

Abstract
Immunotherapy has emerged as one of the most powerful anti-cancer therapy classes but is stymied by the limits of existing preclinical models with respect to disease latency and reproducibility. In addition, the influence of differing immune microenvironments within tumors observed clinically and associated with immunotherapeutic resistance cannot be tuned to facilitate drug testing workflows without changing model system or laborious genetic approaches. To address this testing platform gap in the immune oncology drug development pipeline, we deployed engineered biomaterials as a scaffold to increase tumor formation rate, decrease disease latency, and diminish variability of immune infiltrates into tumors formed from murine mammary carcinoma cell lines implanted into syngeneic mice. By altering synthetic gel formulations that reshaped infiltrating immune cells within the tumor, responsiveness of the same tumor model to varying classes of cancer immunotherapies, including in situ vaccination with a molecular adjuvant and immune checkpoint blockade, diverged. These results demonstrate the significant role the local immune microenvironment plays in immunotherapeutic response. These engineered tumor immune microenvironments therefore improve upon the limitations of current breast tumor models used for immune oncology drug screening to enable immunotherapeutic testing relevant to the variability in tumor immune microenvironments underlying immunotherapeutic resistance seen in human patients.
Keywords: cancer immunotherapy, biomaterials, drug screening, immunotherapy, breast cancer
Graphical Abstract
A system leveraging synthetic material scaffolds that induce reliable tumor formation as well as programmable immune infiltration based on composition, enabling enhanced consistency and relevance to human disease was developed. Various immunotherapeutic strategies were tested, revealing differential efficacy based on local immune milieu, demonstrating the promise for this system in improving the application of immunotherapy to human cancers.
1. Introduction
Breast cancer is the most common cancer among women worldwide, affecting approximately 2.3 million people annually.[1,2] Triple-negative breast cancer (TNBC), which lacks hormone receptor and excess HER2 protein expression, is the most aggressive type of breast cancer, with higher rates of metastasis and shorter overall survival.[3] Immunotherapies have emerged as one of the most promising tools in the fight against breast cancer. However, response rates in the clinic are disappointingly low – only ~16% of patients respond to immune checkpoint blockade (ICB) therapy,[4] and no vaccine therapies have been approved for breast cancer, despite successes in melanoma and prostate cancer.[5,6] These poor outcomes reflect an unmet need in understanding the differences in patient responses and tools to develop new and better immunotherapeutic strategies for patients who are less likely to respond to immunotherapies.
Substantial efforts in the immune oncology field are currently dedicated to unraveling the determinants of the anti-cancer immune response, given its role in disease progression and response to immunotherapy. Immunologically “hot” tumors generally have more immune infiltration, while “cold” tumors harbor fewer immune cells.[7] Additionally, other stratifications have been identified, with the potential to influence immunotherapy responses – among immune-infiltrated (hot) tumors, macrophages and CD8 T cells have been shown to have opposite impacts on patient survival, and in turn on responses to chemotherapy.[8] Likewise, neutrophils and CD8 T cells form unique signatures that induce differing ICB responses, with CD8 T cell-infiltrated tumors demonstrating much higher responsiveness to ICB.[9] Additionally, T cell programmed death-1 (PD1) expression impacts survival opposite to regulatory T cell infiltration,[10] with implications for ICB responses.[11] Thus, even within immunologically “hot” tumors, different immune cell signatures have potential ramifications on patient survival as well as responses to immunotherapy and chemotherapy. This variability in local immune microenvironment is thought to underlie immunotherapeutic resistance, including in the context of TNBC, motivating patient screening approaches to identify therapies with the highest curative potential.[12,13]
Due to their reliance on the coordinated effects of multiple arms of the immune system, immunotherapies are tested primarily using preclinical animal models.[14] Current breast cancer models include patient-derived xenografts, genetically engineered systems, and tumors formed from cell lines implanted into syngeneic animals that vary substantially in their breadth of use, cost, disease latency, and immune physiology.[14] In patient-derived xenografts, a portion of a patient’s tumor is introduced into an immune deficient or humanized animal, typically within Matrigel™ (MT), a mouse sarcoma-derived extracellular matrix extract.[14] However, only ~40% of xenografts form tumors, even in immune deficient animals.[15] Additionally, responses to immunotherapies elicited in humanized mouse xenograft models are often highly variable and do not predict responses seen in patients.[16–18] Alternatively, genetically engineered models have oncogenic and other genetic information inserted in the animal genome,[19] sometimes in tissue specific loci, to induce tumor formation. An example is the mouse mammary tumor virus-polyoma middle tumor-antigen (MMTV-PyMT) mouse model in which the polyoma tumor virus is expressed in the mammary fatpad (MFP) to spontaneously form breast tumors.[20] The spontaneous nature of these responses more closely mimics the variable latency and progression of human tumors.[20,21] However, the rate of tumor development is highly variable,[22,23] which has the potential to result in disparate tumor-localized immune microenvironments, making testing of immunotherapies exceedingly challenging. Lastly, syngeneic tumor models consist of immortalized tumor cell lines injected into syngeneic, immunocompetent animals.[14,24] While this latter model class could be considered perhaps the least sophisticated, it is by far the most commonly implemented in the immunotherapy field given the ease and rapid nature in which tumor-bearing animals can be generated in varying animal cohorts by synchronous implantation of cancerous cells, the wide availability of transgenic rodents to enable mechanistic testing, and because mice are the lowest phylogenetic species that have human-mimicking immune systems. However, given the low rate of tumor formation when injected cells are suspended in saline alone, syngeneic TNBC models often employ MT to increase the rate of tumor formation.[24,25] Unfortunately, this approach is plagued by MT’s batch-to-batch variation in composition[26] and uncharacterized effects on the recipient animal’s immune response. Furthermore, the above listed model classes are limited in their capacity to generate different tumor immune microenvironments without varying the biology of the engrafted tumor cell line, using depleting or modulatory interventions, or use of a genetic knockout system. This is because disease onset and progression are predicated by the host-tumor interaction,[9] which makes the study of the varied microenvironments seen in the tumors of human patients not easily recapitulated in rodent models without changing tumor cell line or animal host. Accelerating and better predicting immunotherapeutic responses in human patient populations are thus severely limited by the gap in the existing repertoire of preclinical tumor models for immunotherapeutic testing.
In this study, well-defined, degradable synthetic hydrogels used in in vivo tissue engineering applications[26–30] were implemented as a scaffold to reproducibly form tumors from TNBC cell lines in syngeneic mice. Achieving a tumor formation rate of 100% with short latency, formed tumors exhibited low variability in tumor growth profiles and infiltrating immune cell repertoires, attributes favorable for immunotherapeutic drug screening. Furthermore, anti-tumor immune responses that mimic various patterns of immune microenvironments seen in human TNBC tumors were programmed based on hydrogel formulation through the incorporation of cell adhesive peptides within the synthetic polymer network that direct local immune response. Strikingly, microenvironments formed from the same polymer scaffold but directing varying immune microenvironments resulted in disparate responses to ICB and cancer vaccine immunotherapies. These engineered tumor immune microenvironments therefore address the limitations of current tumor models used for drug screening in the immune oncology field to enable immunotherapeutic testing relevant to the variability in tumor-infiltrating lymphocyte profiles seen in human patients.
2. Results
2.1. Engineered hydrogel scaffolds for consistent, controllable breast tumor formation in vivo with short latency
Genetically engineered tumor models, including but not limited to the MMTV-PyMT system,[20] are generally considered the most physiologically relevant systems for the study of immune remodeling in cancer.[19] However, within MMTV-PyMT tumors analyzed at various timepoints throughout disease progression, we found wide variability in terms of the rate of tumor development, with cell numbers ranging by >1 order of magnitude, and coefficients of variation ranging from 26 to 131% (Figure 1a), which can make immunotherapeutic testing in synchronous animal cohorts exceedingly difficult. Even when excised tumors from lesions formed in MMTV-PyMT mice were dispersed and reimplanted into the MFP of wildtype (non-transgenic, tumor-naïve, immunocompetent) C57Bl/6 mice, rates of tumor formation varied considerably with suspension composition (Figure 1b). When dispersed in saline alone, implanted cells formed tumors in ~ 60% of injected animals (Figure 1b). The use of MT, a commonly used basement membrane extract product generated from mouse sarcoma cells,[14] increased tumor formation rate, with 100% and ~85% of injected animals forming tumors with 107 and 106 cells implanted, respectively (Figure 1b). However, tumor growth was still highly variable between formed tumors (Figure 1c). Implantation of Py230 cells, a cell line derived from lesions of MMTV-PyMT animals and thought to be less differentiated compared to typical immortalized cell lines,[31] similarly benefited from injection when suspended in MT compared to saline with respect to tumor formation rate, with 100 and ~90% versus 20% or no tumors forming at 106 versus 0.25×106 cells respectively (Figure 1d). However, for different batches of MT, the latency of tumor formation from 0.25×106 Py230 cells (Figure 1e) and animal survival (Figure S1a) varied considerably. Both trends in MT’s benefits with respect to tumor implantation rate and batch effects on tumor growth rate were also seen in the TNBC murine mammary carcinoma line E0771 (Figure 1f–h, Figure S1b–c). Furthermore, the immune microenvironments of E0771 tumors formed in immunocompetent C57Bl/6 mice varied substantially both between animals and matrix vehicle types. Among major immune cell subtypes (total CD45+ cells, macrophages, DCs, and T cells, Figure S2 and S3), the extent of infiltration at day 2 post implantation was initially similar between MT and saline vehicle but subsequently varied substantially at day 7 and 28 post implantation (Figure 1i). Moreover, tumors implanted in saline exhibited vastly varying extents of infiltration amongst identically treated animals, varying by >2 orders of magnitude between tumors at day 7 post implantation (Figure 1j). Tumors formed at day 7 after cells were injected in MT resulted in more consistent tumor infiltrating cell levels, varying by less than 0.5 order of magnitude, with coefficients of variation below 50% (relative to >150% in tumors implanted in saline) (Figure 1j). However, the number of infiltrating T cells was significantly different between different MT batches (Figure 1j). Macrophage M2/M1 ratios (defined as CD206+ versus CD86+), ratios of CD206 to CD86 expressing DCs, and the ratio of Treg (CD4+CD8-FoxP3+CD25+) to total CD3+CD8+CD4- (CD8) T cells also varied substantially amongst mice implanted with tumors using saline (Figure 1k), an effect ameliorated, except for the case of DC phenotype, by MT as the tumor matrix vehicle (Figure 1k). Nevertheless, ratios varied substantially between MT batch (Figure 1k). As a whole, both MT and saline, when used as vehicles for the implantation of breast tumors into immunocompetent animals, induce highly variable immune infiltration, limiting their application for immunotherapeutic testing.
Figure 1. Current preclinical models of breast cancer are insufficient.
