

Abstract
Aerosols play an important yet uncertain role in modulating the radiation balance of the sensitive Arctic atmosphere. Organic aerosol is one of the most abundant, yet least understood, fractions of the Arctic aerosol mass. Here we use data from eight observatories that represent the entire Arctic to reveal the annual cycles in anthropogenic and biogenic sources of organic aerosol. We show that during winter, the organic aerosol in the Arctic is dominated by anthropogenic emissions, mainly from Eurasia, which consist of both direct combustion emissions and long-range transported, aged pollution. In summer, the decreasing anthropogenic pollution is replaced by natural emissions. These include marine secondary, biogenic secondary and primary biological emissions, which have the potential to be important to Arctic climate by modifying the cloud condensation nuclei properties and acting as ice-nucleating particles. Their source strength or atmospheric processing is sensitive to nutrient availability, solar radiation, temperature and snow cover. Our results provide a comprehensive understanding of the current pan-Arctic organic aerosol, which can be used to support modelling efforts that aim to quantify the climate impacts of emissions in this sensitive region.
Subject terms: Atmospheric chemistry, Atmospheric chemistry, Atmospheric chemistry
Organic aerosols in the Arctic are predominantly fuelled by anthropogenic sources in winter and natural sources in summer, according to observations from eight sites across the Arctic
Main
Organic aerosols (OAs) contribute to the Arctic aerosol mass near the surface1–4 and affect the local climate through direct aerosol–radiation interactions and by altering the cloud properties5,6. OAs interact with other aerosol components7–9, for example, black carbon or elemental carbon (EC) and sulfate, and can augment or offset their radiative forcing10. The Arctic OA indirect effect is estimated to be of a similar magnitude as that of the sulfate indirect effect, and much larger than the OA direct effect11. The magnitude of these effects depends on the OA physicochemical properties, sources and formation processes12,13, which are not traceable by satellites14, and hence surface observations are indispensable15. However, OAs have received little attention in the Polar Regions16–18, mainly because of measurement challenges, and hence their complex composition and sources are poorly understood19,20. Nevertheless, an increasing abundance of natural OAs in a warming Arctic is expected21 as a result of northward-expanding vegetation22, intensifying boreal forest fires23,24, decreasing sea-ice extent25 and thawing permafrost26,27. Enhanced OA emissions are also expected from increasing local anthropogenic emissions, which include oil and gas exploration, and shipping activities28,29.
Attempts to model the Arctic OA concentrations have been limited, with typical under-predictions in winter and/or spring, when haze can be omnipresent and persistent30–33. The formation of secondary organic aerosols (SOAs) from anthropogenic or natural sources in different seasons is poorly represented32,34. Of 16 models deployed in a recent AeroCom evaluation of the simulated annual aerosol optical depth in Polar Regions32, only 6 considered biogenic precursors, which can contribute to particle growth and so the size range of cloud condensation nuclei. Only in one of the models was methanesulfonic acid (MSA) considered, which resulted in an outlier Arctic aerosol optical depth in terms of both seasonal variability and year-long magnitude32. A recent study showed that natural OAs other than MSA may account for about half of the summertime Arctic OAs33. Biological emissions, often not considered in Arctic models, may be a missing source of ice-nucleating particles2,35 that is possibly further enhanced with the thawing permafrost36. The contribution of anthropogenic emissions, for example, from gas flaring37, to primary OA and SOA precursors is expected to vary substantially in space and time across the Arctic region but, unlike black carbon38, has been largely overlooked. Long-term ambient measurements of the Arctic OA composition are critically needed to identify its main sources and therefore are a first step for subsequent implementation into models. However, available studies3,39–41 are single-site, short-term3,39 or campaign-based39,40, and too infrequent to reveal seasonal patterns in the main sources of Arctic OAs42. Hence, Arctic aerosol source apportionment studies typically do not consider OAs43,44, or are limited to a few primary markers (for example, levoglucosan from biomass burning or EC) and radiocarbon measurements45,46. Therefore, current efforts have so far been unable to provide an understanding of the sources and formation pathways of the pan-Arctic OAs in different seasons.
Determination of OA sources across the Arctic
Here we fill this critical knowledge gap by determining the spatial and seasonal distribution of individual OA classes (factors) across the Arctic (Fig. 1, Methods and Supplementary Text 1). This was achieved by offline aerosol mass spectrometer (AMS) measurements of nebulized aerosol water extracts47 from filter samples (Methods and Supplementary Text 2), followed by multisite positive matrix factorization (PMF) for OA source apportionment in relative terms, and subsequent quantification using externally measured water-soluble organic carbon (WSOC; see Methods). Factor identification is aided by organic marker, major ion and EC measurements (Methods and Supplementary Text 3). Source apportionment (Methods and Supplementary Text 4) results are also combined with concentration-weighted trajectory (CWT) analysis48 to determine the geographical origin of the OA factors (Methods and Supplementary Text 5). We performed the analysis of 350 (bi-)weekly composite samples from filters collected at eight observatories across the Arctic (Fig. 1a) at roughly overlapping periods in 2014–2019 (Supplementary Table 1), yielding unique results of annual cycles which included the periods of winter darkness and summer daylight (Supplementary Table 2). Using this methodology, we determined the anthropogenic and natural sources that drive the mass of primary OAs and SOAs in the pan-Arctic region in both winter and summer. Our work is the first systematic, concerted and multiseason, multistation effort with advanced analysis techniques, which goes beyond intensive observation periods and case-study work20.
Fig. 1. Sites, OA factors and chemical characteristics.

a, Arctic political map showing the aerosol filter sampling stations (Supplementary Text 1 and Supplementary Table 1). A, Alert; B, Cape Baranova; G, Gruvebadet; P, Pallas-Matorova; T, Tiksi; U, Utqiaġvik; V, Villum Research Station; Z, Zeppelin. Adapted from Hugo Ahlenius/GRID-Arendal (https://www.grida.no/resources/8378). b, Station-specific average total OA mass concentrations (whiskers are 1 standard deviation, s.d., corresponding to sample-to-sample variability) based on a statistical analysis of the water-soluble fraction to obtain factor recoveries (Supplementary Text 4) in the polar night (winter) versus midnight sun (summer) periods (Supplementary Table 2), sorted in descending order of the station annual average, and percent contribution to the particulate mass (including non-sea salt sulfate, nitrate, ammonium, EC and estimated sea salt63). The dashed blue lines connect winter and summer contributions at each station (no winter samples for G and T, and U winter samples were not analysed for ions). c, Van Krevelen plot as a tool for compositional differentiation among samples: atomic O:C ratio versus H:C ratio of the Arctic AMS-PMF-based factors (Supplementary Fig. 2), and individual PMF input bulk samples (colour coded by month: 1, January, through to 12, December). Red and blue dashed curves refer to the triangle reported by Ng et al.85. Grey dashed lines denote two example oxidation states (OS). Error bars correspond to 1 s.d. from a bootstrap analysis (Methods and Supplementary Text 4). d, Spatial distribution and seasonal variability in the average factor percentage contributions to total OA (water-soluble (Supplementary Fig. 7); entire time-series (Supplementary Fig. 10)). Factors are sorted from bottom (Haze) to top (POA) based on their onset (see Fig. 3), starting from late winter for Haze. Primary OAs, POA + PBOA (top). Orange outline, sum of natural-dominated OAs.
The total OA was found to be a major aerosol fraction that typically contributes 10–40% to the total particulate mass at the different stations (Fig. 1b; the dataset range is 3–65%), with higher relative contributions in the summer. Although only water-soluble OA (WSOA) components were measured, the results represent the total OA by scaling the measurements by the water-soluble fraction of each factor (Methods and Supplementary Text 4). On average, WSOAs comprise 82 ± 4% of the total OA. Absolute OA concentrations were generally higher at the more continental stations, ~1.4 µg m−3 in Tiksi (TIK), ~0.4 µg m−3 in Cape Baranova (BAR) and 0.2 µg m−3 and 1.0 µg m−3 in Pallas-Matorova (PAL) in winter and summer, respectively, whereas typical concentrations at the other stations were lower (0.1–0.2 µg m−3). Figure 1c shows the O:C versus H:C ratios for all the samples measured by the AMS, which highlights the large variability in the OA chemical composition across stations and seasons. We determined six major OA factors (Methods and Supplementary Text 4, Supplementary Tables 3 and 4 and Supplementary Figs. 1–10). These include factors related to anthropogenic-dominated emissions, namely, oxygenated organic aerosol (OOA), Haze and primary-anthropogenic organic aerosol (POA), and to natural-dominated emissions, namely, MSA-related organic aerosol (MSA-OA), biogenic secondary organic aerosol (BSOA) and primary biological organic aerosol (PBOA). We could not relate any factor to fresh wildfire emissions in the spring or summer, probably because of multiple reasons such as the rapid aerosol ageing during transport49, emissions remaining aloft on ascent to the middle and/or upper troposphere (if they originate from North America and East and South Asia)2,23,50 and/or because this source becomes important only during short-term events. The atomic O:C versus H:C ratios of the six major OA factors (Fig. 1c) and their relative contributions across stations and seasons (Fig. 1d) are very distinct, and show the large diversity in the sources that drive the OA mass in the Arctic region. In the following, we discuss characteristic features of the factors, their spatial (Fig. 2) and seasonal (Fig. 3) variabilities, as well as their major source regions (Fig. 4).
