

Abstract
p21-activated kinases (PAKs) are a family of cell division control protein 42/ras-related C3 botulinum toxin substrate 1 (Cdc42/Rac1)-activated serine/threonine kinases. Group I PAKs (PAK1–3) have distinct activation mechanisms from group II PAKs (PAK4–6) and are the focus of this review. In transformed cancer cells, PAKs regulate a variety of cellular processes and molecular pathways which are also important for myelin formation and repair in the central nervous system (CNS). De novo mutations in group I PAKs are frequently seen in children with neurodevelopmental defects and white matter anomalies. Group I PAKs regulate virtually every aspect of neuronal development and function. Yet their functions in CNS myelination and remyelination remain incompletely defined. Herein, we highlight the current understanding of PAKs in regulating cellular and molecular pathways and discuss the status of PAK-regulated pathways in oligodendrocyte development. We point out outstanding questions and future directions in the research field of group I PAKs and oligodendrocyte development.
Keywords: p21-activated kinases (PAKs), oligodendrocyte progenitor cells (OPCs), oligodendrocytes (OLs), differentiation, myelination, demyelination, remyelination, multiple sclerosis (MS)
I. INTRODUCTION
In the developing central nervous system (CNS), myelin-forming oligodendrocytes (OLs) are differentiated from oligodendrocyte progenitor cells (OPCs). The differentiation of OPCs into myelinating OLs is characterized by both morphological changes and expression of myelin-associated genes. Compared with OPCs, myelinating OLs undergo as much as a 6,500-fold increase in membrane area (Baron & Hoekstra, 2010) and an 80,000-fold increase in myelin basic protein expression (Zhang et al., 2018). Myelination is characterized by ensheathing and wrapping axons with distal oligodendroglial processes, which also undergo extensive morphological changes. Oligodendrocytes and myelination maintain axonal integrity by providing physical, trophic, and metabolic support and are essential for proper neurological functions. Defects of OPC differentiation and myelination result in axonal dysfunction and neurological deficits in neurological disorders such as periventricular leukomalacia, multiple sclerosis, and leukodystrophy (Bercury & Macklin, 2015). Hence, understanding the mechanisms underlying OL development and myelination provides new insights into devising therapeutic strategies to promote myelin regeneration in CNS dysmyelinating and demyelinating disorders.
In the past three decades, group I p21-activated kinases (PAKs) have been shown to control neuronal precursor proliferation, migration, neuronal polarity and morphological complexity, dendritic and axonal formation, and neuronal plasticity and function (reviewed by Nikolic, 2008). However, there are limited data available on the role of PAKs in CNS myelin formation and repair. Previous data have shown that cell division control protein 42 (Cdc42) and ras-related C3 botulinum toxin substrate 1 (Rac1), the major activators of PAKs, modulate OPC differentiation and myelination both in vitro (Liang, Draghi & Resh, 2004) and in vivo (Thurnherr et al., 2006). In addition to Rho family GTPases, PAKs are also activated by bioactive lipids (Bokoch et al., 1998), which play important roles in oligodendroglial differentiation and regeneration (Podbielska, Krotkiewski & Hogan, 2012; Jana & Pahan, 2010). These data collectively suggest that group I PAKs may play crucial roles in regulating different aspects of oligodendroglial development and CNS myelination.
II. CURRENT UNDERSTANDING OF GROUP I PAKS
(1). Structure and functional domain
PAK1 is the most studied member among PAKs and plays a crucial role in neuronal development (Wang et al., 2018; Asrar et al., 2009; Meng et al., 2005; Huang et al., 2011; Pan et al., 2015; Xia, Zhou & Jia, 2018) and tumorigenesis (Radu et al., 2014). Human PAK1, a 545 amino acid (aa) peptide, harbours an N-terminal regulatory region (aa 1–249) that modulates its kinase activity and interaction with other proteins and a C-terminal catalytic region which binds to ATP and catalyses the phosphorylation of its substrates at serine (Ser, or S) and threonine (Thr or T) residues (Fig. 1A). The N-terminal regulatory region consists of the Cdc42 and Rac1 interactive binding [CRIB; also known as the p21-binding domain (PBD)] domain, the autoinhibition domain (AID) which partially overlaps with CRIB, five proline-rich PxxP motifs which specifically bind to Src homology 3 (SH3) domain-containing adaptor proteins such as non-catalytic region of tyrosine kinase (Nck) (Lu & Mayer, 1999; Lu et al., 1997) and growth factor receptor-bound protein 2 (Grb2) (Puto et al., 2003) (Fig. 1A), and the binding domain for p21-interacting Rac-guanine exchange factor (PIX) (Manser et al., 1998; Mott et al., 2005). The C-terminal catalytic region contains the kinase domain and the binding domain for G-protein beta gamma (Gβγ) subunits which are mediators of the membrane-tethered G protein-coupled receptors (Menard & Mattingly, 2004) (Fig. 1A).
Fig. 1.
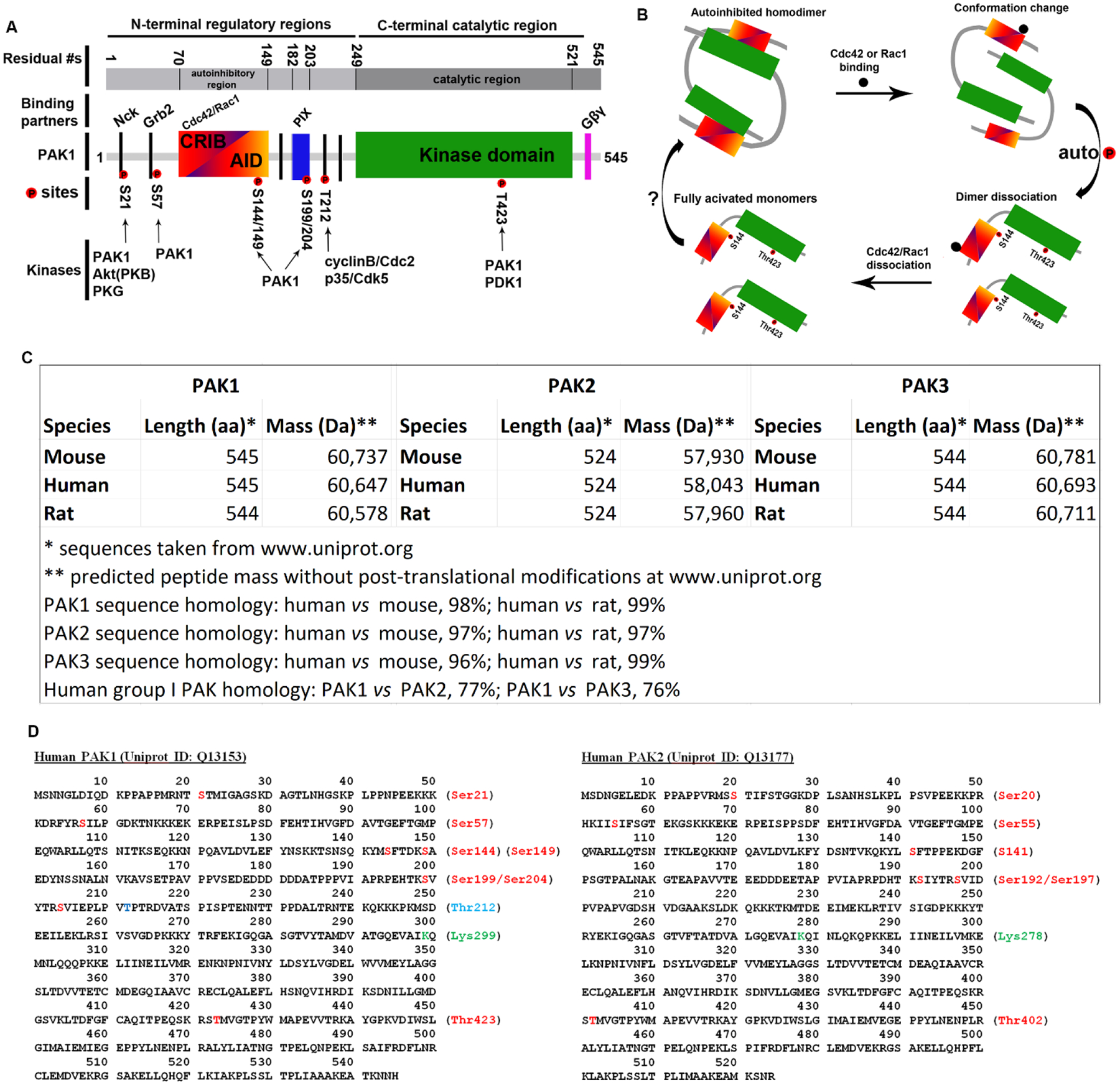
Structure, activation, and homology among group I p21-activated kinases (PAKs). (A) Schematic diagram depicting the major domains and motifs for protein–protein interactions, major phosphorylation sites, and respective kinases using PAK1 as an example. AID, autoinhibitory domain; Akt/PKB, protein kinase B; Cdc2, cell division control 2; CRIB, Cdc42 and Rac1 interactive binding domain; Gβγ, G protein beta gamma; Grb2, growth factor receptor-bound protein 2; Nck, non-catalytic region of tyrosine kinase; p35/Cdk5,; PDK1, 3-phosphoinositide-dependent kinase 1; PIX, p21-interacting Rac-guanine exchange factor; PKG, cGMP-dependent protein kinase. (B) PAK1 is autoinhibited in trans and the binding of GTP-Cdc42 or GTP-Rac1 induces a conformational change, autophosphorylation at Ser144 and Thr423, and kinase activation. Fully activated PAK1 may undergo dephosphorylation by certain phosphatases (Wang & Wang, 2008; Ke et al., 2004) and convert to autoinhibited homodimers or be subject to proteasome-mediated degradation (Weisz Hubsman et al., 2007), thus down-regulating PAK activity. (C) Amino acid (aa) length and predicted molecular mass of PAK1–3. Sequence % homology was retrieved from Uniprot.org and calculated as the number of identical aa residues between species. (D) Amino acid sequences of human PAK1 and PAK2. Residues and numbers highlighted in red are the major autophosphorylation sites during kinase activation. PAK1 Lys299 and PAK2 Lys278 (highlighted in green) are ATP binding sites of the activated PAKs. PAK1 Thr212 (highlighted in blue) is a phosphorylated site present only in PAK1. Mutation of the lysine residue to other residues, for example to arginine (K299R), prevents ATP binding and creates a kinase-inactive PAK1 (kinase-dead PAK1). Therefore, expression of K299R-PAK1 plasmids, in vivo or in vitro, provides a dominant-negative approach to probe the role of PAK1 kinase activity.
(2). Canonical activation mechanisms
Autophosphorylation at several Ser and Thr residues regulates the kinase activation and subcellular localization of PAK1. Previous in vitro study demonstrated that autophosphorylation at Ser144 and Thr423 promotes kinase activation while autophosphorylation at Ser21, Ser57, and Ser199/204 controls the association of PAK1 with focal complexes (Chong et al., 2001) and facilitates kinase activity (Zhao, Manser & Lim, 2000). It is well established that group I PAKs are activated via a different mechanism from that of group II PAKs. Group I PAKs are maintained as inactive heterodimers in which the AID of one PAK molecule contacts and inhibits the kinase domain of the other PAK molecule and vice versa (Lei et al., 2000) (Fig. 1B). In the canonical activation mechanism, binding of one GTP-loaded Cdc42 or Rac1 induces conformational changes of the heterodimers, relieves the trans-inhibition, and leads to subsequent autophosphorylation of the kinase domain at Thr423 and of a serine residue in the regulatory region such as Ser144, Ser57, Ser21, eventually activating two PAK1 monomers (Parrini et al., 2002) (Fig. 1B). It seems that the conformational change is necessary but not sufficient to activate PAK1. Casein kinase 2 (CK2)-dependent PAK1 phosphorylation at Ser223 is required and sufficient to convert monomeric PAK1 into catalytically active PAK1 because a phosphomimetic mutation at PAK1 Ser223 bypasses the requirement for GTPases in PAK1 activation (Shin, Kim & Kim, 2013). Kinase-active PAK1 regulates diverse cellular processes, such as actin and microtubule cytoskeleton reorganization, cell proliferation, migration, motility, and cell survival through phosphorylating and modulating its target substrates and signalling pathways (Hofmann, Shepelev & Chernoff, 2004). Based on the established role of Ser144 and Thr423 in the kinase activation, antibody-based immunodetection of phosphorylated PAK1 at Ser144 and/or Thr423 provides a reliable means to evaluate PAK1 kinase activity.
In addition to autophosphorylation, other protein kinases participate in the cross-phosphorylation of PAK1 and modulate PAK1 activity. For example, PAK1 can be phosphorylated at Ser21 by protein kinase B (PKB, also known as Akt) (Zhou et al., 2003) and cGMP-dependent protein kinase (PKG) (Fryer et al., 2006). Because the adaptor protein Nck binds PAK1 near Ser21 (Fig. 1A), phosphorylation of PAK1 by PKB/Akt or PKG at Ser21 is proposed to disassociate PAK1 from Nck at the plasma membrane (Zhou et al., 2003; Fryer et al., 2006), potentially facilitating the cycling of PAK1 from the plasma membrane to the cytosol and reducing PAK1 activity (Zhao et al., 2000). The conserved Thr423 in the activation loop of the kinase domain (Fig. 1A) is also phosphorylated by 3-phosphoinositide-dependent kinase 1 (PDK1) in the presence of sphingosine and PDK1-mediated Thr423 phosphorylation increases the kinase activity of PAK1 (King et al., 2000).
(3). Sequence identity and common biochemical properties
Human PAK1, PAK2, and PAK3 display >95% sequence identity in amino acid residues to their rodent counterparts. Within the group I PAKs, the PAK1 amino acid sequence displays 77% and 76% identity to that of PAK2 and PAK3, respectively (Fig. 1C). The sequence homology is even greater in the conserved domains and motifs such as CRIB, AID, the proline-rich PxxP motifs, and kinase domain. Because of the high sequence homology and identity among PAK1, PAK2, and PAK3, phosphorylated sites in PAK1 (Fig. 1D) are also conserved in PAK2 and PAK3. Phosphorylation of these conserved sites in PAK1, PAK2, and PAK3 exerts the same biological functions on the downstream targets. It has been demonstrated that Ser144 in PAK1 is conserved to Ser141 and Ser139 in PAK2 and PAK3, respectively, and Thr423 in PAK1 is equivalent to Thr402 and Thr421 in PAK2 and PAK3, respectively, in the process of PAK activation. Similarly, the other phosphorylated sites in PAK1, for example Ser21, Ser57, and Ser199/204, are also conserved in PAK2 (to Ser20, Ser55, and Ser192/197, respectively) (Fig. 1D), and in PAK3 (to Ser20, Ser50, and Ser200/Ser205, respectively). It should be noted that antibodies recognizing PAK1 phospho-Ser144 also recognize PAK2 phospho-Ser141 and PAK3 phospho-Ser139 and those specifically targeting PAK1 phospho-Thr423 also target PAK2 phospho-Thr402 and PAK3 phospho-Thr421.
