

Abstract
Purpose of Review
Transthyretin amyloid cardiomyopathy (ATTR-CM) is a life-threatening disease that disproportionately affects older adults and people of African descent. This review discusses current knowledge regarding racial and ethnic disparities in the diagnosis and management of ATTR-CM.
Recent Findings
Historically, ATTR-CM was thought to be a rare cause of heart failure. Recent evidence has shown that ATTR-CM is more common among older adults, men, and people of African descent. In addition, significant geographic variation exists in the identification of amyloid cardiomyopathy. Despite the high burden of ATTR-CM among Black individuals, most clinical data for ATTR-CM are from North America and Europe. Moreover, only a minority of clinical trial participants thus far have been Black patients. In addition to racial differences, socioeconomic disparities may be further compounded by the potentially prohibitive cost and limited accessibility of disease-modifying ATTR therapies.
Summary
ATTR-CM is an important cause of heart failure that disproportionately affects people of African descent. Efforts to promote earlier identification of ATTR-CM in general practice will likely improve clinical outcomes for all groups. Future trials should strive to enroll a higher proportion of Black patients. Furthermore, enhanced efforts are warranted to improve treatment accessibility among racial and ethnic minority groups that may be more likely to be affected by ATTR-CM.
Keywords: Amyloidosis, Transthyretin, Cardiomyopathy, Racial disparities, Diagnosis, Treatment, Cardiac scintigraphy
Introduction to Amyloid Cardiomyopathy
Amyloid cardiomyopathy (CM) results when misfolded proteins aggregate to form rigid amyloid fibrils that deposit in the myocardium. [1] The amyloid infiltrates can cause damage to the heart via both direct toxicity and disruption of the normal organ architecture. The subsequent restrictive CM can lead to heart failure, arrhythmias, and sudden death. [1–5] The fibril precursor protein defines the subtype of amyloid CM, which differ in prognosis and treatment. The degree of cardiac involvement in systemic amyloidosis is often the most important predictor of prognosis. The most common types of cardiac amyloidosis are transthyretin amyloid CM (ATTR-CM) and immunoglobulin light chain amyloid CM (AL-CM). Other systemic amyloidoses, such as serum amyloid A (AA) and beta-2 microglobulin (ABeta2M), have rare cardiac involvement. [6, 7] Leukocyte cell-derived chemotaxin 2 (ALECT2) is a more recently identified form of amyloidosis that is much more common in Hispanics/Latinxs, particularly Mexican-Americans. [7] ALECT2 is most commonly associated with renal and hepatic involvement; significant cardiac involvement has not been described. [7]
AL-CM occurs in the setting of a plasma cell dyscrasia with deposition of immunoglobulin light chains in virtually any organ; deposition in the cardiovascular, renal, nervous, integumentary, and gastrointestinal/hepatic organ systems are most common. AL-CM is a rapidly progressive disease with a median survival of only 6 months if left untreated. [8–10] Multiple treatments, including chemotherapy, stem cell transplant, immunotherapy, and monoclonal antibodies, are available for its management. [8, 11] Unlike multiple myeloma which is twice as common and has a higher mortality in Black patients compared to White patients, [12] no clear gender, racial, or ethnic predominance has been reported in AL amyloidosis. However, a recent study by Staron et al. found that non-Hispanic Black patients may have more aggressive disease phenotypes. [13] They also identified lower rates of stem cell transplantation in Hispanic patients, which appeared to be driven by socioeconomic and clinical factors rather than race/ethnicity. [13]
ATTR-CM will be the primary focus of this review. This disease is caused by deposition of transthyretin (TTR), a protein secreted by the liver that transports thyroxine and retinol. The native, homotetramer form dissociates into unstable TTR monomers which misfold, aggregate, and form amyloid fibrils. ATTR-CM carries high morbidity and mortality, although the life expectancy is longer than with AL-CM with a median survival of about 3.5 years. [2] ATTR-CM can result either from a genetic mutation [variant ATTR (ATTRv)] or from deposition of wild-type TTR (ATTRwt). ATTRv has an autosomal dominant inheritance pattern with incomplete penetrance; cardiomyopathy and neuropathy are the most common ATTRv manifestations. Because of its genetic distribution, certain racial and ethnic groups, most notably people of African and Portuguese descent, have higher rates of ATTR-CM. [14–17] In the USA, an analysis of two biobanks found that participants of African or Latinx descent who carry the V122I TTR variant have twice the risk of incident heart failure compared to non-carriers. [18] ATTRwt, previously known as senile amyloid cardiomyopathy, usually manifests with cardiac, tendon, and ligament involvement in older men. Extracardiac manifestations, such as bilateral carpal tunnel syndrome, [2, 19–21] distal biceps tendon rupture, [22] and lumbar spinal stenosis, [23, 24] are often present years before significant cardiac dysfunction is apparent. [25]
