

Single-cell transcriptomic techniques have enabled investigation of vascular smooth muscle cell (SMC) phenotype in clinical thoracic aortic aneurysm (TAA)1 and analysis of dynamic, patterned transcriptomic changes associated with aneurysm in Marfan syndrome (MFS), the most common hereditary TAA syndrome2. Despite this progress, monogenic hereditary aortopathies encompass a minority of proximal TAA (affecting the aortic root and ascending aorta) disease,3 and the translatability of MFS findings to the broader spectrum of thoracic aortic disease is uncertain.
Clinically, hypertension remains the greatest risk factor for proximal TAA and dissection (a distinct pathologic entity from atheromatous aneurysm affecting the descending thoracic and abdominal aortic segments), but rigorous assessment of hypertension-induced aortic SMC phenotype modulation is lacking. To establish a central hypertension model, we performed transverse aortic constriction (TAC) to induce aortic arch stenosis to 27-gauge needle diameter. Concurrent left heart catheterization confirmed a mean 56.4 mmHg increase (range 44–66mmHg) in central systolic blood pressure in anesthetized 12-week-old mice (n=3, Figure 1A). To test the hypothesis that hypertension would induce SMC aneurysmal phenotype, we generated a Cre-lox mediated transgenic fluorescent SMC tracing model (TgMyh11-CreERT2/RosatdTomato, ‘SMClin’) then induced permanent SMC tracing via tamoxifen injection at 8 weeks followed by 27G TAC at 12 weeks. Sham group animals underwent identical aortic manipulation and encirclement followed by loose knot tying (no stenosis).
Figure 1.
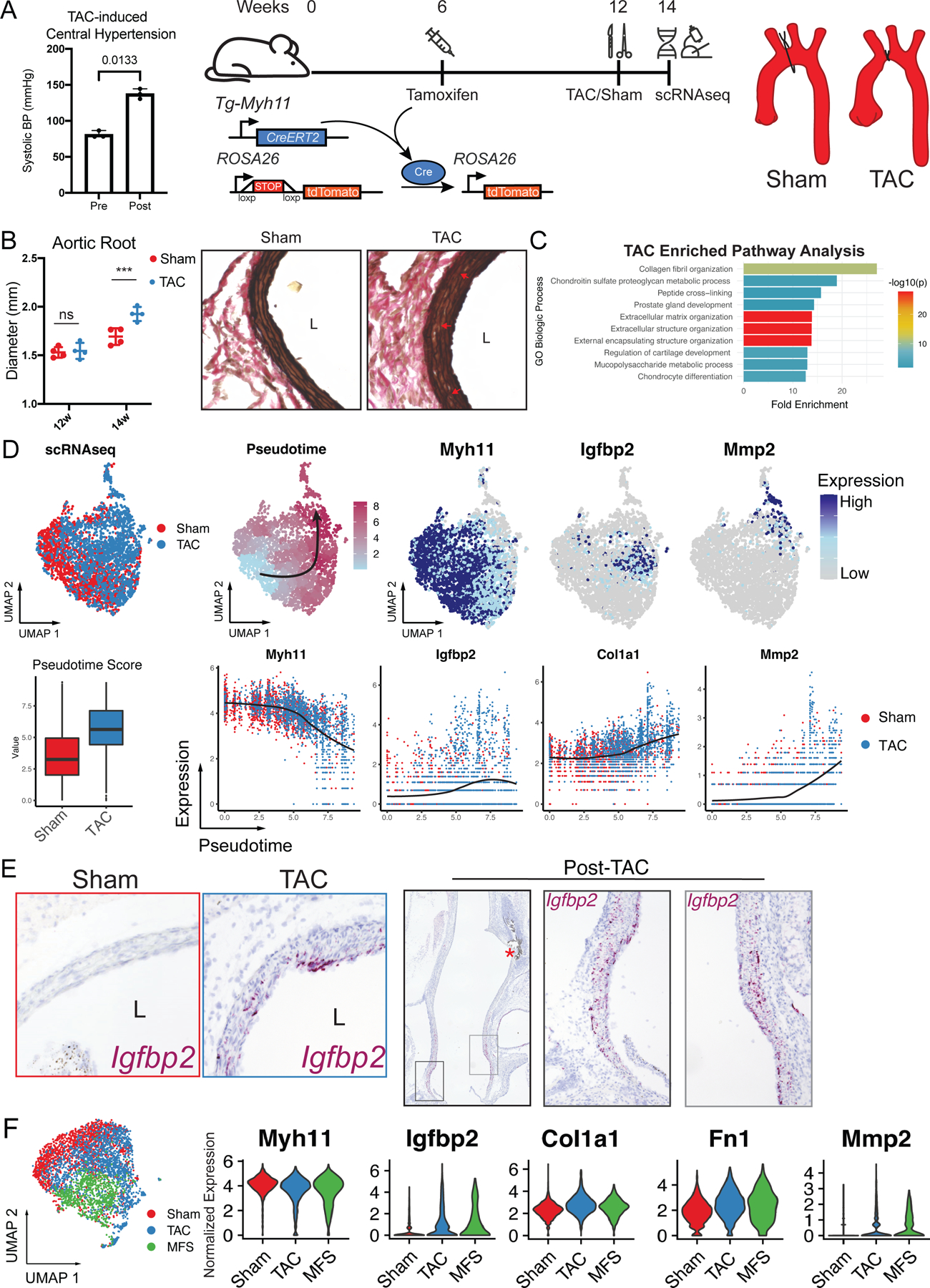
A Experimental model. Invasive left heart catheterization data validating central hypertension induced acutely by 27G TAC in n=3 anesthetized mice pre and post suture tying, p value reflects paired t-test. Cartoon (right) depicts workflow for survival TAC experiment. SMC lineage-traced mice (TgMyh11-CreERT2/RosatdTomato/+) mice were treated with tamoxifen at 6 weeks to induce SMC labeling, followed by either TAC or Sham surgery at 12 weeks and sacrifice two weeks post-surgery. B Post-TAC or Sham transthoracic echocardiography and histology demonstrate mild aortic root aneurysm and elastin fragmentation. *** indicates p < 0.01 using t-test. C Functional annotation for gene ontology (GO) pathways enriched in TAC SMCs using 241 enriched genes as input, ranked by fold enrichment. p value determined using Bonferroni correction for multiple comparisons. D Top row: UMAP distribution of SMCs following single cell RNA sequencing (scRNAseq) labeled by surgical procedure (left) and pseudotemporal ordering values (‘Pseudotime’). Higher pseudotime values correspond with expression of aneurysm markers (reduced Myh11, enriched Igfbp2 and Mmp2) in UMAP space. Bottom row: TAC SMCs demonstrate higher pseudotime scores compared to sham (bar indicates median, box indicates 25th and 75th percentiles, whiskers indicate 1.5 interquartile range Normalized gene expression plots for highlighted genes across the pseudotime spectrum confirm association of pseudotime