be used with total RNA extracted from any biological source. Quantitative PCR (qPCR) following RT is often the method of choice given its extreme sensitivity of detection and accurate comparison of amplification of PCR products during the exponential phase of the PCR reaction[5]. Semi-quantitative methods are less useful for precise quantification of the relative abundance of splice isoforms, however, combined with electrophoresis, these methods allow for the direct visualization and comparison of splice isoform abundance based on size disparity between differentially spliced transcripts.
Quantification of levels of different splice isoforms is especially useful in studying the regulation of alternative splicing. Regulation of alternative splicing is accomplished by the recognition of cis-acting sequences in the pre-mRNA, usually located in the variable exonic or adjacent intronic sequences, by trans-acting RNA-binding proteins known as splicing factors[6]. In this protocol, we make use of a splicing “minigene” construct to study the regulation of alternative splicing of a variable exon within an alternatively spliced gene[7]. This minigene then offers a versatile tool for studying the regulation of splicing of a particular exon as it can be expressed in cell-culture along with potential splicing factors that could influence variable exon inclusion or skipping.
2. Materials
2.1. RNA Source Material
Alternative splicing can be examined using RNA from any source. In this protocol we use RNA extracted from mammalian cells grown in tissue-culture.
2.2. RNA Extraction and RT
(1). E.Z.N.A. ® Total RNA Isolation Kit (Omega Bio-Tek)
(2). GoScript™ Reverse Transcriptase Reagents (Promega)
2.3. qPCR
(1). GoTaq ® Green Master Mix (Promega)
(2). Primers for specific splice isoform detection and a housekeeping gene. As an example, primers for the detection of CD44 splice isoforms and housekeeping gene TBP are included in Table 1
Table 1:
qPCR Primers for the analysis of CD44 alternative splicing
| Primer Name | Forward (5'-3') | Reverse (5'-3') |
|---|---|---|
| CD44v with v5/v6 exons | GTAGACAGAAATGGCACCAC | CAGCTGTCCCTGTTGTCGAA |
| CD44s | TACTGATGATGACGTGAGCA | GAATGTGTCTTGGTCTCTGGT |
| human TBP | GGAGAGTTCTGGGATTGTAC | CTTATCCTCATGATTACCGCAG |
2.4. Semi-Quantitative PCR
(1). HotStarTaq Plus DNA Polymerase Reagents (Qiagen)
(2). Primers for detecting minigene splice isoforms. As an example, primers for the detection of a CD44 variable exon 8 (v8) minigene are included in Table 2
Table 2:
Primers for the analysis of CD44 v8 minigene alternative splicing
| Primer Name | Forward (5'-3') | Reverse (5'-3') |
|---|---|---|
| qPCR v8 minigene inclusion | CAATGACAACGCTGGCACAA | CCAGCGGATAGAATGGCGCCG |
| qPCR v8 minigene skipping | GAGGGATCCGGTTCCTGCCCC | CAGTTGTGCCACTTGTGGGT |
| semi-quantitative PCR v8 | GAGGGATCCGGTTCCTGCCCC | CCAGCGGATAGAATGGCGCCG |
(3). 0.5x TBE buffer (45 mM Tris, 45 mM boric acid, 1 mM EDTA) for electrophoresis
(4). 1.5% Agarose gel prepared with 0.5x Tris-borate-EDTA (TBE) buffer and 0.5 μg/mL ethidium bromide
2.5. Co-Transfection of Splicing Minigene and Splicing Factors
(1). Transfectable mammalian cell line, such as HEK293T (ATCC® CRL-3216™)
(2). Gibco® Dulbecco’s Modified Eagle Medium High Glucose (plain media as well as media supplemented with L-Glutamine and 10% Fetal Bovine Serum)
(3). Lipofectamine 2000 Reagents (Invitrogen)
(4). Mammalian expression plasmids encoding splicing minigene and splicing factors of interest. As an example, we use a CD44 v8 minigene plasmid, splicing factor hnRNPM plasmid, and pcDNA3 control plasmid for co-transfection
2.6. Equipment
(1). Thermo Fisher Scientific Nanodrop 1000 Spectrophotometer for quantification of RNA
(2). A thermal cycler, such as the BioRad DNA Engine Tetrad® 2, for Reverse Transcriptase reaction and semi-quantitative PCR
(3). A real-time thermal cycler such as the Roche Lightcycler® 480 II System with associated LightCycler software for qPCR
(4). Horizontal electrophoresis apparatus
(5). A UV Transilluminator with Camera such as the BioRad Gel Doc XR System with Quantity One 1-D Analysis Software for visualization and densitometric analysis of ethidium-bromide stained DNA
(6). A tabletop centrifuge such as the Eppendorf Centrifuge 5424
3. Methods
Prepare all reactions with molecular grade nuclease-free water to prevent RNA degradation.
