

Abstract
Previously, organophosphate flame retardants (OPFRs) were found to produce intersecting disruptions of energy homeostasis using an adult mouse model of diet-induced obesity (Vail et al. 2020). Using the same mixture consisting of 1 mg/kg/day of each triphenyl phosphate, tricresyl phosphate, and tris(1,3-dichloro-2-propyl)phosphate, the current study aimed to identify the role of estrogen receptor alpha (ERα) in OPFR-induced disruption, utilizing ERα knockout (ERαKO) mice fed either a low-fat diet (LFD) or high-fat diet (HFD). Body weight and composition, food intake patterns, glucose and insulin tolerance, circulating peptide hormones, and expression of hypothalamic genes associated with energy homeostasis were measured. When fed HFD, no marked direct effects of OPFR were observed in mice lacking ERα, suggesting a role for ERα in generating previously reported wildtype (WT) findings. Male ERαKO mice fed LFD experienced decreased feeding efficiency and altered insulin tolerance, whereas their female counterparts displayed less fat mass and circulating ghrelin when exposed to OPFRs. These effects were not noted in the previous WT study, indicating that loss of ERα may sensitize animals fed LFD to alternate pathways of endocrine disruption by OFPRs. Collectively, these data demonstrate both direct and indirect actions of OPFRs on ERα-mediated pathways governing energy homeostasis and support a growing body of evidence urging concern for risk of human exposure.
Keywords: flame retardants, metabolism, ingestive behavior, diet-induced obesity, ER-alpha
Introduction
Endocrine disrupting chemicals (EDCs) are substances capable of disrupting typical endocrine system function (Barouki 2017). The endocrine system plays a vital role in maintaining bodily homeostasis and EDC interference might result in reprotoxic, immunotoxic, neurotoxic, and obesogenic endpoints. It should be noted that EDCs are found in a variety of household products, including toys, furniture, most plastics, nail polish, and foodstuffs (Li et al. 2019; Peng et al. 2020; Yang et al. 2019; Young et al. 2018). The now ubiquitous nature of EDCs within the environment acknowledges EDCs as a substantial human health risk (National Academy of Sciences 2019). In the past, toxicological research motivated regulatory action to limit human exposure for such EDCs such as polybrominated diphenyl ethers (PBDEs) (Zota et al. 2013). PBDEs were a popular variety of flame-retardant (FR) chemicals utilized for their dampening effect on flammability (Cádiz et al. 2011), until data revealed toxicological impacts on reproduction, neurodevelopment, and thyroid homeostasis (Linares et al. 2015; Gilbert et al. 2012; Herbstman et al. 2010; Zota et al. 2011). Subsequent decline in PBDE use was matched with rise of another type of FR known as organophosphate flame retardants (OPFRs) (Israel Chemicals ltd. 2015; van der Veen and de Boer 2012; Yasin et al. 2016; Zota et al. 2013). OPFRs were originally postulated to be a safer alternative to PBDEs since these compounds were expected to exhibit less persistence in the environment (Zhang et al. 2016). Despite this, their overwhelming usage enabled OPFR detection at concerning concentrations in human serum (680-709 ng/ml), breast milk (1-10 ng/ml), and urine (1-10 ng/ml) samples (Butt et al. 2014; Hoffman et al. 2017; Ma et al. 2017; 2019; Meeker et al. 2013). Further, OPFRs are now known to exert similar toxicological impacts as their predecessors, demonstrating neurological, reproductive, immune, obesogenic, and endocrine disruptive effects (Belcher et al. 2014; Dishaw et al. 2011; Hu et al. 2019; Kylie et al. 2018; Liu et al. 2012; 2013; National Academy of Sciences 2019; Patisaul et al. 2013; Pillai et al. 2014; Steves et al. 2018). In combination with their prevalent human exposure, OPFRs present as a strikingly unaddressed human health risk.
It is well known that the ability of OPFR to affect the endocrine system was identified, in part, through an interaction with nuclear hormone receptors such as peroxisome proliferator-activated receptor gamma (PPARγ) and estrogen receptor alpha (ERα) (Kojima et al. 2013; Ji et al. 2020; National Academy of Sciences 2019; Li et al. 2020). This study focused on OPFR interactions with ERα, and three commonly used OPFRs that demonstrate this association are triphenyl phosphate (TPP), tris(1,3-dichloro-2-proply)phosphate (TDCPP), and tricresyl phosphate (TCP). In vitro studies demonstrated that TPP activates ERα (Kojima et al. 2013; Li et al. 2020), and TDCPP is known to upregulate ERα and associated genes (Liu et al. 2013). TCP also demonstrates agonistic activity on ERα (Kojima et al. 2013). However, Liu et al. (2012) noted that TPP, TDCPP, and TCP all acted as antagonists to ERα, blocking receptor binding of estrogen (E2), but produced elevated E2 and testosterone levels in developing zebrafish. The mixed capacity for disruption may be attributed to weak agonistic activity of these compounds competing with endogenous ligand signaling. Regardless, the ability of TPP, TDCPP, and TCP to interact with ERα is the basis for these chemicals to be selected for our current study investigating OPFR-mediated EDC action on ERα associated signaling.
ERα is involved in many homeostatic pathways, one of which is the maintenance of energy homeostasis (Mauvais-Jarvis et al. 2013). ERα and its endogenous ligand 17β-estradiol initiate an overall “catabolic” effect, decreasing energy intake and increasing its expenditure (Mauvais-Jarvis et al. 2013). Disruption of estrogenic regulation of energy balance might result in metabolic syndrome and its symptomatic sequelae obesity, hypertension, inflammation and pre-diabetes (Dabass et al. 2018; Hevener et al. 2015; Kobos et al. 2020). ERα is densely present within adipose tissue, where it acts to regulate fat distribution and storage (Rettberg et al. 2014). Menopause and the resulting decline in circulating E2 is associated with increased bodyweight gain, altered leptin and adiponectin levels, and elevated risk for obesity and type 2 diabetes development (Rettberg et al. 2014). The integrated crosstalk between ERα and insulin-like growth factor 1 receptor (IGF-1R) has been extensively studied and indicates the role of E2 in insulin signaling (Garcia-Segura et al. 2010; Kahlert et al. 2000; Mendez and Garcia-Segura 2006; Shen et al. 2014; Song et al. 2004). Further, ERα knockout (ERαKO) mice exhibit insulin insensitivity and severe intra-abdominal obesity (Heine et al. 2000), the influence of which was potentiated by a high-fat diet (Ribas et al. 2010). ERα is also expressed within the brain, and particularly concentrated in the arcuate (ARC) nucleus of the hypothalamus (Shughrue et al. 1997). The ARC contains neuropeptide Y (NPY) and proopiomelanocortin (POMC) neurons, both of which are integral in central regulation of feeding behavior and are regulated by E2 actions through multiple types of estrogen receptors (Acosta-Martinez et al. 2007; Saito et al. 2016; de Souza et al. 2011; Stincic et al. 2018).
Previously, Vail et al. (2020) demonstrated that sub-chronic OPFR exposure within adult, wildtype mice elicited sex-dependent alterations in feeding behavior and energy homeostasis. These effects interacted with diet-induced obesity, resulting in exposed males gaining more weight and fat mass only when fed a high-fat diet (HFD). In addition, while in exposed females no marked body weight effects were noted, these animals ate less food and consumed fewer HFD meals per day (Vail et al. 2020). Because ERα regulates central and peripheral control of feeding behavior, and since OPFRs are known to interact with each of these receptors, it was postulated that OPFR-initiated dysregulation of energy homeostasis may be associated with disruption of ERα signaling. To further elucidate the association of ERα with the toxicological effects of OPFR on ingestive behavior, this study utilized global ERα knockout mouse models fed either a LFD or HFD.
Materials and Methods
Animals
All animal experiments were conducted with approval by the Rutgers University Institutional Animal Care and Use Committee and followed National Institution of Health standards. Estrogen receptor alpha knockout transgenic mice (ERαKO) were selectively bred as previously described (Yasrebi et al. 2017) and were fed food and water ad libitum and maintained under controlled temperature (23°C) and photoperiod conditions (12/12 hr light/dark cycle). At weaning, animals were number-tagged and ear-clipped for genotyping and fed a standard low-phytoestrogen chow diet (Lab Diets 5V75) until the start of experimentation.
Genotyping
Ear-clippings were removed from all mice for genotyping at weaning, and again upon euthanasia after experimental completion for genotype confirmation. Genotyping for ERαKO mice was determined using previously published protocols using forward (Ex3a-F: CTGTAGGCTTTGTCTTCGCTTT) and reverse primers (Ex3a-R: CAACCAAGGAGAACAGACAGACTTA) (Hewitt et al. 2010; 2014).
Diets
To examine the intersection of adult OPFR exposure and PPARγ and ERα influence on diet-induced obesity, male and female ERαKO mice were fed either a low-fat diet (LFD, 3.85 kcal/g, 10% fat, 20% protein, 70% carbohydrate; D12450H) or high-fat diet (HFD, 4.73 kcal/g, 45% fat, 20% protein, 35% carbohydrate; D12451; Research Diets). Starting at 10 weeks of age, mice were continually fed either LFD or HFD concurrently with OPFR treatment up through study completion.
OPFR Dosing
A singular mixture of three OPFRs were used in this study to emulate the mixed nature of human exposures. OPFRs utilized were tricresyl phosphate (TCP, CAS no. 1330-78-5; purity 99%; purchased from AccuStandard, New Haven, CT), and triphenyl phosphate (TPP, CAS no. 115-86-6; purity 99%) and tris (1,3-dichloro-2-propyl)phosphate (TDCPP, CAS no. 13674-87-8; purity 95.6%) (both purchased from Sigma-Aldrich, St. Louis, MO). One hundred mg of each OPFR were dissolved as a mixture within the same 1 ml acetone (Sigma) for long term storage to generate 1 mg/ml stock mixture of OPFR-acetone. A working solution was generated by transferring 100 μl OPFR-acetone into 10 ml sesame oil (Sigma-Aldrich) to create an oil mixture containing 1 mg/ml OPFR (OPFR-oil). To generate control-oil mixture, 100 μl acetone was added to 10 ml sesame oil (control-oil). OPFR-oil and control-oil mixtures were left stirring for 48-72 hr to evaporate the acetone from the mixture. Based upon body weight, the resulting mixtures were then added to just enough dehydrated peanut butter vehicle (approximately 50 mg) to create an appetizing rehydrated peanut butter mixture with a final concentration of 1 mg/kg bw OPFR or equivalent amount of OPFR-free peanut butter. The resulting 1 mg/kg bw doses were placed on weight paper and supplied to mice daily to be consumed orally. Oral exposure began at 10 weeks of age at 0900-1100 hr each day for a total of 7 weeks in a sub-chronic paradigm.
