

Abstract
The debilitating psychomotor symptoms of Huntington's disease (HD) are linked partly to degeneration of the basal ganglia indirect pathway. At early symptomatic stages, before major cell loss, indirect pathway neurons exhibit numerous cellular and synaptic changes in HD and its models. However, the impact of these alterations on circuit activity remains poorly understood. To address this gap, optogenetic- and reporter-guided electrophysiological interrogation was used in early symptomatic male and female Q175 HD mice. D2 dopamine receptor-expressing striatal projection neurons (D2-SPNs) were hypoactive during synchronous cortical slow-wave activity, consistent with known reductions in dendritic excitability and cortical input strength. Downstream prototypic parvalbumin-expressing external globus pallidus (PV+ GPe) neurons discharged at 2-3 times their normal rate, even during periods of D2-SPN inactivity, arguing that defective striatopallidal inhibition was not the only cause of their hyperactivity. Indeed, PV+ GPe neurons also exhibited abnormally elevated autonomous firing ex vivo. Optogenetic inhibition of PV+ GPe neurons in vivo partially and fully ameliorated the abnormal hypoactivity of postsynaptic subthalamic nucleus (STN) and putative PV– GPe neurons, respectively. In contrast to STN neurons whose autonomous firing is impaired in HD mice, putative PV– GPe neuron activity was unaffected ex vivo, implying that excessive inhibition was responsible for their hypoactivity in vivo. Together with previous studies, these data demonstrate that (1) indirect pathway nuclei are dysregulated in Q175 mice through changes in presynaptic activity and/or intrinsic cellular and synaptic properties; and (2) prototypic PV+ GPe neuron hyperactivity and excessive target inhibition are prominent features of early HD pathophysiology.
SIGNIFICANCE STATEMENT The early symptoms of Huntington's disease (HD) are linked to degenerative changes in the action-suppressing indirect pathway of the basal ganglia. Consistent with this linkage, the intrinsic properties of cells in this pathway exhibit complex alterations in HD and its models. However, the impact of these changes on activity is poorly understood. Using electrophysiological and optogenetic approaches, we demonstrate that the indirect pathway is highly dysregulated in early symptomatic HD mice through changes in upstream activity and/or intrinsic properties. Furthermore, we reveal that hyperactivity of external globus pallidus neurons and excessive inhibition of their targets are key features of early HD pathophysiology. Together, these findings could help to inform the development and targeting of viral-based, gene therapeutic approaches for HD.
Keywords: arkypallidal, electrophysiology, globus pallidus, striatum, subthalamic nucleus
Introduction
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder caused by expansion of trinucleotide CAG repeats (>35) in exon 1 of the huntingtin gene (HTT) (Bates et al., 2015). The cardinal features of HD are progressive dysregulation and degeneration of the basal ganglia and cortex, and the emergence of associated motor, cognitive, and psychiatric symptoms (Bates et al., 2015). Although mutant huntingtin (mHTT) species impair multiple processes that are critical for neuronal function, the mechanisms that underlie the relative vulnerability of cortico-basal ganglia circuit function remain poorly understood (Reddy and Shirendeb, 2012; Seredenina and Luthi-Carter, 2012; Johri et al., 2013; Tong et al., 2014; Martin et al., 2015; Rosas et al., 2018). Under normal conditions, the basal ganglia promote contextually appropriate behavior, in part through the processing of functionally diverse cortical inputs (Mink and Thach, 1993; Nelson and Kreitzer, 2014; Klaus et al., 2019) by complex microcircuits, including the so-called direct and indirect pathways. The results of basal ganglia computation are then broadcast to the thalamus, midbrain, and brainstem, affecting behavior (Albin et al., 1989; Gerfen et al., 1990). D2 dopamine receptor expressing striatal projection neurons (D2-SPNs) that are the source of the so-called action-suppressing indirect pathway are susceptible early in HD (Albin et al., 1992; Richfield et al., 1995; Reiner and Deng, 2018). As a result, loss of indirect pathway function is posited to underlie chorea and impaired behavioral control, in the initial clinical phase of adult-onset HD (Bates et al., 2015; Reiner and Deng, 2018).
Examination of HD and its experimental models at early symptomatic ages has revealed that neurons in the basal ganglia and cortex exhibit complex changes in their intrinsic and synaptic properties (Cepeda et al., 2007; Raymond et al., 2011; Plotkin and Surmeier, 2015). For example, D2-SPNs exhibit loss of cortico-striatal long-term potentiation (LTP), and reductions in axospinous synapse density, miniature excitatory postsynaptic current (mEPSC) amplitude, and dendritic excitability (Plotkin and Surmeier, 2014; Plotkin et al., 2014; Sebastianutto et al., 2017; Carrillo-Reid et al., 2019). These observations suggest that D2-SPNs will be less effectively engaged by cortical excitation in HD mice. Consistent with this view, the activities of cortical neurons and SPNs in vivo are less well correlated in HD mice (Miller et al., 2008; Walker et al., 2008; Estrada-Sanchez et al., 2015). However, downstream external globus pallidus (GPe) neurons exhibit no sign of disinhibition in vivo, that is, they are not hyperactive (Beaumont et al., 2016), possibly because striatopallidal transmission has been upregulated (Perez-Rosello et al., 2019) and autonomous subthalamic nucleus (STN) activity has been downregulated (Atherton et al., 2016) in compensation. Thus, the effects of cortical drive on the indirect pathway cannot be easily inferred from ex vivo observations alone. Furthermore, most of the in vivo electrophysiological studies on HD mice are difficult to interpret because the recorded nuclei are comprised of multiple cell types that were not discriminated. To address these gaps, we used optogenetic- and reporter-guided electrophysiological interrogation of indirect pathway neurons in a well-characterized mouse model of HD at an early symptomatic stage. Specifically, we used Q175 model mice, in which exon 1 of human HTT (containing ∼190 trinucleotide repeats) is knocked into the endogenous Htt gene. This model exhibits many of the progressive molecular, neuropathological, and behavioral abnormalities seen in HD patients (Heikkinen et al., 2012; Menalled et al., 2012). In pilot experiments, we found that stereotyped patterns of cortical activity present under urethane anesthesia were similar in WT and Q175 mice, allowing us to compare firing in the indirect pathway during analogous cortical states. We also used patch-clamp recording in ex vivo brain slices to determine whether the changes in GPe neuron activity that we observed in Q175 mice in vivo were due in part to alterations in autonomous firing. Together with other studies, our data argue that mHTT profoundly dysregulates indirect pathway activity through complex changes in presynaptic activity and/or cellular and synaptic properties.
Materials and Methods
Animals
Procedures were performed in compliance with the policies of the National Institutes of Health and approved by the Institutional Animal Care and Use Committee of Northwestern University. Mice were maintained on a 14 h light/10 h dark cycle with food and water ad libitum, and monitored regularly by animal care technicians, veterinarians, and research staff.
Heterozygous Q175 mice (B6J.129S1-Htttm1.1Mfc/190ChdiJ; RRID:IMSR_JAX:029928; The Jackson Laboratory) were bred with homozygous PT-kj18-cre mice (Tg(Sim1-cre)KJ18Gsat; RRID:MMRRC_036958-UCD; MMRRC Repository), or A2A-cre mice (Tg(Adora2a-cre)KG139Gsat; RRID:MMRRC_036158-UCD; MMRRC Repository), or PV-cre mice (B6.Cg-Pvalbtm1.1(cre)Aibs/J; RRID:IMSR_JAX:012358; The Jackson Laboratory) to generate offspring that were either homozygous for Htt (WT), or heterozygous for Htt and mHtt (Q175), and heterozygous for Cre-recombinase. The following experimental mice were used (median and age range are reported): WT/PT-kj18-cre: age = 233, 225-239 d old, n = 5; Q175/PT-kj18-cre: age = 238, 228-244 d old, n = 4; WT/A2A-cre: age = 210, 210-233 d old, n = 3; Q175/A2A-cre: age = 211, 211-235 d old, n = 3; WT/PV-cre: age = 219.5, 204-232 d old, n = 4; Q175/PV-cre: age = 220, 205-234 d old, n = 5. Experimental mice were male, except for PV-cre mice, where both male (WT/PV-cre: n = 3; Q175/PV-cre: n = 3) and female (WT/PV-cre: n = 1; Q175/PV-cre: n = 2) mice were used. Data from male and female PV-cre mice were overlapping and therefore pooled.
Stereotaxic injection of viral vectors
Anesthesia was induced with vaporized 3%-4% isoflurane (Smiths Medical ASD) followed by an intraperitoneal injection of ketamine (100 mg/kg). After securing the mouse in a stereotaxic instrument (Neurostar), anesthesia was maintained with 1%-2% isoflurane. Adeno-associated viruses (AAVs) diluted in HEPES-buffered synthetic interstitial fluid (HBS SIF: 140 mm NaCl, 23 mm glucose, 15 mm HEPES, 3 mm KCl, 1.5 mm MgCl2, 1.6 mm CaCl2; pH 7.2 with NaOH; 300-310 mOsm/L) were then injected under stereotaxic guidance. AAV injection was conducted over 5-10 min at each site. An additional 5-10 min was then allowed for the injectate to diffuse before syringe retraction. ChR2(H134R)-eYFP was virally expressed in: (1) layer V pyramidal tract (PT) neurons in PT-kj18-cre mice through unilateral injection of AAV9.EF1a.DIO.hChR2(H134R)-eYFP.WPRE.hGH (RRID:Addgene_20298; 1 × 1013 genome copies/ml; AP: 0.6 mm, 1.2 mm, 1.8 mm; ML: 1.5 mm; DV: 1.0 mm; 0.5 μl per injection); and (2) D2-SPNs in A2A-cre mice through unilateral injections of AAV9.EF1a.DIO.hChR2(H134R)-eYFP.WPRE.hGH (RRID:Addgene_20298; 3 × 1011 genome copies/ml; AP: 0.4 mm, 0.9 mm; ML: 2.2 mm; DV: 3.7 mm, 2.7 mm; 0.3 μl per injection). Arch-GFP was virally expressed in parvalbumin-expressing (PV+) GPe neurons in PV-cre mice through unilateral injection of AAV9.CBA.Flex.Arch-GFP.WPRE.SV40 (RRID:Addgene_22222; 5 × 1011 genome copies/ml; AP: −0.27 mm; ML: 1.90 mm; DV: 3.95 mm, 3.45 mm; 0.25 μl per ventral injection and 0.20 μl per dorsal injection). eGFP was virally expressed in PV+ GPe neurons in PV-cre mice through unilateral injection of AAV9.Syn.DIO.eGFP.WPRE.hGH (RRID:Addgene_100043; 3 × 1011 genome copies/ml; AP: −0.27 mm; ML: 1.90 mm; DV: 3.95 mm, 3.45 mm; 0.25 μl per ventral injection and 0.20 μl per dorsal injection).
In vivo electrophysiological recording
Two to 4 weeks following surgery, anesthesia was induced with vaporized 3%-4% isoflurane followed by intraperitoneal injection of urethane (1.5 g/kg; Sigma-Aldrich). Mice were placed back into their home cage for ∼60 min until the toe-pinch withdrawal reflex was abolished. If necessary, additional urethane supplements (0.5 g/kg, i.p.) were administered every 30 min until the withdrawal reflex was eliminated. Mice were then placed into a stereotaxic instrument (David Kopf Instruments) for the duration of the recording session, with urethane supplements administered as required to maintain anesthesia. Craniotomies were drilled over the primary motor cortex (AP: 1.4 mm; ML: 1.5 mm), striatum (AP: 0.65 mm; ML: 1.95 mm), GPe (AP: −0.3 mm; ML: 2.0 mm), and/or STN (AP: −1.9 mm; ML: 1.4 mm) and irrigated with HBS. The intracranial electroencephalogram (EEG) was recorded from a peridural screw electrode (MS-51 960-1; McMaster-Carr) affixed over the ipsilateral primary motor cortex of A2A-cre and PV-cre mice. In PT-kj18-cre mice in which extracellular recordings of motor cortical neurons were made, the EEG screw was implanted over the contralateral primary motor cortex. Extracellular single-unit recordings were acquired using silicon tetrodes/optrodes (A1x4-tet-10 mm-100-121-A16 and A1x4-tet-10 mm-100-121-OA16, respectively; NeuroNexus Technologies) connected to a 64-channel Digital Lynx (Neuralynx) data acquisition system via a unity gain headstage, with a reference wire implanted adjacent to the ipsilateral temporal musculature. Signals were sampled at 40 kHz, with a gain of 14×. Online digital finite impulse response filters were applied. Single-unit activity was bandpass-filtered between 200 and 9000 Hz, and EEG and local field potential signals were bandpass-filtered between 0.1 and 400 Hz. Optogenetic stimulation was delivered using either a 473 nm diode laser (LuxX+ 473-100; Omicron-Laserage Laserprodukte) or a custom 577 nm laser system (Genesis MX STM 577-500 OPSL CW; Coherent). In order to histologically verify recording sites, silicon tetrodes/optrodes were dipped in a lipophilic florescent dye (DiI; 20 mg/ml in 50% acetone/methanol; D282; Thermo Fisher Scientific) before implantation. Sensory-evoked cortical activation (ACT) was generated by pinching the hindpaw for 5 s using a pair of fine forceps (Fine Science Tools). ChR2(H134R)-eYFP-expressing neurons were optogenetically stimulated with 473 nm light (<6 mW) for a duration of 5 ms. Stimulation was repeated at least 5 times with each trial of stimulation separated by a minimum of 2 min. Arch-GFP-expressing neurons were optogenetically inhibited through delivery of 577 nm light (<6 mW) for a duration of 5 s. Optogenetic inhibition was repeated at least 2 times with each trial being separated by a minimum of 2 min. Laser intensity was calibrated as power at the tip of the optrode before probe implantation and verified at the conclusion of each experiment.
Immunohistochemistry
After electrophysiological recording, mice were given a lethal dose of anesthetic and then perfused transcardially with ∼5-10 ml of 0.01 m phosphate buffered saline (PBS) (pH 7.4; P3813, Millipore Sigma) followed by 15-30 ml of 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer (PB), pH 7.4. Each brain was then removed and postfixed overnight in 4% PFA (in 0.1 m PB, pH 7.4) before being washed in PBS, blocked, and sectioned in the coronal plane at 70 µm using a vibratome (VT1000S; Leica Microsystems). Sections were then processed for the immunohistochemical detection of NeuN, an antigen expressed by neurons that is commonly used to delineate brain structures. First, sections were washed in PBS and incubated for 48-72 h at 4°C in anti-NeuN (1:200; clone A60; Millipore Sigma; RRID:AB_2532109) in PBS with 0.5% Triton X-100 (Millipore Sigma) and 2% normal donkey serum (Jackson ImmunoResearch Laboratories). Then, sections were washed in PBS before being incubated for 90 min at room temperature in AlexaFluor-488- or 594-conjugated donkey anti-mouse IgG (1:250; Jackson ImmunoResearch Laboratories; RRID:AB_2313584; RRID:AB_2340621) in PBS with 0.5% Triton X-100 and 2% normal donkey serum. In a subset of PV-cre mice, in which PV+ GPe neurons expressed Arch-GFP or eGFP, adjacent sections of the GPe were processed for the immunohistochemical detection of PV (primary antibody: 1:1000 guinea pig anti-PV; Synaptic Systems; RRID:AB_2156476; secondary antibody: 1:250 AlexaFluor-594 donkey anti-guinea pig IgG; Jackson ImmunoResearch Laboratories; RRID:AB_2340474) or FoxP2 (primary antibody: 1:1000 rabbit anti-FoxP2; Millipore Sigma; RRID:AB_1078909; secondary antibody: 1:250 AlexaFluor-594 donkey anti-rabbit IgG; Jackson ImmunoResearch Laboratories; RRID:AB_2340621). Finally, sections were washed in PBS and mounted on glass slides with ProLong Diamond Antifade Reagent (P36965; Thermo Fisher Scientific). Mountant was allowed to cure for at least 24 h before storage at 4°C or imaging. DiI and immunofluorescent labeling were imaged using a Zeiss Axioskop 2 microscope (Carl Zeiss), equipped with an Axiocam CCD camera (426508-9901-000, Carl Zeiss), and Neurolucida software (MFB Bioscience). DiI and immunohistochemical labeling were used to map sites of recording (Fig. 1). Representative images were also acquired using confocal laser scanning microscopy (A1R; Nikon Instruments).
Figure 1.
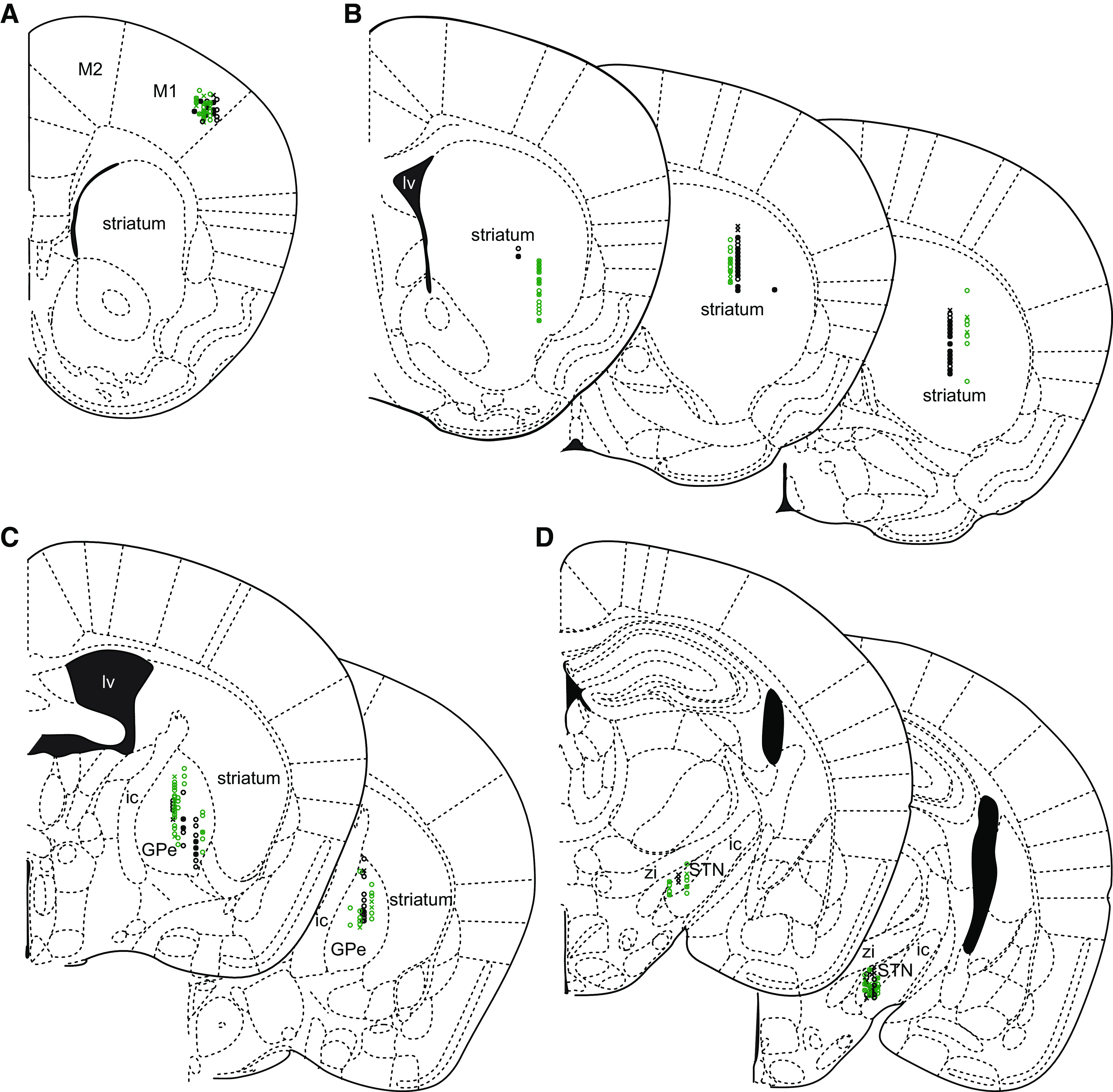
In vivo recording sites in the primary motor cortex, striatum, GPe, and STN. A–D, The locations of optogenetically responsive (O) and nonresponsive (X) neurons in WT (black) and Q175 (green) mice were mapped onto a standard atlas (Paxinos and Franklin, 2001) using the location of DiI-labeled recording tracks relative to cytoarchitectonic boundaries delineated by immunohistochemical labeling of NeuN. A, Recording sites in the primary motor cortex of PT-kj18-cre mice (M1, primary motor cortex; M2, secondary motor cortex). B, Recording sites in the striatum of A2A-cre mice (lv, lateral ventricle). C, Recording sites in the GPe of PV-cre mice (ic, internal capsule). D, Recording sites in the STN of PV-cre mice (zi, zona incerta).
