

Abstract
The mitotic phase is a vital step in cell division and may be involved in cancer progression, but it remains unclear whether genetic variants in mitotic phase-related pathways genes impact the survival of these patients. Here, we investigated associations between 31,032 single nucleotide polymorphisms (SNPs) in 368 mitotic phase-related pathway genes and overall survival (OS) of patients with non-small cell lung cancer (NSCLC). We assessed the associations in a discovery dataset of 1,185 NSCLC patients from the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial and validated the findings in another dataset of 984 patients from the Harvard Lung Cancer Susceptibility Study. As a result, we identified three independent SNPs (i.e., CHEK1 rs76744140 T>C, PRIM2 rs6939623 G>T and CDK6 rs113181986 G>C) to be significantly associated with NSCLC OS with an adjusted hazards ratio of 1.29 [95% confidence interval=1.11–1.49, P = 8.26x10−4), 1.26 (1.12–1.42, 1.10x10−4) and 0.73 (0.63–0.86, 1.63x10−4), respectively. Moreover, the number of combined unfavorable genotypes of these three SNPs was significantly associated with NSCLC OS and disease-specific survival in the PLCO dataset (Ptrend < 0.0001 and 0.0003, respectively). Further expression quantitative trait loci analysis showed that the rs76744140C allele predicted CHEK1 mRNA expression levels in normal lung tissues and that rs113181986C allele predicted CDK6 mRNA expression levels in whole blood tissues. Additional analyses indicated CHEK1, PRIM2 and CDK6 may impact NSCLC survival. Taken together, these findings suggested that these genetic variants may be prognostic biomarkers of patients with NSCLC.9.
Keywords: Non-small cell lung cancer, Single-nucleotide polymorphism, Mitotic phase, Survival
Introduction
Lung cancer is the most common malignancy and the leading cause of cancer-related deaths worldwide 1, 2. It is estimated that there were approximately 228,820 new cases and 135,720 deaths to occur in 2020 in the United States alone 3. Non-small cell lung cancer (NSCLC) accounts for about 80–85% of lung cancer with a dismal 15–25% 5-year overall survival (OS) rate as most patients present with advanced stage disease 4–6. Despite the advances in CT screening, traditional surgery, radiotherapy, and chemotherapy as well as recent targeted molecular therapy and immunotherapy for NSCLC, the 5-year survival is poor 7. However, there is marked variability in response and outcomes, even among patients with the same stage, cell type, and treatment, suggesting an important role of genetic susceptibility 8, 9. Therefore, it is essential to explore potential genetic factors that facilitate more precise diagnosis and treatment strategies for NSCLC patients.
Recently, a number of genome-wide association studies (GWASs) have identified that single nucleotide polymorphisms (SNPs) on susceptibility loci of the chromosome are strongly associated with lung cancer risk 10–13. However, few SNPs were reported to modulate clinical outcomes of patients with NSCLC at a GWAS level. However, post-GWAS pathway-based hypothesis-driven analyses have explored functional SNPs in specific biological pathways, which have moderate but detectable effects NSCLC survival 14, 15; thus, this approach may provide a better understanding of the development and progression of NSCLC.
The cell cycle consists of interphase and mitotic phase 16. The mitotic phase is a process in which the parental cell divides into two daughter cells. There five phases in a cell cycle:prophase, prometaphase, metaphase, anaphase, and telophase 17. Oncogenes-induced DNA replication pressure has been thought to be the driving force for tumorigenesis 18. However, it is recently reported that the rapid replication of tumor cells in the S phase causes DNA damage and instability, which makes DNA more vulnerable to further insults, while DNA replication in M phase is uniqueo tumor cells, and essential for maintaining genomic stability 19. Because abnormalities in different mitotic phase-related genes may cause uncontrolled replication of tumor cells, cancer therapies targeting mitotic phase-related proteins appears to be a powerful strategy. The microtubules, kinases, and polyprotein complexes could be targeted by many compounds, which may lead to mitotic arrest and cell death 20. However, cancer patients’ response to these drugs have varied in clinical trials, suggesting that genetic factors may play a vital role in individual treatment effects.
To date, associations between SNPs in the mitotic phase-related pathway genes and survival of NSCLC are still largely unknown. Therefore, we hypothesize that genetic variants of the mitotic phase-related pathway genes may be associated with NSCLC survival. To test this hypothesis, we performed a pathway gene-set analysis to identify functional SNPs that are associated with NSCLC outcomes by using two independent, previously published NSCLC GWAS datasets.
Materials and methods
Study populations
In the present study, the discovery dataset was the from the Prostate, Lung, Colorectal, and Ovarian cancer screening trial (PLCO), which was a multicenter randomized study of 10 medical centers in the United States between 1993 and 2011, including 1,185 Caucasian patients diagnosed with NSCLC 21. The PLCO trial enrolled 77,500 men and 77,500 women aged 55–74, who were randomized to either the intervention arm with screening or the control arm with standard care. All the individuals were followed up to 13 years after enrollment. Genomic DNA extracted from the whole blood samples of these participants were genotyped with Illumina Human Hap240Sv1.0 and Human Hap550v3.0 (dbGaP accession: phs000093.v2.p2 and phs000336.v1.p1) 22, 23.
Another dataset from the Harvard Lung Cancer Susceptibility (HLCS) study was utilized to validate the significant SNPs identified in the PLCO dataset. The HLCS study, which included 984 histology-confirmed NSCLC patients of Caucasian and extracted DNA by the Auto Pure Large Sample Nucleic Acid Purification System (QIAGEN Company, Venlo, Limburg, Netherlands) from whole blood samples of all participants. Genotyping data was performed by using Illumina Humanhap610-Quad arrays, which was imputed by using MaCHsoftware based on the 1000 Genomes project 24. The details of individuals in the HLCS study have been previously described 25.
The use of the data from both of the PLCO trial (n=1,185) and HLCS study (n=984) was approved by the Internal Review Board of Duke University School of Medicine (Project #Pro00054575) and the National Center for Biological Information for access to the dbGaP database of genotypes and phenotypes (Project #6404). The comparison of the characteristics between the two GWAS datasets is described in Table S1.
Gene selection and SNP imputation
The genes involved in the mitotic phase-related pathway were selected through the Molecular Signatures Database (http://software.broadinstitute.org/gsea/msigdb/index.jsp) with the keyword “mitotic AND phase”. After excluding 60 duplicated genes and one pseudogene, 368 genes remained as the candidate genes for further analysis (Table S2). Imputation was performed by miniMac4 with the reference panel of the 1000 Genomes Project data (phase 3). After imputation, all the SNPs in these genes and their ±2 kb flanking regions were extracted according to the quality criteria: r-square ≥ 0.3, minor allele frequency ≥ 0.05, individual call rate ≥ 95%, and Hardy–Weinberg equilibrium P-value ≥ 1 × 10−5. As a result, a total of 31,032 SNPs (2,820 genotyped and 28,212 imputed) were obtained for further analysis.