Experimental Design
Adult male and female mice (n = 8 per gender, per diet, per treatment) were weight-matched in paired housing and fed either LFD or HFD and dosed orally, daily with OPFR-oil or control-oil for the entirety of the 7-week experimental timeline. Mice started treatment in sequential batches of 8 male and 8 female mice to ensure adequate sample size for metabolic and feeding behavior studies. Body composition (fat and lean mass) were quantified by EchoMRI™ Body composition (Houston, TX) on the first day of dosing (baseline), and again after 4 weeks of treatment with OPFR-oil mixture. During this time, body weight and crude food intake per cage were measured weekly. Subsequently, mice were transferred to the Biological Data Acquisition (BioDAQ, Research Diets, New Brunswick, NJ) chambers for 1 week. Mice were single-housed and continually dosed with OPFR-oil or control-oil during this time. Mice received 94 hr habituation and 72 hr data acquisition for feeding behaviors (meal size, duration, frequency). Touch-sensitive hoppers containing LFD or HFD chow recorded food intake as decreased chow weight within the hoppers. Whenever the hopper was touched for food, the software marked that as a “bout.” The first bout denoted the start of a “meal”, which could consist of any number of bouts, until the inter-bout interval exceeded 300 sec. The next recorded bout after which would then mark the beginning of a new meal. Some mice fed HFD exhibited what is to be referred to as “food grinding” behavior, where chow was removed from the hopper but employed for enrichment chewing, and not actually consumed. When food grinding behavior was observed, feeding data for that day was excluded from analysis for the respective mouse. This accounts for the variation in n within our feeding behavior data. Lastly, all mice were tested for glucose and insulin tolerance. At this point, mice had already undergone 5 weeks of OPFR- or control-oil exposure. Prior to the glucose tolerance test (GTT), mice were fasted for 5 hr and then intraperitoneally (ip) injected with a bolus of 2 g/kg glucose. Blood-glucose was measured from tail bleeds using an AlphaTrak glucometer (Zoetis, Parsippany, NJ) at 0, 15, 30, 60, 90, and 120-min post-injection. Mice were provided a 4-day recovery period before then undergoing the insulin tolerance test (ITT). After a 4 hr fast, mice were injected ip with 0.75 U/kg insulin and blood-glucose was measured from tail bleeds at 0, 15, 30, 60, 90, and 120 min. One week following the ITT, mice were dosed with OPFR- or control-oil at 0900 hr, fasted at 1000 hr, and euthanized at 1100 hr by decapitation after sedation with 100 mg/ml ketamine. Female mice were euthanized during diestrus, as determined by vaginal cytology, to control for circulating ovarian hormone levels. Terminal trunk blood was collected in K+-EDTA coated tubes with proteinase inhibitor 4-(2-aminoethyl) benzene sulfonyl fluoride hydrochloride (1 mg/ml, Sigma-Aldrich) to inhibit peptide degradation. Samples were kept on ice until centrifugation at 1,100 g for 15 min at 4°C. Plasma supernatant was collected and stored at −80°C for analysis of insulin, leptin, and ghrelin levels, using a multiplex assay (MMHMAG-44 K, EMD Millipore, Billerica, MA). In addition, microdissection samples from the arcuate nucleus of the hypothalamus were collected and stored at −80°C for later RNA extraction and conversion to cDNA in preparation for quantification according to Yasrebi et al. (2016). A graphical depiction of the experimental outline is summarized in Figure 1.
Figure 1.
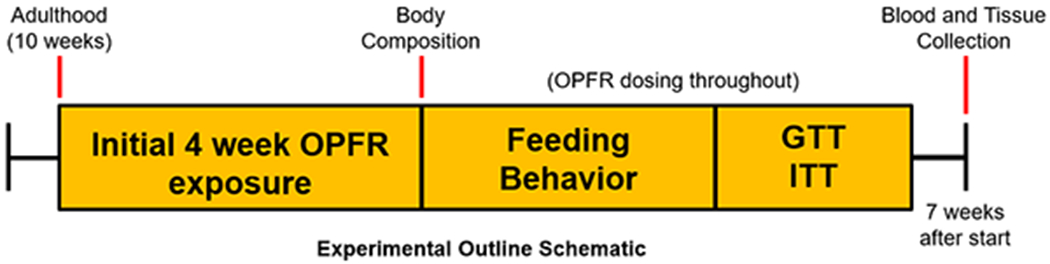
Experimental timeline graphic. OPFR- and control-oil dosing began at 10 weeks of age and continued for the entire duration of experiments. At this time initial bodyweight and body composition data were acquired. During the first 4 weeks, bodyweight and crude food intake were measured weekly. After 4 weeks body composition was measured again. Next, feeding behavior was analyzed, followed by glucose and insulin tolerance tests. After recovery from tolerance tests, animals were euthanized for tissue and blood collection.
Real-time Quantitative PCR
All primers for real-time polymerase chain reaction quantification (rt-qPCR) were designed to span exon-exon junctions using Clone Manager 5 software (Sci Ed Software, Cary, NC) and synthesized by Life Technologies and are listed in Table 1. A CFX-Connect Real-time PCR instrument (BioRad, Hercules, CA) was utilized to amplify 4 μL ARC cDNA using either PowerSYBR Green master mix (Life Technologies) or SsoAdvanced SYBR Green (BioRad). Amplification of all genes used the following protocol: initial denaturing at 95°C for 10 min (PowerSYBR) or 3 min (SsoAdvanced) followed by 40-45 cycles of amplification by alternating 10 sec of denaturing at 94°C and 45 sec of annealing at 60°C. A final dissociation step was incorporated for melting point analysis by 60 cycles of 95°C for 10 sec, 65°C to 95°C (stepping 0.5°C increments each cycle) for 5 sec, and 95°C for 5 sec. Standard curves for each primer pair were generated using serial dilutions of basal hypothalamic cDNA in triplicate to determine efficiency [E = 10(−1/m)–1, m = slope] of each primer pair and are denoted in Table 1.
Table 1.
List of primer pairs for real-time quantitative PCR
| Gene Name | Product Length | Primer Eff. (%) | Primer sequence | Base Pair # | Accession # |
|---|---|---|---|---|---|
| Actβ | 63 | 101 | F:GCCCTGAGGCTCTTTTCCA R:TAGTTTCATGGATGCCACAGGA |
849-867 890-911 |
NM_007393.3 |
| Agrp | 146 | 105 | F:CTCCACTGAAGGGCATCAGAA R:ATCTAGCACCTCCGCCAAA |
287-307 414-432 |
NM_007427.2 |
| Cart | 169 | 95 | F:GCTCAAGAGTAAACGCATTCC R:GTCCCTTCACAAGCACTTCAA |
227-297 425-445 |
NM_013732 |
| Gapdh | 98 | 93 | F:TGACGTGCCGCCTGGAGAAA R:AGTGTAGCCCAAGATGCCCTTCAG |
778-797 852-875 |
NM_008084.2 |
| Ghsr | 122 | 123 | F:CAGGGACCAGAACCACAAAC R:AGCCAGGCTCGAAAGACT |
1003-1022 1107-1124 |
NM_177330 |
| Hprt | 117 | 107 | F:GCTTGCTGGTGAAAAGGACCTCTCGAAG R:CCCTGAAGTACTCATTATAGTCAAGGGCAT |
631-658 718-747 |
NM_013556 |
| Insr | 89 | 114 | F: GTGTTCGGAACCTGATGAC R: GTGATACCAGAGCATAGGAG |
1215-1233 1686-1706 |
NM_010568 |
| Lepr | 149 | 105 | F:AGAATGACGCAGGGCTGTAT R:TCCTTGTGCCCAGGAACAAT |
3056-3075 3185-3204 |
NM_146146.2 |
| Npy | 182 | 100 | F:ACTGACCCTCGCTCTATCTC R:TCTCAGGGCTGGATCTCTTG |
106-125 268-287 |
NM_023456 |
| Pomc | 200 | 103 | F:GGAAGATGCCGAGATTCTGC R:TCCGTTGCCAGGAAACAC |
145-164 327-344 |
NM_008895 |
| Pparg | 113 | 103 | F: CTGCTCAAGTATGGTGTCCATGAG R: GAGGAACTCCCTGGTCATGAATC |
1076-1101 1166-1188 |
NM_011146.3 |
Reference genes: Actβ, Gapdh, Hprt. Actβ = β-actin; Agrp = agouti-related peptide; Cart = cocaine- and amphetamine-regulated transcript; Npy = neuropeptide Y; Pomc = proopiomelanocortin; Gapdh = glyceraldehyde-3-phosphate dehydrogenase; Ghsr = growth hormone secretagogue receptor (ghrelin receptor); Hprt = hypoxanthine-guanine phosphoribosyltransferase; Insr = insulin receptor; Lepr = leptin receptor; Pparg = peroxisome proliferator-activated receptor gamma.
Reference genes used for target gene comparison were Actb (β-actin), Gapdh (glyceraldehyde-3-phosphate dehydrogenase), and Hprt (Hypoxanthine-guanine phosphoribosyltransferase). Diluted (1:20) basal hypothalamic (BH) cDNA from an untreated, intact wild-type male was employed as a positive control, and negative controls consisted of a water blank and the same BH RNA sample, but not converted to cDNA. Quantification data were excluded from samples that did not show a single product at the expected melting point. All gene expression data were calculated using the geometric mean of the reference genes Actb, Gapdh, and Hprt. Relative mRNA expression data were then analyzed using the ΔΔCq method, normalizing to control-LFD samples (Livak and Schmittgen 2001; Pfaffl 2001; Schmittgen and Livak 2008).
Data Analysis
All data are depicted as mean ± SEM. Data were analyzed using either GraphPad Prism software (GraphPad Software, LA Jolla, CA) by a two-way ANOVA (OPFR and Diet) with a post-hoc Newman-Keul’s multiple comparisons test, or with Statistica 7.1 software (StatSoft, Tulsa, OK, USA) using both multi-factorial ANOVA with repeated-measures, and a three-way ANOVA (Diet, OPFR, Time), followed with post-hoc Newman-Keul’s multiple comparisons test. Effects were considered significant at P ≤ 0.05.