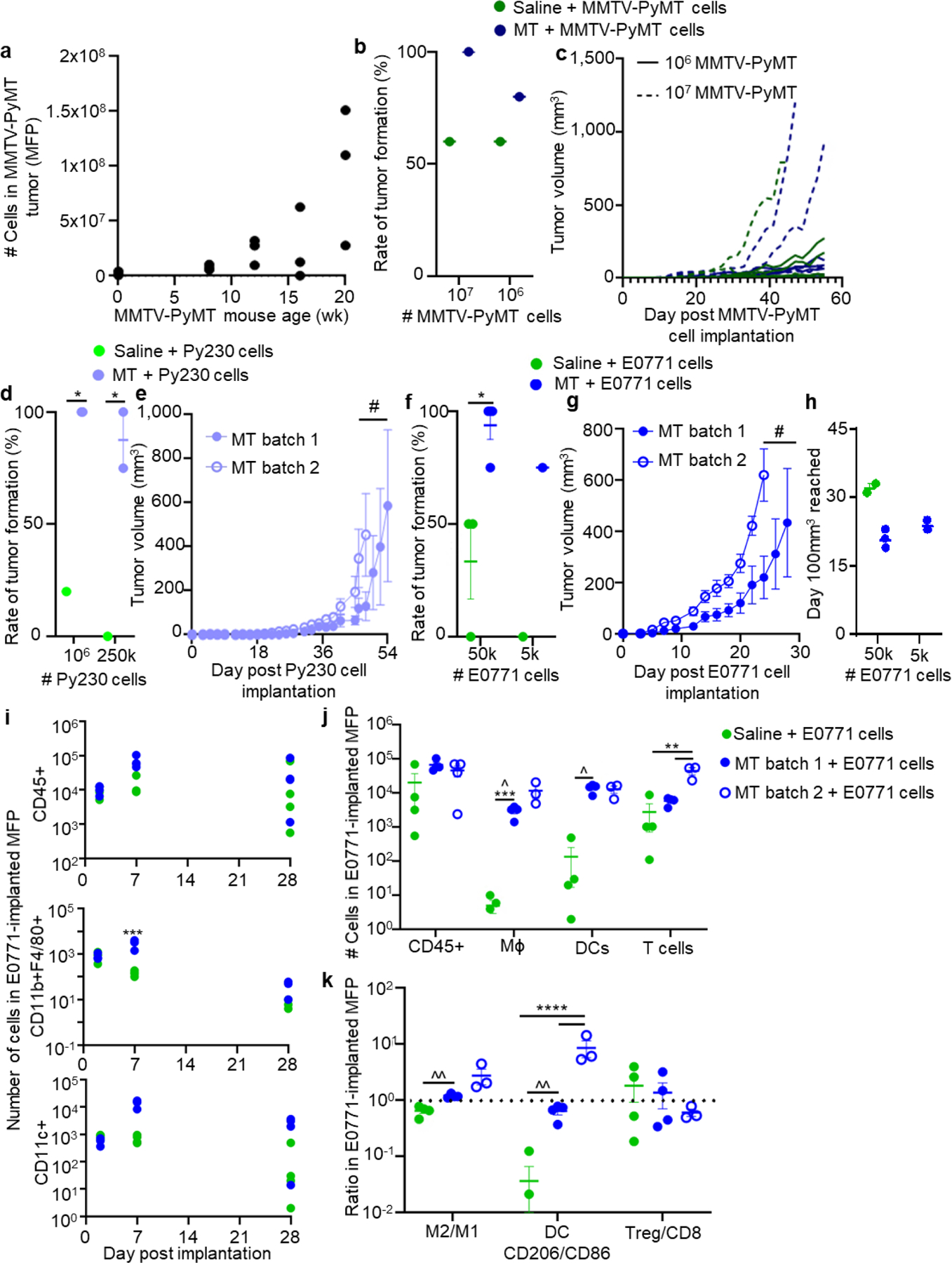
(a) Number of total cells in excised MMTV-PyMT tumors at week 8, 12, 16, and 20, and wild-type animals (week 0). a, each point represents one mouse. Rate of tumor formation (b) and tumor volume (c) after implantation of varying numbers of cells derived from MMTV-PyMT tumors into mammary fatpad (MFP) of C57/Bl6 mice. b, each point represents one experiment with n=5 mice. c, each line represents the growth curve of one tumor implanted in the MFP a mouse, n=5 per group. Rate of tumor formation (d) at two total Py230 cell doses, and tumor growth curves (e) of 250,000 Py230 cells implanted in the MFP of C57Bl6 mice in either saline and Matrigel (MT). d, each point represents one experiment with n=4–10 animals. e, data represents mean ± s.e.m. with n=5 animals implanted with one tumor. Rate of tumor formation (f) at two total E0771 cell doses, and tumor growth curves (g) and time until tumors reached 100 mm3 (h) of 50,000 E0771 cells implanted in the MFP of C57/Bl6 mice in either saline and MT. f, each point represents one experiment with n=4 animals. g, data represents mean ± s.e.m. with n=4 animals implanted with one tumor. h, each data point represent one tumor implanted animal. (i) Number of total lymphocytes (CD45+), macrophages (CD11b+F4/80+), and dendritic cells (CD11c+) at various times post implantation of 50,000 E0771 tumor cells in saline or MT into the MFP of C57Bl6 mice. Each data point represents one tumor implanted animal. Number of total lymphocytes (CD45+), macrophages, dendritic cells, and T cells (j) and phenotype of macrophages, DCs, and T cells (k) at day 7 after 50,000 E0771 cells were implanted in the MFP of C57Bl6 mice in saline or two different batches of MT. Each data point represents one tumor implanted animal. * indicates significance by two-way ANOVA (b) or mixed-effects analysis (d, f, i, j, k) with Tukey’s post-hoc comparison; # indicates significance by RM ANOVA with Tukey’s post-hoc comparison; ^ indicates significant difference in variance by Brown-Forsythe’s test. *, #, ^ indicate p<0.05, **, ^^ indicate p<0.01, *** indicates p<0.005, **** indicates p<0.001.
We hypothesized that well-defined synthetic matrices[30,32,33] could overcome the deficiencies in tumor latency, growth, and immune infiltration intrinsic to existing preclinical tumor model systems. Specifically, synthetic matrices formed from 4-armed poly(ethylene glycol) (PEG) maleimide (PEG-4MAL) macromers were used. The PEG-4MAL platform provides structurally defined hydrogels with stoichiometric incorporation of biological ligands, improved crosslinking efficiency, and excellent in vitro and in vivo cytocompatibility.[30] The PEG-4MAL synthetic system is also reproducible, with highly consistent (CV <5%) rheological values across macromer and peptide batches.[28] To synthesize gels, cysteine-containing RGD adhesive peptide (or its scrambled control peptide RDG) was conjugated to the PEG-4MAL macromer via reaction with the maleimide group to produce a functionalized macromer. RGD was chosen for incorporation to generate a potentially immunomodulatory hydrogel due to this bioadhesive ligand’s capacity to modulate immune infiltration, tissue ingrowth and vascularization, and dendritic cell phenotype via engagement with cell-expressed integrins[34–36]. This functionalized precursor was then crosslinked into a network in the presence of cancer cells by reacting with a bis-cysteine-flanked crosslinker peptide (VPM) susceptible to cleavage by proteases including matrix metalloproteinases (Figure 2a). Based on our extensive experience with this hydrogel platform, we selected 6% w/v PEG-4MAL gels functionalized with 1.0 mM RGD or RDG and crosslinked with VPM. Rheological testing confirmed previous reports64 of no difference in average storage (G’) and loss moduli (G’’) between RGD versus RDG-presenting hydrogels (Figure S1e–f). Also consistent with previous results demonstrating the lack of toxicity of the matrix or its degradation components,[37,38] the platform did not induce toxicity when used as a tumor vehicle, as evidenced by normal animal weight gain (Figure S1g) and the lack of signs of illness or duress, or toxicity as measured by alanine transaminase or aspartate transaminase levels (Figure S1h–i) in animals implanted with the synthetic matrix scaffold.
Figure 2: Engineered hydrogels induce consistent and controllable tumor immune microenvironments.
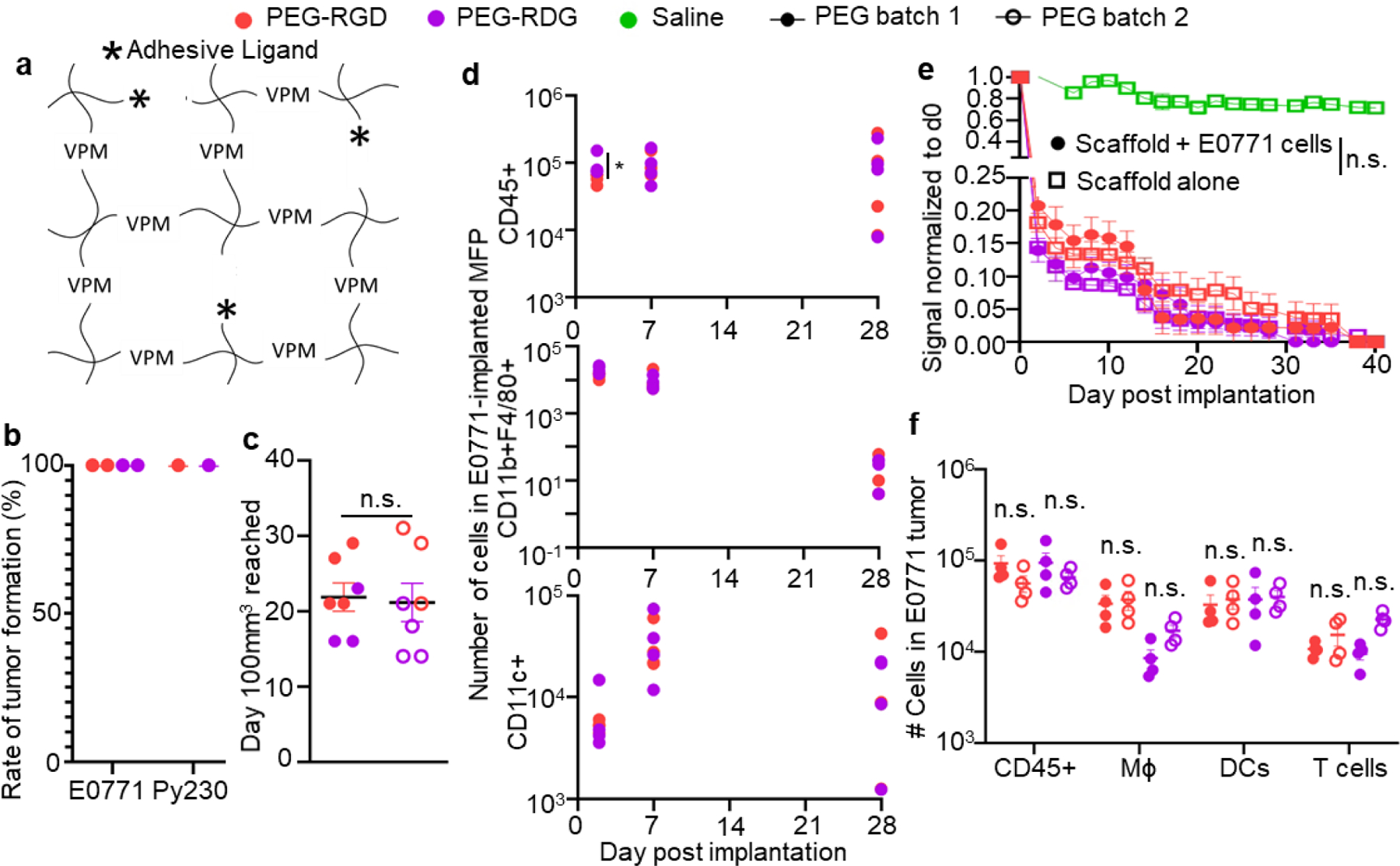
(a) Schematic diagram of hydrogels consisting of PEG-4MAL, with VPM crosslinkers, and adhesive ligands (stars). Tumor formation (b) and growth (c) rate after implantation of 50,000 E0771 cells into C57Bl6 mice in PEG hydrogel matrix vehicle. b, each point represents one experiment with n=4 mice. c, measured as time to 100mm3 in tumor volume. Each data point represents one tumor implanted animal. (d) Number of total lymphocytes (CD45+), macrophages (CD11b+F4/80+), and DCs (CD11c+) within tumor various times post implantation of 50,000 E0771 cells into C57Bl6 mice in PEG hydrogel matrix vehicle. Each point represents one animal. (e) Degradation of AF750-labelled PEG matrix vehicle implanted into C57Bl6 mice relative to d0 signal, measured by IVIS imaging, with or without co-implantation of 50,000 E0771 cells. Data represents mean ± s.e.m. with n=6 animals implanted with one tumor. (f) Number of total lymphocytes (CD45+), macrophages, dendritic cells, and T cells within tumor day 7 post implantation of 50,000 E0771 cells into C57Bl6 mice in two separate PEG hydrogel matrix vehicle batches prepared ~1 year apart. Each data point represents one tumor implanted animal. For all groups, error bars indicate mean ± s.e.m. * indicates significance by one-way ANOVA with Tukey’s post-hoc comparison; * indicates p<0.05, n.s. indicates not significant (p>0.05).
