

Abstract
Ferroptosis is a caspase-independent, iron-dependent form of regulated necrosis extant in traumatic brain injury, Huntington disease, and hemorrhagic stroke. It can be activated by cystine deprivation leading to glutathione depletion, the insufficiency of the antioxidant glutathione peroxidase-4, and the hemolysis products hemoglobin and hemin. A cardinal feature of ferroptosis is extracellular signal-regulated kinase (ERK)1/2 activation culminating in its translocation to the nucleus. We have previously confirmed that the mitogen-activated protein (MAP) kinase kinase (MEK) inhibitor U0126 inhibits persistent ERK1/2 phosphorylation and ferroptosis. Here, we show that hemin exposure, a model of secondary injury in brain hemorrhage and ferroptosis, activated ERK1/2 in mouse neurons. Accordingly, MEK inhibitor U0126 protected against hemin-induced ferroptosis. Unexpectedly, U0126 prevented hemin-induced ferroptosis independent of its ability to inhibit ERK1/2 signaling. In contrast to classical ferroptosis in neurons or cancer cells, chemically diverse inhibitors of MEK did not block hemin-induced ferroptosis, nor did the forced expression of the ERK-selective MAP kinase phosphatase (MKP)3. We conclude that hemin or hemoglobin-induced ferroptosis, unlike glutathione depletion, is ERK1/2-independent. Together with recent studies, our findings suggest the existence of a novel subtype of neuronal ferroptosis relevant to bleeding in the brain that is 5-lipoxygenase-dependent, ERK-independent, and transcription-independent. Remarkably, our unbiased phosphoproteome analysis revealed dramatic differences in phosphorylation induced by two ferroptosis subtypes. As U0126 also reduced cell death and improved functional recovery after hemorrhagic stroke in male mice, our analysis also provides a template on which to build a search for U0126's effects in a variant of neuronal ferroptosis.
SIGNIFICANCE STATEMENT Ferroptosis is an iron-dependent mechanism of regulated necrosis that has been linked to hemorrhagic stroke. Common features of ferroptotic death induced by diverse stimuli are the depletion of the antioxidant glutathione, production of lipoxygenase-dependent reactive lipids, sensitivity to iron chelation, and persistent activation of extracellular signal-regulated kinase (ERK) signaling. Unlike classical ferroptosis induced in neurons or cancer cells, here we show that ferroptosis induced by hemin is ERK-independent. Paradoxically, the canonical MAP kinase kinase (MEK) inhibitor U0126 blocks brain hemorrhage-induced death. Altogether, these data suggest that a variant of ferroptosis is unleashed in hemorrhagic stroke. We present the first, unbiased phosphoproteomic analysis of ferroptosis as a template on which to understand distinct paths to cell death that meet the definition of ferroptosis.
Keywords: brain hemorrhage, cell death, ferroptosis, MAP signaling, phosphoproteomics, stroke
Introduction
Iron dyshomeostasis is observed in many neurologic conditions, including hemorrhagic stroke. The release of red blood cells into the brain following brain bleeding ultimately leads to lysis and the release of hemoglobin and its degradation product heme (Cao et al., 2016; Dang et al., 2017; Liu et al., 2019). Heme is then taken up into neurons where it leads to iron overload and cell death (Robinson et al., 2009). Indeed, a causal role for iron in hemorrhagic stroke is substantiated by some studies which show that nonselective iron chelators reduce death and total iron content (Hua et al., 2008; Gu et al., 2009; Wu et al., 2011; Li et al., 2017a). A major challenge in the therapeutic extension of iron chelators to humans is inhibiting pathologic iron toxicity without affecting normal physiological roles for iron. Indeed, iron is a cofactor for many enzymes in neurons (Mills et al., 2010). Accordingly, there is an urgent need to identify strategies to inhibit iron-mediated toxicity that can preserve the physiological function of iron.
A potential solution to this conundrum has emerged via the increased understanding of a regulated pathway to necrosis called ferroptosis. Ferroptosis is an iron-dependent, caspase-independent form of necrosis that is classically activated by cyst(e)ine deprivation and glutathione. This form of cell death has recast iron, a highly redox active metal, as a mediator of cell death signaling rather than nonspecific oxidation (Ratan, 2020). The importance of signaling to ferroptosis execution was highlighted in the early 2000s by studies from the DeFranco laboratory that demonstrated that ferroptotic death can be completely abrogated by pharmacological or molecular inhibition of the Ras-Raf-mitogen-activated protein (MAP) kinase kinase (MEK)-extracellular signal-regulated kinase (ERK) signaling pathway (Levinthal and Defranco, 2005). As expected from these results, mutations in the Ras protooncogene associated with tumorigenesis have been shown to confer sensitivity to ferroptosis induced by cystine deprivation and glutathione depletion (Dixon et al., 2012). Additionally, GW-5074, an inhibitor of c-Raf, the kinase upstream of MEK has also been shown to inhibit ferroptosis in neurons (Chin et al., 2004). Following these studies, the MEK inhibitor U0126 has emerged as one of several important chemical criteria by which cell death is defined as ferroptotic (Dixon et al., 2012). Hemin-induced and hemoglobin-induced death meet all of these criteria (Zille et al., 2017). As expected, we previously showed that U0126 completely abrogated cell death induced by hemoglobin and hemin consistent with the notion that these stimuli induce ferroptosis in hemorrhagic stroke in vitro and in vivo (Zille et al., 2017).
Here, we extend the protective effects of U0126 to demonstrate that it reduces cell death and improves functional recovery after hemorrhagic stroke in vivo. Armed with its beneficial effects in a disease-relevant model of brain hemorrhage, we used the experimental leverage of the in vitro model to probe the mechanism by which U0126 inhibits hemin-induced ferroptosis. Unexpectedly, we show that in contrast to classical ferroptosis in neurons and cancer cells (Levinthal and Defranco, 2005; Zille et al., 2019), hemin induces ferroptosis independent of Ras-Raf-MEK-ERK signaling. Using unbiased phosphoproteomics, we provide further hypotheses how U0126 is protective in ferroptosis. Taken together with other recent observations, these findings define hemin-induced death as a novel, ERK-independent and transcription-independent, but 5-lipoxygenase-dependent variant of classical ferroptosis.
Materials and Methods
Chemicals and reagents are listed in the Extended Data.
Study approval
All animal procedures were approved by the Weill Cornell Medicine Institutional Animal Care and Use Committee (approval #2011-0019 for in vivo experiments, #0707-633A for in vitro experiments on primary cortical neurons) and conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and ARRIVE guidelines.
Mouse model of intracerebral hemorrhage (ICH) and U0126 administration
Male C57BL/6 mice (8–10 weeks old) were purchased from Charles River Laboratories and housed at 68−72°F, 30–70% humidity, under 12/12 h light/dark cycle, with food (PicoLab Rodent diet 5053, LabDiet) and water freely accessible. Mice were randomly assigned to groups.
We induced ICH in mice using collagenase as previously described (Karuppagounder et al., 2016). Sham animals received saline injections instead of collagenase. For intraperitoneal injections, mice received daily intraperitoneal injections of 25 mg/kg U0126 or vehicle for 7 d (n = 13 for sham+vehicle, n = 17 for ICH+vehicle, n = 16 for ICH + 25 mg/kg U0126). Four ICH and six ICH+U0126 animals died from the surgery. For intracerebroventricular injections, mice received a single intracerebroventricular injection of 12 µg U0126 or vehicle (n = 16 per group) 2 h after ICH. Two animals died from the surgery but were replaced to have a total of n = 16. To assess the hematoma volume and hemispheric swelling, a separate set of mice received 12 µg U0126 (n = 6) or vehicle (n = 6) intracerebroventricularly 2 h after ICH that were killed 22 h later. One animal of the vehicle group died from the surgery (resulting in n = 5 for analysis in the vehicle group). To assess neuronal cell death by Fluoro-jade, a separate set of mice received 12 µg U0126 (n = 6) or vehicle (n = 6) intracerebroventricularly 2 h after ICH and the mice were killed 22 h later.
Behavioral analysis
To assess the sensorimotor function, the corner task and adhesive tape removal task were performed as previously described (Karuppagounder et al., 2016). The experimenter (Y.C.) was blinded to the group allocation. In the intraperitoneal injection study, one animal of the ICH+vehicle group at day 1 and one of the animals of the ICH+U0126 group at day 7 after ICH did not remove the tape from any of their paws within 5 min and were therefore excluded from the analysis of that particular day.
Lesion volume assessment
For the lesion volume assessment, we collected 30-µm-thick cryosections at an interval of 0.2 mm using a Leica CM 3050 S cryostat (Leica). The photographs of whole sections were captured with SteREO Discovery.V12 microscope (Zeiss). The ipsilateral and contralateral hemisphere as well as the hematoma (identified by blood deposition) were manually outlined using ImageJ v.1.49 (http://imagej.nih.gov/ij/) by an experimenter (M.Z.) blinded to the group allocation.
The volume of the hemispheres and hematomas were calculated by integrating the area on each section multiplied by the distance between adjacent sections (0.2 mm). The final infarct volume was reported after subtracting the volume difference between contralateral and ipsilateral side to correct for swelling. The percent hemispheric swelling was calculated using the difference in volume between the two hemispheres divided by the contralateral hemisphere, % hemispheric swelling = [(ipsilateral volume − contralateral volume)/contralateral volume] × 100 (Lin et al., 1993).
Fluoro-jade B staining and NeuN immunofluorescence
Neuronal cell death was assessed using Fluoro-jade B staining and NeuN immunofluorescence. The animals were transcardially perfused with PBS followed by 4% paraformaldehyde. Then, the brains were immersed in 4% paraformaldehyde for 24 h. We collected 30-µm-thick free-floating sections at an interval of 150 µm using a cryostat (Leica CM1800).
We performed Fluoro-jade B staining according to the instructions of the manufacturer. Briefly, the free-floating brain sections were immersed in a graded series of alcohol solutions (50%, 70%, and 100% ethanol for 5 min each), followed by a 0.06% potassium permanganate solution (KMnO4 in distilled water) for 15 min. The sections were washed with double distilled (ddH2O) and then immersed in a 0.001% Fluoro-jade B staining solution (Fluoro-jade B in ddH2O) for 20 min while gently shaking in the dark. Then, we washed the sections with ddH2O and mounted them on coated slides (TruBond 380; Fisher Scientfic).
For NeuN immunofluorescence, the free-floating sections were blocked for 1 h in blocking solution (3% bovine serum albumin and 0.5% Triton X-100 in PBS). We incubated the sections with mouse anti-NeuN in blocking solution overnight at 4°C. The next day, the sections were washed three times in PBS and incubated with the secondary antibody donkey anti-mouse Alexa Fluor 568 in blocking solution. The sections were then rinsed three times in PBS and mounted on coated slides.
Images were taken on a Nikon Eclipse Ti-U confocal microscope. Six pictures were collected from the cortex and perihematomal region and three pictures were acquired from the striatum. The images were quantified using ImageJ v.1.49 (http://imagej.nih.gov/ij/) by an experimenter (Y.C.) blinded to the group allocation.
Cell culture
Primary cortical neurons were obtained from CD-1/ICR mice at embryonic day 14.5 as previously described (Ratan et al., 1994a) and cultured at 37°C in a humidified 5% CO2 atmosphere (density: 105 cells/well in 96-well plates). Immortalized hippocampal neuroblasts (HT22 cells) were cultured in DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin and treated when 70% confluent.