In vivo electrophysiological analysis
Estimates of spectral power density were extracted using the Chronux data analysis toolbox (Bokil et al., 2010) for MATLAB (http://chronux.org/; The MathWorks). The EEG signal was downsampled to 1000 Hz, and spectral power was assessed at a resolution frequency of 0.061 Hz. Putative single-unit activity was discriminated with Plexon Offline Sorter software (version 3, Plexon; RRID:SCR_000012) using a combination of template matching, principal component analysis, and manual clustering. In addition, a threshold of <0.5% of interspike intervals under 2 ms was required for classification as a putative single unit (% interspike interval within 2 ms; cortical neurons: WT: 0.0, 0.0-0.0, n = 26; Q175: 0.0, 0.0-0.0, n = 43; striatal neurons: WT: 0.0, 0.0-0.0, n = 102; Q175: 0.0, 0.0-0.0, n = 65; GPe neurons: WT: 0.08, 0.02-0.29, n = 50; Q175: 0.05, 0.01-0.24, n = 55; STN neurons: WT: 0.23, 0.11-0.38, n = 30; Q175: 0.11, 0.00-0.19, n = 61; values represent median and interquartile range). Electrophysiological data were visually inspected in NeuroExplorer (Nex Technologies; RRID:SCR_001818) and then exported to MATLAB (The MathWorks; RRID:SCR_001622). Epochs with stable and robust slow-wave activity (SWA) or ACT were selected for analysis.
To determine whether neurons were responsive to optogenetic manipulation, peristimulus time histograms (PSTHs) were constructed from either 5 trials of hChR2(H134R)-eYFP stimulation or 2 or 3 trials of Arch-GFP stimulation. Cortical or striatal neurons were considered directly responsive if their activity exceeded the prestimulus mean by 2 SDs within 10 ms (bin size 2.5 ms) of hChR2(H134R)-eYFP stimulation. PV+ GPe neurons were considered directly responsive if their activity fell below 2 SDs of the prestimulus mean within 100 ms of Arch-GFP stimulation (bin size 100 ms). Putative PV– GPe neurons and STN neurons were considered to be disinhibited if their activity exceeded the prestimulus mean by 2 SDs within 100 ms of the optogenetic inhibition of PV+ GPe neurons (bin size 100 ms). To ensure that measurements were made from areas with opsin expression, analysis was restricted to recordings in which there was at least one responsive neuron on a given tetrode array. Direct or indirect responses to optogenetic stimulation were not observed when optrodes were activated in regions or mice without opsin expression (data not shown). Evoked firing was also easily discriminated from photo-electric artifacts, which were either minimal or absent.
Mean firing rates were calculated from the number of spikes divided by epoch length. The coefficient of variation (CV) of the interspike interval was used as a metric of regularity. To examine the relationship between cortical SWA and neuronal firing, phase histograms were generated in MATLAB. The EEG signal was first downsampled to 1000 Hz, and SWA was then extracted by applying a bandpass 0.1-1.5 Hz second-order Butterworth filter in the forward and reverse directions (to avoid phase shifts). The instantaneous phase of the EEG was calculated from the Hilbert transform (Le Van Quyen et al., 2001). In order to correct for the nonsinusoidal nature of slow cortical oscillations, the empirical cumulative distribution function (MATLAB) was applied (Siapas et al., 2005; Mallet et al., 2008b; Abdi et al., 2015; Kovaleski et al., 2020). Thus, each spike was assigned to a phase of the EEG from 0° to 360° (with 0°/360° and 180° corresponding to the peak active and inactive components of the EEG, respectively). Phase histograms were then constructed using 15° bins and plotted as median and interquartile range. In-phase activity was defined as activity occurring within a 180° window centered on 0°/360°. Anti-phase activity was defined as activity occurring within a 180° window centered on 180°.
Ex vivo electrophysiological recording
Mice were first lightly anesthetized with isoflurane, and then deeply anesthetized with ketamine/xylazine (87/13 mg/kg, i.p.), before being perfused transcardially with ∼10 ml of ice-cold sucrose-based ACSF (230 mm sucrose, 2.5 mm KCl, 1.25 mm NaH2PO4, 0.5 mm CaCl2, 10 mm MgSO4, 10 mm glucose, 26 mm NaHCO3, 1 mm sodium pyruvate, 5 μm L-glutathione; equilibrated with 95% O2 and 5% CO2). The brain was then removed, immersed in ice-cold 95% O2/5% CO2-equilibrated sucrose-based ACSF, and sectioned at 250 µm in the sagittal plane with a vibratome (VT1200S; Leica Microsystems). Slices were transferred to a holding chamber, immersed in ACSF (126 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 2 mm CaCl2, 2 mm MgSO4, 10 mm glucose, 26 mm NaHCO3, 1 mm sodium pyruvate, 5 μm L-glutathione; equilibrated with 95% O2 and 5% CO2), and maintained at 35°C for 30 min, and then room temperature. Next, individual slices were transferred to a recording chamber where they were perfused at 4-5 ml/min with SIF (126 mm NaCl, 3 mm KCl, 1.25 mm NaH2PO4, 1.6 mm CaCl2, 1.5 mm MgSO4, 10 mm glucose, 26 mm NaHCO3; equilibrated with 95% O2 and 5% CO2) at 35°C.
Patch-clamp recordings were made using 3-6 mΩ impedance, borosilicate glass electrodes filled with HBS SIF. Electrodes were positioned under visual guidance (Axioskop FS2, Carl Zeiss) using computer-controlled micromanipulators (Luigs and Neumann). Somatic recordings were made in the loose-seal, cell-attached configuration using an amplifier (MultiClamp 700B; Molecular Devices), and an associated digitizer (Digidata 1440A; Molecular Devices) controlled by pCLAMP 10.3 (Molecular Devices). Electrode capacitance was compensated, and signals were low-pass filtered online at 10 kHz and sampled at 25 kHz. Recordings of autonomous action potential generation were made in the presence of 20 μm DNQX, 50 μm D-AP5, 10 μm Gabazine (SR-95531), and 2 μm CGP 55845 to block synaptic transmission at AMPA, NMDA, GABAA, and GABAB receptors, respectively. All drugs used were purchased from Hello Bio and bath-applied.
PV+ GPe neurons were identified through visualization of eGFP under 460 nm light-emitting diode epifluorescent illumination (OptoLED; Cairn Research). To increase the probability that GPe neurons that did not express eGFP were PV– GPe neurons, non-eGFP-expressing GPe neurons were only recorded if eGFP-expressing PV+ GPe neurons were present in the same field of view. The frequency and regularity of eGFP-expressing PV+ GPe neuron activity and non–eGFP-expressing GPe neuron activity were calculated from 30 s recording epochs.
Experimental design and statistical analyses
Data are reported as median and interquartile range. Data are represented graphically as violin (kernel density) plots and overlaid box plots, with the median (central line), interquartile range (box), and 10%-90% range (whiskers) denoted. The number and nature of observations for each parameter are specified throughout. To ensure that the proposed research was adequately powered, sample sizes were estimated using the formulae described by Noether (1987) assuming 80% power (i.e., a 20% probability of a Type 2 error) and a two tailed α level of 0.05. For unpaired data (groups X and Y), and probabilities of X > Y (or X < Y) being 0.7, 0.8, and 0.9, the estimated sample sizes for each group are 33, 15, and 9, respectively. For paired data (where Xi and Xj are independent samples from X, reflecting effect size and sign) and the probabilities of Xi + Xj > 0 being 0.7, 0.8, and 0.9, the estimated sample sizes are 66, 30, and 17, respectively. Probabilities between 0.7 and 0.9 are representative of our historical and pilot data. To minimize assumptions concerning the distribution of data, nonparametric, two-tailed statistical comparisons were made using the Mann–Whitney U (MWU) and Wilcoxon signed-rank (WSR) tests for unpaired and paired comparisons, respectively. In addition, Fisher's exact test was used for contingency analyses. p < 0.05 was considered significant. Where appropriate, p values were adjusted for multiple comparisons using the Holm–Bonferroni method. Plots and statistical comparisons were generated in Prism (GraphPad Software; RRID:SCR_002798) and R (https://www.r-project.org/; RRID:SCR_001905).
Results
Cortical activity is similar in Q175 and WT mice during both SWA and ACT
Under anesthesia, the cortex can exhibit the following: (1) robust ∼1 Hz cortical SWA during which cortical projection neurons, including those projecting to the basal ganglia, exhibit synchronous transitions between hyperpolarized quiescent and depolarized active states, analogous to those during deep sleep (Stern et al., 1997; Steriade, 2000); and (2) spontaneous or somatosensory stimulation-triggered cortical activation ACT, during which cortical projection neurons exhibit desynchronized activity, analogous to that during arousal (Stern et al., 1997; Steriade, 2000). As a result, anesthesia has been routinely used to generate stereotyped cortical activity and probe its impact on the downstream basal ganglia in normal rodents and psychomotor disease models (Magill et al., 2001; Walters et al., 2007; Mallet et al., 2008a,b; Zold et al., 2012; Sharott et al., 2017; Aristieta et al., 2020; Kovaleski et al., 2020; Ketzef and Silberberg, 2021). Therefore, to determine whether indirect pathway activity is dysregulated in 6-month-old Q175 mice relative to WT age-matched controls, we compared neuronal activity during both cortical SWA and sensory-evoked cortical ACT under urethane anesthesia.
Cortical network activity was assessed from the intracranial EEG, which was obtained from a peridural screw “electrode” affixed over primary motor cortex. Cortical SWA was manifest in the EEG as a high-amplitude, low-frequency (∼1 Hz) oscillation on which phase-locked, low-amplitude, high-frequency oscillations were superimposed (Fig. 2A). Cortical ACT occurred spontaneously or could be triggered by somatosensory stimulation, which was signified in the EEG by diminution of the 1 Hz oscillation and persistence of higher-frequency oscillations (Fig. 2A). During cortical SWA, the powers of motor cortical oscillations in frequency bands ranging from 0 to 100 Hz were similar in Q175 and WT mice (Fig. 2B–E; Table 1). In addition, low-frequency and high-frequency cortical oscillations were attenuated and elevated, respectively, to a similar degree in Q175 and WT mice during hind paw pinch-evoked cortical ACT (Fig. 2B–E; Table 1). Together, these data suggest that motor cortical network activity is similar in 6-month-old Q175 and WT mice.
Figure 2.
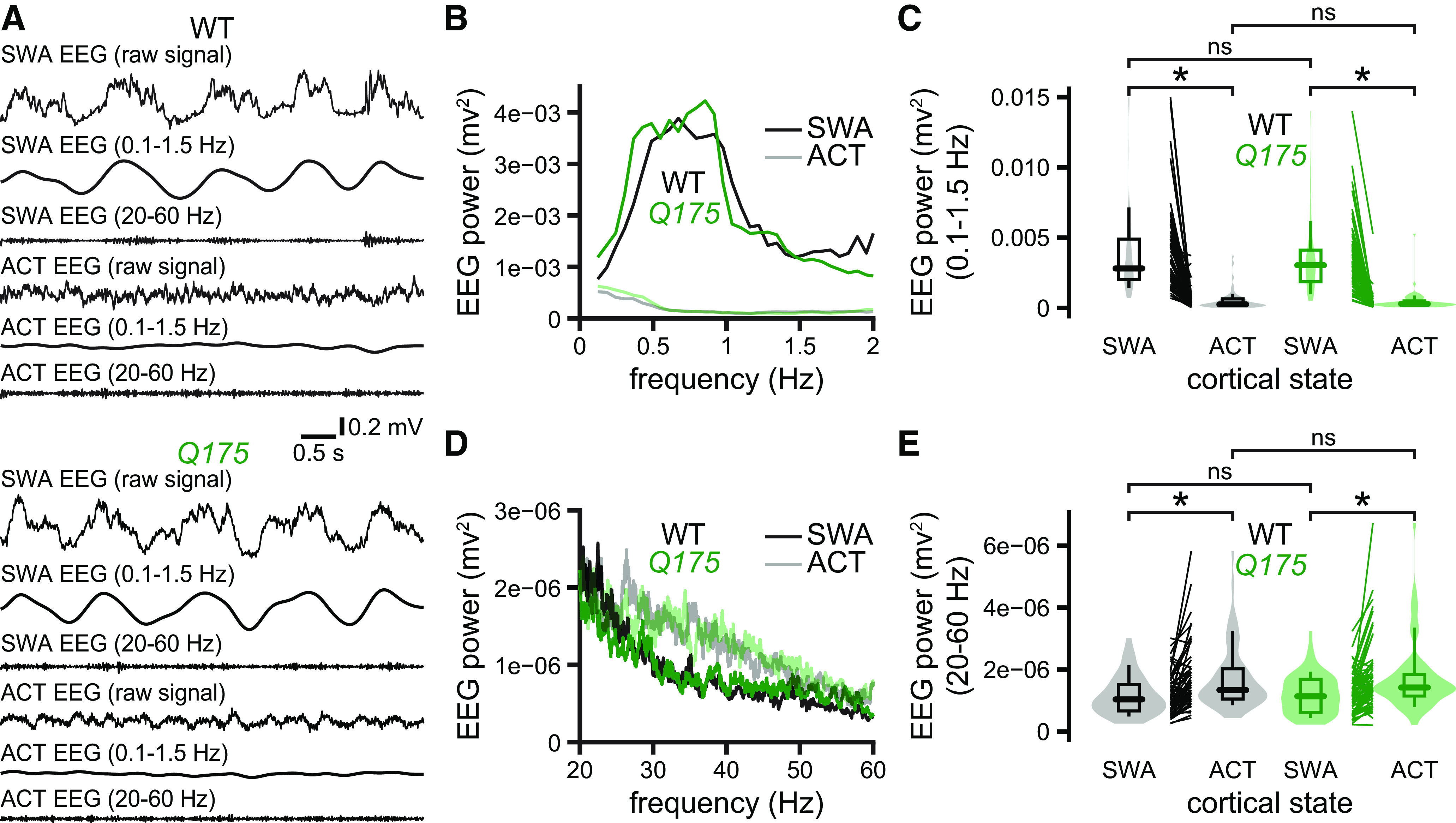
Motor cortical network activity is similar in Q175 and WT mice during both SWA and ACT. A–E, The motor cortical EEGs of urethane-anesthetized Q175 and WT mice were similar during SWA and ACT (A, examples; B–E, population data). Spectral power was similar for both low- (B, C) and high-frequency (D, E) EEG components. *p < 0.05.
Table 1.
Motor cortical EEG measurements during cortical SWA and ACT in Q175 and WT micea
| Figure/text | Measurement | Brain state | Genotype | n (mice/EEG epochs) | Median [IQ range] (mV2) | p (comparison; test) |
|---|---|---|---|---|---|---|
| 2B, C | 0.1-1.5 Hz EEG power | SWA | WT | 12/64 | 2.8e-03 [2.0e-03-4.9e-03] | p = 0.70 (SWA: WT vs Q175; MWU) |
| Q175 | 11/72 | 3.0e-03 [1.9e-03-4.1e-03] | ||||
| 2D, E | 20-60 Hz EEG power | SWA | WT | 12/64 | 1.0e-06 [6.7e-07-1.5e-06] | p = 1.3 (SWA: WT vs Q175; MWU) |
| Q175 | 11/72 | 1.1e-06 [6.2e-07-1.7e-06) | ||||
| text | 4-8 Hz EEG power | SWA | WT | 12/64 | 6.1e-05 [3.5e-05-1.4e-04] | p = 0.062 (SWA: WT vs Q175; MWU) |
| Q175 | 11/72 | 4.8e-05 [3.0e-05-9.2e-05] | ||||
| text | 8-12 Hz EEG power | SWA | WT | 12/64 | 1.4e-05 [8.7e-06-2.3e-05] | p = 0.19 (SWA: WT vs Q175; MWU) |
| Q175 | 11/72 | 1.2e-05 [5.8e-06-1.9e-05] | ||||
| text | 12-40 Hz EEG power | SWA | WT | 12/64 | 2.7e-06 [1.8e-06-3.7e-06] | p = 0.60 (SWA: WT vs Q175; MWU) |
| Q175 | 11/72 | 2.4e-06 [1.5e-06-3.3e-06] | ||||
| text | 40-100 Hz EEG power | SWA | WT | 12/64 | 3.3e-07 [2.4e-07-4.6e-07] | p = 0.11 (SWA: WT vs Q175; MWU) |
| Q175 | 11/72 | 4.1e-07 [2.5e-07-6.8e-07] | ||||
| 2B, C | 0.1-1.5 Hz EEG power | ACT | WT | 12/64 | 2.5e-04 [1.4e-04-6.4e-04] | p = 1.7e-11 (WT: SWA vs ACT; WSR) |
| Q175 | 11/72 | 2.9e-04 [1.6e-04-5.0e-04] | p = 6.8e-13 (Q175: SWA vs ACT; WSR) | |||
| p = 0.76 (ACT: WT vs Q175; MWU) | ||||||
| 2D, E | 20-60 Hz EEG power | ACT | WT | 12/64 | 1.3e-06 [1.1e-06-2.0e-06] | p = 3.1e-06 (WT: SWA vs ACT; WSR) |
| Q175 | 11/72 | 1.4e-06 [1.1e-06-1.8e-06] | p = 7.2e-06 (Q175: SWA vs ACT; WSR) | |||
| p = 0.72 (ACT: WT vs Q175; MWU) | ||||||
| text | 4-8 Hz EEG power | ACT | WT | 12/64 | 4.6e-05 [2.6e-05-6.3e-05] | p = 3.0e-03 (WT: SWA vs ACT; WSR) |
| Q175 | 11/72 | 2.8e-05 [1.8e-05-5.6e-05] | p = 9.0e-05 (Q175: SWA vs ACT; WSR) | |||
| p = 0.083 (ACT: WT vs Q175; MWU) | ||||||
| text | 8-12 Hz EEG power | ACT | WT | 12/64 | 9.0e-06 [5.2e-06-1.6e-05] | p = 4.0e-04 (WT: SWA vs ACT; WSR) |
| Q175 | 11/72 | 8.1e-06 [4.7e-06-1.2e-05] | p = 0.058 (Q175: SWA vs ACT; WSR) | |||
| p = 0.32 (ACT: WT vs Q175; MWU) | ||||||
| text | 12-40 Hz EEG power | ACT | WT | 12/64 | 2.3e-06 [1.7e-06-3.8e-06] | p = 0.87 (WT: SWA vs ACT; WSR) |
| Q175 | 11/72 | 2.3e-06 [1.7e-06-2.9e-06] | p = 1.30 (Q175: SWA vs ACT; WSR) | |||
| p = 1.1 (ACT: WT vs Q175; MWU) | ||||||
| text | 40-100 Hz EEG power | ACT | WT | 12/64 | 5.2e-07 [3.8e-07-7.6e-07] | p = 6.4e-10 (WT: SWA vs ACT; WSR) |
| Q175 | 11/72 | 5.4e-07 [4.0e-07-8.0e-07] | p = 1.2e-03 (Q175: SWA vs ACT; WSR) | |||
| p = 0.59 (ACT: WT vs Q175; MWU) |
aOscillatory activities in the motor cortex of Q175 and WT mice were similar. The power of low-frequency oscillations declined and the power of high-frequency oscillations increased following the transition from cortical SWA to ACT in both Q175 and WT mice.
Layer V cortical pyramidal neurons, some of which innervate the basal ganglia, are comprised of two major cell classes, PT and intratelencephalic type (IT) neurons (Harris and Shepherd, 2015). To compare the activity of PT neurons in Q175 and WT mice, an “opto-tagging” approach was used to identify the firing of PT neurons in vivo. First, ChR2(H134R)-eYFP was virally expressed in PT cortical neurons through injection of an AAV carrying a cre-dependent expression construct into the primary motor cortex of Q175 and WT mice that had been crossed with a PT neuron selective cre-driver line (PT-kj18-cre) (Fig. 3A–D). Two to 4 weeks later, the activity of cortical neurons was compared using an array of tetrodes fiber-coupled to a laser. Construction of peri-optogenetic stimulus time histograms revealed that the latency of firing evoked by optogenetic stimulation was unimodal in distribution (Fig. 3E,F). The majority of cells fired within 10 ms of the start of the 5 ms optogenetic stimulation pulse (latency; WT: 2.5, 2.5-5.0 ms, n = 15; Q175: 2.5, 2.5-3.7 ms, n = 21; values represent median and interquartile range) (Fig. 3E,F). These response latencies are consistent with the direct optogenetic stimulation of ChR2-expressing neurons (Hooks, 2018; Li et al., 2018). Therefore, neurons that fired within 10 ms of optogenetic stimulation were positively identified as PT neurons (Fig. 3D–F). Consistent with this classification, all identified PT neurons exhibited action potential properties that are typical of cortical pyramidal neurons (Mitchell et al., 2007; Kaufman et al., 2010; Takahashi et al., 2015; Lohani et al., 2019) (Fig. 3G,H). The proportion of PT neurons that exhibited short-latency responses to optogenetic activation and the latency of those evoked responses (Table 2) were similar in Q175 and WT mice. Neurons that were recorded on the same tetrode as identified PT neurons but were unresponsive to optogenetic stimulation or exhibited longer-latency presumably indirect, monosynaptic responses (WT: n = 11; Q175: n = 22) were also recorded. The majority of neurons (WT: n = 7; Q175: n = 15) that were unresponsive to optogenetic stimulation or exhibited latencies of discharge > 10 ms also exhibited action potential properties that are typical of cortical pyramidal neurons, consistent with the relative abundance of pyramidal versus interneurons in this cortical layer (Fig. 3G,H). The neurons with pyramidal-like action potential properties were termed unidentified layer V pyramidal neurons and likely comprise IT neurons and PT neurons that failed to express ChR2(H134R)-eYFP. The unidentified layer V pyramidal neurons that exhibited long-latency discharge in response to optogenetic stimulation are likely to be PT neurons because primary motor cortical PT neurons preferentially innervate each other rather than IT neurons (Kiritani et al., 2012). A minority of unidentified layer V neurons that exhibited interneuron action potential properties (Fig. 3G,H) were excluded from further description or analysis here because of their rarity.