Statistical analyses
The follow-up time of the participants in both the PLCO trial and HLCS study was defined as from the diagnosis of NSCLC to the last follow-up or date of death. The OS of patients with NSCLC was chosen as the primary endpoint, and their disease-specific survival (DSS) was also analyzed. In the single-locus analysis, multivariable Cox proportional hazards regression analysis was utilized to assess the association between each of the SNPs in these 368 candidate genes and OS in an additive genetic model with adjustment for age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy, surgery and the top four principal components (Table S3) in the PLCO trial using the GenABEL package of R software 26. Since the majority of SNPs were imputed with a high linkage disequilibrium (LD) (r2 > 0.8), the Bayesian false discovery probability (BFDP) with a cutoff value of 0.80 was used for multiple testing correction to reduce the likelihood of potential false-positive results 27. We then assigned a prior probability of 0.10 to detect a hazards ratio (HR) of 3.0 for an OS-associated variant genotypes or minor alleles of the SNPs with P < 0.05. The identified SNPs in the PLCO trial were further validated in the HLCS study. Subsequently, the multivariable stepwise Cox regression model was performed with adjustment for clinical variables, top four principal components, and 41 previously published survival-associated SNPs from the same PLCO trial to identify additional independent SNPs. Finally, the meta-analysis was performed to combine the results of the PLCO trial and HLCS study by using PLINK 1.90 with Cochran’s Q statistics (Q-test) and heterogeneity statistic (I2). The fixed-effects model was applied, if the Q-test p-value > 0.10 and the I2 < 50%; otherwise, the random-effects model was implemented. In addition, the identified SNPs were also visualized by Manhattan plots and regional association plots.
Then, the Kaplan-Meier (KM) survival curves were constructed to evaluate survival probability associated with the combined unfavorable genotypes of identified SNPs. Meanwhile, we also assessed the heterogeneity between subgroups and possible interaction with a Chi-square-based Q-test in the stratified analysis. Moreover, the receiver operating characteristic (ROC) curves and time-dependent area under the curve (AUC) were utilized to elucidate the ability of the genetic model in predicting the OS and DSS of NSCLC from a clinical perspective using the timeROC package of R software (version 3.6.2).
Subsequently, the three online bioinformatics tools, RegulomeDB 28 (http://www.regulomedb.org), HaploReg 29 (http://archive.broadinstitute.org/mammals/haploreg/haploreg.php), and the Encyclopedia of DNA Elements (ENCODE) project (http://genome.ucsc.edu), were used to predict potential functions of the identified SNPs and their high LD SNPs in the same genes. Then, the expression quantitative trait loci (eQTL) analyses were performed to assess correlations between identified SNPs and the corresponding mRNA expression levels with a linear regression model by using R software (version 3.6.2). The mRNA expression data were obtained from two sources: lymphoblastoid cell lines derived from the 373 European descendants included in the 1,000 Genomes Project 24, and the Genotype-Tissue Expression (GTEx) Project (http://www.gtexportal.org/home) 30. Meanwhile, we also explored the differences in mRNA expression levels of genes between paired tumor tissues and adjacent normal tissues from the Cancer Genome Atlas (TCGA) database by using a paired t-test. Besides, KM survival analysis was constructed to evaluate the correlation between the corresponding genes mRNA expression levels of identified SNPs and NSCLC survival probability from an online database (http://kmplot.com/analysis/). Finally, tumor mutation data of the corresponding genes, where the identified SNPs are located, were also assessed in the publicly available database of the cBioPortal for Cancer Genomics (http://www.cbioportal.org). All statistical analyses were performed with the SAS software (version 9.4; SAS Institute, Cary, NC, USA), unless specified otherwise.
Results
Associations between SNPs in the Mitotic phase-related pathway genes and NSCLC OS in both PLCO trial and HLCS datasets
The flowchart for the present study is shown in Figure 1. The basic characteristics of the discovery dataset from the PLCO trial included 1,185 NSCLC patients, and the validation dataset from the HLCS study included 984 NSCLC patients, both have been described previously (Table S1). In the discovery dataset, a total of 31,032 SNPs (including 2,820 genotyped and 28,212 imputed SNPs) in 368 mitotic phase-related pathway genes were identified, of which 1,286 SNPs were statistically significantly associated with NSCLC OS (both P < 0.05 and BFDP < 0.80). After further replication in the HLCS validation dataset, 35 SNPs remained statistically significant (P < 0.05).
Figure 1.
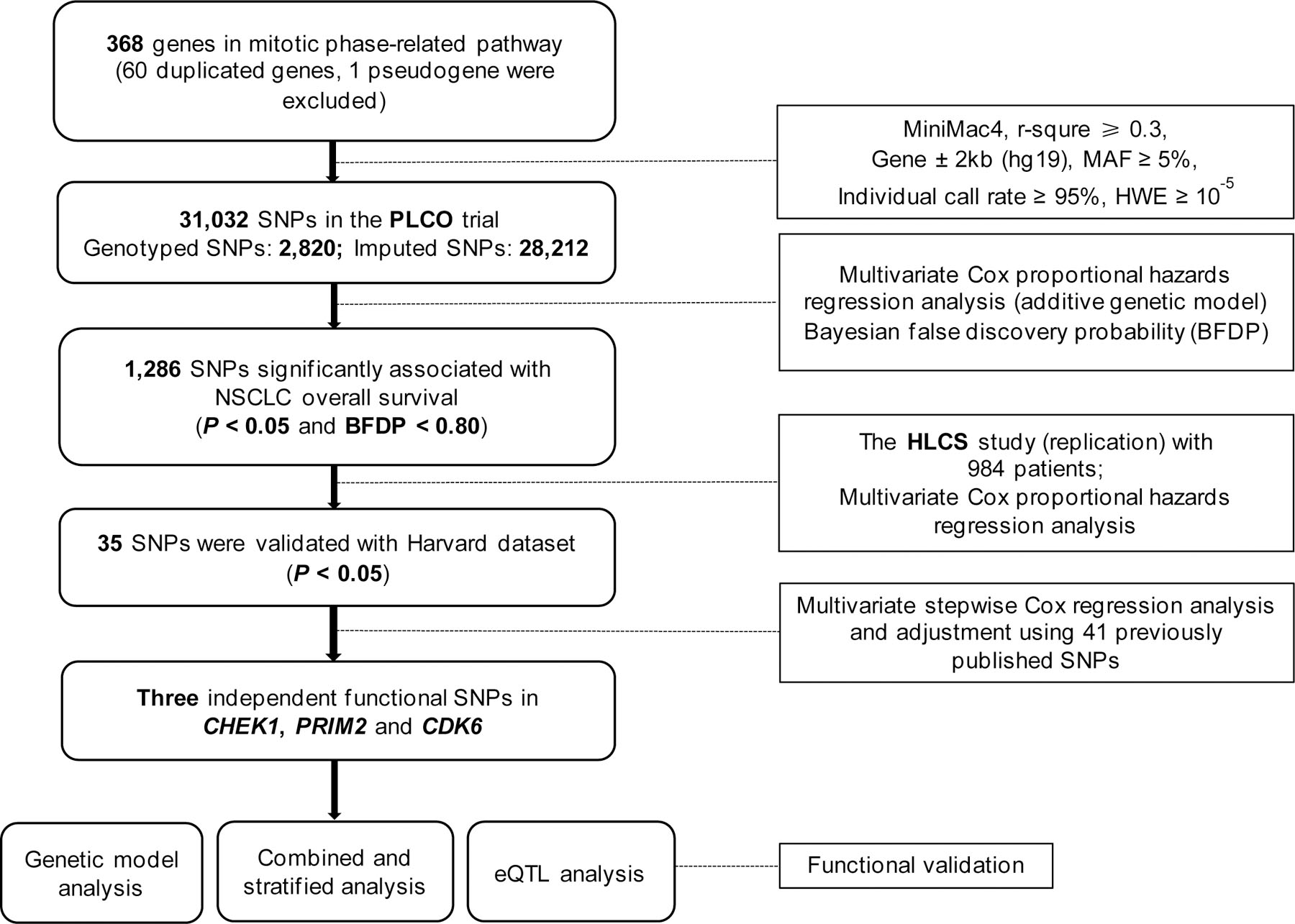
Study flowchart. The overall procedures of the present study.