In vitro models of ferroptosis
To induce ferroptosis, we used the glutamate analog L-homocysteic acid (HCA), erastin, hemin or hemoglobin. Primary neurons were exposed to 5 mm HCA, 5 μm erastin, 100 μm hemin or 1.5 μm hemoglobin and HT22 cells to 25–100 μm hemin or 10 mm HCA as previously established (Zille et al., 2017; Zille et al., 2019). Putative inhibitors were added at the same time as the ferroptotic stimulus. We determined cell viability using 3-(4,5-dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide (MTT) as previously described (Mosmann, 1983), 18–24 h after addition of the ferroptotic stimulus (Zille et al., 2017; Zille et al., 2019). Plates were measured in a SpectraMax Plus Microplate Reader using SoftMax Pro v4.7.1 (both Molecular Devices). We confirmed the results of the MTT assay by LIVE/DEAD assay and fluorescence microscopy at Nikon Eclipse TS100 microscope using Nikon DS-L3 (Nikon Instruments).
Plasmid transfection
HT22 cells were transfected with MAP kinase phosphatase 3 [Mkp3; also known as dual-specificity phosphatase 6 (Dusp6)], Mkp3 C293S or pSG5 (empty vector) using Lipofectamine 2000 according to the manufacturer's instructions (Thermo Fisher Scientific). Twenty-four hours after transfection, we treated the cells with hemin or glutamate analog (HCA) to induce ferroptosis.
Immunoblot analysis
For whole-cell extracts, we prepared protein extracts using 1% Triton buffer (25 mm Tris pH 7.4, 100 mm NaCl, 1 mm EGTA, 1% Triton X-100, 1% protease inhibitors, and 2.5 mm sodium orthovanadate). For cytoplasmic and nuclear extracts, cytoplasmic fractions were obtained in cytoplasmic buffer (10 mm HEPES, 10 mm potassium chloride, 2 mm magnesium chloride, 0.1 mm EDTA, 3 mm phenylmethanesulfonyl fluoride, 3 mm dithiothreitol, and 1% protease inhibitors) and subsequently nuclear fractions were extracted using nuclear buffer (50 mm HEPES, 50 mm potassium chloride, 300 mm magnesium chloride, 0.1 mm EDTA, 3 mm phenylmethanesulfonyl fluoride, 3 mm dithiothreitol, 10% glycerol, and 1% protease inhibitors). Samples were electrophoresed under reducing conditions on NuPAGE gels and transferred to nitrocellulose membrane. Antibodies (dilutions are specified in the Extended Data) against phospho-ERK1/2, total ERK1/2, MKP3, phopsho-5′-AMP-activated protein kinase (AMPK), total AMPK, γ-tubulin, GAPDH, and Histone H4 were incubated overnight at 4°C. Secondary antibodies were incubated for 1 h at room temperature. Proteins were detected using Odyssey infrared imaging system (LI-COR Biosciences).
RNA extraction and real-time PCR
Total RNA was prepared using NucleoSpin RNA isolation kit according to established protocols. We performed Real-time PCR using Taqman RNA-to-CT 1-Step Kit for mouse Dusp1/Mkp1 (#Mm00457274_g1) and Dusp6/Mkp3 (#Mm00518185_m1) at a 7500 Real-Time PCR System (Applied Biosystems). Expression levels were normalized to mouse β-actin endogenous control.
Phosphoproteomics experiments
Primary cortical neurons (107 cells) were treated with (1) vehicle, (2) 5 μm erastin for 7 h, (3) 5 μm erastin + 10 μm U0126 for 7 h, (4) 100 μm hemin for 5 h, (5) 100 μm hemin + 10 μm U0126 for 5 h. Three biological replicates with each two technical replicates for each group were used. After the treatment, the cells were washed with PBS, centrifuged at 150 × g, and stored at −80°C until use.
In-solution digestion
Pellets were thawed and resuspended in 50 µl 8 m urea in 25 mm ammonium bicarbonate buffer with phosphatase inhibitors (2 µl of Sigma Phosphatase Inhibitor Cocktails 2 and 3) at 4°C, and homogenized using a probe sonicator. We estimated the amount of protein in the lysates per triplicate using 1 µl of the lysate, with a bicinchoninic acid protein assay kit (Micro BCA Protein Assay kit, Thermo Scientific). Aliquots containing 230 µg of protein were treated with 8.8 mm dithiothreithol (DTT) at 56°C for 15 min, followed by a 30 min incubation at room temperature in the dark with 15 mm iodoacetamide. We then diluted the samples fourfold with 100 mm ammonium bicarbonate to reduce urea concentration to 2 m, and then added 2% (w/w) modified trypsin (Trypsin Gold, Mass Spectrometry grade; Promega). The pH was adjusted to 8.0 with 250 mm ammonium bicarbonate, and the samples were incubated 12 h at 37°C. After that, another aliquot of trypsin was added (2% w/w) and digested for additional 6 h. After this, we acidified the samples with formic acid to a final concentration of 5%. The digests were then desalted using a MAX-RP Sep Pak classic C18 cartridge (Waters) following the manufacturer's protocol. Sep Pak eluates were dried-evaporated in preparation for labeling with TMTPro-16 label plex reagents.
Tandem mass tag (TMT) labeling
We resuspended the samples in 40 µl 0.5 m triethylammonium bicarbonate pH 8.0. Dried samples were labeled according to TMTPro-16 label plex kit instructions (ThermoFisher Scientific). Briefly, TMT reagents were dissolved in acetonitrile at 25 µg/µl, and 40 µl of these stocks added to the samples (1000 µg reagent, over four times peptide amount). After incubation for 1 h at room temperature, the samples were quenched with hydroxylamine, and all 15 samples were combined over 60 ml 0.1% fomic acid, and desalted using a C18 SepPak. The Sep Pak eluate was dried in preparation for phosphopeptide enrichment.
Enrichment of phosphorylated peptides using titanium dioxide
We performed phosphopeptide enrichment in an AKTA Purifier (GE Healthcare) using 5 µm titanium dioxide (TiO2) beads (GL Sciences) in-house packed into a 2.0 mm × 2 cm analytical guard column (Upchurch Scientific). Combined TMT labeled tryptic digests were resuspended in 240 µl buffer containing 35% MeCN, 200 mm NaCl, 0.4% trifluoroacetic acid (TFA) and loaded onto the TiO2 column at a flow rate of 2 ml/min. The column was then washed for 2 min with 35% MeCN, 200 mm NaCl, 0.4% TFA to remove non phosphorylated peptides. We eluted the phosphopeptides from the column using 1 m potassium phosphate monobasic (KH2PO4) at a flow rate of 0.5 ml/min for 30 min directly onto an on-line coupled C18 macrotrap peptide column (Michrom Bioresources). This column was washed with 5% MeCN, 0.1% TFA for 14 min and the adsorbed material was eluted in 400 µl of 50% MeCN, 0.1% TFA at a flow rate of 0.25 ml/min. The eluate was solvent evaporated and then resuspended in 240 ml 20 mm ammonium formiate pH 10.4 for fractionation of the peptide mixture by high pH reverse phase chromatography.
High pH reverse phase chromatography
We fractionated the phosphopeptides-enriched sample on an AKTA purifier system using a Phenomenex Gemini 5u C18 110A 150 × 4.60 mm column, operating at a flow rate of 0.550 µl/min. Buffer A consisted of 20 mm ammonium formate (pH 10.4), and buffer B consisted of 20 mm ammonium formate in 90% acetonitrile (pH 10.4). Gradient details were as follows: 1% to 9% B in 3.6 min, 9% B to 49% B in 36.3 min, 49% B to 70% B in 2.7 min, 70% B back down to 1% B in 1.8 min. Fifty peptide-containing fractions were collected, evaporated and resuspended in 0.1% formic acid combining early and late chromatographic fractions into 28 samples.
Mass spectrometry analysis
Samples from reverse phase fractionation were run onto a 2 µm, 75 mm ID × 50 cm PepMap RSLC C18 EasySpray column (Thermo Scientific). Three-hour acetonitrile gradients (2–30% in 0.1% formic acid) were used to separate peptides, at a flow rate of 300 nl/min, for analysis in a Orbitrap Lumos Fusion (Thermo Scientific) in positive ion mode. We acquired MS spectra between 375 and 1500 m/z with a resolution of 120,000. For each MS spectrum, multiply charged ions over the selected threshold (2E4) were selected for tandem mass spectrometry (MS/MS) in cycles of 3 s with an isolation window of 0.7 m/z. Precursor ions were fragmented by HCD using stepped relative collision energies of 30, 35, and 40 to ensure efficient generation of sequence ions as well as TMT reporter ions. MS/MS spectra were acquired in centroid mode with resolution 50,000 from m/z = 110. A dynamic exclusion window was applied which prevented the same m/z from being selected for 30 s after its acquisition.
Peptide and protein identification and TMT quantitation
We generated peak lists using PAVA in-house software (Guan et al., 2011a). All generated peak lists were searched against the mouse subset of the SwissProt database (SwissProt.2019.07.31), using Protein Prospector (Clauser et al., 1999) with the following parameters: enzyme specificity was set as trypsin, and up to two missed cleavages per peptide were allowed. Carbamidomethylation of cysteine residues, and TMTPro16plex labeling of lysine residues and N terminus of the protein were allowed as fixed modifications. N-acetylation of the N terminus of the protein, loss of protein N-terminal methionine, pyroglutamate formation from of peptide N-terminal glutamines, oxidation of methionine and phosphorylation on serine, threonine and tyrosine were allowed as variable modifications. Mass tolerance was 10 ppm in MS and 30 ppm in MS/MS. We estimated the false positive rate by searching the data using a concatenated database which contains the original SwissProt database as well as a version of each original entry where the sequence has been randomized. A 1% false discovery rate was permitted at the protein and peptide level. For quantitation, only unique peptides were considered; peptides common to several proteins were not used for quantitative analysis. Relative quantization of peptide abundance was performed via calculation of the intensity of reporter ions corresponding to the different TMT labels, present in MS/MS spectra. Intensities were determined by Protein Prospector. We used summed intensity per sample on each TMT channel for all identified spectra to normalize individual intensity values. Normalizes values were batch-corrected using pyComBat (Behdenna et al., 2021). Relative abundances were calculated as ratios versus the average intensity levels in the three channels corresponding to control samples. Spectra representing replicate measurements of the same peptide were kept and used to calculate the dispersion and the significance threshold for the analysis. For total protein relative levels, peptide ratios were aggregated to the protein levels using median values of the log2 ratios.
Sequences (11 aa) around the phosphorylation sites of interest were aligned so the phosphorylated residue occupied position 6. Aligned sequences were uploaded into the Motif & Logo Analysis Tools in the PhophositePlus website.
Kinase enrichment analysis
Proteins with a phosphopeptide increased (p < 0.001) after erastin or hemin treatment compared with control were separately submitted to KEA3 (Kuleshov et al., 2021). Additionally, proteins with a phosphopeptide decreased (p < 0.001) in the erastin+U0126 compared with erastin samples and in the hemin+U0126 versus hemin samples were submitted to KEA3. The top 10 ranked kinases using the MeanRank score are reported.
Experimental design and statistical analyses
The sample size for functional recovery in vivo was calculated a priori by predicting detectable differences to reach a power of 0.80 at a significance level of <0.05, assuming a 40% difference in the mean and a SD of 25%. Mice were randomly assigned to the groups. For the in vitro experiments, indicated sample sizes refer to biological replicates, i.e., separate preparations of primary cortical neurons from embryos of different mice or different, independent passages of HT22 cells. We randomly assigned the plates to the experiments and experimental groups. Treatment of the chemical inhibitors of the Ras-Raf-MEK-ERK pathway and metformin were applied at the same time as the inducers of ferroptosis (cotreatment).
