

Abstract
Objective
A major challenge in multiple sclerosis (MS) research is the understanding of silent progression and Progressive MS. Using a novel method to accurately capture upper cervical cord area from legacy brain MRI scans we aimed to study the role of spinal cord and brain atrophy for silent progression and conversion to secondary progressive disease (SPMS).
Methods
From a single‐center observational study, all RRMS (n = 360) and SPMS (n = 47) patients and 80 matched controls were evaluated. RRMS patient subsets who converted to SPMS (n = 54) or silently progressed (n = 159), respectively, during the 12‐year observation period were compared to clinically matched RRMS patients remaining RRMS (n = 54) or stable (n = 147), respectively. From brain MRI, we assessed the value of brain and spinal cord measures to predict silent progression and SPMS conversion.
Results
Patients who developed SPMS showed faster cord atrophy rates (−2.19%/yr) at least 4 years before conversion compared to their RRMS matches (−0.88%/yr, p < 0.001). Spinal cord atrophy rates decelerated after conversion (−1.63%/yr, p = 0.010) towards those of SPMS patients from study entry (−1.04%). Each 1% faster spinal cord atrophy rate was associated with 69% (p < 0.0001) and 53% (p < 0.0001) shorter time to silent progression and SPMS conversion, respectively.
Interpretation
Silent progression and conversion to secondary progressive disease are predominantly related to cervical cord atrophy. This atrophy is often present from the earliest disease stages and predicts the speed of silent progression and conversion to Progressive MS. Diagnosis of SPMS is rather a late recognition of this neurodegenerative process than a distinct disease phase. ANN NEUROL 2022;91:268–281
Since its first description more than 150 years ago, neurologists have struggled to predict the onset of progressive disease, a key milestone determining clinical prognosis in relapsing remitting MS (RRMS). 1 The term “progression” in relapse‐onset MS has traditionally been reserved for the second disease phase called secondary progressive MS (SPMS) where inflammation‐related relapses cease, 2 and neurodegenerative processes lead to continuous disability accumulation. However, recently an insidious relapse‐free disability worsening termed silent progression was reported early in the relapsing phase in a subset of patients suggesting that clinically relevant widespread neurodegeneration occurs from early disease stages. 3 , 4 , 5 To date, the pathophysiological and structural correlates underlying this process remain elusive.
As such, brain and cord atrophy from MRI volume changes might be useful correlates of disability worsening and progressive disease. 6 , 7 Of all brain measures, deep gray matter structures show the strongest associations with disability. 6 , 8 When compared with brain measures, however, spinal cord measures have shown the most robust correlates with disability measured by EDSS. 6 , 7 , 9 Due to cost and technical challenges of spinal cord sequences, long‐term data on spinal cord atrophy are currently limited. Recently, methods were developed to measure the most cranial portion of the cervical cord from brain images. 10 Here we advanced previous normalization methods to reduce the variability in cord area measurements using the foramen magnum area. 11 , 12 This innovation enabled us to accurately assess the upper cervical cord area at C1 vertebral level (C1A) from brain scans acquired from a large prospective longitudinal cohort of well‐characterized MS patients.
First, we aimed to study the prognostic value of the currently most promising radiographic brain and spinal cord measures for the conversion to a progressive course. To achieve this, we analyzed the subset of 54 patients (RR → SP) who converted to secondary progressive MS (SPMS) over the 12‐year follow‐up and compared them to 54 matched individuals who had similar baseline characteristics but remained RRMS (RR → RR) during that period. Second, we assessed the prognostic value of these quantitative MRI measures for silent progression, i.e., relapse‐free EDSS worsening. For this purpose, we utilized data from the entire study cohort and stratified all patients who remained RRMS based on whether they remained stable or silently progressed during the 12‐year observation period. In contrast to the previously published definition of silent progression requiring the absence of relapse activity, 3 here we allow inclusion of patients with relapse activity while retaining the requirement of EDSS worsening that is independent from relapses. The definition of silent progression used here derives from the hypothesis that there is clinically silent neurodegeneration (measurable as CNS atrophy on MRI) that precedes and predicts relapse free EDSS worsening, and in some cases eventually an SPMS course, and that this neurodegeneration occurs in patients with and without relapses. This approach builds on previous findings that the presence of relapses does not affect long‐term disability worsening. 3
However, to exclude bias from the dichotomization of the RRMS/SPMS classification, in a third analysis we assessed the prognostic value of spinal cord atrophy for silent progression using the same patient group assignments from the above‐mentioned study 3 on silent progression that included both, RRMS and SPMS patients.
Methods
Study Design and Participants
From an on‐going prospective study on the phenotypic‐genotypic characterization of MS (EPIC: Expression, Proteomics, Imaging, Clinical) at the UCSF MS Center, 13 all patients recruited between 07/2004 and 09/2005 with a diagnosis of clinically isolated syndrome (CIS), RRMS or SPMS at baseline and a diagnosis of RRMS or SPMS at 12‐year follow‐up (n = 484) were screened for this study (Fig 1). Patients with primary progressive MS were excluded from this study. Eighty age‐ and sex‐matched healthy controls (controls) were examined at study baseline and 32 were followed longitudinally under the same MRI protocol as the MS patients.
FIGURE 1.
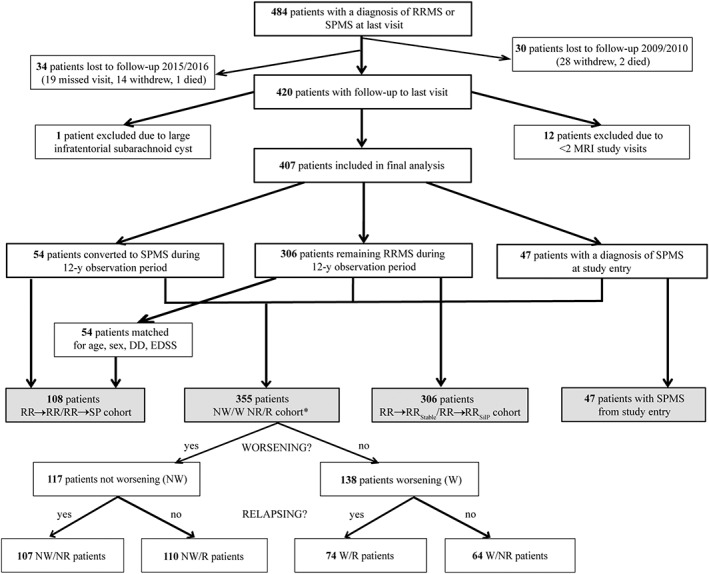
Graphical illustration of the study population and patient subsets. RR → RR = matched patients remaining relapsing remitting multiple sclerosis during the 12‐year observation period. RR → SP = patients who converted to secondary progressive multiple sclerosis during the 12‐year observation period. RR → RRSilP = patients who developed silent progression but retained a clinical diagnosis of RRMS to study end. RR → RRStable = patients who remained stable RRMS to study end. RRMS = relapsing remitting multiple sclerosis. SPMS = secondary progressive multiple sclerosis. NW/W NR/R cohort = silent progression groups as defined by Cree et al. 3 classifying RRMS and SPMS patients solely based on confirmed EDSS worsening (worsening/non‐worsening), i.e., silent progression, and disease activity (relapsing/non‐relapsing), thereby avoiding the dichotomy of the current RRMS/SPMS classification: NR = non‐relapsing. NW = non‐worsening, i.e., stable. R = relapsing. W = worsening.