Fig. 2. Station-specific yearly mass concentration of speciated Arctic OAs.
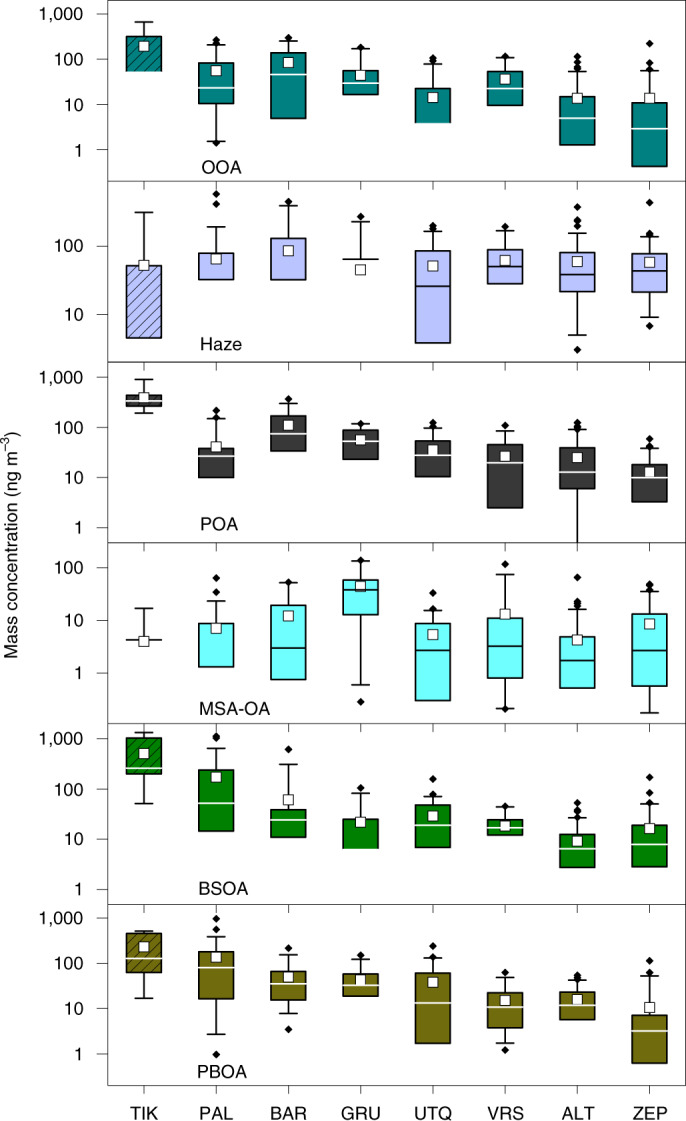
Absolute OA factor concentrations at the different stations shown as box-and-whisker plots. The stations appear in the same order as in Fig. 1b, whereas the factors appear in the same order as discussed in the main text. Note the different range of y-axis values for the different factors. The lower whiskers are missing if associated values are out of scale. Boxes for TIK are hatched to indicate incomplete (inter)annual coverage. For WSOAs, see Supplementary Fig. 8. Horizontal line and box, yearly median and interquartile range; squares and whiskers, yearly mean and range within the 5th and 95th percentiles; diamonds, outliers.
Fig. 3. Seasonal variability of speciated pan-Arctic OAs.
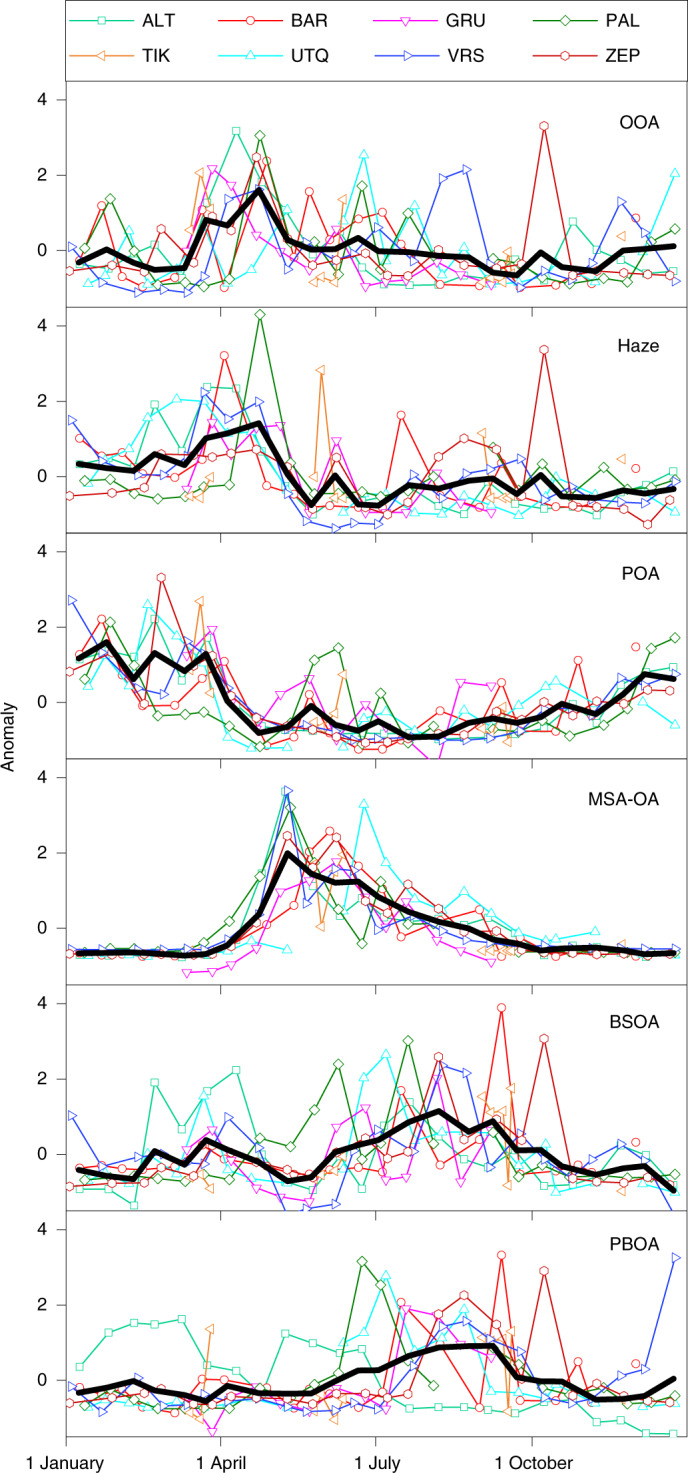
Standardized annual cycles for each OA factor at the different stations. Bi-weekly averaged data from multiple years for each station are merged into a single annual cycle (the sum of individual station values in each panel equals zero). The y-axis values (anomalies) were calculated using the absolute mass concentration values as: (value – station average)/s.d. of station. The thick black lines indicate the average annual cycle of each factor over all the stations (note that here the sum of the yearly values in each panel is not equal to zero).
Fig. 4. Major source regions of long-range transported Arctic OA factors.
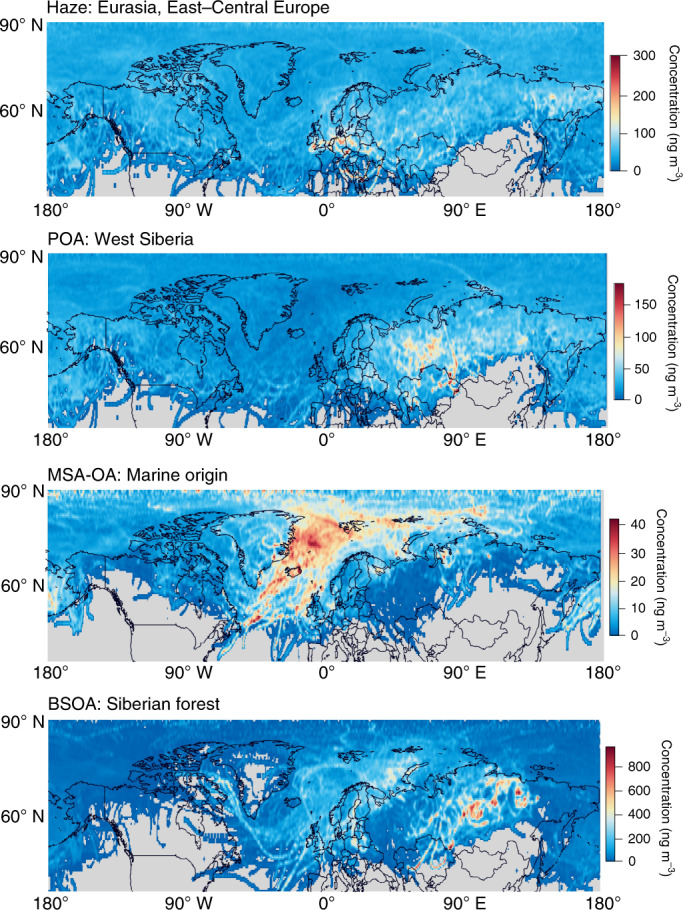
Merged results from the CWT-based back-trajectory (BT) analysis with ZeFir (Methods) at different Arctic stations (Supplementary Text 5 and Supplementary Fig. 11) showing long-term pan-Arctic hot spots of transported anthropogenic-dominated (Haze and POA) and natural-dominated (MSA-OA and BSOA) OA factors. The entire time series of each factor mass concentration at the different stations (time periods shown in Supplementary Table 1 and Supplementary Fig. 10) were used to create the maps (see Supplementary Text 5 for a discussion of the potential uncertainties in the source regions). The trajectories represent 5 days back in time for MSA-OA and (up to) 10 days for the other factors (Supplementary Text 5 and Supplementary Fig. 11). Colour scales indicate the water-soluble factor concentrations linked to the major source regions (‘long-range’ probability heat maps). The individual station results shown in Supplementary Fig. 11 were merged for each factor, except for POA, for which only six stations with winter data were considered here (no GRU and TIK), to indicate specific regions with intense gas-flaring activity during winter (for example, the Komi Republic, Khanty-Mansisk and Yamalo-Nenets autonomous districts in West Siberia). PBOA is expected to reside mainly in the coarse aerosol mode, and thus has a relatively short atmospheric lifetime (and hence more local and/or regional origins), and the formation of OOA might be linked to a prior accumulation of volatile organic compounds (thus probably not directly transported in the particle phase); therefore, the merged results for these factors are shown only in the Supplementary Information (Supplementary Fig. 11). The World Maps available with ZeFir are taken from Natural Earth Data.