Cardiac biomarkers, including troponin and N-terminal (NT)-pro hormone B-type natriuretic peptide (NT-proBNP), are usually elevated in ATTR-CM but are nonspecific. [2, 25, 26] Echocardiogram findings often raise suspicion for the diagnosis of amyloid CM with concentric left ventricular (LV) wall thickening > 12 mL, bi-atrial enlargement, and reduced LV longitudinal strain with apical sparing. [27–29] The diagnosis was historically confirmed via endomyocardial biopsy. However, 99mTc-pyrophosphate (99mTc-PYP) cardiac scintigraphy has become the standard for diagnosis for patients in whom the presence of a monoclonal gammopathy has been excluded. [30–35] This has provided a lower-cost, widely available, non-invasive method of diagnosis. [36]
Management of ATTR-CM previously focused on palliation with symptomatic management of progressive heart failure with diuretics to control congestion and antiarrhythmics to control atrial and ventricular arrhythmias. Recently, tafamidis, a TTR stabilizer that reduces the formation and deposition of amyloid fibrils, was approved by the FDA for ATTR-CM. The landmark, randomized ATTR-ACT trial showed improvements in both mortality and cardiovascular hospitalizations at 30 months for tafamidis compared to placebo. [37, 38] Patisiran and inotersen are two TTR silencers that have been recently approved for ATTR polyneuropathy with or without cardiomyopathy. [39, 40] TTR silencers are currently being tested specifically in patients with ATTR-CM. [36, 41, 42]
Disparities in Heart Failure
Overall, Black individuals are 30% more likely to die from heart disease, have higher rates of myocardial infarction, and worse outcomes after acute coronary syndrome events. [43] With regard to heart failure, Black Americans are more likely to develop congestive heart failure and to do so at an earlier age. Black patients have a 2.5-fold increased risk of hospitalization for heart failure compared to White patients and higher mortality (particularly in patient less than 65 years old). [44, 45] Black patients also have higher rates of dilated and peripartum cardiomyopathy with more severe disease profiles when they occur. [46–48] Black patients with hypertrophic cardiomyopathy are less likely to be referred for sudden cardiac death risk stratification and to have implanted cardioverter-defibrillators when indicated, though without noted differences in clinical outcomes and mortality. [49] The etiologies of these disparities in heart failure are thought to be multifactorial with genetic, comorbid, socioeconomic, systemic, and policy contributions that remain to be fully elucidated. [44]
Genetics of ATTR-CM
ATTR-CM is caused by a number of TTR mutations with over 140 known variants associated with clinical amyloidosis. [50] It is becoming increasingly appreciated that specific TTR mutations have distinct demographic predominance. The V122I (valine to isoleucine) mutation is by far the most common variant in the USA and is carried by approximately 4% of Black Americans. [51] Up to 5% of the West African population carries the V122I variant. In a single referral center analysis in the UK, V122I was the most common TTR mutation, comprising 43% of all ATTRv; nearly 50% of this cohort had Afro-Caribbean or African ancestry. [52] The V30M (valine to methionine) mutation is the best described TTR variant. V30M ATTRv is associated with predominant polyneuropathy and is a common variant in Portugal, Japan, Sweden, and Brazil. [53–56]
The Transthyretin Amyloidosis Outcomes Survey (THAOS) is an ongoing multinational registry that collects observational data on the natural history of ATTR. In a publication by the THAOS investigators in 2016, 2530 patients were included, 390 of whom were in the USA and more than 1000 in Portugal. They found that about one in four enrolled patients in the USA were of African descent compared to 0.5% in other regions of the world. While V122I was the most common pathologic mutation in the USA, V30M was the most common in the other included regions of the world. The THAOS data for Hispanic/Latinx patients were sparse with 256 patients from Latin American countries (Brazil, Mexico, and Argentina). The most common pathologic mutation in this group was V30M possibly resulting from Portuguese colonization in South America. [57] Other pathologic TTR variants have been described in different Hispanic subpopulations. For example, S50A (serine to alanine) is the most common TTR variant in certain regions of Mexico. [58] Given the data suggesting worse clinical outcomes for ATTRv compared to ATTRwt, further comprehensive studies in Black and Hispanic/Latinx populations will be critical.[59]