Normality was evaluated by the Kolmogorov–Smirnov test, variance homogeneity using the Levené test, and sphericity by the Mauchly test. For the comparisons of two groups, the Student's t test was performed when normality and variance homogeneity were met, otherwise the Mann–Whitney U test was done. For multiple comparisons with normal distribution and variance homogeneity, we performed a one-way ANOVA followed by the post hoc Bonferroni test. In case one of the criteria was not met, the Kruskal–Wallis test was performed followed by the post hoc Mann–Whitney U test with α-correction according to Bonferroni–Holm to adjust for the inflation of Type I error because of multiple testing. For the repeated testing with covariates, a repeated measures ANOVA was performed with Greenhouse–Geisser adjustment if sphericity was not given. The data are represented as the mean ± SD except for nonparametric data, where the medians are given. A value of p < 0.05 was considered statistically significant. The detailed statistical analyses can be found in Extended Data Figures 1-2, 1-3, 2-1, 3-1, 3-2, 4-1, 4-2, 5-1, 5-2, 6-1. All statistical analyses were performed with IBM SPSS version 23 (RRID:SCR_002865).
Data availability
The phosphoproteomics data have been deposited to the PRIDE Archive (http://www.ebi.ac.uk/pride/archive/) via the PRIDE partner repository with the data set identifier PXD029318.
Results
The MEK and ferroptosis inhibitor U0126 improves functional recovery after ICH in vivo
Prior studies from our lab showed that the canonical MEK inhibitor U0126 abrogated ferroptosis in cortical neurons induced by hemin or hemoglobin (Zille et al., 2017). Multiple studies have implicated ferroptotic death in mediating impairment after ICH in mice (Li et al., 2017b, 2018; Zille et al., 2017; Karuppagounder et al., 2018; Alim et al., 2019). Accordingly, we wanted to determine whether U0126 promotes improved behavior in mice when delivered after ICH. Contrary to reports in the literature, we did not find that intraperitoneal administration of U0126 improved behavioral outcomes (adhesive removal and corner task) compared with a vehicle control (Extended Data Fig. 1-1). It is likely that, under the conditions of our experiments, U0126 does not cross the blood-brain barrier. Therefore, we examined the effects of U0126 delivered directly into the brain. We observed that 12 μg U0126 delivered via a single dose intracerebroventricularly 2 h after ICH significantly improved measures of spatial neglect (corner task) and sensory neglect (tape removal task) at 3 and 7 d after ICH in mice (Fig. 1A).
Figure 1.
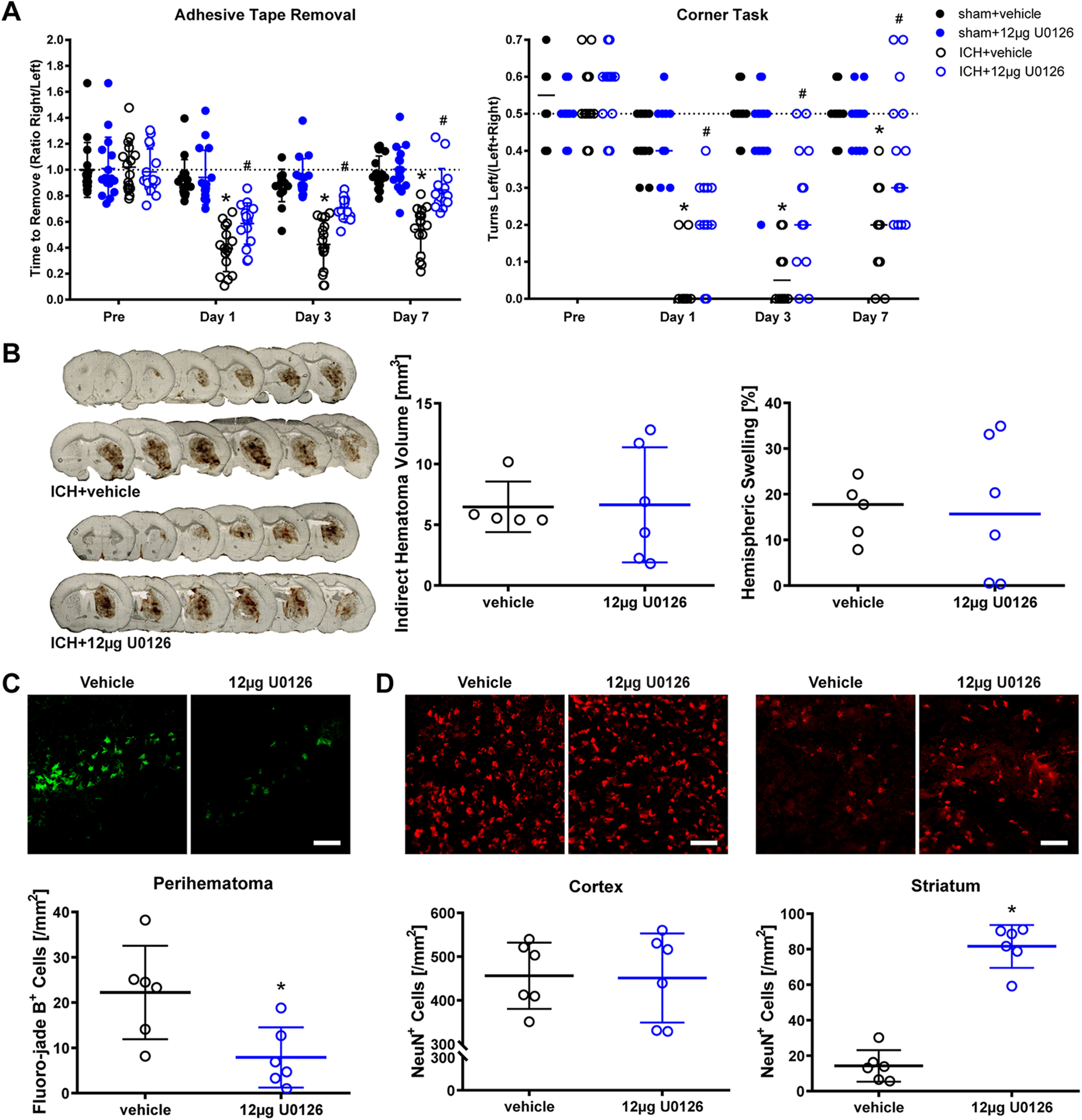
Intracerebroventricular administration of the MEK inhibitor U0126 promotes functional recovery after ICH in mice. A, ICH and sham animals received intracerebroventricular injections of 12 µg U0126 or vehicle at 2 h after ICH surgery, and the functional recovery (adhesive tape removal and corner task) was assessed up to 7 d after ICH. Values show mean ± SD for adhesive tape removal and median for corner task; *p < 0.05 versus baseline, #p < 0.05 versus ICH+vehicle. B, The indirect hematoma volume and hemispheric swelling (edema) were measured at 24 h after ICH. The values show the mean ± SD for hematoma volume and the median for hemispheric swelling. C, D, Neuronal cell survival was assessed using Fluoro-jade B staining in the perihematoma (C) and NeuN immunofluorescence in the cortex and striatum (D) at 24 h after ICH. Scale bar: 100 µm. Mean ± SD is shown. *p < 0.05. For exact p values, refer to Extended Data Figure 1-3.
The intraperitoneal injection of the MEK inhibitor U0126 does not improve functional recovery after ICH in mice. ICH animals received daily intraperitoneal injections of 25 mg/kg U0126 or vehicle starting at 2 h after surgery for 7 d. The functional recovery (adhesive tape removal and corner task) was assessed up to 7 d after ICH. The values show the mean ± SD for adhesive tape removal and the median for corner task; *p < 0.05 versus baseline. For exact p values, refer to Extended Data Figure 1-2. Download Figure 1-1, TIF file (4.1MB, tif) .
Intraperitoneal injection of U0126 after ICH. Download Figure 1-2, DOCX file (15.3KB, docx) .
Intracerebroventricular injection of U0126 after ICH. Download Figure 1-3, DOCX file (18.5KB, docx) .
As we induced ICH in our mouse model using collagenase to break down the basal lamina and induced bleeding throughout the striatum, it is formally possible that U0126 improved recovery by inhibiting collagenase. To exclude this possibility, we measured hematoma volume as well as one of its downstream consequences, brain swelling. As expected, neither parameter was affected by U0126 treatment (Fig. 1B). However, U0126 reduced neuronal cell death as evidenced by reduced numbers of Fluoro-jade B-positive cells (Fig. 1C) and increased numbers of NeuN-positive cells (Fig. 1D).
U0126 prevents hemin-induced ferroptosis independent of MEK activation and ERK phosphorylation
Having established that U0126 can inhibit ICH-induced ferroptosis in vitro and improve functional recovery following experimental ICH in vivo, we wanted to explore the precise mechanism of U0126 in interdicting ferroptosis. As U0126 is known to be a potent and selective MEK inhibitor, we examined MEK inhibitors that are chemically distinct from U0126 to see whether they are also able to abrogate hemin or hemoglobin-induced ferroptosis in cortical neurons. By contrast to classical ferroptosis induced by cystine deprivation and glutathione depletion (Canals et al., 2003; de Bernardo et al., 2004; Basso et al., 2012), MEK1/2 inhibitors chemically distinct from U0126, were not effective (with the exception of primasertib) in reducing hemin-induced ferroptosis as determined by population measures of cell death (MTT assay) or via single cell measures of cell death (Live/Dead assay; Figs. 2A, 3A,B). Similarly, MEK1/2 inhibitors other than U0126 including primasertib were ineffective in preventing hemoglobin-induced ferroptosis (Figs. 2A, 3A,B). The inability of chemically diverse inhibitors of MEK1/2 to inhibit hemin or hemoglobin-induced ferroptosis was not attributed to their inability to inhibit hemin or hemoglobin-induced phosphorylation of ERK (Fig. 2C). Altogether, these data suggest that hemin or hemoglobin-induced ferroptosis is ERK-independent.
Figure 2.