Previous studies have identified Thr212 as a phosphorylation site unique to PAK1 (which is absent from PAK2 and PAK3). PAK1 Thr212 is phosphorylated by cyclinB1/cell division control protein 2 (Cdc2) in mitotic cells (Thiel et al., 2002; Banerjee et al., 2002) and by p35/cyclin-dependent kinase 5 (CDK5) in post-mitotic neurons (Rashid, Banerjee & Nikolic, 2001; Nikolic et al., 1998) (Fig. 1A). PAK1 Thr212 phosphorylation has been shown to negatively regulate PAK1 activity in post-mitotic neurons (Rashid et al., 2001; Nikolic et al., 1998) and to alter protein–protein interactions and post-mitotic spreading in proliferating fibroblasts (Thiel et al., 2002). In the rodent brain, PAK1 phospho-Thr212 is detected only during embryonic and very early postnatal development but is absent from the adult brain (Zhong, Banerjee & Nikolic, 2003), suggesting that it may regulate cell proliferation and/or early-stage neural cell differentiation. Since OPCs are the major rapidly dividing cells in the brain, it is tempting to define the role of PAK1 as functioning in OPC proliferation and population expansion during early postnatal brain development.
(4). Non-canonical activation mechanisms
The well-established canonical activation mechanisms of group I PAKs involve the binding of small GTPases (such as Cdc42 and Rac1). Increasingly, the available data support non-canonical, GTPase-independent activation: some are common to group I PAKs and others are specific to certain PAK members. Sphingosine lipids bind to a site overlapping or identical to the PAK1 CRIB site and this binding activates PAK1 independent of GTPases (Bokoch et al., 1998) presumably by PDK1-mediated phosphorylation of PAK1 at Thr423 (King et al., 2000), an autophosphorylation site crucial for Cdc42/Rac1-mediated PAK1 activation. This observation suggests that group I PAKs may be activated in response to active lipid signals which play important roles in OL biology and pathology (Coelho, Saini & Sato-Bigbee, 2010). It is well established that members of group I PAKs form auto-inhibitory inactive homodimers (PAK1/PAK1, PAK2/PAK2, and PAK3/PAK3). However, PAK1 also has been reported to form heterodimers with PAK2 (PAK1/PAK2) (Grebenova et al., 2019) or with PAK3 (PAK1/PAK3) (Combeau et al., 2012), which provides potential mechanisms for trans-inhibition between PAK1 and PAK2 or PAK3. The auto- and trans-inhibition among group I PAKs suggest that the loss of catalytic activity of PAK1 may be compensated by that of PAK2 and/or PAK3 or vice versa (Huang et al., 2011). It has been reported that calcium and integrin binding protein 1 (CIB1) specifically interacts with PAK1 (but not PAK2 or PAK3) via PAK1 N-terminal regions of aa 50–60 and aa 130–137 (Leisner et al., 2005). This interaction is sufficient to activate PAK1 activity independent of Cdc42/Rac1 (Leisner et al., 2005), suggesting an alternative activation mechanism unique to PAK1.
PAK2 is additionally activated by caspase 3-mediated cleavage in response to stress stimulants (Rudel & Bokoch, 1997; Jakobi et al., 2003); cleavage at Asp212 of PAK2 releases the N-terminal autoregulatory domains (aa 1–212) from the C-terminal kinase domain (aa 213–524), thus creating a constitutively active C-terminal-containing PAK2 which may play a crucial role in cell apoptosis (Huang et al., 2020; Bokoch, 1998). It is yet to be determined whether the mechanism of caspase 3-mediated PAK2 activation exists in oligodendroglial-lineage cells and, if so, whether it plays a role in oligodendroglial cell death under stressful demyelinating conditions.
PAK3 splicing variants may exhibit a different activation mechanism (Kreis et al., 2008). Mammalian PAK3 genes encode four messenger RNA (mRNA) splice variants and the corresponding proteins, which are all detected in the rodent brain and particularly in neurons: PAK3a (without alternative splicing, 544 aa in length; Fig. 1C), PAK3b (with a spliced-in 45-bp exon b, 559 aa) (Rousseau et al., 2003), PAK3c (with a spliced-in 63-bp exon c, 565aa), and PAK3cb (with both exons b and c spliced in, 580aa) (Kreis et al., 2008). It has been shown that PAK3b, PAK3c, and PAK3cb isoforms are constitutively active in terms of their basal kinase function because the inserts of b and/or c are present in the N-terminal autoregulatory domain of PAK3, potentially compromising the auto-inhibitory ability of PAK3 homodimers (Kreis et al., 2008). Interestingly, PAK1 forms heterodimers with all four PAK3 isoforms, but only PAK3a activity was trans-inhibited by PAK1 (Combeau et al., 2012).
III. PAK EXPRESSION IN THE BRAIN AND OLIGODENDROCYTES
PAK1, PAK2, and PAK3 display different tissue specificities in humans and rodents. Human PAK1 and PAK3 proteins are relatively enriched in the brain whereas PAK2 shows a lower level in the brain and is ubiquitously expressed in different tissues (Fig. 2A). In rodents, PAK1 is highly expressed in the brain, lung, intestine, kidney, and stomach during embryonic development and progressively restricted to the brain in adults (Zhong et al., 2003). Similar to humans, rodent PAK3 is specifically expressed in the brain and PAK2 is ubiquitously expressed in different organs during both embryonic development and in adults (Zhong et al., 2003). With technical advances in bulk and single-cell RNA sequencing (RNA-seq), it has become possible quantitatively to compare the mRNA levels of PAK1–3 in different neural cell types of the brain and at different maturation stages of oligodendroglial-lineage cells. Bulk RNA-seq data (Zhang et al., 2014) demonstrate that in the brain PAK1 and PAK2 are expressed at higher levels than PAK3 (Fig. 2B). The expression of PAK1, and to a lesser extent PAK2, is relatively high in oligodendroglial-lineage cells compared with neurons or astrocytes based on the normalized fragments per kilobase of transcript per million mapped reads (FPKM). By contrast, PAK3 is highly enriched in neurons in the rodent brain (Fig. 2B). More recently, oligodendroglial-lineage cells have been divided developmentally into 12 maturation stages from OPCs to fully matured myelinating OLs based on the unique molecular signatures derived from single-cell RNA-seq data (Marques et al., 2016). In contrast to PAK3 which shows downregulation during OL maturation, PAK1 and PAK2 maintain high and relatively stable expression levels in mature OLs (Fig. 2C). PAK1 mRNA expression levels appear comparable to those of the oligodendroglia-specific transcription factor myelin regulatory factor (MYRF) (Fig. 2C) (Emery et al., 2009). Recent data demonstrate that PAK1 protein levels appear to be upregulated in primary mature OLs compared with primary OPCs in vitro (Brown et al., 2021). These expression data suggest that group I PAKs may play a role in modulating oligodendroglial development and maturation.
Fig. 2.
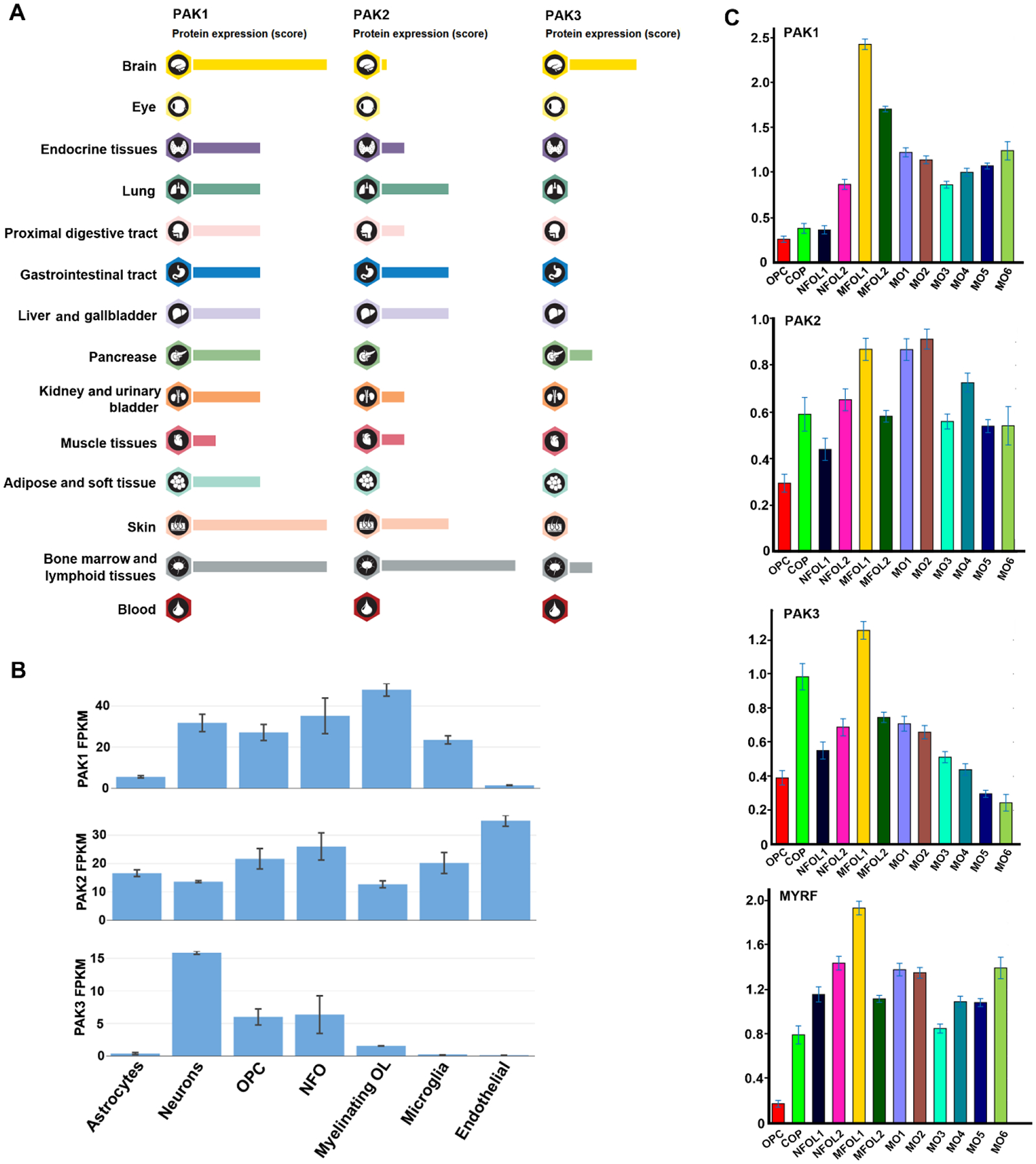
Expression of the p21-activated kinases PAK1, PAK2, and PAK3 in the body, central nervous system (CNS), and in oligodendroglial-lineage cells. (A) Relative levels of human PAK1–3 protein in different organs. Data adapted from the Human Protein Atlas (www.proteinatlas.org). (B) Relative levels of mouse PAK1–3 messenger RNA (mRNA) in different brain cells. Values are means ± standard deviations. Data adapted from the brain RNA-sequencing (RNA-seq) database (www.brainrnaseq.org) (Zhang et al., 2014). FPKM, fragments per kilobase of transcript per million mapped reads; myelinating OL, myelinating oligodendrocytes identified by myelin oligodendrocyte glycoprotein (MOG) expression; NFO, newly formed oligodendrocytes identified by galactosylceramidase (GalC) expression; OPC, oligodendrocyte progenitor cells. (C) Relative levels of mouse PAK1–3 mRNA in different maturation stages from OPC to mature oligodendrocytes stage 6 (MO6) (from left to right). COP, differentiation-committed OPC; NFOL, newly formed oligodendrocytes; MFOL, myelin-forming oligodendrocytes. The oligodendroglial lineage-specific marker myelin regulatory factor (MYRF) (Emery et al., 2009) serves as a control against which the relative level of PAK1–3 can be compared. Data adapted from single-cell RNA-seq (www.linnarssonlab.org/oligodendrocytes) (Marques et al., 2016). The y-axis shows unique molecular identifier (UMI) counts.
IV. MUTATIONS OF GROUP I PAKS IN CHILDREN
Clinical studies have demonstrated that group I PAKs can be mutated in children with diverse neurodevelopmental and white matter defects (Table 1). Three independent groups have reported heterozygous missense mutations of PAK1 in unrelated subjects and these PAK1 mutations significantly reduce dimerization and increase the kinase activity, i.e. kinase gain-of-function mutations (Harms et al., 2018; Horn et al., 2019; Kernohan et al., 2019). Children with PAK1 activation mutations display a very broad range of abnormalities, such as delayed developmental milestones, macrocephaly (bigger brain), seizures, and impaired intellectual and motor ability. Very interestingly, some of the affected brains display hyperintensity in the subcortical white matter on T2-weighted magnetic resonance imaging (T2-MRI), a clinical indication of white matter hypomyelination. These clinical data point to the possibility that PAK1 activity dysregulation may interfere with OL development and/or CNS myelination and brain development.
Table 1.
Group I p21-activated kinase (PAK) mutations and clinical symptoms in humans.
| PAKs | Mutation type / residue changes | Function alterations | Major neurological symptoms | Brain magnetic resonance imaging (MRI) findings | References |
|---|---|---|---|---|---|
| PAK1 | Missense*
Tyr131Cys Tyr429Cys |
Gain of function Activating kinase activity |
Developmental delay, macrocephaly, seizure, and ataxic gait | White matter hyperintensity | Harms et al. (2018) |
| Missense Ser110Thr Ser133Pro Pro121Ser Leu470Arg |
Gain of function Activating kinase activity |
Developmental delay, intellectual disability, macrocephaly, autism, ataxic gait, and seizures | White matter hyperintensity, Abnormal corpus callosum |
Horn et al. (2019) | |
| Missense Pro121Leu |
Gain of function Activating kinase activity |
Autism, intellectual disability, and epilepsy | Not determined | Kernohan et al. (2019) | |
| PAK2 | Nonsense** Arg479Stop Copy-number deletion in the 3q29 region containing 22 genes including PAK2 |
Loss of function Inhibiting kinase activity |
Autism | Not determined | Wang et al. (2018; Willatt et al. (2005) |
| PAK3 | Nonsense Arg419Stop*** | Non-pathogenic mutations | X-linked non-syndromic mental retardation, no specific neurological findings | Not determined | Allen et al. (1998) |
| Missense Arg67Cys | Loss of function Inhibiting kinase activity |
X-linked non-syndromic mental retardation, no specific neurological findings | Not determined | Bienvenu et al. (2000); des Portes et al. (1997b) | |
| Missense Ala365Glu |
Loss of function Inhibiting kinase activity |
X-linked non-syndromic mental retardation, no specific neurological findings, neuropsychiatric problems | Not determined | Gedeon et al. (2003) | |
| Missense Trp446Ser | Loss of function Inhibiting kinase activity |
Both affected males and carrier females display learning problems and mild mental disability Affected males display microcephaly |
Not determined | Peippo et al. (2007) | |
| Missense Cys371Tyr |
Pathogenic mutation | Motor and mental developmental delays | Enlarged lateral ventricles Thin periventricular white matter Corpus callosum dysplasia |
Qian et al. (2020) | |
| Missense Gly424Arg |
Loss of function Inhibiting kinase activity |
Intellectual disability | Short corpus callosum with severe splenium hypoplasia White matter gliosis |
Duarte et al. (2020) |
Missense mutation, a change in DNA sequence that substitutes a different amino acid in the resulting protein (the substitution may have no effect, or it may render the protein non-functional).
Nonsense mutation (or stop mutation): a change in DNA sequence that causes a protein to end its translation earlier than expected, resulting in a dysfunctional protein.
Residue numbers are based on the 544 amino acid (aa) long human PAK3a isoform; the first residue is wildtype whereas the second is mutant.