Anthropogenic-dominated OA factors
The OOA factor (O:C ~0.5) contains large oxygenated fragment ions (Supplementary Table 5). Its time-series shows a pan-Arctic enhancement during and after polar sunrise (Fig. 3), which potentially links this factor to oxidation products of volatile organic compounds that have accumulated during the polar night and form SOAs in the spring25,51–53. In this regard, a strong decrease in volatile organic compound concentrations was reported at PAL for April54. Although the OOA factor might be dominated by wintertime anthropogenic emissions, the contribution from natural sources during the summer cannot be excluded. The Haze factor contains more fragmented oxygenated ions (O:C ~0.75; Fig. 1c), builds up in late winter, peaks in spring (Fig. 3) and correlates with sulfate (Supplementary Table 6), similar to the profile and timing of the Arctic haze phenomenon55. Except for intermittent peaks at BAR, PAL and TIK, its yearly concentration is spatially homogeneous (Fig. 2), but with a certain temporally structured variability56 with the timing of the winter/spring peak occurring a few weeks later in time at most observatories as we move from west to east (Fig. 3), which indicates the effect of regional transport. This factor therefore becomes predominant in relative terms (Fig. 1d) at remote stations with lower OA loadings, that is, ~45% throughout the year at Alert (ALT) and Zeppelin (ZEP) (1st and 3rd quartiles, Q1–Q3 = 38–54%). Haze organics are associated with long-range atmospheric transport from Eurasia (Fig. 4). Therefore, the transport of pollutants from distant urbanized areas to remote environments contributes substantially to the formation of pan-Arctic SOAs (OOA and Haze; Q1–Q3 = 44–56% of the total OA during the polar night).
The POA factor is mainly composed of hydrocarbons (Supplementary Table 5) from primary anthropogenic emissions (O:C ~0.2), most probably related to gas flaring. POA has an H:C ratio of ~1.3 (Fig. 1c), lower than the H:C ratio (~1.8–1.9) of freshly emitted hydrocarbons detected at lower latitudes from traffic emissions57,58, which suggests a high contribution from unsaturated hydrocarbons. This factor is ubiquitous under dark and cold conditions, and peaks in December–March (Fig. 3) with a typical median pan-Arctic contribution to the total OA of 35% during the polar night (Fig. 1d, Q1–Q3 = 28–42%). In winter, POA correlates strongly with EC (Supplementary Fig. 12), which is reported to be of fossil origin45,49. The POA:EC ratio of ~1.1 is similar to the respective organic carbon (OC):EC ratios from oil and gas extraction emission estimates59. We found West Siberian locations (Fig. 4) as a major potential source region of the POA in winter (mainly at ALT, BAR, PAL and ZEP; Supplementary Fig. 11), similar to those previously found for surface black carbon42,60,61. The presence of POA at Gruvebadet (GRU) and TIK in the summer (Figs. 1d and Fig. 2) might indicate a more local origin at urban-type Arctic settlements.
Natural-dominated OA factors
MSA-OA is characterized by the fragmentation pattern of MSA (for example, CH3 and CH3SO; Supplementary Table 5), which is produced from marine dimethylsulfide oxidation. It correlates strongly with MSA, which comprises on average 80% of the factor, as measured by ion chromatography (Supplementary Table 7). This factor consistently originates from marine regions (Fig. 4), and increases with solar radiation (Supplementary Fig. 14). The MSA-OA absolute concentrations (Fig. 2) and clear annual cycles (Fig. 3) are consistent among the stations, with much higher concentrations in May–June, the main season of phytoplankton blooms. The range of maximum concentrations at ALT and Utqiaġvik (UTQ) (~20–30 ng m−3) is comparable to those of previous measurements of MSA from these stations by ion chromatography62,63. The highest weekly averaged MSA-OA levels, which exceed 100 ng m−3, occur at GRU, Villum Research Station (VRS) and ZEP. Although at these stations MSA-OA is typically 22% (Q1–Q3 = 12–34 %) of the total OA during the midnight-sun period (Fig. 1d), this factor is neither the only nor the predominant natural OA component.
BSOA is linked to biogenic emissions, for example, mono- and/or sesquiterpenes and isoprene64,65, from forests, tundra, lakes, wetlands and marine waters. This factor dominates the variability of moderately oxygenated CHO fragments related to isoprene and terpene SOAs or, potentially, to biogenic stress responses (Supplementary Table 5), and correlates strongly (R2 = 0.81; Supplementary Fig. 13) with 3-methyl-1,2,3-butanetricarboxylic acid, a second-generation oxidation product of α-pinene. BSOA exhibits a clear annual cycle with enhanced values in June–September (Fig. 3), consistent with the exponential increase of biogenic precursor emissions with temperature66 (Supplementary Fig. 14). This factor dominates at PAL and TIK where it contributes, on average, 40% to the total OA in the summer (Fig. 1d, Q1–Q3 = 25–53%). At stations less affected by the boreal biome, BSOA appears in smaller but non-negligible amounts in the summer, and contributes 20%, on average, at UTQ and ZEP (Q1–Q3 = 11–28%) and ~9%, on average, at other stations. At PAL, we expect a more local and/or semiregional source67,68, whereas the episodic occurrence of BSOA in both Russian stations is linked to northward air mass transport from the Siberian forest (Fig. 4).
PBOA (O:C ~0.4, H:C ~1.6) is related to biological matter potentially from both terrestrial and marine origins, for example, fungal spores, bacteria, vegetative detritus, phytoplankton or fragments of it, and (ice) algae secretions1,69,70. It correlates with carbohydrate-related AMS fragments typically linked to primary biological compounds (Supplementary Table 5), and with arabitol and mannitol71 (R2 = 0.86; Supplementary Fig. 13), which are sugar-alcohols present in fungal spores and other biological matter. PBOA exhibits substantial concentration increases in July–September, to reach up to 1.0 µg m−3 at PAL (Fig. 2), and a distinct temporal evolution, peaking later than MSA-OA (Fig. 3). Similar to BSOA, PBOA exhibits higher relative contributions at lower latitude Arctic stations (~70° N), averaging one-third of the total OA at PAL and UTQ in the summer (Fig. 1d, Q1–Q3 = 26–47%). PBOA anticorrelates with the snow depth at PAL and VRS (where such data are available) and with the 30-year climate-normal average snowfall at UTQ (Supplementary Fig. 14). These findings indicate that maximum local biological activity is associated with the lack of snow covering the ground72, which causes entrainment of PBOA into the air.