54/360 patients with CIS (n = 5) or RRMS (n = 49) were diagnosed with SPMS (RR → SP) during the 12‐year observation period by the treating physician. For study purposes, two independent investigators (BACC, CJB) ascertained the treating physician's diagnosis of conversion to SPMS based on expert consensus and supported by a recently validated definition 14 : onset of irreversible disability worsening measured by increased EDSS, 15 confirmed over 12 months, and independent from relapses. 14 EDSS worsening was defined based on 3‐strata: an increase in EDSS by 1.5, if the EDSS was 0, an increase by 1.0, if the EDSS was 1.0–5.0, and an increase by 0.5, if the EDSS was 5.5 or higher. 3 , 13 The 54 RR → SP patients were matched at baseline to 54 controls and 54 patients who remained RRMS (RR → RR) for age, sex, disease duration (time from symptom onset) and EDSS. We performed a stepwise increase of ±1 (maximum: 6) year in difference to the RR → SP patient's age and disease duration if a matching RR → RR patient with the exact same parameters could not be found. Likewise, we aimed at matching the two groups for EDSS (Expanded Disability Status Scale); when no matching RR → RR patient within the range of 0.5 points was available, the search was repeated with an EDSS difference increasing stepwise by ±0.5 points per search between the RR → SP patient and the match. To ensure that RR → RR patients were labelled correctly we included only those followed at our center up to present. For the second aim of this study, we stratified all patients in the EPIC cohort who remained RRMS based on whether they showed irreversible disability worsening as defined above (RR → RRSilP) or remained stable (RR → RRStable). Lastly, to assess a potential bias resulting from the dichotomization into RRMS/SPMS, we performed a sensitivity analysis by comparing C1A rates using the same group assignments from a recently published study on silent progression by our group 3 where RRMS and SPMS patients were classified into four groups based on whether they showed disability worsening or not (W/NW) and whether they had relapses or not (R/NR) over a 5‐year observation period and confirmed at 10‐year follow‐up.
Disease‐modifying treatments were prescribed at the discretion of the primary neurologist. To evaluate the potential bias from treatment differences on clinical course and MRI metrics, we extracted the disease‐modifying treatments administered during the study period and categorized them into three tiers according to treatment efficacy reported from clinical trials: low (interferon‐beta, glatiramer acetate, teriflunomide, minocycline, mycophenolate mofetil, 16 methotrexate 17 ), intermediate (dimethyl fumarate, fingolimod), and high (natalizumab, rituximab, mitoxantrone, 18 cyclophosphamide 19 ).
The research protocol was approved by the Committee on Human Research at UCSF and informed consent was obtained by all participants prior to study enrolment.
Procedures
All subjects were scanned on the same 3T Signa scanner (GE) from study inception that was changed to a 3T Skyra scanner (Siemens) 9 years after study initiation (Table S1). 13 , 20 In total, 3,276 patient MRI scans were acquired including 823 scans for the matched RR → RR/RR → SP subset, 479/823 scans were acquired up to and including the year of conversion. 52/54 patients had two or more post‐conversion follow‐up timepoints. The median time interval between MRI acquisitions was 1.0 (IQR 0.6) years. All MRI analyses were performed blinded to clinical data. At each timepoint, the same high‐resolution T1‐weighted sequence was used for both, brain and spinal cord atrophy measurements.
C1A Measurements
All C1A and foramen magnum area (FMA) measurements were performed by two independent raters, respectively. C1A estimates were obtained using the semi‐automatic segmentation method implemented in JIM7 software 11 , 21 that provides high intra‐ and inter‐rater reliability and between‐scanner robustness. 22 , 23 We measured C1A on five consecutive axial slices similar to a method reported recently 10 (Fig 2A) using the obex to determine the measurement level as it is close to the structure of interest. To ensure consistent alignment between patients and over time, images were (1) reconstructed and oriented perpendicular to the long axis of the upper cervical cord, and (2) rotated in the sagittal plane around the center of the cervical cord at the C1A measurement level to correct for differences in angulation from head flexion/extension whereby the angulation with the smallest resulting C1A was used for the final analysis. We imputed missing slices from incomplete caudal coverage using linear mixed‐effects models taking into account the available slices from the respective timepoint and the intra‐individual anatomical shape of this region across all timepoints that we assumed to be stable over time.
FIGURE 2.
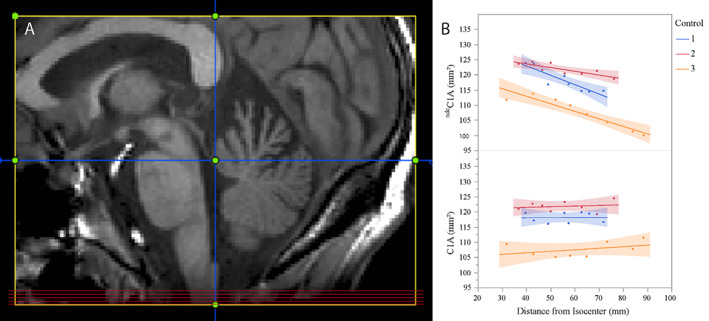
C1A measurement level and effect of foramen magnum normalization on C1A measures. (A) Axial images for C1A measurement (red horizontal lines) were obtained by reformatting the sagittal T1‐weighted image perpendicular to the dorsal surface of the cervical cord in the midline plane (vertical blue line) and selecting five contiguous slices 8–12mm caudal to the obex (horizontal blue line, slice thickness 1mm). (B) C1A measures of three controls (Control 1–3) before (ndcC1A, upper graph) and after (C1A, lower graph) normalization by the foramen magnum area (FMA), depicted as a function of distance of the center C1A slice from the scanner isocenter: Control 1: C1A 118.1 mm2, standard deviation (SD) 3.9 before and 1.7 after normalization; Control 2: C1A 121.8 mm2, SD 2.0/1.7 before/after normalization; Control 3: C1A 107.6 mm2, SD 5.2/2.5 before/after normalization. Note decreasing ndcC1A values with increasing distance from isocenter if correction by FMA is not performed, potentially leading to apparent cord volume changes in longitudinal studies.
In total we imputed 713 of 16,380 (4.4%) slices in the entire cohort (4.8 and 5.3% in the RR → RR and RR → SP patients respectively), which is within the missing rate considered acceptable for valid statistical inferences. 24
Normalization of C1A Measurements
In this study we applied two distinct normalization strategies: (1) normalization for head size using the SIENAX‐derived volume scaling factor (V‐scale) to correct for inter‐individual differences, 21 and (2) normalization by foramen magnum area (FMA) to correct for gradient nonlinearity distortions and other scanner/protocol related differences. Gradient nonlinearities impact upper cervical cord areas from brain images due to their location in the periphery of the field‐of‐view. We recently reported a method to retrospectively reduce gradient non‐linearity effects that explained up to 12% of the variability in upper cervical cord area (UCCA) measurements from brain T1‐weighted images at C2‐3 vertebral level by normalization to adjacent bony structures. 11 To normalize the UCCA as a function of the C2/3‐disc distance from the scanner isocenter ZCI, we previously used a measure of the vertebral body area close to the spinal cord at the same level. 11 The product Vert(ZCI) of the anterior–posterior and right–left C2 vertebral body diameters was calculated using the following equation:
| (1) |
where UCCA(ZCI)′ indicates the normalized and UCCA(ZCI) the uncorrected value, and Vert (ZCImin) the value Vert(ZCI) measured in the acquisition with the table positioned closest to the scanner isocenter ZCImin.
Here, since no vertebra is present at the C1A level, we replicated the method using the foramen magnum as the normalizing bony structure (for clarity, we refer to the raw non‐distortion corrected C1A as ndcC1A and to the foramen magnum corrected C1A as C1A). Instead of multiplying for the position closer to the isocenter as performed in equation (1) above, 11 in the present study we multiplied ndcC1A/FMA by the average value across all timepoints for each individual. Thereby, the inter‐subject variability of the FMA is removed and only the effects of gradient nonlinearity/positioning and scanner/protocol effects over time are corrected:
| (2) |
Importantly, recent research indicates that the foramen magnum remains stable in size throughout adulthood. To validate the foramen magnum normalization, we analyzed a dataset of three controls that was specifically acquired to examine the effects of gradient nonlinearities. Three controls were scanned at eight different head positions with the scanner‐implemented distortion correction algorithm switched off. The distance from isocenter was retrieved as the Z‐position of the center ndcC1A slice. Normalization by the FMA reduced the variability in the ndcC1A measurement in all three controls (Fig 2B).