3.1. RNA Extraction and Purification
(1). Total RNA from mammalian cells in tissue culture is extracted using the E.Z.N.A.® Total RNA Isolation Kit (Omega Bio-Tek) by following the product manual (see Note 1). For RNA isolation via this kit, cells can be directly lysed using the provided lysis buffer.
(2). As per the manufacturer protocol, first collect cells in 350 μL RNA lysis buffer.
(3). Add 350 μL of 70% ethanol to the lysate and mix thoroughly. Then transfer the sample to the RNA purification column.
(4). Centrifuge at 10,000 x g for 1 min and discard flow-through.
(5). Wash the column once with 500 μL of RNA wash buffer I and twice with RNA wash buffer II, centrifuging at 10,000 x g for 1 min between each wash and discarding flow-through.
(6). Remove residual RNA wash buffer II from the column by centrifugation at maximum speed for 2 min.
(7). Transfer the column into a new 1.5 mL tube, add 50 μL nuclease-free water at the center of the column matrix, and elute RNA by centrifugation at the maximum speed for 1 min.
(8). Determine RNA quantity using a UV spectrometer (such as NanoDrop). High quality RNA should have a 260 nm / 280 nm absorbance ratio of approximately 2.0. RNA should be stored at −80°C for long-term storage.
3.2. RT Reaction
(1). In a reaction totaling 20 μL, combine 250-1000 ng of total RNA with 0.05 μg random hexamer primers, 50 pmol MgCl2, 10 pmol dNTPs, 2 μL 5x GoScript™ Buffer, and 1μL of GoScript™ Reverse Transcriptase (see Note 2). Mix by vortex and briefly centrifuge.
(2). Incubate reaction mixture 25°C for 5 minutes for primer annealing, 42°C for 60 minutes for the RT reaction, and 70°C for 5 minutes to inactivate the RT enzyme. These incubations are best performed in a programmable thermocycler. Completed reactions may now be frozen at −20°C for long-term storage.
3.3. Primer Design for qPCR
(1). In this protocol we will provide an example to detect the most common form of alternative splicing, exon skipping. In a hypothetical four-exon pre-mRNA (Fig. 1A), exon 3 is a variable exon that is either included in the mature mRNA to generate a longer isoform or skipped to generate a shorter isoform. Three primers are designed to differentiate one splice isoform from another. A forward primer lies within the variable exon 3 with a reverse primer in the immediately downstream constitutive exon 4. These primers allow the selective amplification of the longer isoform with variable exon 3 included. To detect the shorter isoform with the variable exon 3 skipped, a forward primer is designed that spans the junction between exon 2 and 4. The junction between exon 2 and 4 is disrupted with exon 3 inclusion. Thus, together with the corresponding reverse primer located in constitutive exon 4, this primer set only amplifies mRNA isoforms where exon 3 is skipped. When multiple consecutive variable exons exist in the gene of interest, primers can be designed in two different variable exons for detection of specific variable exon-containing isoforms (Fig. 1B).
Figure 1: Characterization of alternative splicing by qRT-PCR.