Recently, PAK2 nonsense mutation and copy-number deletion in the PAK2 gene-containing chromosomal region have been reported in a large cohort of children displaying autistic spectrum disorders (ASD) and other neurodevelopmental disorders (Table 1) (Wang et al., 2018; Willatt et al., 2005). Unfortunately, no brain-imaging data are available assessing brain white matter abnormalities in children affected by PAK2 loss-of-function mutations. Nevertheless, previous studies employing diffusion tensor imaging (DTI) and diffusion-weighted imaging (DWI) have documented accelerated maturation of the white matter and hypermyelination in young ASD children compared with healthy controls (Ben Bashat et al., 2007). These clinical data suggest that PAK2 may negatively regulate OL development and/or CNS myelination; its deficiency may be associated with precocious white matter maturation and hypermyelination in the autistic brain.
PAK3 is regarded as a mental retardation risk gene because PAK3 mutations were originally reported in children with X-linked non-syndromic mental retardation without any other neurological abnormalities 25 years ago (Allen et al., 1998; des Portes et al., 1997b,a). There is an increasing number of cases reporting PAK3 mutations in children with a diverse panel of neurological and behavioural presentations beyond mental retardation, such as delayed motor development and autism (Qian et al., 2020; Bienvenu et al., 2000; Peippo et al., 2007) (Table 1), suggesting a role of PAK3 beyond mental retardation. Brain images show that PAK3 dysfunctional mutations are associated with thinner white matter tracts and a malformed corpus callosum (Duarte et al., 2020; Qian et al., 2020), suggesting that pathogenic PAK3 mutations may cause malformation of the subcortical white matter.
V. TARGETING GROUP I PAKS FOR RESEARCH
(1). Genetic models for probing neuronal/oligodendroglial development and function
Because PAK3 was the first member among group I PAKs found to be mutated in children with non-syndromic mental retardation, PAK3 knockout (KO) mice (Pak3−/−) were generated to define the role of PAK3 in neuronal development and function (Meng et al., 2005). Surprisingly, PAK3 KO mice display only very subtle changes to brain phenotypes during postnatal development except for impaired hippocampal late-phase long-term potentiation (L-LTP) (Table 2). Likewise, PAK1 KO (Pak1−/−) mice show no noticeable abnormalities in neuronal development and function except for selective deficits in hippocampal L-LTP (Asrar et al., 2009). The subtle neuronal phenotypes of PAK1 or PAK3 KO mice suggest that PAK1 and PAK3 may be functionally redundant under physiological conditions, which is supported by their biochemical properties: there are few downstream enzymatic substrates unique to PAK1, PAK2, or PAK3. Evidence for redundancy between PAK1 and PAK3 in regulating neuronal development and function is further provided by PAK1/PAK3 double-KO mice (Huang et al., 2011) which develop severe and progressive defects during postnatal brain development including neuronal, dendritic, axonal, and spine deficits and behavioural abnormalities (Table 2).
Table 2.
Neural phenotypes in group I p21-activated kinase (PAK)-knockout mouse models.
| Mouse models | Major findings of neural phenotypes | Reference |
|---|---|---|
| Pak3−/− (viable and fertile) | Normal brain development/normal spine and synaptic structure/normal basal synaptic function/normal spatial memory/impaired late-phase hippocampal long-term potentiation (LTP)/reduced CREB-Ser133 phosphorylation/unaltered LIM kinase and cofilin activity | Meng et al. (2005) |
| Pak1−/− (viable and fertile) | Normal brain development/normal synaptic and spine structure but with deficits in spinal actin filament/normal basal synaptic strength and presynaptic function/selective deficits in CA1 LTP/unaltered LIM kinase and cofilin activity | Asrar et al. (2009) |
| Pak1−/−Pak3−/−[mice from Meng et al. (2005) and Asrar et al. (2009)] | Normal brain size and structure at birth/progressive defects in postnatal brain growth (smaller brain)/thinner white matter tract/hyperactivity, increased anxiety, and learning deficits/increased neural and glial cells (presumably due to decreased cortical thickness)/normal neuronal polarity/reduced dendritic arborization and length/reduced axonal growth/enhanced basal synaptic strength and impaired synaptic plasticity/reduced synapse density and enlarged individual synapses/enhanced cofilin activity and reduced F-actin | Huang et al. (2011) |
| Pak1−/− (mice from Asrar et al., 2009) | Defects in neuronal migration and progenitor cell proliferation during embryonic and early postnatal development (by postnatal day 7) | Pan et al. (2015) |
| Pak2−/− | Die at embryonic day 8.5 due to abnormal vascular formation | Kelly & Chernoff (2012) |
| Pak2+/− (viable and fertile) | Normal brain development/normal basal synaptic transmission/normal behaviour in locomotor, anxiety, spatial memory, and acoustic startle response/display autism-related behaviour/reduced density of spines and asymmetric synapse and impaired LTP/altered LIM kinase and cofilin activity and reduced level of F-actin | Wang et al. (2018) |
| Pak3−/− (mice from Meng et al., 2005) | Transiently delayed oligodendroglial proliferation, differentiation, and myelination in restricted white matter tracts, for example corpus callosum in vivo/no effects on oligodendroglial proliferation and migration in vitro/reduced oligodendroglial differentiation in vitro | Maglorius Renkilaraj et al. (2017) |
Abbreviations: CA1, hippocampal cornu ammonis; CREB, cAMP response element-binding protein; LIM kinase, Lin-11, Islet-1, and Mec-3 domain kinase.
In contrast to PAK3, PAK2 is ubiquitously expressed in the CNS and other organs (Fig. 2). PAK2-KO (Pak2−/−) mouse mutants die from severe vasculature defects by embryonic day 8.5 (Kelly & Chernoff, 2012) when neurogenesis and gliogenesis are not yet taking place in the rodent brain. The early embryonic lethality of PAK2-KO mutants clearly demonstrates that the role of PAK2 in vascular development cannot be compensated by PAK1 or PAK3. Recently, Wang et al. (2018) reported that PAK2 haploinsufficiency in heterozygous PAK2-KO (Pak2+/−) mice resulted in reduction of spine density, asymmetric synapse number, hippocampal LTP, and autism-related behaviours. Pak2+/− mice are viable and fertile and display no quantitative abnormalities in brain morphology, cortical lamination, basal synaptic transmission, nor impairment in locomotor, anxiety and spatial memory (Wang et al., 2018). Mechanistically, PAK2 haploinsufficiency reduces the activity of Lin-11, Islet-1, and Mec-3 domain kinase 1 (LIMK1), an established downstream substrate of group I PAKs, which ultimately leads to decreased actin filament assembly and reduced spine and synapse formation (Wang et al., 2018). Given that LIMK1 is also a substrate of PAK1 and PAK3, the downregulation of LIMK1 activity selectively in Pak2+/− mice (Wang et al., 2018) but not in Pak1−/− (Asrar et al., 2009) or Pak3−/− (Meng et al., 2005) mice (Table 2) suggests that PAK2 plays a dominant role in regulating LIMK1 activity among group I PAKs. Nevertheless, it remains to be determined whether the observed phenotypes (Wang et al., 2018) result from a functional effect of PAK2 haploinsufficiency on neurons or from a secondary non-cell-autonomous effect on other neural and vascular cell defects. Neuron-specific knockdown or ablation of PAK2 will help resolve this question.
Oligodendroglial phenotypes in PAK-KO mice have not been determined until a recent study (Maglorius Renkilaraj et al., 2017) reported that PAK3-KO mice displayed subtle and transient delay of myelination selectively in the corpus callosum but not in the anterior commissure at the ultra-structural level. In vitro OPC culture derived from PAK3-KO and control mice suggests that PAK3 may play a cell-autonomous role in promoting OPC differentiation (Maglorius Renkilaraj et al., 2017). Interestingly, the subcortical white matter tracts are significantly thinner in PAK1/PAK3 double-KO mice, but the intensity of myelin staining (indicated by myelin basic protein, MBP) is comparable to that in littermate controls (Huang et al., 2011). This observation suggests that the overall reduction in myelin reactive signal in that study (Huang et al., 2011) may be secondary to white matter tract atrophy resulting from defective axonal outgrowth in the white matter of PAK1/PAK3 double-KO mice (Huang et al., 2011). However, it remains to be defined whether oligodendroglial development is affected in PAK1/PAK3 double-KO mice. Given the expression of group I PAKs in most neural and vascular cells, although at varying levels, in the brain and their potential functional redundancy, it is very important to employ cell-specific PAK1 and/or PAK2 or PAK3 conditional KO to determine the role of group I PAKs in oligodendroglial development and CNS myelination.
(2). Pharmacologically targeting group I PAK in cells and animals
PAK hyperactivity is frequently observed in human cancers, playing an essential role in tumour genesis and metastasis (Ye & Field, 2012). Previous research has pointed to group I PAKs as a potential therapeutic target in cancer treatment due to its role in many oncogenic signalling pathways (Yao et al., 2020). To this end, many small compounds have been identified that inhibit different members of group I PAKs. These inhibitors are classified into two major categories based on their mode of action: ATP-competitive inhibitors and non-ATP-competitive allosteric inhibitors (Table 3).
Table 3.
Small inhibitors for group I p21-activated kinases (PAKs): potency, specificity, and known off-target kinases.
| Small compounds | Group I PAK specificity and IC50 | Examples of off-target kinases | Reference |
|---|---|---|---|
|
FRAX486 ATP-competitive inhibitor |
Greater potency for group I PAKs and PAK4: PAK1 IC50 = 14 nM; PAK2 IC50 = 33 nM; PAK3 IC50 = 39 nM; PAK4 IC50 = 575 nM |
Off-target screening: not determined | Dolan et al. (2013) |
|
G5555 ATP-competitive inhibitor |
Greater potency for group I PAKs: PAK1 Ki = 3.7 nM PAK2 IC50 = 11 nM |
In additional to group I PAKs, only 6 of 235 kinases tested showed an inhibition of >70%: SIK2 (IC50 = 9 nM); MAP4K5 (IC50 = 10 nM); MST4 (IC50 = 20 nM); YSK1 (IC50 = 34 nM); MST3 (IC50 = 43 nM); LCK (IC50 = 52 nM) Exhibit low inhibition against hERG channel activity (<50% inhibition at 10 μM) |
Ndubaku et al. (2015); Rudolph et al. (2016) |
|
IPA3 Allosteric inhibitor |
Greater potency for group I PAKs: PAK1 IC50 = 2.5 μM (at 10μM PAK1 inhibition by >95%) |
At 10 μM significantly inhibited (>50% inhibition) only 9 out of 214 kinases tested (4% of total), for example, SGK3, PLK3, MAPK14 | Deacon et al. (2008); Viaud & Peterson (2009) |
|
NVS (Novartis)-PAK1–1 Allosteric inhibitor |
Greater potency for PAK1 (IC50 = 5 nM) than for PAK2 | Screening against 442 kinases identified 22 off-target kinases with IC50 > 10 μM, for example, phosphodiesterase 4D (IC50 = 13 μM), pregnane X receptor (IC50 = 16 μM), histamine receptor H1 (IC50 > 30 μM), muscarinic receptor M1 (IC50 > 30 μM) | Karpov et al. (2015) |
|
Peptide inhibitor PAK1 AID (aa 83–149) |
Inhibits group I PAKs | Also binding to fragile-X syndrome-related proteins FMR1 and FXR1; may interfere with FMR1/FXR1 function | Thullberg et al. (2007); Say et al. (2010); Chow et al. (2018) |
IC50, concentration of inhibitor needed to reach 50% inhibition; Ki, concentration of inhibitor required to reach 50% enzyme saturation.
Abbreviations: FMR1, fragile X mental retardation 1; FXR1, fragile X mental retardation syndrome-related protein 1; hERG, human Ether-à-go-go-related gene; LCK, lymphocyte-specific protein tyrosine kinase; MAP4K5, mitogen-activated protein kinase kinase kinase kinase 5; MAPK14, mitogen-activated protein kinase 14; MST4, mammalian STE20-like protein kinase 4; PAK1 AID, PAK1 autoinhibitory domain; PLK3, mammalian polo-like kinase 3; SGK3, serum/glucocorticoid regulated kinase family member 3; SIK2, salt inducible kinase 2; YSK1, yeast Sps1/Ste20-related kinase 1.
ATP-competitive inhibitors target the ATP binding site of the PAK kinase domain which is conserved among PAKs and non-PAK kinases. FRAX486, an ATP-competitive inhibitor targeting group I PAKs and PAK4, has been shown to cross the blood–brain barrier to reach the neural parenchyma in live animals. Although its off-target effects are yet to be determined, it has been reported that FRAX486 inhibits brain PAK activity and rescues fragile-X syndrome-like phenotypes in fragile X mental retardation 1 (Fmr1)-KO mice (Dolan et al., 2013) including dendritic spine abnormalities and hyperactive behaviours (Hayashi et al., 2007). G5555, very potent for group I PAKs, is the most specific ATP-competitive inhibitor identified thus far for group I PAKs because in vitro off-target screening against 235 kinases found that only six kinases (2.5% off-target substrates) were significantly inhibited (by 70%) at a concentration of 10 μM (Rudolph et al., 2016; Ndubaku et al., 2015).
By contrast, non-ATP-competitive allosteric inhibitors bind to PAKs outside the kinase domain which is less conserved among PAKs and non-PAK kinases and affect the activation mechanism without inhibiting pre-activation PAKs. IPA3, an allosteric inhibitor for group I PAKs, binds covalently to the regulatory domain and prevents PAK binding to the GTPase Cdc42/Rac1 (Deacon et al., 2008). In vitro off-target assay against 214 kinases identified only nine kinases (4% off-target substrates) as significantly inhibited (by 50%) at a concentration of 10 μM. Another representative potent allosteric inhibitor novartis (NVS) is more potent for PAK1 than PAK2 (Karpov et al., 2015). In vitro cell culture assay demonstrates that NVS selectively inhibits PAK1 at a concentration of ~0.25–1 μM and additionally inhibits PAK2 at a higher concentration of >2 μM. Kinome screening assay against 442 kinases found 22 off-target kinases to be significantly inhibited by NVS with an inhibitor concentration needed to reach 50% inhibition (IC50) >10 μM including muscarinic receptor M1 (Karpov et al., 2015) which is a potent negative regulator of OL differentiation and (re)myelination (Cree et al., 2018; De Angelis et al., 2012; Mei et al., 2016). In conclusion, no pharmacological PAK inhibitors, whether ATP competitive or allosteric, have been shown to target solely to the kinase activity of group I PAKs or to a specific member of group I PAKs. Thus, caution should be used in basic research into their mechanisms of action. The inevitable off-target effects (see Table 3) and potential cellular toxicity at higher inhibitor doses must not be neglected during data interpretation. A recent study employed three different PAK inhibitors (FRAX486, G5555, and NVS) (Table 3) to document that PAK inhibition decreased OL differentiation in zebrafish (Danio rerio) and in primary rodent OPC cultures (Brown et al., 2021). However, the off-target effects of these pharmacological inhibitors cannot be excluded in these systems.
To address the intrinsic caveats of pharmacological inhibitors, cyclic recombinase/locus of X-over P1 (Cre/loxP)-mediated expression of peptide inhibitors in PID (LoxP-STOP-loxP-PAK inhibitory domain transgene) mice has been recently developed (Chow et al., 2018) in an approach leveraging the unique activation mechanism of group I PAKs. PAK1 inhibitory domain (PID) peptide (aa 83–149) has been shown to bind to and inhibit the kinase domain of group I PAKs (Semenova & Chernoff, 2017; Zhao et al., 1998). The PAK1-derived PID peptide is known to bind only two categories of proteins: group I PAKs and the fragile-X syndrome proteins FMR1/fragile-X-related 1 (FXR1) (Say et al., 2010). Notably, the PID transgene encodes a PAK1-derived PID bearing a synthetic E129K mutation (residue 129 of the PAK1 AID, glutamic acid, is required for FMR1/FXR1 binding), thus this mutation prevents FMR1/FXR1 binding (Chow et al., 2018; Say et al., 2010). Cre-mediated expression of the mutant PID thus represents the most specific known PAK inhibitor with no recognized off-target effects yet reported among protein kinases. It would be very interesting to use these PAK peptide inhibitor transgenic mice to study the effects of cell-specific PAK inhibition on the development of oligodendroglial-lineage cells and other neural-lineage cells during development and following injury.