Results
Physiological Parameters
Body weight and crude food intake were measured over the course of 4 weeks in male and female mice lacking expression of estrogen receptor alpha (ERαKO). During this time, mice received either control-oil or OPFR-oil mixture (1 mg/kg each of TCP, TPP, and TDCPP). Feeding efficiency was calculated as the ratio of body weight gain to crude food intake and represented as grams gained to kcal consumed. Subsequently, body composition of lean and fat mass was assessed by EchoMRI™ (Figures 2–3). Baseline body weights were taken at day zero, just prior to treatment and diet initiation (males: control – 23.3 ± 0.4 g, OPFR – 24.2 ± 0.4 g; females: control – 23.2 ± 0.5 g, OPFR – 22.5 ± 0.5g).
Figure 2.
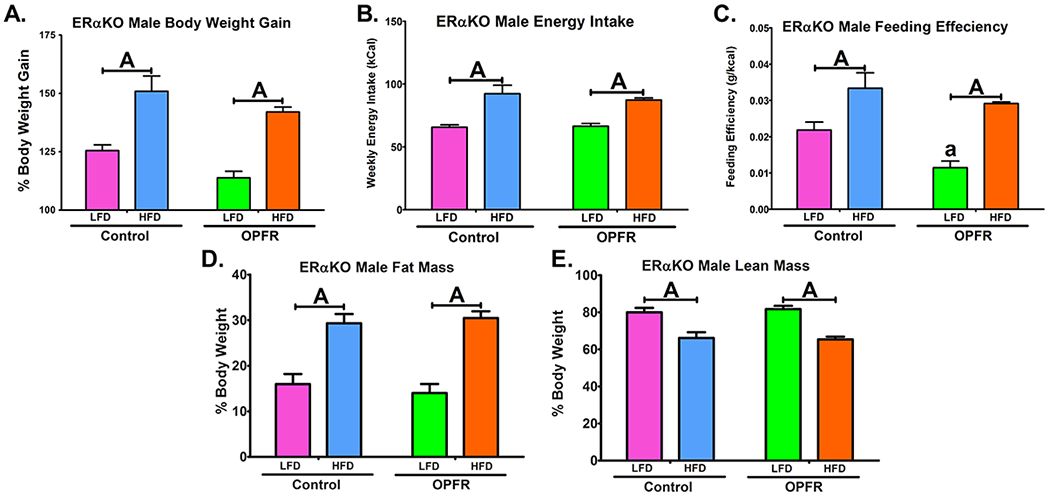
Physiological parameters in ERαKO males orally dosed with an OPFR mixture (1 mg/kg bw) for 4 weeks. (A) % Body Weight Gain over 4 weeks; (B) Energy Intake; (C) Feeding Efficiency; (D) Body composition % Fat Mass; (E) Body composition % Lean Mass. Data were analyzed by a two-way ANOVA with post-hoc Newman-Keuls multiple comparisons test. Uppercase letters denote diet effects within exposure group. Lowercase letters denote OPFR effects within diet group. Data (A, D, E n=6-8 animals; B, C n = 4 pairhoused cages) are presented as mean ± SEM.
Figure 3.
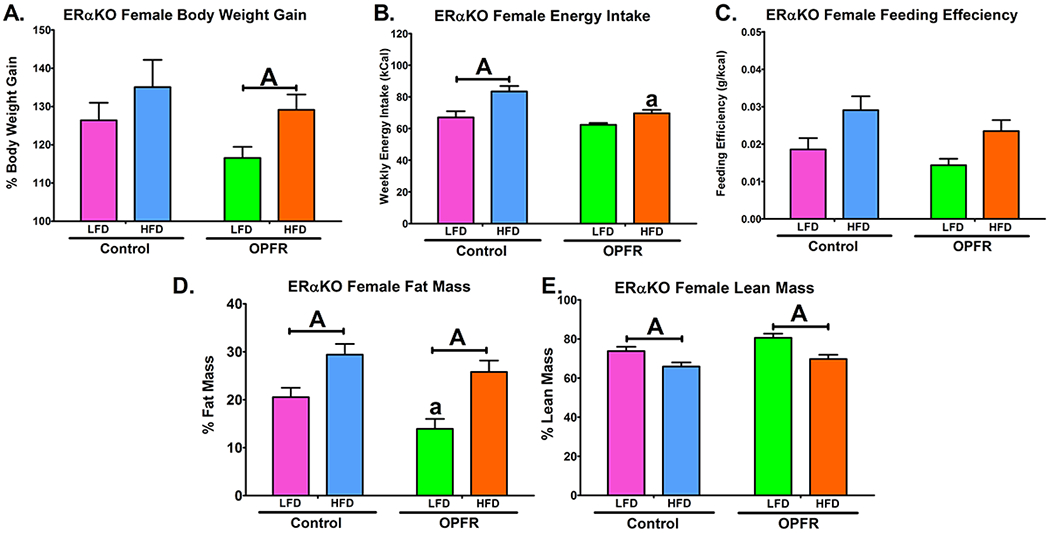
Physiological parameters in ERαKO females orally dosed with an OPFR mixture (1 mg/kg bw) for 4 weeks. (A) % Body Weight Gain over 4 weeks; (B) Energy Intake; (C) Feeding Efficiency; (D) Body composition % Fat Mass; (E) Body composition % Lean Mass. Data were analyzed by a two-way ANOVA with post-hoc Newman-Keuls multiple comparisons test. Uppercase letters denote diet effects within exposure group. Lowercase letters denote OPFR effects within diet group. Data (A, D, E n=6-8 animals; B, C n=4 pairhoused cages) are presented as mean ± SEM.
Male ERαKO mice fed a high fat diet (HFD), compared to when fed a low fat diet (LFD), exhibited an elevation in energy intake, feeding efficiency, bodyweight gain, as well as increased fat mass and reduced lean mass (Figure 3). While OPFR treatment significantly decreased the feeding efficiency of males fed LFD (Figure 3C) this did not result in any marked changes in body mass and OPFR appears to have minimal impact in males. In females, HFD significantly augmented bodyweight gain, but only when fed OPFR-oil mixture (Figure 3A). Interestingly, while OPFR-exposed females gained more weight when fed HFD compared to LFD, there was no marked difference in caloric intake between diets in OPFR-treated females (Figure 3B). This may be attributed to a reduced HFD intake compared to oil-control in female mice (Figure 3B) suggesting altered metabolic processing of HFD. Female OPFR-exposed mice also experienced lowered fat mass when fed LFD (Figure 3D).
Feeding Behaviors
The BioDAQ™ apparatus was utilized for more in-depth analysis of feeding behaviors, expanding on the crude body weight and caloric intake discussed above. Over a 96-hr trial period, hourly and total food intake were measured, and the size, duration, and frequency of meals was calculated. OPFR did not markedly affect either total or hourly patterns of food intake in ERαKO males (Figure 4A and 4B). OPFR did not significantly alter meal duration or size. Meal frequency, while increased by HFD in oil-fed males (Figure 4C), was unaffected by diet in OPFR-fed males. On the other hand, females experienced perturbations to diurnal food intake patterns initiated by OPFR (Figure 5A). From 2000 to 2100 hr, OPFR-treated mice ate half as much HFD as their oil-treated counterparts (Figure 5A). In turn, OPFR-treated mice consumed less HFD than LFD during the same time frame, as well as during 2100-2200 and 2300-2400 hr (Figure 5A). Total food intake over the 96-hr testing period remained unaltered by OPFR exposure, as did meal frequency, meal duration, and meal size.
Figure 4.
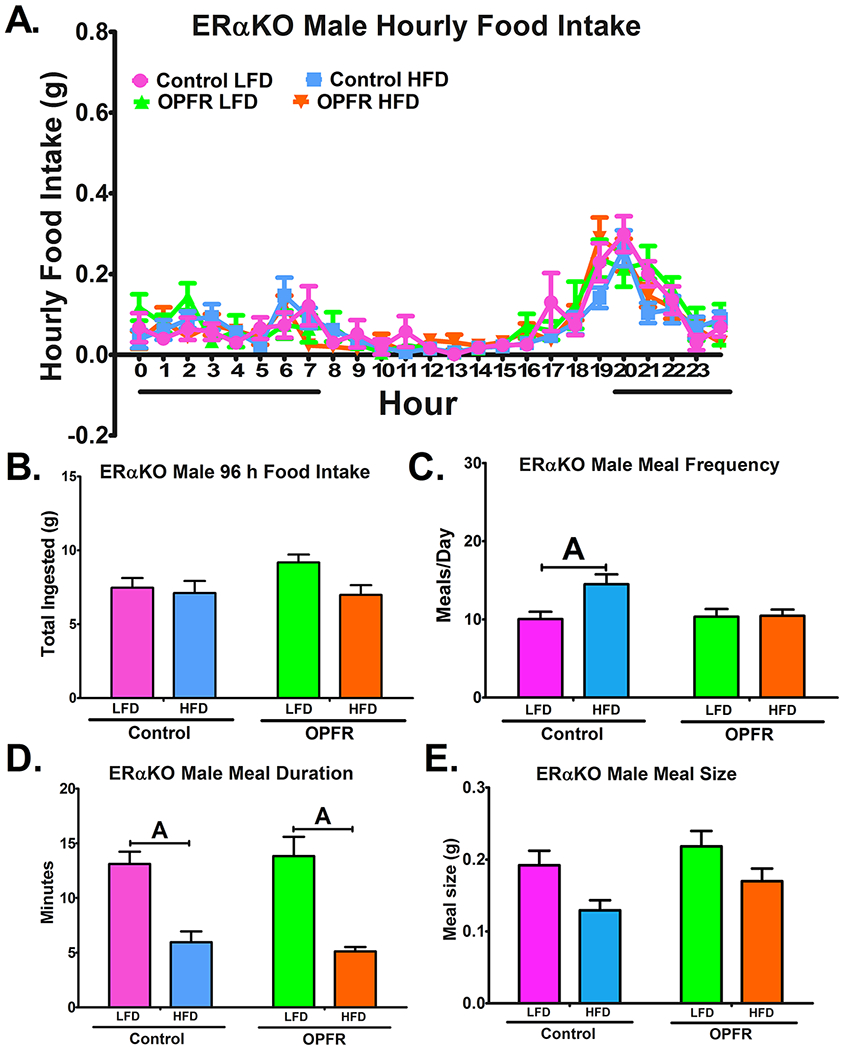
Analysis of feeding behaviors in ERαKO males orally dosed with an OPFR mixture (1 mg/kg bw) for ~5 weeks. (A) hourly food intake; (B) 96 h total food ingested (C) meals/day; (D) meal duration; (E) meal size. Data were analyzed by a two-way ANOVA (B-E) and a repeated-measures three-way ANOVA (A) with post-hoc Newman-Keuls test. Uppercase letters denote diet effects within exposure group; and lowercase letters denote OPFR effects within diet group. Data (n=6-8 for all groups) are presented as mean ± SEM.