Implications of Arctic OA spatiotemporal variability
Overall, the pan‐Arctic OA composition and sources are not uniform. They are largely driven by the stations’ latitude (and altitude; Supplementary Fig. 15), favourable conditions for long-range atmospheric transport and distance to anthropogenic and natural aerosol sources, as well as the presence of light and snow cover. We show that secondary Arctic OAs, separated into various distinct subtypes, typically dominate the OA mass, although the contribution of primary OAs from both natural and anthropogenic emissions is often equally important (Fig. 1d). This finding is in line with lower-latitude OA source apportionment studies12. The current and future abundance of condensable organics could be critical for the cloud condensation nuclei budget and cloud properties. In the Arctic winter, the sea-ice extent is at its maximum, low-level clouds have a pronounced warming effect and surface pollutants can become trapped close to the ground due to temperature inversion55. In this period, the land-surface OA sources are predominantly anthropogenic and linked to both primary emissions and secondary processes. The widespread Haze organics are dominated by aged anthropogenic-dominated emissions transported mainly from Eurasia, peak during polar sunrise and abruptly decrease in May. Under cold and dark conditions, all the stations are affected by OA emissions related to oil or gas extraction activities, mainly in West Siberia. The Arctic amplification of temperature increase73,74 is more intense during winter75, and may be affected by aerosols altering the cloud properties. Hence, the future evolution of anthropogenic-dominated wintertime Arctic OA should be monitored from a climate perspective, especially in response to the development of effective emission control measures at lower latitudes11,29. In the summer, the decreasing anthropogenic pollution is replaced by natural OA emissions, with similar absolute concentrations. These emissions include marine SOAs from dimethylsulfide oxidation and little-explored primary biological emissions and biogenic SOAs. These collectively contribute to an increased importance of natural OA in the summertime aerosol particle mass in the inner Arctic76. We found an overall nearly equal yearly abundance (Fig. 5) of summed anthropogenic-dominated OAs (average of 115 ng m−3, Q1–Q3 = 20–100 ng m−3; 95th percentile, 435 ng m−3) and summed natural-dominated OAs (average of 140 ng m−3, Q1–Q3 = 40–160 ng m−3; 95th percentile, 445 ng m−3). This indicates that the typically lower total aerosol volume/mass in the summer versus winter or spring77 is due to species other than the total OA, which exhibits less of a seasonal cycle across the Arctic (Supplementary Fig. 16). The effects of anthropogenic and natural organics on cloud condensation nuclei and ice-nucleating particles, and potentially on the Arctic climate, have been largely unexplored. The importance of considering land coverage and biosphere–atmosphere exchanges78,79 for the response of natural Arctic OAs to warming is highlighted in Supplementary Fig. 14. A small increase in temperature results in a substantial (exponential) increase of BSOA, which identifies one of the potential feedback mechanisms in the Arctic. Also, extension of the ice-free season and expansion of snow-free areas80 may lead to enhanced airborne organics from biological activity and secondary marine emissions.
Fig. 5. Conceptual overview of anthropogenic-dominated versus natural-dominated Arctic OAs.
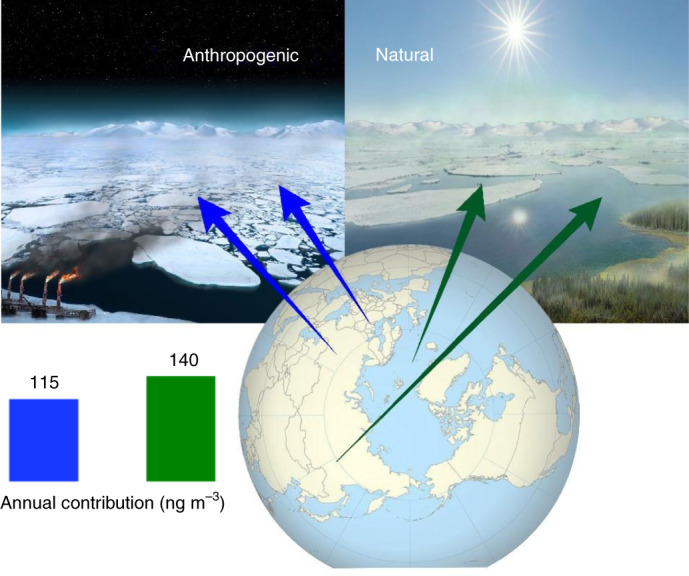
A conceptual image of anthropogenic-dominated and natural-dominated emissions that drive the OA mass in the Arctic in winter and summer, respectively. The most important geographical source regions are indicated by the arrows. Bars show the entire-dataset average contributions of nearly equally contributing summed anthropogenic-dominated (blue) and summed natural-dominated (green) organic components. Credits: Helen Cawley for the landscape drawing; map made using Natural Earth.
Currently, the Arctic system is in transition21, with long-range transported anthropogenic-dominated emissions (including sulfate) continuously decreasing due to better air quality regulations in the lower latitude regions. Meanwhile, natural emissions are expected to increase81,82, which probably enhances the magnitude and relative importance of the composition-dependent OA–cloud effect11. Our results provide the first understanding of the present-day year-long, pan-Arctic OA sources, which can be used for comparisons with past (for example, through ice-core archives83) and future measurements of these changing biogenic and anthropogenic emissions. Given practical difficulties in deploying multiple online AMS instruments for long time periods around the Arctic and the widespread availability of ambient filters, the measurement methodology and analysis techniques employed here are also applicable to emerging Arctic stations (for example, in far East Siberia84).
Methods
Filter sampling
Total suspended particulate matter or PM10 (particulate matter with aerodynamic diameter d < 10 µm) was collected on quartz fibre filters at eight stations around the Arctic (Fig. 1a). This study combined distinct locations affected by anthropogenic versus natural, marine versus terrestrial and local versus long-range transported emission sources at different seasons. PAL stands out from the other stations as it is in a sub-Arctic site influenced substantially by the boreal biome, which highlights differences with higher-latitude sites. Typically, at least one annual cycle is covered per station (Supplementary Table 1). The samples from GRU cover the spring and summer season (Supplementary Table 2) over two consecutive years, whereas the longest temporal coverage is almost four years (ALT). Although the data coverage at TIK is relatively less wide, this site experiences distinctly higher levels of pollution compared with that of other sites and includes the spring, summer and autumn periods over three consecutive years, and so covers a wide range of conditions. More details about the stations and filter sampling are provided in Supplementary Text 1, which includes a description of the conditions under which the filters were handled, transported between stations and/or labs and stored during and/or after sampling.
AMS measurements and data analysis
The offline AMS technique was established by Daellenbach et al.47. Briefly, punches from the quartz fibre filter samples were extracted in ultrapure water (18.2 Mohm cm, with the total organic carbon (TOC) <3 ppb by weight). Teflon filters were tested but not measured (Supplementary Text 2). Typical quartz-fibre-filtered water-extracted organic concentrations were 2–3 µg C ml–1. The extracts were inserted into an ultrasonic bath for 20 min at 30 °C. Each sonicated sample was then filtered through a nylon membrane syringe (0.45 µm; Infochroma AG) and transferred to a ‘Greiner’ sample tube (50 ml). From the obtained solutions, aerosols were generated in synthetic air (80% volume N2, 20% volume O2; Carbagas) via an apex Q nebulizer (Elemental Scientific, Inc.) operated at 60 °C, dried by a Nafion dryer and directed into a long-time-of-flight AMS. Each sample was recorded for 480 s, with a collection time for each spectrum of ~40 s. Ultrapure water was measured for 720 s before and after each sample measurement. The technique was performed on ~370 extracts (samples and field blanks) in total.
For the data analysis, we used Squirrel v1.59B for the m/z calibration and baseline subtraction, and Pika v1.19B for high-resolution (HR) analysis, in the Igor Pro software package 6.37. The HR peak fitting was performed in the m/z range 12–191. After the peak fitting (fragments consisted of C, O, H, N and S), the isotope ions (and the inorganic fragments) were removed, and the average water-blank signal was subtracted from the average signal of the following sample. The resulting data matrix was the organic fraction mass spectra time series (normalized fragment ion intensity) and an error matrix that included the blank variability and measurement uncertainties. The signal-to-noise ratio (was >2.0 for 390 out of 578 fitted organic fragment ions with m/z up to 133, whereas it dropped to 1.9 on average for fragments with m/z between 134 and 180. The inorganic-salt artefact on the AMS CO2 (ref. 86) and CO fragment ion signal was also accounted for (Supplementary Text 2), but the resulting corrections were minor.
Auxiliary measurements
Additional offline analyses were carried out (Supplementary Text 3); these included the measurement of EC and OC by a thermo-optical transmission method using a Sunset analyser, WSOC by a TOC analyser, major water-soluble ions (including MSA) by ion chromatography and organic markers (sugar-alcohols, sugars and organic acids) for selected samples. Several environmental parameters were retrieved as well, such as temperature, solar radiation and snow-depth data, which were averaged to match the time resolution of the filter sample composites measured by AMS (Supplementary Text 3).
AMS-PMF analysis
Concept
The normalized organic mass spectra from the offline AMS measurements were analysed by PMF87,88 using the multilinear engine 289. The aim was to derive source components (factors) that can be linearly combined to reproduce the observed time and chemical (mass spectral profiles) variations90 in the extracted organics. The separation of OA factors was therefore based on differences in their chemical composition and temporal behaviour, regardless of their original mixing state with other particulate matter components when suspended in the atmosphere. We did not use the absolute AMS signals for the quantification of the organic fraction, as they are affected by the AMS collection and transmission efficiencies and by the nebulization efficiency. Instead, we scaled the relative factor contributions determined by PMF using the TOC-based WSOC (see ‘Final AMS-PMF results’). Therefore, the total OA concentration is not affected by the collection efficiency. All extracted aerosol species are expected to be internally mixed in the nebulized particles, so the offline AMS collection efficiency should be the same for all identified OA factors91,92.