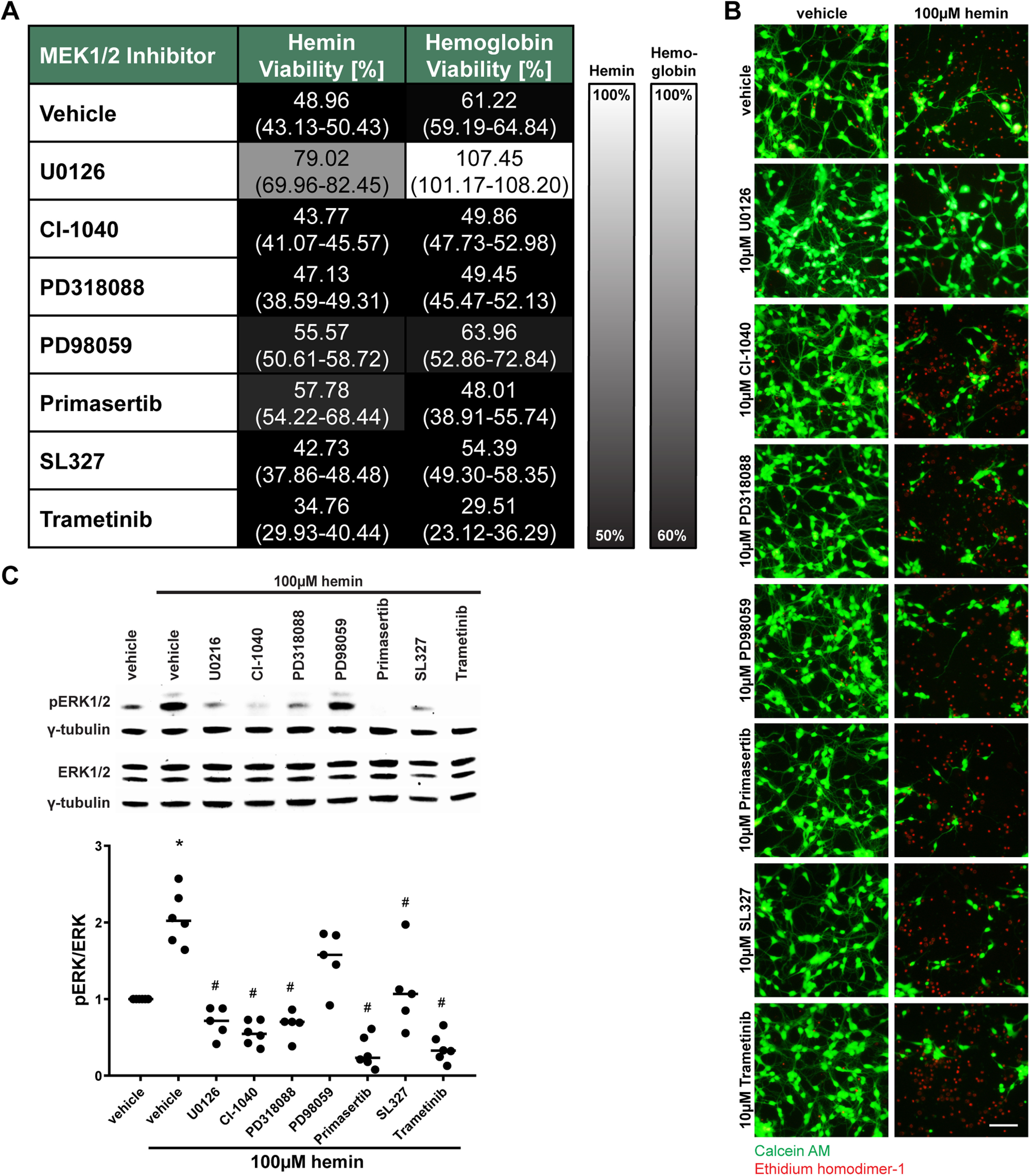
MEK1/2 inhibitors other than U0126 do not protect primary neurons from hemin-induced and hemoglobin-induced neuronal ferroptosis. A, Primary neurons were exposed to 100 μm hemin or 1.5 μm hemoglobin (LD50) and different, structurally diverse MEK1/2 inhibitors. The values reflect the medians (interquartile ranges) at 10 μm of each inhibitor. Grayscale coding reflects a continuum from no protection from hemin or hemoglobin toxicity by a MEK1/2 inhibitor (black) to maximal possible cell viability (white). B, The representative live/dead staining of the inhibitors in hemin-induced neuronal ferroptosis are shown. Scale bar: 100 µm. C, The total and phospho-ERK1/2 protein levels were assessed in primary neurons exposed to hemin and 10 μm different MEK1/2 inhibitors. Medians are given of the ratios of phospho-ERK by total ERK1/2 normalized to γ-tubulin loading control that does not change in response to hemin; *p = 0.017 versus vehicle, #p < 0.05 versus hemin. For exact p values, refer to Extended Data Figure 2-1.
Figure 3.

MEK1/2 inhibitors in hemin-induced and hemoglobin-induced neuronal ferroptosis. The concentration-responses of MEK1/2 inhibitors in primary neurons exposed to (A) hemin-induced and (B) hemoglobin-induced ferroptosis. Medians are indicated; *p < 0.05 versus hemin or hemoglobin + vehicle. For exact p values, refer to Extended Data Figures 3-1, 3-2.
Effect of MEK1/2 inhibitors on pERK/ERK levels in hemin-induced ferroptosis in primary cortical neurons. Download Figure 2-1, DOCX file (14KB, docx) .
MEK1/2 inhibitors in hemin-induced ferroptosis in primary cortical neurons. Download Figure 3-1, DOCX file (16.7KB, docx) .
MEK1/2 inhibitors in hemoglobin (hb)-induced ferroptosis in primary cortical neurons. Download Figure 3-2, DOCX file (16.7KB, docx) .
To verify that phosphorylation of ERK1/2 is not required for the execution of hemin-induced ferroptosis, we overexpressed an ERK-selective MKP3 (Fig. 4A). Phosphatases such as MKP3 counterbalance the enhanced phosphorylation of ERK1/2 that is mediated by the activation of MEK1/2 (Levinthal and Defranco, 2005). As expected from our pharmacological studies described above, the overexpression of Mkp3 led to a decrease in hemin-induced phospho-ERK1/2 (Fig. 4B), but it failed to inhibit hemin-induced ferroptosis (Fig. 4C). In contrast and as expected, it was able to suppress glutathione depletion (HCA or glutamate)-induced neuronal ferroptosis as described previously (Levinthal and Defranco, 2005). While we cannot exclude the possibility that MKP3 leads to the dephosphorylation of an ERK-independent substrate required for inhibiting ferroptosis in the presence of hemin, together with the pharmacological data described above, we conclude that hemin-induced death is independent of ERK phosphorylation.
Figure 4.

The molecular knock-down of phospho-ERK1/2 by overexpressing MKP3 (Mkp3) has distinct effects on hemin-induced and glutathione depletion-induced ferroptosis. A, We overexpressed the highly ERK-specific phosphatase Mkp3 and its catalytic mutant, Mkp3 C293S that enhances ERK1/2 phosphorylation but blocks its nuclear translocation, in HT22 cells. pSG5 served as control (empty vector). B, The levels of phospho-ERK and total ERK (normalized to γ-tubulin) were evaluated after overexpressing MKP3 or MKP3 C293S for 36 h. Medians are given of the ratios of phospho-ERK by total ERK1/2 normalized to γ-tubulin loading control that does not change in response to hemin; *p < 0.001 versus pSG5. For all statistical analyses, refer to Extended Data Figure 4-1. C, The cell viability was assessed after overexpressing Mkp3 or Mkp3 C293S (40 µl/ml lipofectamine) for 24 h followed by treatment with 25–100 μm hemin (hemin-induced ferroptosis) and 10 mm HCA (glutathione depletion-induced ferroptosis) for another 24 h. Medians are given; *p < 0.05 versus 0 μm hemin, #p < 0.05 versus pSG5. For exact p values, refer to Extended Data Figure 4-2.
Effect of Mkp3 overexpression on pERK/ERK levels in HT22 cells. Download Figure 4-1, DOCX file (14.1KB, docx) .
Cell survival after Mkp3 overexpression in hemin-induced versus glutathione depletion (HCA)-induced ferroptosis in HT22 cells. Download Figure 4-2, DOCX file (15KB, docx) .
Prior studies have shown that classical ferroptosis (induced by cystine deprivation and glutathione depletion) involves not only enhanced phosphorylation of ERK, but also its translocation to the nucleus to induce ferroptosis (Levinthal and Defranco, 2005). Indeed, ERK can be phosphorylated by MEK1 on its TXY motif (Payne et al., 1991). This leads to the dissociation of ERK from cytoplasmic scaffolding proteins including MEK, β-arrestin, and SEF1. The dissociation from these scaffolding proteins exposes a SPY motif in the nuclear localization signal of ERK. The phosphorylation of the SPY motif of ERK1/2 allows it to bind to importin-7 to traffic into the nucleus (Wortzel and Seger, 2011).
To determine whether hemin, like classical ferroptotic stimuli (e.g., glutamate, HCA, erastin), induces nuclear translocation of ERK, we performed subcellular fractionation of phospho-ERK1/2. Hemin induced an increase in phospho-ERK1/2 primarily in the cytoplasm, except a smaller amplitude, but statistically significant increase in the nucleus at 8 h. In contrast, HCA led to the elevation of phospho-ERK1/2 in the nucleus (Fig. 5A). As Mkp gene transcription is activated by nuclear ERK1/2 (Cook et al., 1997; Li et al., 2001; Smith et al., 2005), we hypothesized that if phospho-ERK1/2 weakly translocates into the nucleus after hemin exposure, Mkp-1 and Mkp-3 should be weakly transcribed. As expected, we observed that hemin did not induce Mkp-1 and Mkp-3 gene transcription until 8 h after hemin, which coincided with the increase in phospho-ERK1/2 seen in the nuclear fraction. Again, by contrast, ferroptosis induced by the glutamate analog HCA was associated with the translocation of nuclear ERK1/2 soon after HCA addition and a significant increase in both Mkp-1 and Mkp-3 mRNA levels (Fig. 5B). As a more definitive test that nuclear translocation of phospho ERK1/2 is not necessary for hemin-induced ferroptosis, we forced expression of a mutant version Mkp3 with a critical cysteine in the active site of the phosphatase mutated to serine (C293S). This construct acts as a dominant negative and prevents endogenous active MKP3 from binding to ERK1/2. Moreover, it has another functionality in preventing the translocation of ERK1/2 to the nucleus (Levinthal and Defranco, 2005). While the overexpression of Mkp3 C293S protected against glutathione depletion (HCA)-induced neuronal ferroptosis as described previously (Levinthal and Defranco, 2005), it had no effect on ferroptosis induced by hemin (Fig. 4C). Altogether, these findings demonstrate that hemin-induced ferroptosis is MEK-independent and ERK-independent.
Figure 5.

Hyperactivated ERK1/2 remains in the cytoplasm in hemin-induced ferroptosis and the transcription of Mkp1 and Mkp3, its negative regulators, is delayed. A, The time course of the levels of phospho-ERK and total ERK was assessed in the cytoplasmic and nuclear extracts of primary neurons exposed to 100 μm hemin or 5 mm HCA (normalized to γ-tubulin for cytoplasmic fractions or Histone H4 for nuclear fractions). The fractionation was confirmed by evaluating the Histone H4 expression in the cytoplasmic fractions and the GAPDH expression in the nuclear fractions. Medians are given, except for the nuclear fractions in hemin that are means ± SD; *p < 0.05 versus 0 h. For exact p values, refer to Extended Data Figure 5-1. B, The Mkp1 and Mkp3 mRNA expression was measured in primary neurons exposed to 100 μm hemin or 5 mm HCA. The values represent the medians for hemin and the means ± SD for HCA; *p < 0.05 versus 0 h. For exact p values, refer to Extended Data Figure 5-2.
pERK/ERK levels in the cytoplasm and nucleus in hemin-induced versus glutathione depletion (HCA)-induced ferroptosis in primary cortical neurons Download Figure 5-1, DOCX file (15.8KB, docx) .
Mkp1 and Mkp3 mRNA expression in hemin-induced versus glutathione depletion (HCA)-induced ferroptosis in primary cortical neurons. Download Figure 5-2, DOCX file (15.6KB, docx) .
Upstream of MEK in the Ras-Raf-MEK-ERK signaling pathway is the first step of the MAP kinase cascade, Raf. C-Raf is the principal member of the Raf kinase family of serine/threonine specific protein kinases. Prior studies have shown that the selective Raf inhibitor GW5074 prevented glutamate analog (HCA)-induced ferroptosis completely at 50 nm (Chin et al., 2004). In contrast and consistent with our findings with MEK and ERK inhibitors, GW5074 had no effect on hemin-induced ferroptosis (Fig. 6A). Together, these studies show that inhibitors of Raf, MEK, or ERK all inhibit canonical ferroptosis induced by the glutamate analog HCA but fail to inhibit hemin or hemoglobin-induced ferroptosis.
Figure 6.