Identification of independent SNPs associated with NSCLC OS in the PLCO dataset
Since the HLCS dataset did not have detailed genotyping data and clinical covariates for further analyses, we performed the stepwise multivariable Cox regression analysis to identify independent SNPs associated with NSCLC OS using the PLCO dataset. In the stepwise Cox regression analysis with adjustment for clinical variables, top four principal components, as well as the 41 additional previously published survival-associated SNPs for NSCLC from the same dataset, three SNPs (rs76744140 in CHEK1, rs6939623 in PRIM2, and rs113181986 in CDK6) remained independently associated with NSCLC OS (P = 0.006, 0.021, and 0.046, respectively) (Table 1), and 28 SNPs of the 41 published survival-associated SNPs for NSCLC remained significant (Table S4). The meta-analysis results for these three independent SNPs indicated that no heterogeneity was observed across these two datasets (Table 2). In addition, we also depicted these three identified SNPs in Manhattan (Figure S1) and regional association plots (Figure S2).
Table 1.
Three independent SNPs in a multivariate Cox proportional hazards regression analysis with adjustment for other covariates and 41 previously published SNPs for NSCLC in the PLCO Trial
| Variables | Category | Frequency | HR (95% CI)a | P a | HR (95% CI)b | P b |
|---|---|---|---|---|---|---|
| Age | Continuous | 1,185 | 1.03 (1.02–1.05) | <0.0001 | 1.05 (1.03–1.06) | <0.0001 |
| Sex | Male | 698 | 1.00 | 1.00 | ||
| Female | 487 | 0.79 (0.67–0.92) | 0.002 | 0.70 (0.59–0.83) | <0.0001 | |
| Smoking status | Never | 115 | 1.00 | 1.00 | ||
| Current | 423 | 1.73 (1.28–2.32) | 0.0003 | 2.10 (1.53–2.87) | <0.0001 | |
| Former | 647 | 1.70 (1.29–2.34) | 0.0002 | 2.06 (1.53–2.76) | <0.0001 | |
| Histology | Adenocarcinoma | 577 | 1.00 | 1.00 | ||
| Squamous cell | 285 | 1.16 (0.96–1.40) | 0.114 | 1.18 (0.96–1.44) | 0.109 | |
| others | 323 | 1.27 (1.08–1.51) | 0.005 | 1.42 (1.18–1.71) | 0.0003 | |
| Tumor stage | I-IIIA | 655 | 1.00 | 1.00 | ||
| IIIB-IV | 528 | 2.93 (2.41–3.55) | <0.0001 | 3.91 (3.16–4.85) | <0.0001 | |
| Chemotherapy | No | 639 | 1.00 | 1.00 | ||
| Yes | 538 | 0.55 (0.46–0.66) | <0.0001 | 0.52 (0.43–0.63) | <0.0001 | |
| Radiotherapy | No | 762 | 1.00 | 1.00 | ||
| Yes | 415 | 0.94 (0.79–1.10) | 0.418 | 1.12 (0.94–1.34) | 0.211 | |
| Surgery | No | 637 | 1.00 | 1.00 | ||
| Yes | 540 | 0.20 (0.16–0.26) | <0.0001 | 0.20 (0.15–0.26) | <0.0001 | |
| CHEK1 rs76744140 | TT/TC/CC | 1,028/152/5 | 1.29 (1.06–1.56) | 0.009 | 1.35 (1.09–1.66) | 0.006 |
| PRIM2 rs6939623 | GG/GT/TT | 981/199/5 | 1.35 (1.14–1.59) | 0.001 | 1.24 (1.03–1.50) | 0.021 |
| CDK6 rs113181986 | GG/GC/CC | 1,052/131/2 | 0.76 (0.60–0.97) | 0.025 | 0.77 (0.60–1.00) | 0.046 |
Abbreviations: SNP: single-nucleotide polymorphisms; NSCLC, non-small cell lung cancer; PLCO, the Prostate, Lung, Colorectal and Ovarian cancer screening trial; HLCS, Harvard Lung Cancer Susceptibility Study; HR: hazards ratio; CI: confidence interval
Adjusted for age, sex, tumor stage, histology, smoking status, chemotherapy, radiotherapy, surgery, and PC1, PC2, PC3, PC4.
Other 41 published SNPs were included for further adjustment:rs779901, rs3806116, rs199731120, rs10794069, rs1732793, rs225390, rs3788142, rs73049469, rs35970494, rs225388, rs7553295, rs1279590, rs73534533, rs677844, rs4978754, rs1555195, rs11660748, rs73440898, rs13040574, rs469783, rs36071574, rs7242481, rs1049493, rs1801701, rs35859010, rs1833970, rs254315, rs425904, rs35385129, rs4487030, rs60571065, rs13213007, rs115613985, rs9673682, rs2011404, rs7867814, rs2547235, rs4733124, rs11225211, rs11787670, rs67715745
Table 2.
Associations of three significant SNPs with of NSCLC overall survival in both discovery and validation datasets from two published GWASs
| SNPs | Allelea | Gene | PLCO (n=1,185) |
HLCS (n=984) |
Meta-analysis |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EAF | HR (95% CI)b | P b | EAF | HR (95% CI)C | P C | P hetd | I 2 | HR (95% CI)e | P e | |||
| rs76744140 | T>C | CHEK1 | 0.07 | 1.28 (1.06–1.55) | 0.010 | 0.05 | 1.30 (1.02–1.64) | 0.033 | 0.920 | 0 | 1.29 (1.11–1.49) | 8.26x10−4 |
| rs6939623 | G>T | PRIM2 | 0.09 | 1.34 (1.13–1.58) | 0.001 | 0.09 | 1.19 (1.01–1.40) | 0.039 | 0.324 | 0 | 1.26 (1.12–1.42) | 1.10x10−4 |
| rs113181986 | G>C | CDK6 | 0.06 | 0.75 (0.59–0.94) | 0.014 | 0.07 | 0.72 (0.58–0.90) | 0.004 | 0.804 | 0 | 0.73 (0.63–0.86) | 1.63x10−4 |
Abbreviations: SNPs, single-nucleotide polymorphisms; NSCLC, non-small cell lung cancer; GWAS, genome-wide association study; PLCO, the Prostate, Lung, Colorectal and Ovarian cancer screening trial; HLCS, Harvard Lung Cancer Susceptibility Study; EAF, effect allele frequency; HR, hazards ratio; CI, confidence interval.
Reference/effect allele.
Adjusted for age, sex, stage, histology, smoking status, chemotherapy, radiotherapy, surgery, identified SNPs, PC1, PC2, PC3 and PC4.
Adjusted for age, sex, stage, histology, smoking status, chemotherapy, radiotherapy, surgery, PC1, PC2 and PC3.
Phet: P value for heterogeneity by Cochrane’s Q test.
Meta-analysis in the fix-effects model.
As shown in Table 3, both CHEK1 rs76744140 C and PRIM2 rs6939623 T alleles were unfavorable for survival of patients with NSCLC (Ptrend = 0.0101 and 0.0006 for OS, respectively, and Ptrend = 0.0133 and 0.0072 for DSS, respectively), while patients with the CDK6 rs113181986 C allele had a significantly better OS (Ptrend = 0.0146) but a borderline improved DSS (Ptrend = 0.0554 for DSS). Comparing with the reference genotype in a dominant genetic model, patients had a significantly poorer survival, if they had CHEK1 rs76744140 TC+CC genotypes (OS: HR = 1.25, 95% CI = 1.03–1.52, and P = 0.0267; DSS: 1.25, 1.02–1.53, 0.0357) or had PRIM2 rs6939623 GT+TT genotypes (OS: 1.41, 1.17–1.68, and 0.0002; DSS: 1.36, 1.12–1.64, 0.0017), whereas patients had a significantly better OS, if they had CDK6 rs113181986 GC+CC genotypes (0.74, 0.58–0.94, and 0.0150) or had a borderline improved DSS (0.78, 0.61–1.01, and 0.0568).
Table 3.