Disparities in the Diagnosis of ATTR-CM
Early diagnosis of ATTR-CM is crucial to slow amyloid deposition and delay disease progression. [37] While diagnoses appear to be increasing over time, [16, 59–61] early recognition and diagnosis remain challenging perhaps in part due to limited physician awareness of the condition and its management. [62, 63] In fact, in the USA, there are large geographic variations in reported mortality due to amyloidosis. The highest mortality rates were reported in counties near amyloid referral centers. Notably, the southern states, despite having the highest proportions of Black individuals, reported the lowest mortality due to amyloidosis, [16] likely reflective the disparities in diagnosis (Fig. 1).
Fig. 1.
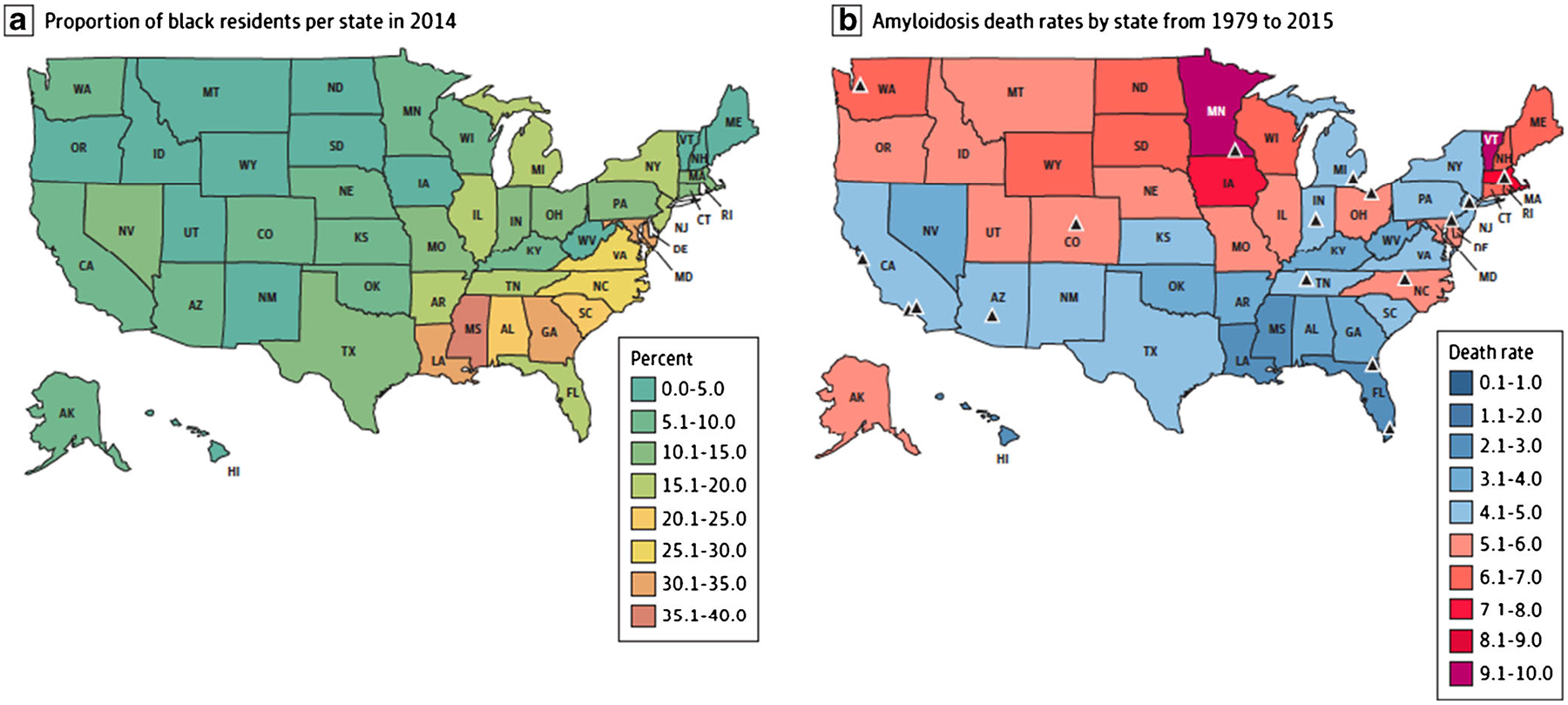
Geographic disparities in reported age-adjusted death rates for amyloidosis in the USA. The triangles represent the location of established amyloidosis referral centers. Reproduced with permission from Alexander, K.M., et al., Geographic Disparities in Reported US Amyloidosis Mortality From 1979 to 2015: Potential Underdetection of Cardiac Amyloidosis. JAMA Cardiol, 2018. 3(9): p. 865–870
For patients with ATTR-CM that are identified, the median time from onset of cardiac symptoms requiring hospitalization to diagnosis appears to be approximately 2–3 years. [59] The V122I mutation results in almost exclusively cardiac involvement; mortality appears to be higher in patients with V122I ATTRv, with a median survival of 25.6 months versus 43 months in ATTRwt. [64] A multivariable analysis of 1034 patients with ATTR-CM who were referred to an amyloid center in the UK showed that advanced disease and lower ejection fraction at the time of diagnosis and the V122I mutation were factors associated with a higher mortality. [59] In the case of V122I ATTRv, in a study composed of 96% Black participants, women were, on average, about 7 years older at the time of diagnosis with similar clinical characteristics and mortality despite being older at the time of diagnosis. It is unclear whether this difference in age at the time of diagnosis is attributable to contextual issues like decreased access to care and clinician bias or an intrinsic feature of the disease presentation in women with later development of clinical symptoms.[60]
Several ongoing studies seek to identify the prevalence of ATTR-CM in specific populations. [65, 66] In the ongoing Screening for Cardiac Amyloidosis in Using Nuclear Imaging for Minority Populations (SCAN-MP), investigators are using cardiac scintigraphy to screen and diagnose patients of Black or Hispanic Caribbean origin with heart failure not caused by valvular or ischemic heart disease in an attempt to improve ATTR-CM in these populations. [67] While more asymptomatic carriers are likely to be identified via genotyping, guidelines for screening and management of these patients are not currently available. Some families may choose to forego genetic screening to avoid the influence of pre-existing conditions on acquiring life insurance. [68]
Disparities in ATTR-CM Trials and Treatment
The ATTR clinical trials thus far have studied either patients with predominant polyneuropathy or with primarily cardiac involvement. The phase 3 ATTR-ACT trial tested the efficacy of the TTR stabilizer, tafamidis, compared to placebo in patients with ATTR-CM. [37] The primary endpoint was mortality and cardiovascular hospitalizations. In this multi-center, international study including 441 participants, approximately 14% (n = 63) were Black patients. Notably, no site or center from the African continent participated in the trial. Two participants were included from Brazil (the sole South or Central American site). The majority of patients were recruited from the USA (n = 279), Germany (n = 49), Italy (n = 29), France (n = 28), and Japan (n = 17). [37] The APOLLO trial tested the efficacy of patisiran in 225 patients with ATTRv polyneuropathy. [40] Because this trial recruited patients with ATTR polyneuropathy, only 1% had a V122I mutation. Similarly, NEURO-TTR, which tested the efficacy of inotersen compared to placebo had only 2% of patients with the V122I mutation (Fig. 2). [39] Although there are several ongoing randomized trials focused on patients with ATTR-CM, they do not appear to include African countries as recruitment centers or state explicit goals for recruitment of Black patients or patients with V122I ATTRv (Table 1).
Fig. 2.