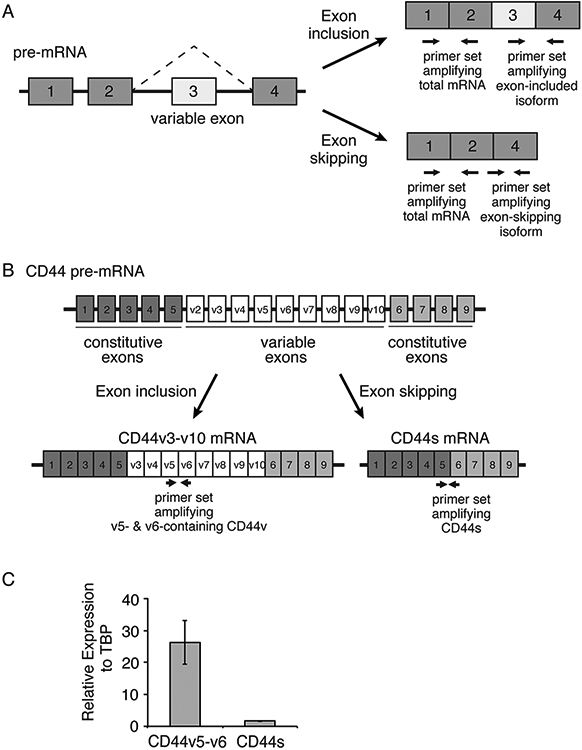
(A) A schematic illustrating a hypothetical four-exon gene with a variable exon 3 that undergoes exon-inclusion or exon-skipping. Three primer sets for qPCR indicated by the paired arrows are designed to amplify the total mRNA, the variable exon 3 inclusion isoform, or the exon-skipping isoform. (B) A schematic depicting the exon structure of the transmembrane protein CD44, with the 9 constitutive and 9 variable exons labeled. CD44v3-v10 and CD44s are predominant splice isoforms and are depicted. Paired arrows indicate primer sets to amplify CD44v v5-v6 containing isoforms and the CD44s isoform which lacks all variable exons. (C) Representative qRT-PCR results using primers in Table 1 to show the relative expression of CD44v5/v6 and CD44s, normalized to TBP, in an immortalized human mammary epithelial cell line (HMLE).
(2). It is useful to design qPCR primers that can detect all isoforms of the mRNA of interest. In this way the total expression of the gene as well as the expression of individual isoforms can be compared. In the hypothetical four-exon pre-mRNA (Fig. 1A), exons 1 and 2 are constitutive exons immediately adjacent to one another without any variable exons in between. By designing a forward primer in exon 1 and a reverse primer in exon 2, all mature mRNA isoforms generated from this pre-mRNA can be detected via PCR.
(3). Primers should also be designed to amplify a reference “housekeeping” gene that is expected to be uniformly expressed in all samples. In this protocol we use primers amplifying a segment of human TATA-binding protein (TBP) (Table 1).
(4). Primers should be designed with an ideal melting temperature (Tm) of approximately 55°C. The primer length is generally 18-22 nucleotides. Tm of each primer can be estimated using online tools such as the oligoanalyzer of Integrative DNA Technologies (http://www.idtdna.com/analyzer/applications/oligoanalyzer). The amplicon for each primer pair should be between 80-150 base pairs.
(5). As an example, we have depicted primer design to detect alternatively spliced isoforms of the CD44 gene that encodes a family of CD44 transmembrane proteins (Fig. 1B, Table 1). CD44 contains 9 constitutive exons and 9 variable exons, and in our example, primers have been designed to detect isoforms containing CD44 variable exons 5-6 (CD44v5-6) and isoforms in which all variable exons have been skipped (CD44s).
3.4. Quantification of splicing via qPCR
(1). Prepare qPCR reactions in a total volume of 20 μL by combining 0.1-1.0 μL of cDNA generated from the RT reaction with 10 μL GoTaq® Green Master Mix and 5 pmol each of forward and reverse primers. Be sure to include reactions for the reference gene.
(2). Perform triplicate qPCR reactions with 40-50 cycles of the following steps: 95°C denaturation for 10 seconds, 58°C annealing for 10 seconds, 60°C extension for 30 seconds. Include a thermal denaturing step to generate dissociation curves that can be used to verify specific amplification of PCR products (see Note 3).
(3). After the PCR is completed, the amplified signal from each reaction is presented as a threshold cycle (Ct) determined using an arbitrary detection threshold, usually set by the qPCR software to reside within the exponential range of PCR amplification. Relative quantification of splice isoforms is accomplished via comparison to expression of a reference gene such as TBP. Relative quantification assumes that the PCR amplification efficiency for each primer pair is perfect, resulting in doubling of PCR amplicons with every cycle. Since the reference gene expression remains constant between experimental samples, the quantity of mRNA for each splice isoform relative to the reference gene is calculated using the following formula: 2−ΔCt, where ΔCt = (Ct splice isoform – Ct reference mRNA). As an example, quantification of two splice isoforms of CD44 was conducted via qPCR using RNA extracted from the immortalized human mammary epithelial cell line HMLE (Fig. 1C). The primers used are in Table 1.