Methodology
The interface for the data processing was provided by the source finder toolkit93 (SoFi version 6.86) for Igor Pro (WaveMetrics, Inc.). PMF attempts to solve the bilinear matrix equation, Xij = ∑n (GinFnj + Eij), by following the weighted least-squares approach. In the case of AMS, i represents the time index, j is the fragment and n is the factor number. If Xij is the matrix of the organic mass spectral data and sij the corresponding error matrix (including the blank variability and measurement uncertainties), Gin is the matrix of the factor time series, Fnj the matrix of the factor profiles and Eij the model residual matrix, then PMF determines Gin and Fnj such that the ratio of the Frobenius norm of Eij over sij is minimized. The allowed Gin and Fnj are always non-negative. The PMF input matrices (Xij and sij) here included data from all the stations (~350 samples) and 578 HR fragments with m/z up to 133 (or 1,029 HR fragments with m/z up to 191). All fragments with a signal-to-noise ratio below 0.2 were removed from the matrices, and those with a signal-to-noise ratio below 2.0 were down-weighted according to the recommendations of Paatero and Hopke (2003)94. For factor identification90, we used a combination of criteria where applicable. These include the factor seasonal cycle, fragmentation pattern, characteristic fragments, time series correlation with external markers, time series correlation with environmental parameters and the BT analysis.
Number of factors
Unconstrained PMF was performed for n = 3–15 factors to choose a ‘base case’ solution before the uncertainty analysis. Five random seed runs were performed for each n (65 runs in total). Preliminary diagnostics were then produced (Supplementary Fig. 1) to investigate the optimum n based on the explained variability of the input matrix (Q/Qexp (exp, expected), scaled residuals) and the stability and/or interpretability of preliminary solutions among different runs for each n. All random seed runs provided essentially identical results (that is, the lowest Q/Qexp relative s.d.) only for the 11-factor solution (the factor profiles are shown in Supplementary Fig. 2). The stability of this preliminary solution only (versus other n) among the different random seed runs is detailed in Supplementary Table 3. The Q/Qexp of this average (‘base case’) 11-factor solution exhibited a random pattern in both dimensions (time series and variables) of the reconstructed PMF output matrix (350 samples and 578 HR fragments up to m/z 133). With regard to a partial exploration of the rotational ambiguity, the 11-factor solution was the most robust (Supplementary Text 4), as other factor solutions were less stable among the different random seed runs compared with that of n = 11 for certain factors (Supplementary Table 3).
PMF errors
The PMF model statistical and rotational uncertainty was assessed via a bootstrapping (BS) approach (Supplementary Text 4) applied to the entire dataset (by including PMF samples from the all stations and seasons). The BS approach generated new input matrices by randomly resampling samples from the original input matrix. Each newly generated PMF input matrix had a total number of samples equal to those of the original matrices, although some of the original filter samples were represented several times, and others were not represented at all. The resulting error interval represents temporal variations of the 11-factor profiles, random errors and errors in the modelling process, such as rotational ambiguity and a mis-specified number of factors95. Note that a preliminary BS analysis was performed also for the 10-factor solution, but the results were less stable than those of the 11-factor solution.
Retention of PMF factors related to ambient organic aerosols
Although the optimum PMF solution contains 11 factors, only the 6 factors presented in the main text and figures were interpreted to be related to the sampled OA. The remaining factors were associated with carbonate and (field) blanks (Supplementary Text 4). The carbonate-related (inorganic) factor may originate from sea spray and/or transported dust from lower latitudes or inner-Arctic sources. It exhibited an unusually high O:C of ~1.9 due to a very high of around 0.8 (Supplementary Fig. 2). We confirmed the presence of carbonate by HCl fumigation and offline AMS experiments for selected samples96,97. All the data presented are corrected for the carbonate (Supplementary Text 4). We separated a factor that was systematically associated with the filter substrate in terms of both the mass spectral fingerprint (factor profile) and the absolute mass concentrations at the different stations (Supplementary Text 4) and three other factors with contributions that could not be decoupled from the background signal of the filter (Supplementary Text 4 and Supplementary Fig. 4).
Final AMS-PMF results
The atomic ratios (O:C, H:C, N:C and S:C) and OM:OC ratios shown in Supplementary Fig. 2 were calculated for all the PMF input samples and for each AMS-PMF factor (for m/z up to 133), using the Analytical Procedure for Elemental Separation version 1.05 within Igor. The error bars in Fig. 1c correspond to 1 s.d. from the 100 BS runs. The median and interquartile range of the factor-specific (k) fractional contribution (Fck) time series that resulted from the BS AMS-PMF analysis were converted into absolute WSOA mass concentrations by multiplying with the AMS-based (WS)OA:(WS)OC ratios of the different PMF-input samples (on carbonate subtraction) times the corresponding TOC-analyser-based WSOC values (total dissolved OC mass without any field blank subtraction): WSOAk = Fck × WSOC × (WSOA/WSOC).
Recovery analysis performed using PMF
Recovery analysis was performed using PMF, following a simplified version of the approach of D. Bhattu et al. (personal communication; see Supplementary Text 4). The lowest water solubility was ~60–80% for POA, PBOA, Haze and OOA, whereas MSA-OA and BSOA can be considered fully water-soluble (Supplementary Text 4 and Supplementary Fig. 9). The AMS-PMF-based WSOA (x, the sum of six WSOA factors) versus the recovery-corrected total OA mass (y, the sum of six OA factors) time series were correlated with an R2 of 0.99 and a slope of 1.16, which indicated that the majority of OAs are water-soluble (the remaining factors were subtracted from both the x and y variables). The reconstructed AMS-PMF-based OC mass (x, the sum of all factors) versus the measured sunset OC (y) were correlated with an R2 of 0.87 and a slope of 1.14, which indicated a sufficient closure within the uncertainties. Although the elemental ratios shown in Supplementary Fig. 2 apply to water-extracted factors, the water solubility of Arctic OAs in this work was high (average ± 1 s.d. = 82 ± 4%), in agreement with previous estimates98, and does not vary significantly between locations and seasons. As a result, the data for the total OA shown in Figs. 1b,c and 2 are similar to those obtained for WSOA (Supplementary Figs. 7 and 8). The corresponding (median) total OA factor time series (Supplementary Fig. 10) were provided on conversion from WSOA values by applying median factor-specific recoveries, and were used as the result with regard to the factor time series. The median time series correlated highly with the base-case-solution time series (Supplementary Table 4).
Source-marker AMS fragments
We provide in Supplementary Table 5 specific fragments identified in our dataset as characteristic of specific sources, which were also identified in previous studies. These fragments were selected based on their highest contribution to these factors and the dominant contribution of these factors to these fragments.
Uncertainties
We briefly discuss here the PMF errors, the errors on the recovery, the errors on the relative ionization efficiency and the errors due to negative artefacts. The BS analysis indicated relative errors below 30% on average for median factor mass concentrations >50 ng m−3, with MSA-OA clearly being the least uncertain factor (Supplementary Text 4). The interquartile range of the median time series corresponds to errors from the BS runs without contingency for errors from the recovery analysis, by applying constant WSOA-to-OA factor-specific (median) conversion factors. We used the same relative ionization efficiency for all OA fractions. Given the recently reported relative ionization efficiencies for organic compounds of 1.2–1.8 (refs. 99–101), we expect the uncertainties that result from this simplification to be on the order of 20–30%, comparable to our modelling error estimates. Although the positive filter artefact is probably captured in this work through the identification of the field blank-related factor, the negative filter artefact (loss of semivolatile organics) could not be extensively assessed due to the current routine filter sampling strategies. In future analyses, it would be useful to address the negative filter artefact and other effects related to sample treatment, such as the effect of filter storage and revolatilization on water extraction, and to compare the offline results with concurrent online measurements.
BT analysis for OA factor geographical origin assessment
BTs show the air mass history (origins and transport paths) and thus can provide information on the geographical location of potentially advected emissions at large geographical scales. Here, BT analysis (Supplementary Text 5) was performed to assess potential source locations of individual Arctic organic components or their precursors over the entire time period covered at each station. The trajectories were calculated backward for up to ten days using the HYSPLIT4 model with meteorological data from the Global Data Assimilation System with 1° resolution (GDAS1; ftp://arlftp.arlhq.noaa.gov/pub/archives/gdas1). The station coordinates correspond to the arriving location of each BT. We weighted the calculated BTs with the AMS-PMF-based factor time series using the CWT model to localize the air parcels responsible for high measured concentrations at the receptor site. For the CWT analysis, the Igor-based user interface ‘ZeFir’ was used48. Although the HYSPLIT trajectory module within ZeFir is currently limited to the use of regular GDAS meteorological field data files, it offers the option to run the model at any date and/or time (for example, an entire year). The average mass concentration of each factor in each sample was enlarged in ZeFir for 2, 7 or 14 days back in time (resolution of 6 h, except for the Russian stations at 4 h), depending on the average sampling time resolution at each station (for example, a 14 day enlargement for a bi-weekly sampling time resolution), and the corresponding enlarged BT results were averaged (within the software) to represent each individual sample. Air parcels rising at altitudes above 3 km a.g.l. were considered less likely to arrive at the receptor site and hence were discarded (a threshold altitude of 1.5 km did not produce substantially different results unless otherwise noted). We assessed the sensitivity of our results to the BT timescales showing that the resulting maps are robust and not too sensitive beyond certain lengths of time (Supplementary Text 5). Multisite merging is a powerful option in ZeFir to explore the bigger picture of source–receptor approaches, as combining several sites together leads to higher trajectory density values. Merged results (without normalization) are presented in Fig. 4 to indicate pan-Arctic hot spots with a greater accuracy than that of single-site results. No weighting function was applied to include the influence of less-frequent air mass trajectories from distant regions (‘long-range’ probability heat maps102). The main observations from individual station results are described in Supplementary Text 5.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41561-021-00891-1.