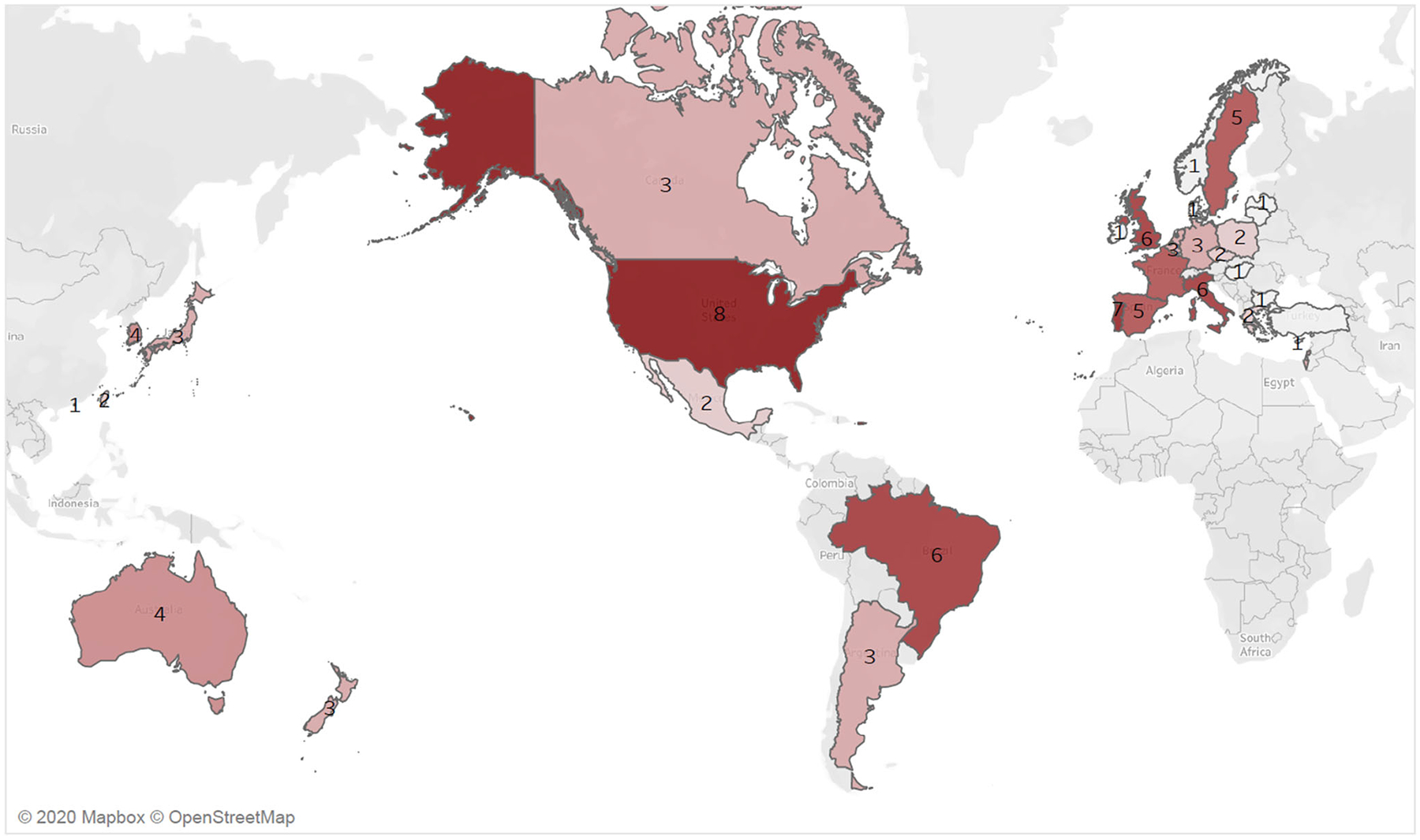
Total number of transthyretin amyloidosis (ATTR) clinical trials by country. The heatmap shows the number of ATTR clinical trials, with darker reds reflecting greater numbers of ongoing trials. Eight recent trials for ATTR cardiomyopathy or neuropathy from a total of 33 countries were included. [37, 39–42, 71–73] There were no trial sites on the continent of Africa
Table 1.
Demographics of notable transthyretin amyloidosis (ATTR) clinical trials
| Drug name Trial name |
Number of patients | Route | Main findings and primary outcomes | Percent women participants | Percent Black participants | Countries included |
|---|---|---|---|---|---|---|
| Transthyretin stabilizers | ||||||
| Tafamidis ATTR-ACT |
441 | Oral | Decreased mortality and cardiovascular-related hospitalizations. | 10% | 14% | Belgium, Brazil, Canada, Czech Republic, France, Germany, Italy, Japan, Netherlands, Spain, Sweden, UK, USA |
| AG10 ATTRibute-CM |
510 | Oral | Results pending: Primary outcomes will be change in 6-minute walk test and the composite of all-cause mortality and cardiovascular hospitalizations. | Recruitment ongoing | Recruitment ongoing | Australia, Belgium, Brazil, Canada, Greece, Hungary, Ireland, Israel, Italy, Korea, Netherlands, New Zealand, Poland, Portugal, Spain, USA |
| Transthyretin silencers | ||||||
| Patisiran APOLLO |
225 (with neuropathy ± cardiomyopathy) | IV | Vastly improved neurological function vs. placebo. Subanalysis showed decreased LV wall thickness, decreased NT-ProBNP, and improved global longitudinal strain. |
26% | 2% | Argentina, Brazil, Canada, Bulgaria, Cyprus, France, Italy, Japan, Mexico, Netherlands, Portugal, South Korea, Spain, Sweden, Taiwan, Turkey, UK, USA |
| Inotersen NEURO-TTR |
172 (with neuropathy ± cardiomyopathy) | SQ | Greatly improved neurological function vs. placebo. No significant difference in cardiac parameters. |
31% | 4%. | Argentina, Brazil, France, Germany, Italy, New Zealand, Portugal, UK, USA |
| Patisiran APOLLO-B |
300 | IV | Recruitment ongoing: primary outcome will be change from baseline at month 12 in Six-Minute Walk Test | Recruitment ongoing | Recruitment ongoing | Argentina, Australia, Brazil, Czechia, Denmark, France, Hong Kong, Italy, Japan, Republic of Korea, Mexico, Poland, Portugal, Sweden, Taiwan, UK, USA |
| Vutrisiran HELIOS-B |
600 | SQ | Recruitment ongoing: primary outcome will be the composite of all-cause mortality and cardiovascular-related hospitalizations. | Recruitment ongoing | Recruitment ongoing | Australia, France, Germany, Israel, Republic of Korea, Latvia, Lithuania, Norway, Portugal, Spain, Sweden, UK, USA |
| AKCEA-TTR-LRx CARDIO-TTRansform |
750 | SQ | Recruitment ongoing: The primary outcome will be the change in 6-minute walk test and the composite of cardiovascular mortality and clinical cardiovascular events. | Pending | Pending | Australia, Belgium, Brazil, Greece, Israel, Italy, Portugal, Spain, Sweden, USA |
| Amyloid clearance | ||||||
| PRX004 | 36 | IV | Results pending: maximum tolerated dose and treatment-emergent adverse events | Pending | Phase I trial | Portugal, Spain, Sweden, USA |
ATTR transthyretin amyloidosis, ATTRv variant ATTR, ATTRwt wild-type ATTR, IV intravenous, SQ subcutaneous, TTR transthyretin
Beyond the need for targeted inclusion of Black patients in trials for ATTR-CM, questions remain regarding the accessibility, affordability, and ethics of the pricing of novel therapeutics. [69] Tafamidis has an estimated yearly cost of $225,000. [70] Gurwitz and Maurer recently called attention to affordability issues for the uninsured and those with high deductible health plans and/or coverage gaps. They also called into question the drug’s designation as an orphan drug given that, thus far, ATTR-CM has likely been grossly underdiagnosed and a true prevalence in the USA of greater than 200,000 people would invalidate the drug’s market exclusivity provisions. [69] The multiple deficiencies in identification and accessibility create a leaky pipeline likely to leave many patients undiagnosed and create barriers to accessibility for those who are diagnosed (Fig. 3).