V. GROUP I PAKS AND OLIGODENDROCYTE DEVELOPMENT
(1). Insights from cytoskeleton regulation
PAKs have been studied extensively in cancer cells, whereas data regarding the role of PAKs in oligodendroglial development are very limited. Below, we review PAK-regulated tumorigenic processes and signalling pathways that are also crucial for OL development and propose hypothetical roles and future research directions for understanding how PAKs function in OPC proliferation, differentiation, and CNS (re)myelination.
Compared with neuron-enriched PAK3, PAK1 and PAK2 are expressed at a relatively high level in oligodendroglial lineage cells. The role of PAK1 and PAK2 in OL development and brain myelination remains incompletely understood. Two clinical observations suggest that PAK1 and PAK2 may regulate OL development: (1) PAK1 activation mutations in children are associated with a variable degree of white matter hyperintensity in brain T2-MRI (Harms et al., 2018; Horn et al., 2019), an indication of white matter hypomyelination (Steenweg et al., 2010), and (2) PAK2 loss-of-function mutations in ASD children (Wang et al., 2018) are associated with accelerated white matter maturation and hypermyelination based on brain imaging analyses (Ben Bashat et al., 2007). Thus, the clinical evidence indicates that PAK1 and PAK2 may inhibit myelination during postnatal brain development.
Oligodendrocyte development from OPCs to myelinating OLs involves dramatic morphological changes (Fig. 3A) during which dynamic cytoskeleton reorganization provides physical support for morphological complexity. The cytoskeleton comprises three major components: actin filaments (microfilaments), intermediate filaments, and microtubule (MT) filaments. Oligodendroglial-lineage cells lack intermediate filaments and only contain an actin and MT cytoskeleton (Fig. 3B) (Bauer, Richter-Landsberg & Ffrench-Constant, 2009). It has been shown that actin filament (F-actin) assembly from globular actin (G-actin) drives oligodendroglial lamellipodia-like protrusion, process extension and arborization, and initial axon ensheathment while subsequent myelin sheath growth, wrapping, and compaction require F-actin disassembly into G-actin (Zuchero et al., 2015; Nawaz et al., 2015). The role of the MT cytoskeleton in oligodendroglial morphological differentiation and myelination remains incompletely defined. Recent data show that MT bundle assembly along oligodendroglial processes mediated by the Golgi outpost protein tubulin polymerization promoting protein (TPPP) is required for CNS myelination (Roll-Mecak, 2019), as TPPP-KO mice display shorter myelin sheath length (i.e. shorter internodal length) and a thinner myelin sheath (Fu et al., 2019), indicating that MT assembly in oligodendroglial processes may regulate proper morphological differentiation and myelination. To support this idea, a more recent in vitro study using MT-targeting pharmacological agents reported that MT-destabilizing agents (such as Nocodazole) promoted OL process branching and MBP expression whereas MT-stabilizing agents (such as Taxol) exerted the opposite effects (Lee & Hur, 2020). Both MT-destabilizing and stabilizing agents inhibited in vitro myelination in an OL–neuron co-culture assay system (Lee & Hur, 2020), indicating that MT dynamics is essential for proper myelination. Previous studies reported that non-muscle myosin II, an actin-associated motor protein participating in stress fibre formation (Fig. 3B), regulates OL morphological differentiation (Wang et al., 2012; Kippert et al., 2009). Together, these data suggest that actin and MT cytoskeleton dynamics play an indispensable role in regulating morphological changes and complexity during OL differentiation, maturation, and myelination.
Fig. 3.
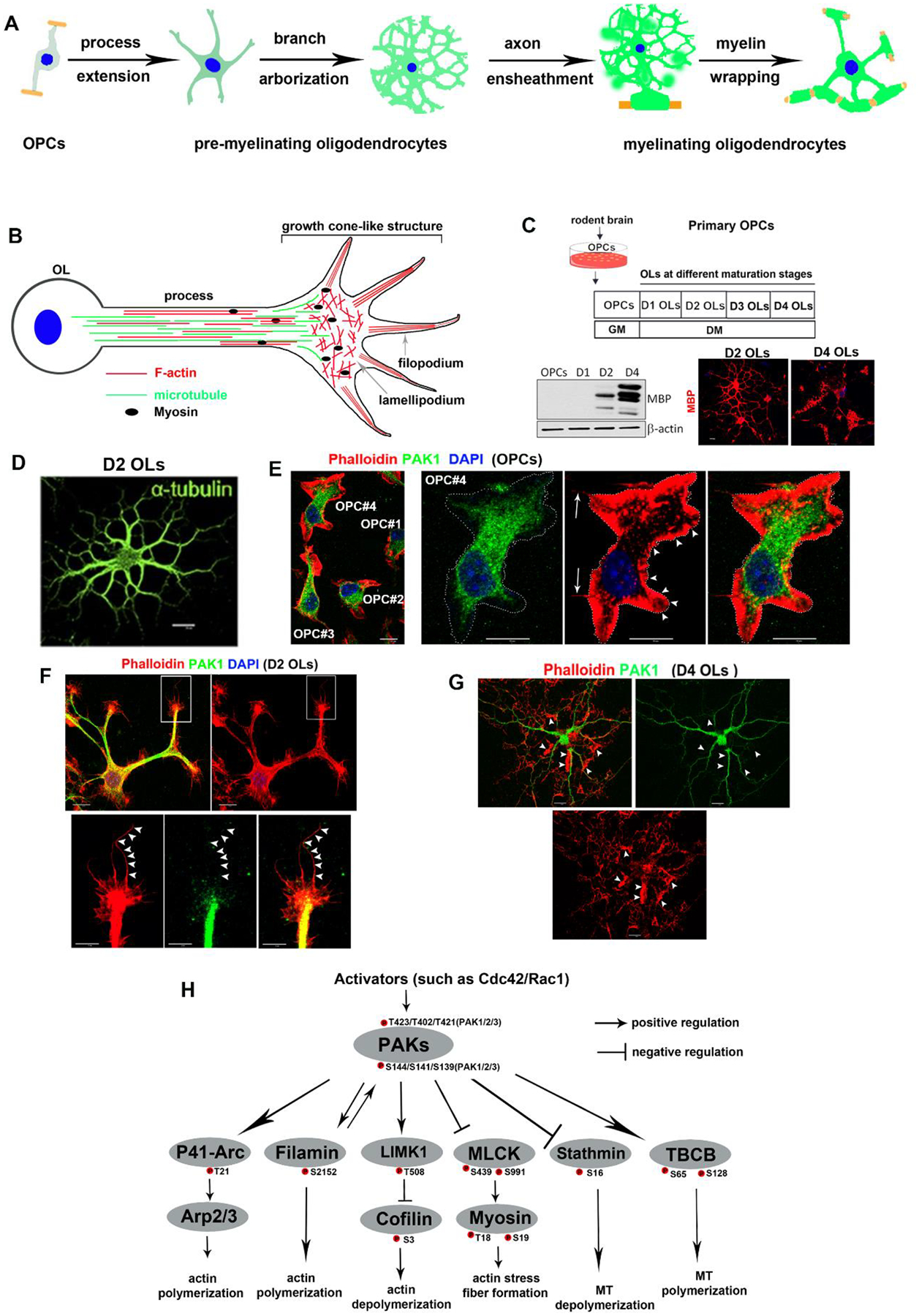
p21-activated kinases (PAKs) and cytoskeleton in oligodendrocyte progenitor cells (OPCs) and oligodendrocytes (OLs). (A) Major steps of morphological differentiation from OPCs to myelinating OLs. (B) Simplified diagram depicting actin and microtubule (MT) cytoskeleton distribution in OL processes which are analogues to neuronal growth cones. The F-actin network is concentrated at the leading edge of the growth cone-like structures whereas the MT network is distributed in parallel orientation along the processes. Non-muscle myosin II participates in actin stress fibre formation, which provides contractile forces for migrating mammalian cells. (C) Primary OPC culture and validation of OPC differentiation (Zhang et al., 2018). D1–D4, days 1–4; DM, differentiation medium; GM, growth medium; MBP, myelin basic protein. (D) Representative image showing MTs revealed by α-tubulin immunostaining in a D2 immature OL. Image adapted from Lang et al. (2013). Scale bar, 10μm. (E–G) Actin cytoskeleton (indicated by phalloidin staining; red) and PAK1 distribution (green) in OPCs (E) D2 immature OLs (F), and D4 mature OLs (G). DAPI, 4,6-diamidine-2-phenylindole is blue-fluorescent DNA stain. Scale bars: 10μm. In E, OPC#4 is shown at a higher magnification in the images on the right. Dotted outline in the second image from the left indicates the outermost rim of the F-actin network. Arrows point to the leading filopodial-like spikes positive for F-actin. Arrowheads point to the outermost edge of the F-actin-rich lamellipodia-like protrusions. In F, the boxed area is shown at a higher magnification in the lower panels. The arrowheads indicate the F-actin+ filopodia-like spikes at the end of oligodendroglial processes that appear negative for PAK1. (G) Distribution of PAK1 and F-actin in D4 mature OLs. F-actin is localized in distal processes and myelin sheets whereas PAK1 is restricted to the primary and secondary processes and cell bodies. F-actin is often concentrated in the F-actin-rich lamellipodia-like protrusions which are negative for PAK1 (arrowheads). The PAK1 antibody used was a validated antibody (#223849; Abcam) (Grebenova et al., 2019). Note that the Abcam PAK1 antibody #131522 previously used for immunostaining of primary OLs (Brown et al., 2021) was shown to recognize an unknown antigen but not endogenous PAK1 (Grebenova et al., 2019). (H) PAKs regulate cytoskeleton dynamics by phosphorylating and modulating the activity of their substrates. The best studied substrate is Lin-11, Islet-1, and Mec-3 domain kinase 1 (LIMK1), which is phosphorylated at residue T508 and subsequently activated by PAKs. Activated LIMK1 phosphorylates cofilin and inhibits the actin depolymerizing and severing activity of cofilin (Delorme et al., 2007). PAKs phosphorylate filamin (Barnes et al., 2003) and actin-related protein 2/3 complex 41 kDa subunit (p41/Arc) (Vadlamudi et al., 2004b), the regulatory component of the actin-related protein 2/3 complex (Arp2/3), to promote actin polymerization. PAKs regulate actin stress fibre formation by phosphorylating and inhibiting myosin light chain kinase (MLCK) (Goeckeler et al., 2000; Sanders et al., 1999). PAKs regulate MT dynamics by phosphorylating and modulating the activity of the MT-destabilizing protein stathmin (Wittmann et al., 2004) and the MT polymerizing protein tubulin cofactor B (TBCB) (Vadlamudi et al., 2005a).
One of the best-known functions of PAKs is regulation of actin and MT dynamics through modulating the activity of a variety of downstream substrates that are crucial for cytoskeleton reorganization (Fig. 3H) in a cell type-dependent manner. It should be noted that most, if not all, of the downstream substrates are shared by the catalytically active PAK1, PAK2, and PAK3. In oligodendroglial-lineage cells, Cdc42 and Rac1 (the canonical activators of PAKs) were thought to act as positive regulators for OPC differentiation as demonstrated by plasmid-mediated overexpression of dominant negative or constitutively active Cdc42/Rac1 (Liang et al., 2004). Subsequent conditional gene-KO data have instead demonstrated that Cdc42/Rac1-deficient OPCs differentiate normally in culture conditions (Thurnherr et al., 2006). Interestingly, Cdc42 and Rac1 act synergistically to regulate appropriate formation of myelin sheath in the CNS, as conditional depletion of Cdc42 and/or Rac1 results in abnormal myelin outfoldings and enlargement of the inner tongue of the myelin sheath (Thurnherr et al., 2006). These in vivo myelination defects indicate that myelin sheath formed by Cdc42/Rac1-deficient myelinating OLs may undergo excessive growth and/or insufficient wrapping presumably resulting from dysregulated cytoskeletal dynamics (Zuchero et al., 2015; Nawaz et al., 2015; Lee & Hur, 2020). Given that PAKs are the major molecules linking Cdc42/Rac1 to Cdc42/Rac1-regulated cytoskeleton dynamics (Fig. 3D), the in vivo myelination-specific phenotypes in Cdc42/Rac1 conditional knockout (cKO) mice (Thurnherr et al., 2006) indicate that PAKs may control myelin sheath growth and wrapping (the final step of myelination) during development.
The role of group I PAKs in OPC proliferation, differentiation and myelination has not been assessed until a recent study reported that myelination was transiently delayed in very restricted white matter tracts of PAK3 systemic KO mice (Maglorius Renkilaraj et al., 2017) in which potential developmental defects of other types of neural cells and vascular cells cannot be excluded. A more recent study employed pharmacological inhibition and overexpression of dominant negative and constitutively active PAK1 plasmids and concluded that PAK1 is a positive regulator for OL morphological differentiation and myelination in the zebrafish CNS (Brown et al., 2021). This important and interesting study provided the first evidence supporting a potential role of PAK1 in OL morphological differentiation and myelination (Brown et al., 2021). The proposed positive role of PAK1 activity in OL differentiation and myelination (Brown et al., 2021) is appealing, however it seems difficult to reconcile with the clinical observation that PAK1 activation mutations are associated with variable degrees of T2-MRI signal hyperintensity in affected human brain (Table 1), which is an indication (but not a conclusion) of white matter hypomyelination (Merino, 2019; Steenweg et al., 2010). These data suggest that PAK1 may have a species-dependent role in regulating OL differentiation and myelination. Alternatively, an appropriate level of PAK activity may be crucial for CNS myelination; its inactivation or overactivation then resulting in myelination impairment.
Mechanistically, Brown et al. (2021) showed that PAK1 regulates OL morphological differentiation through phosphorylating and modulating the activity of the LIMK1/cofilin pathway, a crucial regulator of actin dynamics (Fig. 3H). A previous in vivo study demonstrated that PAK1 KO alone did not affect the activity of LIMK1 and cofilin in the mammalian brain (Asrar et al., 2009) (Table 2) due to functional redundancy between group I PAK members (Huang et al., 2011). It is possible that the morphological changes arising from short-hairpin RNA (shRNA)-mediated PAK1 knockdown in primary OPCs (Brown et al., 2021) might result from activity-independent function of PAK1 (Higuchi et al., 2008), or from an intrinsic difference between in vivo and in vitro requirements for PAK1 in LIMK1/cofilin regulation. Another interpretation is that plasmid-mediated overexpression of dominant negative and constitutively active PAK1 may initiate certain ‘toxic’ gain-of-functions which are otherwise absent from physiological conditions. Moreover, plasmid-mediated overexpression of dominant negative and constitutively active PAK1 alters the function of not only PAK1, but also PAK2 or PAK3, thus making data interpretation more complicated. It would be interesting and straightforward to employ oligodendroglial-specific PAK cKO animals to determine the in vivo function of PAKs in OL differentiation and CNS myelination.