Supplementary information
Supplementary Text 1–5, Tables 1–7, Figs. 1–16 and references.
Acknowledgements
This project received funding from the European Union’s Horizon 2020 Framework Programme via the ERA-PLANET (The European Network for observing our changing Planet) project iCUPE (Integrative and Comprehensive Understanding on Polar Environments) under grant agreement no. 689443, and the Swiss State Secretariat for Education, Research and Innovation (no. 15.0159-1). J.S. holds the Ingvar Kamprad chair for extreme environments research. V.M., J.S., I.E.H. and O.P., and K.D. and A.S.H.P., acknowledge the SNSF Scientific Exchanges grants ‘Source apportionment of Russian Arctic aerosol’ (SARAA, no. 187566) and SAFICA (no. 189495), respectively. We acknowledge the contribution by the Sino-Swiss Science and Technology Cooperation (SSSTC) within the project HAZECHINA (IZLCZ2_169986). L.H., S.S. and W.Z. acknowledge the A-base fund for supporting climate related long-term observations and research by Environment & Climate Change Canada. We thank the personnel at ECCC, including D. Ernst for technical support for the EC/OC analysis, A. Chivulescu for the ion chromatography analysis, D. Veber for HiVol calibrations, all the operators and coop students for collecting the filters, A. Platt for managing the ALT station and Canadian Forces Station Alert for maintaining the ALT station infrastructure. Sample collections at BAR and TIK were performed under the financial support of the RFBR project no. 18-60084. O.P. and J.S.K. thank RFBR-DFG project no. 20-55-12001 for the support of wildfire ageing impact analyses. The methodology of chemical speciation analyses for BAR and TIK was validated and improved in the framework of the Russian Government grant #075-15-2021-574. UTQ received financial and technical support for this campaign from the US Department of Energy (ARM Field Campaign nos. 2013-6660 and 2014-6694), NOAA (award no. NA14OAR4310150), the Ukpeaġvik Iñupiat Corporation and the C. Gus Glasscock, Jr, Endowed Fund for Excellence in Environmental Sciences. UTQ thanks W. Brower and J. Ivanoff for sample collection and field assistance. Financial support is also acknowledged from the Villum Foundation to finance the establishment of VRS, the Danish EPA and the Danish Energy Agency with means from MIKA/DANCEA funds (Danish EPA project no. MST-113-00-140 and Ministry of Climate, Energy, and Utilities project no. 2018-3767), the iGOSP project and the Graduate School of Science and Technology, Aarhus University. Thanks to the Royal Danish Air Force and the Arctic Command for providing logistic support to the project; C. Christoffersen, B. Jensen, and K. Mortensen are gratefully acknowledged for their technical support. The observations at ZEP, funded by the Norwegian Environment Agency, are part of the national monitoring programme. The Institut des Géosciences de l’Environnement (IGE) thanks financial support for some of the equipment by Labex OSUG@2020 (ANR10 LABX56). A. Amore is acknowledged for taking care of the filters from GRU 2017. V. Hyöky, P. Pietikäinen and M. Hatanpää are acknowledged for regular filter exchanges at PAL. The work of engineers in the lab at IGE (C. Vérin and A. Vella) for auxiliary analyses is also acknowledged.
Author contributions
I.E.H. and V.M. conceptualized the study. V.M. and H.L. performed the AMS measurements. V.M. performed the AMS, PMF and CWT analyses. K.D., D.B. and R.C. assisted with AMS data analysis. F.C. and K.R.D. contributed codes to the offline analysis and SoFi. P.R. assisted with sample extractions for AMS measurements. L.H., W.Z. and S.S. provided EC, OC and ion chromatography data for ALT. O.P. coordinated the filter sampling and provided ion data from BAR and TIK. M.V. set up the filter sampling system at PAL. R.J.S. and C.E.M. organized filter collection at UTQ. C.E.M. performed ion chromatography for UTQ. A.M. and H.S. coordinated the filter sampling at VRS. J.K.N. analysed the OC and EC data from VRS. K.E.Y. and W.A. are responsible for the collection of aerosol filter samples at ZEP. J.-L.J. conducted analyses for EC, OC and dissolved organic carbon, an carried out ion chromatography, high performance liquid chromatography–pulsed amperometric detection and liquid chromatography–mass spectrometry measurements on filters from the different stations that provided the concentrations for tracers, which included MSA, sugar-alcohols and organic acids. I.E.H., J.S., A.S.H.P. and U.B. supported and supervised the research. V.M., I.E.H., J.S. and U.B. interpreted the results. V.M., I.E.H. and J.S. prepared the figures and wrote the manuscript. All the co-authors commented on the manuscript.
Peer review
Peer review information
Nature Geoscience thanks Megan Willis and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Xujia Jiang, Thomas Richardson, in collaboration with the Nature Geoscience team.
Data availability
The data used in this study103 is openly available at Zenodo (10.5281/zenodo.5775070). Please contact the corresponding authors when using the data.
Code availability
Datalystica Ltd is the official distributor of SoFi Pro licenses. Origin was used for graph plotting. The open-source Igor-based package ZeFir is free to download at https://sites.google.com/site/zefirproject/download. The World Maps available with ZeFir are taken from Natural Earth Data.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Julia Schmale, Email: julia.schmale@epfl.ch.
Imad El Haddad, Email: imad.el-haddad@psi.ch.
Supplementary information
The online version contains supplementary material available at 10.1038/s41561-021-00891-1.
References
- 1.Russell LM, Hawkins LN, Frossard AA, Quinn PK, Bates TS. Carbohydrate-like composition of submicron atmospheric particles and their production from ocean bubble bursting. Proc. Natl Acad. Sci. USA. 2010;107:6652–6657. doi: 10.1073/pnas.0908905107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willis MD, Leaitch WR, Abbatt JPD. Processes controlling the composition and abundance of Arctic aerosol. Rev. Geophys. 2018;56:621–671. [Google Scholar]
- 3.Nielsen IE, et al. Biogenic and anthropogenic sources of aerosols at the High Arctic site Villum Research Station. Atmos. Chem. Phys. 2019;19:10239–10256. [Google Scholar]
- 4.Moschos, V. et al. Elucidating the present-day chemical composition, seasonality and source regions of climate-relevant aerosols across the Arctic land surface. Environ. Res. Lett.10.1088/1748-9326/ac444b (2022).
- 5.IPCC Climate Change 2013: The Physical Science Basis (eds Stocker, T. F. et al.) (Cambridge Univ. Press, 2013).