The inhibition of Raf, MEK5/ERK5, MEK4, and the activation of AMPK do not protect primary neurons from hemin-induced neuronal ferroptosis. A, Primary neurons were exposed to vehicle or 100 μm hemin and different concentrations of the selective Raf inhibitor GW5074. B, The concentration-responses of BIX02189 (MEK5 inhibitor) and XMD17-109 (ERK5 inhibitor) in primary neurons exposed to hemin-induced ferroptosis. C, The concentration-responses of the MEK4 inhibitors in primary neurons exposed to hemin-induced ferroptosis. D, Concentration response for metformin (AMPK activator) in primary neurons exposed to hemin-induced ferroptosis. AMPK activation is shown by western blotting of the total and phospho-AMPK protein levels assessed in primary neurons exposed to vehicle or 5 mm metformin. The values represent the medians. For exact p values, refer to Extended Data Figure 6-1.
Inhibitors of Raf, MEK5, ERK5, MEK4, and an activator of AMPK in hemin-induced ferroptosis in primary cortical neurons. Download Figure 6-1, DOCX file (16.8KB, docx) .
The protection of U0126 is not mediated by its known off-target effects
In the absence of evidence that U0126 prevents hemin-induced ferroptosis by inhibiting the Raf-MEK-ERK signaling pathway, it is possible that a known off-target effect of the drug may explains its antiferroptotic effects in brain hemorrhage models. U0126 was originally described as a specific inhibitor of MEK1 (IC50 = 0.072 ± 0.02 μm) and MEK2 (IC50 = 0.058 ± 0.02 μm) that does not affect other kinases below 10 μm (Favata et al., 1998). Since then, U0126 has also been shown to inhibit the MEK5-ERK5 signaling pathway below a concentration of 10 μm (Kamakura et al., 1999; Mody et al., 2001). We therefore investigated whether inhibitors of MEK5 and ERK5 protect neurons from hemin-induced ferroptosis. However, both the selective ERK5 inhibitor BIX02189 and the MEK5 inhibitor XMD17-109 failed to show any protection (Fig. 6B), making MEK5-ERK5 an unlikely target for the protective effects of U0126.
Furthermore, U0126 is a weak inhibitor of the MEK4 (Favata et al., 1998), also known as MEK4. We therefore assessed MEK4 inhibitors in their ability to abrogate hemin-induced ferroptosis. The three selective MEK4 inhibitors investigated, NUCC-201167, NUCC-202360, and NUCC-226297, did not improve cell survival in hemin-induced neuronal ferroptosis (Fig. 6C).
Phosphorylation of ERK1/2 has also been associated with an overall decrease in the phosphorylation of AMPK that is involved in controlling cell growth, transcription, and responses to nutrient limitation and stress (Kodiha et al., 2007). Moreover, another study showed that U0126 and PD98059, chemically diverse MEK1/2 inhibitors, can activate AMPK by increasing the cellular AMP:ATP ratio independent of ERK1/2 inhibition (Dokladda et al., 2005). We therefore investigated whether activating AMPK mimics the ability of U0126 to block hemin-induced ferroptosis. The AMPK activator metformin did not abrogate hemin-induced cell death (Fig. 6D).
Unbiased phosphoproteomics in search of the mechanism underlying the protective effect of U0126 in hemin-induced ferroptosis
Given the robust ability of U0126 to prevent hemin-induced ferroptosis in vitro and reduce ICH-induced impairment in vivo, we sought to identify the underlying mechanism using an unbiased phosphoproteomics approach. We assessed canonical ferroptosis using erastin treatment with and without U0126 for 7 h, the latest time that U0126 can be added and protect all the neurons, and hemin-treated cortical neurons with and without U0126 for 5 h.
We compared phosphorylation levels across these groups in 28,022 phosphopeptides from 4871 proteins (Extended Data Fig. 7-1). Of these, 452 peptides from 369 proteins showed altered levels over the significance threshold (p < 0.001) after hemin treatment. A total of 51 peptides from 49 proteins showed altered levels after erastin treatment. Overall, both patterns were remakably distinct with only nine peptides significantly changed in both sets, of which eight were altered in the same direction (Jaccard similarity coefficient = 0.016; Fig. 7A; Extended Data Fig. 7-1).
Figure 7.
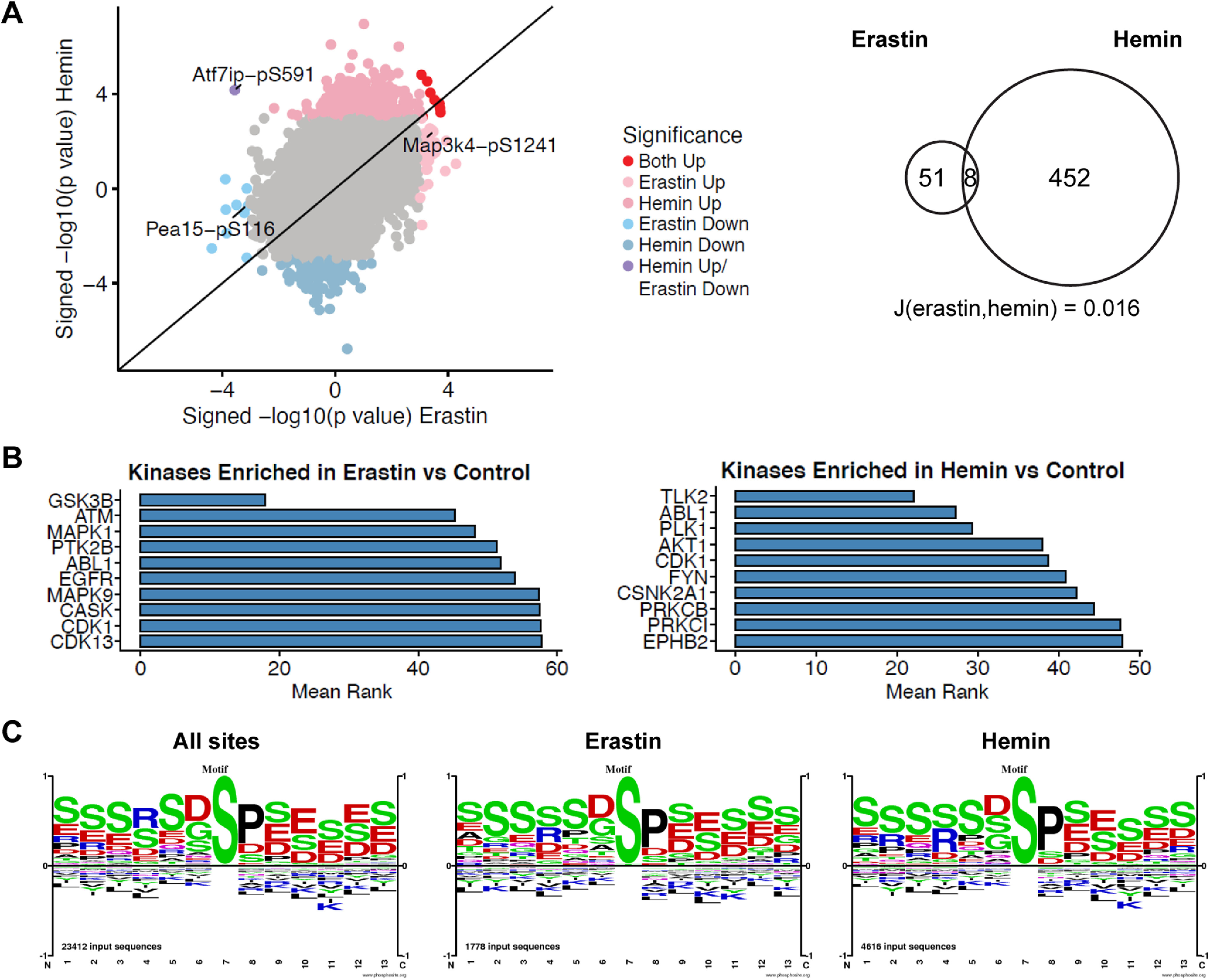
Unbiased phosphoproteomics of ferroptotic cell death induced by erastin and hemin. A, Primary neurons were treated with 5 μm erastin for 7 h or 100 μm hemin for 5 h. Scatter plots compared with control are shown where p < 0.001 was used as a threshold. A total of 51 peptides from 49 proteins after erastin treatment and 452 peptides from 369 proteins after hemin treatment showed altered levels. Eight peptides were similarly increased or decreased. The Jaccard similarity coefficient = 0.016 (for full list of phosphopeptides, refer to Extended Data Fig. 7-1). B, Top 10 ranked enriched kinases in erastin-treated or hemin-treated cells compared with control based on the mean rank of enrichment over 11 gene set libraries (see also Extended Data Fig. 7-2). For top 10 ranked kinases downregulated in hemin-treated cells, see Extended Data Figure 7-3. C, Motif analysis around the phosphorylation sites. Size of characters are informative of amino acid abundances on each position (in a scale from 1 to −1; positive values, overrepresented; negative values, undrerrepresented) and are normalized to background amino acid frequencies calculated from all aminoacids within ±7 residues of the central modified residues of the specified type (here, S) that are identified in PhosphoSitePlus.Rxxs (CAMK2, Akt, PKA, DMPK1/2) was overrepresented in hemin versus control compared with erastin versus control (ratio 1.18), whereas (D/E)xx(s/t) (casein kinase I) was underrepresented (ratio 0.85; see also Extended Data Fig. 7-4).
Top 10 ranked kinases downregulated by hemin. Full phosphoset of the unbiased phosphoproteomics of ferroptotic cell death induced by erastin and hemin and treated with U0126. Download Figure 7-1, XLSX file (24.6MB, xlsx) .
Mean rank of enrichment over 11 gene set libraries from a KEA3 kinase enrichment analysis of erastin-treated, hemin-treated, or U0126-treated cells compared to their respective controls. Download Figure 7-2, XLSX file (147.9KB, xlsx) .
Motifs surrounding the phosphorylation sites in single phosphorylated peptides with abundances altered by erastin and hemin treatments. Download Figure 7-3, TIF file (19KB, tif) .
Motifs surrounding the phosphorylation sites in single phosphorylated peptides with abundances altered by erastin and hemin treatments. Download Figure 7-4, XLSX file (10.4KB, xlsx) .
To assess altered kinase activity, we submitted proteins with increased phosphopeptides (p < 0.001) in erastin-treated or hemin-treated cells to KEA3 (Kuleshov et al., 2021). KEA3 ranks kinases based on the enrichment of substrate and interacting proteins (low rank suggests greater enrichment). As expected, MAPK1 was among the top ten ranked kinases increased after erastin but not hemin treatment (Fig. 7B; Extended Data Fig. 7-2). Top ranked kinases upregulated by hemin treatment, but not erastin treatment, included TLK2 and AKT1 (Fig. 7B). To note, AKT1 was also among the top ranked kinases downregulated by hemin (Extended Data Figs. 7-2, 7-3), suggesting that hemin drives AKT1 phosphorylation at a specific subset of substrates. On the other hand, the analysis of the kinases downregulated by erastin treatment did not reveal sufficient gene symbols for enrichment.
We further investigated the motifs surrounding the phosphorylation sites in the two sets. They included sP, where the small case S represents phosphorylated serine, a substrate for proline-directed protein kinases such as MAP kinases (Fig. 7C). Rxxs (CAMK2, Akt, PKA, DMPK1/2) was overrepresented in hemin versus control compared with erastin set versus control (ratio 1.18), while (D/E)xx(s/t) (casein kinase I) was underrepresented (ratio 0.85). Furthermore, we detected an increase in the frequency of R in position 5 in the differentially modulated set of phosphopeptides of the hemin-treated samples (ratio 1.18; Extended Data Fig. 7-4).