(4). Relative levels of splice isoforms of the same gene can also be expressed as a ratio of one splice isoform to another, for example a variable exon inclusion / variable exon skipping ratio. In such a scenario, a reference gene is not required because splice isoforms of the same gene from the same sample are compared. The ratio of splice isoforms is calculated using the following formula: 2−ΔCt, where ΔCt = (Ct inclusion splice isoform mRNA – Ct skipping splice isoform mRNA in the same sample). Relative comparison of this ratio between different experimental samples is accomplished via the formula: 2−ΔCt(experiment) / 2−ΔCt(control), where ΔCt(experiment) = (Ct inclusion splice isoform mRNA in experimental sample – Ct skipping splice isoform mRNA in experimental sample) and ΔCt(control) = (Ct inclusion splice isoform mRNA in control sample – Ct skipping splice isoform mRNA in control sample). In this way the fold change in inclusion/skipping ratio compared to the control sample is obtained.
3.5. Primer design for detection of splice isoforms via semi-quantitative PCR
Through careful primer and PCR design, differences in splice isoform expression in cDNA generated from total RNA can be visualized using agarose gel electrophoresis. Returning to a hypothetical example of a four-exon pre-mRNA, a forward primer in constitutive exon 2 and a reverse primer in constitutive exon 4 will amplify differently sized PCR amplicons depending on whether or not the template includes variable exon 3 (Fig. 2). Using semi-quantitative PCR, these primers facilitate the amplification, separation via electrophoresis, and approximate quantification of different splice isoforms. Primers should be designed with an ideal Tm of 55°C, and ideal amplicons should be no more than 300 base pairs long (see Note 4).
Figure 2: Semi-quantitative PCR for examination of exon skipping.
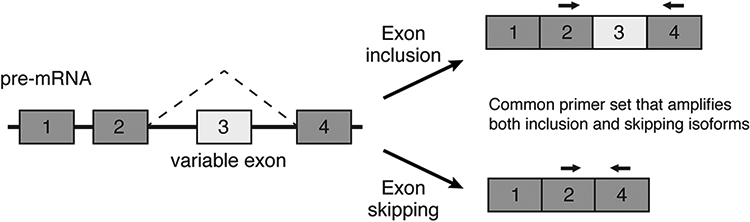
A schematic is shown depicting a hypothetical four-exon gene with variable exon 3 undergoing alternative splicing. The paired arrows indicate primer sets that flank the variable exon 3 and thus amplify both variable exon-inclusion and skipping isoforms from a cDNA template. The different splice isoforms can be distinguished by size using semi-quantitative PCR and electrophoresis.
3.6. Performing and analyzing semi-quantitative PCR
(1). In a 20 μL reaction, combine 0.1-1.0 μL cDNA with 2 μL 10x PCR Buffer (Qiagen), 4 μL 5x Q-Solution (Qiagen), 0.5 μL HotStarTaq DNA Polymerase (Qiagen), and 5 pmol each of forward and reverse primers.
(2). Complete PCR reaction using the following “hot start” program: 95°C denaturation for 5 minutes, approximately 30 cycles of 95°C for 30 seconds, 55°C for 30 seconds, 72°C for 30 seconds, and lastly 72°C for 10 minutes. Ensure that PCR is completed in the exponential range (see Note 5).
(3). Visualize PCR reactions via horizontal gel electrophoresis using a 1.5% agarose-TBE gel prepared with 0.5 μg/mL ethidium bromide (see Note 6). Detection of two distinct bands differing in size by exactly the length of the variable exon indicates a successful PCR amplification. The approximate quantity of each splice isoform present in the original RNA sample can be inferred using densitometric analysis by comparing the fluorescence intensity of the inclusion and skipping PCR amplicons using a UV transilluminator with camera and image intensity quantitation software (such as the BioRad Gel Doc XR system with Quantity One 1-D Analysis Software). Brighter intensity of the band is equivalent to higher expression of the splice isoform in the original RNA.