Figure 3.

Optogenetic identification of layer V motor cortical projection neurons. A, B, Cre-dependent viral expression of ChR2(H134R)-eYFP (green) in layer V PT neurons (M1, primary motor cortex; DV, dorsoventral axis; ML, mediolateral axis). B, ChR2(H134R)-eYFP expression was present in a subset of layer V, presumably PT neurons (white arrows), and absent in other layer V neurons (magenta arrows), which presumably comprise a mixture of nonexpressing PT, IT, and interneurons. Cortical neurons were also immunohistochemically labeled for the pan neuronal marker NeuN (red). C, Schematic of experimental setup illustrating placement of the EEG screw electrode (gray) and optrode (blue). D, An optogenetically stimulated layer V PT neuron and an adjacent unresponsive, unidentified (UI) neuron (blue, optogenetic stimulation; inset, spontaneous [black] and optogenetically evoked [blue] action potentials). E, F, Cortical neurons were considered to be directly responsive if their activity exceeded the prestimulus mean (solid red line) by 2 SDs (dotted red line) within 10 ms (bin size, 2.5 ms) of the start of the optogenetic stimulation pulse (E, PSTHs from examples in D; F, population latency to first spike histograms after the onset of stimulation). G, H, Action potential properties in responsive, opto-tagged PT (O) and non-responsive, UI (X) layer V cortical neurons in WT (black) and Q175 (green) mice. Spike waveforms were measured from the average extracellular waveform of each unit and classified by their peak-to-valley width [1] and valley width at its half-maximum [2]. Broad and narrow waveforms were used to discriminate pyramidal neurons from interneurons, respectively.
Table 2.
Layer V motor cortical projection neuron activity during cortical SWA and ACT in Q175 and WT micea
| Figure/text | Measurement | Brain state | Genotype | n (mice/neurons) | Median [IQ range] | p (comparison; test) |
|---|---|---|---|---|---|---|
| text | Optogenetically responsive | — | WT | 5/15 | 57.7% | p = 0.62 (WT vs Q175; Fisher's Exact) |
| Q175 | 4/21 | 48.8% | ||||
| text | Latency of evoked response | — | WT | 5/15 | 2.5 [2.5-5.0] ms | p = 0.97 (WT vs Q175; MWU) |
| Q175 | 4/21 | 2.5 [2.5-3.7] ms | ||||
| 4 C | PT frequency | SWA | WT | 5/15 | 1.6 [0.6-2.4] Hz | p = 0.48 (SWA: WT vs Q175; MWU) |
| Q175 | 4/21 | 0.8 [0.4-1.5] Hz | ||||
| 4 C | UI frequency | SWA | WT | 4/7 | 1.8 [1.2-3.4] Hz | p = 0.64 (SWA: WT vs Q175; MWU) |
| Q175 | 4/15 | 1.2 [0.4-3.0] Hz | ||||
| 4 C | PT frequency | SWA | WT | 5/15 | 1.6 [0.6-2.4] Hz | p = 0.42 (SWA: PT vs IT; MWU) |
| UI frequency | 4/7 | 1.8 [1.2-3.4] Hz | ||||
| 4 C | PT frequency | SWA | Q175 | 4/21 | 0.8 [0.4-1.5] Hz | p = 0.87 (SWA: PT vs IT; MWU) |
| UI frequency | 4/15 | 1.2 [0.4-3.0] Hz | ||||
| text | PT CV | SWA | WT | 5/13 | 1.4 [0.99-2.0] | p = 0.80 (SWA: WT vs Q175; MWU) |
| Q175 | 4/13 | 1.3 [0.85-2.0] | ||||
| text | UI CV | SWA | WT | 4/7 | 1.1 [1.0-1.7] | p = 0.92 (SWA: WT vs Q175; MWU) |
| Q175 | 4/10 | 1.7 [1.2-2.3] | ||||
| text | PT CV | SWA | WT | 5/13 | 1.4 [0.99-2.0] | p = 1.3 (SWA: PT vs IT; MWU) |
| UI CV | 4/7 | 1.1 [1.0-1.7] | ||||
| text | PT CV | SWA | Q175 | 4/13 | 1.3 [0.85-2.0] | p = 1.0 (SWA: PT vs IT; MWU) |
| UI CV | 4/10 | 1.7 [1.2-2.3] | ||||
| 4 D | PT frequency | SWA | WT | 5/15 | 1.6 [0.6-2.4] Hz | p = 0.25 (SWA: WT vs Q175; MWU) |
| Q175 | 4/20 | 0.8 [0.4-1.5] Hz | ||||
| 4 D | PT frequency | ACT | WT | 5/15 | 0.0 [0.0-1.0] Hz | p = 0.01 (WT: SWA vs ACT; WSR) |
| Q175 | 4/20 | 0.0 [0.0-0.15] Hz | p = 4.0e-04 (Q175: SWA vs ACT; WSR) | |||
| p = 0.28 (ACT: WT vs Q175; MWU) | ||||||
| 4 E | UI frequency | SWA | WT | 4/7 | 1.8 [1.2-3.4] Hz | p = 0.64 (SWA: WT vs Q175; MWU) |
| Q175 | 4/15 | 1.2 [0.4-3.0] Hz | ||||
| 4 E | UI frequency | ACT | WT | 4/7 | 0.0 [0.0-1.2] Hz | p = 0.13 (WT: SWA vs ACT; WSR) |
| Q175 | 4/15 | 0.4 [0.0-4.4] Hz | p = 0.38 (Q175: SWA vs ACT; WSR) | |||
| p = 0.78 (ACT: WT vs Q175; MWU) | ||||||
| text | PT frequency | ACT | WT | 5/15 | 0.0 [0.0-1.0] Hz | p = 0.65 (ACT: PT vs UI; MWU) |
| UI frequency | 4/7 | 0.0 [0.0-1.2] Hz | ||||
| text | PT frequency | ACT | Q175 | 4/20 | 0.0 [0.0-0.15] Hz | p = 0.18 (ACT: PT vs UI; MWU) |
| UI frequency | 4/15 | 0.4 [0.0-4.4] Hz |
aThe frequency and pattern of layer V motor cortical projection neuron activity during cortical SWA and ACT were similar in Q175 and WT mice.
Consistent with previous studies, PT and unidentified layer V pyramidal neurons fired preferentially during the active component of cortical SWA (Steriade et al., 1993; Amzica and Steriade, 1998; Beltramo et al., 2013) (Fig. 4A). During cortical SWA, the frequency and regularity of PT and unidentified layer V pyramidal neuron activity were similar in Q175 and WT mice (Fig. 4A,C–E; Table 2). In addition, pinch-evoked cortical desynchronization led to a similar reduction in PT and unidentified layer V pyramidal neuron activities in both Q175 and WT mice (Fig. 4B,D,E; Table 2). Together, the EEG and single-unit data suggest that 6-month-old Q175 and WT mice exhibit similar patterns and levels of motor cortical activity under urethane anesthesia.
Figure 4.
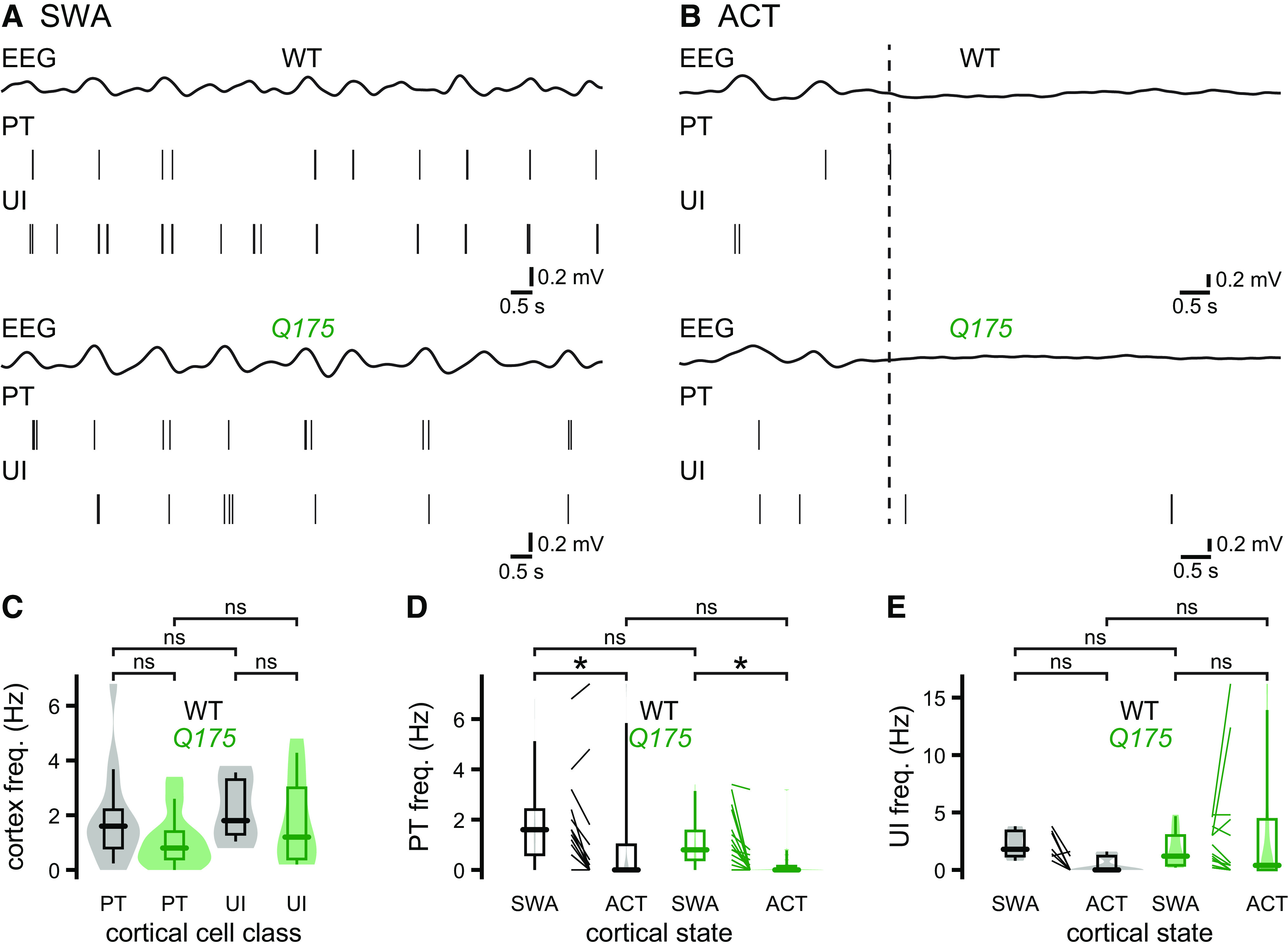
Layer V motor cortical projection neuron activity in Q175 and WT mice. A–E, The activities of layer V PT and unidentified pyramidal neurons in Q175 and age-matched WT mice were similar during cortical SWA and ACT. A, B, Representative examples of concurrent EEG (bandpass-filtered at 0.1-1.5 Hz) and cortical neuron activity during SWA (A) and ACT (B). The frequencies of firing of PT and unidentified pyramidal neurons were similar in both genotypes during cortical SWA (A, examples; C–E, population data). B, D, E, Hind paw pinch-evoked cortical ACT (dotted line) similarly affected PT and unidentified layer V pyramidal neuron activity in Q175 and WT mice (B, examples; D, E, population data). *p < 0.05.
D2-SPNs are hypoactive in Q175 mice during cortical SWA
The striatum is largely composed of similar numbers of direct and indirect pathway SPNs that express D1 or D2 dopamine receptors, respectively (Albin et al., 1989; Gerfen et al., 1990). As their names suggest, direct pathway D1-SPNs directly innervate basal ganglia output neurons, whereas indirect pathway D2-SPNs regulate basal ganglia output indirectly via the GPe and STN (Mink and Thach, 1993; Maurice et al., 1999; Tachibana et al., 2008). Appropriate, cortical patterning of D1- and D2-SPN activity is critical for the regulation of psychomotor function by the basal ganglia (Cui et al., 2013; Keeler et al., 2014; Nelson and Kreitzer, 2014; Tecuapetla et al., 2014; Sippy et al., 2015; Barbera et al., 2016; Lambot et al., 2016; Lemos et al., 2016; Tecuapetla et al., 2016; Klaus et al., 2019; LeBlanc et al., 2020). Although D2-SPNs are a key locus of dysregulation and degeneration in HD and its models, precisely how their activity is perturbed in vivo is unknown. To address this question, ChR2(H134R)-eYFP was virally expressed in D2-SPNs through injection of an AAV carrying a cre-dependent expression construct into the striatum of Q175 and WT mice that had been crossed with a D2-SPN selective cre-driver line (A2A-cre). Two to 3 weeks later, the activities of striatal neurons in Q175 and WT mice were compared using silicon optrodes. Consistent with the successful targeting of D2-SPNs in both Q175 and WT mice, ChR2(H134R)-eYFP was expressed in the subset of striatal neurons projecting to the GPe but not to the substantia nigra pars reticulata (Fig. 5A–D). Therefore, optogenetic activation was used to positively identify D2-SPNs (Fig. 5E–G). Construction of peri-optogenetic stimulus time histograms revealed that the latency of firing evoked by optogenetic stimulation was unimodal in distribution (Fig. 5F,G). Consistent with the direct optogenetic stimulation of ChR2 in D2-SPNs, the majority of neurons discharged within 10 ms of the start of the 5 ms optogenetic stimulation pulse (latency; WT: 5.0, 2.5-5.0 ms, n = 54; Q175: 5.0, 2.5-5.0 ms, n = 42) (Fig. 5F,G) and exhibited SPN-like rather than striatal interneuron-like action potential properties (Berke et al., 2004; Gage et al., 2010; Cayzac et al., 2011; Kim et al., 2014; Shin et al., 2018) (Fig. 5H).
Figure 5.

Optogenetic identification of D2-SPNs. A–C, Viral-mediated, cre-dependent expression of hChR2(H134R)-eYFP (green) in D2-receptor expressing striatopallidal neurons in WT/Q175 X A2A-cre mice in the striatum (A, B, lv, lateral ventricle; DV, dorsoventral axis; ML, mediolateral axis) and GPe (C, ic, internal capsule; same orientation as in A). B, hChR2(H134R)-eYFP expression was present (white arrows) or absent (magenta arrows) in striatal neurons that were coimmunoreactive for the pan neuronal marker NeuN (red). C, Axon-terminal expression of hChR2(H134R)-eYFP in the GPe but not the substantia nigra pars reticulata (SNr), consistent with selective expression in D2- but not D1-SPNs (ic, internal capsule). D, Schematic of the experimental setup, illustrating striatal placement of the optrode. E, Example of optogenetic activation of a D2-SPN but not a neighboring unidentified (UI) striatal neuron (blue, optogenetic stimulation; inset, spontaneous [black] and optogenetically evoked [blue] action potentials). F, G, Striatal neurons were considered to be directly responsive if their activity exceeded the prestimulus mean (solid red line) by 2 SDs (dotted red line) within 10 ms (bin size, 2.5 ms) of the start of the optogenetic stimulation pulse (F, PSTHs from examples in E; G, population latency to first spike histograms after the onset of stimulation). H, Action potential properties of responsive, opto-tagged D2-SPNs and non-responsive, UI striatal neurons in WT (black) and Q175 (green) mice. Broad and narrow waveforms were used to discriminate SPNs from interneurons, respectively.
The proportion of striatal neurons that exhibited short-latency excitatory responses to optogenetic stimulation was similar in Q175 and WT mice (Table 3). Neurons that were recorded on the same tetrode as identified D2-SPNs but were unresponsive to optogenetic stimulation (WT: n = 48; Q175: n = 23) were also recorded. The majority of neurons that were unresponsive to optogenetic stimulation exhibited action potential properties that are typical of SPNs (WT: n = 45; Q175: n = 19) and likely comprise D1-SPNs and a small fraction of D2-SPNs that failed to express ChR2(H134R)-eYFP (Fig. 5H). Thus, these neurons were termed unidentified, putative D1-SPNs. A minority of unresponsive neurons exhibited interneuron-like action potential properties (Fig. 5H) and were excluded from further description or analysis here because of their rarity. Optogenetic responses were not observed when optrodes were activated in regions or mice where ChR2(H134R)-eYFP expression was absent (data not shown).
Table 3.
D2-SPN and unidentified, putative D1-SPN activity during cortical SWA and ACT in Q175 and WT micea
| Figure/text | Measurement | Brain state | Genotype | n (mice/neurons) | Median [IQ range] | p (comparison; test) |
|---|---|---|---|---|---|---|
| text | Optogenetically responsive | — | WT | 3/54 | 52.9% | p = 0.15 (WT vs Q175; Fisher's Exact) |
| Q175 | 3/42 | 64.6% | ||||
| text | Latency of evoked response | — | WT | 3/54 | 5.0 [2.5-5.0] ms | p = 0.49 (WT vs Q175; MWU) |
| Q175 | 3/42 | 5.0 [2.5-5.0] ms | ||||
| 6 B | D2-SPN frequency | SWA | WT | 3/54 | 1.0 [0.40-1.9] Hz | p = 0.039 (SWA: WT vs Q175; MWU) |
| Q175 | 3/42 | 0.40 [0.20-0.85] Hz | ||||
| 6 B | UI frequency | SWA | WT | 3/45 | 0.80 [0.20-1.2] Hz | p = 0.67 (SWA: WT vs Q175; MWU) |
| Q175 | 3/19 | 0.40 [0.20-1.6] Hz | ||||
| 6 B | D2-SPN frequency | SWA | WT | 3/54 | 1.0 [0.40-1.9] Hz | p = 0.43 (SWA: D2 vs UI; MWU) |
| UI frequency | 3/45 | 0.80 [0.20-1.2] Hz | ||||
| 6 B | D2-SPN frequency | SWA | Q175 | 3/42 | 0.40 [0.20-0.85] Hz | p = 0.99 (SWA: D2 vs UI; MWU) |
| UI frequency | 3/19 | 0.40 [0.20-1.6] Hz | ||||
| 6 C | D2-SPN CV | SWA | WT | 3/39 | 1.4 [0.91-1.7] | p = 1.9 (SWA: WT vs Q175; MWU) |
| Q175 | 3/18 | 1.4 [0.84-1.8] | ||||
| 6 C | UI CV | SWA | WT | 3/27 | 1.3 [1.0-1.7] | p = 2.6 (SWA: WT vs Q175; MWU) |
| Q175 | 3/8 | 1.3 [1.0-1.8] | ||||
| 6 C | D2-SPN CV | SWA | WT | 3/39 | 1.4 [0.91-1.7] | p = 2.6 (SWA: D2 vs UI; MWU) |
| UI CV | 3/27 | 1.3 [1.0-1.7] | ||||
| 6 C | D2-SPN CV | SWA | Q175 | 3/18 | 1.4 [0.84-1.8] | p = 0.9783 (SWA: D2 vs UI; MWU) |
| UI CV | 3/8 | 1.3 [1.0-1.8] | ||||
| 6 D-F | D2-SPN anti-:in-phase ratio | SWA | WT | 3/43 | 0.0 [0.0-0.25] | p = 0.98 (SWA: WT vs Q175; MWU) |
| Q175 | 3/25 | 0.0 [0.0-0.02] | ||||
| 6 D-F | UI anti-:in-phase ratio | SWA | WT | 3/30 | 0.0 [0.0-0.33] | p = 1.3 (SWA: WT vs Q175; MWU) |
| Q175 | 3/13 | 0.0 [0.0-0.11] | ||||
| 6 D-F | D2-SPN anti-:in-phase ratio | SWA | WT | 3/43 | 0.0 [0.0-0.25] | p = 0.81 (SWA: D2 vs UI; MWU) |
| UI anti-:in-phase ratio | 3/30 | 0.0 [0.0-0.33] | ||||
| 6 D-F | D2-SPN anti-:in-phase ratio | SWA | Q175 | 3/25 | 0.0 [0.0-0.02] | p = 1.4 (SWA: D2 vs UI; MWU) |
| UI anti-:in-phase ratio | 3/13 | 0.0 [0.0-0.11] | ||||
| 6 H | D2-SPN frequency | SWA | WT | 3/52 | 1.0 [0.45-2.1] Hz | p = 0.013 (SWA: WT vs Q175; MWU) |
| Q175 | 3/42 | 0.40 [0.20-0.85] Hz | ||||
| 6 H | D2-SPN frequency | ACT | WT | 3/52 | 0.0 [0.0-0.0] Hz | p = 3.9e-07 (WT: SWA vs ACT; WSR) |
| Q175 | 3/42 | 0.0 [0.0-0.20] Hz | p = 0.001 (Q175: SWA vs ACT; WSR) | |||
| p = 0.028 (ACT: WT vs Q175; MWU) | ||||||
| 6 I | UI frequency | SWA | WT | 3/43 | 0.80 [0.20-1.2] Hz | p = 0.61 (SWA: WT vs Q175; MWU) |
| Q175 | 3/19 | 0.40 [0.20-1.6] Hz | ||||
| 6 I | UI frequency | ACT | WT | 3/43 | 0.0 [0.0-0.0] Hz | p = 1.2e-04 (WT: SWA vs ACT; WSR) |
| Q175 | 3/19 | 0.0 [0.0-1.2] Hz | p = 0.10 (Q175: SWA vs ACT; WSR) | |||
| p = 4.0e-08 (ACT: WT vs Q175; MWU) | ||||||
| text | D2-SPN frequency | ACT | WT | 3/52 | 0.0 [0.0-0.0] Hz | p = 0.18 (ACT: D2 vs UI; MWU) |
| UI frequency | 3/43 | 0.0 [0.0-0.0] Hz | ||||
| text | D2-SPN frequency | ACT | Q175 | 3/42 | 0.0 [0.0-0.20] Hz | p = 0.33 (ACT: D2 vs UI; MWU) |
| UI frequency | 3/19 | 0.0 [0.0-1.2] Hz |
aDuring cortical SWA, D2-SPNs were hypoactive in Q175 mice. During cortical ACT, SPNs were hyperactive in Q175 mice.