Associations between the number of unfavorable genotypes of three independent SNPs with NSCLC OS and DSS in the PLCO Trial
| Genotype | Frequencya | OSb |
DSSb |
||||
|---|---|---|---|---|---|---|---|
| Death (%) | HR (95% CI) | P | Death (%) | HR (95% CI) | P | ||
| CHEK1 rs76744140 T>C | |||||||
| TT | 1018 | 664 (65.23) | 1.00 | -- | 595 (58.45) | 1.00 | -- |
| TC | 152 | 120 (78.95) | 1.21 (0.99–1.47) | 0.0622 | 109 (71.71) | 1.20 (0.98–1.48) | 0.0835 |
| CC | 5 | 5 (100.00) | 4.44 (1.81–10.90) | 0.0011 | 5 (100.00) | 4.50 (1.83–11.05) | 0.0011 |
| Trend test | 0.0101 | 0.0133 | |||||
| Dominant | |||||||
| TT | 1018 | 664 (65.23) | 1.00 | -- | 595 (58.45) | 1.00 | -- |
| TC+CC | 157 | 125 (79.62) | 1.25 (1.03–1.52) | 0.0267 | 114 (72.61) | 1.25 (1.02–1.53) | 0.0357 |
| PRIM2 rs6939623 G>T | |||||||
| GG | 973 | 635 (65.26) | 1.00 | -- | 575 (59.10) | 1.00 | -- |
| GT | 197 | 150 (76.14) | 1.43 (1.19–1.71) | 0.0001 | 132 (67.01) | 1.40 (1.16–1.70) | 0.0006 |
| TT | 5 | 4 (80.00) | 0.88 (0.33–2.38) | 0.7992 | 2 (40.00) | 0.46 (0.11–1.85) | 0.2712 |
| Trend test | 0.0006 | 0.0072 | |||||
| Dominant | |||||||
| GG | 973 | 635 (65.26) | 1.00 | -- | 575 (59.10) | 1.00 | -- |
| GT+TT | 202 | 154 (76.24) | 1.41 (1.17–1.68) | 0.0002 | 134 (66.34) | 1.36 (1.12–1.64) | 0.0017 |
| CDK6 rs113181986 G>C | |||||||
| GG | 1044 | 710 (68.01) | 1.00 | -- | 637 (61.02) | 1.00 | -- |
| GC | 129 | 78 (60.47) | 0.75 (0.59–0.95) | 0.0175 | 71 (55.04) | 0.79 (0.61–1.01) | 0.0630 |
| CC | 2 | 1 (50.00) | 0.56 (0.08–4.02) | 0.5663 | 1 (50.00) | 0.62 (0.09–4.45) | 0.6353 |
| Trend test | 0.0146 | 0.0554 | |||||
| Dominant | |||||||
| GG | 1044 | 710 (68.01) | 1.00 | -- | 637 (61.02) | 1.00 | -- |
| GC+CC | 131 | 79 (60.31) | 0.74 (0.58–0.94) | 0.0150 | 72 (54.96) | 0.78 (0.61–1.01) | 0.0568 |
| Reserved | |||||||
| GC+CC | 131 | 79 (60.31) | 1.00 | 72 (54.96) | 1.00 | -- | |
| GG | 1044 | 710 (68.01) | 1.35 (1.06–1.71) | 0.0150 | 637 (61.02) | 1.28 (0.99–1.64) | 0.0568 |
| NUG c | |||||||
| 0 | 98 | 54 (55.10) | 1.00 | -- | 50 (51.02) | 1.00 | -- |
| 1 | 774 | 503 (64.99) | 1.36 (1.03–1.81) | 0.0332 | 453 (58.53) | 1.28 (0.95–1.72) | 0.1012 |
| 2 | 280 | 210 (75.00) | 1.79 (1.32–2.42) | 0.0002 | 186 (66.43) | 1.65 (1.20–2.26) | 0.0021 |
| 3 | 23 | 22 (95.65) | 2.41 (1.45–4.00) | 0.0007 | 20 (86.96) | 2.21 (1.30–3.75) | 0.0034 |
| Trend test | <0.0001 | <0.0001 | |||||
| Dichotomized NUG | |||||||
| 0–1 | 872 | 557 (63.88) | 1.00 | -- | 503 (57.68) | 1.00 | -- |
| 2–3 | 303 | 232 (76.57) | 1.39 (1.19–1.62) | <0.0001 | 206 (67.99) | 1.35 (1.15–1.60) | 0.0003 |
Abbreviations: SNP, single nucleotide polymorphism; NSCLC, non-small cell lung cancer; OS, overall survival; DSS, disease-specific survival. PLCO, Prostate, Lung, Colorectal and Ovarian cancer screening trial; HR, hazards ratio; CI, confidence interval; NUG, number of unfavorable genotypes.
10 with missing data were excluded.
Adjusted for age, sex, smoking status, histology, tumor stage, chemotherapy, surgery, radiotherapy and principal components.
Unfavorable genotypes were CHEK1 rs76744140_TC+CC, PRIM2 rs6939623_GT+TT, and CDK6 rs113181986 GG.
Combined and stratified analyses of the three independent SNPs associated with NSCLC survival in the PLCO dataset
To evaluate the effect of the three independent SNPs on NSCLC survival, we combined unfavorable genotypes (NUG) (i.e., CHEK1 rs76744140 TC+CC, PRIM2 rs6939623 GT+TT, and CDK6 rs113181986 GG) into a genetic score and then divided the patients into four groups (i.e., 0, 1, 2, and 3). As shown in Table 3, multivariable Cox analysis found that an increased genetic score was associated with a higher risk of death or poorer survival (Ptrend: P < 0.0001 for both OS and DSS). We then dichotomized patients into low-risk (0–1 NUG score) and high-risk groups (2–3 NUGs score). Patients in the high-risk group had a significant higher risk of death (OS: HR = 1.39, 95% CI = 1.19–1.62, and P < 0.0001; DSS: 1.35, 1.15–1.60, and 0.0003) as compared to those in the low-risk group. Moreover, we also generated the KM survival curves to assess associations between unfavorable genotypes and death risk (Figure 2).
Figure 2.