Figure 5.
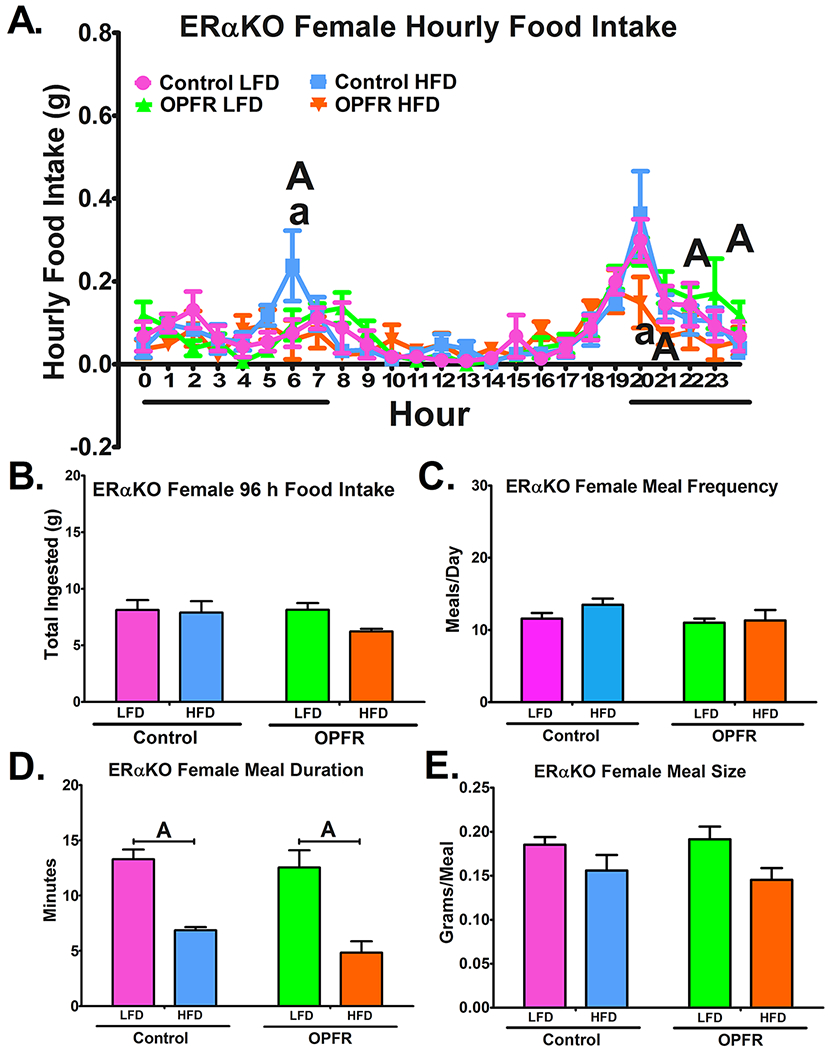
Analysis of feeding behaviors in ERαKO females orally dosed with an OPFR mixture (1 mg/kg bw) for ~5 weeks. (A) hourly food intake; (B) 96 h total food ingested (C) meals/day; (D) meal duration; (E) meal size. Data were analyzed by a two-way ANOVA (B-E) and a repeated-measures three-way ANOVA (A) with post-hoc Newman-Keuls test. Uppercase letters denote diet effects within exposure group; and lowercase letters denote OPFR effects within diet group. Data (n=5-8 for all groups) are presented as mean ± SEM.
Glucose and Insulin Tolerance
Glucose and insulin tolerance tests (Figures 6 and 7) were performed to examine OPFR’s impact on the body’s response to sudden changes in glucose homeostasis. OPFR exerted no direct effect on glucose tolerance compared to oil-control counterparts. However, while diet did not significantly alter glucose tolerance in ERαKO males (Figure 6A–6C), female mice exhibited greater area under the curve (AUC) when fed HFD than when fed LFD (Figure 6F). Data indicate that for female mice, consuming a HFD impeded the uptake of glucose after being administered as a bolus injection. This diet effect was not observed in OPFR-treated female mice (Figure 6F), indicating that OPFR is reducing the effect HFD exerted on glucose clearance. This is further supported by the significant difference noted in HFD blood-glucose at the last time point in the tolerance test. By this point, OPFR-treated females nearly returned to baseline glucose levels, whereas oil-control mice remain significantly elevated (Figure 6E).
Figure 6.
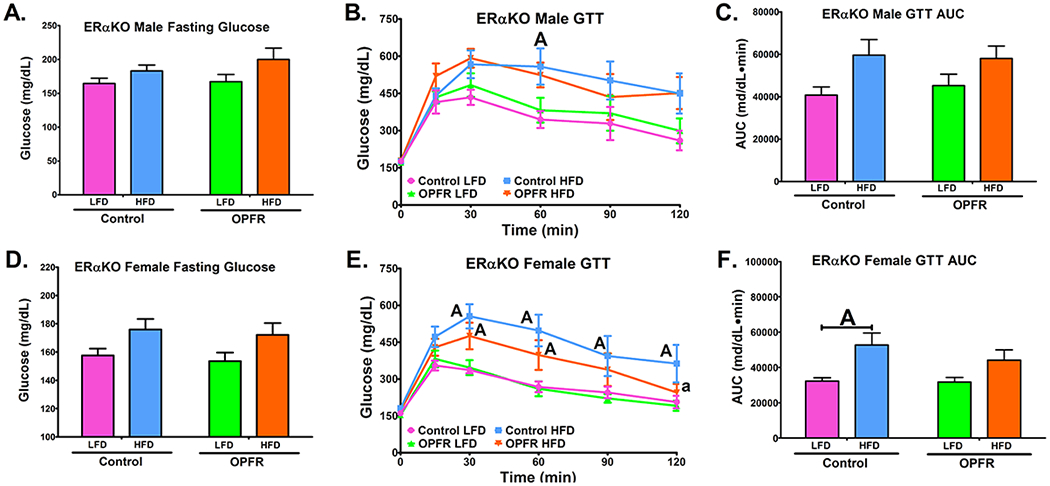
Glucose tolerance tests in ERαKO mice orally dosed with an OPFR mixture (1 mg/kg bw) for ~6 weeks. (A) Male fasting glucose; (B) Male GTT; (C) Area under the curve (AUC) of Male GTT; (D) Female fasting glucose; (E) Female GTT; (F) Area under the curve (AUC) of Female GTT. Data were analyzed by a two-way ANOVA (A, C, D, F) or a repeated-measures, three-way ANOVA (B, E) with post-hoc Newman-Keuls multiple comparisons test. Uppercase letters denote diet effects within exposure group and lowercase letters denote OPFR effect within diet group. Data (n=5-8 for all groups) are presented as mean ± SEM.
Figure 7.
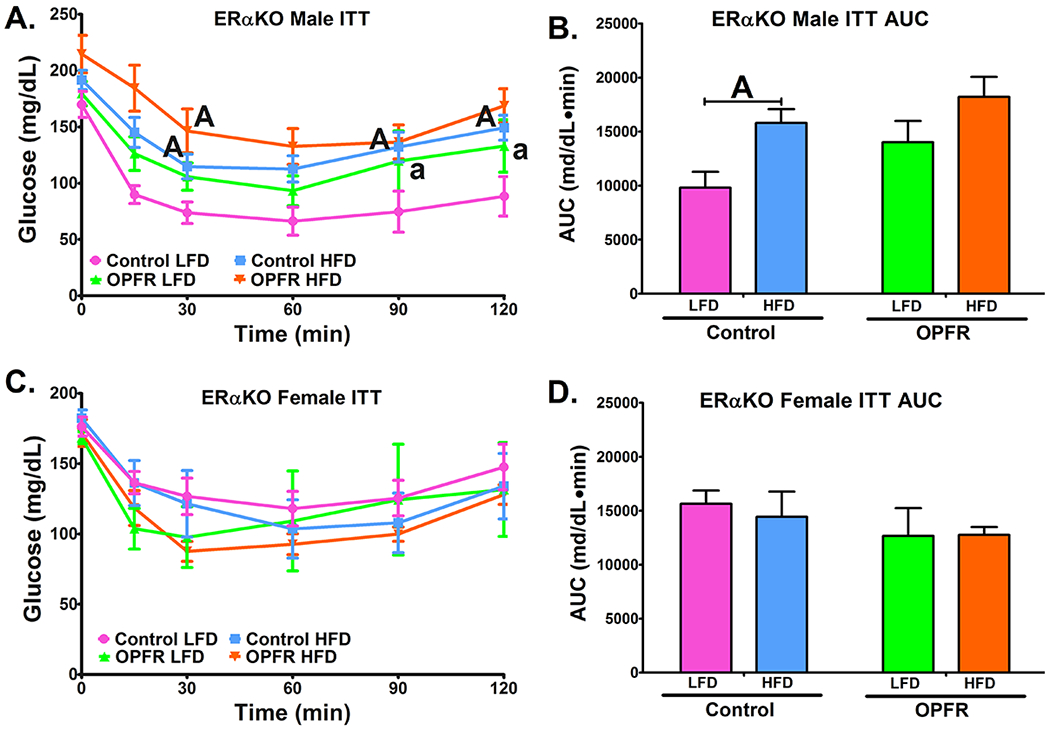
Insulin tolerance tests in ERαKO mice orally dosed with an OPFR mixture (1 mg/kg bw) for ~6 weeks. (A) Male ITT; (B) Area under the curve (AUC) of Male ITT; (C) Female GTT; (D) Area under the curve (AUC) of Female ITT. Data were analyzed by a two-way ANOVA (B,D) or a repeated-measures, three-way ANOVA (A, C) with post-hoc Newman-Keuls multiple comparisons test. Uppercase letters denote diet effects within exposure group and lowercase letters denote OPFR effect within diet group. Data (n=5-8 for all groups) are presented as mean ± SEM.