Consistent with direct cortical driving, D2-SPN and unidentified putative D1-SPN activity were phase-locked to the active component of cortical SWA in both Q175 and WT mice (Fig. 6A). The frequency of D2-SPN firing in Q175 mice was approximately one-half to two-thirds lower than that in WT control mice during cortical SWA, consistent with reductions in axospinous synapse density, mEPSC amplitude, and dendritic excitability in D2-SPNs (Fig. 6B,C; Table 3). In contrast, the frequency of unidentified, putative D1-SPN activity was similar in Q175 and WT mice during cortical SWA (Fig. 6B,C; Table 3). The phase-locking of D2-SPN and unidentified striatal neuron activity to cortical SWA was similar in WT and Q175 mice (Fig. 6D–F; Table 3).
Figure 6.
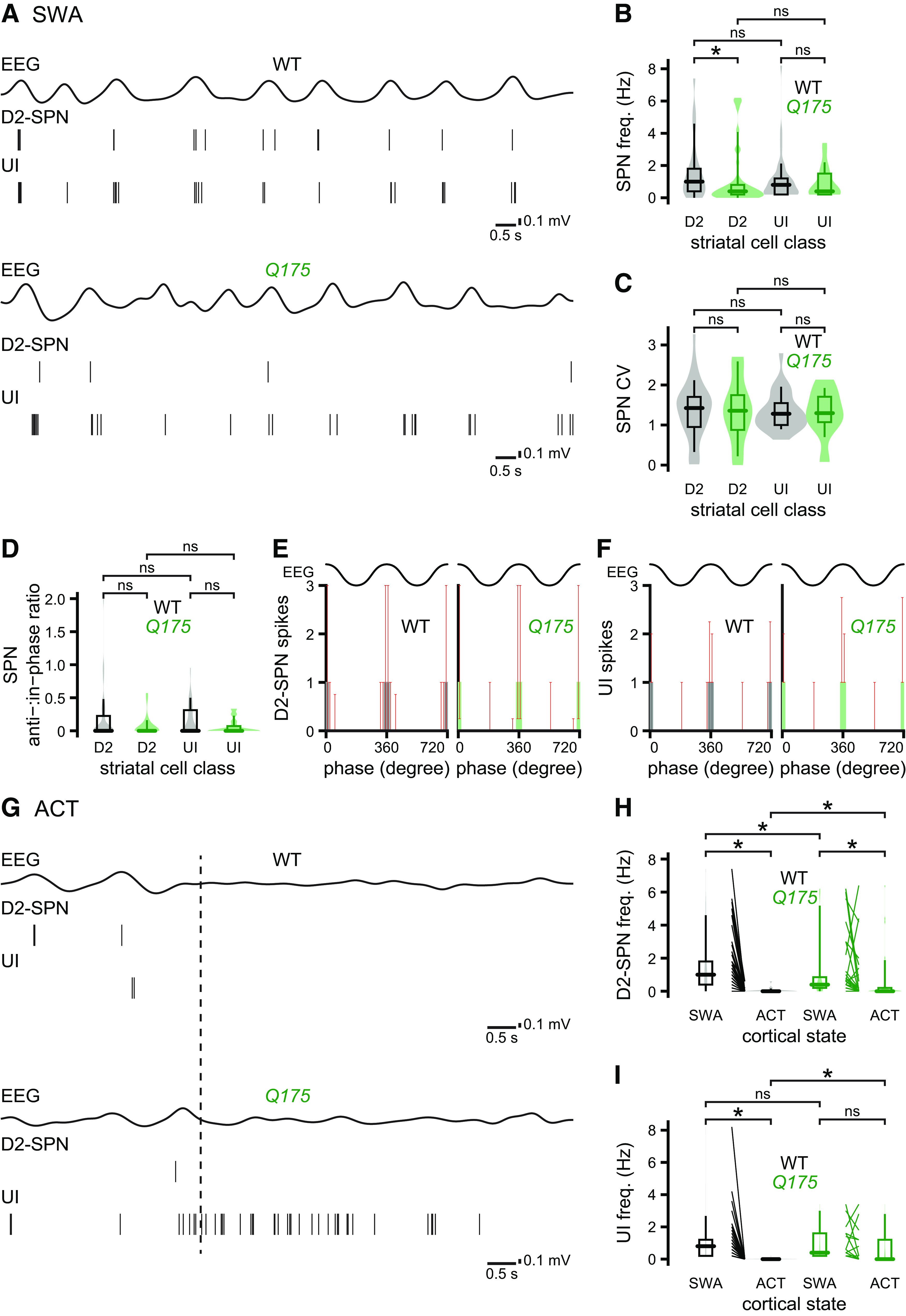
D2-SPNs are hypoactive in Q175 mice during cortical SWA. A–C, The frequency of D2-SPN firing was lower in Q175 mice relative to age-matched WT mice during cortical SWA (A, examples; B, C, population data). The frequency of unidentified (UI), putative D1-SPN activity during SWA was similar in Q175 and age-matched WT mice (A, examples; B, C, population data). D–F, D2-SPN activity and UI, putative D1-SPN activity were similarly phase-locked to the active component of cortical SWA in Q175 and WT mice (D, population data; E, F, population spike phase histograms). G–I, Effect of pinch-evoked cortical ACT (dotted line) on both D2-SPNs and UI, putative D1-SPNs in Q175 and WT mice (G, examples; H, I, population data; an outlier data point from the WT D2-SPN group was not plotted for legibility). During cortical ACT, the frequencies of D2-SPN and UI, putative D1-SPN activity were higher in Q175 mice (H, I). *p < 0.05.
Pinch-evoked cortical ACT decreased D2-SPN firing in both genotypes (Fig. 6G,H; Table 3). However, the frequency of D2-SPN discharge was modestly but significantly elevated in Q175 relative to WT mice during cortical ACT (Fig. 6G,H; Table 3). During pinch-evoked cortical ACT, the frequency of unidentified, putative D1-SPN activity declined in WT but not in Q175 mice. As a result, the activity of unidentified, putative D1-SPN neurons was elevated in Q175 mice (Fig. 6G,I; Table 3). Together, these data demonstrate that (1) during cortical SWA, D2-SPNs are hypoactive in Q175 mice; and (2) during cortical ACT, both D2-SPNs and unidentified putative D1-SPNs are more active in Q175 mice.
Prototypic PV+ GPe neurons are hyperactive in Q175 mice
To determine whether there are changes in the activity of GPe neurons downstream of D2-SPNs, we compared the in vivo firing of GPe neurons in Q175 and age-matched WT control mice. The GABAergic projection neurons of the GPe have been divided into two major types, so-called prototypic and arkypallidal neurons (Mallet et al., 2012; Mastro et al., 2014; Abdi et al., 2015; Dodson et al., 2015; Hernandez et al., 2015). Prototypic neurons comprise approximately three-fourths of all GPe neurons, are preferentially innervated by D2-SPNs, and innervate arkypallidal and downstream basal ganglia neurons (Mastro et al., 2014; Abdi et al., 2015; Dodson et al., 2015; Hernandez et al., 2015; Aristieta et al., 2020; Ketzef and Silberberg, 2021). A subset of prototypic neurons also innervates the striatum and/or cortex (Bevan et al., 1998; Mastro et al., 2014; Saunders et al., 2016; Abecassis et al., 2020). The majority of prototypic GPe neurons express the calcium-binding protein PV (Mastro et al., 2014; Abdi et al., 2015; Dodson et al., 2015; Hernandez et al., 2015). In contrast, arkypallidal neurons comprise approximately one-fourth of GPe neurons, are preferentially innervated by D1-SPNs rather than D2-SPNs, innervate the striatum rather than prototypic GPe neurons or the downstream basal ganglia, and express the transcription factor forkhead box protein 2 (FoxP2) but not PV (Mallet et al., 2012; Mastro et al., 2014; Abdi et al., 2015; Dodson et al., 2015; Hernandez et al., 2015; Aristieta et al., 2020; Ketzef and Silberberg, 2021). Although arkypallidal neurons are more weakly innervated by D2-SPNs compared to prototypic neurons, D2-SPNs can powerfully regulate arkypallidal activity indirectly via their effects on prototypic GPe neurons (Aristieta et al., 2020; Ketzef and Silberberg, 2021).
Because the majority of GPe neurons fire tonically, we used optogenetic silencing to identify and manipulate GPe neurons in vivo. The inhibitory opsin Arch-GFP was virally expressed in prototypic PV+ GPe neurons through injection of an AAV vector carrying a cre-dependent expression construct into the GPe of Q175 and WT mice that had been crossed with a PV-cre driver line (Fig. 7A–D). Consistent with the selective expression of Arch-GFP in prototypic GPe neurons, the majority of Arch-GFP-expressing GPe neurons were immunoreactive for PV, and all were immunonegative for FoxP2 (Fig. 7B). In addition, Arch-GFP was expressed in the axon terminal fields of PV+ GPe neurons in the downstream basal ganglia, including the STN, consistent with their prototypic identity (Fig. 7C). Optogenetic inhibition was therefore used to positively identify PV+ GPe neurons. Optogenetic activation of Arch-GFP rapidly and persistently inhibited a similar proportion of neurons in the GPe of Q175 and WT mice (Fig. 7E–H; Table 4). Optogenetic responses were not observed when optrodes were activated in regions or mice where Arch-GFP expression was absent (data not shown).
Figure 7.
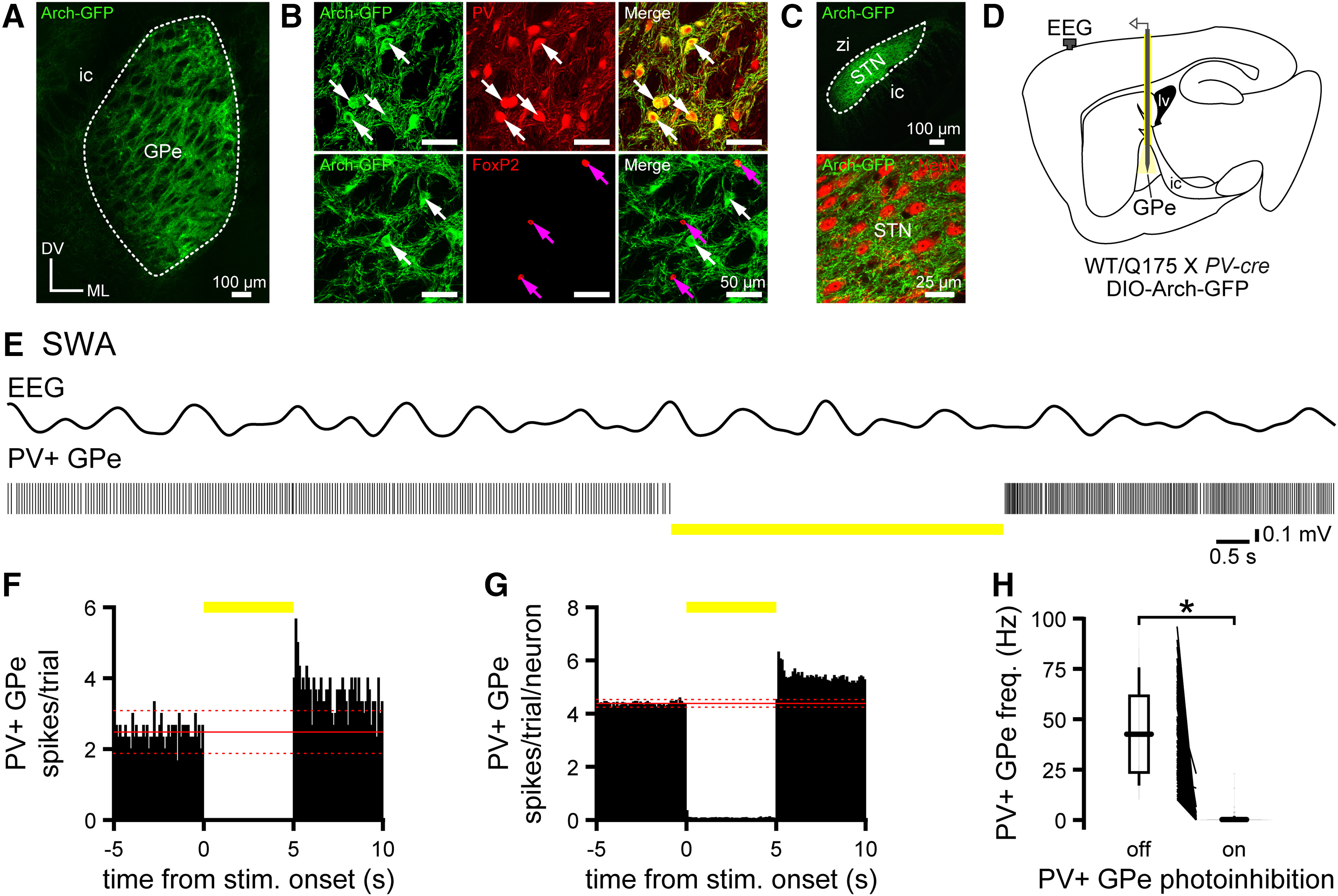
Optogenetic identification of prototypic PV+ GPe neurons. A–C, Viral cre-dependent expression of Arch-GFP (green) in prototypic PV+ GPe neurons in Q175/WT X PV-cre mice. A, Representative confocal micrograph of the GPe (ic, internal capsule; DV, dorsoventral axis; ML, mediolateral axis). B, Arch-GFP expression was present in PV-immunoreactive GPe neurons (white arrows; top) and FoxP2-immunonegative neurons (white arrows; bottom) and absent in FoxP2-immunoreactive arkypallidal GPe neurons (magenta arrows; bottom). C, Arch-GFP-labeled axon terminal fields in the STN (top; zi, zona incerta; same orientation as in A). Arch-GFP-labeled axon terminal fields in the vicinity of NeuN-immunoreactive STN neurons (red; bottom). D, Schematic of the experimental setup illustrating optrode placement in the GPe. E–H, Optogenetic activation of Arch-GFP rapidly and persistently inhibited PV+ GPe neuron activity. E, Representative example of concurrent EEG (bandpass-filtered at 0.1-1.5 Hz) and PV+ GPe neuron activity before, during, and after activation of Arch-GFP (yellow bar). F, G, PSTHs of PV+ GPe neuron activity in the absence and presence (yellow bar) of optogenetic inhibition (bin size, 100 ms; prestimulus mean, solid red line; ±2 SDs of prestimulus mean, dotted red line; F, PSTH from neuron in E; G, population PSTH). H, Direct responses of all PV+ GPe neurons to activation of Arch-GFP.
Table 4.
PV+ GPe neuron activity during cortical SWA and ACT in Q175 and WT micea
| Figure/text | Measurement | Brain state | Genotype | PV+ GPe inhibition | n (mice/neurons) | Median [IQ range] | p (comparison; test) |
|---|---|---|---|---|---|---|---|
| text | Optogenetically responsive | — | WT | Off | 4/40 | 80.0% | p = 1.0 (WT vs Q175; Fisher's Exact) |
| Q175 | Off | 5/43 | 78.2% | ||||
| text | PV+ GPe frequency | SWA | WT | Off | 4/40 | 29.3 [22.8-45.1] Hz | p = 8.8e-03 (SWA: WT Off vs Q175 Off; MWU) |
| Q175 | 5/43 | 55.8 [30.6-68.8] Hz | |||||
| text | PV+ GPe frequency | SWA | WT | On | 4/40 | 0.20 [0.0-1.1] Hz | p = 1.1e-07 (SWA: WT Off vs On; WSR) |
| Q175 | 5/43 | 0.20 [0.0-0.20] Hz | p = 9.1e-13 (SWA: Q175 Off vs On; WSR) | ||||
| p = 0.39 (SWA: WT On vs Q175 On; MWU) | |||||||
| 8 B | PV+ GPe frequency | SWA | WT | Off | 4/40 | 29.3 [22.8-45.1] Hz | p = 4.4e-03 (SWA: WT vs Q175; MWU) |
| Q175 | Off | 5/43 | 55.8 [30.6-68.8] Hz | ||||
| 8 C | PV+ GPe CV | SWA | WT | Off | 4/40 | 0.33 [0.24-0.48] Hz | p = 0.11 (SWA: WT vs Q175; MWU) |
| Q175 | Off | 5/43 | 0.48 [0.25-0.72] Hz | ||||
| 8D, E | PV+ GPe anti-:in-phase ratio | SWA | WT | Off | 4/40 | 0.94 [0.87-1.0] | p = 4.7e-07 (SWA: WT vs Q175; MWU) |
| Q175 | Off | 5/43 | 1.1 [1.0-1.3] | ||||
| 8 G | PV+ GPe frequency | SWA | WT | Off | 4/36 | 28.2 [21.7-43.5] Hz | p = 0.015 (SWA: WT vs Q175; MWU) |
| Q175 | Off | 5/26 | 56.4 [28.5-70.5] Hz | ||||
| 8 G | PV+ GPe frequency | ACT | WT | Off | 4/36 | 39.8 [30.9-59.4] Hz | p = 3.7e-04 (WT: SWA vs ACT; WSR) |
| Q175 | Off | 5/26 | 59.1 [35.8-76.3] Hz | p = 0.013 (Q175: SWA vs ACT; WSR) | |||
| p = 0.048 (ACT: WT vs Q175; MWU) |
aIn Q175 mice, PV+ GPe neurons were hyperactive during cortical SWA and ACT.
As previously reported (Abdi et al., 2015; Kovaleski et al., 2020), PV+ GPe neurons discharged tonically in WT mice during cortical SWA (Fig. 8A). Although PV+ GPe neurons also discharged tonically in Q175 mice during cortical SWA, their median activity was more than double that in WT mice (Fig. 8B; Table 4). The regularity of PV+ GPe neuron firing, as assessed from the CV of the interspike interval, was similar in Q175 and WT mice (Fig. 8C; Table 4). PV+ GPe neuron firing was elevated during the active component of cortical SWA in Q175 mice (Fig. 8D; Table 4), consistent with the hypoactivity of upstream D2-SPNs, as described above. However, PV+ GPe neurons were also hyperactive in Q175 mice during the inactive component of cortical SWA (Fig. 8D; Table 4). Indeed, the ratio of PV+ GPe neuron activity during the inactive versus active component of cortical SWA was significantly higher in Q175 mice (Fig. 8D,E; Table 4). Given that D2-SPNs discharge only during the active phase of cortical SWA in WT and Q175 mice (Fig. 6D), the relative hyperactivity of PV+ GPe neurons in Q175 mice during the inactive phase of cortical SWA cannot be due to D2-SPN hypoactivity. During pinch-evoked cortical ACT, the firing rate of PV+ GPe neurons increased in both genotypes (Fig. 8F,G; Table 4). However, PV+ GPe neurons remained strongly hyperactive in Q175 mice relative to WT controls (Fig. 8F,G; Table 4), despite the fact that D2-SPNs are hyperactive in Q175 mice during cortical ACT (Fig. 6G,H). Together, these data demonstrate that in Q175 mice prototypic PV+ GPe neuron activity is highly elevated during both cortical SWA and ACT.
Figure 8.