3.7. Generating a splicing minigene to analyze regulation of alternative splicing of a variable exon
Splicing minigenes are useful tools to interrogate the regulation of splicing of a variable exon of interest. A splicing minigene is an expression plasmid consisting of a variable exon of interest flanked by two constitutive exons. It is important to include the immediately upstream and downstream endogenous intronic sequences flanking the variable exon where potential cis-acting sequences important for determining the inclusion or skipping of the exon may be located [7] (see Note 7). As an example, we have included a CD44 v8 minigene with variable exon 8 (v8) of CD44 bounded by the adjacent 250 base pairs of its upstream and downstream introns. This sequence is flanked by two constitutive exons from the human insulin gene with functioning 5’ and 3’ splice sites. Primers for detection of exon inclusion and exon skipping were designed in accordance with the principles mentioned in Methods Sections 3.3 and 3.5 (Fig. 3A, Table 2).
Figure 3: A splicing minigene assay for characterization of exon skipping.

(A) A schematic of the CD44 v8 minigene is shown. The CD44 v8 variable exon flanked by approximately 250 bp of upstream and downstream intronic sequence is cloned between constitutive exons c1 and c2. Exons are shown as boxes and introns as lines. Primer sets in black are used for qRT-PCR reactions. The primer set in white is designed for semi-quantitative methods. (B) A schematic of a fluorescent splicing reporter containing an intronic regulatory element I-8. dsRED and EGFP are cloned after constitutive exon c2, where dsRED is in frame with c2, while the EGFP open reading frame has a +1 bp frameshift compared to dsRED. Inclusion of the variable exon produces EGFP while skipping produces dsRED. (C) qRT-PCR data after cotransfection of 100 ng CD44 v8 minigene with 400 ng of splicing factor hnRNPM using v8 inclusion and skipping primers in Table 2. A relative Inclusion/Skipping ratio (Incl/Skip) is plotted indicating that the Incl/Skip ratio decreases when hnRNPM is transfected. (D) Semi-quantitative PCR image showing decreased percent inclusion (% inclusion) after transfection with 400 ng hnRNPM using semi-quantitative v8 primers in Table 2. Both images were from the same gel with uniform exposure time. % inclusion was calculated using densitometric image analysis software to divide the pixel intensity of the inclusion band by the combined pixel intensity of both inclusion and exclusion bands. Figures 3C and 3D were modified from Figure 4 in our previous publication under Creative Commons License CC-BY-NC 4.0.
Splicing minigenes incorporating fluorescent signals can also be used for splicing analysis, especially in the context of large-scale alternative splicing screens. The RG6 minigene [8] is constructed so that either EGFP or dsRED is expressed by including or excluding an exon with a length of 3n+1 bp, respectively. Exon skipping results in the production of an in frame dsRED, while exon inclusion creates a +1 bp frameshift, producing EGFP in frame. The mutually exclusive nature of dsRED and EGFP expression provides a sensitive and quantitative readout for alternative splicing, suitable for large-scale screening. As an example shown in Fig 3B, the splicing minigene contains a 28-bp artificial exon. To determine the splicing effect of an intronic regulatory element I-8, originally located within the downstream intron of CD44 v8 exon, the I-8 sequence element was inserted into the intron downstream of the alternative exon [9, 10]. Exon skipping or inclusion leads to mutually exclusive expression of dsRED or EGFP, respectively, which can then be quantified through imaging or flow cytometry.
3.8. Investigating the regulation of alternative splicing via cotransfection of a splicing minigene and splicing factors
(1). As the splicing minigene contains native cis-acting splicing regulatory sequences within or near the variable exon, trans-acting RNA binding proteins are capable of modulating inclusion or skipping of the variable exon in the minigene in a transfectable cell model. As an example we will describe co-transfection of the CD44 v8 minigene plasmid and a plasmid expressing splicing factor hnRNPM in the HEK293T mammalian cell line. In this case, hnRNPM promotes exon-skipping of the CD44 v8 minigene [11] (Fig. 3C and 3D).
(2). In a tissue culture hood using aseptic technique, plate 2.25 x 105 HEK293T cells in 24 well tissue culture plates using DMEM containing 10% FBS (see materials).
(3). 24 hours after seeding cells, at which time the cells are approximately 90% confluent, conduct transfections using Lipofectamine 2000 (Invitrogen) (see Note 8). Per well of a 24 well plate, combine 1.5 μL Lipofectamine 2000 with 50 μL plain DMEM (see Note 9) and incubate 5 minutes at room temperature.