Both Py230 and E0771 cells implanted in these PEG gels induced tumors in 100% of animals (Figure 2b) at total implanted cell amounts of 0.25×106 and 0.05×106 cells, respectively, where low tumor formation rates were seen for saline and variable for MT at similar, or higher, cell amounts of 0.25–1×106 or 1–10×106 Py230 or E0771 cells, respectively (Figure 1f). Likewise, the time to reach 100 mm3 in tumor volume, a size at which E0771 tumors grow exponentially,[39] was consistent between batches of PEG scaffolds (Figure 2c). This high success of tumor formation was not dependent on adhesive peptide as the non-adhesive RDG peptide also yielded 100% tumor formation (Figure 2b). Total leukocyte infiltration was higher into PEG-RGD tumors at day 2 after implantation, but otherwise, overall infiltration of leukocytes, macrophages, and DCs were equivalent between hydrogels presenting RGD and RDG over 28 d (Figure 2d). We examined in vivo scaffold degradation using near infrared dye-labeled hydrogels and IVIS imaging and no difference in matrix vehicle signal loss was observed with inclusion of tumor cells compared to the tumor-cell free matrix vehicle, nor between PEG gels presenting RGD and RDG peptides (Figure 2e). In contrast to that seen in MT, tumor growth rate as well as immune infiltration into tumors at day 7 post implantation by injection in PEG hydrogels were highly consistent between polymer batch (Figure 2f, Figure S1d), despite the polymer mixtures being prepared >1 year apart and high degrees of variability in infiltration levels into tumors formed in saline or MT at this tumor stage (Figure 1i–k). This engineered hydrogel system enables the consistent and reproducible growth of tumors formed from TNBC cells that are immunologically consistent for in vivo disease modeling.
2.2. Hydrogel adhesive peptide directs immune infiltration into breast tumor microenvironments
Given the potential for adhesive ligands such as RGD to impact vascularization within scaffolds,[40,41] we next investigated the vascularization response of tumors formed from different hydrogel formulations. First, vascular endothelial growth factor (VEGF)-A levels within E0771 tumors implanted within PEG gels differed substantially based on adhesive ligand at early but not later tumor stages (Figure 3a). Tumors formed from E0771 cells in RGD-functionalized PEG hydrogel (PEG-RGD) matrices also exhibited higher degrees of vascularization compared to RDG-functionalized PEG hydrogel (PEG-RDG)-formed tumors as measured by microcomputed tomography (μCT) of Microfil-perfused animals at early (d7) but not later (d28) tumor stages (Figure 3b–c). Nevertheless, tumor growth rate did not reflect the expected effect of tumor vascularization induced by adhesive ligand incorporation into the scaffold, as PEG-RDG tumors grew more rapidly than PEG-RGD tumors. In fact, PEG-RDG tumors exhibited faster tumor growth, decreased time until a tumor volume of 100 mm3 was reached, and shorter animal survival (Figure 3d–f). Histological analysis of these tumors revealed that PEG matrix vehicles remained largely intact at day 2 post tumor implantation and by day 7 had more tissue integration (Figure 3g). Qualitatively, PEG-RDG tumors also contained higher cell densities compared to PEG-RGD tumors (Figure 3g). In order to delineate the immunological and structural effects of tumor implantation within a scaffold, we implanted E0771 tumors within saline, MT, and both engineered hydrogels, into NOD-SCID-gamma (NSG) mice (Figure 3h–i), which are immune deficient in B cells, T cells, and natural killer cells.[42] This analysis revealed that differences in tumor latency and growth rate between scaffold type were lost. This striking result suggests that the immune competency of the host, and as a result, infiltration by host immune cells into the tumor cell-laden scaffold, impacts scaffold-dependent effects on tumor formation.
Figure 3: Adhesive ligands alter immune responses in tumors formed using engineered hydrogel matrix vehicles.
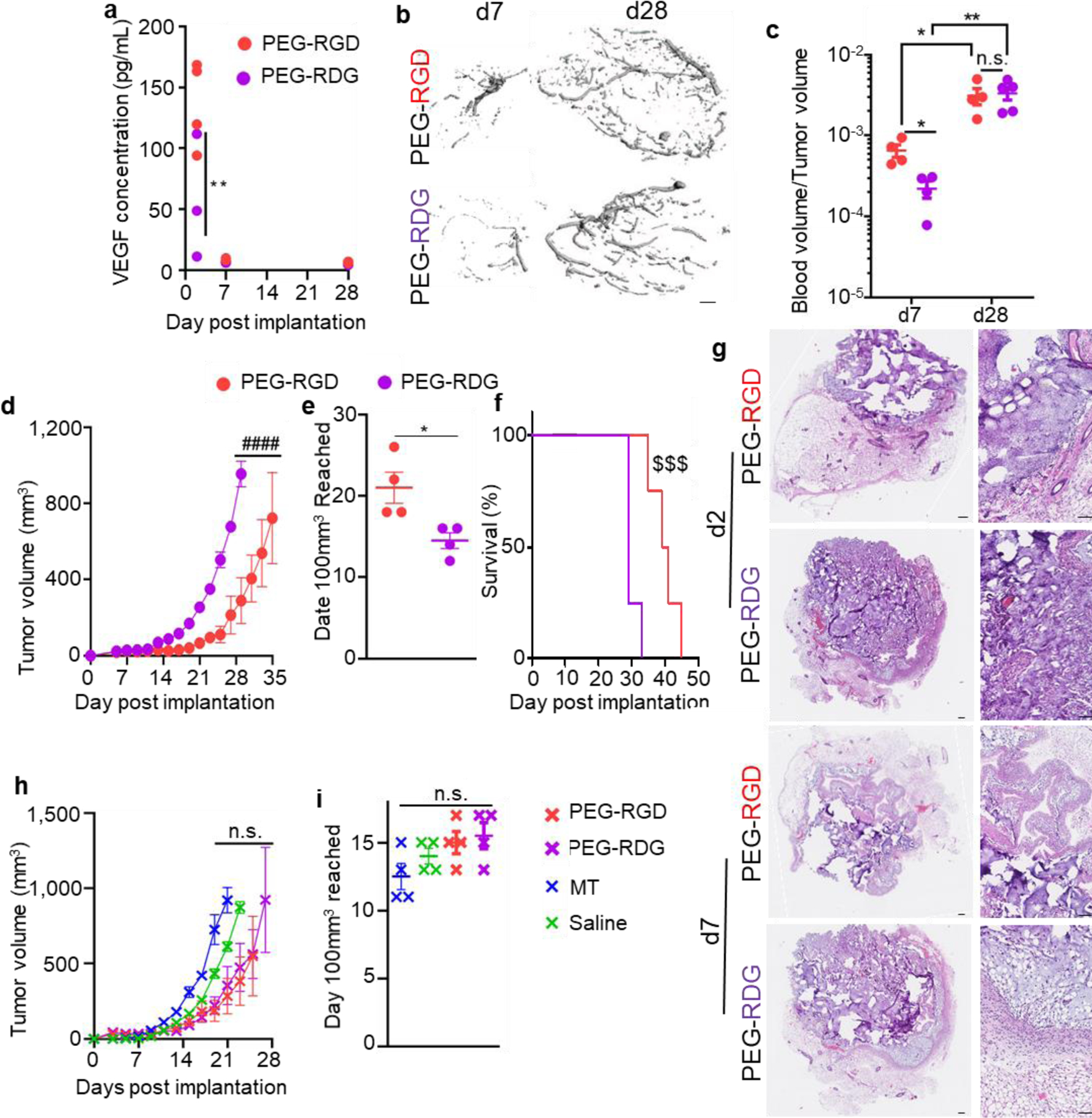
(a) VEGF-A concentration within tumors formed from 50,000 E0771 cells implanted in C57Bl6 mice in PEG hydrogel matrix vehicles at d2, 7, and 28 after implantation. Each data point represents one tumor implanted animal. Vasculature within tumors formed from 50,000 E0771 cells implanted in C57Bl6 mice in PEG hydrogel matrix vehicles, as measured by micro-computed tomography, visualized in (b) and quantified in (c). b, scale bar indicates 100μm. c, each data point represents one tumor implanted animal. Tumor growth curves (d), days until a tumor volume of 100mm3 was reached (e) and animal survival (f) after 50,000 E0771 cells were implanted in C57/Bl6 mice in PEG hydrogel matrix vehicles. d, data represent mean ± s.e.m. of n=4 tumors implanted into individual animals. e, each data point represents one tumor implanted animal. f, n=4. (g) Histological sections of tumors 2 and 7 days after implantation of 50,000 E0771 cells into C57/Bl6 mice in either PEG hydrogel matrix vehicle formulation. Images are representative of n=4 animals per group, scale bars represent 100μm. Tumor growth curves (h) and days until a tumor volume of 100mm3 was reached (i) after 50,000 E0771 cells were implanted into NSG mice in various matrix vehicle types. h, data represent mean ± s.e.m. of n=4 tumors implanted into individual animals. i, each data point represents one tumor implanted animal. * indicates significance by one-way ANOVA (i) or mixed-effects analysis (a, c) with Tukey’s post-hoc comparison, or Mann-Whitney test (e); # indicates significance by RM ANOVA with Tukey’s post-hoc test; $ indicates significance by Log-Rank test; * indicates p<0.05, ** indicates p<0.01, $ $ $ indicates p<.005, #### indicates p<0.001, n.s. indicates not significant (p>0.05).