In Q175 mice, PV+ GPe neurons are hyperactive during cortical SWA and ACT. A, Representative examples of concurrent EEG (bandpass-filtered at 0.1-1.5 Hz) and PV+ GPe neuron activity in the absence and presence of optogenetic inhibition (yellow bar). A–C, During cortical SWA, the median frequency of PV+ GPe neuron firing in Q175 mice was more than twice that in WT mice, but the variability of firing was unaltered (A, representative examples; B,C, population data). D, E, In Q175 mice, the firing of PV+ GPe neurons was relatively antiphasic to cortical SWA (D, population spike phase histograms; E, population box and violin plots). F, G, Pinch-evoked cortical ACT (hind paw pinch, dotted line) increased PV+ GPe neuron firing in both genotypes. However, the frequency of PV+ GPe neuron discharge remained elevated in Q175 mice relative to WT mice (F, representative examples; G, population data). *p < 0.05.
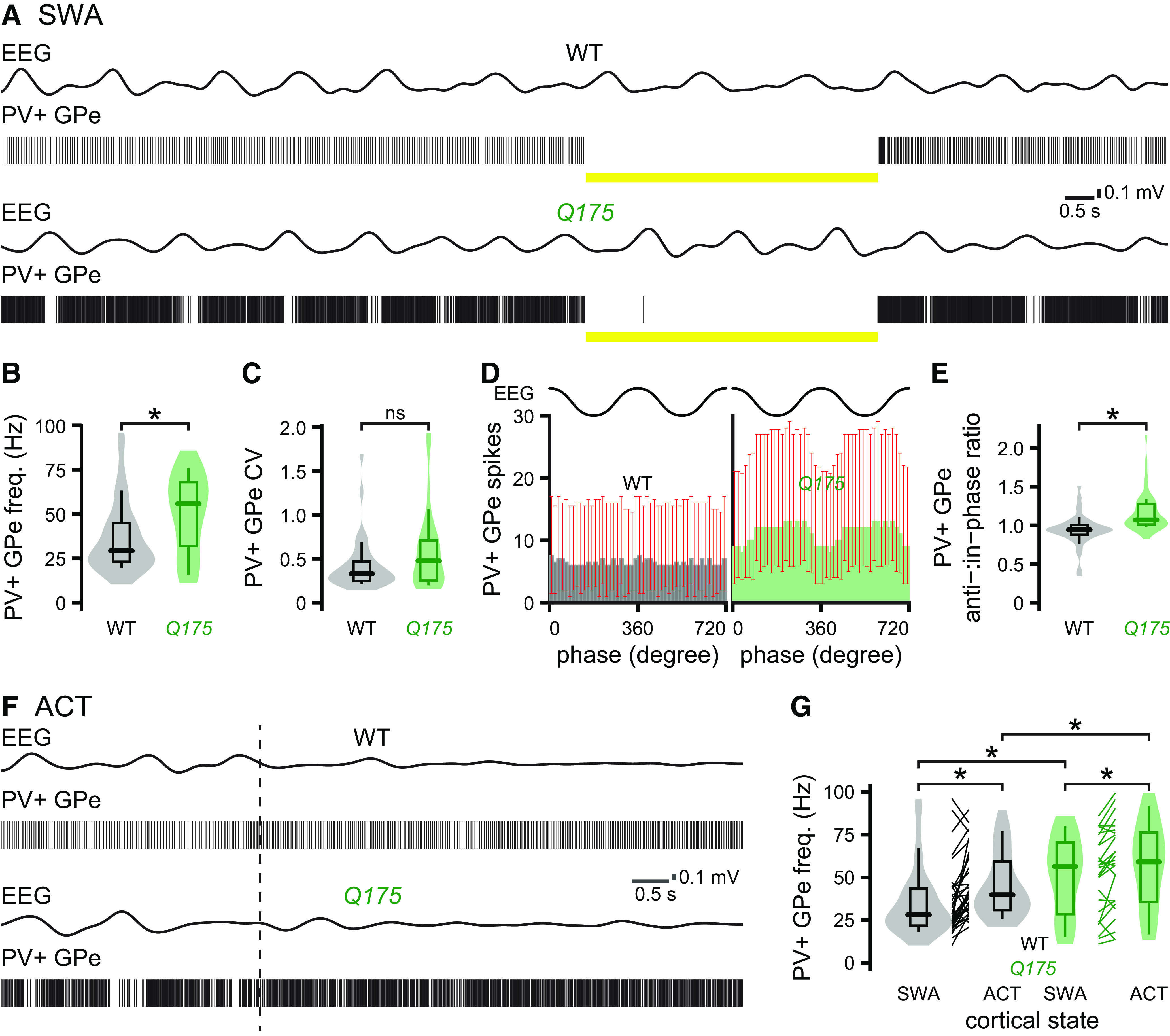
The abnormal hypoactivity of PV– GPe neurons in Q175 mice is reversed by optogenetic inhibition of PV+ GPe neurons
We next analyzed the activity of GPe neurons that were not directly inhibited during optogenetic activation of Arch-GFP in PV-cre mice (Fig. 9; Table 5). These most likely comprise PV– prototypic GPe and arkypallidal neurons but could also include a small fraction of PV+ GPe neurons that failed to express Arch-GFP. During cortical SWA, unidentified, putative PV– GPe neurons were less active than PV+ GPe neurons in both WT and Q175 mice, consistent with previous reports in WT rodents (Fig. 9A,D; Table 5) (Abdi et al., 2015; Dodson et al., 2015; Mallet et al., 2016). Putative PV– GPe neurons also discharged more slowly in Q175 than WT mice (Fig. 9A,D,F; Table 5). In contrast, the precision of putative PV– GPe neuron activity was similar in WT and Q175 mice (Fig. 9E; Table 5). Recent studies suggest that prototypic PV+ and PV– GPe neurons inhibit each other and arkypallidal neurons through powerful local connections (Aristieta et al., 2020; Ketzef and Silberberg, 2021). In contrast, arkypallidal neuron innervation of prototypic GPe neurons is minimal (Aristieta et al., 2020; Ketzef and Silberberg, 2021). If elevated inhibition emanating from hyperactive prototypic PV+ GPe neurons is responsible for the hypoactivity of putative PV– GPe neurons in Q175 mice, then their activity should be rescued by optogenetic inhibition of PV+ GPe neurons. Consistent with this hypothesis, optogenetic silencing of PV+ GPe neurons during cortical SWA rapidly and persistently disinhibited putative PV– GPe neuron activity in both WT and Q175 mice, confirming that PV+ GPe neurons actively inhibit PV– GPe neuron activity in vivo (Fig. 9A–C,F; Table 5). Furthermore, during optogenetic inhibition of PV+ GPe neurons, the activities of putative PV– GPe neurons in WT and Q175 mice were no longer significantly different (Fig. 9A–C,F; Table 5), arguing that increased inhibition from hyperactive PV+ GPe neurons was indeed responsible for their relative hypoactivity in Q175 mice. There were no consistent differences in the phase of putative PV– GPe neuron activity (relative to cortical SWA) with or without optogenetic inhibition of PV+ GPe neurons in WT and Q175 mice (Fig. 9G,H; Table 5).
Figure 9.
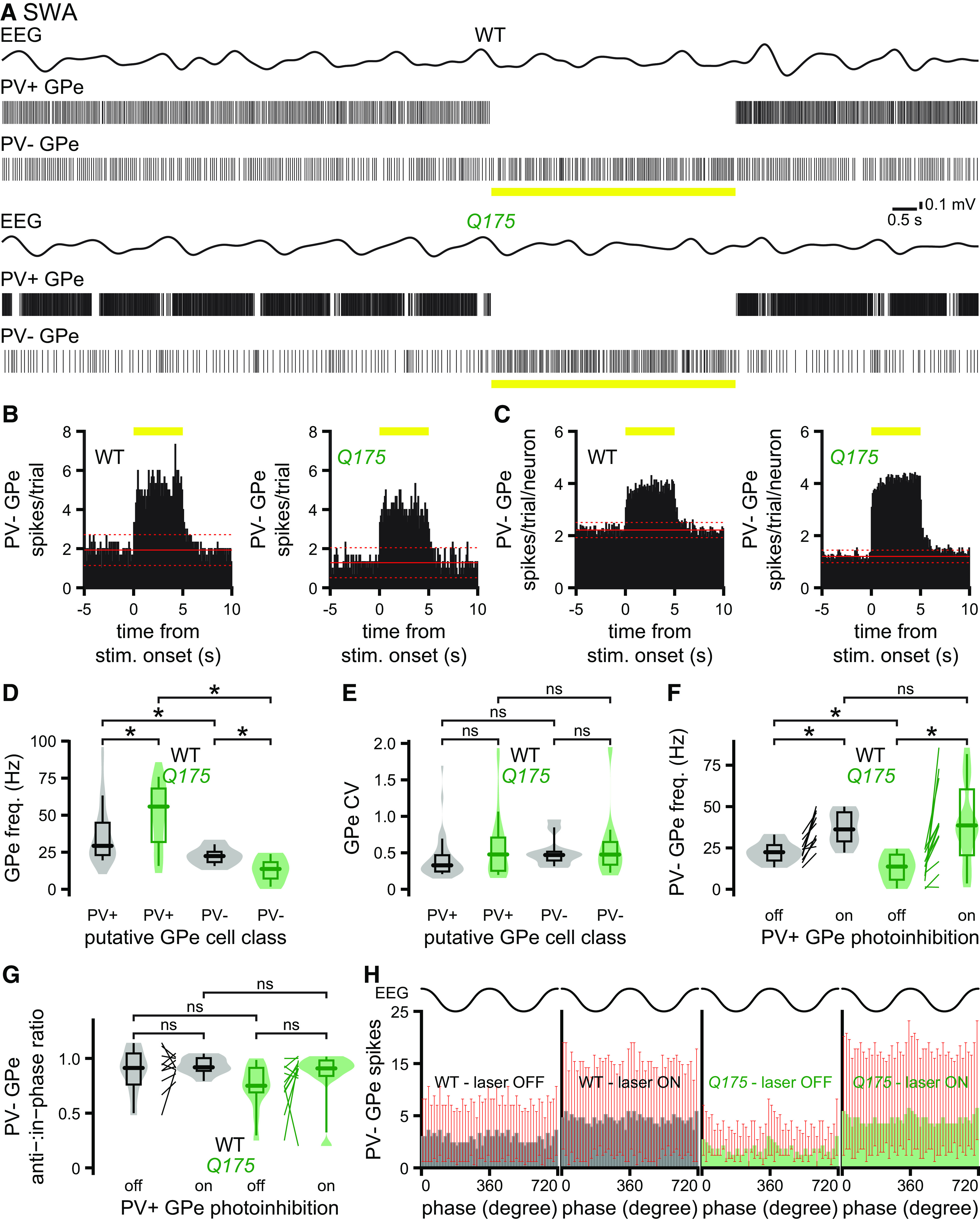
The hypoactivity of putative PV– GPe neurons in Q175 mice is alleviated by optogenetic inhibition of hyperactive PV+ GPe neurons. A, Representative examples of concurrent EEG (bandpass-filtered at 0.1-1.5 Hz) and GPe neuronal activity in Q175/WT X PV-cre mice during optogenetic inhibition (yellow bar) of PV+ GPe neurons. A–C, Optogenetic inhibition of PV+ GPe neuron activity rapidly and persistently disinhibited putative PV– GPe neurons in Q175 and WT mice. B, C, PSTHs of putative PV– GPe neuron activity in the absence and presence of PV+ GPe neuron optogenetic inhibition (yellow bar) (bin size, 100 ms; prestimulus mean, solid red line; ±2 SDs of prestimulus mean, dotted red line; B, PSTHs from representative neurons in A; C, population PSTHs). A–F, During cortical SWA, putative PV– GPe neurons were less active than PV+ GPe neurons in both WT and Q175 mice. Furthermore, the frequency of putative PV– GPe neuron activity in Q175 mice was lower than in WT mice (A, representative examples; D, population data). E, The precision of PV– GPe neuron activity was similar in WT and Q175 mice. Optogenetic inhibition of PV+ GPe neurons disinhibited putative PV– GPe neurons in WT and Q175 mice and eliminated the difference in firing frequencies between the two genotypes, arguing that GABAergic inhibition emanating from abnormally hyperactive PV+ GPe neurons is responsible for the relative hypoactivity of PV– GPe neurons in Q175 mice (A, examples; F, population data). G, H, Relative to cortical SWA, there were no differences in the phase of putative PV– GPe neuron activity in WT and Q175 mice, with or without optogenetic inhibition of PV+ GPe neurons. *p < 0.05.
Table 5.
Impact of optogenetic inhibition of PV+ GPe neurons on putative PV– GPe neuron activity in Q175 and WT micea
| Figure/text | Measurement | Brain state | Genotype | PV+ GPe inhibition | n (mice/neurons) | Median [IQ range] | p (comparison; test) |
|---|---|---|---|---|---|---|---|
| 9 D | PV+ GPe frequency | SWA | WT | Off | 4/40 | 29.3 [22.8-45.1] Hz | p = 0.013 (SWA: WT vs Q175; MWU) |
| Q175 | 5/43 | 55.8 [30.6-68.8] Hz | |||||
| 9 D | PV– GPe frequency | SWA | WT | Off | 4/10 | 22.4 [17.4-26.7] Hz | p = 0.029 (SWA: WT vs Q175; MWU) |
| Q175 | 4/12 | 13.7 [5.8-20.9] Hz | |||||
| 9 D | PV+ GPe frequency | SWA | WT | Off | 4/40 | 29.3 [22.8-45.1] Hz | p = 0.022 (SWA: PV+ vs PV–; MWU) |
| PV– GPe frequency | 4/10 | 22.4 [17.4-26.7] Hz | |||||
| 9 D | PV+ GPe frequency | SWA | Q175 | Off | 5/43 | 55.8 [30.6-68.8] Hz | p = 3.6e-05 (SWA: PV+ vs PV–; MWU) |
| PV– GPe frequency | 4/12 | 13.7 [5.8-20.9] Hz | |||||
| 9 E | PV+ GPe CV | SWA | WT | Off | 4/40 | 0.33 [0.24-0.48] | p = 0.33 (SWA: WT vs Q175; MWU) |
| Q175 | 5/43 | 0.48 [0.25-0.72] | |||||
| 9 E | PV– GPe CV | SWA | WT | Off | 4/10 | 0.47 [0.36-0.60] | p = 1.5 (SWA: WT vs Q175; MWU) |
| Q175 | 4/11 | 0.48 [0.28-0.67] | |||||
| 9 E | PV+ GPe CV | SWA | WT | Off | 4/40 | 0.33 [0.24-0.48] | p = 0.15 (SWA: PV+ vs PV–; MWU) |
| PV– GPe CV | 4/10 | 0.47 [0.36-0.60] | |||||
| 9 E | PV+ GPe CV | SWA | Q175 | Off | 5/43 | 0.48 [0.25-0.72] | p = 0.93 (SWA: PV+ vs PV–; MWU) |
| PV– GPe CV | 4/11 | 0.48 [0.28-0.67] | |||||
| 9 F | PV– GPe frequency | SWA | WT | Off | 4/10 | 22.4 [17.4-26.7] Hz | p = 0.029 (SWA: WT Off vs Q175 Off; MWU) |
| Q175 | 4/12 | 13.7 [5.8-20.9] Hz | |||||
| 9 F | PV– GPe frequency | SWA | WT | On | 4/10 | 36.2 [29.0-46.6] Hz | p = 6.0e-03 (SWA: WT Off vs On; WSR) |
| Q175 | 4/12 | 38.6 [20.6-60.4] Hz | p = 4.0e-03 (SWA: Q175 Off vs On; WSR) | ||||
| p = 0.97 (SWA: WT On vs Q175 On; MWU) | |||||||
| 9 G | PV– GPe anti-:in-phase ratio | SWA | WT | Off | 4/10 | 0.91 [0.76-1.0] | p = 0.35 (SWA: WT Off vs Q175 Off; MWU) |
| Q175 | 4/11 | 0.75 [0.69-0.91] | |||||
| 9 G | PV– GPe anti-:in-phase ratio | SWA | WT | On | 4/10 | 0.92 [0.89-1.0] | p = 0.86 (SWA: WT Off vs On; WSR) |
| Q175 | 4/11 | 0.91 [0.84-0.98] | p = 0.39 (SWA: Q175 Off vs On; WSR) | ||||
| p = 0.67 (SWA: WT On vs Q175 On; MWU) |
aIn Q175 mice, putative PV– GPe neurons were hypoactive; and this hypoactivity was fully alleviated by optogenetic inhibition of PV+ GPe neurons.
The autonomous activity of PV+ GPe neurons but not putative PV– GPe neurons is increased in brain slices from Q175 mice
The high rates of discharge of extrastriatal basal ganglia neurons in vivo are generated in part by their intrinsic autonomous activity (Wilson, 2013). Alterations in the autonomous firing of PV+ and PV– GPe neurons could therefore contribute to their abnormal in vivo activity in Q175 mice. To determine whether the autonomous firing of GPe neurons is altered in Q175 mice, an AAV vector carrying a cre-dependent eGFP-expression construct was injected into the GPe of WT or Q175 PV-cre mice. Two to 3 weeks later, brain slices were prepared and visually guided somatic patch-clamp recordings of GPe neurons were conducted. Expression of eGFP was used to identify prototypic PV+ GPe neurons. PV– GPe neurons in the vicinity of eGFP-expressing PV+ GPe neurons were identified putatively by their absence of eGFP expression. GPe neurons were recorded in the loose-seal, cell-attached, current-clamp configuration and recordings were made in the presence of AMPA, NMDA, GABAA, and GABAB receptor antagonists to minimize the impact of synaptic inputs on autonomous firing. As for previous studies (Abdi et al., 2015; Hernandez et al., 2015), we found that the frequency and precision of autonomous firing were greater and autonomously generated action potentials were briefer in prototypic PV+ GPe neurons than putative PV– GPe neurons in WT mice (Fig. 10A–F; Table 6). Thus, these data are consistent with the appropriate identification of PV+ and PV– GPe neurons rather than the failure of reporter expression in PV+ GPe neurons. In Q175 mice, the frequency and precision of autonomous PV+ GPe neuron activity were greater than in WT mice (Fig. 10A,E,F; Table 6). In contrast, there were no differences in the frequency or precision of autonomous putative PV– GPe neuron firing in Q175 and WT mice (Fig. 10B,E,F; Table 6). Together, these data argue that the autonomous firing of prototypic PV+ GPe neurons is upregulated in Q175 mice and that this alteration may contribute to the hyperactivity of these cells in vivo. In contrast, the similarity of autonomous putative PV– GPe neuron firing in Q175 and WT mice further supports the conclusion that excessive inhibition emanating from hyperactive PV+ GPe neurons is responsible for their hypoactivity in Q175 mice in vivo.
Figure 10.