(4). Combine Lipofectamine and media with transfecting plasmids diluted in 50 μL plain DMEM and mix well. As an example, transfect all wells with 100 ng CD44 v8 minigene, one well with a control plasmid such as pcDNA3, and other wells with splicing factors of interest such as hnRNPM. Each well should be transfected with a total of 800 ng of plasmid, so add in the remaining DNA with a control vector such as pcDNA3 (for example, a well could be transfected with 100 ng CD44 v8 minigene, 400 ng hnRNPM, and 300 ng pcDNA3). Incubate mixture 20 minutes at room temperature.
(5). During the above incubation, replace media on HEK293T cells with fresh DMEM containing 10% FBS.
(6). Add lipofectamine-transfectant mixture dropwise slowly over cells and incubate at 37°C for 24 hours (see Note 10).
(7). Collect cells using RNA extraction protocol detailed in Methods section 3.1.
(8). As an example, qRT-PCR data (Fig. 3C) and semi-quantitative RT-PCR data (Fig. 3D) from the CD44 v8 splicing minigene experiments were collected using primers in Table 2 [11] (see Note 11).
Acknowledgement
This work was supported by research grants from the US National Institutes of Health (R01 CA182467, R35GM131876) to CC. C.C. is a Cancer Prevention and Research Institute of Texas Scholar in Cancer Research (RR160009).
Footnotes
Using the E.Z.N.A.® Total RNA Isolation Kit (Omega Bio-Tek), genomic DNA contamination is minimal. In addition, as described in the Methods section, the primers are designed in different exons. This would eliminate PCR products amplified from genomic DNA which contains long intronic sequences. Therefore, the RNA may be used directly for RT-PCR analysis. If genomic DNA contamination is a concern, the manufacturer provides instructions for an on-column DNase treatment protocol. Also, during the elution step, we elute with RNAse-free H2O. If DEPC-treated H2O is used as an eluent, it can interfere with quantification of RNA via UV spectrometry [12].
Depending on the expression level of the gene of interest and the amount of different splice isoforms in the mRNA, the total input RNA into the Reverse Transcriptase reaction can be adjusted. Usually 250 ng of total RNA is sufficient, however up to 1 μg of RNA may be added to the reaction to increase the cDNA concentration without saturating the Reverse Transcriptase during cDNA synthesis.
The thermal denaturing step in qPCR is critical to determine the specific amplification of the target PCR product. The thermal denaturation curve for each pair of primers per reaction should show a single isolated peak with a uniform melting temperature. If multiple peaks with different melting temperatures are observed, then non-specific amplification is occurring and the PCR reaction must be optimized before qPCR results can be analyzed[5]. Redesigning primers and adjusting the annealing temperature of the reaction may be necessary. We also recommend electrophoresing the PCR products on an agarose gel and visualizing the product using ethidium-bromide under UV. If amplification is specific, a single band of the appropriate size should be observed.
Designing primers so that the PCR amplicons for each splice isoform are relatively small (ideally 80-300 base pairs) ensures that both the large and small amplicons are amplified with similar efficiency during the PCR reaction. Automated tools to design primers include PrimerSeq (primerseq.sourceforge.net) for semiquantitative PCR design and Primer-Blast for splice junction design (https://www.ncbi.nlm.nih.gov/tools/primer-blast/)[13, 14].
Accuracy of semi-quantitative PCR for visualizing the relative amounts of splice isoforms is dependent on the number of PCR cycles. The relative quantities of splice isoforms in a sample of mRNA remain proportional during the exponential phase of a PCR reaction, but this proportional relationship is less accurate in later PCR cycles once the reaction reaches the linear and plateau phases as PCR reagents and the polymerase are exhausted[5]. This is the reason qPCR offers more accurate quantification of expression compared to semi-quantitative PCR. It is important to perform a series of PCR cycles to identify the range of cycles where the proportional intensity of the different splice isoform bands remains constant [15]. This range also depends on the expression level of the gene in the original RNA sample. It is also recommended to conduct 32P-labeled PCR by adding 32P-labeled dCTP to the PCR reaction to increase the sensitivity of detecting bands on a gel. In 32P-labeled PCR, the total PCR cycle number can be reduced to approximately 20 cycles to ensure that results are captured during the exponential phase of the reaction.