To interrogate the apparent immune association of differences in tumor latency and growth rate directed by adhesive ligand incorporation into the matrix vehicle injected with E0771 cells, immune phenotyping of formed breast tumors during development and growth was performed, focusing on cell subsets previously implicated in tumor latency and disease progression.[9,10,43–47] In particular, DCs within tumors were found to exhibit higher activation states when PEG-RGD compared to PEG-RDG scaffolds were used at early times post implantation (d 2), an effect lost at later (d 7 and 28) tumor stages (Figure 4a). Whereas M2/M1 ratios were similar at all measured tumor stages (Figure 4b), infiltration of CD11b+Ly6C+Ly6G+ cells, a cell type that in a cancer context is likely a neutrophil subset,[9,48–51] into tumors formed from PEG-RGD was higher to that of tumors formed with PEG-RDG at day 2 and 7 but not at day 28 post implantation (Figure 4c). Tumors formed from PEG-RGD scaffolds also exhibited higher ratios of CD8+ to regulatory T cells compared to tumors formed from PEG-RDG at day 2 post implantation, a difference that, like DC phenotype, was not seen at day 7 and 28 post implantation (Figure 4d). CD44−, naïve, CD8+ T cells were higher in day 2 tumors in PEG-RGD tumors, and this difference similarly dissipated in later stage tumors (Figure 4e). Taken together, PEG-RGD tumors induced higher infiltration of CD8+ T cells and CD86+ DCs, while PDG-RDG tumors contained more neutrophils (Figure 4f). No differences were noted in immune cell contents of the spleen and draining lymph node, except for higher numbers of CD11b+Ly6C+Ly6G+ cells in spleens at day 28 post implantation of E0771 cells within PEG-RDG scaffolds (Figure S5). Together, these results demonstrate that engineered hydrogels have the potential to modulate immune infiltration locally throughout tumor development and progression.
Figure 4: Hydrogel adhesive ligands alter immune responses in tumors formed within synthetic hydrogel matrix vehicles.
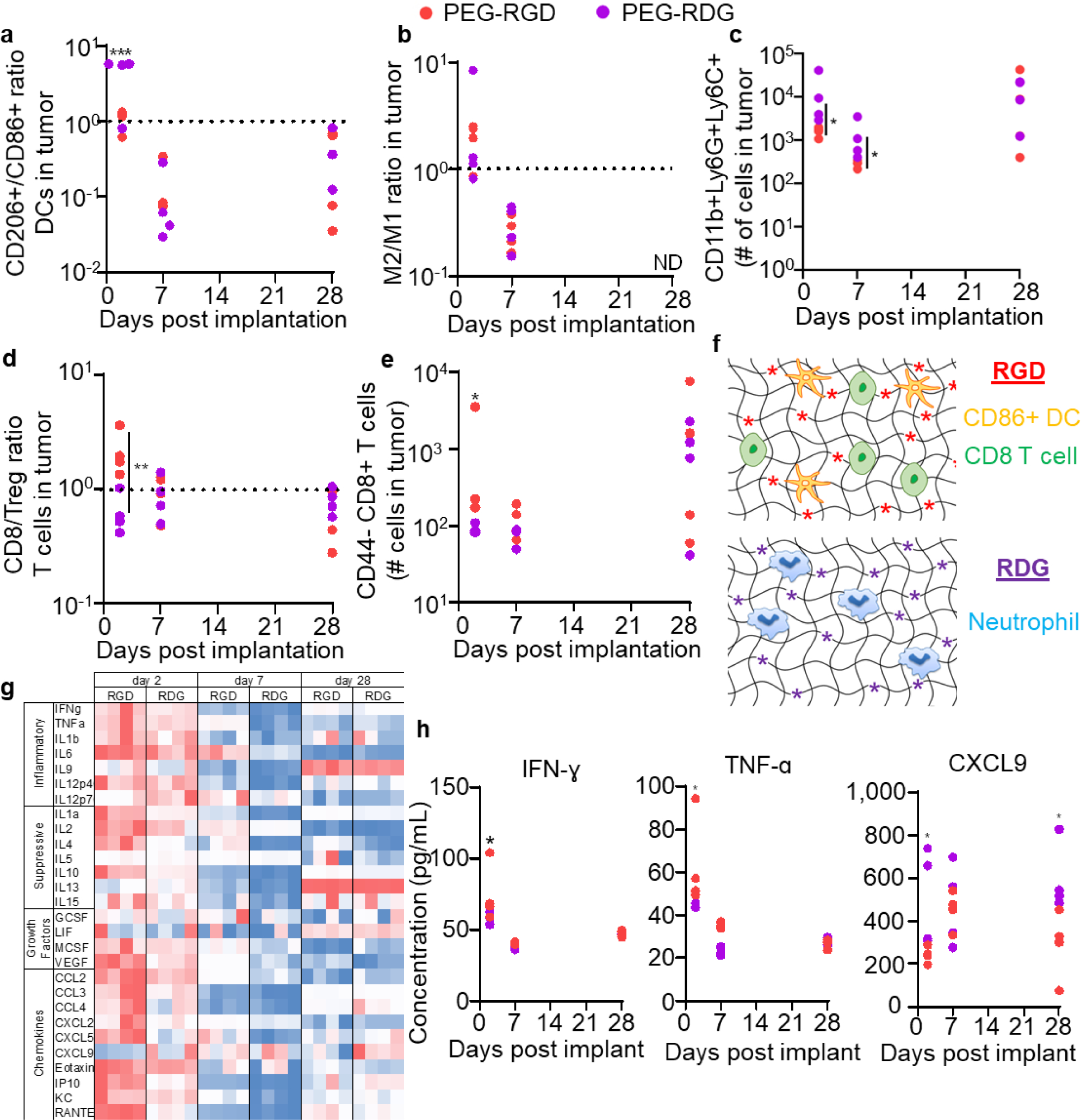
CD206/CD86 ratio among DCs (a), M2/M1 ratio among macrophages (b), number of CD11b+Ly6C+Ly6G+ cells (c), CD8/Treg ratio among T cells (d), and number of CD44+PD1+CD8+ T cells (e) infiltrating tumors at d2, 7, and 28 after implantation of 50,000 E0771 cells into C56Bl6 mice in varying PEG hydrogel matrix vehicle formulations. Each data point represents one tumor implanted animal. (f) Resulting immune infiltration from RGD (top) and RDG (bottom) ligands. (g-h) Tumor cytokine levels at d2, 7, and 28 after implantation of 50,000 E0771 cells into C57Bl6 mice within PEG hydrogel matrix vehicles. g, each column represents an individual animal, and levels are scaled from minimum (blue) to maximum (red) in each row. h, each data point represents one tumor implanted animal. * indicates significance by one-way ANOVA (a, b, e) or mixed-effects analysis (c, d, h) with Tukey’s post-hoc comparison; statistics for g in Table S1; * indicates p<0.05, ** indicates p<0.01, *** indicates p<0.005, ND indicates no data (not enough cells counted for reliable result).
Cytokine production within these tumors was also assessed over time. As a whole, higher levels of cytokines were present within PEG-RGD tumors relative to PEG-RDG tumors at day 2 and 7 post tumor implantation, with the exception of C-X-C motif chemokine ligand (CXCL) 9, an effect that dissipated by day 28 (Figure 4g). In particular, concentrations of interferon (IFN) γ and tumor necrosis factor (TNF) α, cytokines associated with a Th1 response,[52] were enhanced at day 2 in PEG-RGD tumors relative to PEG-RDG tumors (Figure 4h), implying changes in local immune cells discussed above (Figure 4f) as inducing functional cytokine responses within the formed tumors. Similarly, interleukin (IL) 1a, IL4, IL10, IL13, CXCL10, and RANTES were elevated in PEG-RGD tumors at day 2; and IL6, IL13, KC, and RANTES were elevated in PEG-RGD tumors at day 7 (Figure 4g, Table S1), indicative of overall increase in inflammatory response. It should be noted that both immunostimulatory and immunosuppressive cytokines were higher within PEG-RGD compared to PEG-RDG tumors, suggestive of an overall inflammatory response engendered by the former matrix vehicle. Notably, type I interferon (IFN-α and IFN-β) levels were equivalent in tumors formed with either PEG-RGD or PEG-RDG matrix scaffolds (Figure S4), suggestive of type I interferons not driving the oberserved differences in immune microenvironments of tumors formed from synthetic matrix vehicles of differing compositions. Interestingly, CXCL9, a chemokine which can be attributed to neutrophils,[53] showed opposite trends compared to all other cytokines assessed here (Figure 4g, h), however, with higher concentrations in PEG-RDG tumors relative to PEG-RGD tumors at day 2 and 28 of tumors growth, implicating neutrophil infiltration as being functional. These cytokine responses correlate with cellular responses above, with PEG-RGD tumors inducing a CD8+ T cell and CD86+ DC, Th1 type response, while PEG-RDG tumors induce a neutrophil-driven response. Of note, human TNBC tumors have been shown develop one of three immune signatures: (1) immune-infiltrated responses driven by neutrophils; (2) immune-infiltrated tumors driven by M1 macrophages and CD8+ T cells; and (3) immunologically “cold” tumors, generally devoid of immune cells.[9,40] The two immune-infiltrated tumor subsets relevant to human TNBC appear recapitulated using the engineered scaffold system presented here, with PEG-RGD tumors demonstrating a CD8+ T cell-driven response, and PEG-RDG tumors demonstrating a neutrophil-driven response (Figure 4f).