The downstream substrates of PAK kinase activity are likely to be cell-region and cell-type dependent. For example, PAK1 exhibits a region-dependent functionality in regulating F-actin dynamics in kidney epithelial cells: in the lamellipodia, PAK1 promotes F-actin turnover by regulating the activity of cofilin whereas in the lamella immediately following the lamellipodia, PAK1-regulated myosin (but not cofilin) plays a role in F-actin turnover (Delorme et al., 2007; Delorme-Walker et al., 2011). In this regard, it is important to assess the anatomical distribution of PAK1 and F-actin during OL development. The spatial relationship between PAK1 expression and the F-actin network has not been reported in oligodendroglial lineage cells. Because the actin cytoskeleton is present ubiquitously and densely in all type of neural and vascular cells in the brain, primary culture of OPCs and OLs (Fig. 3C) provides a valuable system for studying the spatial distribution of PAK1 and F-actin. In OPCs, F-actin was concentrated in the lamellipodium-like structures whereas PAK1 seems restricted to F-actin-negative areas in the cell body (Fig. 3E). In immature OLs undergoing active process extension and arborization, many F-actin+ lamellipodium-like and filopodium-like protrusions were frequently seen along and at the tip of oligodendroglial processes whereas PAK1 expression is primarily located in the cell bodies and proximal processes and downregulated in F-actin+ lamellipodium-like and filopodium-like structures (Fig. 3F). In mature OLs, F-actin was substantially downregulated within the process network (Fig. 3G) and restricted primarily to the outermost rim of the process network (Zuchero et al., 2015; Nawaz et al., 2015). By contrast, PAK1 was restricted to the primary and secondary processes and seemed absent from MBP+ myelin sheets (Fig. 3G). Together, high PAK1 expression appears to be restricted to the subcellular areas with little F-actin network in OPCs, and immature and mature OLs in vitro. Notably, the exclusion of PAK1 expression from MBP+ and F-actin+ distal processes (Fig. 3G) suggests that downregulation of PAK1 may be permissive for the final steps of morphological maturation of OLs. Based on the dynamic distribution of PAK1 in the cell body and processes, it is plausible to hypothesize that PAK1 may play a stage-dependent role in OPC proliferation, OPC differentiation, OL maturation, and myelination via regulating actin and MT cytoskeleton dynamics. It will be important to determine the spatial distribution of kinase-active PAK1 in OPCs, immature OLs, and mature OLs.
(2). Insights from signalling cross-regulation
(a). Regulation between PAKs and Wnt signalling
Both PAK activity and β-catenin-mediated signalling activity are dysregulated in cancer cells and exert oncogenic potential in promoting cancer cell proliferation and survival. Accumulating evidence obtained from the cancer research field has demonstrated that PAK1 positively regulates β-catenin-mediated canonical Wingless-related integration site (Wnt) signalling (Fig. 4A). PAK1 interacts with and directly phosphorylates β-catenin, the key shuttling molecule linking upstream Wnt signalling to downstream T cell factor/lymphoid enhancer factor (TCF/LEF)-mediated transcriptional activity, at the residues Ser675 (Zhu et al., 2012; Sun et al., 2009; He, Shulkes & Baldwin, 2008; Arias-Romero et al., 2013; Park et al., 2012) and Ser663 (Park et al., 2012). Phosphorylation of Ser675 and S663 stabilizes β-catenin and prevents it from proteasome-mediated degradation. The phosphorylated β-catenin is accumulated in the cytoplasm and translocated into the nucleus where it binds to the Wnt effectors TCF/LEF and activates the Wnt/β-catenin/TCF/LEF signalling pathway. PAK1 may also enhance Wnt/β-catenin/TCF signalling indirectly by potentiating Akt/PKB-mediated β-catenin phosphorylation (Fang et al., 2007) (Fig. 4A).
Fig. 4.
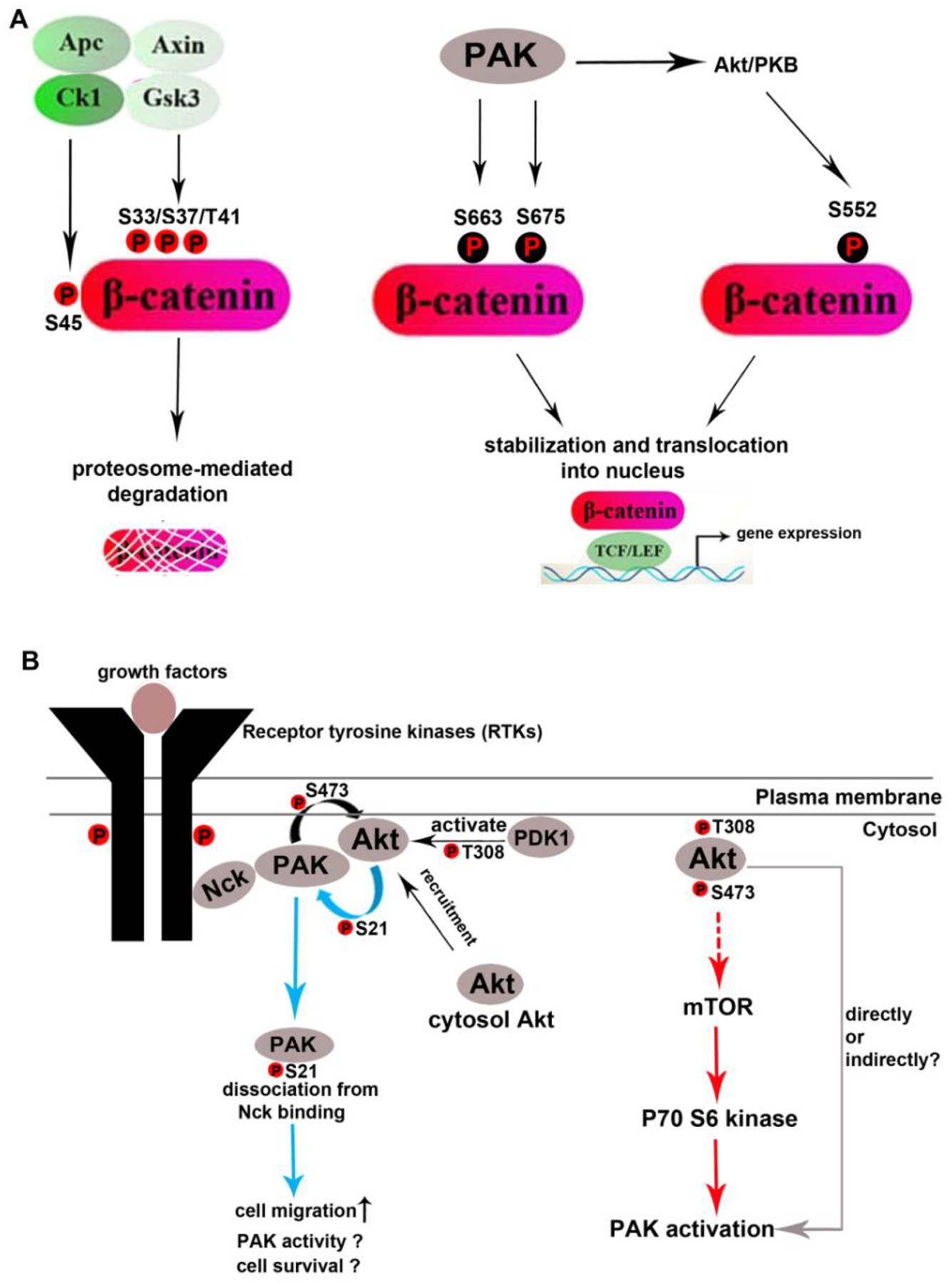
p21-activated kinases (PAKs) regulate Wnt/β-catenin (A) and protein kinase B (Akt/PKB) (B) signalling pathways. (A) PAK activates the β-catenin-mediated signalling pathway both directly and indirectly. In the absence of upstream Wnt signalling, β-catenin undergoes rapid turnover, a proteasome-mediated degrading process involving the destruction complex consisting of adenomatous polyposis coli (Apc), Axin2, casein kinase I (Ck1), and glycogen synthesis kinase 3 (Gsk3). Ck1-mediated phosphorylation at Ser45 and subsequent Gsk3-mediated phosphorylation at Ser33/Ser37/Thr41 mark β-catenin for proteasome-mediated degradation. Kinase-active PAKs directly interact with and phosphorylate β-catenin at Ser663 and S675, both of which stabilize β-catenin and activate β-catenin/T cell factor (TCF)/lymphoid enhancer factor (LEF)-mediated signalling. PAKs also indirectly enhance β-catenin transcriptional activity by potentiating serine/threonine kinase Akt/PKB activation (Higuchi et al., 2008), which, in turn, phosphorylates β-catenin at Ser552 and increases its stability and transcriptional activity. (B) Reciprocal regulation between PAK and Akt/PKB signalling pathways. PAK1 is re-localized to the cell membrane by binding to adaptor proteins (such as non-catalytic region of tyrosine kinase, Nck) through its proline-rich PxxP motifs (see Fig. 1A) in response to growth factor-activated receptor tyrosine kinases (RTKs). PAK1 stimulates Akt/PKB through its kinase-independent scaffolding function by recruiting Akt/PKB from the cytosol to the cell membrane where Akt/PKB is activated by the membrane-associated 3-phosphoinositide-dependent protein kinase-1 (PDK1) (Higuchi et al., 2008). PAK1 also positively regulates Akt/PKB by directly phosphorylating Akt/PKB at Ser473 at the C-terminal regulatory domain (Mao et al., 2008), an essential phosphorylation event for Akt/PKB activation. Reciprocally, activated Akt/PKB disassociates PAK1 from the cell membrane by phosphorylating PAK1 at Ser21 (Zhou et al., 2003), a residue that was originally identified as an autophosphorylation site during PAK activation. Akt/PKB also promotes PAK activation (Tang et al., 2000) possibly through the mammalian target of rapamycin (mTOR)/P70S6 kinase signalling pathway (Ishida et al., 2007).
Canonical Wnt/β-catenin signalling plays important roles in OL development and CNS myelination (Guo et al., 2015). While Wnt/β-catenin signalling may exert developmental stage-dependent effects on OPC generation, differentiation, maturation, and myelination (Guo et al., 2015; Dai et al., 2014), consensus has been reached that Wnt/β-catenin overactivation inhibits OPC differentiation, maturation, and CNS myelination not only during normal development but also during remyelination (Lang et al., 2013; Feigenson et al., 2009; Ye et al., 2009; Lee et al., 2015b,a; Fancy et al., 2014; Fancy et al., 2011b). PAK1 (or group I PAKs) may control OPC differentiation and (re)myelination through modulating Wnt/β-catenin activity. In this regard, it would be interesting to determine whether Wnt/β-catenin signalling activity is dysregulated in oligodendroglial-lineage cells with constitutive PAK1 activation, which has been reported in the brain of PAK1-activation mutations.
(b). Reciprocal regulation of PAK1 and AKT signalling
Like PAKs, the Akt/PKB signalling pathway is commonly dysregulated in various cancer cells. The published data have unveiled a reciprocal regulation between the PAK and Akt pathways. PAK1, acting through its kinase-independent scaffold function, recruits Akt from the cytosol to the plasma membrane where PDK1 phosphorylates Akt at Thr308 in the kinase domain (Higuchi et al., 2008). Additionally, PAK1 directly phosphorylates Akt at Ser 473 in the regulatory domain (Mao et al., 2008) (Fig. 4B). Since Akt phosphorylation at Thr308 and Ser473 is essential and sufficient for the full kinase activation, PAK1 is functionally placed upstream of Akt activation in this regard. Reciprocally, activated Akt was reported to stimulate PAK1 through a GTPase-independent manner in rat fibroblasts (Tang et al., 2000). Knockdown of the mammalian target of rapamycin (mTOR) substrate p70 S6 kinase abrogates PAK1 phosphorylation at Thr423 and inhibits PAK1 activity in human hepatocellular carcinoma cell lines (Ishida et al., 2007), suggesting that Akt may potentiate PAK1 activation via Akt downstream mTOR signalling in a cell-dependent manner (Fig. 4B). Furthermore, Akt may regulate PAK1 subcellular localization and possibly PAK1 activity by trans-phosphorylating PAK1 at Ser21, an event that dissociates PAK1 from the cell membrane and re-locates it to the cytosol (Zhou et al., 2003). The net outcome of PAK1 activity upon Akt-mediated membrane disassociation is unclear but it probably involves sequestration of PAK activity from regulating cytoskeleton organization at the leading edge of migrating cells because membrane-associated PAK1 is exposed to high concentrations of active GTPases, the upstream activators of PAKs (Lu & Mayer, 1999).
The role of the Akt/PKB pathway in OPC differentiation and myelination has been well documented. The current conclusion is that the Akt pathway is a positive regulator for oligodendroglial myelination. A series of studies demonstrated that constitutive Akt hyperactivity driven by the proteolipid protein (Plp) promoter enhances CNS myelination without affecting OPC proliferation, survival, and differentiation (Flores et al., 2008) and the myelination-promoting effect appears to act through mTOR signalling, one of the downstream substrates of Akt (Narayanan et al., 2009; Wahl et al., 2014), specifically through Raptor-containing mTOR complex 1 (mTORC1) (Bercury et al., 2014). It is conceivable that Plp promoter-driven Akt overexpression (Flores et al., 2008) may create non-physiological Akt hyperactivity in the later stages of OL differentiation, hence it remains uncertain whether physiological levels of Akt activity play a role in OPC proliferation, differentiation, and myelination and whether other downstream targets [such as forehead transcription factor of the O class (FoxO), glycogen synthase kinase 3 β (GSK3β), Wnt/β-catenin] mediate Akt-regulated oligodendroglial development during CNS myelination. Conditional Akt-deletion experimental systems may help answer these questions. It should be noted that Akt consists of three isoforms (encoded by Akt1, Akt2, and Akt3, respectively) which may be functionally redundant in regulating oligodendroglial development. It is possible that oligodendroglial development is unaffected in conditional KO of each one of the isoforms but severely perturbed in triple Akt1/2/3 KO.
One outstanding question is whether PAK1 functions upstream or downstream of Akt in the context of OL development. Interestingly, recent data demonstrated that transgene-mediated constitutive Akt hyperactivation increased the percentage of OLs that were positive for cytoplasmic PAK1 (Brown et al., 2021), suggesting that Akt may upregulate PAK1 expression. It is also possible that constitutively active Akt in OLs relocates PAK1 from the plasma membrane to the cytosol through Akt-mediated PAK1 Ser21 phosphorylation (Fig. 4B). Further studies are needed to determine whether constitutively active Akt increases the level of PAK1 Ser21 phosphorylation and regulates PAK1 kinase activity in the cytosol and, reciprocally, whether PAK1 activation regulates Akt-mediated signalling pathways.
(c). Fine-tuning of the ERK/MAPK pathway by PAKs
The mitogen-activated protein kinases (MAPKs) are a family of serine/threonine protein kinases consisting of at least three subfamilies: extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs), and p38 MAPKs, among which the ERK/MAPK pathways are most studied in OL development and CNS myelination.
The rat sarcoma virus (Ras) family genes are the most common oncogenes in human cancers. The canonical Ras/rapidly accelerated fibrosarcoma 1(Raf1)/MEK/ERK signalling axis transduces extracellular signals to gene expression during cell proliferation and survival. PAK1 has been shown to stimulate the Raf1/MEK/ERK signalling axis by directly phosphorylating Raf1 at Ser338 and MEK1 (also known as ERK kinase) at Ser298, thus facilitating signal transduction from Ras to ERK (Fig. 5A). It has been also reported that PAK1 attenuates ERK-mediated signalling through activating the phosphatase activity of protein phosphatase 2A (PP2A), a negative regulator that dephosphorylates ERK and inhibits ERK activity (Fig. 5A) at least in mast cells and cardiomyocytes. It is likely that PAK1 may potentiate or attenuate the ERK/MAPK-mediated signalling pathway in a context-dependent manner according to cell type, level of ERK activation, and/or developmental stage.