scores with markers of SMC modulation. E RNA in situ hybridization for Igfbp2, a marker for early SMC phenotype modulation in the scRNAseq dataset. Left panel depicts 20X magnification at the aortic root level, right panel depicts stitched long-axis photomicrographs of post -TAC aorta revealing highest Igfbp2 mRNA density in the aortic root and proximal ascending aorta. F Analysis of combined TAC/Sham and Marfan syndrome (MFS) SMC scRNAseq dataset (left) identifies similar core gene expression changes between models including heterogeneously reduced contractile gene expression, Igfbp2 activation, and concomitant extracellular matrix component overexpression.
After two weeks, TAC evoked mild aortic root dilation via echocardiography (1.93±0.07 mm vs. 1.69±0.09 mm, n=4, p<0.01, Figure 1B) and histologic adventitial hyperplasia with mild elastin fiber fragmentation compared to sham, consistent with prior reports4. To comprehensively analyze hypertension-induced SMC phenotype, we performed enzymatic aortic digestion and FACS purification of fluorescent (tdTomato+) SMCs from n=4 TAC and sham mice followed by single cell RNA sequencing (scRNAseq) library preparation. Global comparison of TAC vs. sham SMCs identified 388 differentially expressed genes between groups. Gene ontology (GO) analysis for the 241 genes overexpressed in TAC SMCs highlighted extracellular matrix modulatory pathway enrichment (Figure 1C). To fully harness scRNAseq analysis of heterogeneous cell behavior, we dimensionally reduced individual cell transcriptomic identities into a two-dimensional uniform manifold approximation and projection (UMAP) rendering, identifying distinct distributions for TAC and sham SMCs suggesting disparate phenotypes between these groups. To quantitatively analyze this heterogeneity, we performed unsupervised pseudotemporal ordering, which utilizes the continuous nature of cell phenotype transitions to construct a trajectory of inferred temporal changes and assign numerical ‘pseudotime’ values to each cell along this continuum. TAC SMCs had higher pseudotime values confirming greater phenotypic variation from the quiescent SMC baseline. To determine whether TAC induces a MFS-like SMC modulatory phenotype, we analyzed expression patterns of previously identified aneurysm-associated genes2 throughout the pseudotemporal transition. Consistent with MFS, we found contractile gene expression silencing (e.g., Myh11) associated with higher pseudotime, consistent with phenotypic switch and identified insulin-like growth factor binding protein (Igfbp2) as an ‘early’ marker of this switch (mid pseudotime) suggesting that this gene is a transitional marker in TAA. The extreme end of the phenotypic spectrum was defined by enriched ECM component (e.g., Col1a1, Fn1) and proteolytic matrix metalloproteinase (e.g., Mmp2/Mmp14) gene expression (Figure 1D). To validate the enrichment of these genes within the aorta, we performed RNA in situ hybridization, localizing Igfbp2-expressing SMCs (cells in phenotypic transition) within the tunica media layer of TAC proximal thoracic aortas (Figure 1E).