Fig. 3.
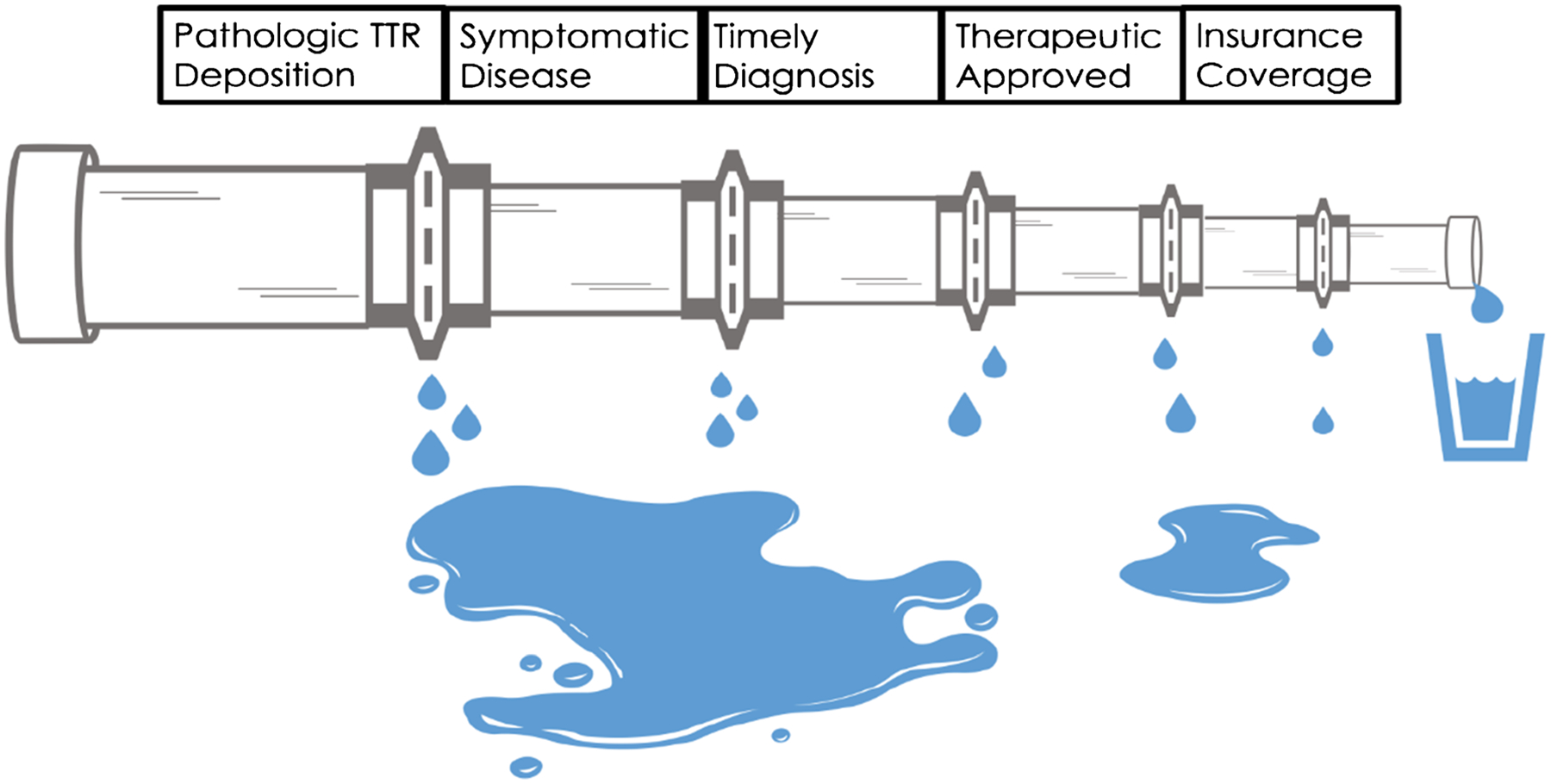
The leaky pipeline for transthyretin amyloidosis (ATTR) diagnosis and treatment. Only a fraction of patients with ATTR are treated. Cardiomyopathy does not develop in all patients with pathologic mutations due to incomplete penetrance. ATTR cardiomyopathy is frequently identified post-mortem, an untimely diagnosis. Until May 2019, there were no approved therapeutics specifically approved for ATTR cardiomyopathy. At a cost of over $200,000, tafamidis remains unaffordable for some patients with high deductibles and coverage gaps
Future Directions
Although there is a growing appreciation and acknowledgement of the prevalence of ATTR-CM and the current underdiagnosis of this condition, there still remains a significant gap in diagnosis of Black and Hispanic/Latinx patients. Current data, though limited, demonstrate that these cohorts may have worse outcomes and prognosis, yet ongoing trials of ATTR-CM overlook these forgotten and likely high-risk groups. To mitigate future disparities, investigators should acknowledge this critical lack in data and be purposeful in recruiting Black and Hispanic/Latinx patients. Studies such as the SCAN-MP trial can serve as an example. This study also highlights that a number of Black patients who carry a diagnosis of heart failure with preserved ejection fraction (HFpEF) have not had thorough evaluation for secondary etiologies and often are assumed to be secondary to hypertensive heart disease. [17] This study will also help answer the question about the utility of using cardiac scintigraphy as a standard evaluation for patients with HFpEF and its utility in screening asymptomatic carriers of clinically significant TTR mutations for early signs of ATTR-CM. In addition to the strong demographic correlation of certain ATTR-CM variants, such as V122I, V30M, and S50A, proteomic and epigenetic evaluations may provide more information to guide diagnosis and treatment among Black and Hispanic/Latinx patients and should be a focus for future research.