Correlation of C1A with Brain and Cord Measures
A recent study demonstrated that cord area measured at C1 vertebral level correlates better with upper cervical cord area measured at the standard C2/3 vertebral level (UCCAC2/3) than with brain measures. 10 To replicate this finding in our dataset we analyzed brain volumes, C1A and UCCAC2/3 as described previously 25 in a subset of 90 randomly selected patients on the same T1‐weighted MPRAGE image that was used for C1A measurements.
Brain Volumetry
Brain T2/FLAIR and T1 lesion masks were created using LST:Lesion Segmentation Tool 26 and in‐house semi‐automatic segmentation pipelines using a mixture model brain lesion segmentation algorithm and T1 lesion contouring (TLC) algorithm based on symmetric diffeomorphic image registration (SyN, part of the Advanced Normalization Tools, ANTs package 27 ; https://github.com/zxh2135645/TLC/releases). Two independent investigators, a neurologist (AB) and a neuroradiologist (SS), edited the lesion masks. SIENAX with lesion mask inputs and optiBET were used to estimate brain gray, cortical gray, white matter and lateral ventricular volumes, and baseline whole brain volume. 28 , 29 SIENA was used to estimate annual whole brain volume change. 28 Thalamus volumes were estimated using FIRST with lesion mask inputs which was previously shown to perform similar to the longitudinal FreeSurfer algorithm on our dataset. 30 , 31 , 32
MRI measures were normalized by subject head size using the SIENAX‐derived volumetric scaling factor to minimize inter‐subject variability. Brain volumes were calibrated to account for differences in imaging technique over the 12‐year observation period. 33
Statistical Analysis
Continuous variables were transformed to normal quantiles. MRI measures were log‐transformed to directly estimate annual percentage change from the slope over time. Visual inspection of residuals and model predictions demonstrated homoscedasticity of all data. Atrophy rates were calculated using multivariable linear mixed‐effects models that are well suited for datasets with variable numbers of scans between subjects which is of particular importance as time to conversion/silent progression varied between two and 12 years. MRI volumes were set as the response variable while subject intercepts and slopes over time were treated as random effects. We entered potentially confounding variables as fixed effects including demographic (age, sex) and clinical characteristics (disease duration, EDSS, disease‐modifying treatment, number of patients with spinal cord onset) and inflammatory activity (annualized relapse rate, spinal cord relapses, new/enlarging brain lesions). Lognormal accelerated failure time (AFT) models were used to evaluate associations of clinical and MRI variables (baseline volumes and longitudinal changes of whole brain, white, grey, cortical grey matter, lateral ventricular, thalamus, T1 and T2 lesions) with time to silent progression and conversion, respectively. Raw atrophy rates were recalculated from baseline to silent progression and conversion, respectively. The proportional hazards assumption was violated based on Schoenfeld residuals. Akaike Information Criterion was used to select lognormal as the most appropriate distribution for the AFT model. Variable selection was performed using LASSO (least absolute shrinkage and selection operator) regression analyses. 34
To assess generalizability and validity of our novel C1A measurement methodology we applied the same Cox model (disease progression, time to disease progression ~ disease type + baseline EDSS + baseline spinal cord volume + annual spinal cord volume rate + baseline whole brain volume) reported from an independent European MS cohort study 35 to a comparable subset of the EPIC cohort including RRMS and SPMS patients (n = 345, mean follow‐up time: 5.9 (SD 1.3) years, mean number of follow‐ups: 5.1 (SD 1.4)). The reader is referred to the Supplement for further details.
Statistical analyses were performed using R survival, flexsurv and survreg packages (https://www.R-project.org/, v4.0.0) and JMP Statistics (www.jmp.com, v14.3.0, SAS Institute, Cary, NC) software. Significance levels were set to p < 0.05. We applied the false discovery rate correction for multiple comparisons. 36
Results
Study Population
Four hundred and eighty‐four patients completed the follow‐up in 2009/2010 and 454 patients in 2015/2016 (Fig 1). 3 Two or more timepoints were available in 408/419 patients. One RRMS patient was excluded from further analyses as spinal cord analyses could not be performed due to a large arachnoid cyst deforming brainstem and cord. Thirty‐nine scans were excluded due to acquisition or segmentation errors.
Validation of Intra‐ and Interrater Reliability of C1A and Foramen Magnum Measurements
Intra‐ and inter‐rater reproducibility for C1A and FMA measurements was assessed in 20 controls using intra‐class correlation coefficients (ICC). 37 Specifically, for intra‐rater reproducibility one rater (AB) performed the measurements three times for each subject. For inter‐rater reproducibility, measurements were performed once by two different raters (C1A: JMM/AB, FMA: CA/AB). ICC for intra‐rater reliability of C1A and foramen magnum measurements were 1.000 and 0.999, respectively. ICC for inter‐rater reliability C1A and foramen magnum measurements were 0.999 and 0.966, respectively.
Correlations of C1A Measurements with Brain and UCCAC2 /3 Measures
Correlation of C1A with brain volumes and UCCAC2/3 demonstrated that C1A correlates better with spinal cord area at C2/3 level than with brain volumes. Pearson's Correlation Coefficients were 0.75 with UCCAC2/3 (p < 0.001), 0.52 with WBV (p < 0.001), 0.57 with WMV (p < 0.001), 0.41 with GMV (p < 0.001), and 0.42 with cGMV (p < 0.001).
Validation of C1A Calibration across Different Scanners
To demonstrate calibration effects of the foramen magnum normalization for our current study we compared the overall log(C1A) in a patient subset (n = 295) that was scanned at least twice at both scanners during the observation period. We found that the C1A atrophy rates are −0.63 (95% CI: −0.91 to – 0.35) %/yr at the Signa (GE) scanner at the beginning of the study and −0.58 (95% CI: −0.74 to −0.42) %/yr at the Skyra (Siemens) scanner at the end of the study, consistent with our observation of decreasing rates over time. The intercepts of the model fits for the two scanners were identical (4.56, 95% CI of the intercept: 4.55 to 4.57 for the Signa scanner and 4.56, 95% CI of the intercept: 4.54 to 4.58 for the Skyra scanner), confirming the absence of bias for the normalized C1A across the scanner change.
Patients
Demographic, clinical and MRI characteristics of the matched RR → RR and RR → SP, RR → RRStable and RR → RRSilP and SPMS groups are shown in Table S6. RR → RR and RR → SP patients had similar age, sex and disease duration by design. However, RR → SP patients had higher baseline EDSS than RR → RR patients. A subset of the matched pairs (n = 90) with matched baseline EDSS was identified (median EDSS 2, IQR (interquartile range) 1.5 in both groups, p = 0.057). The RR → RRStable and RR → RRSilP patients were incidentally of similar age, sex and disease duration; these subgroups were earlier in the disease (median disease duration = 5 and 6 years, respectively) compared to the RR → RR and RR → SP groups. 159/306 RRMS patients developed silent progression during the observation period. Interestingly, RR → RRSilP patients had a lower EDSS at study entry than those who remained stable. SPMS patients from study entry demonstrated advanced disease with high baseline disability levels (median EDSS = 5.0) and the longest disease duration (17 years) of all patient groups.