Finally, to directly compare SMC phenotype between syndromic and pressure-induced aortopathies, we analogously generated SMC scRNAseq libraries from 4-month-old SMClin male mice harboring the Fbn1C1041G/+ MFS mutation (n=4) and integrated this data into the TAC/Sham dataset. (Figure 1F). Direct comparison of SMCs between these models identified similar heterogeneous contractile gene silencing (e.g., Myh11) and enriched expression of Igfbp2 and critical mediators of ECM disarray in MFS aneurysm (Col1a1, fibronectin (Fn1), and Mmp2). Taken together, we conclude that acute pressure overload activates a similar choreographed gene regulatory pathway as primary Fbn1 loss-of-function, ultimately promoting aortic ECM remodeling.
This data confirms that acute central hypertension is sufficient to recapitulate the ECM-modulatory SMC pathology observed in MFS. This finding has multiple ramifications for future studies. First, TAC may serve as a functional model for testing potential modulators of SMC phenotype derangement during TAA development (e.g., genetic knockouts of upstream transcription factors or novel therapies) in an acute experimental setting. Second, by studying the effects of pressure overload on SMCs on a healthy mouse background, we confirm that the patterned transcriptomic shift observed in MFS is more broadly applicable to hypertension-induced aortic degeneration.
This study is limited by the artificial mechanism of TAC-induced hypertension; however TAC is free from confounding cell signaling activities of alternative models such as angiotensin II infusion, enabling a more direct assessment of SMC response to pressure overload. Furthermore, bioinformatic pseudotemporal ordering implies, but does not prove the temporal nature of ‘early’ and ‘late’ transcriptomic changes without empiric longitudinal study. Finally, while core SMC transcriptomic changes in hypertension-induced aortopathy correspond with those in MFS, rigorous characterization of the distinctions between these models and identification of cohesive upstream triggering molecular events, such as altered mechanosensing or disrupted elastin-contractile units5, represents an important area of future investigation.
Acknowledgments:
We thank the Stanford Genome Sequencing Service Center for assistance with single-cell RNA library preparation and sequencing.
Sources of Funding:
This work was supported by grants from National Institute of Health F32HL154681 (AJP), F32HL160058 (ARD), R01AR066629 (MPF), R01HL157949 (MPF), and from the American Heart Association #834986 (RS). The NovaSeq 6000 device used in this study was purchased using NIH grant S10OD025212 funds.
Footnotes
Disclosures: None
References
- 1.Li Y et al. Single-Cell Transcriptome Analysis Reveals Dynamic Cell Populations and Differential Gene Expression Patterns in Control and Aneurysmal Human Aortic Tissue. Circulation 142, 1374–1388 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pedroza AJ et al. Single-Cell Transcriptomic Profiling of Vascular Smooth Muscle Cell Phenotype Modulation in Marfan Syndrome Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol 40, 2195–2211 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Isselbacher EM, Lino Cardenas CL & Lindsay ME Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation 133, 2516–2528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuang S-Q et al. Aortic remodeling after transverse aortic constriction in mice is attenuated with AT1 receptor blockade. Arterioscler. Thromb. Vasc. Biol 33, 2172–2179 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milewicz Dianna M et al. Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections. Arterioscler. Thromb. Vasc. Biol 37, 26–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]