As both the tumor cells and adhesive ligands tethered to the hydrogel have the potential to alter the immune status of developing tumors, we compared the infiltrating immune cells in scaffolds implanted with or without tumor cells. The phenotype of DCs (CD206:CD86 ratio) and type of T cell (by CD8+ T cell:Treg ratio), which defined the cellular phenotype within PEG-RGD tumors (Figure 4f), were identical within PEG-RGD scaffolds implanted with or without tumor cells, indicating that presentation of RGD within the scaffold (i.e., not the cancer cells) drove this Th1 response (Figure 5a–b). However, within PEG-RDG scaffolds, both DC phenotype and type of T cell were initially (at day 2) higher in tumor cell-containing scaffolds compared to scaffold vehicles implanted alone before equalizing at day 7 and day 28 (Figure 5a–b), indicating that the initial Th1 response in PEG-RDG scaffolds was tumor cell- and not adhesive ligand-driven. For neutrophils, which infiltrated PEG-RDG scaffolds to greater extents, initial infiltration (at day 2) was not different between scaffolds implanted with or without tumor cells when implanted with hydrogels presenting either RGD or RDG (Figure 5c). However, at day 7 and 28, scaffolds implanted with tumor cells had higher neutrophil infiltration (Figure 5c), indicating a primarily tumor-driven response. As a whole, the Th1 response in PEG-RGD scaffolds appears associated with the adhesive ligand engineered within the system. In contrast, the neutrophil response in PEG-RDG scaffolds was tumor cell-driven. Thus, scaffold-directed host immune response within the tumor, but not tumor vascularization, correlate with tumor growth rate. Taken together, these results show that the engineered delivery scaffold for the tumor cells can direct the local immune microenvironment within the tumor.
Figure 5: Immune status is affected by both tumor- and adhesive ligand-specific responses.
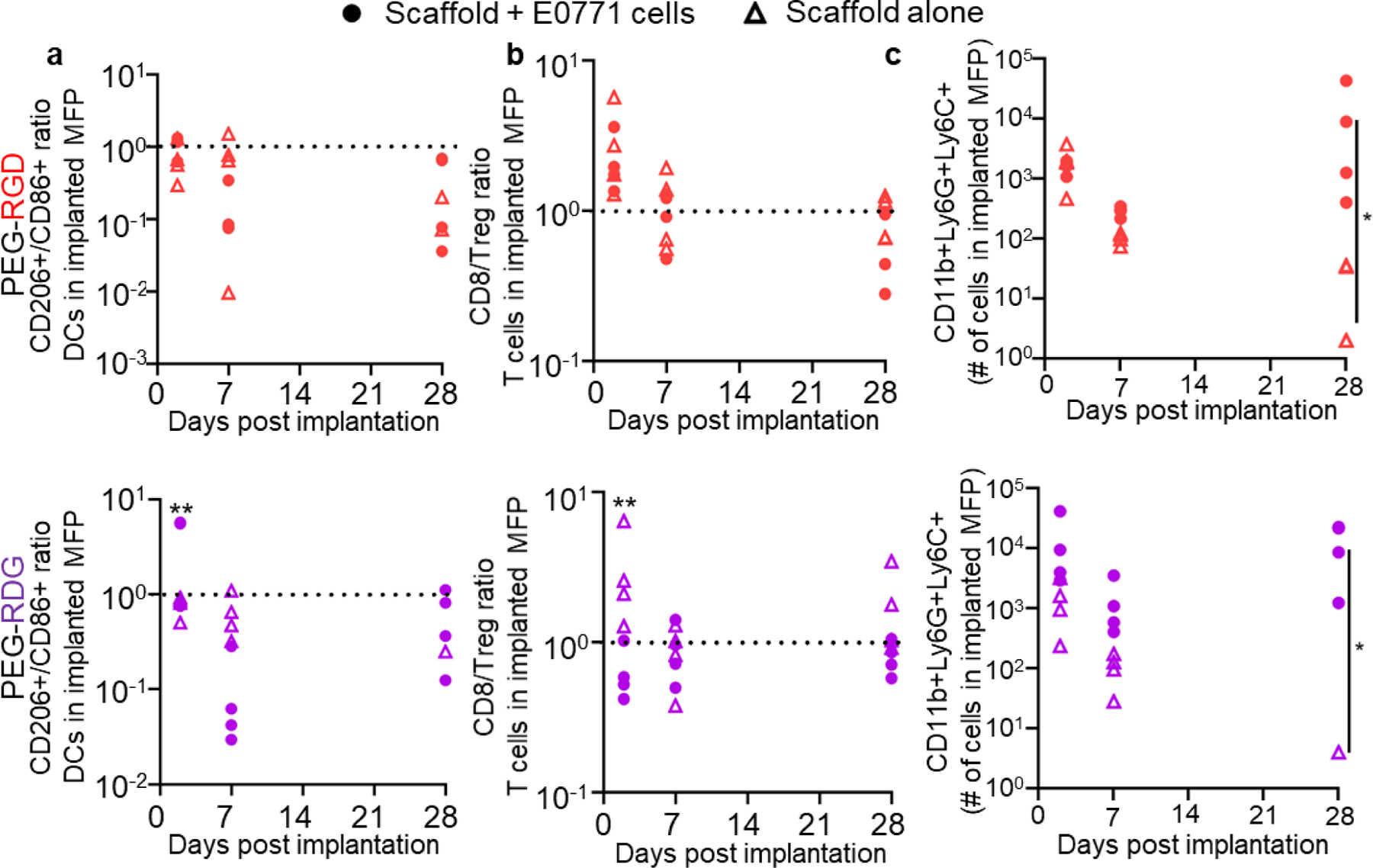
CD206+/CD86+ ratio of CD11c+ cells (a), CD8+ T cell: Treg ratio (b), and (c) number of CD11b+Ly6C+Ly6G+ cells in PEG-RGD and PEG-RDG scaffolds in C57Bl6 mice with or without co-implantation of 50,000 E0771 cells. Each data point represents one scaffold implanted animal. * indicates significance by mixed-effects analysis with Tukey’s post-hoc comparison; n=4 mice; * indicates p<0.05, ** indicates p<0.01.
2.3. Matrix scaffold-directed tumor immunophenotype dictates immunotherapeutic responses
The response of tumors formed from TNBC E0771 cells implanted within different matrix vehicles to immunotherapy was assessed. Immunotherapies evaluated included an in situ cancer vaccine, in the form of intratumorally (i.t.) injected Toll-like receptor 9 agonist CpG,[54] and ICB in the form of intraperitoneally (i.p.) administered monoclonal antibodies (mAb) recognizing cytotoxic T lymphocyte antigen (CTLA) 4 and PD1. Responses to isotype control mAb also administered i.p. were also evaluated. Therapies were administered to individual animals once tumors reached 100 mm3 in ellipsoidal volume in order to account for the highly variable differences in tumor formation and growth rate between matrix vehicle types.
With i.t. CpG treatment, tumors implanted in saline, MT, and PEG-RGD matrices did not respond to the i.t. vaccine, demonstrating similar growth curves as the untreated animals (Figure 6a, Figure S6, S7). In contrast, tumors implanted using PEG-RDG scaffold exhibited significant slowing of tumor growth (Figure 6a) that resulted in prolonged animal survival as a result of i.t. vaccination (Figure 6b). These results are consistent with a reversal of the poor Th1 response previously seen in these tumors (Figure 4f). We note, however, that the physicochemical characteristics of intratumorally administered vaccine could alter the responsiveness of these tumors. Contrastingly, after ICB treatment, therapeutic responses were minimal in mice bearing tumors formed from PEG-RDG, saline, and MT matrices, but robust in animals formed from PEG-RGD matrices (Figure 6c, Figure S6, S7). As a result, the overall survival of PEG-RGD tumor-bearing animals was prolonged relative to all other groups (Figure 6d). This result suggests that ICB therapy is beneficial in the context of tumor immune microenvironments that have a heightened local CD8+ T cell and CD86+ DC response. Of note, due to the differential blood vasculature within these tumors (Figure 3b), it is possible that the efficiency of delivery of monoclonal antibodies within the tumor microenvironment may be altered between these two tumor subsets. However, we previously showed that the dose of monoclonal antibody administered intraperitoneally used here has little effect on survival in this mouse model when saline was used as a vehicle of tumor cell implantation.[55] Importantly, no matrix vehicle-specific differences in tumor growth or survival were observed for animals receiving isotype control mAb (Figure 6e–f), nor were survival benefits that varied by therapy class associated with tumor growth rates in untreated animals (Figure S7). These results demonstrate that local immune microenvironments within the tumor programmed by the engineered scaffold direct the resulting sensitivity versus resistance to differing classes of immunotherapy.
Figure 6: Tumor response to immunotherapy is dependent upon matrix vehicle.
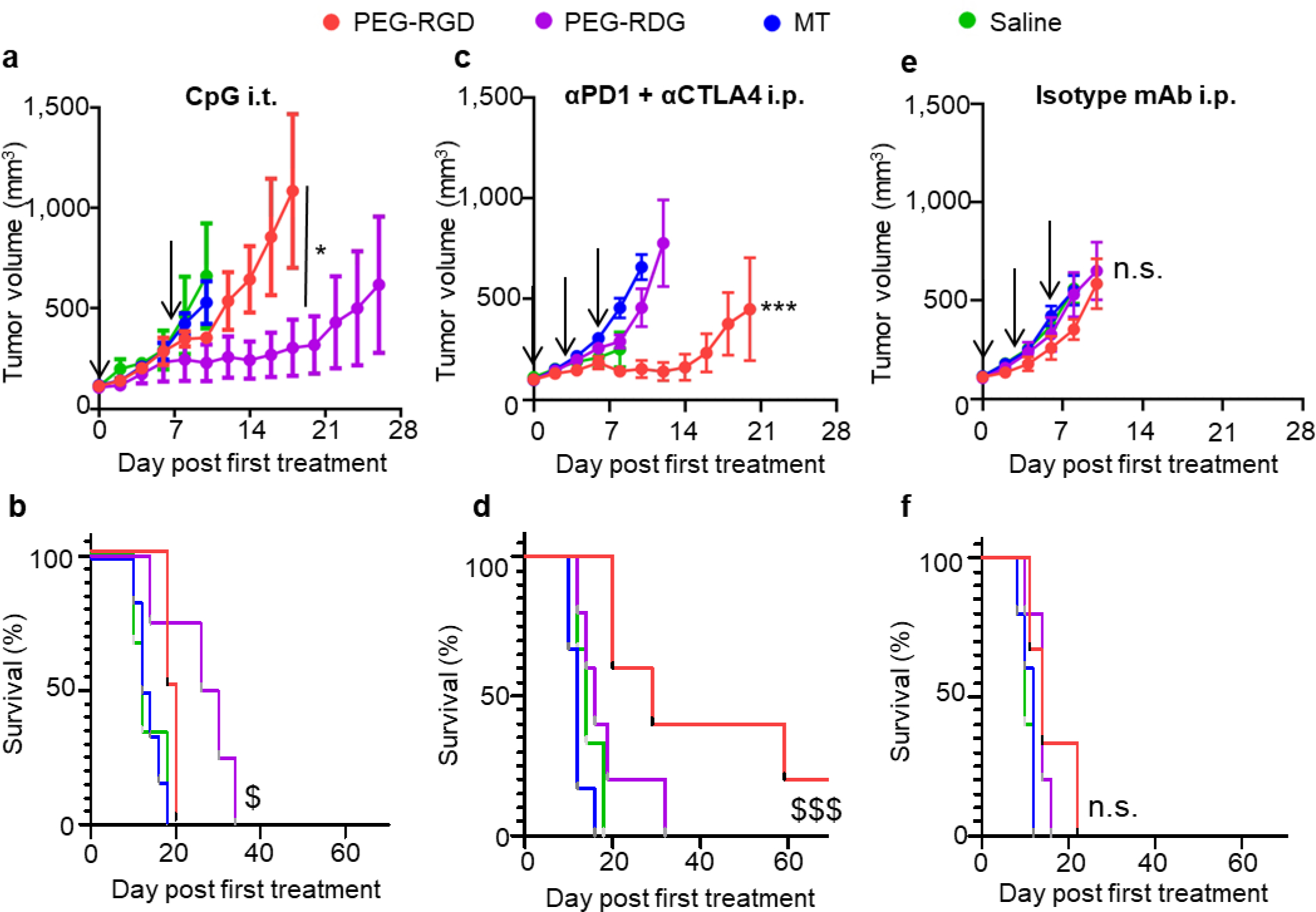
Tumor growth curves (a, c, e) and animal survival (b, d, f) of C57Bl6 mice implanted with E0771 cells after CpG vaccine administered i.t. on day 0 and 7 (a-b), combination αPD1 + αCTLA4 mAb treatment administered i.p. on days 0, 3, and 6 (c-d), and isotype control mAb administered i.p. on days 0, 3, and 6 (e-f). a, c, e, data represent mean ± s.e.m. of n=5–7 tumors implanted into individual animals. d0 signifies the first day of treatment when tumors reached 100 mm3. Arrows indicate days therapy is given. * indicates significance by repeated measures ANOVA with Tukey’s post-hoc test, against all other groups if not specified; $ indicates significance against all other groups by log-rank test. a, b, e, and f are representative of two independent experiments; *, $ indicate p<0.05, ***, $ $ $ indicate p<0.005; n.s. indicates not significant (p>0.05).