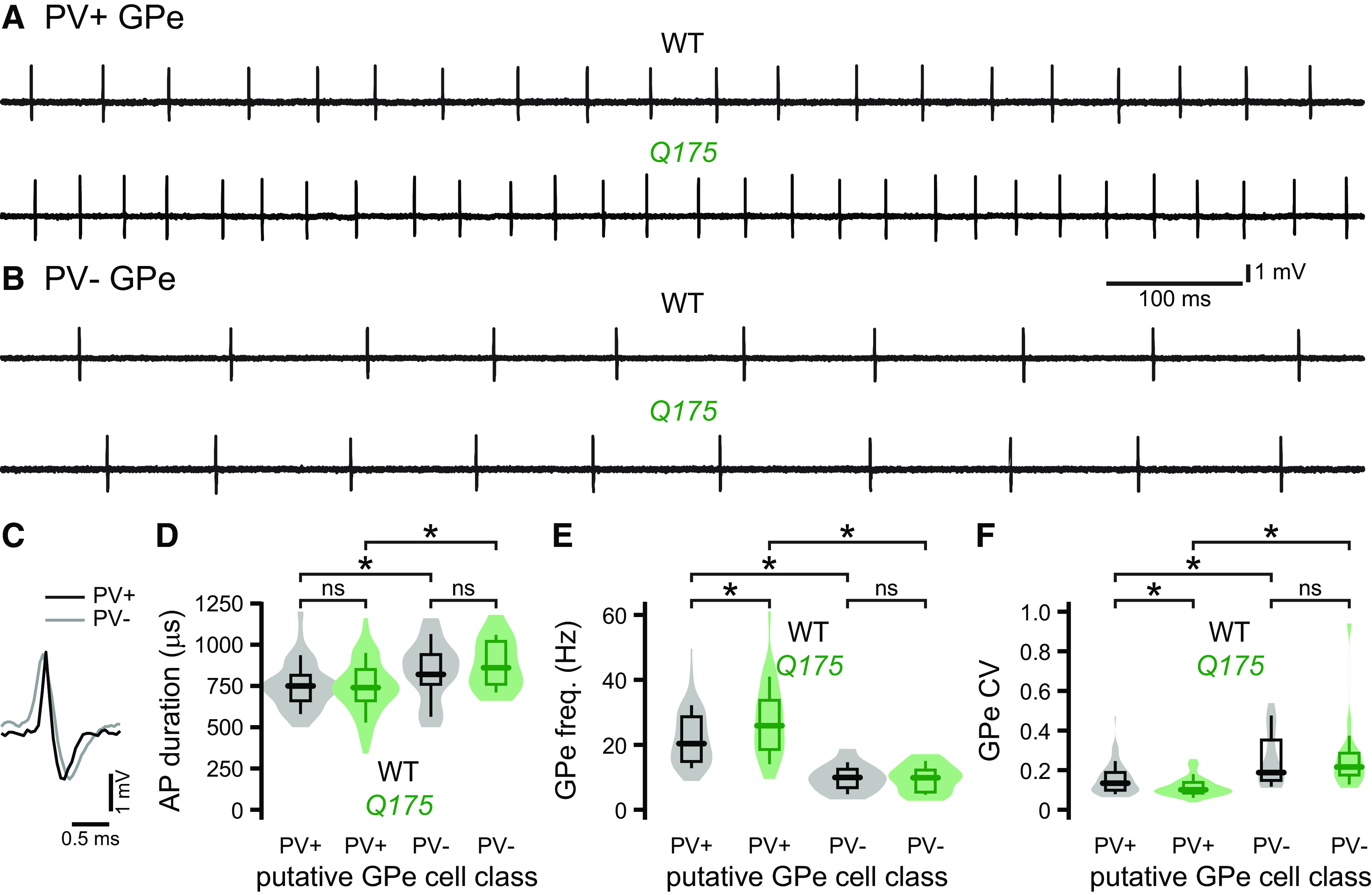
The autonomous activity of PV+ GPe neurons is elevated in Q175 mice. A, B, Representative examples of autonomous GPe neuron activity recorded in the loose-seal, cell-attached, current-clamp configuration in brain slices from Q175/WT X PV-cre mice. C, D, Average spike waveforms were significantly shorter in duration in PV+ GPe neurons than putative PV– GPe neurons (C, representative examples; D, population data). A, E, F, The frequency and precision of autonomous PV+ GPe neuron activity were greater in slices from Q175 mice (A, representative examples; E, F, population data). B, E, F, In contrast, the frequency and precision of autonomous PV– GPe neuron activity were similar in Q175 and WT mice (B, representative examples; E, F, population data). *p < 0.05.
Table 6.
The autonomous activity of GPe neurons in ex vivo brain slices from Q175 and WT micea
| Figure | Measurement | Genotype | n (mice/neurons) | Median [IQ range] | p (comparison; test) |
|---|---|---|---|---|---|
| 10 D | PV+ GPe AP duration | WT | 7/82 | 750 [657.5-825] µs | p = 0.90 (WT vs Q175; MWU) |
| Q175 | 8/73 | 740 [650-850] µs | |||
| 10 D | PV– GPe AP duration | WT | 3/27 | 820 [740-940] µs | p = 1.3 (WT vs Q175; MWU) |
| Q175 | 3/16 | 860 [760-1020] µs | |||
| 10 D | PV+ GPe AP duration | WT | 7/82 | 750 [657.5-825] µs | p = 0.02 (PV+ vs PV–; MWU) |
| PV– GPe AP duration | 3/27 | 820 [740-940] µs | |||
| 10 D | PV+ GPe AP duration | Q175 | 8/73 | 740 [650-850] µs | p = 0.02 (PV+ vs PV–; MWU) |
| PV– GPe AP duration | 3/16 | 860 [760-1020] µs | |||
| 10 E | PV+ GPe frequency | WT | 7/82 | 20.4 [14.8-28.8] Hz | p = 3.6e-03 (WT vs Q175; MWU) |
| Q175 | 8/73 | 25.9 [18.5-33.8] Hz | |||
| 10 E | PV– GPe frequency | WT | 3/27 | 9.9 [6.6-12.8] Hz | p = 0.99 (WT vs Q175; MWU) |
| Q175 | 3/16 | 9.9 [5.3-13.0] Hz | |||
| 10 E | PV+ GPe frequency | WT | 7/82 | 20.4 [14.8-28.8] Hz | p = 1.9e-10 (PV+ vs PV–; MWU) |
| PV– GPe frequency | 3/27 | 9.9 [6.6-12.8] Hz | |||
| 10 E | PV+ GPe frequency | Q175 | 8/73 | 25.9 [18.5-33.8] Hz | p = 3.0e-08 (PV+ vs PV–; MWU) |
| PV– GPe frequency | 3/16 | 9.9 [5.3-13.0] Hz | |||
| 10 F | PV+ GPe CV | WT | 7/82 | 0.13 [0.10-0.19] | p = 1.2e-03 (WT vs Q175; MWU) |
| Q175 | 8/73 | 0.10 [0.082-0.14] | |||
| 10 F | PV– GPe CV | WT | 3/27 | 0.19 [0.14-0.37] | p = 0.79 (WT vs Q175; MWU) |
| Q175 | 3/16 | 0.22 [0.17-0.29] | |||
| 10 F | PV+ GPe CV | WT | 7/82 | 0.13 [0.10-0.19] | p = 4.4e-04 (PV+ vs PV–; MWU) |
| PV– GPe CV | 3/27 | 0.19 [0.14-0.37] | |||
| 10 F | PV+ GPe CV | Q175 | 8/73 | 0.10 [0.082-0.14] | p = 2.1e-06 (PV+ vs PV–; MWU) |
| PV– GPe CV | 3/16 | 0.22 [0.17-0.29] |
aThe frequency and precision of autonomous firing were greater in PV+ GPe neurons than putative PV– GPe neurons in both genotypes. The frequency and precision of autonomous firing in PV+ GPe neurons were greater in Q175 mice than WT mice.
The hypoactivity of the STN in Q175 mice is partially alleviated by optogenetic inhibition of prototypic PV+ GPe neurons
The glutamatergic STN is a key component of the indirect pathway and forms a reciprocally connected network with the GPe (Mink and Thach, 1993; Maurice et al., 1999; Plenz and Kital, 1999; Nambu et al., 2002; Tachibana et al., 2008). Previous research has demonstrated that autonomous STN activity is downregulated in HD mice (Atherton et al., 2016). Furthermore, we demonstrate here that GABAergic prototypic PV+ GPe neurons, which potently inhibit STN activity through their activation of postsynaptic GABAA and GABAB receptors (Bevan et al., 2002; Hallworth and Bevan, 2005; Baufreton et al., 2009; Atherton et al., 2013; Kovaleski et al., 2020), are hyperactive in Q175 mice in vivo. Given the loss of intrinsic STN activity and increased frequency of GABAergic GPe-STN transmission, we predicted that STN neurons will be hypoactive in Q175 mice in vivo compared with WT mice. To test this prediction, we recorded the activity of STN neurons with silicon tetrodes during cortical SWA and ACT in Q175 and WT mice crossed with PV-cre mice. To isolate the effect of PV+ GPe neuron inhibition on the STN, we virally expressed Arch-GFP in PV+ GPe neurons and inhibited their activity optogenetically, as described above (Fig. 11A). As for previous studies in WT rodents, STN neurons fired in phase with cortical SWA in WT and Q175 mice (Magill et al., 2000, 2001; Mallet et al., 2008a; Callahan and Abercrombie, 2015a,b; Kovaleski et al., 2020) (Fig. 11B; Table 7). As predicted, the frequency of STN activity was significantly lower in Q175 mice (Fig. 11B,C; Table 7). The regularity of STN activity and the proportion of spikes generated during the inactive component of cortical SWA relative to those during the active component were similar in Q175 and WT mice (Fig. 11B–F; Table 7). Pinch-evoked cortical ACT elevated the frequency of STN activity in both WT and Q175 mice (Fig. 11G,H; Table 7). However, the frequency of STN activity remained lower in Q175 mice relative to that in WT mice during cortical ACT (Fig. 11G,H; Table 7).
Figure 11.
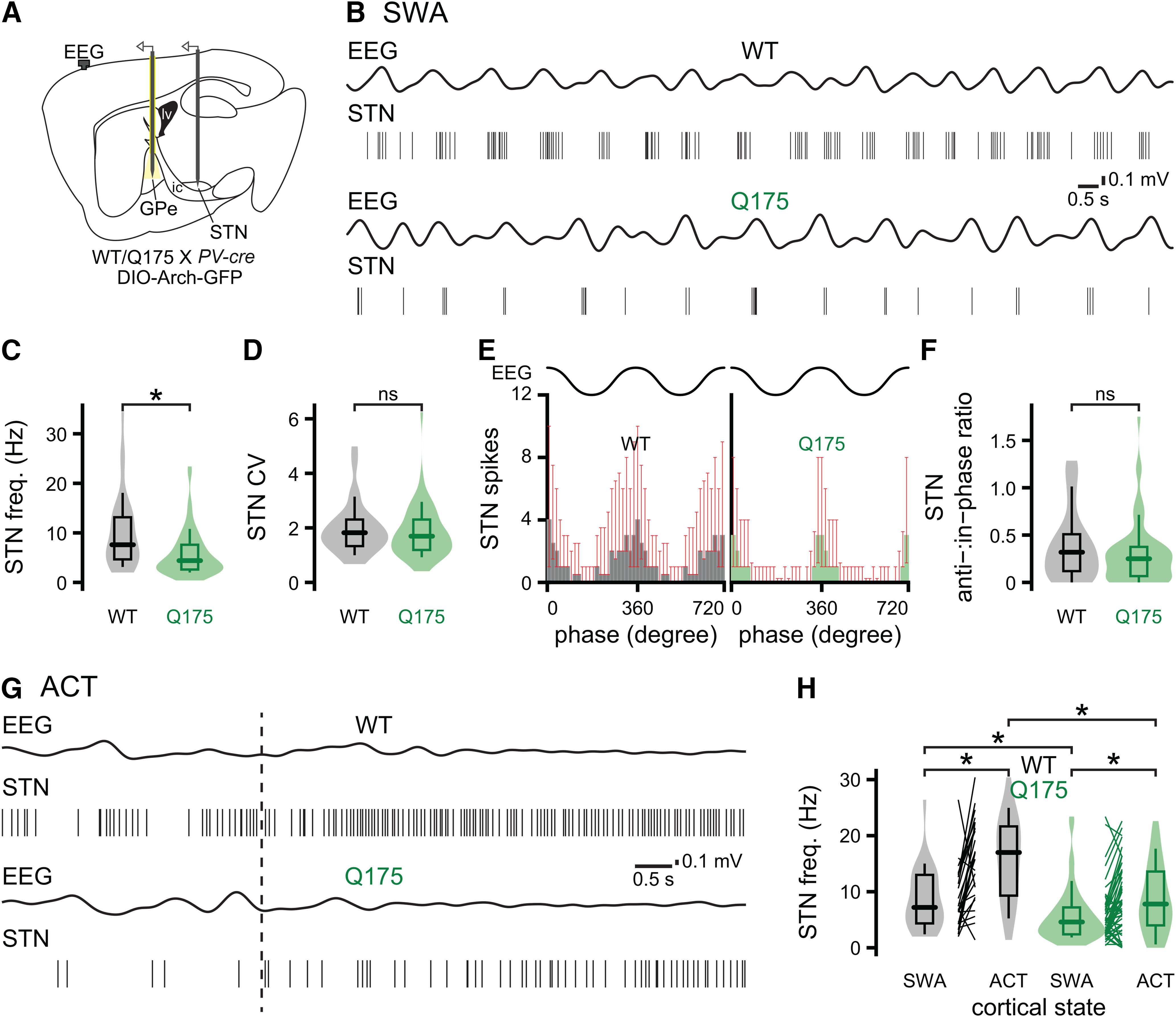
STN neurons are hypoactive in Q175 mice. A–D, During cortical SWA, the frequency (but not the regularity) of STN activity was lower in Q175 mice (A, schematic of experimental setup, illustrating placement of an optrode in the GPe and a tetrode array in the STN; B, representative examples of concurrent 0.1-1.5 Hz bandpass-filtered EEG and STN unit activity; C, D, population data). E, F, STN activity was similarly phase-locked to the active component of cortical SWA in Q175 and WT mice (E, population spike phase histograms; F, population data). G, H, Pinch-evoked cortical ACT (hind paw pinch, dotted line) increased STN neuron activity in both genotypes. However, during cortical ACT, STN neurons remained hypoactive in Q175 mice relative to WT (G, representative examples; H, population data). *p < 0.05.
Table 7.
STN neuron activity during cortical SWA and ACT in Q175 and WT micea
| Figure/text | Measurement | Brain state | Genotype | n (mice/neurons) | Median [IQ range] | p (comparison; test) |
|---|---|---|---|---|---|---|
| 11 C | STN frequency | SWA | WT | 4/30 | 7.6 [4.6-13.3] Hz | p = 7.0e-04 (SWA: WT vs Q175; MWU) |
| Q175 | 5/61 | 4.4 [2.5-7.6] Hz | ||||
| 11 D | STN CV | SWA | WT | 4/30 | 1.8 [1.3-2.3] | p = 0.41 (SWA: WT vs Q175; MWU) |
| Q175 | 5/60 | 1.7 [1.2-2.4] | ||||
| 11E, F | STN anti-:in-phase ratio | SWA | WT | 4/30 | 0.32 [0.11-0.53] | p = 0.25 (SWA: WT vs Q175; MWU) |
| Q175 | 5/61 | 0.25 [0.064-0.38] | ||||
| 11 H | STN frequency | SWA | WT | 4/26 | 7.2 [4.4-13.0] Hz | p = 5.8e-03 (SWA: WT vs Q175; MWU) |
| Q175 | 5/51 | 4.6 [2.4-7.2] Hz | ||||
| 11 H | STN frequency | ACT | WT | 4/26 | 17.0 [9.3-21.7] Hz | p = 4.0e-04 (WT: SWA vs ACT; WSR) |
| Q175 | 5/51 | 7.8 [4.0-13.6] Hz | p = 3.0e-04 (Q175: SWA vs ACT; WSR) | |||
| p = 3.2e-04 (ACT: WT vs Q175; MWU) |
aSTN neurons were relatively hypoactive in Q175 mice during both cortical SWA and ACT.
To determine how PV+ GPe neurons regulate STN activity, the response of STN neurons to optogenetic inhibition of PV+ GPe neurons was recorded. The proportion of STN neurons that responded to inhibition of PV+ GPe neurons was similar in Q175 and WT mice (Table 8). Optogenetic inhibition of PV+ GPe neurons rapidly and persistently increased the frequency and regularity of STN activity in both WT and Q175 mice (Fig. 12A–E; Table 8). However, during optogenetic inhibition, STN neurons remained less active in Q175 mice during cortical SWA (Fig. 12A–D; Table 8) and ACT (Table 8). The ratio of anti- to in-phase STN activity was also significantly elevated by inhibition of PV+ GPe neurons in Q175 and WT mice (Fig. 12F,G; Table 8). Together, with data from this and previous studies, these findings argue that excessive inhibition arising from hyperactive PV+ GPe neurons and autonomous firing deficits contribute to the relative hypoactivity of STN neurons in Q175 mice.
Table 8.
Impact of optogenetic inhibition of PV+ GPe neurons on STN activity in Q175 and WT micea
| Figure/text | Measurement | Brain state | Genotype | PV+ GPe inhibition | n (mice/neurons) | Median [IQ range] | p (comparison; test) |
|---|---|---|---|---|---|---|---|
| text | % optogenetic activation | — | WT | — | 4/16 | 53.3% | p = 0.37 (WT vs Q175; Fisher's Exact) |
| Q175 | 5/39 | 63.9% | |||||
| 12 D | STN frequency | SWA | WT | Off | 4/16 | 7.4 [3.5-14.3] Hz | p = 0.026 (SWA: WT Off vs Q175 Off; MWU) |
| Q175 | 5/39 | 4.4 [2.2-6.8] Hz | |||||
| 12 D | STN frequency | SWA | WT | On | 4/16 | 21.7 [16.3-43.2] Hz | p = 8.0e-04 (SWA: WT Off vs On; WSR) |
| Q175 | 5/39 | 10.0 [7.0-17.8] Hz | p = 1.0e-06 (SWA: Q175 Off vs On; WSR) | ||||
| p = 6.0e-04 (SWA: WT On vs Q175 On; MWU) | |||||||
| 12 E | STN CV | SWA | WT | Off | 4/16 | 1.9 [1.5-3.0] | p = 0.087 (SWA: WT Off vs Q175 Off; MWU) |
| Q175 | 5/39 | 1.7 [1.2-2.5] | |||||
| 12 E | STN CV | SWA | WT | On | 4/16 | 0.62 [0.46-0.91] | p = 9.2e-05 (SWA: WT Off vs On; WSR) |
| Q175 | 5/39 | 0.80 [0.54-0.93] | p = 1.1e-08 (SWA: Q175 Off vs On; WSR) | ||||
| p = 0.30 (SWA: WT On vs Q175 On; MWU) | |||||||
| 12F, G | STN anti-:in-phase ratio | SWA | WT | Off | 4/16 | 0.19 [0.059-0.35] | p = 0.93 (SWA: WT Off vs Q175 Off; MWU) |
| Q175 | 5/39 | 0.23 [0.0-0.38] | |||||
| 12F, G | STN anti-:in-phase ratio | SWA | WT | On | 4/16 | 0.78 [0.61-0.84] | p = 9.2e-05 (SWA: WT Off vs On; WSR) |
| Q175 | 5/39 | 0.60 [0.50-0.85] | p = 5.4e-05 (SWA: Q175 Off vs On; WSR) | ||||
| p = 0.28 (SWA: WT On vs Q175 On; MWU) | |||||||
| text | STN frequency | ACT | WT | Off | 3/14 | 17.0 [9.3-22.4] Hz | p = 2.4e-03 (ACT: WT Off vs Q175 Off; MWU) |
| Q175 | 4/30 | 7.9 [4.1-10.7] Hz | |||||
| text | STN frequency | ACT | WT | On | 3/14 | 27.5 [21.1-44.4] Hz | p = 6.0e-04 (ACT: WT Off vs On; WSR) |
| Q175 | 4/30 | 16.9 [9.8-27.6] Hz | p = 1.3e-04 (ACT: Q175 Off vs On; WSR) | ||||
| p = 5.8e-03 (ACT: WT On vs Q175 On; MWU) |
aOptogenetic inhibition of PV+ GPe neurons disinhibited STN neurons in WT and Q175 mice. However, optogenetic inhibition of PV+ GPe neurons only partially alleviated the hypoactivity of STN neurons in Q175 mice relative to that in WT mice during both cortical SWA and ACT.
Figure 12.