Funding
Dr. Alexander is supported by the American Heart Association-Amos Medical Faculty Development Program (19AMFDP34990036) and the National Center for Advancing Translational Sciences of the National Institutes of Health under award number KL2TR003143.
Conflict of Interest
Dr. Spencer-Bonilla, Dr. Pearson, Dr. Njoroge, and Dr. Aras do not have any conflicts of interest to disclose. Dr. Witteles has received consulting fees (modest) from Pfizer, Alnylam, Eidos, and Ionis/Akcea. Dr. Alexander has received an investigator-initiated research grant from Pfizer and has received consulting fees (modest) from Alnylam, Eidos, and Pfizer.
Footnotes
Human and Animal Rights and Informed Consent All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
References
- 1.Rapezzi C, Lorenzini M, Longhi S, Milandri A, Gagliardi C, Bartolomei I, et al. Cardiac amyloidosis: the great pretender. Heart Fail Rev. 2015;20(2):117–24. [DOI] [PubMed] [Google Scholar]
- 2.Grogan M, Scott CG, Kyle RA, Zeldenrust SR, Gertz MA, Lin G, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol. 2016;68(10):1014–20. [DOI] [PubMed] [Google Scholar]
- 3.Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–29. [DOI] [PubMed] [Google Scholar]
- 4.Liepnieks JJ, Zhang LQ, Benson MD. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology. 2010;75(4):324–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holmgren G, Steen L, Ekstedt J, Groth CG, Ericzon BG, Eriksson S, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet. 1991;40(3):242–6. [DOI] [PubMed] [Google Scholar]
- 6.Sipe JD, Benson MD, Buxbaum JN, Ikeda SI, Merlini G, Saraiva MJM, et al. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid. 2014;21(4):221–4. [DOI] [PubMed] [Google Scholar]
- 7.Nasr SH, Dogan A, Larsen CP. Leukocyte cell-derived chemotaxin 2-associated amyloidosis: a recently recognized disease with distinct clinicopathologic characteristics. Clin J Am Soc Nephrol. 2015;10(11):2084–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Falk RH, Alexander KM, Liao R, Dorbala S. AL (Light-Chain) Cardiac amyloidosis: a review of diagnosis and therapy. J Am Coll Cardiol. 2016;68(12):1323–41. [DOI] [PubMed] [Google Scholar]
- 9.Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O’Fallon WM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79(7):1817–22. [PubMed] [Google Scholar]
- 10.Grogan M, Dispenzieri A. Natural history and therapy of AL cardiac amyloidosis. Heart Fail Rev. 2015;20(2):155–62. [DOI] [PubMed] [Google Scholar]
- 11.Mohty D, Damy T, Cosnay P, Echahidi N, Casset-Senon D, Virot P, et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis. 2013;106(10):528–40. [DOI] [PubMed] [Google Scholar]
- 12.Marinac CR, Ghobrial IM, Birmann BM, Soiffer J, Rebbeck TR. Dissecting racial disparities in multiple myeloma. Blood Cancer J. 2020;10(2):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Staron A, Connors LH, Zheng L, Doros G, Sanchorawala V. Race/ethnicity in systemic AL amyloidosis: perspectives on disease and outcome disparities. Blood Cancer J. 2020;10(11):118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobson DR, Alexander AA, Tagoe C, Buxbaum JN. Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14 333 African-Americans. Amyloid. 2015;22(3):171–4. [DOI] [PubMed] [Google Scholar]
- 15.Quarta CC, Buxbaum JN, Shah AM, Falk RH, Claggett B, Kitzman DW, et al. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med. 2015;372(1):21–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexander KM, Orav J, Singh A, Jacob SA, Menon A, Padera RF, et al. Geographic disparities in reported US amyloidosis mortality from 1979 to 2015: potential underdetection of cardiac amyloidosis. JAMA Cardiol. 2018;3(9):865–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shah KB, Mankad AK, Castano A, Akinboboye OO, Duncan PB, Fergus IV, et al. Transthyretin cardiac amyloidosis in Black Americans. Circ Heart Fail. 2016;9(6):e002558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Damrauer SM, Chaudhary K, Cho JH, Liang LW, Argulian E, Chan L, et al. Association of the V122I hereditary transthyretin amyloidosis genetic variant with heart failure among individuals of African or Hispanic/Latino ancestry. JAMA. 2019;322(22): 2191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sperry BW, Reyes BA, Ikram A, Donnelly JP, Phelan D, Jaber WA, et al. Tenosynovial and cardiac amyloidosis in patients undergoing carpal tunnel release. J Am Coll Cardiol. 2018;72(17):2040–50. [DOI] [PubMed] [Google Scholar]
- 20.Nakagawa M, Sekijima Y, Yazaki M, Tojo K, Yoshinaga T, Doden T, et al. Carpal tunnel syndrome: a common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid. 2016;23(1):58–63. [DOI] [PubMed] [Google Scholar]
- 21.Sekijima Y, Uchiyama S, Tojo K, Sano K, Shimizu Y, Imaeda T, et al. High prevalence of wild-type transthyretin deposition in patients with idiopathic carpal tunnel syndrome: a common cause of carpal tunnel syndrome in the elderly. Hum Pathol. 2011;42(11): 1785–91. [DOI] [PubMed] [Google Scholar]