- 6.Bennartz R, et al. July 2012 Greenland melt extent enhanced by low-level liquid clouds. Nature. 2013;496:83–86. doi: 10.1038/nature12002. [DOI] [PubMed] [Google Scholar]
- 7.Kirpes RM, et al. Secondary sulfate is internally mixed with sea spray aerosol and organic aerosol in the winter Arctic. Atmos. Chem. Phys. 2018;18:3937–3949. [Google Scholar]
- 8.Lohmann U, et al. Future warming exacerbated by aged-soot effect on cloud formation. Nat. Geosci. 2020;13:674–680. [Google Scholar]
- 9.Moschos V, et al. Source-specific light absorption by carbonaceous components in the complex aerosol matrix from yearly filter-based measurements. Atmos. Chem. Phys. 2021;21:12809–12833. [Google Scholar]
- 10.Yang Q, Bitz CM, Doherty SJ. Offsetting effects of aerosols on Arctic and global climate in the late 20th century. Atmos. Chem. Phys. 2014;14:3969–3975. [Google Scholar]
- 11.Sand M, et al. Response of Arctic temperature to changes in emissions of short-lived climate forcers. Nat. Clim. Change. 2015;6:286–289. [Google Scholar]
- 12.Jimenez JL, et al. Evolution of organic aerosols in the atmosphere. Science. 2009;326:1525–1529. doi: 10.1126/science.1180353. [DOI] [PubMed] [Google Scholar]
- 13.Moschos V, et al. Source apportionment of brown carbon absorption by coupling ultraviolet–visible spectroscopy with aerosol mass spectrometry. Environ. Sci. Technol. Lett. 2018;5:302–308. [Google Scholar]
- 14.Tomasi C, et al. Aerosol remote sensing in polar regions. Earth Sci. Rev. 2015;140:108–157. [Google Scholar]
- 15.Uttal T, et al. International Arctic systems for observing the atmosphere: an international polar year legacy consortium. Bull. Am. Meteorol. Soc. 2016;97:1033–1056. [Google Scholar]
- 16.Quinn PK, et al. A 3-year record of simultaneously measured aerosol chemical and optical properties at Barrow, Alaska. J. Geophys. Res. Atmos. 2002;107:8–15. [Google Scholar]
- 17.Hirdman D, et al. Long-term trends of black carbon and sulphate aerosol in the Arctic: changes in atmospheric transport and source region emissions. Atmos. Chem. Phys. 2010;10:9351–9368. [Google Scholar]
- 18.Petäjä T, et al. Overview: integrative and comprehensive understanding on polar environments (iCUPE)—concept and initial results. Atmos. Chem. Phys. 2020;20:8551–8592. [Google Scholar]
- 19.Tjernström M, et al. The Arctic Summer Cloud Ocean Study (ASCOS): overview and experimental design. Atmos. Chem. Phys. 2014;14:2823–2869. [Google Scholar]
- 20.Abbatt JPD, et al. Overview paper: new insights into aerosol and climate in the Arctic. Atmos. Chem. Phys. 2019;19:2527–2560. [Google Scholar]
- 21.Schmale J, Zieger P, Ekman AML. Aerosols in current and future Arctic climate. Nat. Clim. Change. 2021;11:95–105. [Google Scholar]
- 22.Pearson RG, et al. Shifts in Arctic vegetation and associated feedbacks under climate change. Nat. Clim. Change. 2013;3:673–677. [Google Scholar]
- 23.Warneke C, et al. An important contribution to springtime Arctic aerosol from biomass burning in Russia. Geophys. Res. Lett. 2010;37:L01801. [Google Scholar]
- 24.Brock CA, et al. Characteristics, sources, and transport of aerosols measured in spring 2008 during the Aerosol, Radiation, and Cloud Processes Affecting Arctic Climate (ARCPAC) Project. Atmos. Chem. Phys. 2011;11:2423–2453. [Google Scholar]
- 25.Mungall EL, et al. Microlayer source of oxygenated volatile organic compounds in the summertime marine Arctic boundary layer. Proc. Natl Acad. Sci. USA. 2017;114:6203–6208. doi: 10.1073/pnas.1620571114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kramshoj M, et al. Biogenic volatile release from permafrost thaw is determined by the soil microbial sink. Nat. Commun. 2018;9:3412. doi: 10.1038/s41467-018-05824-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, et al. Overlooked organic vapor emissions from thawing Arctic permafrost. Environ. Res. Lett. 2020;15:104097. [Google Scholar]
- 28.Corbett JJ, et al. Arctic shipping emissions inventories and future scenarios. Atmos. Chem. Phys. 2010;10:9689–9704. [Google Scholar]
- 29.Schmale J, et al. Local Arctic air pollution: a neglected but serious problem. Earth Future. 2018;6:1385–1412. [Google Scholar]
- 30.Wang Q, et al. Sources of carbonaceous aerosols and deposited black carbon in the Arctic in winter–spring: implications for radiative forcing. Atmos. Chem. Phys. 2011;11:12453–12473. [Google Scholar]
- 31.Browse J, et al. The complex response of Arctic aerosol to sea-ice retreat. Atmos. Chem. Phys. 2014;14:7543–7557. [Google Scholar]
- 32.Sand M, et al. Aerosols at the poles: an AeroCom Phase II multi-model evaluation. Atmos. Chem. Phys. 2017;17:12197–12218. [Google Scholar]
- 33.Croft B, et al. Arctic marine secondary organic aerosol contributes significantly to summertime particle size distributions in the Canadian Arctic Archipelago. Atmos. Chem. Phys. 2019;19:2787–2812. [Google Scholar]
- 34.Mann GW, et al. Intercomparison and evaluation of global aerosol microphysical properties among AeroCom models of a range of complexity. Atmos. Chem. Phys. 2014;14:4679–4713. [Google Scholar]
- 35.Wilson TW, et al. A marine biogenic source of atmospheric ice-nucleating particles. Nature. 2015;525:234–238. doi: 10.1038/nature14986. [DOI] [PubMed] [Google Scholar]
- 36.Creamean JM, et al. Thawing permafrost: an overlooked source of seeds for Arctic cloud formation. Environ. Res. Lett. 2020;15:084022. [Google Scholar]
- 37.Shah, T. Composition of organic gas emissions from flaring natural gas (Ramboll Environ, 2017); https://www.epa.gov/sites/production/files/2017-11/documents/organic_gas.pdf
- 38.Eleftheriadis K, Vratolis S, Nyeki S. Aerosol black carbon in the European Arctic: measurements at Zeppelin station, Ny-Ålesund, Svalbard from 1998–2007. Geophys. Res. Lett. 2009;36:L02809. [Google Scholar]
- 39.Frossard AA, et al. Springtime Arctic haze contributions of submicron organic particles from European and Asian combustion sources. J. Geophys. Res. Atmos. 2011;116:D05205. [Google Scholar]
- 40.Chang RYW, et al. Aerosol composition and sources in the central Arctic Ocean during ASCOS. Atmos. Chem. Phys. 2011;11:10619–10636. [Google Scholar]
- 41.Leaitch WR, et al. Organic functional groups in the submicron aerosol at 82.5° N, 62.5° W from 2012 to 2014. Atmos. Chem. Phys. 2018;18:3269–3287. [Google Scholar]
- 42.AMAP Assessment 2015: Black Carbon and Ozone as Arctic Climate Forcers (AMAP, 2015).
- 43.Polissar AV, Hopke PK, Paatero P, Malm WC, Sisler JF. Atmospheric aerosol over Alaska: 2. Elemental composition and sources. J. Geophys. Res. Atmos. 1998;103:19045–19057. [Google Scholar]
- 44.Nguyen QT, et al. Source apportionment of particles at Station Nord, North East Greenland during 2008–2010 using COPREM and PMF analysis. Atmos. Chem. Phys. 2013;13:35–49. [Google Scholar]
- 45.Winiger P, et al. Source apportionment of circum-Arctic atmospheric black carbon from isotopes and modeling. Sci. Adv. 2019;5:eaau8052. doi: 10.1126/sciadv.aau8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodríguez BT, et al. Seasonal cycle of isotope‐based source apportionment of elemental carbon in airborne particulate matter and snow at Alert, Canada. J. Geophys. Res. Atmos. 2020;125:e2020JD033125. [Google Scholar]
- 47.Daellenbach KR, et al. Characterization and source apportionment of organic aerosol using offline aerosol mass spectrometry. Atmos. Meas. Tech. 2016;9:23–39. [Google Scholar]
- 48.Petit JE, Favez O, Albinet A, Canonaco F. A user-friendly tool for comprehensive evaluation of the geographical origins of atmospheric pollution: wind and trajectory analyses. Environ. Model. Softw. 2017;88:183–187. [Google Scholar]
- 49.Barrett TE, Robinson EM, Usenko S, Sheesley RJ. Source contributions to wintertime elemental and organic carbon in the Western Arctic based on radiocarbon and tracer apportionment. Environ. Sci. Technol. 2015;49:11631–11639. doi: 10.1021/acs.est.5b03081. [DOI] [PubMed] [Google Scholar]
- 50.Stohl A. Characteristics of atmospheric transport into the Arctic troposphere. J. Geophys. Res. Atmos. 2006;111:D11306. [Google Scholar]
- 51.Kawamura K, et al. Source and reaction pathways of dicarboxylic acids, ketoacids and dicarbonyls in Arctic aerosols: one year of observations. Atmos. Environ. 1996;30:1709–1722. [Google Scholar]
- 52.Asmi E, et al. Aerosol size distribution seasonal characteristics measured in Tiksi, Russian Arctic. Atmos. Chem. Phys. 2016;16:1271–1287. [Google Scholar]
- 53.Kolesar KR, et al. Effect of Prudhoe Bay emissions on atmospheric aerosol growth events observed in Utqiaġvik (Barrow), Alaska. Atmos. Environ. 2017;152:146–155. [Google Scholar]
- 54.Hakola H, Hellén H, Laurila T. Ten years of light hydrocarbons (C2–C6) concentration measurements in background air in Finland. Atmos. Environ. 2006;40:3621–3630. [Google Scholar]
- 55.Shaw G. The Arctic haze phenomenon. Bull. Am. Meterol. Soc. 1995;76:2403–2414. [Google Scholar]
- 56.Stone RS, et al. A characterization of Arctic aerosols on the basis of aerosol optical depth and black carbon measurements. Elementa Sci. Anthrop. 2014;2:000027. [Google Scholar]
- 57.Crippa M, et al. Identification of marine and continental aerosol sources in Paris using high resolution aerosol mass spectrometry. J. Geophys. Res. Atmos. 2013;118:1950–1963. [Google Scholar]