Fig. 5.
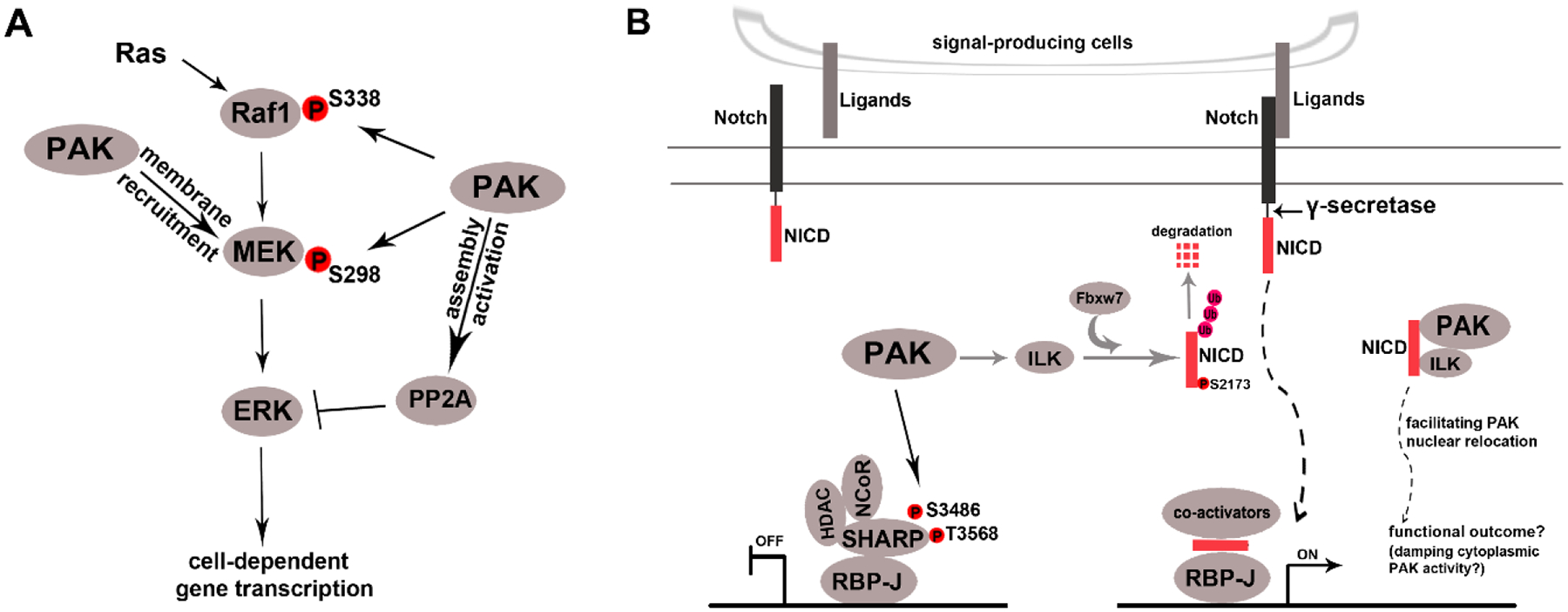
Regulation of rapidly accelerated fibrosarcoma 1 (Raf1)/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) (A) and Notch (B) signalling pathways by p21-activated kinases (PAKs). (A) PAKs positively and negatively regulate the Raf1/MEK/ERK signalling pathway in different contexts. PAK1 activates ERK signalling pathways by directly phosphorylating the upstream activator Raf1 at Ser338 and MEK at S298 and augmenting the kinase activity of Raf1 and MEK (Zang et al., 2002; Chaudhary et al., 2000; Shrestha et al., 2012; Coles & Shaw, 2002). PAK1 promotes ERK activation independent of its kinase activity and presumably by PAK’s scaffold function which recruits MEK to the cell membrane where MEK can be activated by membrane-bound activated Raf1 (Wang et al., 2013). PAK1 also inhibits ERK signalling by activating protein phosphatase 2A (PP2A) (Taglieri et al., 2011; Staser et al., 2013), a negative regulator of ERK activity (Miglietta et al., 2006; Van Kanegan et al., 2005). (B) Hypothetical model of PAK regulation of Notch signalling. Notch receptors are membrane-anchored proteins consisting of an extracellular ligand-binding domain and an intracellular domain (NICD, Notch intracellular domain). Notch ligands are membrane-associated glycoproteins (such as Jagged1, Delta, and F3/contactin) expressed in Notch signal-producing cells. PAK1 interacts with and phosphorylates the Notch repressor component SHARP (SMRT/HDAC1 associated repressor protein) at Ser3486 and Thr3568 within its repression domain, enhancing SHARP-mediated repression of Notch target genes (Vadlamudi et al., 2005b). PAK1 also controls Notch signalling through phosphorylating and activating integrin-linked kinases (ILKs) (Acconcia et al., 2007) which, in turn, phosphorylates the NICD at Ser2173 and promotes proteosome-mediated NICD degradation, a process in which the E3 ligase F-box/WD repeat-containing protein 7 (Fbxw7) [which tags NICD with ubiquitin (Ub)] plays an essential role (Mo et al., 2007). ILK, which plays an essential role in tumorigenesis, has been shown to regulate OPC proliferation and differentiation (Hussain & Macklin, 2017). In addition to ILK, NICD is also reported to interact directly with PAK1 and facilitate PAK1 (and/or ILK) nuclear translocation (Yoon et al., 2016). HDAC, histone deacetylase; NCoR, nuclear receptor co-repressor 2; RBP-J, recombination signal binding protein for immunoglobulin kappa J region.
The role of the ERK/MAPK pathway in OPC proliferation, differentiation, myelination initiation, and myelin growth is still controversial and may be OL developmental-stage dependent. A study employing Olig2-Cre:Erk1−/−:Erk2fl/fl mice (Olig2:ERK1/2 cKO; died after birth) concluded that ERK1/2 promotes OPC proliferation yet inhibits OPC differentiation and myelination initiation (Newbern et al., 2011). Consistent with this idea, Ishii, Furusho & Bansal (2013) showed that ERK1/2 depletion in Olig1-Cre:Erk1−/−:Erk2fl/fl mice (Olig1:ERK1/2 cKO, died after birth) resulted in diminished OPC proliferation. The same group used Cnp-Cre:Erk1−/−:Erk2fl/fl mice (Cnp:ERK1/2 cKO, viable) and documented that ERK1/2 depletion resulted in a reduction in myelin sheath thickness without affecting OPC proliferation and differentiation (Ishii et al., 2012). The expression of oligodendrocyte lineage transcription factor 2 (Olig2) (or Olig1) commences in pre-OPCs and continues in OPCs and OLs whereas 2´,3´-cyclic nucleotide-3´-phosphodiesterase (Cnp) is upregulated primarily in the later stages of OLs (O4+ immature OLs, and mature OLs) (Zhang et al., 2018), therefore, the discrepant results obtained from Olig2 (Olig1)-Cre versus Cnp-Cre-driven ERK1/2-deletion mice suggest that ERK/1/2 promotes OPC proliferation and population expansion but inhibits subsequent OPC differentiation and myelination initiation. The inhibitory role of ERK1/2 in OPC differentiation and myelination initiation is further supported by recent in vitro OPC cultures showing that dampening the ERK pathway using a variety of small compounds remarkably promotes OPC differentiation in a cell-autonomous manner and enhances myelination initiation in OPC–neuron co-cultures (Suo et al., 2019). ERK1/2 gain-of-function via Olig1-Cre-driven expression of constitutively active MEK (see Fig. 5A) resulted in OPC hyperproliferation and OPC overproduction whereas Cnp-Cre-mediated ERK1/2 gain-of-function increased myelin sheath thickness without affecting OPC proliferation and myelination initiation (Ishii, Furusho & Bansal, 2013), which was confirmed by recent data from the same group (Ishii et al., 2019). Taken together, these ERK1/2 loss- and gain-of-function data collected from Olig2 (Olig1)+ and Cnp+ cells suggest that the ERK pathway may play a stage-dependent role in oligodendroglial development: it promotes OPC proliferation and population expansion, inhibits OPC differentiation and myelination initiation, and enhances myelin sheath growth (the final step of myelination). Future studies leveraging stage-specific ERK loss- and gain-of-function are needed to support this likely stage-dependent role. Considering the potential role of PAK1 in modulating ERK signalling activity (Fig. 5A), the hypothetical stage-dependent role of ERK1/2 in OL development suggests that PAK1 may coordinate different steps of OL development through fine-tuning the activity level of ERK1/2. Future studies are needed to determine whether PAK1 regulates ERK signalling activity at different developmental stages of oligodendroglial-lineage cells.
(d). Crosstalk between PAK and Notch signalling
Conserved Notch signalling plays important roles in oligodendroglial development. In the absence of ligand binding, the Notch effector and transcriptional factor recombination signal binding protein for immunoglobulin kappa J region (RBP-J) recruit a co-repressor complex consisting of histone deacetylase (HDAC), SMRT/HDAC1 associated repressor protein (SHARP) (Oswald et al., 2002), and nuclear receptor co-repressor 2 (NCoR), and inhibit Notch target gene expression (Fig. 5B). The binding of ligands to Notch receptors triggers gamma-secretase-mediated cleavage of Notch intracellular domain (NICD) from the plasma membrane, which subsequently translocates into the nucleus where it activates Notch/RBP-J target-gene expression by recruiting transcriptional co-activators (Fig. 5B). Previous studies conducted in non-oligodendroglial-lineage cells have demonstrated that PAK could suppress Notch signalling by enhancing the function of the signalling co-repressors and or enhance Notch signalling by modulating the stability of NICD, the key shuttling factor linking extracellular signals to downstream gene expression (Fig. 5B). Interestingly, recent data showed that NICD directly complexes with PAK1, which promotes the translocation of PAK1 from the cytosol to the nucleus. A previous study reported that PAK1 complexes and phosphorylates integrin-linked kinase (ILK) which promotes ILK retention in the cytoplasm (ILK is predominantly cytoplasmic) (Acconcia et al., 2007). In the cytoplasm, ILK negatively modulates a variety of its downstream substrates including NICD itself (Mo et al., 2007) (Fig. 5B) and GSK3β (Naska et al., 2006). In this regard, NICD-mediated PAK1 nuclear relocation provides a positive feedback to Notch signalling activity by reducing NICD degradation. NICD-mediated PAK1 nuclear relocation may also regulate the nuclear activity of PAK1 (Rayala, Molli & Kumar, 2006a; Rayala et al., 2006b) and affects cell mitosis and gene expression in proliferating cells (Li et al., 2002) (see Section VI.3).
Notch signalling has been proposed as a molecular rheostat controlling OL development (Popko, 2003). The inhibitory or promoting effect of Notch signalling on OL development may be both stage dependent and Notch ligand dependent which is suggested by discrepant observations from different contexts (for example development versus regeneration) (Brosnan & John, 2009; Stidworthy et al., 2004; Zhang et al., 2009). Delta-Notch signalling promotes OPC generation from neural precursor cells in the embryonic spinal cord of zebrafish (Park & Appel, 2003). By contrast, Jagged1-Notch signalling inhibits OPC differentiation in the CNS of neonatal rodents (Wang et al., 1998; Genoud et al., 2002). Jagged1 expression is sharply downregulated by the first postnatal weeks in rodents (Wang et al., 1998), suggesting that Jagged1-activated Notch signalling in OPCs diminishes over time during postnatal development. Interestingly, F3/contactin is identified as a physiological axon-derived ligand for Notch receptors and F3/contactin-initiated Notch signalling in OPCs promotes OPC differentiation (Hu et al., 2003). It is plausible that, during active myelination, axon-derived F3/contactin predominates over Jagged1 in activating Notch signalling in OPCs and the activation of F3/contactin–Notch signalling promotes OPC differentiation and facilitates axonal ensheathment. Given the importance of PAK in controlling Notch signalling (Fig. 5B), we hypothesize that PAK may regulate different steps of OL development through fine tuning the activity level of Notch signalling in OPCs and OLs.
(3). PAKs in the nucleus: a potential role in oligodendrocyte development?
PAK1 is reported to primarily located in the cytoplasm and at the leading plasma membrane of migrating cells (Lu & Mayer, 1999; Kichina et al., 2010). PAK1 is also present, albeit in a low amount, in the nucleus during mitosis in proliferating cells and has been proposed to play a role in mitotic spindle formation during breast cancer cell division (Li et al., 2002). In the nucleus, PAK1 interacts with and phosphorylates histone H3 at Ser10, likely modulating the function of histone H3 in gene expression (Li et al., 2002). PAK1 has also been observed to translocate into the nucleus in response to extracellular-signalling molecules such as prolactin (Oladimeji & Diakonova, 2016) and epithelial growth factor (EGF) (Singh et al., 2005) in breast cancer cells. It has been shown that PAK1 (and PAK2) consists of three nuclear localization sequences (NLSs) whose mutation eliminates the nuclear translocation of PAK1 (Singh et al., 2005). Recent data demonstrated that PAK1’s interacting partner dynein light chain (LC8) (Lightcap et al., 2008; Vadlamudi et al., 2004a), a protein well known for its role in dynein motor complex assembly, facilitates PAK1 nuclear import in human breast cancer cells (Lightcap et al., 2009). In particular, LC8-mediated PAK nuclear transport is unique to PAK1 due to the absence of the LC8-interacting domain in other PAKs (Lightcap et al., 2009). Knockdown of PAK1 in zebrafish resulted in a decreased rate of embryo survival and morphological alterations in surviving embryos, however, overexpression of mutant PAK1 deficient for NLS or LC8-binding failed to rescue the embryo death or morphological alterations (Lightcap et al., 2009), suggesting that PAK1 nuclear localization is crucial for cell survival and zebrafish embyo development.
As discussed in Section VI.2d, PAK1 directly complexes with NICD which contains one canonical NLS (Huenniger et al., 2010). Therefore, it is plausible that PAK1 nuclear translocation is facilitated by NICD binding in response to Notch signals. Reciprocally, PAK1 may also facilitate NICD nuclear translocation to enhance its downstream Notch target because PAK1 contains three NLSs in its N-termimal domain.
The biological significance of PAK1 nuclear translocation remains enigmatic. It was traditionally thought that kinase-active PAK1 is primarily located near the plasma membrane where the canonical activators Cdc42 and Rac1 are concentrated (Lu & Mayer, 1999). It is possible that nuclear PAK1 is shunted from Cdc42/Rac1-mediated activation. However, previous data suggest that the kinase activity of nuclear-localized PAK1 is maintained (Oladimeji & Diakonova, 2016; Oladimeji et al., 2016; Lightcap et al., 2009; Li et al., 2002). In addition to regulating mitotic spindle formation (Li et al., 2002), one likely outcome of PAK1 nuclear localization is to regulate gene expression by targeting regulatory elements of chromatin (Singh et al., 2005). It seems that nuclear PAK1 can promote or inhibit its downstream target gene expression in a context-dependent manner (Singh et al., 2005), Together, nuclear PAK1 may have physiological roles in regulating cell division and influencing gene expression, at least in transformed cancer cells.