While female mice experienced diet related effects in glucose tolerance, the opposite was true for insulin tolerance (Figure 7). Insulin tolerance in females was unaffected by both diet and OPFR (Figure 7C, 7D). Although in control males HFD resulted in greater AUC than did LFD (Figure 7B). However, this effect was not seen in OPFR-treated males. Examining Figure 7A, it is notable that OPFR exposure resulted in significantly elevated blood-glucose in male mice fed LFD during the last two time points. Evidence indicates that the reason why a diet effect was not detected in OPFR-treated males may be due to impaired insulin tolerance in OPFR-treated males fed LFD. This is supported by a statistical trending overall effect of OPFR on male mice (Figure 7A).
Peptide Hormones
Terminal plasma hormone levels were analyzed for leptin, insulin, and ghrelin. In male mice, HFD exhibited an overall effect to increase insulin and leptin plasma levels (Figure 8A; 8B), but no marked OPFR effect was observed. Conversely, ghrelin levels were reduced in HFD-fed males (Figure 8C); however, post-hoc analysis revealed significance only between LFD- and HFD-fed animals that were treated with OPFRs (Figure 8C). Data indicate that OPFR exposure may be amplifying the effects of diet on ghrelin levels. Indeed, this is supported by a significant interaction between diet and OPFR exposure (Figure 8C), as well as significant difference in ghrelin levels between OPFR-exposed and control animals fed LFD (Figure 8C).
Figure 8.
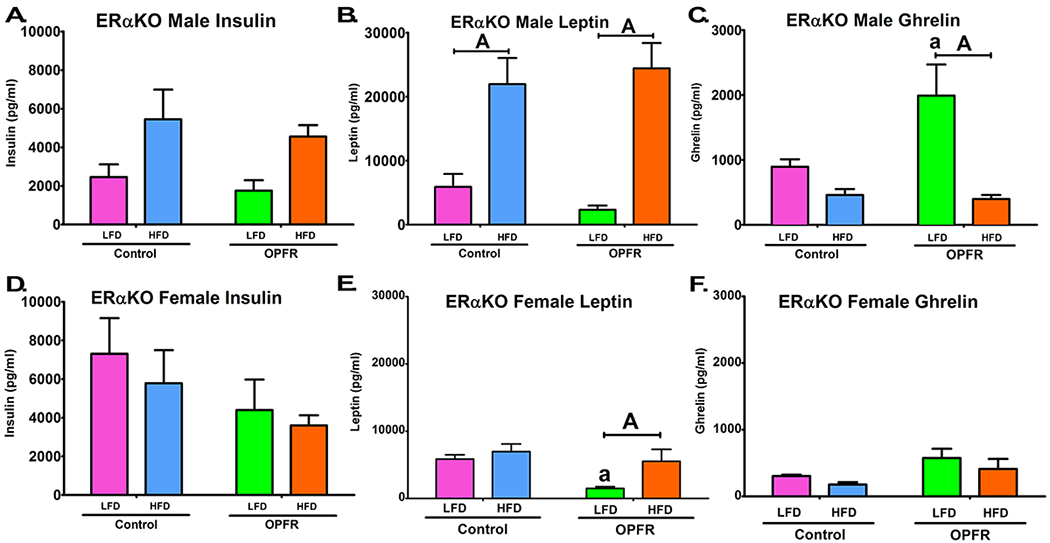
Terminal plasma peptide hormone levels in ERαKO mice orally dosed with an OPFR mixture (1 mg/kg bw) for 7 weeks. (A) Male insulin; (B) Male leptin; (C) Male ghrelin; (D) Female insulin; (E) Female leptin; (F) Female ghrelin. Data were analyzed by a two-way ANOVA (B,D) or a repeated-measures, three-way ANOVA (A, C) with post-hoc Newman-Keuls multiple comparisons test. Uppercase letters denote diet effects within exposure group and lowercase letters denote OPFR effect within diet group. Data (n=6-8 for all groups) are presented as mean ± SEM.
In female mice, insulin levels were unaffected by either diet or OPFR exposure. An overall effect of HFD to lower plasma ghrelin was noted (Figure 8F), similar to males. However, whereas male mice exhibited an interaction diet and OPFR treatment on plasma ghrelin, this effect was not observed in female mice. Evidence indicates that ERα may play a greater role in males than in females in determining OPFR impact on ghrelin signaling. Lastly, while leptin levels were also elevated by HFD in female mice (Figure 8E), significance was only returned when comparing LFD- and HFD-fed animals that were exposed to OPFRs (Figure 8E). This may be attributed to the overall effect of OPFR to diminish circulating leptin (Figure 8E), specifically within LFD-fed females (Figure 8E). Taken together, these data suggest that in the absence of ERα, female mice are more sensitive to OPFR perturbation of leptin signaling, whereas male mice are more sensitive to OPFR alterations of ghrelin.
Arcuate (ARC) Gene Expression
ARC microdissections were analyzed for Pparg expression as well ARC expression of receptors for peripheral signaling peptides insulin (Insr), leptin (Lepr), and ghrelin (Ghsr). In addition, select ARC neuropeptide transcripts governing energy homeostasis Npy, Agrp, Pomc, and Cart were also analyzed. In male animals, no significant effect of diet or OPFR was observed for Agrp, Cart, or Pparg. Analysis of Npy expression revealed an overall significant effect of both diet and OPFR exposure (Table 2). Post-hoc testing did not attribute these observations to any specific comparisons. Further, an interaction between diet and OPFR treatment was seen in Pomc expression (Table 2). This was associated with a significant effect of HFD to elevate Pomc expression by approximately 70%, compared to LFD in OPFR-exposed males (Table 2). Whereas HFD increased Pomc in OPFR-exposed males, Pomc expression was unaffected by diet in controls. This discrepancy is attributed to a significant elevation of Pomc in HFD-fed males exposed to OPFR compared to non-exposed animals (Table 2). Lastly, significant overall effects of OPFR were noted in expression of both leptin and insulin receptors in ARC tissue, indicating a potential increased sensitivity to peripheral anorexigenic signals.
Table 2.
Arcuate expression of neuropeptides and receptors from ERαKO males and females
| Males | Females | |||||||
|---|---|---|---|---|---|---|---|---|
| Gene | Control-LFD | Control-HFD | OPFR-LFD | OPFR-HFD | Control-LFD | Control-HFD | OPFR-LFD | OPFR-HFD |
| Agrp | 1.07 ± 0.19 | 0.64 ± 0.19 | 0.82 ± 0.15 | 0.99 ± 0.19 | 1.04 ± 0.14 | 0.88 ± 0.14 | 1.42 ± 0.29 | 0.73 ± 0.06A |
| Cart | 1.08 ± 0.19 | 0.93 ± 0.11 | 1.00 ± 0.11 | 1.42 ± 0.25 | 1.01 ± 0.08 | 1.08 ± 0.07 | 0.86 ± 0.09 | 1.07 ± 0.06 |
| Npy | 1.03 ± 0.11 | 0.64 ± 0.12 | 1.64 ± 0.28 | 0.95 ± 0.12 | 1.02 ± 0.11 | 0.85 ± 0.10 | 1.51 ± 0.24 | 1.11 ± 0.13 |
| Pomc | 1.10 ± 0.24 | 0.92 ± 0.14 | 0.93 ± 0.05A | 1.66 ± 0.14a | 1.02 ± 0.10 | 0.96 ± 0.11 | 0.85 ± 0.06 | 0.93 ± 0.05 |
| Ghsr | 1.08 ± 0.17 | 0.90 ± 0.13 | 1.14 ± 0.18 | 1.14 ± 0.08 | 1.02 ± 0.09 | 0.94 ± 0.08 | 1.18 ± 0.14 | 1.15 ± 0.09 |
| Insr | 1.05 ± 0.14 | 1.44 ± 0.15 | 1.54 ± 0.17 | 1.65 ± 0.15 | 1.01 ± 0.06 | 1.21 ± 0.12 | 1.67 ± 0.17 | 1.38 ± 0.24 |
| Lepr | 1.04 ± 0.13 | 0.99 ± 0.11 | 1.54 ± 0.23 | 1.25 ± 0.06 | 1.01 ± 0.06 | 1.00 ± 0.08 | 1.37 ± 0.13 | 1.15 ± 0.10 |
| Pparg | 1.03 ± 0.11 | 0.83 ± 0.18 | 0.83 ± 0.09 | 0.98 ± 0.13 | 1.08 ± 0.22 | 1.30 ± 0.13 | 2.72 ± 0.85 | 1.70 ± 0.22 |
Arcuate expression of neuropeptides and receptors. Data were analyzed by a two-way ANOVA with post-hoc Newman-Keuls multiple comparisons test. Uppercase letters denote diet effects within exposure group and lowercase letters denote OPFR effects within diet group. All data were normalized to Control-LFD within each sex. Data (n = 5-6 per group) are presented as mean ± SEM. Significant overall ANOVA effects are as follows. Male: Npy F(1,18)Diet= 8.457, P < .05, F(1,18)OPFR= 6.150, P < .05; Pomc F(1,18)Diet*OPFR= 7.517, P < .05; Insr F(1,18)-OPFR= 4.915, P < .05); Lepr F(1,18)OPFR= 5.668, P < .05. Female: Agrp F(1,19)Diet= 5.422, P < .05; Npy F(1,19)OPFR= 5.462, P < .05; Pparg F(1,19)OPFR= 4.612, P < .05; Insr F(1,19)OPFR= 5.859, P < .05; Lepr F(1,19)OPFR= 6.596, P < .05.
For female ERαKO mice, both diet and OPFR did not markedly affect Pomc and Cart expression. This is interesting in of itself, as their male counterparts showed marked interactions between these categories for Pomc expression, perhaps indicating that ERα plays a greater role in protection against OPFR-induced alterations to Pomc in males than in females. In females, rt-qPCR analysis revealed an overall significant effect of OPFR to elevate Npy expression (Table 2), as well as Pparg (Table 2). An overall effect of diet was also recorded for Agrp, where expression was lower across exposure groups in HFD-fed females (Table 2). However, post-hoc testing only highlighted a significant reduction in OPFR-exposed females, indicating a potential for OPFR exposure to enhance the impact of HFD on Agrp expression in females lacking ERα. As in males, a significant overall effect of OPFR was reported for both insulin and leptin receptor expression (Table 2). This observation suggests that the potential for OPFRs to increase sensitivity to peripheral leptin and insulin is not impacted by gender.