- 22.Geller HI, Singh A, Alexander KM, Mirto TM, Falk RH. Association between ruptured distal biceps tendon and wild-type transthyretin cardiac amyloidosis. JAMA. 2017;318(10):962–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Westermark P, Westermark GT, Suhr OB, Berg S. Transthyretin-derived amyloidosis: probably a common cause of lumbar spinal stenosis. Ups J Med Sci. 2014;119(3):223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73(22):2872–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Witteles RM, Bokhari S, Damy T, Elliott PM, Falk RH, Fine NM, et al. Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC Heart Fail. 2019;7(8):709–16. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez-Lopez E, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585–94. [DOI] [PubMed] [Google Scholar]
- 27.Quarta CC, Solomon SD, Uraizee I, Kruger J, Longhi S, Ferlito M, et al. Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation. 2014;129(18): 1840–9. [DOI] [PubMed] [Google Scholar]
- 28.Chacko L, Martone R, Cappelli F, Fontana M. Cardiac amyloidosis: updates in imaging. Curr Cardiol Rep. 2019;21(9):108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pagourelias ED, Mirea O, Duchenne J, van Cleemput J, Delforge M, Bogaert J, et al. Echo parameters for differential diagnosis in cardiac amyloidosis: a head-to-head comparison of deformation and nondeformation parameters. Circ Cardiovasc Imaging. 2017;10(3):e005588. [DOI] [PubMed] [Google Scholar]
- 30.Castano A, Haq M, Narotsky DL, Goldsmith J, Weinberg RL, Morgenstern R, et al. Multicenter study of planar technetium 99m pyrophosphate cardiac imaging: predicting survival for patients with ATTR cardiac amyloidosis. JAMA Cardiol. 2016;1(8):880–9. [DOI] [PubMed] [Google Scholar]
- 31.Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–12. [DOI] [PubMed] [Google Scholar]
- 32.Treglia G, Glaudemans AWJM, Bertagna F, Hazenberg BPC, Erba PA, Giubbini R, et al. Diagnostic accuracy of bone scintigraphy in the assessment of cardiac transthyretin-related amyloidosis: a bivariate meta-analysis. Eur J Nucl Med Mol Imaging. 2018;45(11): 1945–55. [DOI] [PubMed] [Google Scholar]
- 33.Perugini E, Guidalotti PL, Salvi F, Cooke RMT, Pettinato C, Riva L, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46(6):1076–84. [DOI] [PubMed] [Google Scholar]
- 34.Hanna M, Ruberg FL, Maurer MS, Dispenzieri A, Dorbala S, Falk RH, et al. Cardiac scintigraphy with technetium-99m-labeled bone-seeking tracers for suspected amyloidosis. J Am Coll Cardiol. 2020;75(22):2851–62. [DOI] [PubMed] [Google Scholar]
- 35.Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: Part 2 of 2-Diagnostic criteria and appropriate utilization. J Nucl Cardiol. 2020;27(2):659–73. [DOI] [PubMed] [Google Scholar]
- 36.Spencer-Bonilla G, Alexander K, Witteles R. Advances in the Diagnosis and Management of Transthyretin Amyloid Cardiomyopathy. Curr Treat Options Cardiovasc Med. 2020. IN PRESS;22. [Google Scholar]
- 37.Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–16. [DOI] [PubMed] [Google Scholar]
- 38.Maurer MS, et al. Design and Rationale of the Phase 3 ATTR-ACT Clinical Trial (Tafamidis in Transthyretin Cardiomyopathy Clinical Trial). Circ: Heart Fail. 2017;10(6). [DOI] [PubMed] [Google Scholar]
- 39.Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31. [DOI] [PubMed] [Google Scholar]
- 40.Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. [DOI] [PubMed] [Google Scholar]
- 41.Shilling R, et al. Study Design and Rational of HELIOS-B: a Phase 3 Study to Evaluate the Clinical Efficacy and Safety of Vutrisiran in Patients with ATTR Amyloidosis with Cardiomyopathy. J Am Coll Cardiol. 2020;75(11 Supplement 1):3579. [Google Scholar]
- 42.Falk RH, et al. , Rationale and Design of a Phase 3 Study to Evaluate the Efficacy and Safety of ION-682884 in Patients with Transthyretin-Mediated Amyloid Cardiomyopathy (ATTR-CM). 2019, American Society of Hematology; Washington, DC. [Google Scholar]
- 43.Graham G. Disparities in cardiovascular disease risk in the United States. Curr Cardiol Rev. 2015;11(3):238–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nayak A, Hicks AJ, Morris AA. Understanding the complexity of heart failure risk and treatment in Black patients. Circ Heart Fail. 2020;13(8):e007264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glynn P, Lloyd-Jones DM, Feinstein MJ, Carnethon M, Khan SS. Disparities in cardiovascular mortality related to heart failure in the United States. J Am Coll Cardiol. 2019;73(18):2354–5. [DOI] [PubMed] [Google Scholar]
- 46.Irizarry OC, Levine LD, Lewey J, Boyer T, Riis V, Elovitz MA, et al. Comparison of Clinical Characteristics and Outcomes of Peripartum Cardiomyopathy Between African American and Non-African American Women. JAMA Cardiol. 2017;2(11): 1256–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gentry MB, Dias JK, Luis A, Patel R, Thornton J, Reed GL. African-American women have a higher risk for developing peripartum cardiomyopathy. J Am Coll Cardiol. 2010;55(7):654–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dries DL, Exner DV, Gersh BJ, Cooper HA, Carson PE, Domanski MJ. Racial differences in the outcome of left ventricular dysfunction. N Engl J Med. 1999;340(8):609–16. [DOI] [PubMed] [Google Scholar]