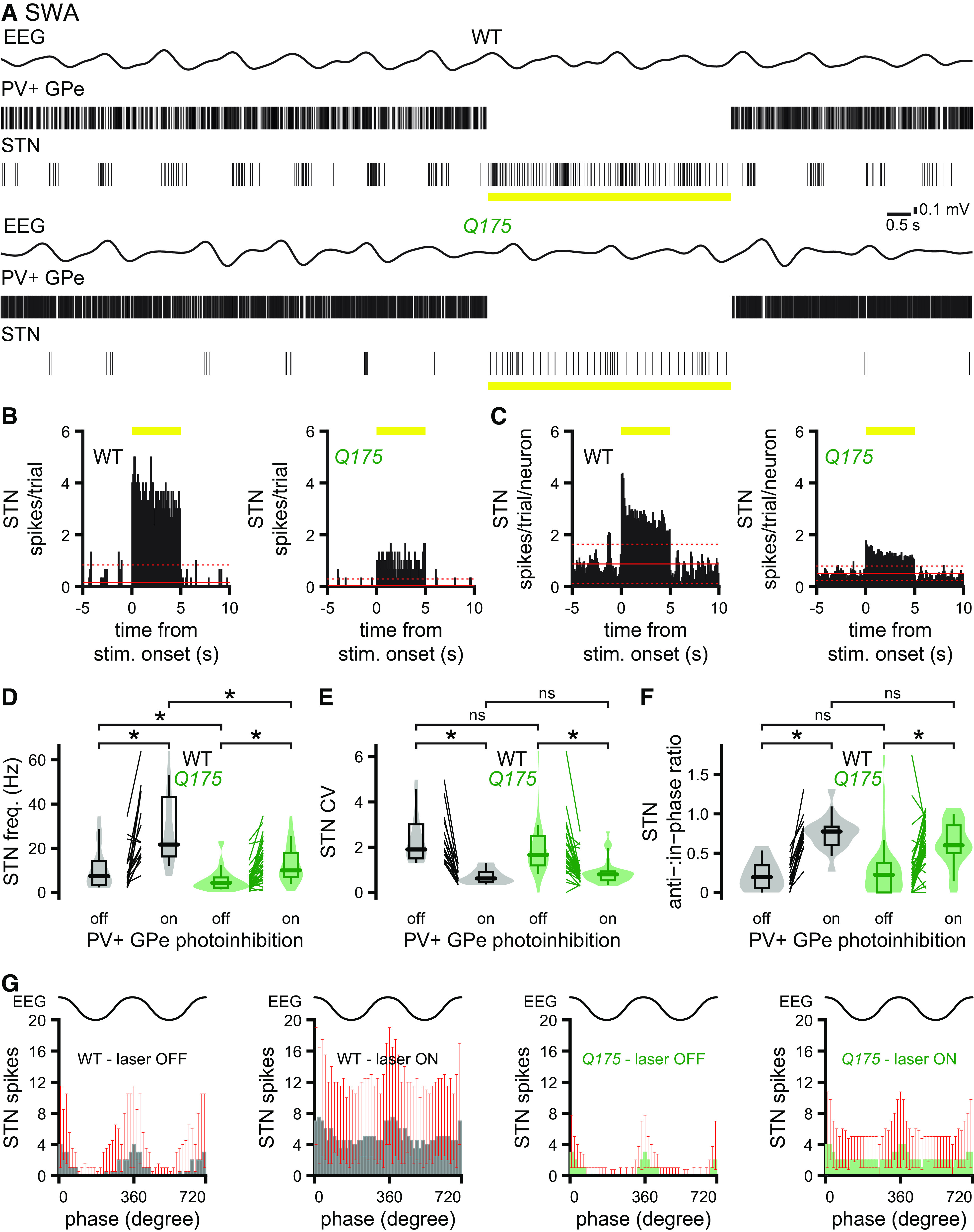
STN hypoactivity is partially rescued by optogenetic inhibition of PV+ GPe neurons in Q175 mice. A, Representative examples of the EEG, bandpass-filtered at 0.1-1.5 Hz, and concurrent PV+ GPe and responsive STN neuron activity in WT and Q175 mice before, during, and after optogenetic inhibition (yellow bar) of PV+ GPe neurons. A–C, Optogenetic inhibition of PV+ GPe neuron activity rapidly and persistently disinhibited STN neuron activity. B, C, PSTHs of STN neuron activity in the absence and presence of PV+ GPe neuron optogenetic inhibition (yellow bar) (bin size, 100 ms; prestimulus mean, solid red line; ±2 SDs of prestimulus mean, dotted red line; B, representative examples from A; C, population data). A–E, Optogenetic inhibition of PV+ GPe neurons disinhibited and regularized STN activity in both genotypes. However, the relative hypoactivity of STN neurons in Q175 mice was not fully reversed by optogenetic inhibition of PV+ GPe neurons (D, E, population data). F, G, The ratio of anti- to in-phase STN activity was increased to a similar extent by inhibition of PV+ GPe neurons in WT and Q175 mice (F, population data; G, population spike phase histograms). *p < 0.05.
Discussion
Cortical network and layer V projection neuron activity
Although the cortex of symptomatic Q175 mice exhibits a variety of pathologic changes, including nuclear aggregates of mHTT (Smith et al., 2014), reduced expression of TrkB (Smith et al., 2014) and mGlu receptors (Bertoglio et al., 2018), a reduction in excitation:inhibition balance (Indersmitten et al., 2015), and cortical volume loss (Heikkinen et al., 2012), the EEG, and the firing rates and patterns of layer V PT and unidentified cortical pyramidal neurons were similar in WT and Q175 mice. The firing rate of unidentified cortical neurons is also unaltered in other HD models (Rebec, 2018).
Striatal projection neuron activity
Consistent with synaptic driving by cortical inputs, D2-SPNs and unidentified, putative D1-SPNs primarily discharged during the active component of cortical SWA. No difference in the firing of identified D2-SPNs and putative D1-SPNs was observed in WT mice during cortical SWA, consistent with some (Sharott et al., 2017) but not other (Mallet et al., 2005) studies that used a similar anesthetic regimen. Given that stimulation was not used to elicit firing in putative D1-SPNs, only neurons with spontaneous activity were detected, which may underlie the higher firing rate of D1-SPNs reported here versus the Mallet et al. (2005) study.
During cortical SWA, the frequency of D2-SPN activity in Q175 mice was half that in WT mice. In contrast, putative D1-SPN activity was similar in Q175 and WT mice. Given similar cortical activity in WT and Q175 mice, the hypoactivity of D2-SPNs in Q175 mice during SWA is consistent with reductions in cortico-striatal inputs, LTP, and strength, and dendritic hypoexcitability in D2-SPNs (Plotkin and Surmeier, 2015; Sebastianutto et al., 2017; Carrillo-Reid et al., 2019; but see Goodliffe et al., 2018). In contrast, the retention of these properties in D1-SPNs (Plotkin and Surmeier, 2015; Carrillo-Reid et al., 2019) may have contributed to their normal activity in Q175 mice. Consistent with impaired cortical driving of D2-SPNs in vivo, firing in response to cortical stimulation, burst firing, and correlated cortico-striatal activity are diminished in unidentified striatal neurons in HD mice (Beaumont et al., 2016; Rebec, 2018).
During cortical desynchronization, D2-SPN activity decreased to a lesser degree in Q175 than WT mice and unidentified/putative D1-SPN activity decreased in WT but not Q175 mice. As a result, SPN activity was elevated in Q175 mice relative to WT during ACT. While depolarization of the resting membrane potential and increased axosomatic excitability of SPNs in Q175 mice (Heikkinen et al., 2012; Indersmitten et al., 2015; Beaumont et al., 2016) do not compensate for reduced cortical driving of D2-SPNs during cortical SWA, they may contribute to elevated SPN activity during cortical desynchronization. Another possibility is that somatosensory stimulation used to trigger cortical desynchronization engages more powerful thalamo-striatal excitation in Q175 mice. Given that SPN activity is sculpted by striatal interneurons (Tepper et al., 2018), alterations in interneuron excitability and transmission (Holley et al., 2019a,b) may also contribute to aberrant SPN activity in HD mice. The spontaneous activity of unidentified striatal neurons has been reported to be elevated, reduced, or unchanged in anesthetized knock-in or awake full-length HD mice (Miller et al., 2008; Estrada-Sanchez et al., 2015; Beaumont et al., 2016). However, an absence of cell-identification approaches and/or different recording conditions in those studies make comparisons with this study problematic (Heikkinen et al., 2012; Indersmitten et al., 2015; Beaumont et al., 2016).
Prototypic PV+ GPe and putative PV– GPe neuron activity
Prototypic PV+ GPe neurons were hyperactive in Q175 mice during cortical SWA, consistent with upstream D2-SPN hypoactivity. However, additional factors must contribute because PV+ GPe neurons were also hyperactive in Q175 mice when (1) D2-SPNs were silent (in both Q175 and WT mice) during the inactive phase of cortical SWA; and (2) D2-SPNs were hyperactive in Q175 mice during cortical ACT. Furthermore, striatopallidal transmission strength increases in HD and its models through upregulation of postsynaptic GABAA receptor expression (Glass et al., 2000; Waldvogel et al., 2015; Perez-Rosello et al., 2019). Indeed, during cortical SWA, PV+ GPe neurons in Q175 mice more commonly exhibited pauses/reductions in firing that were concomitant with bouts of D2-SPN activity. Thus, PV+ GPe neuron hyperactivity may in part stem from elevated autonomous firing, as demonstrated here in Q175 mouse brain slices. Given the widespread nature and potency of prototypic GPe neuron projections in the basal ganglia (Bevan et al., 1998; Mallet et al., 2012; Mastro et al., 2014; Abdi et al., 2015), it will be critical to determine which alterations underlie their upregulated autonomous firing, and whether hyperactivity results from the cell-autonomous effects of mHTT and/or homeostatic compensation.
The hypoactivity of unidentified, putative PV– GPe neurons in Q175 mice in vivo appears to be caused by excessive inhibition because it was alleviated by optogenetic inhibition of hyperactive prototypic PV+ GPe neurons. The powerful regulation of putative PV– prototypic and arkypallidal neuron activity by prototypic PV+ GPe neurons is consistent with recent studies (Aristieta et al., 2020; Cui et al., 2021; Ketzef and Silberberg, 2021). However, optogenetic silencing of PV+ GPe neurons also disinhibited the STN, which feeds back onto GPe neurons (Aristieta et al., 2020; Pamukcu et al., 2020). Thus, STN disinhibition may have contributed to the rescue of putative PV– GPe neuron activity in Q175 mice. The finding that the autonomous firing of putative PV– GPe neurons was unaffected in Q175 mice further suggests that synaptic mechanisms underlie their hypoactivity in vivo. The low rate of putative PV– GPe neuron autonomous firing and longer duration of action potentials compared with PV+ GPe neurons in WT (and Q175) mice are consistent with previous observations in WT rodents (Abdi et al., 2015; Hernandez et al., 2015; Cui et al., 2021). Finally, our data contrast with another study that reported no change in GPe neuron activity in Q175 mice (Beaumont et al., 2016) and WT GPe activity that was substantially lower than for prototypic GPe neurons (Aristieta et al., 2020; Kovaleski et al., 2020). The identities of GPe neurons were not determined in the Beaumont (2016) study; and although a similar anesthetic was used, the depth of anesthesia was not tracked. Thus, their measurements of GPe activity were likely derived from a variety of GPe cell classes during SWA and ACT.
STN activity
In Q175 mice, STN activity was half that in WT mice during both cortical SWA and ACT. Thus, the hypoactivity of STN neurons observed here is consistent with that reported in Q175 mice in which the EEG was not measured and cortical activity state was unknown (Beaumont et al., 2016), and more robust than in YAC128 (Callahan and Abercrombie, 2015a) and R6/2 mice (Callahan and Abercrombie, 2015b), where only a subset of STN neurons exhibited diminished activity. The hypoactivity of STN neurons in Q175 mice could arise from hyperactivity of upstream prototypic PV+ GPe neurons, loss of autonomous STN activity (Atherton et al., 2016), and reduced cortical excitation (Beaumont et al., 2016). Indeed, STN hypoactivity was greatly (but not completely) alleviated in Q175 mice by optogenetic inhibition of prototypic PV+ GPe neurons, consistent with excessive inhibition from PV+ GPe neurons, but also other factors, including loss of intrinsic firing, (Atherton et al., 2016) and disinhibition of prototypic PV– GPe neurons that project to the STN (Mallet et al., 2012; Mastro et al., 2014; Hernandez et al., 2015; Aristieta et al., 2020; Cui et al., 2021). Phase-locking of STN activity to cortical SWA was unaffected in Q175 mice both before and during optogenetic inhibition of prototypic GPe neurons, arguing that excessive inhibition from prototypic GPe neurons and impaired intrinsic excitability, rather than reduced cortical drive, underlie STN hypoactivity. Because optogenetic inhibition of PV+ GPe neurons and disinhibition of the STN rescued putative PV– GPe neuron activity, STN hypoactivity may contribute to PV– GPe neuron hypoactivity in Q175 mice. In contrast, STN hypoactivity did not prevent hyperactivity in PV+ GPe neurons, presumably because their hyperactivity resulted from alterations in autonomous firing and striatopallidal transmission.
Functional implications
Together, our and other studies argue that the basal ganglia indirect pathway is profoundly dysregulated in HD mice through a complex, cell class-specific combination of state-dependent changes in presynaptic activity and aberrant intrinsic and synaptic properties. According to the classic rate model, PV+ GPe neuron hyperactivity and STN (and possibly arkypallidal) neuron hypoactivity should promote hyperkinesia (Albin et al., 1989). However, HD mice typically exhibit bradykinesia/akinesia and subtle kinematic abnormalities (Heikkinen et al., 2020). Therefore, consistent with their large number of trinucleotide repeats, HD mice may better represent juvenile-onset HD in which the initial hyperkinetic phase seen in adult-onset HD is absent (Fusilli et al., 2018; Tereshchenko et al., 2019). Because the indirect pathway is critical for normal volitional movement (Dodson et al., 2015; Tecuapetla et al., 2016; Pasquereau and Turner, 2017; Markowitz et al., 2018; Aristieta et al., 2020), dysregulation of this pathway may actively contribute to motor dysfunction in HD mice.
Footnotes
This work was supported by CHDI Foundation; National Institutes of Health, National Institute of Neurological Disorders and Stroke Grant R37 NS041280; and the Feinberg School of Medicine, Northwestern University. Confocal imaging was performed at the Northwestern University Center for Advanced Microscopy, which is supported by National Institutes of Health NCI Grant CCSG P30 CA060553 (awarded to the Robert H. Lurie Comprehensive Cancer Center). We thank Sasha Ulrich, Danielle Schowalter, and Bonnie Erjavec for the maintenance and supply of transgenic mice for this study. We also thank Drs. Vahri Beaumont, Ignacio Muñoz-Sanjuán, and Roger Cachope for their scientific input throughout this study.
The authors declare no competing financial interests.
References
- Abdi A, Mallet N, Mohamed FY, Sharott A, Dodson PD, Nakamura KC, Suri S, Avery SV, Larvin JT, Garas FN, Garas SN, Vinciati F, Morin S, Bezard E, Baufreton J, Magill PJ (2015) Prototypic and arkypallidal neurons in the dopamine-intact external globus pallidus. J Neurosci 35:6667–6688. 10.1523/JNEUROSCI.4662-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abecassis ZA, Berceau BL, Win PH, García D, Xenias HS, Cui Q, Pamukcu A, Cherian S, Hernández VM, Chon U, Lim BK, Kim Y, Justice NJ, Awatramani R, Hooks BM, Gerfen CR, Boca SM, Chan CS (2020) Npas1(+)-Nkx2.1(+) neurons are an integral part of the cortico-pallido-cortical loop. J Neurosci 40:743–768. 10.1523/JNEUROSCI.1199-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB (1989) The functional anatomy of basal ganglia disorders. Trends Neurosci 12:366–375. 10.1016/0166-2236(89)90074-x [DOI] [PubMed] [Google Scholar]
- Albin RL, Reiner A, Anderson KD, Dure LS, Handelin B, Balfour R, Whetsell WO Jr, Penney JB, Young AB (1992) Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington's disease. Ann Neurol 31:425–430. 10.1002/ana.410310412 [DOI] [PubMed] [Google Scholar]
- Amzica F, Steriade M (1998) Electrophysiological correlates of sleep delta waves. Electroencephalogr Clin Neurophysiol 107:69–83. 10.1016/S0013-4694(98)00051-0 [DOI] [PubMed] [Google Scholar]
- Aristieta A, Barresi B, Lindi SA, Barriere G, Courtand G, de la Crompe B, Guilhemsang L, Gauthier S, Fioramonti S, Baufreton J, Mallet N (2020) A disynaptic circuit in the globus pallidus controls locomotion inhibition. Curr Biol 31:707–721.e7. [DOI] [PubMed] [Google Scholar]
- Atherton JF, Menard A, Urbain N, Bevan MD (2013) Short-term depression of external globus pallidus-subthalamic nucleus synaptic transmission and implications for patterning subthalamic activity. J Neurosci 33:7130–7144. 10.1523/JNEUROSCI.3576-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton JF, McIver EL, Mullen MR, Wokosin DL, Surmeier DJ, Bevan MD (2016) Early dysfunction and progressive degeneration of the subthalamic nucleus in mouse models of Huntington's disease. Elife 5:e21616. 10.7554/Elife.21616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbera G, Liang B, Zhang L, Gerfen CR, Culurciello E, Chen R, Li Y, Lin DT (2016) Spatially compact neural clusters in the dorsal striatum encode locomotion relevant information. Neuron 92:202–213. 10.1016/j.neuron.2016.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R, Wild EJ, Tabrizi SJ (2015) Huntington disease. Nat Rev Dis Primers 1:15005. 10.1038/nrdp.2015.5 [DOI] [PubMed] [Google Scholar]
- Baufreton J, Kirkham E, Atherton JF, Menard A, Magill PJ, Bolam JP, Bevan MD (2009) Sparse but selective and potent synaptic transmission from the globus pallidus to the subthalamic nucleus. J Neurophysiol 102:532–545. 10.1152/jn.00305.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont V, Zhong S, Lin H, Xu WJ, Bradaia A, Steidl E, Gleyzes M, Wadel K, Buisson B, Padovan-Neto FE, Chakroborty S, Ward KM, Harms JF, Beltran J, Kwan M, Ghavami A, Häggkvist J, Tóth M, Halldin C, Varrone A, et al. (2016) Phosphodiesterase 10A inhibition improves cortico-basal ganglia function in Huntington's disease models. Neuron 92:1220–1237. 10.1016/j.neuron.2016.10.064 [DOI] [PubMed] [Google Scholar]
- Beltramo R, D'Urso G, Dal Maschio M, Farisello P, Bovetti S, Clovis Y, Lassi G, Tucci V, De Pietri Tonelli D, Fellin T (2013) Layer-specific excitatory circuits differentially control recurrent network dynamics in the neocortex. Nat Neurosci 16:227–234. 10.1038/nn.3306 [DOI] [PubMed] [Google Scholar]
- Berke JD, Okatan M, Skurski J, Eichenbaum HB (2004) Oscillatory entrainment of striatal neurons in freely moving rats. Neuron 43:883–896. 10.1016/j.neuron.2004.08.035 [DOI] [PubMed] [Google Scholar]
- Bertoglio D, Kosten L, Verhaeghe J, Thomae D, Wyffels L, Stroobants S, Wityak J, Dominguez C, Mrzljak L, Staelens S (2018) Longitudinal characterization of mGluR5 using (11)C-ABP688 PET imaging in the Q175 mouse model of Huntington disease. J Nucl Med 59:1722–1727. 10.2967/jnumed.118.210658 [DOI] [PubMed] [Google Scholar]
- Bevan MD, Booth PA, Eaton SA, Bolam JP (1998) Selective innervation of neostriatal interneurons by a subclass of neuron in the globus pallidus of the rat. J Neurosci 18:9438–9452. 10.1523/JNEUROSCI.18-22-09438.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan MD, Magill PJ, Hallworth NE, Bolam JP, Wilson CJ (2002) Regulation of the timing and pattern of action potential generation in rat subthalamic neurons in vitro by GABA-A IPSPs. J Neurophysiol 87:1348–1362. 10.1152/jn.00582.2001 [DOI] [PubMed] [Google Scholar]
- Bokil H, Andrews P, Kulkarni JE, Mehta S, Mitra PP (2010) Chronux: a platform for analyzing neural signals. J Neurosci Methods 192:146–151. 10.1016/j.jneumeth.2010.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan JW, Abercrombie ED (2015a) Age-dependent alterations in the cortical entrainment of subthalamic nucleus neurons in the YAC128 mouse model of Huntington's disease. Neurobiol Dis 78:88–99. 10.1016/j.nbd.2015.03.006 [DOI] [PubMed] [Google Scholar]
- Callahan JW, Abercrombie ED (2015b) Relationship between subthalamic nucleus neuronal activity and electrocorticogram is altered in the R6/2 mouse model of Huntington's disease. J Physiol 593:3727–3738. 10.1113/JP270268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Reid L, Day M, Xie Z, Melendez AE, Kondapalli J, Plotkin JL, Wokosin DL, Chen Y, Kress GJ, Kaplitt M, Ilijic E, Guzman JN, Chan CS, Surmeier DJ (2019) Mutant huntingtin enhances activation of dendritic Kv4 K(+) channels in striatal spiny projection neurons. Elife 8:e40818. 10.7554/Elife.40818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayzac S, Delcasso S, Paz V, Jeantet Y, Cho YH (2011) Changes in striatal procedural memory coding correlate with learning deficits in a mouse model of Huntington disease. Proc Natl Acad Sci USA 108:9280–9285. 10.1073/pnas.1016190108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Wu N, Andre VM, Cummings DM, Levine MS (2007) The corticostriatal pathway in Huntington's disease. Prog Neurobiol 81:253–271. 10.1016/j.pneurobio.2006.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, Costa RM (2013) Concurrent activation of striatal direct and indirect pathways during action initiation. Nature 494:238–242. 10.1038/nature11846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Pamukcu A, Cherian S, Chang IY, Berceau BL, Xenias HS, Higgs MH, Rajamanickam S, Chen Y, Du X, Zhang Y, McMorrow H, Abecassis ZA, Boca SM, Justice NJ, Wilson CJ, Chan CS (2021) Dissociable roles of pallidal neuron subtypes in regulating motor patterns. J Neurosci 41:4036–4059. 10.1523/JNEUROSCI.2210-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson PD, Larvin JT, Duffell JM, Garas FN, Doig NM, Kessaris N, Duguid IC, Bogacz R, Butt SJ, Magill PJ (2015) Distinct developmental origins manifest in the specialized encoding of movement by adult neurons of the external globus pallidus. Neuron 86:501–513. 10.1016/j.neuron.2015.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada-Sanchez AM, Burroughs CL, Cavaliere S, Barton SJ, Chen S, Yang XW, Rebec GV (2015) Cortical efferents lacking mutant huntingtin improve striatal neuronal activity and behavior in a conditional mouse model of Huntington's disease. J Neurosci 35:4440–4451. 10.1523/JNEUROSCI.2812-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusilli C, Migliore S, Mazza T, Consoli F, De Luca A, Barbagallo G, Ciammola A, Gatto EM, Cesarini M, Etcheverry JL, Parisi V, Al-Oraimi M, Al-Harrasi S, Al-Salmi Q, Marano M, Vonsattel JG, Sabatini U, Landwehrmeyer GB, Squitieri F (2018) Biological and clinical manifestations of juvenile Huntington's disease: a retrospective analysis. Lancet Neurol 17:986–993. 10.1016/S1474-4422(18)30294-1 [DOI] [PubMed] [Google Scholar]
- Gage GJ, Stoetzner CR, Wiltschko AB, Berke JD (2010) Selective activation of striatal fast-spiking interneurons during choice execution. Neuron 67:466–479. 10.1016/j.neuron.2010.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ Jr, Sibley DR (1990) D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250:1429–1432. 10.1126/science.2147780 [DOI] [PubMed] [Google Scholar]