Discussion
The current focus on sensitive developmental exposure windows has resulted in a lack of understanding for how OPFR impacts energy homeostasis throughout adult life. In particular, there are key knowledge gaps in the mechanisms of endocrine disruption. ERα is the most commonly proposed OPFR target, and this study further implicates its role in OPFR-mediated endocrine disruption. This discussion heavily references our previous study using the same concentration and methodology of OPFR exposure in wildtype mice (Vail et al. 2020). While it is understood that the need for previous research as a reference point is a significant limitation of this study, a table summarizing the direct effects of OPFRs in the wildtype (WT) study compared to the results found in the present KO study is provided to help visualize the conclusions drawn from the data (Table 3).
Table 3.
Summary of ERαKO findings compared to WT findings by Vail et al. (2020a)
| Endpoint | Males | Females | ||
|---|---|---|---|---|
| LFD WT / ERαKO |
HFD WT / ERαKO |
LFD WT / ERαKO |
HFD WT / ERαKO |
|
| Bodyweight Gain | n.s. / n.s. | ↑ / n.s. | n.s. / n.s. | n.s. / n.s. |
| Feeding Efficiency | n.s. / ↓ | n.s. / n.s. | n.s. / n.s. | n.s. / n.s. |
| Fat Mass | n.s. / n.s. | ↑ / n.s. | n.s. / ↓ | n.s. / n.s. |
| Lean Mass | n.s. / n.s. | ↓ / n.s. | n.s. / n.s. | n.s. / n.s. |
| 96 hr Energy Intake | n.s. / n.s. | n.s. / n.s. | n.s. / n.s. | ↓ / ↓ |
| Hourly Food Intake | n.s. / n.s. | ↓↑ / n.s. | n.s. / n.s. | ↓↑ / ↓↑ |
| Meal Frequency | n.s. / n.s. | n.s. / n.s. | n.s. / n.s. | ↓ / n.s. |
| Meal Duration | n.s. / n.s. | n.s. / n.s. | n.s. / n.s. | n.s. / n.s. |
| Meal Size | n.s. / n.s. | n.s. / n.s. | n.s. / n.s. | n.s. / n.s. |
| Fasting Glucose | n.s. / n.s. | n.s. / n.s. | n.s. / n.s. | n.s. / n.s. |
| Glucose Tolerance | n.s. / n.s. | n.s. / n.s. | n.s. / n.s. | n.s. / n.s. |
| Insulin Tolerance | n.s. / ↑ | n.s. / n.s. | n.s. / n.s. | n.s. / n.s. |
| Plasma Insulin | n.s. / n.s. | n.s. / n.s. | ↑ / n.s. | n.s. / n.s. |
| Plasma Leptin | n.s. / n.s. | n.s. / n.s. | n.s. / n.s. | ↑ / n.s. |
| Plasma Ghrelin | ↓ / ↑ | n.s. / n.s. | n.s. / ↓ | n.s. / n.s. |
Summary of direct effects of OPFR in ERαKO animals compared to WT findings by Vail et al. (2020a). Only measures that were recorded in both studies are listed. Up arrows denote an OPFR-induced increase and down arrows denote an OPFR-induced decrease. One up and one down arrow indicates a mixed effect dependent on time of day. N.S. = not significant.
In agreement with previous findings (Heine et al. 2000; Bian et al. 2019), complete knockout of ERα resulted in an overall increased weight gain of both male and female mice compared to WT mice (Table 3; Vail et al. 2020). ERαKO males responded to HFD with significantly enhanced adiposity and bodyweight gain compared to mice fed LFD, but no marked effect of OPFR-treatment was observed. However, Vail et al. (2020; Table 3) previously noted that WT males exposed to OPFRs exhibited an elevated fat mass and bodyweight gain when fed HFD, compared to their control-treated counterparts. The absence of these observed effects in ERαKO males suggests that ERα plays a necessary role in facilitating OPFR effects on fat deposition and bodyweight gain. ERα is an important regulator of energy homeostasis, and the observed lack of OPFR action may be due to its inability to target and disrupt ERα regulation of adiposity and bodyweight. Further, OPFR-treated ERαKO male mice experienced decreased feeding efficiency when fed LFD, compared to control mice. This effect was not seen in WT mice (Table 3; Vail et al. 2020), indicating that aspects of OPFR homeostatic disruption are acting through pathways not directly influenced by ERα, but perhaps associated with them and serving as alternate estrogen pathways which were more heavily targeted in the absence of ERα. A novel effect in a knockout model suggests that the loss of the target gene resulted in compensatory alterations to signaling pathways that sensitized the animal to OPFR-mediated actions. In the case of ERα, compensatory pathways such as ERβ (also known to interact with OPFRs {Kojima et al. 2013}), or membrane estrogen receptors such as G protein-coupled estrogen receptor (GPER) or Gq--coupled membrane estrogen receptor (Gq-mER) may be upregulated and might be more sensitive to OPFR action, all of which are capable of regulating energy homeostasis (Mauvais-Jarvis et al. 2013; Prossnitz and Hathaway 2015; Roepke et al. 2010; Shi et al. 2013). It is also possible that, in the absence of ERα, OPFRs may be acting preferentially upon another proposed nuclear receptor target – PPARγ. ERβ−null mice were found to exhibit upregulated PPARγ signaling (Foryst-Ludwig et al. 2008), but ERαKO models are not known to exert the same effect. However, this study reports that ARC expression of Pparg was significantly upregulated in OPFR-exposed female ERαKO mice, indicating that female mice lacking ERα may experience enhanced interaction of OPFRs with PPARγ.
Both ERα and PPARγ are nuclear receptors that, upon activation, bind to respective response elements to initiate transcription of target genes (Bjornstrom and Sjoberg 2005; Janani and Ranjitha Kumari 2015). Importantly, both receptors demonstrated reciprocal ability to bind to and activate response elements associated with the other receptor (Keller et al. 1995; Wang and Kilgore 2002). The apparent crosstalk between ERα and PPARγ may be contributing to the novel observations recorded in ERα knockout mice. ERαKO mice only lack the receptor, and the associated estrogen response elements remain intact. It is therefore possible that activation of intact PPARγ in ERαKO mice may result in transactivation of estrogen response elements and may play a role in the appearance of novel OPFR effects in knockout mice.
Vail et al. (2020) reported that all control WT animals fed HFD experienced elevated weight gain, and the same is reported in ERαKO males, but not within ERαKO females (Figure 3A). Female mice lacking ERα did not demonstrate a significant difference in bodyweight between LFD and HFD-fed groups. This may be considered a genotype effect. It is interesting, however, that OPFR exposure restored the diet-effect in female ERαKO mice (Figure 3A). This might be explained as an estrogenic effect of OPFRs on alternative estrogen receptors (ERs) to restore WT patterns. This idea is based upon the thought that without the “catabolic” actions of ERα, body physiology shifts towards a more “anabolic” pattern. However, alternative ERs may be recruited to compensate for the loss of ERα, though at lesser efficacy. OPFRs may be inducing increased activity of these alternative pathways, aiding their ability to compensate for the lack of ERα. This postulation is further supported by our findings that OPFRs decreased caloric intake of HFD chow, as well as reduced fat mass in female ERαKO mice fed LFD. These OPFR-initiated effects were not seen in WT mice, and again, may therefore be a result of alternative ER activation by OPFR.
While control ERαKO males fed a HFD exhibited the WT pattern of increased meal frequency compared to LFD, OPFR treatment eliminated this difference. Potentially, this may be resulting from elevated levels of ghrelin in OPFR-treated LFD males. Ghrelin is a potent orexigenic hormone and higher signal in LFD animals may confer an enhanced stimulus to feed specific to LFD animals exposed to OPFRs. This may account for how the diet effect was eliminated in OPFR-exposed males. Interestingly, Vail et al. (2020; Table 3) previously found that WT males exhibited lower ghrelin levels on a LFD. This is indicative of interaction between OPFR endocrine disruption and ERα, which is known to stimulate ghrelin signaling (Sakata et al. 2006; Kellokoski et al. 2005). Further, OPFR induced an enhanced expression of Npy in the ARC from male mice (Table 2). Npy is the transcript for neuropeptide Y, a neuropeptide that stimulates food intake, and might be contributing to the noted effects on LFD meal frequency and circulating ghrelin.
Female ERαKO mice also displayed only minor effects of OPFRs in ingestive behavior. In the previous WT study, OPFR exposure significantly impacted the temporal intake of food in female mice, lowering HFD intake during the night (Table 3; Vail et al. 2020). These effects were replicated in ERαKO mice, suggesting that the mechanism of OPFR disruption of diurnal feeding patterns may not involve ERα. However, while WT females experienced a significant decrease in meal frequency and total intake of HFD compared to control (Table 3; Vail et al. 2020), significant effects were not detected within ERαKO mice. Data indicate a potential role for ERα as an OPFR target in these parameters.
Lastly, ERαKO mice were tested for both glucose and insulin tolerance. ERαKO males displayed no significant effect of OPFRs on glucose tolerance similar to WT experiments (Vail et al. 2020). However, insulin tolerance appears altered by OPFR in ERαKO male mice fed LFD. The LFD tolerance curve is significantly elevated in the latter half of the test, and nearly matches HFD observations. This is further represented in a significant difference in AUC between diets in control-treated males, whereas no marked effect was found within OPFR-treated animals. This effect was not seen in WT males and might be attributed to OPFR action on targets that were perhaps sensitized in the absence of ERα. Glucose tolerance in ERαKO females was equivalent to that reported in the WT study (Vail et al. 2020). Some subtle differences are present, but none notable enough to conclude OPFR disruption of glucose tolerance. No observable effects of OPFR were found in the insulin tolerance test. Plasma insulin levels were also unaffected by OPFR exposure; however, a significant reduction was noted in LFD females for circulating leptin, another marker associated with satiety. This was not consistent with WT data, which showed no marked effect on leptin for LFD, but increased leptin for HFD WT females (Vail et al. 2020), implying that this novel effect of OPFR results from the loss of ERα.