Using primary OPC and OL cultures, we observed that PAK1 is primarily distributed in the cytoplasm and present at a low amount in the nucleus in undifferentiated OPCs and gradually increases in the nuclei of maturing OLs (Fig. 3E–G). It appears that PAK1 is also concentrated in OL nuclei in the rodent brain at early postnatal ages (Brown et al., 2021). The potential role of nuclear PAK1 in regulating mitotic spindle formation during OPC proliferation has not been revealed yet. In addition to regulating morphological differentiation (Brown et al., 2021), it remains to be defined whether nuclear PAK1 regulates the expression of certain genes required for OPC molecular differentiation. Understanding the upstream signals driving PAK1 nuclear localization during oligodendroglial-lineage progression and maturation will provide novel insights into the molecular mechanisms underlying PAK-regulated OL development. Moreover, unbiased proteomic identification of PAK’s interacting partners in OPCs and OLs may also provide novel insights into these outstanding questions.
VII. PAKs IN DEMYELINATION: FRIENDS OR FOES IN MYELIN REPAIR?
Blocked OPC differentiation and incomplete remyelination (or myelination failure) are pathological characteristics in the multiple sclerosis (MS) brain (Franklin, 2002), the most common inflammatory demyelinating CNS disorder and the most common cause of non-traumatic disability in young adults (Frischer et al., 2015). The molecular programs during remyelination usually recapitulate those during developmental myelination (Fancy et al., 2011a). It has been proposed that tightly controlled molecular programs during developmental myelination are severely dysregulated and imbalanced in MS brain lesions, causing inefficient or failed remyelination. Searching for such molecular targets is instructive for therapeutic designs to promote myelin regeneration in MS.
PAK inhibition has been shown to exert beneficial effects in other neurodevelopmental disorders with potential myelination impairment. Previous preclinical studies demonstrate that genetically or pharmacologically inhibiting PAK activity rescues neuronal phenotypes and symptoms of fragile-X syndrome (FXS) (Hayashi et al., 2007; Dolan et al., 2013) in FMR1-KO animal models (Kazdoba et al., 2014) in which delayed myelination is also observed (Pacey et al., 2013). These functional data indicate that PAK hyperactivity is detrimental to postnatal brain development. It would be very interesting to determine whether inhibiting PAK activity also rescues myelination delay in the FMR1-KO mouse model of FXS. Such data would provide insights into the function of PAK activity in OL development and myelination.
The conversion of relapse-remitting MS (RRMS), the major form of MS, to secondary progressive MS (SPMS) is commonly seen in MS patients. It has been proposed that this conversion coincides with the change of adaptive immunity-driven pathogenesis to chronic innate immunity-driven neurodegeneration in the CNS of MS patients (Weiner, 2008). The potential for OPC differentiation and remyelination declines with the disease course conversion and, therefore, it is common to see many chronic plaques characterized by remyelination failure in SPMS patients (Frischer et al., 2015). It remains elusive whether PAK activity is upregulated in chronic plaques and if so, whether dysregulated PAK activity is a molecular culprit that blocks OPC differentiation and remyelination. Previous proteomics data demonstrated that the expression level of PAK1 was upregulated fourfold in chronic plaques compared to non-MS white matter controls (Fig. 6). PAK2, albeit at a much lower level than PAK1 (greater than 15-fold difference), displays mild upregulation (Fig. 6). These data suggest that PAK1 and PAK2 dysregulation may inhibit OPC differentiation and remyelination in chronic MS plaques. Given that spontaneous remyelination occurs in some lesions of MS patients (called shadow plaques) (Patrikios et al., 2006), as an alternative interpretation, upregulation of PAK1 (or PAK2) may be an indication of spontaneous remyelination occurrence, in line with a positive role of PAK1 in OL differentiation and developmental myelination proposed recently (Brown et al., 2021). It remains to be determined whether PAK1 or PAK2 upregulation occurs in oligodendroglial lineage cells and whether the kinase activity of PAKs is upregulated in chronic plaques. Such studies would offer new insights into targeting PAK activity for promoting remyelination in MS patients and preclinical animal models. Together, the currently available data suggest that PAK1 dysregulation may be a potential therapeutic target for myelin regeneration in MS. Additional preclinical animal studies are needed to support or falsify this hypothesis.
Fig. 6.
Expression of group I p21-activated kinases (PAKs) in different types of white matter plaques and control white matter of the multiple sclerosis (MS) brain. Different types of MS plaques were isolated by laser capture microdissection and the resulting protein extract was subjected to proteomic identification (Han et al., 2008). Active plaques (AP) are histologically characterized by indistinct margins of demyelination and dense infiltration of myelin debris-loaded macrophages/microglia. Chronic active plaques (CAP), also known as smouldering plaques or slowly expanding plaques (Frischer et al., 2015) are characterized by defined demarcation of demyelination with no or few activated macrophages/microglia within the plaque but with many surrounding plaques. Chronic plaques (CP, or chronic inactive plaques) are characterized by a sharply demarcated demyelination edge with no or few myelin debris-loaded activated macrophages/microglia. ctrl, control. Data adapted from Supplemental Table 3 of Han et al. (2008).
VIII. CONCLUSIONS
(1) PAK1, acting in a kinase-dependent and kinase-independent manner, regulates a plethora of downstream substrates and signalling pathways such as LIMK1/cofilin, Wnt/β-catenin, Akt, Raf1/MEK/ERK, and Notch signalling pathways, all of which have been reported to play important roles in oligodendroglial development and (re)myelination. Consensus has been reached that group I PAKs are required for CNS neuronal development and function and inhibiting PAK hyperactivity is beneficial in animal models of neurodevelopmental disorders. However, the role of CNS myelination and remyelination remains incompletely understood, and the direct demonstration of the role of group I PAKs in mammalian CNS myelination and remyelination is still lacking.
(2) OL development consists of multiple sequential steps (or stages): OPC proliferation and population expansion, OL differentiation and maturation, myelin ensheathment (myelination initiation), and myelin wrapping and compaction. Defects in the upstream steps invariably result in impairments of the downstream steps. The dynamics of group I PAK kinase activity along the different steps of oligodendroglial development has yet to be defined. Since many PAK-controlled oncogenic signalling pathways regulate oligodendroglial development in a stage-dependent and/or dose-dependent manner, it is likely that PAKs may also play a stage (or dose)-dependent role in OPC proliferation versus OPC differentiation and in myelin ensheathment versus myelin wrapping. For example, as highlighted in Fig. 3H, PAK1 activation leads to F-actin assembly, which has been demonstrated to promote OL morphological differentiation and initial axon ensheathment. However, subsequent myelin wrapping and compaction require F-actin disassembly (Zuchero et al., 2015; Nawaz et al., 2015). In this sense, the role of group I PAKs in OL development may not be binary (positive or negative) or uniform along the different steps. Screening and characterizing potential binding partners and downstream targets of PAKs at different stages of oligodendroglial development will provide novel insights into the hypothetical stage-dependent regulation of oligodendroglial development.
(3) Previous studies demonstrate functional redundancy of group I PAKs in regulating neuronal development and function. It remains to be determined whether such functional redundancy occurs during oligodendroglial development and function. It is also noteworthy that PAK1 is uniquely phosphorylated at Thr212 during the mitotic phase by Cyclin B/Cdc2 (no equivalent phosphorylation exists in PAK2 and PAK3) in proliferating cells. PAK1 phospho-T212 is present in the embryonic and early postnatal murine brain but absent from the adult brain (Zhong et al., 2003). Since OPCs are highly proliferating cells in the embryonic and early postnatal brain, it is plausible to hypothesize that PAK1 may modulate OPC proliferation and population number through T212 phosphorylation. Future studies specifically mutating PAK1 T212 in OPCs are needed to prove or disprove this hypothesis.
(4) The kinase-independent functions of PAKs in regulating cellular processes and signalling pathways are not uncommon and may also play important roles in OPC proliferation, OL differentiation, and myelination. Previous data have shown that plasmid-mediated overexpression of a kinase-dead PAK1 mutant (PAK1-K299R) activates the ERK1/2-MAPK (Wang et al., 2013) and Akt/PKB (Higuchi et al., 2008) signalling pathways and that cell cycle arrest induced by expression of the peptide inhibitor PAK1-AID (aa83–149) cannot be rescued by the enforced expression of a kinase-active PAK1 mutant (PAK1-T423E) (Thullberg et al., 2007). Plasmid-mediated expression of these PAK mutants may create a ‘molecular bridge’ by which PAK’s interacting partners are recruited to different subcellular locations, presumably exerting their biological effects through PAK1 kinase-independent scaffold function. Alternatively, aberrantly high levels of the mutant ‘molecular bridges’ via plasmid-mediated overexpression could provide potential ‘toxic’ gain-of-functions that are otherwise absent from cells under physiological conditions. Future in vivo studies employing PAK loss-of-function approaches will help define the physiological role of PAKs in oligodendroglial development.
(5) The functions and molecular mechanisms of PAKs in OL regeneration and remyelination remain enigmatic. PAK1 may play a different role in CNS remyelination from that reported during normal development (Brown et al., 2021) considering its dysregulation in chronic demyelination MS lesions (Fig. 6). Immunohistological data are needed to validate the dysregulation of PAK1 in MS demyelination lesions (Han et al., 2008) and to determine the cell types with PAK1 dysregulation. At the functional level, time-conditional and cell-specific PAK-disruption systems may help to elucidate the potential role of PAKs in demyelination and remyelination. From a translational perspective, it is equally important to determine whether PAK enzymatic activity is dysregulated in MS lesions considering that a large number of PAK inhibitors have been developed and some are in clinial trials in cancer treatment. In this regard, it will be interesting to determine the effect of pharmacologically or genetically inhibiting PAKs on OL regeneration and myelin repair in preclinical animal models of CNS demyelination/remyelination.
IX. ACKNOWLEDGEMENTS AND AUTHOR CONTRIBUTIONS
The authors declare no competing interests. This work was supported by the National Institute of Neurological Disorders and Stroke (NINDS) (R01 NS123080, R01 NS094559R21, and R21 NS109790 to F.G.) and by Shriners Hospitals for Children (85107-NCA-19 and 85200-NCA-16 to F.G., 87400-NCA-21 and 84553-NCA-18 to Y.W.). We thank the authors whose work provides the basis for this review, and apologize to those whose important work was not included due to space limitations.
X. REFERENCES
- Acconcia F, Barnes CJ, Singh RR, Talukder AH & Kumar R (2007). Phosphorylation-dependent regulation of nuclear localization and functions of integrin-linked kinase. Proceedings of the National Academy of Sciences USA 104(16), 6782–6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen KM, Gleeson JG, Bagrodia S, Partington MW, MacMillan JC, Cerione RA, Mulley JC & Walsh CA (1998). PAK3 mutation in nonsyndromic X-linked mental retardation. Nature Genetics 20(1), 25–30. [DOI] [PubMed] [Google Scholar]
- Arias-Romero LE, Villamar-Cruz O, Huang M, Hoeflich KP & Chernoff J (2013). Pak1 kinase links ErbB2 to beta-catenin in transformation of breast epithelial cells. Cancer Research 73(12), 3671–3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asrar S, Meng Y, Zhou Z, Todorovski Z, Huang WW & Jia Z (2009). Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1). Neuropharmacology 56(1), 73–80. [DOI] [PubMed] [Google Scholar]
- Banerjee M, Worth D, Prowse DM & Nikolic M (2002). Pak1 phosphorylation on t212 affects microtubules in cells undergoing mitosis. Current Biology 12(14), 1233–1239. [DOI] [PubMed] [Google Scholar]
- Barnes CJ, Vadlamudi RK, Mishra SK, Jacobson RH, Li F & Kumar R (2003). Functional inactivation of a transcriptional corepressor by a signaling kinase. Nature Structural Biology 10(8), 622–628. [DOI] [PubMed] [Google Scholar]
- Baron W & Hoekstra D (2010). On the biogenesis of myelin membranes: sorting, trafficking and cell polarity. FEBS Letters 584(9), 1760–1770. [DOI] [PubMed] [Google Scholar]
- Bauer NG, Richter-Landsberg C & Ffrench-Constant C (2009). Role of the oligodendroglial cytoskeleton in differentiation and myelination. Glia 57(16), 1691–1705. [DOI] [PubMed] [Google Scholar]
- Ben Bashat D, Kronfeld-Duenias V, Zachor DA, Ekstein PM, Hendler T, Tarrasch R, Even A, Levy Y & Ben Sira L (2007). Accelerated maturation of white matter in young children with autism: a high b value DWI study. Neuroimage 37(1), 40–47. [DOI] [PubMed] [Google Scholar]
- Bercury KK, Dai J, Sachs HH, Ahrendsen JT, Wood TL & Macklin WB (2014). Conditional ablation of raptor or rictor has differential impact on oligodendrocyte differentiation and CNS myelination. Journal of Neuroscience 34(13), 4466–4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bercury KK & Macklin WB (2015). Dynamics and mechanisms of CNS myelination. Developmental Cell 32(4), 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu T, des Portes V, McDonell N, Carrie A, Zemni R, Couvert P, Ropers HH, Moraine C, van Bokhoven H, Fryns JP, Allen K, Walsh CA, Boue J, Kahn A, Chelly J, et al. (2000). Missense mutation in PAK3, R67C, causes X-linked nonspecific mental retardation. American Journal of Medical Genetics 93(4), 294–298. [DOI] [PubMed] [Google Scholar]
- Bokoch GM (1998). Caspase-mediated activation of PAK2 during apoptosis: proteolytic kinase activation as a general mechanism of apoptotic signal transduction? Cell Death and Differentiation 5(8), 637–645. [DOI] [PubMed] [Google Scholar]
- Bokoch GM, Reilly AM, Daniels RH, King CC, Olivera A, Spiegel S & Knaus UG (1998). A GTPase-independent mechanism of p21-activated kinase activation. Regulation by sphingosine and other biologically active lipids. Journal of Biological Chemistry 273(14), 8137–8144. [DOI] [PubMed] [Google Scholar]
- Brosnan CF & John GR (2009). Revisiting Notch in remyelination of multiple sclerosis lesions. Journal of Clinical Investigation 119(1), 10–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TL, Hashimoto H, Finseth LT, Wood TL & Macklin WB (2021). PAK1 positively regulates oligodendrocyte morphology and myelination. Journal of Neuroscience 41(9), 1864–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary A, King WG, Mattaliano MD, Frost JA, Diaz B, Morrison DK, Cobb MH, Marshall MS & Brugge JS (2000). Phosphatidylinositol 3-kinase regulates Raf1 through Pak phosphorylation of serine 338. Current Biology 10(9), 551–554. [DOI] [PubMed] [Google Scholar]
- Chong C, Tan L, Lim L & Manser E (2001). The mechanism of PAK activation. Autophosphorylation events in both regulatory and kinase domains control activity. Journal of Biological Chemistry 276(20), 17347–17353. [DOI] [PubMed] [Google Scholar]