- 58.Qin YM, et al. Impacts of traffic emissions on atmospheric particulate nitrate and organics at a downwind site on the periphery of Guangzhou, China. Atmos. Chem. Phys. 2017;17:10245–10258. [Google Scholar]
- 59.Peters GP, et al. Future emissions from shipping and petroleum activities in the Arctic. Atmos. Chem. Phys. 2011;11:5305–5320. [Google Scholar]
- 60.Popovicheva O, et al. East Siberian Arctic background and black carbon polluted aerosols at HMO Tiksi. Sci. Total Environ. 2019;655:924–938. doi: 10.1016/j.scitotenv.2018.11.165. [DOI] [PubMed] [Google Scholar]
- 61.Zhu C, et al. FLEXPART v10.1 simulation of source contributions to Arctic black carbon. Atmos. Chem. Phys. 2020;20:1641–1656. [Google Scholar]
- 62.Sharma S, et al. A factor and trends analysis of multidecadal lower tropospheric observations of Arctic aerosol composition, black carbon, ozone, and mercury at Alert, Canada. J. Geophys. Res. Atmos. 2019;124:14133–14161. [Google Scholar]
- 63.Moffett CE, et al. Long‐term trends for marine sulfur aerosol in the Alaskan Arctic and relationships with temperature. J. Geophys. Res. Atmos. 2020;125:e2020JD033225. [Google Scholar]
- 64.Hu QH, et al. Secondary organic aerosols over oceans via oxidation of isoprene and monoterpenes from Arctic to Antarctic. Sci. Rep. 2013;3:2280. doi: 10.1038/srep02280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fu PQ, et al. Organic molecular composition of marine aerosols over the Arctic Ocean in summer: contributions of primary emission and secondary aerosol formation. Biogeosciences. 2013;10:653–667. [Google Scholar]
- 66.Guenther A, et al. Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature) Atmos. Chem. Phys. 2006;6:3181–3210. [Google Scholar]
- 67.Ricard V, et al. Two years of continuous aerosol measurements in northern Finland. J. Geophys. Res. Atmos. 2002;107:10–17. [Google Scholar]
- 68.Hellén H, et al. Sesquiterpenes dominate monoterpenes in northern wetland emissions. Atmos. Chem. Phys. 2020;20:7021–7034. [Google Scholar]
- 69.Orellana MV, et al. Marine microgels as a source of cloud condensation nuclei in the high Arctic. Proc. Natl Acad. Sci. USA. 2011;108:13612–13617. doi: 10.1073/pnas.1102457108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leck C, Bigg EK. Biogenic particles in the surface microlayer and overlaying atmosphere in the central Arctic Ocean during summer. Tellus B. 2017;57:305–316. [Google Scholar]
- 71.Bozzetti C, et al. Size-resolved identification, characterization, and quantification of primary biological organic aerosol at a European rural site. Environ. Sci. Technol. 2016;50:3425–3434. doi: 10.1021/acs.est.5b05960. [DOI] [PubMed] [Google Scholar]
- 72.Bokhorst S, et al. Changing Arctic snow cover: a review of recent developments and assessment of future needs for observations, modelling, and impacts. Ambio. 2016;45:516–537. doi: 10.1007/s13280-016-0770-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Najafi MR, Zwiers FW, Gillett NP. Attribution of Arctic temperature change to greenhouse-gas and aerosol influences. Nat. Clim. Change. 2015;5:246–249. [Google Scholar]
- 74.Acosta Navarro JC, et al. Amplification of Arctic warming by past air pollution reductions in Europe. Nat. Geosci. 2016;9:277–281. [Google Scholar]
- 75.Box JE, et al. Key indicators of Arctic climate change: 1971–2017. Environ. Res. Lett. 2019;14:045010. [Google Scholar]
- 76.Becagli S, et al. Relationships linking primary production, sea ice melting, and biogenic aerosol in the Arctic. Atmos. Environ. 2016;136:1–15. [Google Scholar]
- 77.Croft B, et al. Processes controlling the annual cycle of Arctic aerosol number and size distributions. Atmos. Chem. Phys. 2016;16:3665–3682. [Google Scholar]
- 78.Arneth A, et al. Terrestrial biogeochemical feedbacks in the climate system. Nat. Geosci. 2010;3:525–532. [Google Scholar]
- 79.Boy M, et al. Interactions between the atmosphere, cryosphere, and ecosystems at northern high latitudes. Atmos. Chem. Phys. 2019;19:2015–2061. [Google Scholar]
- 80.Callaghan TV, et al. The changing face of Arctic snow cover: a synthesis of observed and projected changes. Ambio. 2012;40:17–31. [Google Scholar]
- 81.Lindwall F, Svendsen SS, Nielsen CS, Michelsen A, Rinnan R. Warming increases isoprene emissions from an arctic fen. Sci. Total Environ. 2016;553:297–304. doi: 10.1016/j.scitotenv.2016.02.111. [DOI] [PubMed] [Google Scholar]
- 82.Gali M, Devred E, Babin M, Levasseur M. Decadal increase in Arctic dimethylsulfide emission. Proc. Natl Acad. Sci. USA. 2019;116:19311–19317. doi: 10.1073/pnas.1904378116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.O’Dwyer J, et al. Methanesulfonic acid in a Svalbard ice core as an indicator of ocean climate. Geophys. Res. Lett. 2000;27:1159–1162. [Google Scholar]
- 84.Petäjä T, et al. Research agenda for the Russian far East and utilization of multi-platform comprehensive environmental observations. Int. J. Digit. Earth. 2020;14:311–337. [Google Scholar]
- 85.Ng NL, et al. Changes in organic aerosol composition with aging inferred from aerosol mass spectra. Atmos. Chem. Phys. 2011;11:6465–6474. [Google Scholar]
- 86.Pieber SM, et al. Inorganic salt interference on CO2+ in Aerodyne AMS and ACSM organic aerosol composition studies. Environ. Sci. Technol. 2016;50:10494–10503. doi: 10.1021/acs.est.6b01035. [DOI] [PubMed] [Google Scholar]
- 87.Paatero P, Tapper U. Positive matrix factorization: a non-negative factor model with optimal utilization of error estimates of data values. Environmetrics. 1994;5:111–126. [Google Scholar]
- 88.Ulbrich IM, et al. Interpretation of organic components from positive matrix factorization of aerosol mass spectrometric data. Atmos. Chem. Phys. 2009;9:2891–2918. [Google Scholar]
- 89.Paatero P. The multilinear engine—a table-driven, least squares program for solving multilinear problems, including the n-way parallel factor analysis model. J. Comput. Graph. Stat. 1999;8:854–888. [Google Scholar]
- 90.Zhang Q, et al. Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: a review. Anal. Bioanal. Chem. 2011;401:3045–3067. doi: 10.1007/s00216-011-5355-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bozzetti C, et al. Argon offline-AMS source apportionment of organic aerosol over yearly cycles for an urban, rural, and marine site in northern Europe. Atmos. Chem. Phys. 2017;17:117–141. [Google Scholar]
- 92.O’Brien RE, et al. Ultrasonic nebulization for the elemental analysis of microgram-level samples with offline aerosol mass spectrometry. Atmos. Meas. Techn. 2019;12:1659–1671. [Google Scholar]
- 93.Canonaco F, Crippa M, Slowik JG, Baltensperger U, Prévôt ASH. SoFi, an IGOR-based interface for the efficient use of the generalized multilinear engine (ME-2) for the source apportionment: ME-2 application to aerosol mass spectrometer data. Atmos. Meas. Techn. 2013;6:3649–3661. [Google Scholar]
- 94.Paatero P, Hopke PK. Discarding or downweighting high-noise variables in factor analytic models. Anal. Chim. Acta. 2003;490:277–289. [Google Scholar]
- 95.Reff A, Eberly SI, Bhave PV. Receptor modeling of ambient particulate matter data using positive matrix factorization: review of existing methods. J. Air Waste Manag. Assoc. 2007;57:146–154. doi: 10.1080/10473289.2007.10465319. [DOI] [PubMed] [Google Scholar]
- 96.Karanasiou A, et al. On the quantification of atmospheric carbonate carbon by thermal/optical analysis protocols. Atmos. Meas. Techn. 2011;4:2409–2419. [Google Scholar]
- 97.Vlachou A, et al. Development of a versatile source apportionment analysis based on positive matrix factorization: a case study of the seasonal variation of organic aerosol sources in Estonia. Atmos. Chem. Phys. 2019;19:7279–7295. [Google Scholar]
- 98.Tomasi C, et al. Aerosols in polar regions: a historical overview based on optical depth and in situ observations. J. Geophys. Res. Atmos. 2007;112:D16205. [Google Scholar]
- 99.Canagaratna MR, et al. Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications. Atmos. Chem. Phys. 2015;15:253–272. [Google Scholar]
- 100.Jimenez JL, et al. Comment on “The effects of molecular weight and thermal decomposition on the sensitivity of a thermal desorption aerosol mass spectrometer”. Aerosol Sci. Technol. 2016;50:i–xv. [Google Scholar]
- 101.Xu W, et al. Laboratory evaluation of species-dependent relative ionization efficiencies in the Aerodyne Aerosol Mass Spectrometer. Aerosol Sci. Technol. 2018;52:626–641. [Google Scholar]
- 102.Potier E, et al. Characterizing the regional contribution to PM10 pollution over northern France using two complementary approaches: chemistry transport and trajectory-based receptor models. Atmos. Res. 2019;223:1–14. [Google Scholar]
- 103.Moschos, V. Equal abundance of summertime natural and wintertime anthropogenic Arctic organic aerosols. Zenodo10.5281/zenodo.5775070 (2021). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Text 1–5, Tables 1–7, Figs. 1–16 and references.
Data Availability Statement
The data used in this study103 is openly available at Zenodo (10.5281/zenodo.5775070). Please contact the corresponding authors when using the data.
Datalystica Ltd is the official distributor of SoFi Pro licenses. Origin was used for graph plotting. The open-source Igor-based package ZeFir is free to download at https://sites.google.com/site/zefirproject/download. The World Maps available with ZeFir are taken from Natural Earth Data.