Conclusions
Overall, there appears to be no sex-specificity for OPFR action through ERα or proposed alternative ER pathways. Male and female ERαKO mice each experienced their own share of OPFR perturbations with predominant effects on bodyweight gain, body composition and relevant homeostatic hormones, and on ingestive behaviors. Data demonstrate that ERα appears to be involved in mediating some, but not all, of ingestion dysregulation elicited by OFPR exposure. Interestingly, OPFR alterations directly attributed to ERα (present in WT but absent in ERαKO) were noted almost exclusively in HFD-fed animals, whereas novel OPFR effects in ERαKO mice were observed in LFD-fed animals (refer to Table 3). This hints at an intriguing notion that ERα is more directly involved in the exacerbation of diet-induced obesity by OPFR exposure, and plays a lesser, perhaps more nuanced role within non-obese conditions.
This study represents new insight into the role of ERα in OPFR-initiated disruption of ingestive behaviors and energy homeostasis. Dysregulation of energy homeostasis might result in metabolic disorders such as obesity, diabetes, and metabolic syndrome. Therefore, it is important to understand the underlying toxicological mechanisms involved following OPFR exposure that may be pre-disposing human populations to such conditions. Future studies should use these data as a foundation with which to build upon by the addition of further mechanistic examinations of metabolism and energy expenditure. OPFR disruption of electrophysiological activity of NPY and POMC neurons has been previously demonstrated (Vail and Roepke. 2020), and future use of selective ERα knockout in these neuronal populations might provide valuable insight into how OPFR exposure interacts with estrogenic regulation of neuronal homeostatic hubs.
Ultimately, the purpose of this study and studies alike is to provide the necessary data to assess the hazard and the extrapolated risk of allowing continued human exposure to endocrine disrupting chemicals such as OPFRs. Continued research into the mechanisms underlying OPFR-induced endocrine disruption may give a better understanding of its long-term potential for adverse effects on human health, but as it stands, the literature already possesses sufficient evidence to indicate reasonable caution for the continued widespread use of organophosphate flame retardants.
Acknowledgements
This work was supported by the US Department of Agriculture–National Institute of Food and Agriculture (NJ06195, TAR) and the National Institutes of Health (R21ES027119, R01MH123544, and P30ES005022, TAR). SNW was funded by R21ES027119-S1 and GMV was funded, in part, by T32ES007148.
Footnotes
Declaration of Interest Statement
No potential competing interest was reported by the authors.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, T.A. Roepke, upon reasonable request.
References
- Acosta-Martinez M, Horton T, and Levine JE. 2007. Estrogen receptors in neuropeptide Y neurons: At the crossroads of feeding and reproduction. Trends Endocrinol Metab 18: 48–50. [DOI] [PubMed] [Google Scholar]
- Barouki R 2017. Endocrine disruptors: Revisiting concepts and dogma in toxicology. Comptes Rendus Biol. 340:410–413. [DOI] [PubMed] [Google Scholar]
- Belcher SM, Cookman CJ, Patisaul HB, and Stapleton HM. 2014. In vitro assessment of human nuclear hormone receptor activity and cytotoxicity of the flame retardant mixture FM 550 and its triarylphosphate and brominated components. Toxicol Lett 228:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian X, Liu T, Zhou M, He G, Ma Y, Shi Y, Wang Y, Tang H, Kang X, Yang M, Gustafsson JA, Fan X, and Tang K. 2019. Absence of estrogen receptor beta leads to abnormal adipogenesis during early tendon healing by an up-regulation of PPARgamma signalling. J Cell Mol Med 23:7406–7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornstrom L, and Sjoberg M. 2005. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol 19 (4):833–42. [DOI] [PubMed] [Google Scholar]
- Butt CM, Congleton J, Hoffman K, Fang M, and Stapleton HM. 2014. Metabolites of organophosphate flame retardants and 2-ethylhexyl tetrabromobenzoate in urine from paired mothers and toddlers. Environ Sci Technol 48 :10432–10438. [DOI] [PubMed] [Google Scholar]
- Cádiz V, Ronda JC, Lligadas G, and Galià M. 2011. Polybenzoxazines with enhanced flame retardancy. In Handbook of Benzoxazine Resins, editor Ishida H and Agag T, pp. 556–576. Amsterdam: Elsevier. [Google Scholar]
- Dabass A, Talbott EO, Rager JR, Marsh GM, Venkat A, Holguin F, and Sharma RK. 2018. Systemic inflammatory markers associated with cardiovascular disease and acute and chronic exposure to fine particulate matter air pollution (PM2.5) among US NHANES adults with metabolic syndrome. Environ Res 161:485–491. [DOI] [PubMed] [Google Scholar]
- de Souza FS, Nasif S, Lopez-Leal R, Levi DH, Low MJ, and Rubinsten M. 2011. The estrogen receptor alpha colocalizes with proopiomelanocortin in hypothalamic neurons and binds to a conserved motif present in the neuron-specific enhancer nPE2. Eur J Pharmacol 660: 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dishaw LV, Powers CM, Ryde IT, Roberts SC, Seidler FJ, Slotkin TA, and Stapleton HM. 2011. Is the PentaBDE replacement, tris (1,3-dichloro-2-propyl) phosphate (TDCPP), a developmental neurotoxicant? Studies in PC12 cells. Toxicol Appl Pharmacol 256: 281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foryst-Ludwig A, Clemenz M, Hohmann S, Hartge M, Sprang C, Frost N, Krikov M, Bhanot S, Barros R, Morani A, A Gustafsson J, Unger T, and Kintscher U. 2008. Metabolic actions of estrogen receptor beta (ERbeta) are mediated by a negative cross-talk with PPARgamma. PLoS Geneti 4 :e1000108–e1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Segura LM, Arevalo MA, and Azcoitia I. 2010. Interactions of estradiol and insulin-like growth factor-I signalling in the nervous system: new advances. Prog Brain Res 181:251–272. [DOI] [PubMed] [Google Scholar]
- Gilbert ME, Rovet J, Chen Z, and Koibuchi N. 2012. Developmental thyroid hormone disruption: prevalence, environmental contaminants and neurodevelopmental consequences. Neurotoxicology 33:842–852. [DOI] [PubMed] [Google Scholar]
- Gupte AA, Pownall HJ, and Hamilton DJ. 2015. Estrogen: An emerging regulator of insulin action and mitochondrial function. J Diabetes Res 2015:916585–916585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, and Cooke PS. 2000. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Nat Acad Sci USA 97:12729–12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbstman JB, Sjodin A, Kurzon M, Lederman SA, Jones RS, Rauh V, Needham LL, Tang D, Niedzwiecki M, Wang RY, and Perera F. 2010. Prenatal exposure to PBDEs and neurodevelopment. Environ Health Perspect 118:712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevener AL, Clegg DJ, and Mauvais-Jarvis F 2015. Impaired estrogen receptor action in the pathogenesis of the metabolic syndrome. Mol Cell Endocrinol 418:306–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SC, Kissling GE, Fieselman KE, Jayes FL, Gerrish KE, and Korach KS. 2010. Biological and biochemical consequences of global deletion of exon 3 from the ER alpha gene. FASEB J 24 :4660–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SC, Li L, Grimm SA, Winuthayanon W, Hamilton KJ, Pockette B, Rubel CA, Pedersen LC, Fargo D, Lanz RB, DeMayo FJ, Schutz G, and Korach KS. 2014. Novel DNA motif binding activity observed in vivo with an estrogen receptor alpha mutant mouse. Mol Endocrinol 28:899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman K, Butt CM, Webster TF, Preston EV, Hammel SC, Makey C, Lorenzo AM, Cooper EM, Carignan C, Meeker JD, Hauser R, Soubry A, Murphy SK, Price TM, Hoyo C, Mendelsohn E, Congleton J, Daniels JL, and Stapleton HM. 2017. Temporal trends in exposure to organophosphate flame retardants in the United States. Environ Sci Technol Lett 4:112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Jia Y, Kang Q, Peng H, Ma H, Zhang S, Hiromori Y, Kimura T, Nakanishi T, Zheng L, Qiu Y, Zhang Z, Wan Y, and Hu J. 2019. Screening of house dust from Chinese homes for chemicals with liver X receptors binding activities and characterization of atherosclerotic activity using an in vitro macrophage cell line and ApoE−/− mice. Environ Health Perspect 127:117003–117003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel Chemicals ltd. 2015. Worldwide flame retardants market to reach 2.8 million tonnes in 2018. Additives for Polymers 2015:11. [Google Scholar]
- Janani C, and Ranjitha Kumari BD. 2015. PPAR gamma gene – A review. Diabetes Metab Syndrome: Clin Res Rev 9: 46–50. [DOI] [PubMed] [Google Scholar]
- Ji X, Li N, Ma M, Rao K, and Wang Z. 2020. In vitro estrogen-disrupting effects of organophosphate flame retardants. Sci Total Environ 727:138484. [DOI] [PubMed] [Google Scholar]
- Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R, and Grohe C. 2000. Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J Biol Chem 275:18447–18453. [DOI] [PubMed] [Google Scholar]
- Keller H, Givel F, Perroud M, and Wahli W. 1995. Signaling cross-talk between peroxisome proliferator-activated receptor/retinoid X receptor and estrogen receptor through estrogen response elements. Mol Endocrinol 9:794–804. [DOI] [PubMed] [Google Scholar]
- Kellokoski E, Pöykkö SM, Karjalainen AH, Ukkola O, Heikkinen J, Kesäniemi YA, and Hörkkö S. 2005. Estrogen replacement therapy increases plasma ghrelin levels. J Clin Endocrinol Metab 90: 2954–2963. [DOI] [PubMed] [Google Scholar]
- Kobos L, Alqahtani S, Xia L, Coltellino V, Kishman R, McIlrath D, Perez-Torres C, and Shannahan. 2020. Comparison of silver nanoparticle-induced inflammatory responses between healthy and metabolic syndrome mouse models. J Toxicol Environ Health A 83: 249–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima H, Takeuchi S, Itoh T, Iida M, Kobayashi S, and Yoshida T. 2013. In vitro endocrine disruption potential of organophosphate flame retardants via human nuclear receptors. Toxicology 314: 76–83. [DOI] [PubMed] [Google Scholar]