Cervical Cord Atrophy before Conversion
At study entry, C1A was reduced compared to controls but similar between the two groups (p = 0.296, Table S7, Table S3). However, over time the C1A during the pre‐conversion period (median 6.0, IQR 5.7 years) declined at a rate of −2.19%/yr in RR → SP patients but at a slower rate of −0.88%/yr in RR → RR patients (mean difference − 1.30%/yr, −1.84 to −0.80, p < 0.001, Fig 3A, Table S7). Importantly, this difference remained significant when increasing the time interval up to 4 years before conversion in the patient subset with at least five consecutive annual scans available for this period (Table 1). Mixed‐effects models yielded similar results (mean difference − 1.30%/yr, −1.87 to −0.72, p < 0.001) when analyzing the subset (n = 90) of matched RR → RR (−0.93%/yr) and RR → SP (−2.23%/yr) patients with no differences in baseline EDSS (Fig 3B). Likewise, mixed‐effects models yielded similar results (mean difference −1.22%/yr, −1.88 to −0.56, p < 0.001) when analyzing a subset (n = 54) including only RR → SP patients (and their RR → RR matches) with a baseline EDSS≤2.0 (Fig 3C). Differences in C1A atrophy rates between RR → RR and RR → SP patients during the pre‐conversion period remained unchanged when excluding patients with focal white matter lesions at C1A measurement level (Table 1). Results were similar when comparing pre‐conversion C1A atrophy rates of RR → SP (−1.86%/yr) to all RRMS patients (n = 297) excluding the matched RR → RR group (−0.57%/yr, mean difference 1.29%/yr, 0.92 to 1.65, p < 0.001). Interestingly, the C1A atrophy rate decelerated after conversion from −2.24%/yr to −1.63%/yr towards that of patients with SPMS from study entry (−1.04%/yr, Table S7).
FIGURE 3.
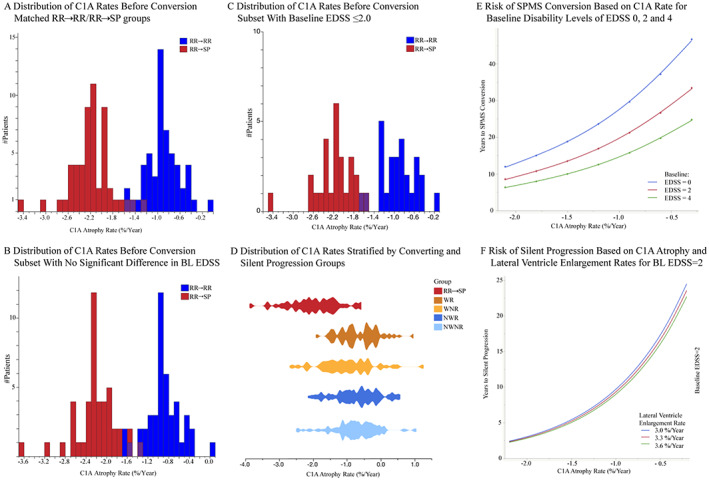
C1A atrophy rates and risk for conversion to SPMS and silent progression. Distribution of estimated annual C1A atrophy rates before conversion for the RR → RR and RR → SP groups (A, n = 108) and in the subsets with no significant difference in baseline EDSS (B, n = 90) and with baseline EDSS≤2.0 (C, n = 54); (D) Comparison of C1A rates over the first 5 study years between RR → SP and silent progression groups stratified by worsening (W)/non‐worsening (NW) and relapsing (R)/non‐relapsing (NR), modified from Cree et al. 3 by extracting the group who converted to SPMS; (E/F) Risk of SPMS Conversion/Silent Progression: Time to SPMS conversion based on C1A atrophy rates (%/yr) for patients with an EDSS of 0 (blue), 2 (red) and 4 (green) at baseline. (F) Time to silent progression based on C1A atrophy (%/yr) and lateral ventricle enlargement rates (%/yr) for patients with a baseline EDSS of 2. BL = baseline. C1A = cervical cord area at C1 vertebral level; EDSS = expanded disability status scale; RR → RR = matched patients remaining relapsing remitting multiple sclerosis during the 12‐year observation period; RR → SP = patients who converted to secondary progressive multiple sclerosis during the 12‐year observation period; SPMS = secondary progressive multiple sclerosis.
TABLE 1.
Annualized cervical cord atrophy rates of patient subsets before conversion/silent progression
| Patient Subset | RR→RR | RR→SP | Diff (95% CI) | p |
|---|---|---|---|---|
| RR→RR/RR→SP | ||||
| Subset without C1 Lesions (n = 60) | −1.04 | −2.28 | −1.24 (−1.77; −0.70) | <0001 |
| By time interval before conversion | ||||
| 0 yr (334 Visits) | −0.89 | −2.14 | −1.25 (−1.80; −0.70) | <0.001 |
| −1 yr (304 Visits) | −0.85 | −2.18 | −1.33 (−2.00; −0.67) | <0.001 |
| −2 yr (281 Visits) | −0.73 | −2.21 | −1.48 (−2.23; −0.72) | <0.001 |
| −3 yr (253 Visits) | −0.72 | −2.17 | −1.45 (2.40; −0.49) | 0.004 |
| −4 yr (219 Visits) | −0.73 | −2.23 | −1.50 (−2.69; −0.32) | 0.015 |
| Relative to conversion | Before | After | Diff (95% CI) | p |
| RR→SP group | −2.24 | −1.63 | 0.61 (0.16; 1.05) | 0.010 |
| Minimum observational time needed to detect difference in atrophy rates | ||||
| Scans before conversion | RR→RR | RR→SP | Diff (95% CI) | p |
| 2 (216 visits) | −1.31 | −2.33 | −1.02 (−2.28; 0.25) | 0.103 |
| 3 (300 visits) | −1.14 | −2.29 | −1.15 (−1.92; −0.39) | 0.004 |
| 4 (372 visits) | −1.20 | −2.30 | −1.10 (−1.69; −0.51) | <0.001 |
| 5 (420 visits) | −1.11 | −2.36 | −1.25 (−1.75; −0.75) | <0.001 |
| Silent progression groups as defined in Cree et al. 3 | ||||
| Worsening | Atrophy Rate † | vs.* | Diff (95% CI) | p |
| Non‐relapsing (W/NR) | −1.10 | NW/NR | 0.38 (0.15; 0.60) | 0.004 |
| Relapsing (W/R) | −0.89 | W/NR | −0.21 (−0.46; 0.02) | 0.112 |
| Stable | ||||
| Non‐relapsing (NW/NR) | −0.72 | W/R | −0.17 (−0.38;0.05) | 0.137 |
| Relapsing (NW/R) | −0.80 | W/NR | −0.30 (−0.52; −0.07) | 0.020 |
Subsets: Subset without C1 lesions = subset of patients without focal white matter lesions at the C1A measurement level; by time interval before conversion (−4 to 0 yr): patient subset with scans available for all intervals listed; relative to conversion: comparison of atrophy rate before and after conversion to SPMS; minimum observational time needed to detect difference in atrophy rates: minimum observational time, ie, number of scans, to detect a meaningful difference in atrophy rate between RR→RR and RR→SP groups; of note, the difference between rates is detectable within 1 yr of follow up and reaching statistical significance within 2 yr of follow up; silent progression groups as defined by Cree et al. 3 classifying RRMS and SPMS patients solely based on confirmed EDSS worsening (worsening/non‐worsening), ie, silent progression, and disease activity (relapsing/non‐relapsing), thereby avoiding the dichotomy of the current RRMS/SPMS classification. Atrophy rates are annual percentage change. p‐values are corrected for multiple comparisons. *Between group comparison between the two groups mentioned in the respective row. Comparisons not mentioned in the table: NW/NR vs. NW/R: −0.08 (95% CI: −0.28; 0.11), p = 0.416; NW/R vs. W/R: −0.09 (95% CI: −0.30; 0.13), p = 0.448. C1A rates were calculated based on mixed‐effects models, adjusted for age, sex and disease duration.
C1A = cervical cord area at C1 vertebral level; Control = healthy control; Diff (95% CI) = mean difference (lower and upper 95% confidence interval) between the respective patient groups; NR = non‐relapsing; NW = non‐worsening, ie, stable; R = relapsing; RR→RR = patients remaining relapsing remitting multiple sclerosis during the 12‐yr observation period; RR→SP = patients converting to secondary progressive multiple sclerosis during the 12‐yr observation period; W = worsening.