3. Conclusion
The demand for a modernized immune oncology drug development testing pipeline has mushroomed with the advent of the cancer immunotherapy era. Such improvements include radical advancements in in vitro bioassays and organoid technologies.[56–59] Yet despite their attractiveness with respect to cost, speed, scale, and mechanistic insights, such approaches necessarily oversimplify the spatial and temporal complexity of the adaptive immune response in cancer and are unable to predict therapeutic benefit. Seemingly in complete opposition to this, there is a push within the in vivo disease modelling space for the use of increasingly complex genetic models with the goal of better recapitulating human disease.[21] Despite their advantages in modeling the heterogeneity of human cancer and adaptive immune response, however, these systems are prohibitively expensive, highly variable, slow, and as a result poorly scalable. An ideal platform for screening the therapeutic efficacy of immune oncology drugs would instead be tumor immune microenvironments that can be generated with deterministic reproducibility at scale that do not sacrifice the dynamic complexity of an in vivo adaptive immune response.
Here, we describe a robust and scalable platform for the in vivo modeling of immunologically defined tumors inspired by but improving upon existing syngeneic rodent tumor models. A well-defined synthetic hydrogel scaffold system previously established in a variety of tissue engineering applications was deployed. In so doing, this system ameliorates the limitations with patient-derived xenografts, as it is much less expensive and tumors form 100% of the time across multiple independent experiments; genetically engineered mouse models, as both the number of tumors and immune responses against those tumors are consistent and controllable; and current syngeneic models, as the immune response is consistent between batches and can be modulated to more closely mimic different immune responses observed in clinical samples.
The concept of immunologically defined tumors that are programmable based on scaffold composition offers numerous advantages with respect to tumor immunotherapy drug development. Such an outcome has been demonstrated here with growth trajectories being shaped by the local immune microenvironment. More provocatively, immunotherapeutic sensitivity versus resistance vary with therapy class and scaffold composition, without changing the tumor cell used that would necessarily change the underlying biology. We hypothesize that RGD is able to modulate immune cell adhesion and differentiation in response to the local matrix, altering the local immune milieu,[34–36] and implicating the incorporated adhesive ligand as important in inducing the immunological effects seen in these tumors. As this benefit is afforded by the modular nature of the material system, other adhesive ligands or immunomodulatory agents could also be tethered to the scaffold to further direct immune responses against the developing tumor. Additionally, this could be implemented for other solid tumor types for which current immunotherapeutic strategies are lacking, in order to better predict immune responses and develop novel, more effective anti-cancer treatments. Finally, synthetic scaffolds are well-suited to engineering and further manipulation of the local microenvironment. This would be of particular interest for the modeling of heterogeneity of human tumors[9,60–62] that could be achieved without the need for co-implantation of multiple differing cell subtypes whose sensitivity to implantation may vary, by ensuring ensuring consistent cell engraftment using this synthetic matrix vehicle system. Some limitations in this system, however, are the expanded vasculature within PEG-RGD tumors, which may not wholly replicate human disease; the speed in which tumors develop; and the lack of heterogeneity in the current model, each of which could be ameliorated with further engineering of the system utilized here. The concept of a tissue engineered tumor thus resembles strategies that have long been explored in the context of regenerative engineering and in in vitro screening and organoid systems, but heretofore have been underutilized as enabling in vivo tumor immunotherapy drug development.
4. Experimental Section/Methods
Cell culture:
E0771 breast cancer cells, derived from C57Bl/6 mice, were cultured in Dulbecco’s modified eagle’s medium (Gibco, Thermo Fisher Scientific, Inc., Waltham, MA) with 10% heat-inactivated fetal bovine serum (Gibco, Thermo Fisher Scientific, Inc.) and 1% penicillin/streptomycin/amphotericin B (Life Technologies, Carlsbad, CA). Py230 murine mammary tumor cells were cultured in F-12K medium (Corning, VWR International, Inc.) with 5% heat-inactivated fetal bovine serum (Gibco, Thermo Fisher Scientific Inc.), 1% penicillin/streptomycin/amphotericin B (Life Technologies), and 0.1% MITO+ serum extender (Corning, VWR International, Inc.). Cells were maintained at 37°C and 5% CO2 and passaged at ~70–80% confluency using 0.05% (E0771) or 0.25% (Py230) Trypsin-EDTA (Thermo Fisher Scientific, Inc.).
Animal tumor models:
All protocols were approved by Georgia Tech’s Institutional Animal Care and Use Committee, performed on animal protocol number A100305. C57Bl/6 or Nod-Scid-Gamma (NSG) mice were purchased at 6 wk of age from Jackson Laboratory (Bar Harbor, ME). MMTV-PyMT mice backcrossed onto C57Bl6 background were bred in house. MMTV-PyMT mice were monitored on a weekly basis throughout tumor development and progression. For MMTV-PyMT transplant studies, tumors were excised around 200 mm3 in ellipsoid volume. MMTV-PyMT tumors were separated using 18G needles and incubated with 1mg/mL collagenase D (Sigma Aldrich) in Dulbecco’s phosphate-buffered saline (D-PBS) for 60 min at 37°C with 5% CO2. Tumors were then dissociated by pushing through a 70 μm cell strainer (Greiner Bio-One, Monroe, NC) twice and washed with D-PBS, and counted for implantation. 5–500 × 103 E0771 cells, 0.25–1 × 106 Py230 cells, or 1–10 × 106 PyMT tumor cells in 30 μL of appropriate scaffold were injected in the fourth (inguinal) mammary fatpad. Animals were monitored every 1–3 days during tumor growth. Tumor dimensions were measured with calipers in three dimensions and reported as ellipsoidal volume. Animals were euthanized if they displayed signs of rodent illness (weight loss >10%, hunched, ungroomed appearance) or if the tumor reached 15 mm in any dimension.
Flow cytometry:
Tumor, lymph node, and spleen samples were excised from animals after CO2 asphyxiation. Tumor samples were broken up using 18G needles and incubated with 1 mg/mL collagenase D (Sigma Aldrich) in D-PBS for 4 h at 37°C. LN samples were incubated with 1 mg/mL collagenase D (Sigma Aldrich) in D-PBS for 75 min at 37°C. Following collagenase incubation, samples were pushed through 70 μm cell strainers (Greiner Bio-One), washed with D-PBS, pelleted, and plated at appropriate dilutions in a 96-well U-bottom plate (VWR International, Inc.). Spleen capsules were disrupted using 18G needles, pushed through 70 μm cell strainers (Greiner Bio-One), washing with D-PBS, pelleted, and resuspended in 1 mL red blood cell lysis buffer (Sigma Aldrich) for 7 min at room temperature. Samples were quenched with ~35 mL D-PBS, pelleted, and plated at appropriate dilutions. Cells were blocked with CD16/CD32 antibody (clone 2.4G2, Tonbo Biosciences, San Diego, CA) for 5 min on ice, washed, and stained with a fixable viability dye Zombie Aqua (1:100 dilution, Biolegend, Inc.) for 30 min at room temperature, before quenching with 0.1% bovine serum albumin in D-PBS (flow cytometry buffer). Antibodies were obtained from Biolegend, Inc. unless otherwise specified, and prepared at the following dilutions on the basis of preliminary titrations: APC-Cy7 anti-mouse CD45 (0.625:100), AF700 anti-mouse CD11b (1.25:100), BV605 anti-mouse CD64 (2.5:100), BV711 anti-mouse Ly6C (2.5:100), FITC anti-mouse MerTK (1:100), PerCP anti-mouse Ly6G (eBioscience, Thermo Fisher Scientific, Inc.; 2.5:100), PE-Cy7 anti-mouse CD11c (1.25:100), BV421 anti-mouse MHC-II (1.25:100), PE anti-mouse CD86 (5:100), and BV786 anti-mouse F4/80 (2.5:100) for APC panel; or PerCP anti-mouse CD45 (0.625:100), BV711 anti-mouse CD3 (1.25:100), APC-Cy7 anti-mouse CD4 (0.15625:100), FITC anti-mouse CD8 (0.3125:100), BV786 anti-mouse PD1 (1.25:100), AF700 anti-mouse CD25 (1:100), and BV421 anti-mouse CD44 (5:100) for T cell panel. APC panel samples were then washed and incubated in IC fixation buffer (eBioscience, Thermo Fisher Scientific, Inc.) for 60 min at room temperature in the dark. Cells were then incubated with APC anti-mouse CD206 (2.5:100) in IC permeabilization buffer (eBioscience, Thermo Fisher Scientific, Inc.) for 60 min at room temperature in the dark. T cell panel samples were washed and resuspended in FoxP3/Transcription factor fixation/permeablization solution (eBioscience, Thermo Fisher Scientific, Inc.) for 60 min on ice in the dark. Cells were then incubated with PE anti-mouse FoxP3 (5:100) in FoxP3/Transcription factor fixation/permeabilization buffer (eBioscience, Thermo Fisher Scientific, Inc.) for 75 min on ice in the dark. Both APC and T cell panel samples were resuspended in flow cytometry buffer and kept at 4°C for a maximum of 48 h before analysis using a customized BD LSRFortessa (BD Biosciences). Compensation was performed using ArC (for live/dead) or UltraComp (for antibodies) compensation beads (Thermo Fisher Scientific, Inc.) and data analyzed using FlowJo software version 10 (FlowJo LLC, Ashland, OR).