- Kylie DR, Horman B, Phillips LA, McRitchie LS, Watson S , Deese-Spruill J, Jima D, Sumner S, Stapleton MH, and Patisaul BH. 2018. EDC IMPACT: Molecular effects of developmental FM 550 exposure in Wistar rat placenta and fetal forebrain. Endocr Connect 7: 305–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao L, Letcher RJ, Zhang Y, Jian K, Zhang J, and Su G. 2019. A review on organophosphate Ester (OPE) flame retardants and plasticizers in foodstuffs: Levels, distribution, human dietary exposure, and future directions. Environ Int 127: 35–51 [DOI] [PubMed] [Google Scholar]
- Li J, Cao H, Mu Y, Qu G, Zhang A, Fu J, and Jiang G. 2020. Structure-oriented research on the antiestrogenic effect of organophosphate esters and the potential mechanism. Environ Sci. Technol 54:14525–14534. [DOI] [PubMed] [Google Scholar]
- Linares V, Belles M, and Domingo JL. 2015. Human exposure to PBDE and critical evaluation of health hazards. Arch Toxicol 89:335–356. [DOI] [PubMed] [Google Scholar]
- Liu X, Ji K, and Choi K. 2012. Endocrine disruption potentials of organophosphate flame retardants and related mechanisms in H295R and MVLN cell lines and in zebrafish. Aquat Toxicol 114-115:173–181. [DOI] [PubMed] [Google Scholar]
- Liu X, Ji K, Jo A, Moon HB, and Choi K. 2013. Effects of TDCPP or TPP on gene transcriptions and hormones of HPG axis, and their consequences on reproduction in adult zebrafish (Danio rerio). Aquat Toxicol 134-135: 104–111. [DOI] [PubMed] [Google Scholar]
- Livak KJ and Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25:402–408. [DOI] [PubMed] [Google Scholar]
- Ma J, Zhu H, and Kannan K. 2019. Organophosphorus flame retardants and plasticizers in breast milk from the United States. Environ Sci Technol Lett 6:525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Jin J, Li P, Xu M, Sun Y, Wang Y, and Yuan H. 2017. Organophosphate ester flame retardant concentrations and distributions in serum from inhabitants of Shandong, China, and changes between 2011 and 2015. Environ Toxicol Chem 36:414–421. [DOI] [PubMed] [Google Scholar]
- Mauvais-Jarvis F, Clegg DJ, and Hevener AL. 2013. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev 34:309–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeker JD, Cooper EM, Stapleton HM, and Hauser R. 2013. Urinary metabolites of organophosphate flame retardants: Temporal variability and correlations with house dust concentrations. Environ Health Perspect 121:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez P, and Garcia-Segura LM. 2006. Phosphatidylinositol 3-kinase and glycogen synthase kinase 3 regulate estrogen receptor-mediated transcription in neuronal cells. Endocrinology 147 :3027–3039. [DOI] [PubMed] [Google Scholar]
- National Academy of Sciences (NAS) 2019. A Class Approach to Hazard Assessment of Organohalogen Flame Retardants. National Academies Press 2019, Washington, DC. 10.17226/25412 [DOI] [PubMed] [Google Scholar]
- Patisaul HB, Roberts SC, Mabrey N, McCaffrey KA, Gear RB, Braun J, Belcher SM, and Stapleton HM. 2013. Accumulation and endocrine disrupting effects of the flame retardant mixture Firemaster(R) 550 in rats: An exploratory assessment. J Biochem Mol Toxicol 27: 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng B, Yu ZM, Wu CC, Liu LY, Zeng L, and Zeng EY. 2020. Polybrominated diphenyl ethers and organophosphate esters flame retardants in play mats from China and the exposure risks for children. Environ Int 135:105348. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. 2001.A new mathematical model for relative quantification in real-time RT-PCR. Nucl Acids Res. 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai HK, Fang M, Beglov D, Kozakov D, Vajda S, Stapleton HM, Webster TF, and Schlezinger JJ. 2014. Ligand binding and activation of PPARγ by Firemaster® 550: effects on adipogenesis and osteogenesis in vitro. Environ Health Perspect 122:1225–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER, and Hathaway HJ. 2015. What have we learned about GPER function in physiology and disease from knockout mice? J Steroid Biochem Mol Biol 153:114–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempe D, Vangeison G, Hamilton J, Li Y, Jepson M, and Federoff HJ. 2006. Synapsin I Cre transgene expression in male mice produces germline recombination in progeny. Genesis 44: 44–49. [DOI] [PubMed] [Google Scholar]
- Rettberg JR, Yao J, and Brinton RD. 2014. Estrogen: a master regulator of bioenergetic systems in the brain and body. Front Neuroendocrinol 35:8–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas V, Nguyen MT, Henstridge DC, Nguyen AK, Beaven SW, Watt MJ, and Hevener AL. 2010. Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERalpha-deficient mice. Am J Physiol Endocrinol Metab 298: E304–E319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepke TA, Bosch MA, Rick EA, Lee B, Wagner EJ, Seidlova-Wuttke D, Wuttke W, Scanlan TS, Rønnekleiv OK, and Kelly MJ. 2010. Contribution of a membrane estrogen receptor to the estrogenic regulation of body temperature and energy homeostasis. Endocrinology 151:4926–4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, He Y, Yan X, Yang Y, Wang C, Xu P, Hinton AO Jr., Shu G, Yu L, Tong Q, and Xu Y. 2016. Visualizing estrogen receptor-α-expressing neurons using a new ERα-ZsGreen reporter mouse line. Metab Clin Exp 65:522–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata I, Tanaka T, Yamazaki M, Tanizaki T, Zheng Z, and Sakai T. 2006. Gastric estrogen directly induces ghrelin expression and production in the rat stomach. J Endocrinol 190:749–757. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD and Livak KJ.2008. Analyzing real-time PCR data by the comparative C(T) method. Nature Protoc. 3:1101–1108. [DOI] [PubMed] [Google Scholar]
- Shen M, Senthil Kumar SPD, and Shi H. 2014. Estradiol regulates insulin signaling and inflammation in adipose tissue. Horm Mol Biol Clin Invest 17:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Senthil Kumar SPD, and Liu X. 2013. G protein-coupled estrogen receptor in energy homeostasis and obesity pathogenesis. Prog Mol Biol Transl Sci 114:193–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shughrue PJ, Lane MV, and Merchenthaler I. 1997. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol 388 :507–525. [DOI] [PubMed] [Google Scholar]
- Song RX, Barnes CJ, Zhang Z, Bao Y, Kumar R, and Santen RJ. 2004. The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor alpha to the plasma membrane. Proc Nat Acad Sci USA 101:2076–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steves AN, Bradner JM, Fowler KL, Clarkson-Townsend D, Gill BJ, Turry AC, Caudle WM, Miller GW, Chan AWS, and Easley CA. 2018. Ubiquitous flame-retardant toxicants impair spermatogenesis in a human stem cell model. Science 3:161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stincic TL, Rønnekleiv OK, and Kelly MJ. 2018. Diverse actions of estradiol on anorexigenic and orexigenic hypothalamic arcuate neurons. Horm Behav 104:146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veen Ike, and Jacob de Boer. 2012. Phosphorus flame retardants: Properties, production, environmental occurrence, toxicity and analysis. Chemosphere 88:1119–1153. [DOI] [PubMed] [Google Scholar]
- Vail GM, Walley SN, Yasrebi A, Maeng A, Conde KM, Roepke TA. 2020. The interactions of diet-induced obesity and organophosphate flame retardant exposure on energy homeostasis in adult male and female mice. J Toxicol Environ Health A 83: 438–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vail GM. And Roepke TA. 2020. Organophosphate flame retardants excite arcuate melanocortin circuitry and increase neuronal sensitivity to ghrelin in adult mice. Endocrinology 161:bqaa168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, and Kilgore MW. 2002. Signal cross-talk between estrogen receptor alpha and beta and the peroxisome proliferator-activated receptor gamma1 in MDA-MB-231 and MCF-7 breast cancer cells. Mol Cell Endocrinol 194:123–133. [DOI] [PubMed] [Google Scholar]
- Wang Y-X 2010. PPARs: Diverse regulators in energy metabolism and metabolic diseases. Cell Res 20:124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zhao Y, Li M, Du M, Li X, and Li Y. 2019. A review of a class of emerging contaminants: The classification, distribution, intensity of consumption, synthesis routes, environmental effects and expectation of pollution abatement to organophosphate flame retardants (OPFRs). Int J Mol Sci 20:2874–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasin S, Behary N, Curti M, and Rovero G. 2016. Global consumption of flame retardants and related environmental concerns: A study on possible mechanical recycling of flame retardant textiles. FIBER 4:16. [Google Scholar]
- Yasrebi A, Hsieh A, Mamounis KJ, Krumm EA, Yang JA, Magby J, Pu H, and Roepke TA. 2016. Differntial gene regulation of GHSR signaling pathway in the arcuate nucleus and NPY neuros by fasting, diet-induced obestiy, and 17β-estradiol. Mol Endocrinol 422:42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasrebi A, Rivera JA, Krumm EA, Yang JA, and Roepke TA. 2017. Activation of estrogen response element-independent ERα signaling protects female mice from diet-induced obesity. Endocrinology 158:319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AS, Allen JG, Kim UJ, Seller S, Webster TF, Kannan K, and Ceballos DM. 2018. Phthalate and organophosphate plasticizers in nail polish: Evaluation of labels and ingredients. Environ Sci Technol 52:12841–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Suhring R, Serodio D, Bonnell M, Sundin N, and Diamond ML. 2016. Novel flame retardants: Estimating the physical-chemical properties and environmental fate of 94 halogenated and organophosphate PBDE replacements. Chemosphere 144:2401–2407. [DOI] [PubMed] [Google Scholar]
- Zota AR, Linderholm L, Park JS, Petreas M, Guo T, Privalsky ML, Zoeller RT, and Woodruff TJ. 2013. Temporal comparison of PBDEs, OH-PBDEs, PCBs, and OH-PCBs in the serum of second trimester pregnant women recruited from San Francisco General Hospital, California. Environ Sci Technol 47:11776–11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zota AR, Park JS, Wang Y, Petreas M, Zoeller RT, and Woodruff TJ. 2011. Polybrominated diphenyl ethers, hydroxylated polybrominated diphenyl ethers, and measures of thyroid function in second trimester pregnant women in California. Environ Sci Technol 45:7896–7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, T.A. Roepke, upon reasonable request.