We did not detect significant effects of sex (female: 0.06%/yr, −0.21 to 0.33, p = 0.651), baseline EDSS (−0.16%/yr, −0.50 to 0.17, p = 0.334), disease‐modifying treatments (−0.09%/yr, −0.37 to 0.19, p = 0.538), annualized relapse rate (−0.59%/yr, −1.77 to 0.59, p = 0.321), spinal cord relapses (−0.58%/yr, −5.70 to 6.85, p = 0.856), spinal cord onset (−0.28%/yr, −0.26 to 0.83, p = 0.298) or new or enlarging lesions (−0.11%/yr, −0.10 to 0.32, p = 0.307) on C1A atrophy rates during the pre‐conversion period. To demonstrate that the different number of timepoints available per patient does not influence the C1A atrophy rates reported in our main analysis we conducted an additional subgroup analysis where we selected those patients who had at least five consecutive annual scans before conversion. We found 48 matched patients (240 scans) who fulfilled these criteria. The C1A atrophy rate in the RR → RR group was −0.89%/yr (95% CI −1.72 to −0.05) and −2.12%/yr (95% CI −3.03 to −1.22) in the RR → SP group, with a mean difference between the groups of −1.23%/yr (95% CI −1.89 to −0.57, p < 0.001), similar to our main result (Table 1).
C1A atrophy rate was the strongest predictor among all MRI measures using survival analyses, with each 1% faster C1A atrophy rate being associated with a 53% shorter time to SPMS conversion (p < 0.0001, Table 2, Fig 3E).
TABLE 2.
Risk of silent progression and SPMS conversion
| Clinical/MRI measures | Time to event | 95%CI time to event | %Change time to event | p |
|---|---|---|---|---|
| Silent progression over 12 yr (n = 306) | ||||
| C1A atrophy rate | 3.19 | 2.31; 4.39 | 69% | <0.001 |
| Baseline EDSS | 1.24 | 1.13; 1.35 | 19% | <0.001 |
| Lateral ventricular rates | 0.87 | 0.78; 0.96 | −16% | 0.007 |
| Loglik (Model): 1035; χ2 = 85.62, p < 0.001; Number of Events: 159/306. | ||||
| SPMS Conversion over 12 yr (Matched Subset, n = 108) | ||||
| Baseline EDSS | 0.86 | 0.76; 0.97 | −17% | 0.013 |
| C1A Atrophy Rate | 2.13 | 1.71; 2.65 | 53% | <0.001 |
| Loglik (Model): 321.0; χ2 = 85.18, p < 0.001; Number of Events: 54/108. | ||||
Disability was measured by EDSS where higher EDSS scores correspond to higher disability, range 0 (no disability) to 10 (death from multiple sclerosis). The final models for the risk of silent progression (time to progression, censor) ~ Baseline EDSS + C1A Atrophy Rate + Lateral Ventricular Atrophy Rate) and SPMS conversion ((time to conversion, censor) ~ C1A atrophy rate + Baseline EDSS) were based on accelerated failure time models with an underlying lognormal distribution using Wald Tests, where “censor” refers to whether the patient reached the event of interest (silent progression, SPMS conversion) during the observation period or not. Time to event = time to silent progression/SPMS conversion. %Change time to event = percentage change in time to event with every 1% increase in the measure for those who silently progress/convert. Please note that for atrophy rates, an increase in the rate, i.e., a more positive value, corresponds to a deceleration, ie, slowing down of the atrophy rate.
95% CI = lower and upper limits of the Wald 95% confidence interval of the time to event (silent progression/SPMS conversion); C1A = cervical cord area at C1 vertebral level; EDSS = expanded disability status scale; Loglik = logistic maximum likelihood estimate; SE = standard error; SPMS = secondary progressive multiple sclerosis.
Brain Atrophy before Conversion
By contrast, least squares regression models showed decreased brain volumes in patients compared to controls at baseline, but no association with future development of a progressive disease course (Fig 4). Of all examined brain structures, only the thalamus demonstrated faster atrophy rates in RR → SP compared to RR → RR patients (Table S7), but differences were less pronounced than for C1A rates. Global and regional brain measures were not significant during the model selection process for the prediction of SPMS conversion and were excluded from the final survival analysis.
FIGURE 4.
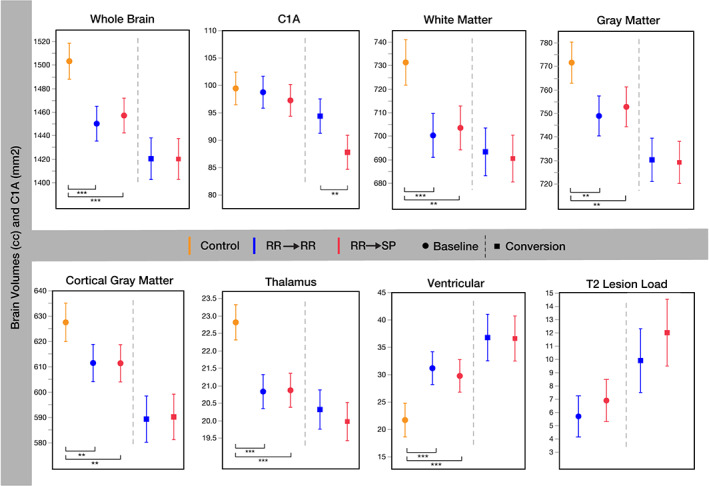
Comparison of Brain Volumes and C1A between the Matched Control, RR→RR and RR→SP Groups at Baseline and Conversion. C1A = cervical cord area at C1 vertebral level (mm2); Control = healthy control; RR → RR = patients remaining relapsing remitting multiple sclerosis during the 12‐year observation period; RR → SP = patients who converted to secondary progressive multiple sclerosis during the 12‐yr observation period. Baseline volumes are cc (least squares mean), whiskers indicate the 95% lower and upper confidence interval. *0.005 < p ≤ 0.05. **0.0001 < p ≤ 0.005. ***p ≤ 0.0001.
Cervical Cord Atrophy before Silent Progression
C1A was similar at study entry in RR → RRSilP compared to RR → RRStable patients (p = 0.269, Table S7). During the period before silent progression C1A atrophy rates were slower in the RR → RRStable group (−0.54%/yr) compared to RR → RRSilP patients (−0.84%/yr) but the difference did not reach significance (−0.30%/yr, p = 0.110). Again, there were no significant effects of sex (female: 0.04%/yr, −0.22 to 0.31, p = 0.752), disease duration (−0.02%/yr, −0.16 to 0.13, p = 0.819), baseline EDSS (−0.05%/yr, −0.21 to 0.11, p = 0.520), disease‐modifying treatments (−0.10%/yr, −0.23 to 0.30, p = 0.133) or annualized relapse rate (0.23%/yr, −0.47 to 0.92, p = 0.516) on C1A atrophy rates before silent progression. C1A atrophy rates were faster in patients with silent progression (W/NR) compared to those who remained stable (NW/R, p = 0.020, NW/NR, p = 0.004), thereby corroborating our results (Table 1, Fig 3D). However, this is a somewhat artificial separation since subjects with long time to silent progression are grouped with those with a short time to silent progression. The survival analyses account for this difference.
Using AFT models, C1A atrophy rate was the strongest MRI predictor, with each 1% faster C1A atrophy rate resulting in a 69% shorter time to silent progression (p < 0.0001, Table 2, Fig 3F).
Brain Atrophy before Silent Progression
Of all brain measures, lateral ventricular enlargement showed the greatest difference (0.58% increase/yr) between the two groups (RR → RRStable: 3.12%/yr, RR → RRSilP: 3.70%/yr) though this difference did not reach statistical significance (p = 0.089 before multiple comparison correction). However, in survival analyses ventricular enlargement was the second strongest MRI and the strongest brain measure to predict silent progression with each 1% enlargement of the lateral ventricles being associated with a 16% shorter time to silent progression (Table 2, Fig 3F).