PEG hydrogels:
PEG-4MAL hydrogels were prepared as described previously [27]. Briefly, PEG-4MAL macromer (molecular mass of 22,000 Da; Laysan Bio, Inc., Arab, AL) was dissolved in 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acis (HEPES) buffer (10 mM in D-PBS, pH 7.4) at 15% w/v (2.5x macromer density, for a final 6% w/v PEG-4MAL concentration). Cell adhesive and crosslinking peptides were custom synthesized by GenScript Biotech (Piscataway, NJ). Cell adhesive peptide RGD (GRGDSPC) and its scrambled control RDG (GRDGSPC) were dissolved in HEPES buffer at 5.0 mM (5x ligand density, for a final 1.0 mM ligand concentration and mixed with PEG-4MAL at a 2:1 PEG-4MAL/ligand ratio to generate functionalized PEG-4MAL precursor. Bis-cysteine crosslinking peptide (GCRDVPMSMRGGDRCG) was dissolved in HEPES at a density corresponding to 1:1 maleimide to cysteine ratio after accounting for maleimide groups reacted with adhesive peptide. Cells were resuspended at 5 x final density in sterile saline and kept on ice. A final density of 50,000 cells were encapsulated in all hydrogels. For all the studies listed, the engineered synthetic hydrogel formulation used was 6% w/v PEG-4MAL functionalized with 1mM RGD or RDG and crosslinked with VPM (storage modulus, G’ = 300 Pa). The functionalized PEG-4MAL (PEG-4MAL:adhesive peptide) and cell mixture was combined with the crosslinker at 2:1:1:1 volume ratio (PEG-4MAL:adhesive peptide:cells:crosslinker) immediately before injection. Based on previous work,[63,64] >98% of input adhesive ligand is incorporated into the network.
For in vivo hydrogel degradation experiments, both RGD and RDG were conjugated with AlexaFluor750 dye through NHS ester reaction kit following manufacturer instructions (A37575, Molecular Probes by Life Technology).
IVIS imaging:
Animals were anesthetized using isoflurane anesthesia and placed in a PerkinElmer IVIS (in vivo imaging system) Spectrum CT (Waltham, MA). AF750 signal was collected every other day until signal was at or below signal from saline-injected animals or animals reached endpoint due to tumor growth.
Micro-computed tomography imaging:
Animals were perfused with D-PBS at the heart followed by neutral buffered formalin (Thermo Fisher Scientific, Inc.) for 5 min, then with saline to rinse, and MicroFil contrast agent (Flow Tech Inc., Carver, MA) catalyzed at a viscosity appropriate for small vessels (5 mL lead-based contrast agent, 2.5 mL diluent, 0.25 mL curing agent). Perfused mice were carefully stored at 4°C overnight to cure. The following day, tumor samples were excised, and imaged using a SCANCO Medical μCT50 (Scanco USA, Inc., Wayne, PA). μCT image slices were constrained using manual selection of the sample outline and processed with a Gaussian filter at a consistent global threshold via the Scanco Medical μCT Evaluation Program before 3-dimensional reconstruction.[65]
Cytokine analysis:
Tumor, LN, and spleen samples were excised from animals, flash frozen using liquid nitrogen, and stored at −80°C. BioPlex lysis buffer (Bio-Rad) was prepared and 100 μL added to each sample. Tissues were mashed using P200 pipette tips until a smooth solution formed. Samples were oscillated at 4°C for 20 min, spun down, and tissue lysate collected and frozen at −80°C. The following day, total protein content was measured using Pierce bicinchoninic acid assay (Thermo Fisher Scientific, Inc.). Milliplex MAP 32-plex mouse cytokine/chemokine magnetic bead panel (Millipore Sigma) was used to assess cytokine and chemokine content in samples. In brief, samples were added to plate at appropriate dilutions with premixed magnetic beads (Milliplex) in assay buffer (Milliplex) and incubated on a plate shaker overnight at 4°C. Samples were then placed in magnetic plates, decanted, and washed. Detection antibodies (Milliplex) were added to each sample and incubated for 1 h at 4°C on plate shaker. Streptavidin-phycoerythrin (Milliplex) was added directly to each well, and incubated for 30 min at 4°C on plate shaker. Samples were then placed in magnetic plate, decanted, and washed. Samples were resuspended in 100 μL drive fluid and analyzed using a MagPix system (Luminex, Austin, TX).
Therapeutic studies:
E0771 tumor cells (50,000 cells) were implanted into immunocompentent C57/Bl6 mice in saline, MT, PEG-RGD, and PEG-RDG, and tumors measured every 48 h. Animals were randomized among cages and researchers were blinded to groups. Animals were randomly pre-assigned to a therapeutic group. Once tumors reached ~100 mm3 in ellipsoidal volume (treatment day 0 [d0]), animals received first treatment of CpG, ICB, or isotype mAb based on pre-assigned therapeutic group. Animals assigned to receive CpG therapy received 3 μg ODN 1826 (CpG, InvivoGen, Inc., San Diego, CA) in 30 μL sterile saline intratumorally on d0 and d7. Animals assigned to receive ICB therapy were given 100 μg each of αPD1 (BioXCell) and αCTLA-4 (clone 9H10, BioXCell) in 100 μL sterile saline intraperitoneally on day 0, 3, and 6. Animals assigned to isotype mAb group were given 100 μg each of rat IgG2a anti-trinitrophenol (BioXCell) and polyclonal Armenian hamster IgG (BioXCell) intraperitoneally on day 0, 3, and 6. Tumor volume and animal weight was monitored every 48 h until animals reached the predetermined endpoint (tumor size of 15 mm in any dimension or if the animal displayed signs of illness or distress).
Rheological testing:
Hydrogels were cast in a parafilm-covered glass slide. Upon gelation at 37°C, hydrogels were extracted and swelled in 1X PBS at 4°C overnight. Fully swollen hydrogels were tested using a rheometer (MCR-302, AntonPaar; CP10–2) at 37°C. Frequency sweep (10 to 0.1 rad/s) was performed at a constant strain of 2%. G’ and G’’ were determined by averaging all data points acquired from 10 to 0.1 rad/s interval.
Statistical analysis:
Data are represented as the mean accompanied by SEM, and statistics were calculated using GraphPad Prism 6 and 8 software (GraphPad Software, Inc.). Statistical significance was defined as p<0.05, 0.01, 0.005, and 0.001 based on ANOVA with Tukey’s post-hoc test unless otherwise specified.
Supplementary Material
Acknowledgements
We thank Paul Archer, Claire McClain, Margaret Manspeaker, Rebecca Parker, and Trayvon Baxter for technical assistance. This work was supported by U.S. National Institutes of Health grants R01CA207619 (SNT), U01CA214354 (SNT), R01AR062920 (AJG), R01AR062368 (AJG), S10OD016264 (AJG), T32GM008433 (MJO), and T32EB006343 (AMR).
Footnotes
Tumor growth dynamics and reproducibility of preclinical breast cancer models are improved via biomaterial scaffolds. Different immune milieus within the biomaterial-conditioned tumor microenvironment resulted in immunotherapeutic resistances that differed by immunotherapy class, demonstrating the potential for this system as an in vivo immunotherapy screening platform.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Meghan J. O’Melia, Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30308, USA
Adriana Mulero-Russe, Parker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GA 30308, USA; School of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30308, USA.
Jihoon Kim, Parker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GA 30308, USA; George W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30308, USA.
Alyssa Pybus, Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30308, USA; Parker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GA 30308, USA.
Deborah DeRyckere, Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta and Department of Pediatrics, Emory University School of Medicine, Atlanta, GA 30308, USA; Winship Cancer Institute, Emory University, Atlanta, GA 30308, USA.
Levi Wood, Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30308, USA; Parker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GA 30308, USA; George W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30308, USA.
Douglas K. Graham, Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta and Department of Pediatrics, Emory University School of Medicine, Atlanta, GA 30308, USA Winship Cancer Institute, Emory University, Atlanta, GA 30308, USA.
Edward Botchwey, Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30308, USA; Parker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GA 30308, USA.
Andrés J. García, Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30308, USA Parker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GA 30308, USA; George W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30308, USA.
Susan N. Thomas, Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30308, USA Parker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GA 30308, USA; Winship Cancer Institute, Emory University, Atlanta, GA 30308, USA; George W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30308, USA.