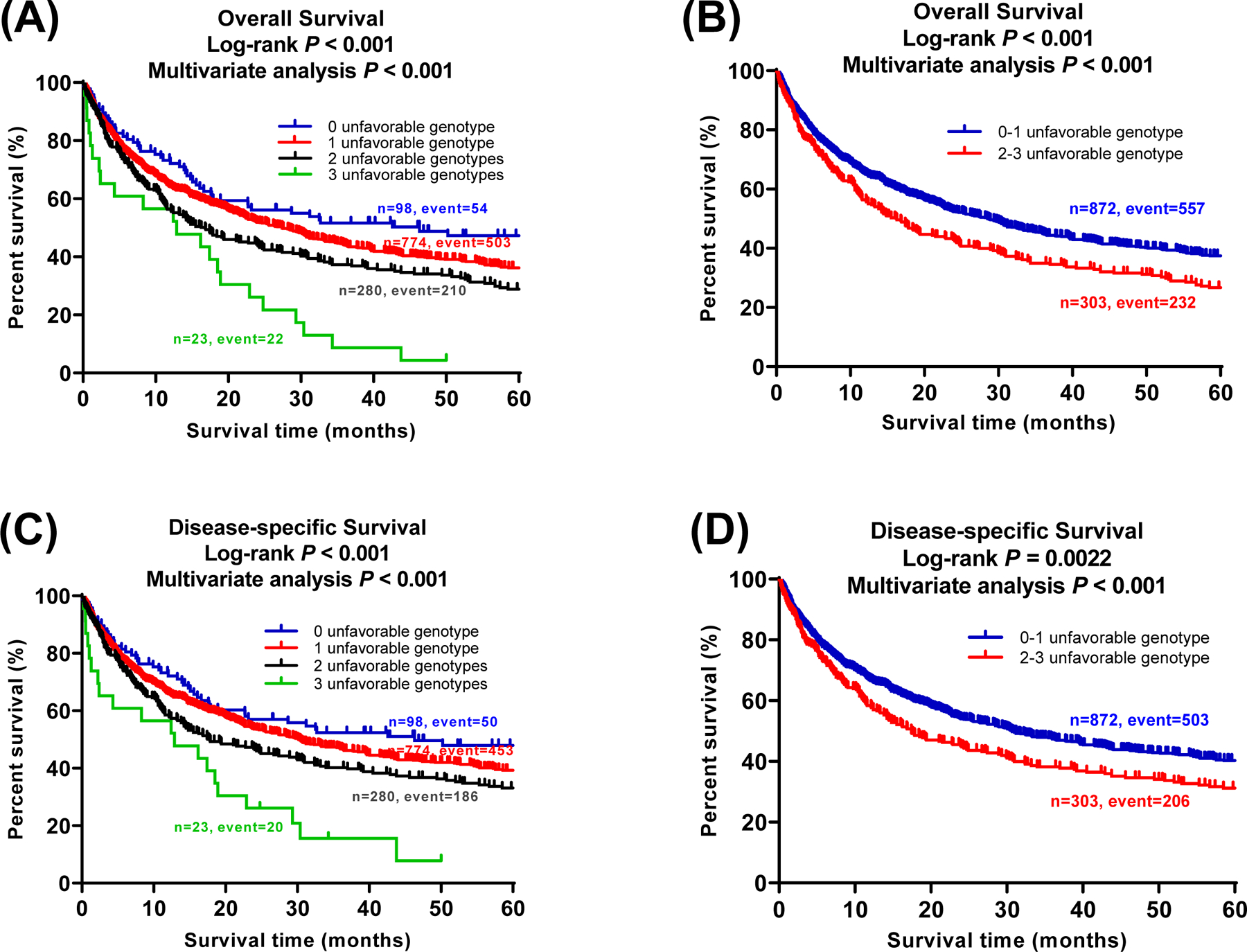
Three independent SNPs in mitotic phase-related pathway genes predict NSCLC survival. Kaplan-Meier (KM) survival curves for NSCLC patients of three identified SNPs and combined unfavorable genotypes in the PLCO trial: (A) 0, 1, 2, and 3 unfavorable genotypes in OS; (B) Dichotomized groups of the unfavorable genotypes into 0–1 and 2–3 in OS; (C) 0, 1, 2, and 3 unfavorable genotyes in DSS; (D) Dichotomized groups of the unfavorable genotypes into 0–1 and 2–3 in DSS.
We then compared the time-dependent AUC and ROC curves derived from the model for clinical covariates with or without the SNPs to quantify the predictive ability of genotypeon NSCLC OS and DSS. We also compared the AUC and ROC curves from the model for clinical covariates and 41 previously published SNPs with or without the three new SNPs to assess the predictive ability of new SNPs on NSCLC OS and DSS. The addition of the three new SNPs to the prediction model without the 41 previously published SNPs for 5-year survival rate significantly extended AUC from 87.00% to 88.13% for OS (P = 0.027) (Figure S3A–B) and from 88.54% to 89.11% for DSS (P = 0.047) (Figure S3C–D). The addition of the three new SNPs to the prediction model with the 41 previously published SNPs improved the efficiency of the model by reducing the 41 previously published SNPs to 28 SNPs; therefore, the current model with the three new SNPs is statistically more efficient, although the AUC for the 5-year survival rate with three new SNPs aloneas non-significantly increased from 90.70% to 90.98% for OS (P = 0.248) (Figure S3E–F) and from 90.66% to 90.95% for DSS (P = 0.284) (Figure S3G–H). We next performed stratified analysis to estimate whether the effects of combined unfavorable genotypes on NSCLC OS and DSS were modified by age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy, and surgery. For both 0–1 and 2–3 NUG groups, there were no significant interactions between unfavorable genotypes and clinical covariates, such as age, sex, and smoking status, in NSCLC OS and DSS (P > 0.05). However, interactions of unfavorable genotypes with histology, tumor stage, chemotherapy, radiotherapy, and surgery in modifying both OS and DSS were statistically significant (P < 0.05 for both) (Table S5). Besides, as non-proportional hazards were observed for several clinical covariates (i.e., stage, chemotherapy, and surgery; Figure S4), we also re-estimated the effects of these covariates and the three independent SNPs by using stratified Cox proportional hazards model by the combination of stage and chemotherapy. The proportion hazards assumption was satisfied for all covariates and the three independent SNPs in this model. As shown in Table S6, the risk effects of two risk SNPs rs76744140 and rs6939623 were significant (P = 0.026 and 0.001, respectively), and the SNP rs113181986 with a protective effect showed a marginal significance (P = 0.077).
In silico functional analysis
To assess biological functions of the three identified SNPs and their high-LD SNPs, we utilized three online bioinformatics tools (i.e., HaploReg, RegulomeDB, and the ENCODE project) to predict function. As shown in Table S7, we found that both CHEK1 rs76744140 T > C and CDK6 rs113181986 G > C might alter protein motifs and have an effect on enhancer histone marks, and a G > T change in PRIM2 rs6939623 might alter protein motifs. In addition, we found an additional 24 SNPs in high LD with the representative SNP rs113181986 in CDK6 have vrious potential functions (Table S8), while no SNPs are in high LD with rs76744140 in CHEK1 and rs6939623 in PRIM2. According to experimental data from the ENCODE project, rs76744140 is probably located on the substantial region of the H3K4Me1 layer and possibly affects transcriptional activities, but no obvious effects were observed for rs6939623 and rs113181986 (Figure S5). Ten of the 24 SNPs in high LD with rs113181986 are also located on potential functional regions (Figure S6).
eQTL analysis
The eQTL analysis was performed to further explore potential functions of the three identified SNPs. We found that the CHEK1 rs76744140 C allele significantly correlated with decreasing mRNA expression levels in normal lung tissues (P = 0.029, NES = −0.13) (Figure 3A) but not in whole blood tissues (Figure S7A) from the GTEx (V8) Project. Meanwhile, the CDK6 rs113181986 C allele also showed a significant correlation with the decreased mRNA expression levels of the gene in whole blood tissues (P = 0.048, NES = −0.08) (Figure 3B) but not in normal lung tissues (Figure S7B); nineteen of the 24 SNPs are in high LD with the representative SNP rs113181986 in CDK6 were also significantly correlated with the decreased mRNA expression levels of CDK6 in whole blood tissues (Figure S8). However, there was no significant correlation between the PRIM2 rs6939623 T allele and its corresponding mRNA expression levels in either normal lung (Figure S7C) or whole blood tissues (Figure S7D). Additionally, we also performed the eQTL analysis using data of the 373 European descendants in the 1000 Genomes Project, which indicated that these minor alleles of the identified SNPs were not significantly correlated with the mRNA expression levels of their corresponding genes (Figure S9).
Figure 3.