Discussion
This is the first study to demonstrate a prognostic relationship of MRI for conversion to an SP course. We found that spinal cord atrophy rates during the relapsing phase were markedly faster in patients who later converted to SPMS compared to those who did not, with strikingly little overlap and detectable up to 4 years before conversion to SPMS. Among all studied brain and cord measures, the C1A atrophy rate was the strongest predictor of both impending conversion and silent progression with 53 and 69% shorter time to these events, respectively, for each 1% increase (ie, faster) in atrophy rate. Thus, it appears that both types of progression share a common pathological substrate. C1A atrophy rates in patients with silent progression were faster even when combining RRMS and SPMS patients. These findings question the current dichotomic classification of RRMS and SPMS suggesting that MRI biomarkers including the C1A atrophy rate could be useful in RRMS to classify and stratify patients for therapeutic decisions early in the disease. Importantly, the cervical cord is depicted on sagittal T1‐weighted images as part of many state‐of‐the‐art MS protocols 38 and can now be measured by fully automated algorithms. 35 , 39 With improved precision, eg, by the acquisition of dedicated spinal cord images, these advances can facilitate the implementation of C1A measures as a biomarker in the clinical setting to eventually enable precision medicine.
C1A atrophy rates decelerated after conversion (−2.24/yr to −1.63%/yr) towards those of SPMS patients from study entry (−1.04%/yr). To date, little is known regarding the dynamics of CNS atrophy rates across disease stages in MS. Our finding suggests that spinal cord atrophy is faster during the relapsing phase in SPMS patients. By contrast, clinical worsening does not become obvious before a certain threshold of tissue loss has been reached, leading to the clinical recognition of SPMS conversion. Another contribution to volume loss might be related to edema that has been postulated to be present in the inflamed spinal cord of RRMS patients. 40 , 41 Along with the gradual decline of acute inflammatory processes, resolution of edema may contribute to the fast cord atrophy rates around the time of conversion while atrophy rates decelerate after conversion once edema has resolved. 2 Furthermore, the observed slowing of cord volume loss is remarkably consistent with the post‐mortem observation of similar spinal cord area reduction (~20%) in SPMS patients after a disease duration of 29 years. 42 Compared to controls (99.4 mm2), we found a reduction of 11% in our SPMS patients at baseline (88.7 mm2), ie, after 17 years of disease duration. Assuming a 1% loss per year as estimated here in SPMS, this would lead to a 22% cord area loss at 29 years of disease duration, which is very comparable to the post‐mortem data. Intriguingly, around the time of conversion (17 years since disease onset) the RR‐ > SP patients also showed a 10% reduction in cord area (89.1 mm2).
Contrary to recent work demonstrating an association between the T2 lesion load expansion and disability accumulation based on a single measurement or short‐term observations 43 , 44 we did not find such an association for the development of secondary or silent progression in our long‐term study. This is in line with recently reported findings that EDSS worsening studied over a long‐term observational period in RRMS and SPMS patients was independent of T2 lesion load accumulation whereas inflammatory disease activity, ie, relapses, did contribute to short‐term disability accumulation. 3
As expected from previous studies we found marked thalamic and whole brain atrophy at study entry but surprisingly no differences between RR → RR and RR → SP patients. 45 , 46 Though thalamic atrophy rates were faster before SPMS conversion compared to RR → RR patients, thalamic volumes remained similar between the groups around the time of conversion. This finding is in line with a recent study suggesting that thalamic atrophy plays a minor role for the conversion to SPMS. 47 Of note, the stringent definition for EDSS worsening requiring confirmation of worsening and excluding any disability resulting from relapses corroborates the notion that spinal cord atrophy is related to neurodegeneration. Interestingly, lateral ventricular enlargement emerged as an MRI predictor for silent progression but not for SPMS conversion indicating that neurodegenerative processes in the brain seem to play a more important role in silent progression than for SPMS conversion.
The atrophy rates found here corroborate those found in previous studies. In a recent meta‐analysis spinal cord atrophy rates at C2‐3 vertebral level ranged between −0.7%/yr and − 4.4%/yr in progressive MS with a pooled rate of −2.1%/yr, similar to what we found in RR → RR (−0.88%/yr) and RR → SP (−2.2%/yr) patients. 48 Studies using the same active‐surface method at higher vertebral levels (C1‐3) reported RRMS rates between −0.2%/yr and − 3.4%/yr, 49 , 50 , 51 , 52 and a similar rate of −0.07%/yr in controls compared to our study (−0.07%/yr). 50
We replicated results on the predictive value of spinal cord atrophy for non‐confirmed EDSS worsening from a recent 6‐year follow‐up study 35 supporting the validity of our novel method and generalizability to a larger population.
Whereas higher baseline EDSS was associated with an increased risk of SPMS conversion in the matched subset, we found the inverse association in the silent progression and replication analysis (including RRMS and SPMS patients), similar to previous reports. 35 This might result from patients with the same spinal cord atrophy rate having a lower probability of worsening at higher EDSS. Here, we did not find relevant treatment effects on C1A atrophy rates in sensitivity analyses. However, whether disease‐modifying treatments slow down spinal cord atrophy as has been demonstrated for the brain 53 cannot be addressed in this heterogeneously treated observational cohort.
Several limitations must be acknowledged. The number of patients in the RR → RR/RR → SP subset was limited by the conversion rate of RR → SP during the 12‐year observation period. Furthermore, few individuals in RR → RR/RR → SP subset had cervical atrophy rates more characteristic of the other group. We speculate that these individuals might include RRMS patients who are approaching but have not crossed a threshold for conversion to SPMS, or RR → SP patients with a distinct pathophysiological mechanism underlying the conversion to SPMS. Another limitation resulting from the longitudinal aspect of our study is a potential confounding variability from hard−/software differences. However, the large longitudinal cohort supports calibration of MRI volume metrics across protocol changes and building on our previous validation of our calibration model, we demonstrate the consistency of our calibration method to preserve atrophy rates across protocols. Lastly, the current definition of SPMS is based on EDSS worsening and thereby strongly weighted towards locomotor disability. Given the predominant role of the spinal cord for locomotor dysfunction this could have led to an overestimation of the importance of the spinal cord in the prediction of silent progression and SPMS conversion. 14 We have adhered to this convention as there exists no validated definition of progression and SPMS conversion yet that includes other important domains like cognition.
Taken together, our findings add to the current knowledge of the value of brain and spinal cord neuroimaging markers in MS suggesting that spinal cord atrophy is a promising prognostic marker for silent progression and SPMS conversion. They extend the concept of silent progression demonstrating that spinal cord atrophy present from early disease stages appears to primarily determine the speed of disability progression. Importantly, our findings challenge the traditional dichotomy of an RRMS and subsequent SPMS phenotype and suggest that relapse‐onset MS should be considered a continuum stratified by early quantitative measures related to disability worsening including spinal cord atrophy. C1A atrophy rate as prognostic biomarker is likely to have utility for many types of studies in MS. Of particular advantage is its measurement from brain scans, allowing the application in the clinical setting and clinical trials without adding scan time. Furthermore, this method enables the retrospective analysis of well‐curated legacy datasets from clinical trials and observational cohorts worldwide to study the role of genetic, epidemiologic, and immune variables on disease phenotypes, and the impact of disease‐modifying therapies on long‐term disease course.
Author Contributions
A.B., N.P., S.L.H., R.G.H.: study concept and design.
A.B., N.P., A.K., A.R., G.K., X.Z., J.M.M., C.A., S.S., T.J.G., C.Z., W.A.S., E.C., Y.Z., R.G., N.R.R., A.S., A.H.Z., J.J., C.J.B., R.M.B., E.C., J.M.G., D.S.G., J.S.G., A.J.G., J.R.O., E.W., M.R.W., S.S.Z., B.A.C.C., S.L.H., R.G.H.: data acquisition and analysis.