To improve the resolution of PCR product bands, it may be necessary to increase to the concentration of the agarose gel to 2%. Polyacrylamide gel electrophoresis (6%) may also be used to resolve even smaller PCR products or resolve splice isoform bands that differ by a small size.
Splicing minigenes using constitutive exons not derived from the gene of interest offer a convenient tool for cloning different variable exons of interest into the same minigene construct. However, the actual exons flanking the variable exon may also be included in the minigene to better approximate the genomic cis-acting sequences present for native splicing of the gene. These cis-elements are located in both the variable exon of interest as well as adjacent introns[6].
When using the transfection reagent Lipofectamine 2000 (Invitrogen), it is essential that cells be at a high confluency of at least 90%. This is to ensure that cells are still dividing to maximize uptake of transfected DNA while minimizing the impact of cell death that can occur during transfection.
Do not use DMEM containing serum when preparing transfection mixtures with the transfection reagent and transfected DNA. Serum proteins can interfere with the formation of the DNA-lipid transfection complexes and reduce transfection efficiency.
Although 24 hours is usually sufficient to observe efficient expression of transfected DNA, cells can be incubated for up to 48 hours to obtain even higher transgene expression.
To confirm protein transgene expression, for example transfected splicing factor expression constructs, Western blot analysis using cell lysates and an antibody specific for the protein of interest can be conducted. Efficient expression should result in an intense band on Western blot compared to control.
References
- 1.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB (2008) Alternative isoform regulation in human tissue transcriptomes. Nature 456 (7221):470–476. doi: 10.1038/nature07509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature Genetics 40:1413–1415 . 10.1038/ng.259 [DOI] [PubMed] [Google Scholar]
- 3.Liu S, Cheng C (2013) Alternative RNA splicing and cancer. Wiley Interdisciplinary Reviews: RNA 4:547–566 . 10.1002/wrna.1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keren H, Lev-Maor G, Ast G (2010) Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 11:345–355 . 10.1038/nrg2776 [DOI] [PubMed] [Google Scholar]
- 5.Freeman WM, Walker SJ, Vrana KE (1999) Quantitative RT-PCR: pitfalls and potential. BioTechniques 26:112–122, 124–125 . 10.2144/99261rv01 [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Burge CB (2008) Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 14:802–813 . 10.1261/rna.876308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper TA (2005) Use of minigene systems to dissect alternative splicing elements. Methods 37:331–340 . 10.1016/j.ymeth.2005.07.015 [DOI] [PubMed] [Google Scholar]
- 8.Orengo JP, Bundman D, Cooper TA (2006) A bichromatic fluorescent reporter for cell-based screens of alternative splicing. Nucleic Acids Res 34:e148 . 10.1093/nar/gkl967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang H, Zhang J, Harvey SE, Hu X, Cheng C (2017) RNA G-quadruplex secondary structure promotes alternative splicing via the RNA-binding protein hnRNPF. Genes Dev 31:2296–2309 . 10.1101/gad.305862.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Harvey SE, Cheng C (2019) A high-throughput screen identifies small molecule modulators of alternative splicing by targeting RNA G-quadruplexes. Nucleic Acids Res 47:3667–3679 . 10.1093/nar/gkz036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Y, Gao XD, Lee J-H, Huang H, Tan H, Ahn J, Reinke LM, Peter ME, Feng Y, Gius D, Siziopikou KP, Peng J, Xiao X, Cheng C (2014) Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev 28:1191–1203 . 10.1101/gad.241968.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okamoto T, Okabe S (2000) Ultraviolet absorbance at 260 and 280 nm in RNA measurement is dependent on measurement solution. Int J Mol Med 5:657–659 . 10.3892/ijmm.5.6.657 [DOI] [PubMed] [Google Scholar]
- 13.Tokheim C, Park JW, Xing Y (2014) PrimerSeq: Design and visualization of RT-PCR primers for alternative splicing using RNA-seq data. Genomics Proteomics Bioinformatics 12:105–109 . 10.1016/j.gpb.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL (2012) Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13:134 . 10.1186/1471-2105-13-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horner RM (2006) Relative RT-PCR: Determining the Linear Range of Amplification and Optimizing the Primers:Competimers Ratio. CSH Protoc 2006: . 10.1101/pdb.prot4109 [DOI] [PubMed] [Google Scholar]