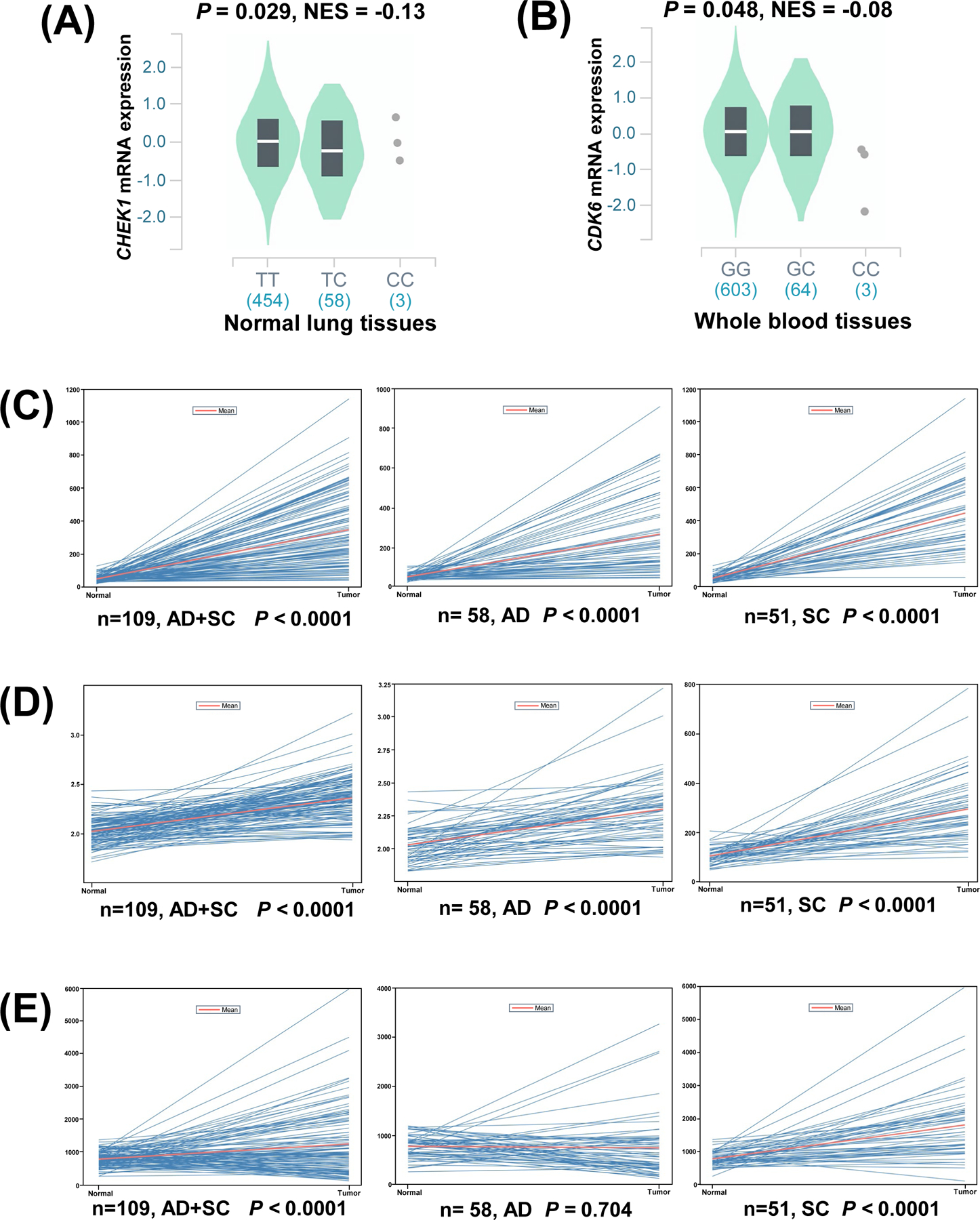
The eQTL analysis for CHEK1 rs76744140 and CDK6 rs113181986 from the GTEx (V8) database and comparison of mRNA expression levels of CHEK1, PRIM2 and CDK6 between lung cancer tissue and adjacent normal lung tissues in the TCGA dataset. (A) The correlation of rs76744140 genotypes and CHEK1 mRNA expression in normal lung tissues; (B) The correlation of rs113181986 genotypes and CDK6 mRNA expression in whole blood tissues; (C) Higher expression of CHEK1 were found both in the LUAD and LUSC tumor tissues compared to the normal tissues; (D) Higher expression of PRIM2 were found both in the LUAD and LUSC tumor tissues compared to the normal tissues; (E) Higher expression of CDK6 were found in the LUSC tumor tissues compared to the normal tissues.
Differential expression analysis in the TCGA dataset
We then explored the differences in mRNA expression levels of the three identified SNP-related genes between paired tumor tissues and adjacent normal tissues from the TCGA dataset. The mRNA expression levels of CHEK1 were significantly higher in lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), and the combined LUAD+LUSC tissues than that in adjacent normal tissues (P < 0.0001, 0.0001, and 0.0001, respectively) (Figure 3C), and the higher mRNA expression levels of CHEK1 were associated with a higher risk of lung cancer death or poorer survival (P < 0.0001) (Figure S10A). As compared with the adjacent normal tissues, the mRNA expression levels of PRIM2 were also significantly higher in LUAD, LUSC, and the combined LUAD+LUSC tissues (P < 0.0001, 0.0001, and 0.0001, respectively) (Figure 3D), while the higher mRNA expression levels of PRIM2 were not associated with a higher risk of death (P = 0.78) (Figure S10B). The mRNA expression levels of CDK6 were significantly higher in LUSC (P < 0.0001) and the combined LUAD+LUSC tissues (P < 0.0001) but not in LUAD tissues (P = 0.704) than that in adjacent normal tissues (Figure 3E), and the higher mRNA expression levels of CDK6 were also associated with a higher risk of dying of lung cancer (P = 0.0049) (Figure S10C).
Mutation analysis
It is likely that gene mutations in tumor tissues may also affect tumor progression. Therefore, we explored mutation status of CHEK1, PRIM2, and CDK6 in NSCLC tissues by using a publicly available database, cBioPortal for Cancer Genomics (Figure S11). We found that CHEK1 had an extremely low somatic mutation rate in different NSCLC datasets (e.g., 0.33% in the MSK 2017, 0.42% in the MSK PD1, 0.61% in the TSP, and 0.99% in the OncoSG 2020, respectively). Similarly, PRIM2 also had a relatively low somatic mutation rate in different NSCLC datasets (e.g., 0.45% in the TRACERx 2017, 2.17% in the TCGA LUAD, 2.27% in the TCGA 2016, and 2.79% in the TCGA LUSC, respectively). Additionally, CDK6 also displayed a low somatic mutation rate in different NSCLC datasets (e.g., 0.22% in the MSK 2017, 0.43% in the TCGA LUAD, 0.61% in the TCGA 2016, 0.67% in the TRACERx 2017, and 0.83% in the MSK PD1, respectively). Therefore, these low mutation frequencies of CHEK1, PRIM2, and CDK6 suggest that these rare mutations may not have a substantial effect on the mRNA expressiolevels of these three genes in NSCLC, if any.
Discussion
Recent studies in lung cancer found that the mitotic cell cycle is probably contributing to NSCLC progression. Mitotic phase-related genes are highly expressed in metastatic lung cancer tissues, affecting the migration, invasion, and epithelial-mesenchymal transition in lung cancer 31, 32. In the present study, we identified three genetic variants (i.e., CHEK1 rs76744140 T > C, PRIM2 rs6939623 G > T, and CDK6 rs113181986 G > C) in the mitotic phase-related pathway, which were significantly associated with NSCLC survival in Caucasian patients. Notably, an increased number of unfavorable genotypes of these three independent SNPs were significantly correlated with poor NSCLC OS and DSS. The combined unfavorable genotypes of these three independent SNPs in the model also predicted a significantly decreased 5-year survival of patients with NSCLC, suggesting that these three independent SNPs may be effective biomarkers for their clinical outcomes. Further analyses for functional relevance of identified SNPs and mRNA expression levels indicating that both the rs76744140 C and rs113181986 C variant alleles appeared to cause a decrease in mRNA expression levels of CHEK1 and CDK6, respectively. However, these correlations were not found for the PRIM2 rs6939623 T variant allele. Furthermore, the mRNA expression levels of CHEK1, PRIM2, and CDK6 were significantly lower in adjacent normal lung tissues than in lung tumor tissues, and higher mRNA expression levels of CHEK1 and CDK6 were significantly associated with poor outcomes in NSCLC. Therefore, our findings provided further support for theassociation between the genetic variants in the mitotic phase-related genes and NSCLC survival.