A.B., N.P., G.K., R.M.B., J.M.G., M.R.W., B.A.C.C., S.L.H., R.G.H.: drafting the manuscript and figures
Potential Conflicts of Interest
The following companies that have had financial relationships with authors manufacture disease modifying drugs for MS that were mentioned in this study: Bayer Schering, Biogen, EMD Serono, Genentech, Genzyme, F. Hoffmann‐La Roche Ltd, Mylan Pharmaceuticals, Novartis, Sanofi, Teva Pharmaceuticals.
R.M.B. has received personal compensation for medical legal consulting and for consulting or serving on the advisory boards of F. Hoffmann‐La Roche Ltd, Sanofi‐Genzyme, and Novartis.
J.M.G. reports consulting fees from Biogen and research support from Genentech. D.S.G. has received research support and given lectures on MS and its treatment that have been sponsored by Biogen Idec, Bayer Schering, Novartis, E.M.D. Serono, Genzyme, and Teva pharmaceuticals. J.S.G. reports personal fees from Novartis and Genentech and grants from Biogen, and E.M.D. Serono. A.J.G. reports personal fees from Mylan Pharmaceuticals, and grants from Novartis, and payments for serving on committees for Biogen and Novartis. E.W. has received research support from Novartis and Roche. MRW receives research support from Roche/Genentech. S.S.Z. has served as a consultant and received honoraria from Biogen‐Idec, EMD‐Serono, Genzyme, Novartis, Roche/Genentech, and Teva Pharmaceuticals, Inc., and has served on Data Safety Monitoring Boards for Teva. B.A.C.C. has received personal compensation for consulting from Biogen, E.M.D. Serono, and Novartis. R.G.H. received grants from Hoffmann La Roche, and consultancy honoraria from Roche/Genentech, Sanofi/Genzyme, and Novartis.
S.L.H. has received travel reimbursement and writing assistance from F. Hoffman‐La Roche Ltd. and Novartis Pharma.
A.G., A.B., N.P., A.K., A.R., G.K., J.M.M., X.Z., C.A., S.S., T.J.G., C.Z., W.A.S., E.C., Y.Z., R.G., N.R.R., A.S., A.H.Z., J.J., C.J.B., E.C. and J.R.O. have nothing to disclose.
Supporting information
Table S1 Details on MRI acquisition and correlations between C1A and other brain and cord MRI measures.
Table S2: Replication Analysis
Table S3. Comparison of brain and cervical cord measures at baseline and before conversion/silent progression between patients and controls.
Table S4. Comparison of baseline brain volumes and cervical cord area between silent progression groups and controls.
Table S5. Comparison of baseline and longitudinal brain and cervical cord measures between secondary progressive multiple sclerosis patients and controls.
Table S6. Demographic, clinical and MRI characteristics at baseline and for the period before conversion/silent progression.
Table S7. Brain and cervical cord measures at baseline and before conversion/silent progression
Acknowledgements
We are deeply grateful to the patients for their long‐term participation in this ambitious study. This study was supported by a gift from the Ostby Foundation (RGH), the National Institute of Neurological Diseases and Stroke (R35NS111644, SLH; RO1NS26799, SLH, JRO; K23 NS048869, BACC), the National Multiple Sclerosis Society (RG‐1707‐28775, RGH), the Valhalla Foundation, and gifts from Friends of the Multiple Sclerosis Research Group at UCSF. AB was supported by the Freiwillige Akademische Gesellschaft, Switzerland, and the Gottfried and Julia Bangerter‐Rhyner Stiftung, Switzerland. We would like to thank Whitaker Evans, UCSF library, for assistance with the literature search and all UCSF researchers and staff who contributed to this study.
References
- 1. Landtblom AM, Fazio P, Fredrikson S, Granieri E. The first case history of multiple sclerosis: Augustus d'Este (1794–1848). Neurol Sci. 2010;31:29–33. [DOI] [PubMed] [Google Scholar]
- 2. Mc AD, Compston N. Some aspects of the natural history of disseminated sclerosis. Q J Med 1952;21:135–167. [PubMed] [Google Scholar]
- 3. University of California SFMSET , Cree BAC, Hollenbach JA, et al. Silent progression in disease activity‐free relapsing multiple sclerosis. Ann Neurol 2019;85:653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bergsland N, Horakova D, Dwyer MG, et al. Gray matter atrophy patterns in multiple sclerosis: a 10‐year source‐based morphometry study. Neuroimage Clin. 2018;17:444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kremenchutzky M, Rice GP, Baskerville J, et al. The natural history of multiple sclerosis: a geographically based study 9: observations on the progressive phase of the disease. Brain 2006;129:584–594. [DOI] [PubMed] [Google Scholar]
- 6. Rocca MA, Comi G, Filippi M. The role of T1‐weighted derived measures of Neurodegeneration for assessing disability progression in multiple sclerosis. Front Neurol 2017;8:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ciccarelli O, Cohen JA, Reingold SC, et al. Spinal cord involvement in multiple sclerosis and neuromyelitis optica spectrum disorders. Lancet Neurol 2019;18:185–197. [DOI] [PubMed] [Google Scholar]
- 8. Eshaghi A, Prados F, Brownlee WJ, et al. Deep gray matter volume loss drives disability worsening in multiple sclerosis. Ann Neurol 2018;83:210–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lukas C, Knol DL, Sombekke MH, et al. Cervical spinal cord volume loss is related to clinical disability progression in multiple sclerosis. J Neurol Neurosurg Psychiatry 2015;86:410–418. [DOI] [PubMed] [Google Scholar]
- 10. Liu Z, Yaldizli O, Pardini M, et al. Cervical cord area measurement using volumetric brain magnetic resonance imaging in multiple sclerosis. Mult Scler Relat Disord 2015;4:52–57. [DOI] [PubMed] [Google Scholar]
- 11. Papinutto N, Bakshi R, Bischof A, et al. Gradient nonlinearity effects on upper cervical spinal cord area measurement from 3D T1 ‐weighted brain MRI acquisitions. Magnetic Resonance in Medicine. 2018;79:1595‐1601. 10.1002/mrm.26776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Papinutto N, Asteggiano C, Bischof A, et al. Intersubject variability and normalization strategies for spinal cord total cross‐sectional and gray matter areas. J Neuroimaging 2019;30:110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. University of California SFMSET , Cree BA, Gourraud PA, et al. Long‐term evolution of multiple sclerosis disability in the treatment era. Ann Neurol 2016;80:499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lorscheider J, Buzzard K, Jokubaitis V, et al. Defining secondary progressive multiple sclerosis. Brain. 2016;139:2395–2405. [DOI] [PubMed] [Google Scholar]
- 15. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983;33:1444–1452. [DOI] [PubMed] [Google Scholar]
- 16. Michel L, Vukusic S, De Seze J, et al. Mycophenolate mofetil in multiple sclerosis: a multicentre retrospective study on 344 patients. J Neurol Neurosurg Psychiatry 2014;85:279–283. [DOI] [PubMed] [Google Scholar]
- 17. Ashtari F, Savoj MR. Effects of low dose methotrexate on relapsing–remitting multiple sclerosis in comparison to interferon beta‐1alpha: a randomized controlled trial. J Res Med Sci 2011;16:457–462. [PMC free article] [PubMed] [Google Scholar]
- 18. Hartung HP, Gonsette R, Konig N, et al. Mitoxantrone in progressive multiple sclerosis: a placebo‐controlled, double‐blind, randomised, multicentre trial. Lancet 2002;360:2018–2025. [DOI] [PubMed] [Google Scholar]