In search for the target of U0126 in hemin-induced ferroptosis compared with erastin-induced ferroptosis, we compared hemin-treated cells with U0126 to erastin-treated cells with U0126. We identified 34 phosphopeptides from 34 proteins with significantly altered levels in hemin+U0126-treated versus hemin-treated cells and 61 phosphopeptides from 58 proteins in erastin+U0126-treated versus erastin-treated cells, with no overlap between both groups (p < 0.001; Fig. 8A; Extended Data Fig. 7-1). A KEA3 analysis of proteins with downregulated phosphopeptides after U0126 treatment showed the expected enrichment of MAPK1/3 for erastin+U0126 (Fig. 8B; Extended Data Fig. 7-2), suggesting a decrease in MAPK1/3 activity with erastin+U0126 compared with erastin alone. However, hemin+U0126 had a decrease in kinases such as CHEK2, PAK1/2, CSNK2A1, and EGFR (Fig. 8B; Extended Data Fig. 7-2). To note, the analysis of the kinases upregulated by hemin+U0126 treatment did not reveal sufficient gene symbols for enrichment, but erastin+U0126 treatment upregulated AKT1, CDK1/2/4, and CSNK2A1/2/3 (Extended Data Figs. 7-2, 8-1).
Figure 8.
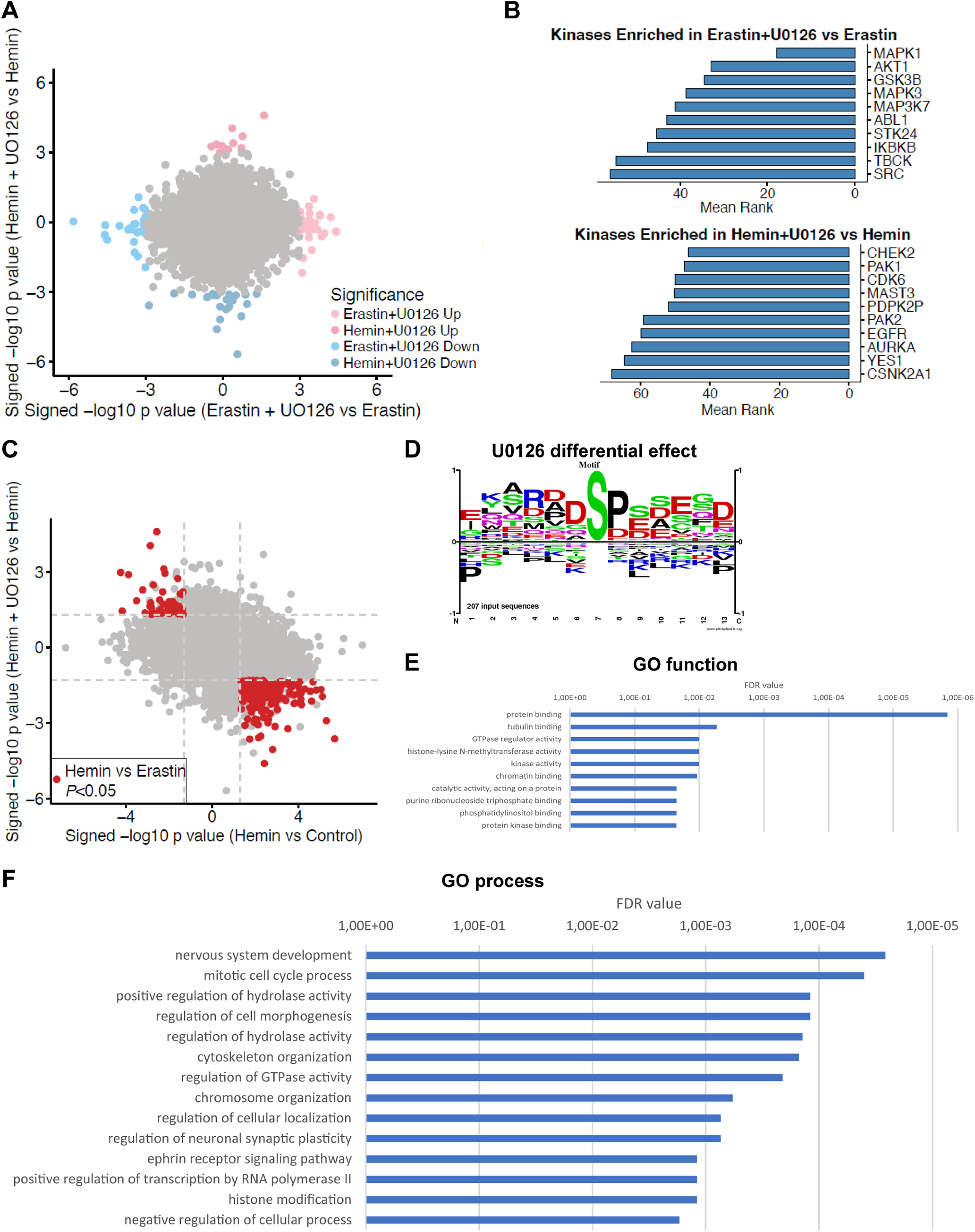
Unbiased phosphoproteomics of U0126 treatment in ferroptotic cell death. A, Primary neurons were treated with 10 μm U0126 for 7 or 5 h in erastin-treated or hemin-treated cells. A total of 34 phosphopeptides from 34 proteins were significantly altered in hemin+U0126-treated versus hemin-treated cells and 61 phosphopeptides from 58 proteins in erastin+U0126-treated versus erastin-treated cells, with no overlap between both groups (p < 0.001, for full list of phosphopeptides, refer to Extended Data Fig. 7-1). B, Kinases enriched for proteins with phosphopeptides that were downregulated by U0126 compared with either erastin or hemin treatment alone. The top 10 kinases are ordered by the mean rank of enrichment over 11 gene set libraries (see also Extended Data Fig. 7-2). For top 10 ranked kinases upregulated in erastin+U0126-treated cells, see Extended Data Figure 8-1. C, Analysis of phosphopeptides that were significantly changed in hemin versus control treatment, that also had significantly different levels between the hemin-treated and erastin-treated cells, and where U0126 treatment led to changes in the opposite direction of the hemin treatment identified 247 phosphopeptides with significantly altered levels (p < 0.05, see also Extended Data Fig. 8-2). Dashed lines indicate a p value threshold of 0.05. D, Motif analysis shows that Rxxs (CAMK2, Akt, PKA, DMPK1/2), (s/t)xx(E/D) (CK2), and sP (MAPK and other kinases) were enriched versus their abundance in the proteome. sxxD/E was overrepresented (ratio 2.06 or 2.05, respectively, see also Extended Data Fig. 8-3). E, F, Network analysis of the proteins revealed some GO cellular processes (E) and functions (F) that provide the basis for a future search for the target of U0126 in hemin-induced ferroptosis.
Top 10 ranked kinases upregulated in erastin+U0126 treatment. Download Figure 8-1, TIF file (20.2KB, tif) .
Phosphopeptides that were significantly changed in hemin versus control treatment, that also had also significantly different levels between the hemin-treated and erastin-treated cells, and where U0126 treatment led to changes in the opposite direction of the hemin treatment. Download Figure 8-2, XLSX file (9.9MB, xlsx) .
Motifs surrounding the phosphorylation sites in single phosphorylated peptides with abundances altered by hemin that have also significantly different levels between the hemin-treated and erastin-treated cells and that are affected by the U0126 treatment in hemin-treated cells. Download Figure 8-3, XLSX file (10.4KB, xlsx) .
Next, we looked at hemin-treated cells compared with untreated controls, that had also significantly different levels of phosphopeptides between the hemin-treated and erastin-treated cells, and where U0126 treatment led to changes in the opposite direction of the hemin treatment. We identified 247 altered phosphopeptides (p < 0.05; Fig. 8C; Extended Data Fig. 8-2). In the motifs surrounding the phosphorylation sites, we observed that Rxxs (CAMK2, Akt, PKA, DMPK1/2), (s/t)xx(E/D) (CK2), and sP (MAPK and other kinases) were enriched versus their abundance in the proteome (Fig. 8D). sxxD/E was overrepresented (ratio 2.06 or 2.05, respectively; Extended Data Fig. 8-3). Finally, network analysis of the proteins revealed some GO cellular processes and functions that provide the basis for a future search for the target of U0126 in hemin-induced ferroptosis (Fig. 8E,F).
Discussion
The exposure of cultured neurons to hemoglobin and hemin has been shown to be a model that is reliably predictive of therapeutic targets in rodent models of hemorrhagic stroke (Karuppagounder et al., 2016, 2018; Zille et al., 2017; Alim et al., 2019). Several years ago, we demonstrated that hemoglobin-induced and hemin-induced cell death in neurons can be prevented by a cassette of classical inhibitors of ferroptosis including N-acetylcysteine (a glutathione prodrug), deferoxamine (an iron chelator), ferrostatin (a lipid peroxidation inhibitor) and U0126 (a MEK inhibitor; Zille et al., 2017). Here, we show, for the first time, that U0126 improves functional recovery after ICH in mice. Additionally, we used the experimental leverage of an in vitro model of brain hemorrhage to probe the mechanism by which U0126 prevents ferroptosis. Unexpectedly, and in contrast to classical ferroptosis induced by glutamate or erastin, our studies revealed that U0126 protects against hemin-induced ferroptosis independent of MEK1/2. Together with other emerging evidence, these findings define hemin-induced death as a novel, bona fide variant of classical neuronal ferroptosis. Finally, we also present a novel, unbiased phosphoproteomic analysis of ferroptosis that will help to better understand these variants of ferroptosis and to identify the protective mechanism of U0126 in hemin-induced ferroptosis.
Classically, ferroptosis is induced by cystine transport blockade leading to the depletion of the antioxidant glutathione (Dixon et al., 2012). The depletion of glutathione results in the hypoactivity of glutathione peroxidase 4 (GPX4) and an unopposed generation of reactive lipid species via 12/15-lipoxygenase (Ratan, 2020). Reactive lipid species downstream of 12/15-lipoxygenase are believed to lead to the oxidative inactivation of MKP3 that in turn permits the constitutive activation of the Ras-Raf-MEK-ERK1/2 pathway (Levinthal and Defranco, 2005). This constitutive activation leads to translocation of ERK1/2 to the nucleus where it stabilizes c-Myc, Elk-1 and likely other transcription factors (Davis, 1995; Sleiman et al., 2011; Tsai et al., 2012) and enhances the transactivation of a host of genes including those involved in iron uptake into the cell (Wu et al., 1999; Ratan, 2020). This may be one of a number of mechanisms by which cells undergoing ferroptotic oxidative death paradoxically load up with iron.
The ability of Raf, MEK, and ERK nuclear translocation inhibition to abrogate classical ferroptosis is well established (Dixon et al., 2012; Xie et al., 2016). Here, we show that neither Raf inhibition (GW5074; Fig. 6A), nor pharmacological MEK inhibition (Extended Data Fig. 2-3), nor reduced ERK phosphorylation (via MKP overexpression; Fig. 4) affect hemin-induced ferroptosis. The findings show that despite the ability of U0126 to protect against hemin-induced ferroptosis, this variant of ferroptosis is Ras-Raf-MEK-ERK-independent.
The ability of U0126 to protect independent of MEK1/2 raises several questions. First, is hemin-induced death ferroptotic? Based on our prior statistical analysis using a host of accepted criteria for ferroptosis, hemin-induced death meets the definition of ferroptosis (Zille et al., 2017). Like classical ferroptosis, hemin-induced death is abrogated by forced expression of GPX4 (Alim et al., 2019). GPX4 is unique among glutathione peroxidases in that it is monomeric and can therefore insert into membranes to neutralize oxidized lipids. Hemin has been shown to intercalate into membranes where it can directly influence their composition (Wyse and Butterfield, 1989) and function, providing a plausible scheme by which hemin-induced death is linked to membrane derived reactive lipid species. As expected, like classical ferroptosis, hemin can also be abrogated by glutathione enhancing agents (Karuppagounder et al., 2018) and chelators of iron that target specific metalloenzymes (Karuppagounder et al., 2016).