- Glass M, Dragunow M, Faull RL (2000) The pattern of neurodegeneration in Huntington's disease: a comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington's disease. Neuroscience 97:505–519. 10.1016/S0306-4522(00)00008-7 [DOI] [PubMed] [Google Scholar]
- Goodliffe JW, Song H, Rubakovic A, Chang W, Medalla M, Weaver CM, Luebke JI (2018) Differential changes to D1 and D2 medium spiny neurons in the 12-month-old Q175+/- mouse model of Huntington's disease. PLoS One 13:e0200626. 10.1371/journal.pone.0200626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallworth NE, Bevan MD (2005) Globus pallidus neurons dynamically regulate the activity pattern of subthalamic nucleus neurons through the frequency-dependent activation of postsynaptic GABAA and GABAB receptors. J Neurosci 25:6304–6315. 10.1523/JNEUROSCI.0450-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KD, Shepherd GM (2015) The neocortical circuit: themes and variations. Nat Neurosci 18:170–181. 10.1038/nn.3917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkinen T, Lehtimaki K, Vartiainen N, Puolivali J, Hendricks SJ, Glaser JR, Bradaia A, Wadel K, Touller C, Kontkanen O, Yrjanheikki JM, Buisson B, Howland D, Beaumont V, Munoz-Sanjuan I, Park LC (2012) Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington's disease. PLoS One 7:e50717. 10.1371/journal.pone.0050717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkinen T, Bragge T, Bhattarai N, Parkkari T, Puoliväli J, Kontkanen O, Sweeney P, Park LC, Munoz-Sanjuan I (2020) Rapid and robust patterns of spontaneous locomotor deficits in mouse models of Huntington's disease. PLoS One 15:e0243052. 10.1371/journal.pone.0243052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez VM, Hegeman DJ, Cui Q, Kelver DA, Fiske MP, Glajch KE, Pitt JE, Huang TY, Justice NJ, Chan CS (2015) Parvalbumin+ neurons and Npas1+ neurons are distinct neuron classes in the mouse external globus pallidus. J Neurosci 35:11830–11847. 10.1523/JNEUROSCI.4672-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley SM, Galvan L, Kamdjou T, Cepeda C, Levine MS (2019a) Striatal GABAergic interneuron dysfunction in the Q175 mouse model of Huntington's disease. Eur J Neurosci 49:79–93. 10.1111/ejn.14283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley SM, Galvan L, Kamdjou T, Dong A, Levine MS, Cepeda C (2019b) Major contribution of somatostatin-expressing interneurons and cannabinoid receptors to increased GABA synaptic activity in the striatum of Huntington's disease mice. Front Synaptic Neurosci 11:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooks BM (2018) Dual-channel photostimulation for independent excitation of two populations. Curr Protoc Neurosci 85:e52. 10.1002/cpns.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indersmitten T, Tran CH, Cepeda C, Levine MS (2015) Altered excitatory and inhibitory inputs to striatal medium-sized spiny neurons and cortical pyramidal neurons in the Q175 mouse model of Huntington's disease. J Neurophysiol 113:2953–2966. 10.1152/jn.01056.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johri A, Chandra A, Flint Beal M (2013) PGC-1α, mitochondrial dysfunction, and Huntington's disease. Free Radic Biol Med 62:37–46. 10.1016/j.freeradbiomed.2013.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman MT, Churchland MM, Santhanam G, Yu BM, Afshar A, Ryu SI, Shenoy KV (2010) Roles of monkey premotor neuron classes in movement preparation and execution. J Neurophysiol 104:799–810. 10.1152/jn.00231.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeler JF, Pretsell DO, Robbins TW (2014) Functional implications of dopamine D1 vs. D2 receptors: a 'prepare and select' model of the striatal direct vs. indirect pathways. Neuroscience 282:156–175. 10.1016/j.neuroscience.2014.07.021 [DOI] [PubMed] [Google Scholar]
- Ketzef M, Silberberg G (2021) Differential synaptic input to external globus pallidus neuronal subpopulations in vivo. Neuron 109:516–529. [DOI] [PubMed] [Google Scholar]
- Kim N, Barter JW, Sukharnikova T, Yin HH (2014) Striatal firing rate reflects head movement velocity. Eur J Neurosci 40:3481–3490. 10.1111/ejn.12722 [DOI] [PubMed] [Google Scholar]
- Kiritani T, Wickersham IR, Seung HS, Shepherd GM (2012) Hierarchical connectivity and connection-specific dynamics in the corticospinal-corticostriatal microcircuit in mouse motor cortex. J Neurosci 32:4992–5001. 10.1523/JNEUROSCI.4759-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus A, Alves da Silva J, Costa RM (2019) What, if, and when to move: basal ganglia circuits and self-paced action initiation. Annu Rev Neurosci 42:459–483. 10.1146/annurev-neuro-072116-031033 [DOI] [PubMed] [Google Scholar]
- Kovaleski RF, Callahan JW, Chazalon M, Wokosin DL, Baufreton J, Bevan MD (2020) Dysregulation of external globus pallidus-subthalamic nucleus network dynamics in parkinsonian mice during cortical slow-wave activity and activation. J Physiol 598:1897–1927. 10.1113/JP279232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambot L, Chaves Rodriguez E, Houtteman D, Li Y, Schiffmann SN, Gall D, de Kerchove d'Exaerde A (2016) Striatopallidal neuron NMDA receptors control synaptic connectivity, locomotor, and goal-directed behaviors. J Neurosci 36:4976–4992. 10.1523/JNEUROSCI.2717-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Van Quyen M, Foucher J, Lachaux J, Rodriguez E, Lutz A, Martinerie J, Varela FJ (2001) Comparison of Hilbert transform and wavelet methods for the analysis of neuronal synchrony. J Neurosci Methods 111:83–98. 10.1016/s0165-0270(01)00372-7 [DOI] [PubMed] [Google Scholar]
- LeBlanc KH, London TD, Szczot I, Bocarsly ME, Friend DM, Nguyen KP, Mengesha MM, Rubinstein M, Alvarez VA, Kravitz AV (2020) Striatopallidal neurons control avoidance behavior in exploratory tasks. Mol Psychiatry 25:491–505. 10.1038/s41380-018-0051-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JC, Friend DM, Kaplan AR, Shin JH, Rubinstein M, Kravitz AV, Alvarez VA (2016) Enhanced GABA transmission drives bradykinesia following loss of dopamine D2 receptor signaling. Neuron 90:824–838. 10.1016/j.neuron.2016.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Yamawaki N, Barrett JM, Körding KP, Shepherd GM (2018) Scaling of optogenetically evoked signaling in a higher-order corticocortical pathway in the anesthetized mouse. Front Syst Neurosci 12:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohani S, Martig AK, Deisseroth K, Witten IB, Moghaddam B (2019) Dopamine modulation of prefrontal cortex activity is manifold and operates at multiple temporal and spatial scales. Cell Rep 27:99–114.e116. 10.1016/j.celrep.2019.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magill PJ, Bolam JP, Bevan MD (2000) Relationship of activity in the subthalamic nucleus-globus pallidus network to cortical electroencephalogram. J Neurosci 20:820–833. 10.1523/JNEUROSCI.20-02-00820.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magill PJ, Bolam JP, Bevan MD (2001) Dopamine regulates the impact of the cerebral cortex on the subthalamic nucleus-globus pallidus network. Neuroscience 106:313–330. 10.1016/S0306-4522(01)00281-0 [DOI] [PubMed] [Google Scholar]
- Mallet N, Le Moine C, Charpier S, Gonon F (2005) Feedforward inhibition of projection neurons by fast-spiking GABA interneurons in the rat striatum in vivo. J Neurosci 25:3857–3869. 10.1523/JNEUROSCI.5027-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Pogosyan A, Marton LF, Bolam JP, Brown P, Magill PJ (2008a) Parkinsonian beta oscillations in the external globus pallidus and their relationship with subthalamic nucleus activity. J Neurosci 28:14245–14258. 10.1523/JNEUROSCI.4199-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Pogosyan A, Sharott A, Csicsvari J, Bolam JP, Brown P, Magill PJ (2008b) Disrupted dopamine transmission and the emergence of exaggerated beta oscillations in subthalamic nucleus and cerebral cortex. J Neurosci 28:4795–4806. 10.1523/JNEUROSCI.0123-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Micklem BR, Henny P, Brown MT, Williams C, Bolam JP, Nakamura KC, Magill PJ (2012) Dichotomous organization of the external globus pallidus. Neuron 74:1075–1086. 10.1016/j.neuron.2012.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Schmidt R, Leventhal D, Chen F, Am N, Boraud T, Berke JD (2016) Arkypallidal cells send a stop signal to striatum. Neuron 89:308–316. 10.1016/j.neuron.2015.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz JE, Gillis WF, Beron CC, Neufeld SQ, Robertson K, Bhagat ND, Peterson RE, Peterson E, Hyun M, Linderman SW, Sabatini BL, Datta SR (2018) The striatum organizes 3D behavior via moment-to-moment action selection. Cell 174:44–58.e17. 10.1016/j.cell.2018.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DD, Ladha S, Ehrnhoefer DE, Hayden MR (2015) Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci 38:26–35. 10.1016/j.tins.2014.09.003 [DOI] [PubMed] [Google Scholar]
- Mastro KJ, Bouchard RS, Holt HA, Gittis AH (2014) Transgenic mouse lines subdivide external segment of the globus pallidus (GPe) neurons and reveal distinct GPe output pathways. J Neurosci 34:2087–2099. 10.1523/JNEUROSCI.4646-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Deniau JM, Glowinski J, Thierry AM (1999) Relationships between the prefrontal cortex and the basal ganglia in the rat: physiology of the cortico-nigral circuits. J Neurosci 19:4674–4681. 10.1523/JNEUROSCI.19-11-04674.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Kudwa AE, Miller S, Fitzpatrick J, Watson-Johnson J, Keating N, Ruiz M, Mushlin R, Alosio W, McConnell K, Connor D, Murphy C, Oakeshott S, Kwan M, Beltran J, Ghavami A, Brunner D, Park LC, Ramboz S, Howland D (2012) Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington's disease: zQ175. PLoS One 7:e49838. 10.1371/journal.pone.0049838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Walker AG, Shah AS, Barton SJ, Rebec GV (2008) Dysregulated information processing by medium spiny neurons in striatum of freely behaving mouse models of Huntington's disease. J Neurophysiol 100:2205–2216. 10.1152/jn.90606.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mink JW, Thach WT (1993) Basal ganglia intrinsic circuits and their role in behavior. Curr Opin Neurobiol 3:950–957. 10.1016/0959-4388(93)90167-w [DOI] [PubMed] [Google Scholar]
- Mitchell JF, Sundberg KA, Reynolds JH (2007) Differential attention-dependent response modulation across cell classes in macaque visual area V4. Neuron 55:131–141. 10.1016/j.neuron.2007.06.018 [DOI] [PubMed] [Google Scholar]
- Nambu A, Tokuno H, Takada M (2002) Functional significance of the cortico-subthalamo-pallidal 'hyperdirect' pathway. Neurosci Res 43:111–117. 10.1016/s0168-0102(02)00027-5 [DOI] [PubMed] [Google Scholar]
- Nelson AB, Kreitzer AC (2014) Reassessing models of basal ganglia function and dysfunction. Annu Rev Neurosci 37:117–135. 10.1146/annurev-neuro-071013-013916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noether GE (1987) Sample size determination for some common nonparametric tests. J Am Statist Assoc 82:645–647. 10.1080/01621459.1987.10478478 [DOI] [Google Scholar]
- Pamukcu A, Cui Q, Xenias HS, Berceau BL, Augustine EC, Fan I, Chalasani S, Hantman AW, Lerner TN, Boca SM, Chan CS (2020) Parvalbumin(+) and Npas1(+) pallidal neurons have distinct circuit topology and function. J Neurosci 40:7855–7876. 10.1523/JNEUROSCI.0361-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquereau B, Turner RS (2017) A selective role for ventromedial subthalamic nucleus in inhibitory control. Elife 6:e31627. 10.7554/Elife.31627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KB (2001) The mouse brain in stereotaxic coordinates, Ed 2. San Diego: Academic. [Google Scholar]
- Perez-Rosello T, Gelman S, Tombaugh G, Cachope R, Beaumont V, Surmeier DJ (2019) Enhanced striatopallidal gamma-aminobutyric acid (GABA)A receptor transmission in mouse models of Huntington's disease. Mov Disord 34:684–696. 10.1002/mds.27622 [DOI] [PubMed] [Google Scholar]
- Plenz D, Kital ST (1999) A basal ganglia pacemaker formed by the subthalamic nucleus and external globus pallidus. Nature 400:677–682. 10.1038/23281 [DOI] [PubMed] [Google Scholar]
- Plotkin JL, Day M, Peterson JD, Xie Z, Kress GJ, Rafalovich I, Kondapalli J, Gertler TS, Flajolet M, Greengard P, Stavarache M, Kaplitt MG, Rosinski J, Chan CS, Surmeier DJ (2014) Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington's disease. Neuron 83:178–188. 10.1016/j.neuron.2014.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JL, Surmeier DJ (2014) Impaired striatal function in Huntington's disease is due to aberrant p75NTR signaling. Rare Dis 2:e968482. 10.4161/2167549X.2014.968482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JL, Surmeier DJ (2015) Corticostriatal synaptic adaptations in Huntington's disease. Curr Opin Neurobiol 33:53–62. 10.1016/j.conb.2015.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond LA, Andre VM, Cepeda C, Gladding CM, Milnerwood AJ, Levine MS (2011) Pathophysiology of Huntington's disease: time-dependent alterations in synaptic and receptor function. Neuroscience 198:252–273. 10.1016/j.neuroscience.2011.08.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebec GV (2018) Corticostriatal network dysfunction in Huntington's disease: deficits in neural processing, glutamate transport, and ascorbate release. CNS Neurosci Ther 24:281–291. 10.1111/cns.12828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Shirendeb UP (2012) Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington's disease. Biochim Biophys Acta 1822:101–110. 10.1016/j.bbadis.2011.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Deng YP (2018) Disrupted striatal neuron inputs and outputs in Huntington's disease. CNS Neurosci Ther 24:250–280. 10.1111/cns.12844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richfield EK, Maguire-Zeiss KA, Cox C, Gilmore J, Voorn P (1995) Reduced expression of preproenkephalin in striatal neurons from Huntington's disease patients. Ann Neurol 37:335–343. 10.1002/ana.410370309 [DOI] [PubMed] [Google Scholar]
- Rosas HD, Wilkens P, Salat DH, Mercaldo ND, Vangel M, Yendiki AY, Hersch SM (2018) Complex spatial and temporally defined myelin and axonal degeneration in Huntington disease. Neuroimage Clin 20:236–242. 10.1016/j.nicl.2018.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Huang KW, Sabatini BL (2016) Globus pallidus externus neurons expressing parvalbumin interconnect the subthalamic nucleus and striatal interneurons. PLoS One 11:e0149798. 10.1371/journal.pone.0149798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastianutto I, Cenci MA, Fieblinger T (2017) Alterations of striatal indirect pathway neurons precede motor deficits in two mouse models of Huntington's disease. Neurobiol Dis 105:117–131. 10.1016/j.nbd.2017.05.011 [DOI] [PubMed] [Google Scholar]
- Seredenina T, Luthi-Carter R (2012) What have we learned from gene expression profiles in Huntington's disease? Neurobiol Dis 45:83–98. 10.1016/j.nbd.2011.07.001 [DOI] [PubMed] [Google Scholar]
- Sharott A, Vinciati F, Nakamura KC, Magill PJ (2017) A population of indirect pathway striatal projection neurons is selectively entrained to parkinsonian beta oscillations. J Neurosci 37:9977–9998. 10.1523/JNEUROSCI.0658-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Kim D, Jung MW (2018) Differential coding of reward and movement information in the dorsomedial striatal direct and indirect pathways. Nat Commun 9:404. 10.1038/s41467-017-02817-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siapas AG, Lubenov EV, Wilson MA (2005) Prefrontal phase locking to hippocampal theta oscillations. Neuron 46:141–151. 10.1016/j.neuron.2005.02.028 [DOI] [PubMed] [Google Scholar]
- Sippy T, Lapray D, Crochet S, Petersen CC (2015) Cell-type-specific sensorimotor processing in striatal projection neurons during goal-directed behavior. Neuron 88:298–305. 10.1016/j.neuron.2015.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GA, Rocha EM, McLean JR, Hayes MA, Izen SC, Isacson O, Hallett PJ (2014) Progressive axonal transport and synaptic protein changes correlate with behavioral and neuropathological abnormalities in the heterozygous Q175 KI mouse model of Huntington's disease. Hum Mol Genet 23:4510–4527. 10.1093/hmg/ddu166 [DOI] [PubMed] [Google Scholar]
- Steriade M (2000) Corticothalamic resonance, states of vigilance and mentation. Neuroscience 101:243–276. 10.1016/s0306-4522(00)00353-5 [DOI] [PubMed] [Google Scholar]
- Steriade M, Nunez A, Amzica F (1993) Intracellular analysis of relations between the slow (< 1 Hz) neocortical oscillation and other sleep rhythms of the electroencephalogram. J Neurosci 13:3266–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern EA, Kincaid AE, Wilson CJ (1997) Spontaneous subthreshold membrane potential fluctuations and action potential variability of rat corticostriatal and striatal neurons in vivo. J Neurophysiol 77:1697–1715. 10.1152/jn.1997.77.4.1697 [DOI] [PubMed] [Google Scholar]
- Tachibana Y, Kita H, Chiken S, Takada M, Nambu A (2008) Motor cortical control of internal pallidal activity through glutamatergic and GABAergic inputs in awake monkeys. Eur J Neurosci 27:238–253. 10.1111/j.1460-9568.2007.05990.x [DOI] [PubMed] [Google Scholar]
- Takahashi K, Kim S, Coleman TP, Brown KA, Suminski AJ, Best MD, Hatsopoulos NG (2015) Large-scale spatiotemporal spike patterning consistent with wave propagation in motor cortex. Nat Commun 6:7169. 10.1038/ncomms8169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Jin X, Lima SQ, Costa RM (2016) Complementary contributions of striatal projection pathways to action initiation and execution. Cell 166:703–715. 10.1016/j.cell.2016.06.032 [DOI] [PubMed] [Google Scholar]
- Tecuapetla F, Matias S, Dugue GP, Mainen ZF, Costa RM (2014) Balanced activity in basal ganglia projection pathways is critical for contraversive movements. Nat Commun 5:4315. 10.1038/ncomms5315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, Koós T, Ibanez-Sandoval O, Tecuapetla F, Faust TW, Assous M (2018) Heterogeneity and diversity of striatal GABAergic interneurons: update 2018. Front Neuroanat 12:91. 10.3389/fnana.2018.00091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tereshchenko A, Magnotta V, Epping E, Mathews K, Espe-Pfeifer P, Martin E, Dawson J, Duan W, Nopoulos P (2019) Brain structure in juvenile-onset Huntington disease. Neurology 92:e1939–e1947. 10.1212/WNL.0000000000007355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, Khakh BS (2014) Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat Neurosci 17:694–703. 10.1038/nn.3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldvogel HJ, Kim EH, Tippett LJ, Vonsattel JP, Faull RL (2015) The neuropathology of Huntington's disease. Curr Top Behav Neurosci 22:33–80. [DOI] [PubMed] [Google Scholar]
- Walker AG, Miller BR, Fritsch JN, Barton SJ, Rebec GV (2008) Altered information processing in the prefrontal cortex of Huntington's disease mouse models. J Neurosci 28:8973–8982. 10.1523/JNEUROSCI.2804-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters JR, Hu D, Itoga CA, Parr-Brownlie LC, Bergstrom DA (2007) Phase relationships support a role for coordinated activity in the indirect pathway in organizing slow oscillations in basal ganglia output after loss of dopamine. Neuroscience 144:762–776. 10.1016/j.neuroscience.2006.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ (2013) Active decorrelation in the basal ganglia. Neuroscience 250:467–482. 10.1016/j.neuroscience.2013.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zold CL, Escande MV, Pomata PE, Riquelme LA, Murer MG (2012) Striatal NMDA receptors gate cortico-pallidal synchronization in a rat model of Parkinson's disease. Neurobiol Dis 47:38–48. 10.1016/j.nbd.2012.03.022 [DOI] [PubMed] [Google Scholar]