CHEK1 commonly referred to as CHK1, located on chromosome 11q24.2, is a serine/threonine-specific protein kinase in humans 33, which is required for the initiation of DNA damage checkpoints and has been shown to play a role in the normal (unperturbed) cell cycle 34. Recently, it has been reported that CHEK1 may be a critical gene in the development and prognosis of NSCLC 35. Studies also showed that CHEK1 expression was increased in NSCLC, compared with adjacent normal tissues 36, and higher expression of CHEK1 in NSCLC was associated with a poor overall survival 37. These results suggested that CHEK1 may play a potential oncogenic role in NSCLC. Consistent with these studies, our results indicated that CHEK1 might also have a possible oncogenic effect on NSCLC. Analysis of data from the ENCODE project, rs76744140 is located on an important region of the H3K4Me1 layer and possibly affects the transcriptional activities, which may modify CHEK1 mRNA expression by regulating histones and transcriptional activities. However, the CHEK1 rs76744140 C allele was found to be correlated with a decrease in mRNA expression of CHEK1 in normal lung tissues but with a poor survival in NSCLC. This inconsistency may be due to thecomplexity and uncertainty of tumor progression associated with unknown genetic changes in the tumors.
PRIM2, located on chromosome 6p11.2, also called DNA primase large subunit 38, which plays an essential role in the initiation of DNA synthesis, and knockdown of PRIM2decreased the viability of lung cancer cells and enhanced cell death 39. In the present study, the PRIM2 rs6939623 T allele was associated with an increased risk of death in patients with NSCLC. PRIM2 mRNA expression was significantly higher in lung cancer tissues than in adjacent normal tissues from TCGA data. However, we did not have data to support the correlation between the rs6939623 T allele and mRNA expression levels of PRIM2, and there was no significant difference in survival between higher and lower expression levels of PRIM2 in patients with NSCLC. Thus, the abnormal expression levels of PRIM2 in NSCLC may be correlated with other molecular mechanisms, which need to be further explored.
CDK6 is located on chromosome 7q21.2, and alterations of CDK6 could directly or indirectly affect the following hallmarks: cellular energy disturbance, maintaining proliferation signals, evading growth suppressors, and inducing angiogenesis 40. Besides, CDK6 might be altered through genomic instability 41. It was previously reported that LncRNA AWPPH could accelerate the progression of NSCLC by upregulating CDK6 42. Similarly, the nicotine-induced proliferative effects were rescued by the recovery of the expression levels of CDK6 in NSCLC 43. These findings also indicated that CDK6 might also have an oncogenic role in NSCLC, which is consistent with our results that the expression of CDK6 was increased in lung cancer tissues than in paired adjacent normal tissues, especially in the LUSC, and that higher expression of CDK6 was associated with a worse survival of patients with NSCLC. Furthermore, the CDK6 rs113181986 C allele correlated with decreased mRNA expression of CDK6 and a better outcome in patients with NSCLC, which supports the biological plausibility of the findings.
Although the present study identified three independent SNPs associated with NSCLC outcomes, there are still some limitations. First, both the two GWAS datasets were from Caucasian patients; thus, the results may not be generalized to other ethnic populations. Second, clinical information was limited, as some clinical variables (e.g., immunotherapy, nutrition status) were not available for analysis. Third, although the PLCO trial has a relatively large sample size, the number of patients in the subgroup was relatively small. Finally, the accurate molecular mechanisms underlying the observed associations between these three identified SNPs and NSCLC survival should be further investigated.
Supplementary Material
Novelty and impact.
The mitotic phase is a vital step in cell division and thus may be involved in cancer progression. Here, we investigated the role of genetic variants in the mitotic phase-related pathway genes in survival of patients with non-small cell lung cancer (NSCLC). We identified that three genetic variants located in CHEK1, PRIM2 and CDK6, respectively, were independently associated with the survival, which suggested that these genetic variants may be prognostic biomarkers for survival of patients with NSCLC.
Acknowledgments
The authors thank all the participants of the PLCO Cancer Screening Trial; and the National Cancer Institute for providing access to the data collected by the PLCO trial. The statements contained herein are solely those of the authors and do not represent or imply concurrence or endorsement by the National Cancer Institute. The authors would also like to acknowledge the dbGaP repository for providing cancer genotyping datasets. This work was supported by the National Institute of Health [CA090578, CA074386, CA092824, and CA209414]; the Duke Cancer Institute as part of the P30 Cancer Center Support Grant [NIH/NCI CA014236]; and the V Foundation for Cancer Research [D2017-19].
Abbreviations:
- AUC
area under the curve
- BFDP
Bayesian false discovery probability
- DSS
disease-specific survival
- ENCODE
Encyclopedia of DNA Elements
- eQTL
expression quantitative trait loci
- GTEx
Genotype-Tissue Expression
- GWAS
genome-wide association study
- HLCS
Harvard Lung Cancer Susceptibility
- HR
hazard ratio
- LD
linkage disequilibrium
- LUAD
lung adenocarcinoma
- LUSC
lung squamous cell carcinoma
- NSCLC
non-small cell lung cancer
- NUG
unfavorable genotypes
- OS
overall survival
- PLCO
Prostate, Lung, Colorectal, and Ovarian cancer screening trial
- ROC
receiver operating characteristic
- SNP
single nucleotide polymorphism
- TCGA
The Cancer Genome Atlas
Footnotes
Disclosure of conflict of interest
None.
Ethics Statement
Each of the original studies with the approval by the Institutional Review Boards of the Participating institutions received written informed consent from the participants.
Data Availability Statement
The datasets used in this study were obtained from dbGaP at http://www.ncbi.nlm.nih.gov/gap through the dbGaP accession numbers phs000336.v1.p1 and phs000093.v2.p2 and the Cancer Genome Atlas (TCGA) database (dbGaP Study Accession: phs000178.v11.p8). Further data that support the findings of the present study are available from the corresponding author upon reasonable request.