In addition to the results presented in this study, there is growing appreciation that hemin-induced death is distinct from classical ferroptosis. First, classical ferroptosis is abrogated by pharmacological inhibitor of 12/15-lipoxgenase inhibition (Tang et al., 1996; Li et al., 1997; Khanna et al., 2003; Seiler et al., 2008), while 12/15-lipoxygenase inhibitors have no effect on hemin-induced ferroptosis (Karuppagounder et al., 2018). Hemin-induced death is 5-lipoxygenase-dependent (Karuppagounder et al., 2018). Second, classical ferroptosis is inhibited by inhibitors of macromolecular synthesis (Ratan et al., 1994a, b; Zille et al., 2019), whereas hemin-induced ferroptosis is not (Zille et al., 2017). It is tempting to speculate that the inability of transcriptional inhibitors to block hemin-induced death reflects the absence of ERK translocation to the nucleus where it is able to activate and stabilize several transcriptional targets. Altogether, these data suggest that while hemin induces ferroptosis, it does so via a pathway that is 12/15-lipoxygenase-independent, transcription-independent, and Ras-Raf-MEK-ERK-independent (Table 1).
Table 1.
Similarities and differences between classical, glutathione depletion-induced ferroptosis and hemin-induced ferroptosis
| Criterion | Classical, glutathione depletion-induced ferroptosis | Hemin-induced ferroptosis |
|---|---|---|
| Reactive lipid species-dependent | + | + |
| Glutathione enhancing agents are protective | + | + |
| GPX4 forced expression is protective | + | + |
| Iron chelators are protective | + | + |
| ERK1/2 hyperactivation | + | + |
| 12/15-lipoxygenase-dependent | + | - |
| Nuclear translocation of phospho-ERK1/2 | + | - |
| MKP3 forced expression is protective | + | - |
| Transcription-dependent | + | - |
ERK1/2, extracellular signal-regulated kinase 1/2; GPX4, glutathione peroxidase 4; MKP3, MAP kinase phosphatase 3.
Another question raised by the data is the following: if hemin induces a variant of ferroptosis, how does hemin differ from erastin and glutamate as an inducer of ferroptosis? The precise answer is unknown, but there is currently no evidence that, like glutamate or erastin, hemin or hemoglobin can interact directly with the xCT cystine dependent transporting agency. Given the ability of hemin to directly interact with membranes (Wyse and Butterfield, 1989), it could indirectly affect the function of the xCT transporter. However, if that were the case, one would expect the mechanisms of hemin-induced death to be identical to that of glutamate and erastin. Accordingly, we speculate that hemin-induced ferroptosis occurs as a result of direct generation of reactive lipid species and consumption of glutathione rather than of the decreased synthesis of glutathione (Table 1).
The ability of hemin to drive a unique path to ferroptosis amplifies the importance of identifying the target for U0126 in preventing hemin-induced death. Our results demonstrating the ability of U0126 to improve functional recovery in a rodent model of ICH (Fig. 1) further point to the therapeutic importance of this exercise. What we can say is that it is unlikely that the salutary effects of U0126 relate to its direct effects as an antioxidant as its inactive kinase analog U0124 is ineffective (Zille et al., 2017). U0124 shares the potency of U0126 as an antioxidant (Ong et al., 2015).
Using unbiased phosphoproteomics, we here provide further insights into the similarities and differences of hemin-induced and erastin-induced ferroptosis and a basis to understand the off-target effects of U0126 in interdicting a variant of ferroptosis. While hemin altered 452 and erastin altered 51 phosphopeptides, only eight of them were changed in the same direction (Fig. 7A; Extended Data Fig. 7-1), resulting in a low Jaccard index (0.016) suggesting that both subtypes of neuronal ferroptosis have different phosphopeptide signatures. In erastin-treated cells, we found Map3k4 S1241 increased, which may increase the activity of Map3k4 (Bullard et al., 2016). The phosphorylation of Pea15 S116 was decreased, which has been demonstrated to sensitize cells for apoptosis (Eckert et al., 2008).
Among the phosphopeptides that were changed in (1) hemin-treated cells compared with untreated controls, (2) between the hemin-treated and erastin-treated cells, and (3) were affected by the U0126 treatment in the opposite direction to hemin treatment, we identified 247 phophopeptides with significantly altered levels (Extended Data Fig. 8-3). Motif analyses surrounding the phosphorylation sites in the two sets revealed that Rxxs (CAMK2, Akt, PKA, DMPK1/2), (s/t)xx(E/D) (CK2), and sP (MAPK and other kinases) were altered in hemin versus control compared with erastin set versus control and U0126 treatment in hemin-treated cells (Figs. 7C, 8C). This may suggest the involvement of CK-II/IKK type kinases based on the sxxE/D motifs in triggering ferroptosis as they have been suggested to play a role in the regulation of cell death (Guan et al., 2011b).
Taken together, this unbiased phosphoproteomic dataset provides a template on which to understand distinct paths to cell death that meet the definition of ferroptosis.
Footnotes
This work was supported by the German Research Foundation Grant DFG Zi 1613/1-1 (to M.Z.), funding from the Sheldon G and Dr. Miriam Adelson Medical Research Foundation and the Sperling Center for Hemorrhagic Stroke Recovery, and the National Institutes of Health Grant P01 NIA AG014930, project 1 (to R.R.R.).
The authors declare no competing financial interests.
References
- Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, Seravalli J, Ai Y, Sansing LH, Ste Marie EJ, Hondal RJ, Mukherjee S, Cave JW, Sagdullaev BT, Karuppagounder SS, Ratan RR (2019) Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177:1262–1279.e25. 10.1016/j.cell.2019.03.032 [DOI] [PubMed] [Google Scholar]
- Basso M, Berlin J, Xia L, Sleiman SF, Ko B, Haskew-Layton R, Kim E, Antonyak MA, Cerione RA, Iismaa SE, Willis D, Cho S, Ratan RR (2012) Transglutaminase inhibition protects against oxidative stress-induced neuronal death downstream of pathological ERK activation. J Neurosci 32:6561–6569. 10.1523/JNEUROSCI.3353-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behdenna A, Haziza J, Azencott C-A, Nordor A (2021) pyComBat, a Python tool for batch effects correction in high-throughput molecular data using empirical Bayes methods. bioRxiv 995431. doi: 10.1101/2020.03.17.995431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard SA, Seo S, Schilling B, Dyle MC, Dierdorff JM, Ebert SM, DeLau AD, Gibson BW, Adams CM (2016) Gadd45a protein promotes skeletal muscle atrophy by forming a complex with the protein kinase MEKK4. J Biol Chem 291:17496–17509. 10.1074/jbc.M116.740308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals S, Casarejos MJ, de Bernardo S, Solano RM, Mena MA (2003) Selective and persistent activation of extracellular signal-regulated protein kinase by nitric oxide in glial cells induces neuronal degeneration in glutathione-depleted midbrain cultures. Mol Cell Neurosci 24:1012–1026. 10.1016/j.mcn.2003.08.004 [DOI] [PubMed] [Google Scholar]
- Cao S, Zheng M, Hua Y, Chen G, Keep RF, Xi G (2016) Hematoma changes during clot resolution after experimental intracerebral hemorrhage. Stroke 47:1626–1631. 10.1161/STROKEAHA.116.013146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin PC, Liu L, Morrison BE, Siddiq A, Ratan RR, Bottiglieri T, D'Mello SR (2004) The c-Raf inhibitor GW5074 provides neuroprotection in vitro and in an animal model of neurodegeneration through a MEK-ERK and Akt-independent mechanism. J Neurochem 90:595–608. 10.1111/j.1471-4159.2004.02530.x [DOI] [PubMed] [Google Scholar]
- Clauser KR, Baker P, Burlingame AL (1999) Role of accurate mass measurement (+/- 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem 71:2871–2882. 10.1021/ac9810516 [DOI] [PubMed] [Google Scholar]
- Cook SJ, Beltman J, Cadwallader KA, McMahon M, McCormick F (1997) Regulation of mitogen-activated protein kinase phosphatase-1 expression by extracellular signal-related kinase-dependent and Ca2+-dependent signal pathways in Rat-1 cells. J Biol Chem 272:13309–13319. 10.1074/jbc.272.20.13309 [DOI] [PubMed] [Google Scholar]
- Dang G, Yang Y, Wu G, Hua Y, Keep RF, Xi G (2017) Early erythrolysis in the hematoma after experimental intracerebral hemorrhage. Transl Stroke Res 8:174–182. 10.1007/s12975-016-0505-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ (1995) Transcriptional regulation by MAP kinases. Mol Reprod Dev 42:459–467. 10.1002/mrd.1080420414 [DOI] [PubMed] [Google Scholar]
- de Bernardo S, Canals S, Casarejos MJ, Solano RM, Menendez J, Mena MA (2004) Role of extracellular signal-regulated protein kinase in neuronal cell death induced by glutathione depletion in neuron/glia mesencephalic cultures. J Neurochem 91:667–682. 10.1111/j.1471-4159.2004.02744.x [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072. 10.1016/j.cell.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokladda K, Green KA, Pan DA, Hardie DG (2005) PD98059 and U0126 activate AMP-activated protein kinase by increasing the cellular AMP: ATP ratio and not via inhibition of the MAP kinase pathway. FEBS Lett 579:236–240. 10.1016/j.febslet.2004.11.084 [DOI] [PubMed] [Google Scholar]
- Eckert A, Böck BC, Tagscherer KE, Haas TL, Grund K, Sykora J, Herold-Mende C, Ehemann V, Hollstein M, Chneiweiss H, Wiestler OD, Walczak H, Roth W (2008) The PEA-15/PED protein protects glioblastoma cells from glucose deprivation-induced apoptosis via the ERK/MAP kinase pathway. Oncogene 27:1155–1166. 10.1038/sj.onc.1210732 [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM (1998) Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem 273:18623–18632. 10.1074/jbc.273.29.18623 [DOI] [PubMed] [Google Scholar]
- Gu Y, Hua Y, Keep RF, Morgenstern LB, Xi G (2009) Deferoxamine reduces intracerebral hematoma-induced iron accumulation and neuronal death in piglets. Stroke 40:2241–2243. 10.1161/STROKEAHA.108.539536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan S, Price JC, Prusiner SB, Ghaemmaghami S, Burlingame AL (2011a) A data processing pipeline for mammalian proteome dynamics studies using stable isotope metabolic labeling. Mol Cell Proteomics 10:M111 010728. 10.1074/mcp.M111.010728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan YJ, Zhang Z, Yu C, Ma L, Hu W, Xu L, Gao JS, Chung CS, Wang L, Yang ZF, Fast LD, Chung AS, Kim M, Ayala A, Zhuang S, Zheng S, Chin YE (2011b) Phospho-SXXE/D motif mediated TNF receptor 1-TRADD death domain complex formation for T cell activation and migration. J Immunol 187:1289–1297. 10.4049/jimmunol.1003399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Keep RF, Hoff JT, Xi G (2008) Deferoxamine therapy for intracerebral hemorrhage. Acta Neurochir Suppl 105:3–6. [DOI] [PubMed] [Google Scholar]
- Kamakura S, Moriguchi T, Nishida E (1999) Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J Biol Chem 274:26563–26571. 10.1074/jbc.274.37.26563 [DOI] [PubMed] [Google Scholar]