- 19. Krishnan C, Kaplin AI, Brodsky RA, et al. Reduction of disease activity and disability with high‐dose cyclophosphamide in patients with aggressive multiple sclerosis. Arch Neurol. 2008;65:1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schlaeger R, Papinutto N, Panara V, et al. Spinal cord gray matter atrophy correlates with multiple sclerosis disability. Ann Neurol 2014;76:568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horsfield MA, Sala S, Neema M, et al. Rapid semi‐automatic segmentation of the spinal cord from magnetic resonance images: application in multiple sclerosis. Neuroimage 2010;50:446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Papinutto N, Schlaeger R, Panara V, et al. 2D phase‐sensitive inversion recovery imaging to measure in vivo spinal cord gray and white matter areas in clinically feasible acquisition times. J Magn Reson Imaging 2015;42:698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weeda MM, Middelkoop SM, Steenwijk MD, et al. Validation of mean upper cervical cord area (MUCCA) measurement techniques in multiple sclerosis (MS): high reproducibility and robustness to lesions, but large software and scanner effects. Neuroimage Clin. 2019;24:101962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dong Y, Peng CY. Principled missing data methods for researchers. Springerplus 2013;2:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Papinutto N, Bakshi R, Bischof A, et al. Gradient nonlinearity effects on upper cervical spinal cord area measurement from 3D T1 ‐weighted brain MRI acquisitions. Magn Reson Med 2018;79:1595–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schmidt P. Bayesian inference for structured additive regression models for large‐scale problems with applications to medical imaging. Dissertation, LMU München: Fakultät für Mathematik, Informatik und Statistik 2017. [Google Scholar]
- 27. Avants BB, Epstein CL, Grossman M, Gee JC. Symmetric diffeomorphic image registration with cross‐correlation: evaluating automated labeling of elderly and neurodegenerative brain. Med Image Anal 2008;12:26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smith SM, Zhang Y, Jenkinson M, et al. Accurate, robust, and automated longitudinal and cross‐sectional brain change analysis. Neuroimage 2002;17:479–489. [DOI] [PubMed] [Google Scholar]
- 29. Lutkenhoff ES, Rosenberg M, Chiang J, et al. Optimized brain extraction for pathological brains (optiBET). PLoS One 2014;9:e115551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Patenaude B, Smith SM, Kennedy DN, Jenkinson M. A Bayesian model of shape and appearance for subcortical brain segmentation. Neuroimage 2011;56:907–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Azevedo CJ, Cen SY, Khadka S, et al. Thalamic atrophy in multiple sclerosis: a magnetic resonance imaging marker of neurodegeneration throughout disease. Ann Neurol 2018;83:223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reuter M, Schmansky NJ, Rosas HD, Fischl B. Within‐subject template estimation for unbiased longitudinal image analysis. Neuroimage 2012;61:1402–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Keshavan A, Paul F, Beyer MK, et al. Power estimation for non‐standardized multisite studies. Neuroimage 2016;01:281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heinze G, Wallisch C, Dunkler D. Variable selection ‐ a review and recommendations for the practicing statistician. Biom J 2018;60:431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsagkas C, Magon S, Gaetano L, et al. Spinal cord volume loss: a marker of disease progression in multiple sclerosis. Neurology 2018;91:e349–e358. [DOI] [PubMed] [Google Scholar]
- 36. Benjamini Y, Drai D, Elmer G, et al. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 2001;125:279–284. [DOI] [PubMed] [Google Scholar]
- 37. Koo TK, Li MY. A guideline of selecting and reporting intraclass correlation coefficients for reliability research. J Chiropr Med 2016;15:155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wattjes MP, Ciccarelli O, Reich DS, et al. 2021 MAGNIMS–CMSC–NAIMS consensus recommendations on the use of MRI in patients with multiple sclerosis. Lancet Neurol. 2021;20:653–670. [DOI] [PubMed] [Google Scholar]
- 39. Sastre‐Garriga J, Pareto D, Battaglini M, et al. MAGNIMS consensus recommendations on the use of brain and spinal cord atrophy measures in clinical practice. Nat Rev Neurol 2020;16:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Klein JP, Arora A, Neema M, et al. A 3T MR imaging investigation of the topography of whole spinal cord atrophy in multiple sclerosis. AJNR Am J Neuroradiol. 2011;32:1138–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Prados F, Cardoso MJ, Yiannakas MC, et al. Fully automated grey and white matter spinal cord segmentation. Sci Rep 2016;27:36151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Petrova N, Carassiti D, Altmann DR, et al. Axonal loss in the multiple sclerosis spinal cord revisited. Brain Pathol 2018;28:334–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Eshaghi A, Young AL, Wijeratne PA, et al. Identifying multiple sclerosis subtypes using unsupervised machine learning and MRI data. Nat Commun 2021;12:2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tsagkas C, Naegelin Y, Amann M, et al. CNS atrophy predicts future dynamics of disability progression in a real‐world multiple sclerosis cohort. Eur J Neurol 2021;28:4153–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Henry RG, Shieh M, Okuda DT, et al. Regional grey matter atrophy in clinically isolated syndromes at presentation. J Neurol Neurosurg Psychiatry 2008;79:1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Azevedo CJ, Cen SY, Jaberzadeh A, et al. Contribution of normal aging to brain atrophy in MS. Neurol Neuroimmunol Neuroinflamm 2019;6:e616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Haider L, Prados F, Chung K, et al. Cortical involvement determines impairment 30 years after a clinically isolated syndrome. Brain 2021;144:1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Casserly C, Seyman EE, Alcaide‐Leon P, et al. Spinal cord atrophy in multiple sclerosis: a systematic review and meta‐analysis. J Neuroimaging 2018;13:556–586. [DOI] [PubMed] [Google Scholar]
- 49. Dupuy SL, Khalid F, Healy BC, et al. The effect of intramuscular interferon beta‐1a on spinal cord volume in relapsing–remitting multiple sclerosis. BMC Med Imaging 2016;16:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Valsasina P, Rocca MA, Horsfield MA, et al. A longitudinal MRI study of cervical cord atrophy in multiple sclerosis. J Neurol 2015;262:1622–1628. [DOI] [PubMed] [Google Scholar]
- 51. Hagstrom IT, Schneider R, Bellenberg B, et al. Relevance of early cervical cord volume loss in the disease evolution of clinically isolated syndrome and early multiple sclerosis: a 2‐year follow‐up study. J Neurol 2017;264:1402–1412. [DOI] [PubMed] [Google Scholar]
- 52. Rashid W, Davies GR, Chard DT, et al. Increasing cord atrophy in early relapsing–remitting multiple sclerosis: a 3 year study. J Neurol Neurosurg Psychiatry 2006;77:51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kappos L, Radue EW, O'Connor P, et al. A placebo‐controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010;362:387–401. [DOI] [PubMed] [Google Scholar]
- 54. Lakshmi AS. Sagittal diameter of foramen magnum in normal population: An mri study. Journal of Evolution of Medical and Dental Sciences. 2015;4:16045‐16047. 10.14260/jemds/2015/2342 [DOI] [Google Scholar]
- 55. Papinutto N, Bakshi R, Bischof A, et al. Gradient nonlinearity effects on upper cervical spinal cord area measurement from 3D T1 ‐weighted brain MRI acquisitions. Magnetic Resonance in Medicine. 2018;79:1595‐1601. 10.1002/mrm.26776 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Details on MRI acquisition and correlations between C1A and other brain and cord MRI measures.
Table S2: Replication Analysis
Table S3. Comparison of brain and cervical cord measures at baseline and before conversion/silent progression between patients and controls.
Table S4. Comparison of baseline brain volumes and cervical cord area between silent progression groups and controls.
Table S5. Comparison of baseline and longitudinal brain and cervical cord measures between secondary progressive multiple sclerosis patients and controls.
Table S6. Demographic, clinical and MRI characteristics at baseline and for the period before conversion/silent progression.
Table S7. Brain and cervical cord measures at baseline and before conversion/silent progression