References
- [1].Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F, CA. Cancer J. Clin. 2021, 71, 209. [DOI] [PubMed] [Google Scholar]
- [2].Siegel RL, Miller KD, Jemal A, CA. Cancer J. Clin. 2019, 69, 7. [DOI] [PubMed] [Google Scholar]
- [3].Li X, Yang J, Peng L, Sahin AA, Huo L, Ward KC, O’Regan R, Torres MA, Meisel JL, Breast Cancer Res. Treat. 2017, 161, 279. [DOI] [PubMed] [Google Scholar]
- [4].Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im S-A, Wright GS, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S, Emens LA, N. Engl. J. Med. 2018, 379, 2108. [DOI] [PubMed] [Google Scholar]
- [5].Andtbacka RHI, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, Milhem M, Cranmer L, Curti B, Lewis K, Ross M, Guthrie T, Linette GP, Daniels GA, Harrington K, Middleton MR, Miller WHJ, Zager JS, Ye Y, Yao B, Li A, Doleman S, Vanderwalde A, Gansert J, Coffin RS, J. Clin. Oncol. 2015, 33, 2780. [DOI] [PubMed] [Google Scholar]
- [6].Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF, N. Engl. J. Med. 2010, 363, 411. [DOI] [PubMed] [Google Scholar]
- [7].Galon J, Bruni D, Nat. Rev. Drug Discov. 2019, 18, 197. [DOI] [PubMed] [Google Scholar]
- [8].DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, Rugo HS, Hwang ES, Jirström K, West BL, Coussens LM, Cancer Discov. 2011, 1, 0F54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, Sheng K, Niu Y, Goldstein A, Zhao N, Bado I, Lo H-C, Toneff MJ, Nguyen T, Bu W, Jiang W, Arnold J, Gu F, He J, Jebakumar D, Walker K, Li Y, Mo Q, Westbrook TF, Zong C, Rao A, Sreekumar A, Rosen JM, Zhang XHF, Nat. Cell Biol. 2019, 21, 1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sun S, Fei X, Mao Y, Wang X, Garfield DH, Huang O, Wang J, Yuan F, Sun L, Yu Q, Jin X, Wang J, Shen K, Cancer Immunol. Immunother. 2014, 63, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, Vitaly K, Itahashi K, Maeda Y, Matsui S, Shibahara T, Yamashita Y, Irie T, Tsuge A, Fukuoka S, Kawazoe A, Udagawa H, Kirita K, Aokage K, Ishii G, Kuwata T, Nakama K, Kawazu M, Ueno T, Yamazaki N, Goto K, Tsuboi M, Mano H, Doi T, Shitara K, Nishikawa H, Nat. Immunol. 2020, 21, 1346. [DOI] [PubMed] [Google Scholar]
- [12].Zhou Y, Fei M, Zhang G, Liang W-C, Lin W, Wu Y, Piskol R, Ridgway J, McNamara E, Huang H, Zhang J, Oh J, Patel JM, Jakubiak D, Lau J, Blackwood B, Bravo DD, Shi Y, Wang J, Hu H-M, Lee WP, Jesudason R, Sangaraju D, Modrusan Z, Anderson KR, Warming S, Roose-Girma M, Yan M, Immunity 2020, 52, 357. [DOI] [PubMed] [Google Scholar]
- [13].Deng Y, Yang Y, Yao B, Ma L, Wu Q, Yang Z, Zhang L, Liu B, Exp. Cell Res. 2018, 364, 208. [DOI] [PubMed] [Google Scholar]
- [14].Breitenbach M, Hoffmann J, Front. Oncol. 2018, 8, 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stewart E, Federico SM, Chen X, Shelat AA, Bradley C, Gordon B, Karlstrom A, Twarog NR, Clay MR, Bahrami A, Freeman BBI, Xu B, Zhou X, Wu J, Honnell V, Ocarz M, Blankenship K, Dapper J, Mardis ER, Wilson RK, Downing J, Zhang J, Easton J, Pappo A, Dyer MA, Nature 2017, 549, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Capasso A, Lang J, Pitts TM, Jordan KR, Lieu CH, Davis SL, Diamond JR, Kopetz S, Barbee J, Peterson J, Freed BM, Yacob BW, Bagby SM, Messersmith WA, Slansky JE, Pelanda R, Eckhardt SG, J. Immunother. Cancer 2019, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang M, Yao LC, Cheng M, Cai D, Martinek J, Pan CX, Shi W, Ma AH, De Vere White RW, Airhart S, Liu ET, Banchereau J, Brehm MA, Greiner DL, Shultz LD, Palucka K, Keck JG, FASEB J. 2018, 32, 1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Snowden E, Porter W, Hahn F, Ferguson M, Tong F, Parker JS, Middlebrook A, Ghanekar S, Dillmore WS, Blaesius R, Mol. Cancer Res. 2017, 15, 429. [DOI] [PubMed] [Google Scholar]
- [19].Sharpless NE, DePinho RA, Nat. Rev. Drug Discov. 2006, 5, 741. [DOI] [PubMed] [Google Scholar]
- [20].Guy CT, Cardiff RD, Muller WJ, Mol. Cell. Biol. 1992, 12, 954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, Pollard JW, Am. J. Pathol. 2003, 163, 2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Qiu TH, Chandramouli GVR, Hunter KW, Alkharouf NW, Green JE, Liu ET, Cancer Res. 2004, 64, 5973. [DOI] [PubMed] [Google Scholar]
- [23].Lifsted T, Le Voyer T, Williams M, Muller W, Klein-Szanto A, Buetow KH, Hunter KW, Int. J. Cancer 1998, 77, 640. [DOI] [PubMed] [Google Scholar]
- [24].Bao L, Matsumura Y, Baban D, Sun Y, Tarin D, Br. J. Cancer 1994, 70, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Benton G, Arnaoutova I, George J, Kleinman HK, Koblinski J, Adv. Drug Deliv. Rev. 2014, 79, 3. [DOI] [PubMed] [Google Scholar]
- [26].Aisenbrey EA, Murphy WL, Nat. Rev. Mater. 2020, DOI 10.1038/s41578-020-0199-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cruz-Acuña R, Quirós M, Huang S, Siuda D, Spence JR, Nusrat A, García AJ, Nat. Protoc. 2018, 13, 2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Han WM, Anderson SE, Mohiuddin M, Barros D, Nakhai SA, Shin E, Amaral IF, Pêgo AP, García AJ, Jang YC, Sci. Adv. 2018, 4, eaar4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cruz-Acuña R, Mulero-Russe A, Clark AY, Zent R, García AJ, J. Cell Sci. 2019, 132, DOI 10.1242/jcs.226639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Phelps EA, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, Sulchek TA, Barker TH, García AJ, Adv. Mater. 2012, 24, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Biswas T, Gu X, Yang J, Ellies LG, Sun L-Z, Cancer Lett. 2014, 346, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Weaver JD, Headen DM, Aquart J, Johnson CT, Shea LD, Shirwan H, García AJ, Sci. Adv. 2017, 3, e1700184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cruz-Acuña R, García AJ, Matrix Biol. 2017, 57–58, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lynn AD, Kyriakides TR, Bryant J, J Biomed. Mater. Res. A. 2010, 93A, 941. [DOI] [PubMed] [Google Scholar]
- [35].Cheng AY, García AJ, Curr. Opin. Biotechnol. 2013, 24, 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Weaver JD, Headen DM, Hunckler MD, Coronel MM, Stabler CL, García AJ, Biomaterials. 2018, 172, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Phelps EA, Headen DM, Taylor WR, Thulé PM, García AJ, Biomaterials 2013, 34, 4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Headen DM, Woodward KB, Coronel MM, Shrestha P, Weaver JD, Zhao H, Tan M, Hunckler MD, Bowen WS, Johnson CT, Shea L, Yolcu ES, García AJ, Shirwan H, Nat. Mater. 2018, 17, 732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].O’Melia MJ, Manspeaker MP, Thomas SN, Cancer Immunol. Immunother. 2021, DOI 10.1007/s00262-020-02792-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lee TT, García JR, Paez JI, Singh A, Phelps EA, Weis S, Shafiq Z, Shekaran A, Del Campo A, García AJ, Nat. Mater. 2015, 14, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Miroshnikova YA, Rozenberg GI, Cassereau L, Pickup M, Mouw JK, Ou G, Templeman KL, Hannachi EI, Gooch KJ, Sarang-Sieminski AL, García AJ, Weaver VM, Mol. Biol. Cell 2017, 28, 2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K, Heike T, Nakahata T, Bone 2002, 100, 3175. [DOI] [PubMed] [Google Scholar]
- [43].Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, Kirkling ME, Reizis B, Ghosh S, D’Amore NR, Bhardwaj N, Rothlin CV, Wolf A, Flores R, Marron T, Rahman AH, Kenigsberg E, Brown BD, Merad M, Nature 2020, 580, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Najafi M, Hashemi Goradel N, Farhood B, Salehi E, Nashtaei MS, Khanlarkhani N, Khezri Z, Majidpoor J, Abouzaripour M, Habibi M, Kashani IR, Mortezaee K, J. Cell. Biochem. 2019, 120, 2756. [DOI] [PubMed] [Google Scholar]
- [45].Santoni M, Romagnoli E, Saladino T, Foghini L, Guarino S, Capponi M, Giannini M, Cognigni PD, Ferrara G, Battelli N, Biochim. Biophys. Acta - Rev. Cancer 2018, 1869, 78. [DOI] [PubMed] [Google Scholar]
- [46].DeNardo DG, Andreu P, Coussens LM, Cancer Metastasis Rev. 2010, 29, 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Huang Y, Ma C, Zhang Q, Ye J, Wang F, Zhang Y, Hunborg P, Varvares MA, Hoft DF, Hsueh EC, Peng G, Oncotarget 2015, 6, 17462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ostrand-Rosenberg S, Fenselau C, J. Immunol. 2018, 200, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, Rodriguez PC, Sica A, Umansky V, Vonderheide RH, Gabrilovich DI, Nat. Commun. 2016, 7, 12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jahchan NS, Mujal AM, Pollack JL, Binnewies M, Sriram V, Reyno L, Krummel MF, Front. Immunol. 2019, 10, 1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Damuzzo V, Pinton L, Desantis G, Solito S, Marigo I, Bronte V, Mandruzzato S, Cytom. Part B - Clin. Cytom. 2015, 88, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Raphael I, Physiol. Behav. 2016, 176, 100. [Google Scholar]
- [53].Scapini P, Laudanna C, Pinardi C, Allavena P, Mantovani A, Sozzani S, Cassatella MA, Eur. J. Immunol. 2001, 31, 1981. [DOI] [PubMed] [Google Scholar]
- [54].Maeyama J, Takatsuka H, Suzuki F, Kubota A, Horiguchi S, Komiya T, Shimada I, Murata E, Osawa Y, Kitagawa H, Matsuki T, Isaka M, Yamamoto S, Iho S, PLoS One 2014, 9, e88846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Francis DM, Manspeaker MP, Schudel A, Sestito LF, O’Melia MJ, Kissick HT, Pollack BP, Waller EK, Thomas SN, Sci. Transl. Med. 2020, 12, eaay3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Neal RA, McClugage SGI, Link MC, Sefcik LS, Ogle RC, Botchwey EA, Tissue Eng. - Part C 2009, 15, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tuveson D, Clevers H, Science (80-. ). 2019, 364, 952. [DOI] [PubMed] [Google Scholar]
- [58].Xu H, Lyu X, Yi M, Zhao W, Song Y, Wu K, J. Hematol. Oncol. 2018, 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Drost J, Clevers H, Nat. Rev. Cancer 2018, 18, 407. [DOI] [PubMed] [Google Scholar]
- [60].Mani NL, Schalper KA, Hatzis C, Saglam D, Tavasol F, Butler M, Chagpar AB, Pusztal L, Rimm DL, Breast Cancer Res. 2016, 18, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gordon S, Taylor PR, Nat. Rev. Immunol. 2005, 12, 953. [DOI] [PubMed] [Google Scholar]
- [62].Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, Salgado R, Byrne DJ, Teo ZL, Dushyanthen S, Byrne A, Wein L, Luen SJ, Poliness C, Nightingale SS, Skandarajan AS, Gyorki DE, Thornton CM, Bearis PA, Fox SB, Jarcy PK, Speed TP, Mackay LK, Weeson PJ, Loi S, Nat. Med. 2018, 24, 986. [DOI] [PubMed] [Google Scholar]
- [63].Enemchukwu ND, Cruz-Acuña R, Bongiorno T, Johnson CT, García JR, Sulchek T, García AJ, J. Cell Biol. 2016, 212, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Clark AY, Martin KE, García JR, Johnson CT, Theriault HS, Han WM, Zhou DW, Botchwey EA, García AJ, Nat. Commun. 2020, 11, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Duvall CL, Taylor WR, Weiss D, Guldberg RE, Am. J. Physiol. - Hear. Circ. Physiol. 2004, 287, H302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.