- Karuppagounder SS, Alim I, Khim SJ, Bourassa MW, Sleiman SF, John R, Thinnes CC, Yeh TL, Demetriades M, Neitemeier S, Cruz D, Gazaryan I, Killilea DW, Morgenstern L, Xi G, Keep RF, Schallert T, Tappero RV, Zhong J, Cho S, et al. (2016) Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates ATF4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci Transl Med 8:e328ra29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuppagounder SS, Alin L, Chen Y, Brand D, Bourassa MW, Dietrich K, Wilkinson CM, Nadeau CA, Kumar A, Perry S, Pinto JT, Darley-Usmar V, Sanchez S, Milne GL, Pratico D, Holman TR, Carmichael ST, Coppola G, Colbourne F, Ratan RR (2018) N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann Neurol 84:854–872. 10.1002/ana.25356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR, Sen CK (2003) Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J Biol Chem 278:43508–43515. 10.1074/jbc.M307075200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodiha M, Rassi JG, Brown CM, Stochaj U (2007) Localization of AMP kinase is regulated by stress, cell density, and signaling through the MEK–>ERK1/2 pathway. Am J Physiol Cell Physiol 293:C1427–C1436. 10.1152/ajpcell.00176.2007 [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Xie Z, London ABK, Yang J, Evangelista JE, Lachmann A, Shu I, Torre D, Ma'ayan A (2021) KEA3: improved kinase enrichment analysis via data integration. Nucleic Acids Res 49:W304–W316. 10.1093/nar/gkab359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinthal DJ, Defranco DB (2005) Reversible oxidation of ERK-directed protein phosphatases drives oxidative toxicity in neurons. J Biol Chem 280:5875–5883. 10.1074/jbc.M410771200 [DOI] [PubMed] [Google Scholar]
- Li J, Gorospe M, Hutter D, Barnes J, Keyse SM, Liu Y (2001) Transcriptional induction of MKP-1 in response to stress is associated with histone H3 phosphorylation-acetylation. Mol Cell Biol 21:8213–8224. 10.1128/MCB.21.23.8213-8224.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Wan J, Lan X, Han X, Wang Z, Wang J (2017a) Neuroprotection of brain-permeable iron chelator VK-28 against intracerebral hemorrhage in mice. J Cereb Blood Flow Metab 37:3110–3123. 10.1177/0271678X17709186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, Cheng T, Yang J, Wang Z, Jiang C, Ying M, Koehler RC, Stockwell BR, Wang J (2017b) Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2:e90777. 10.1172/jci.insight.90777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Weiland A, Chen X, Lan X, Han X, Durham F, Liu X, Wan J, Ziai WC, Hanley DF, Wang J (2018) Ultrastructural characteristics of neuronal death and white matter injury in mouse brain tissues after intracerebral hemorrhage: coexistence of ferroptosis, autophagy, and necrosis. Front Neurol 9:581. 10.3389/fneur.2018.00581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D (1997) A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron 19:453–463. 10.1016/S0896-6273(00)80953-8 [DOI] [PubMed] [Google Scholar]
- Lin TN, He YY, Wu G, Khan M, Hsu CY (1993) Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke 24:117–121. 10.1161/01.STR.24.1.117 [DOI] [PubMed] [Google Scholar]
- Liu R, Li H, Hua Y, Keep RF, Xiao J, Xi G, Huang Y (2019) Early hemolysis within human intracerebral hematomas: an MRI study. Transl Stroke Res 10:52–56. 10.1007/s12975-018-0630-2 [DOI] [PubMed] [Google Scholar]
- Mills E, Dong XP, Wang F, Xu H (2010) Mechanisms of brain iron transport: insight into neurodegeneration and CNS disorders. Future Med Chem 2:51–64. 10.4155/fmc.09.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody N, Leitch J, Armstrong C, Dixon J, Cohen P (2001) Effects of MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett 502:21–24. 10.1016/S0014-5793(01)02651-5 [DOI] [PubMed] [Google Scholar]
- Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63. 10.1016/0022-1759(83)90303-4 [DOI] [PubMed] [Google Scholar]
- Ong Q, Guo S, Zhang K, Cui B (2015) U0126 protects cells against oxidative stress independent of its function as a MEK inhibitor. ACS Chem Neurosci 6:130–137. 10.1021/cn500288n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne DM, Rossomando AJ, Martino P, Erickson AK, Her JH, Shabanowitz J, Hunt DF, Weber MJ, Sturgill TW (1991) Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J 10:885–892. 10.1002/j.1460-2075.1991.tb08021.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratan RR (2020) The chemical biology of ferroptosis in the central nervous system. Cell Chem Biol 27:479–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM (1994a) Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem 62:376–379. 10.1046/j.1471-4159.1994.62010376.x [DOI] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM (1994b) Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci 14:4385–4392. 10.1523/JNEUROSCI.14-07-04385.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SR, Dang TN, Dringen R, Bishop GM (2009) Hemin toxicity: a preventable source of brain damage following hemorrhagic stroke. Redox Rep 14:228–235. 10.1179/135100009X12525712409931 [DOI] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Rådmark O, Wurst W, Bornkamm GW, Schweizer U, Conrad M (2008) Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 8:237–248. 10.1016/j.cmet.2008.07.005 [DOI] [PubMed] [Google Scholar]
- Sleiman SF, Langley BC, Basso M, Berlin J, Xia L, Payappilly JB, Kharel MK, Guo H, Marsh JL, Thompson LM, Mahishi L, Ahuja P, MacLellan WR, Geschwind DH, Coppola G, Rohr J, Ratan RR (2011) Mithramycin is a gene-selective Sp1 inhibitor that identifies a biological intersection between cancer and neurodegeneration. J Neurosci 31:6858–6870. 10.1523/JNEUROSCI.0710-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TG, Sweetman D, Patterson M, Keyse SM, Münsterberg A (2005) Feedback interactions between MKP3 and ERK MAP kinase control scleraxis expression and the specification of rib progenitors in the developing chick somite. Development 132:1305–1314. 10.1242/dev.01699 [DOI] [PubMed] [Google Scholar]
- Tang DG, Chen YQ, Honn KV (1996) Arachidonate lipoxygenases as essential regulators of cell survival and apoptosis. Proc Natl Acad Sci USA 93:5241–5246. 10.1073/pnas.93.11.5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, Kuo MT (2012) Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res 72:2622–2633. 10.1158/0008-5472.CAN-11-3605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortzel I, Seger R (2011) The ERK cascade: distinct functions within various subcellular organelles. Genes Cancer 2:195–209. 10.1177/1947601911407328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wu T, Xu X, Wang J, Wang J (2011) Iron toxicity in mice with collagenase-induced intracerebral hemorrhage. J Cereb Blood Flow Metab 31:1243–1250. 10.1038/jcbfm.2010.209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KJ, Polack A, Dalla-Favera R (1999) Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science 283:676–679. 10.1126/science.283.5402.676 [DOI] [PubMed] [Google Scholar]
- Wyse JW, Butterfield DA (1989) Interaction of hemin with erythrocyte membranes: alterations in the physical state of the major sialoglycoprotein. Biochim Biophys Acta 979:121–126. 10.1016/0005-2736(89)90531-2 [DOI] [PubMed] [Google Scholar]
- Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D (2016) Ferroptosis: process and function. Cell Death Differ 23:369–379. 10.1038/cdd.2015.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zille M, Kumar A, Kundu N, Bourassa MW, Wong VSC, Willis D, Karuppagounder SS, Ratan RR (2019) Ferroptosis in neurons and cancer cells is similar but differentially regulated by histone deacetylase inhibitors. eNeuro 6:ENEURO.0263-18.2019. 10.1523/ENEURO.0263-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J, Milner TA, Jonas EA, Ratan RR (2017) Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke 48:1033–1043. 10.1161/STROKEAHA.116.015609 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The intraperitoneal injection of the MEK inhibitor U0126 does not improve functional recovery after ICH in mice. ICH animals received daily intraperitoneal injections of 25 mg/kg U0126 or vehicle starting at 2 h after surgery for 7 d. The functional recovery (adhesive tape removal and corner task) was assessed up to 7 d after ICH. The values show the mean ± SD for adhesive tape removal and the median for corner task; *p < 0.05 versus baseline. For exact p values, refer to Extended Data Figure 1-2. Download Figure 1-1, TIF file (4.1MB, tif) .
Intraperitoneal injection of U0126 after ICH. Download Figure 1-2, DOCX file (15.3KB, docx) .
Intracerebroventricular injection of U0126 after ICH. Download Figure 1-3, DOCX file (18.5KB, docx) .
Effect of MEK1/2 inhibitors on pERK/ERK levels in hemin-induced ferroptosis in primary cortical neurons. Download Figure 2-1, DOCX file (14KB, docx) .
MEK1/2 inhibitors in hemin-induced ferroptosis in primary cortical neurons. Download Figure 3-1, DOCX file (16.7KB, docx) .
MEK1/2 inhibitors in hemoglobin (hb)-induced ferroptosis in primary cortical neurons. Download Figure 3-2, DOCX file (16.7KB, docx) .
Effect of Mkp3 overexpression on pERK/ERK levels in HT22 cells. Download Figure 4-1, DOCX file (14.1KB, docx) .
Cell survival after Mkp3 overexpression in hemin-induced versus glutathione depletion (HCA)-induced ferroptosis in HT22 cells. Download Figure 4-2, DOCX file (15KB, docx) .
pERK/ERK levels in the cytoplasm and nucleus in hemin-induced versus glutathione depletion (HCA)-induced ferroptosis in primary cortical neurons Download Figure 5-1, DOCX file (15.8KB, docx) .
Mkp1 and Mkp3 mRNA expression in hemin-induced versus glutathione depletion (HCA)-induced ferroptosis in primary cortical neurons. Download Figure 5-2, DOCX file (15.6KB, docx) .
Inhibitors of Raf, MEK5, ERK5, MEK4, and an activator of AMPK in hemin-induced ferroptosis in primary cortical neurons. Download Figure 6-1, DOCX file (16.8KB, docx) .
Top 10 ranked kinases downregulated by hemin. Full phosphoset of the unbiased phosphoproteomics of ferroptotic cell death induced by erastin and hemin and treated with U0126. Download Figure 7-1, XLSX file (24.6MB, xlsx) .
Mean rank of enrichment over 11 gene set libraries from a KEA3 kinase enrichment analysis of erastin-treated, hemin-treated, or U0126-treated cells compared to their respective controls. Download Figure 7-2, XLSX file (147.9KB, xlsx) .
Motifs surrounding the phosphorylation sites in single phosphorylated peptides with abundances altered by erastin and hemin treatments. Download Figure 7-3, TIF file (19KB, tif) .
Motifs surrounding the phosphorylation sites in single phosphorylated peptides with abundances altered by erastin and hemin treatments. Download Figure 7-4, XLSX file (10.4KB, xlsx) .
Top 10 ranked kinases upregulated in erastin+U0126 treatment. Download Figure 8-1, TIF file (20.2KB, tif) .
Phosphopeptides that were significantly changed in hemin versus control treatment, that also had also significantly different levels between the hemin-treated and erastin-treated cells, and where U0126 treatment led to changes in the opposite direction of the hemin treatment. Download Figure 8-2, XLSX file (9.9MB, xlsx) .
Motifs surrounding the phosphorylation sites in single phosphorylated peptides with abundances altered by hemin that have also significantly different levels between the hemin-treated and erastin-treated cells and that are affected by the U0126 treatment in hemin-treated cells. Download Figure 8-3, XLSX file (10.4KB, xlsx) .
Data Availability Statement
The phosphoproteomics data have been deposited to the PRIDE Archive (http://www.ebi.ac.uk/pride/archive/) via the PRIDE partner repository with the data set identifier PXD029318.