- 49.Wells S, Rowin EJ, Bhatt V, Maron MS, Maron BJ. Association between race and clinical profile of patients referred for hypertrophic cardiomyopathy. Circulation. 2018;137(18):1973–5. [DOI] [PubMed] [Google Scholar]
- 50.Ueda M, Yamashita T, Misumi Y, Masuda T, Ando Y. Origin of sporadic late-onset hereditary ATTR Val30Met amyloidosis in Japan. Amyloid. 2018;25(3):143–7. [DOI] [PubMed] [Google Scholar]
- 51.Jacobson DR, Alexander AA, Tagoe C, Garvey WT, Williams SM, Tishkoff S, et al. The prevalence and distribution of the amyloidogenic transthyretin (TTR) V122I allele in Africa. Mol Genet Genomic Med. 2016;4(5):548–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rowczenio D, Quarta CC, Fontana M, Whelan CJ, Martinez-Naharro A, Trojer H, et al. Analysis of the TTR gene in the investigation of amyloidosis: A 25-year single UK center experience. Hum Mutat. 2019;40(1):90–6. [DOI] [PubMed] [Google Scholar]
- 53.Winburn I, Ishii T, Sumikawa T, Togo K, Yasunaga H. Estimating the prevalence of transthyretin amyloid cardiomyopathy in a large in-hospital database in Japan. Cardiol Ther. 2019;8(2):297–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soares ML, Coelho T, Sousa A, Holmgren G, Saraiva MJ, Kastner DL, et al. Haplotypes and DNA sequence variation within and surrounding the transthyretin gene: genotype-phenotype correlations in familial amyloid polyneuropathy (V30M) in Portugal and Sweden. Eur J Hum Genet. 2004;12(3):225–37. [DOI] [PubMed] [Google Scholar]
- 55.Hellman U, Alarcon F, Lundgren HE, Suhr OB, Bonaiti-PelliÉ C, Planté-Bordeneuve V. Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid. 2008;15(3):181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda SI, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016;68(2):161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gonzalez-Duarte A, et al. Amyloidosis due to TTR mutations in Mexico with 4 distincts genotypes in the index cases. Orphanet J Rare Dis. 2018;13(1):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lane T, Fontana M, Martinez-Naharro A, Quarta CC, Whelan CJ, Petrie A, et al. Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation. 2019;140(1):16–26. [DOI] [PubMed] [Google Scholar]
- 60.Batra J, et al. Sex Differences in the phenotype of transthyretin cardiac amyloidosis due to Val122Ile mutation: insights from non-invasive pressure-volume analysis. J Card Fail. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sperry BW, Saeed IM, Raza S, Kennedy KF, Hanna M, Spertus JA. Increasing rate of hospital admissions in patients with amyloidosis (from the national inpatient sample). Am J Cardiol. 2019;124(11): 1765–9. [DOI] [PubMed] [Google Scholar]
- 62.Mircsof D. Diagnosis of amyloidosis: a survey of current awareness and clinical challenges among cardiologists in Switzerland. Cardiol Ther. 2020;9(1):127–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hauptman PJ, Wall JS, Maurer MS. Disparities, uncertainties, and societal cost: precision medicine and transthyretin amyloidosis. Am J Med. 2020;133(8):892–4. [DOI] [PubMed] [Google Scholar]
- 64.Ruberg FL, Maurer MS, Judge DP, Zeldenrust S, Skinner M, Kim AY, et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J. 2012;164(2):222–8 e1. [DOI] [PubMed] [Google Scholar]
- 65.Prevalence of wild type transthyretin cardiac amyloidosis in patients operated for idiopathic carpal tunnel syndrome. [cited 2020 June 15]; Available from: https://ClinicalTrials.gov/show/NCT03996382.
- 66.Cardiac amyloidosis screening at trigger finger release. [cited 2020 June 15]; Available from: https://ClinicalTrials.gov/show/NCT03886155. [DOI] [PubMed]
- 67.Screening for cardiac amyloidosis using nuclear imaging for minority populations. [cited 2020 June 13]; Available from: https://ClinicalTrials.gov/show/NCT03812172.
- 68.Consortium, A.R., Cece - hATTR amyloidosis patient. 2019: youtube.com.
- 69.Gurwitz JH and Maurer MS, Tafamidis-a pricey therapy for a not-so-rare condition. JAMA Cardiol, 2020 [DOI] [PubMed] [Google Scholar]
- 70.Kazi DS, Bellows BK, Baron SJ, Shen C, Cohen DJ, Spertus JA, et al. Cost-effectiveness of tafamidis therapy for transthyretin amyloid cardiomyopathy. Circulation. 2020;141(15):1214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gillmore JD, et al. ATTRibute-CM: a randomized, double-blind, placebo-controlled, multi-center, global phase 3 study of AG10 in patients with transthyretin amyloid cardiomyopathy (ATTR-CM). Circulation. 2019;140(Suppl_1):A14214. [Google Scholar]
- 72.A study of PRX004 in subjects with amyloid transthyretin (ATTR) amyloidosis. June 14 2020]; Available from: https://ClinicalTrials.gov/show/NCT03336580.
- 73.CARDIO-TTRansform: a study to evaluate the efficacy and safety of AKCEA-TTR-LRx in participants with transthyretin-mediated amyloid cardiomyopathy (ATTR CM). [cited 2020 July 4]; Available from: https://ClinicalTrials.gov/show/NCT04136171.