Reference
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018; 68: 394–424. [DOI] [PubMed] [Google Scholar]
- 2.Torre LA, Siegel RL, Jemal A. Lung Cancer Statistics. Adv Exp Med Biol 2016; 893: 1–19. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020; 70: 7–30. [DOI] [PubMed] [Google Scholar]
- 4.Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, Znaor A, Bray F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 2019; 144: 1941–53. [DOI] [PubMed] [Google Scholar]
- 5.Goldstraw P, Chansky K, Crowley J, Rami-Porta R, Asamura H, Eberhardt WE, Nicholson AG, Groome P, Mitchell A, Bolejack V. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer. J Thorac Oncol 2016; 11: 39–51. [DOI] [PubMed] [Google Scholar]
- 6.Akhurst T Staging of Non-Small-Cell Lung Cancer. PET Clin 2018; 13: 1–10. [DOI] [PubMed] [Google Scholar]
- 7.Vrankar M, Stanic K. Long-term survival of locally advanced stage III non-small cell lung cancer patients treated with chemoradiotherapy and perspectives for the treatment with immunotherapy. Radiol Oncol 2018; 52: 281–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pérez-Ramírez C, Cañadas-Garre M, Molina M, Robles AI, Faus-Dáder MJ, Calleja-Hernández M. Contribution of genetic factors to platinum-based chemotherapy sensitivity and prognosis of non-small cell lung cancer. Mutat Res 2017; 771: 32–58. [DOI] [PubMed] [Google Scholar]
- 9.Schrodi SJ, Mukherjee S, Shan Y, Tromp G, Sninsky JJ, Callear AP, Carter TC, Ye Z, Haines JL, Brilliant MH, Crane PK, Smelser DT, et al. Genetic-based prediction of disease traits: prediction is very difficult, especially about the future. Front Genet 2014; 5: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hung RJ, McKay JD, Gaborieau V, Boffetta P, Hashibe M, Zaridze D, Mukeria A, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 2008; 452: 633–7. [DOI] [PubMed] [Google Scholar]
- 11.Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, Manolescu A, Thorleifsson G, Stefansson H, Ingason A, Stacey SN, Bergthorsson JT, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature 2008; 452: 638–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, Dong Q, Zhang Q, Gu X, Vijayakrishnan J, Sullivan K, Matakidou A, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet 2008; 40: 616–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKay J, Hung R, Han Y, Zong X, Carreras-Torres R, Christiani D. Large scale genetic analysis identifies novel loci and histological variability in susceptibility to lung cancer. Nat Genet 2017; 49: 1126–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen K, Liu H, Liu Z, Luo S, Patz EF Jr., Moorman PG, Su L, Shen S, Christiani DC, Wei Q. Genetic variants in RUNX3, AMD1 and MSRA in the methionine metabolic pathway and survival in nonsmall cell lung cancer patients. Int J Cancer 2019; 145: 621–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sepesi B, Ye Y, Mitchell KG, Zhang L, Gu J, Ji L, Antonoff MB, Hofstetter WL, Rice DC, Mehran RJ, Walsh GL, Vaporciyan AA, et al. Genetic variants in cytokine signaling pathways and clinical outcomes in early-stage lung cancer patients. J Thorac Cardiovasc Surg 2018; 155: 2635–45 e15. [DOI] [PubMed] [Google Scholar]
- 16.Frade JM, Ovejero-Benito MC. Neuronal cell cycle: the neuron itself and its circumstances. Cell Cycle 2015; 14: 712–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wieser S, Pines J. The biochemistry of mitosis. CSH PERSPECT BIOL 2015; 7: a015776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science 2008; 319: 1352–5. [DOI] [PubMed] [Google Scholar]
- 19.Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, Hickson ID. Replication stress activates DNA repair synthesis in mitosis. Nature 2015; 528: 286–90. [DOI] [PubMed] [Google Scholar]
- 20.Penna LS, Henriques JAP, Bonatto D. Anti-mitotic agents: Are they emerging molecules for cancer treatment? Pharmacol Ther 2017; 173: 67–82. [DOI] [PubMed] [Google Scholar]
- 21.Weissfeld JL, Schoen RE, Pinsky PF, Bresalier RS, Doria-Rose VP, Laiyemo AO, Church T, Yokochi LA, Yurgalevitch S, Rathmell J, Andriole GL, Buys S, et al. Flexible sigmoidoscopy in the randomized prostate, lung, colorectal, and ovarian (PLCO) cancer screening trial: added yield from a second screening examination. J Natl Cancer Inst 2012; 104: 280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tryka KA, Hao L, Sturcke A, Jin Y, Wang ZY, Ziyabari L, Lee M, Popova N, Sharopova N, Kimura M, Feolo M. NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res 2014; 42: D975–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, Hao L, Kiang A, Paschall J, Phan L, Popova N, Pretel S, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet 2007; 39: 1181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lappalainen T, Sammeth M, Friedländer MR, t Hoen PA, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, Barann M, Wieland T, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013; 501: 506–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhai R, Yu X, Wei Y, Su L, Christiani DC. Smoking and smoking cessation in relation to the development of co-existing non-small cell lung cancer with chronic obstructive pulmonary disease. Int J Cancer 2014; 134: 961–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics 2007; 23: 1294–6. [DOI] [PubMed] [Google Scholar]
- 27.Park JH, Geum DI, Eisenhut M, van der Vliet HJ, Shin JI. Bayesian statistical methods in genetic association studies: Empirical examination of statistically non-significant Genome Wide Association Study (GWAS) meta-analyses in cancers: A systematic review. Gene 2019; 685: 170–78. [DOI] [PubMed] [Google Scholar]
- 28.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, Cherry JM, Snyder M. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 2012; 22: 1790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res 2016; 44: D877–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Consortium G Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015; 348: 648–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu X, Liu X, Li J, Ren F. Identification and Integrated Analysis of Key Biomarkers for Diagnosis and Prognosis of Non-Small Cell Lung Cancer. Med Sci Monit 2019; 25: 9280–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang H, He X, Yu W, Yue B, Yu Z, Qin Y. Mitotic Arrest-Deficient Protein 2B Overexpressed in Lung Cancer Promotes Proliferation, EMT, and Metastasis. Oncol Res 2019; 27: 859–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 1997; 277: 1497–501. [DOI] [PubMed] [Google Scholar]
- 34.Patil M, Pabla N, Dong Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol Life Sci 2013; 70: 4009–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L, Qu J, Liang Y, Zhao D, Rehman FU, Qin K, Zhang X. Identification and validation of key genes with prognostic value in non-small-cell lung cancer via integrated bioinformatics analysis. Thorac Cancer 2020; 11: 851–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu X, Zhang Y, Ma X, Pertsemlidis A. miR-195 potentiates the efficacy of microtubule-targeting agents in non-small cell lung cancer. Cancer Lett 2018; 427: 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu B, Qu J, Xu F, Guo Y, Wang Y, Yu H, Qian B. MiR-195 suppresses non-small cell lung cancer by targeting CHEK1. Oncotarget 2015; 6: 9445–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shiratori A, Okumura K, Nogami M, Taguchi H, Onozaki T, Inoue T, Ando T, Shibata T, Izumi M, Miyazawa H, et al. Assignment of the 49-kDa (PRIM1) and 58-kDa (PRIM2A and PRIM2B) subunit genes of the human DNA primase to chromosome bands 1q44 and 6p11.1-p12. Genomics 1995; 28: 350–3. [DOI] [PubMed] [Google Scholar]
- 39.Yuan B, Liao F, Shi ZZ, Ren Y, Deng XL, Yang TT, Li DY, Li RF, Pu DD, Wang YJ, Tan Y, Yang Z, et al. Dihydroartemisinin Inhibits the Proliferation, Colony Formation and Induces Ferroptosis of Lung Cancer Cells by Inhibiting PRIM2/SLC7A11 Axis. Onco Targets Ther 2020; 13: 10829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, Cascante M. Targeting cell cycle regulation in cancer therapy. Pharmacol Ther 2013; 138: 255–71. [DOI] [PubMed] [Google Scholar]
- 41.Negrini S, Gorgoulis V, Halazonetis T. Genomic instability an evolving hallmark of cancer. NAT REV MOL CELL BIO 2010; 11: 220–28. [DOI] [PubMed] [Google Scholar]
- 42.Wu D, Qin BY, Qi XG, Hong LL, Zhong HB, Huang JY. LncRNA AWPPH accelerates the progression of non-small cell lung cancer by sponging miRNA-204 to upregulate CDK6. Eur Rev Med Pharmacol Sci 2020; 24: 4281–87. [DOI] [PubMed] [Google Scholar]
- 43.Liu Z, Lu C, Zhao G, Han X, Dong K, Wang C, Guan JZ, Wang Z. Downregulation of miR-218 by nicotine promotes cell proliferation through targeting CDK6 in non-small cell lung cancer. J Cell Biochem 2019; 120: 18370–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used in this study were obtained from dbGaP at http://www.ncbi.nlm.nih.gov/gap through the dbGaP accession numbers phs000336.v1.p1 and phs000093.v2.p2 and the Cancer Genome Atlas (TCGA) database (dbGaP Study Accession: phs000178.v11.p8). Further data that support the findings of the present study are available from the corresponding author upon reasonable request.