- Chow HY, Dong B, Valencia CA, Zeng CT, Koch JN, Prudnikova TY & Chernoff J (2018). Group I Paks are essential for epithelial- mesenchymal transition in an Apc-driven model of colorectal cancer. Nature Communications 9(1), 3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho RP, Saini HS & Sato-Bigbee C (2010). Sphingosine-1-phosphate and oligodendrocytes: from cell development to the treatment of multiple sclerosis. Prostaglandins and Other Lipid Mediators 91(3–4), 139–144. [DOI] [PubMed] [Google Scholar]
- Coles LC & Shaw PE (2002). PAK1 primes MEK1 for phosphorylation by Raf-1 kinase during cross-cascade activation of the ERK pathway. Oncogene 21(14), 2236–2244. [DOI] [PubMed] [Google Scholar]
- Combeau G, Kreis P, Domenichini F, Amar M, Fossier P, Rousseau V & Barnier JV (2012). The p21-activated kinase PAK3 forms heterodimers with PAK1 in brain implementing trans-regulation of PAK3 activity. Journal of Biological Chemistry 287(36), 30084–30096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cree BAC, Niu J, Hoi KK, Zhao C, Caganap SD, Henry RG, Dao DQ, Zollinger DR, Mei F, Shen YA, Franklin RJM, Ullian EM, Xiao L, Chan JR & Fancy SPJ (2018). Clemastine rescues myelination defects and promotes functional recovery in hypoxic brain injury. Brain 141(1), 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai ZM, Sun S, Wang C, Huang H, Hu X, Zhang Z, Lu QR & Qiu M (2014). Stage-specific regulation of oligodendrocyte development by Wnt/beta-catenin signaling. The Journal of Neuroscience 34(25), 8467–8473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis F, Bernardo A, Magnaghi V, Minghetti L & Tata AM (2012). Muscarinic receptor subtypes as potential targets to modulate oligodendrocyte progenitor survival, proliferation, and differentiation. Developmental Neurobiology 72(5), 713–728. [DOI] [PubMed] [Google Scholar]
- Deacon SW, Beeser A, Fukui JA, Rennefahrt UE, Myers C, Chernoff J & Peterson JR (2008). An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chemistry & Biology 15(4), 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme-Walker VD, Peterson JR, Chernoff J, Waterman CM, Danuser G, DerMardirossian C & Bokoch GM (2011). Pak1 regulates focal adhesion strength, myosin IIA distribution, and actin dynamics to optimize cell migration. Journal of Cell Biology 193(7), 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme V, Machacek M, DerMardirossian C, Anderson KL, Wittmann T, Hanein D, Waterman-Storer C, Danuser G & Bokoch GM (2007). Cofilin activity downstream of Pak1 regulates cell protrusion efficiency by organizing lamellipodium and lamella actin networks. Developmental Cell 13(5), 646–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- des Portes V, Billuart P, Carrie A, Bachner L, Bienvenu T, Vinet MC, Beldjord C, Ponsot G, Kahn A, Boue J & Chelly J (1997a). A gene for dominant nonspecific X-linked mental retardation is located in Xq28. American Journal of Human Genetics 60(4), 903–909. [PMC free article] [PubMed] [Google Scholar]
- des Portes V, Soufir N, Carrie A, Billuart P, Bienvenu T, Vinet MC, Beldjord C, Ponsot G, Kahn A, Boue J & Chelly J (1997b). Gene for nonspecific X-linked mental retardation (MRX 47) is located in Xq22.3-q24. American Journal of Medical Genetics 72(3), 324–328. [DOI] [PubMed] [Google Scholar]
- Dolan BM, Duron SG, Campbell DA, Vollrath B, Shankaranarayana Rao BS, Ko HY, Lin GG, Govindarajan A, Choi SY & Tonegawa S (2013). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proceedings of the National Academy of Sciences USA 110(14), 5671–5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte K, Heide S, Poea-Guyon S, Rousseau V, Depienne C, Rastetter A, Nava C, Attie-Bitach T, Razavi F, Martinovic J, Moutard ML, Cherfils J, Mignot C, Heron D & Barnier JV (2020). PAK3 mutations responsible for severe intellectual disability and callosal agenesis inhibit cell migration. Neurobiology of Disease 136, 104709. [DOI] [PubMed] [Google Scholar]
- Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB, Ibrahim A, Ligon KL, Rowitch DH & Barres BA (2009). Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 138(1), 172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Chan JR, Baranzini SE, Franklin RJ & Rowitch DH (2011a). Myelin regeneration: a recapitulation of development? Annual Review of Neuroscience 34, 21–43. [DOI] [PubMed] [Google Scholar]
- Fancy SP, Harrington EP, Baranzini SE, Silbereis JC, Shiow LR, Yuen TJ, Huang EJ, Lomvardas S & Rowitch DH (2014). Parallel states of pathological Wnt signaling in neonatal brain injury and colon cancer. Nature Neuroscience [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, Franklin RJ & Rowitch DH (2011b). Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nature Neuroscience 14(8), 1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T & Lu Z (2007). Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. Journal of Biological Chemistry 282(15), 11221–11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenson K, Reid M, See J, Crenshaw EB 3rd & Grinspan JB (2009). Wnt signaling is sufficient to perturb oligodendrocyte maturation. Molecular and Cellular Neuroscience 42(3), 255–265. [DOI] [PubMed] [Google Scholar]
- Flores AI, Narayanan SP, Morse EN, Shick HE, Yin X, Kidd G, Avila RL, Kirschner DA & Macklin WB (2008). Constitutively active Akt induces enhanced myelination in the CNS. Journal of Neuroscience 28(28), 7174–7183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJ (2002). Why does remyelination fail in multiple sclerosis? Nature Reviews Neuroscience 3(9), 705–714. [DOI] [PubMed] [Google Scholar]
- Frischer JM, Weigand SD, Guo Y, Kale N, Parisi JE, Pirko I, Mandrekar J, Bramow S, Metz I, Bruck W, Lassmann H & Lucchinetti CF (2015). Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Annals of Neurology 78(5), 710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer BH, Wang C, Vedantam S, Zhou GL, Jin S, Fletcher L, Simon MC & Field J (2006). cGMP-dependent protein kinase phosphorylates p21-activated kinase (Pak) 1, inhibiting Pak/Nck binding and stimulating Pak/vasodilator-stimulated phosphoprotein association. Journal of Biological Chemistry 281(17), 11487–11495. [DOI] [PubMed] [Google Scholar]
- Fu MM, McAlear TS, Nguyen H, Oses-Prieto JA, Valenzuela A, Shi RD, Perrino JJ, Huang TT, Burlingame AL, Bechstedt S & Barres BA (2019). The Golgi outpost protein TPPP nucleates microtubules and is critical for myelination. Cell 179(1), 132–146 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedeon AK, Nelson J, Gecz J & Mulley JC (2003). X-linked mild non-syndromic mental retardation with neuropsychiatric problems and the missense mutation A365E in PAK3. American Journal of Medical Genetics A 120A(4), 509–517. [DOI] [PubMed] [Google Scholar]
- Genoud S, Lappe-Siefke C, Goebbels S, Radtke F, Aguet M, Scherer SS, Suter U, Nave KA & Mantei N (2002). Notch1 control of oligodendrocyte differentiation in the spinal cord. Journal of Cell Biology 158(4), 709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeckeler ZM, Masaracchia RA, Zeng Q, Chew TL, Gallagher P & Wysolmerski RB (2000). Phosphorylation of myosin light chain kinase by p21-activated kinase PAK2. Journal of Biological Chemistry 275(24), 18366–18374. [DOI] [PubMed] [Google Scholar]
- Grebenova D, Holoubek A, Roselova P, Obr A, Brodska B & Kuzelova K (2019). PAK1, PAK1Delta15, and PAK2: similarities, differences and mutual interactions. Scientific Reports 9(1), 17171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Lang J, Sohn J, Hammond E, Chang M & Pleasure D (2015). Canonical Wnt signaling in the oligodendroglial lineage--puzzles remain. Glia 63(10), 1671–1693. [DOI] [PubMed] [Google Scholar]
- Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, Fernald GH, Gerlitz B, Robinson WH, Baranzini SE, Grinnell BW, Raine CS, Sobel RA, Han DK & Steinman L (2008). Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 451(7182), 1076–1081. [DOI] [PubMed] [Google Scholar]
- Harms FL, Kloth K, Bley A, Denecke J, Santer R, Lessel D, Hempel M & Kutsche K (2018). Activating mutations in PAK1, encoding p21-activated kinase 1, cause a neurodevelopmental disorder. American Journal of Human Genetics 103(4), 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S & Tonegawa S (2007). Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proceedings of the National Academy of Sciences USA 104(27), 11489–11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Shulkes A & Baldwin GS (2008). PAK1 interacts with beta-catenin and is required for the regulation of the beta-catenin signalling pathway by gastrins. Gastroenterology 134(4), A249–A250. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Onishi K, Kikuchi C & Gotoh Y (2008). Scaffolding function of PAK in the PDK1-Akt pathway. Nature Cell Biology 10(11), 1356–1364. [DOI] [PubMed] [Google Scholar]
- Hofmann C, Shepelev M & Chernoff J (2004). The genetics of Pak. Journal of Cell Science 117(Pt 19), 4343–54. [DOI] [PubMed] [Google Scholar]
- Horn S, Au M, Basel-Salmon L, Bayrak-Toydemir P, Chapin A, Cohen L, Elting MW, Graham JM, Gonzaga-Jauregui C, Konen O, Holzer M, Lemke J, Miller CE, Rey LK, Wolf NI, et al. (2019). De novo variants in PAK1 lead to intellectual disability with macrocephaly and seizures. Brain 142(11), 3351–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu QD, Ang BT, Karsak M, Hu WP, Cui XY, Duka T, Takeda Y, Chia W, Sankar N, Ng YK, Ling EA, Maciag T, Small D, Trifonova R, Kopan R, et al. (2003). F3/contactin acts as a functional ligand for Notch during oligodendrocyte maturation. Cell 115(2), 163–175. [DOI] [PubMed] [Google Scholar]
- Huang J, Huang A, Poplawski A, DiPino F Jr., Traugh JA & Ling J (2020). PAK2 activated by Cdc42 and caspase 3 mediates different cellular responses to oxidative stress-induced apoptosis. Biochimica et Biophysica Acta – Molecular Cell Research 1867(4), 118645. [DOI] [PubMed] [Google Scholar]
- Huang W, Zhou Z, Asrar S, Henkelman M, Xie W & Jia Z (2011). p21-Activated kinases 1 and 3 control brain size through coordinating neuronal complexity and synaptic properties. Molecular and Cellular Biology 31(3), 388–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huenniger K, Kramer A, Soom M, Chang I, Kohler M, Depping R, Kehlenbach RH & Kaether C (2010). Notch1 signaling is mediated by importins alpha 3, 4, and 7. Cellular and Molecular Life Sciences 67(18), 3187–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain R & Macklin WB (2017). Integrin-linked kinase (ILK) deletion disrupts oligodendrocyte development by altering cell cycle. Journal of Neuroscience 37(2), 397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida H, Li K, Yi M & Lemon SM (2007). p21-activated kinase 1 is activated through the mammalian target of rapamycin/p70 S6 kinase pathway and regulates the replication of hepatitis C virus in human hepatoma cells. Journal of Biological Chemistry 282(16), 11836–11848. [DOI] [PubMed] [Google Scholar]
- Ishii A, Furusho M & Bansal R (2013). Sustained activation of ERK1/2 MAPK in oligodendrocytes and schwann cells enhances myelin growth and stimulates oligodendrocyte progenitor expansion. Journal of Neuroscience 33(1), 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Furusho M, Macklin W & Bansal R (2019). Independent and cooperative roles of the Mek/ERK1/2-MAPK and PI3K/Akt/mTOR pathways during developmental myelination and in adulthood. Glia 67(7), 1277–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Fyffe-Maricich SL, Furusho M, Miller RH & Bansal R (2012). ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. Journal of Neuroscience 32(26), 8855–8864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobi R, McCarthy CC, Koeppel MA & Stringer DK (2003). Caspase-activated PAK-2 is regulated by subcellular targeting and proteasomal degradation. Journal of Biological Chemistry 278(40), 38675–38685. [DOI] [PubMed] [Google Scholar]
- Jana A & Pahan K (2010). Sphingolipids in multiple sclerosis. NeuroMolecular Medicine 12(4), 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpov AS, Amiri P, Bellamacina C, Bellance MH, Breitenstein W, Daniel D, Denay R, Fabbro D, Fernandez C, Galuba I, Guerro-Lagasse S, Gutmann S, Hinh L, Jahnke W, Klopp J, et al. (2015). Optimization of a Dibenzodiazepine hit to a potent and selective allosteric PAK1 inhibitor. ACS Medicinal Chemistry Letters 6(7), 776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazdoba TM, Leach PT, Silverman JL & Crawley JN (2014). Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable & Rare Diseases Research 3(4), 118–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y, Wang L, Pyle WG, de Tombe PP & Solaro RJ (2004). Intracellular localization and functional effects of P21-activated kinase-1 (Pak1) in cardiac myocytes. Circulation Research 94(2), 194–200. [DOI] [PubMed] [Google Scholar]
- Kelly ML & Chernoff J (2012). Mouse models of PAK function. Cellular Logistics 2(2), 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernohan KD, McBride A, Hartley T, Rojas SK, Care4Rare Canada, C., Dyment DA, Boycott KM & Dyack S (2019). p21 protein-activated kinase 1 is associated with severe regressive autism, and epilepsy. Clinical Genetics 96(5), 449–455. [DOI] [PubMed] [Google Scholar]
- Kichina JV, Goc A, Al-Husein B, Somanath PR & Kandel ES (2010). PAK1 as a therapeutic target. Expert Opinion on Therapeutic Targets 14(7), 703–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CC, Gardiner EM, Zenke FT, Bohl BP, Newton AC, Hemmings BA & Bokoch GM (2000). p21-activated kinase (PAK1) is phosphorylated and activated by 3-phosphoinositide-dependent kinase-1 (PDK1). Journal of Biological Chemistry 275(52), 41201–41209. [DOI] [PubMed] [Google Scholar]
- Kippert A, Fitzner D, Helenius J & Simons M (2009). Actomyosin contractility controls cell surface area of oligodendrocytes. BMC Cell Biology 10, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis P, Rousseau V, Thevenot E, Combeau G & Barnier JV (2008). The four mammalian splice variants encoded by the p21-activated kinase 3 gene have different biological properties. Journal of Neurochemistry 106(3), 1184–1197. [DOI] [PubMed] [Google Scholar]
- Lang J, Maeda Y, Bannerman P, Xu J, Horiuchi M, Pleasure D & Guo F (2013). Adenomatous polyposis coli regulates oligodendroglial development. The Journal of Neuroscience 33(7), 3113–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BY & Hur EM (2020). A role of microtubules in oligodendrocyte differentiation. International Journal of Molecular Sciences 21(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Chaboub LS, Zhu W, Zollinger D, Rasband MN, Fancy SP & Deneen B (2015a). Daam2-PIP5K is a regulatory pathway for Wnt signaling and therapeutic target for remyelination in the CNS. Neuron 85(6), 1227–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Laug D, Zhu W, Patel JM, Ung K, Arenkiel BR, Fancy SP, Mohila C & Deneen B (2015b). Apcdd1 stimulates oligodendrocyte differentiation after white matter injury. Glia 63(10), 1840–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M, Lu W, Meng W, Parrini MC, Eck MJ, Mayer BJ & Harrison SC (2000). Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell 102(3), 387–397. [DOI] [PubMed] [Google Scholar]
- Leisner TM, Liu M, Jaffer ZM, Chernoff J & Parise LV (2005). Essential role of CIB1 in regulating PAK1 activation and cell migration. Journal of Cell Biology 170(3), 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Adam L, Vadlamudi RK, Zhou H, Sen S, Chernoff J, Mandal M & Kumar R (2002). p21-activated kinase 1 interacts with and phosphorylates histone H3 in breast cancer cells. EMBO Reports 3(8), 767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Draghi NA & Resh MD (2004). Signaling from integrins to Fyn to Rho family GTPases regulates morphologic differentiation of oligodendrocytes. Journal of Neuroscience 24(32), 7140–7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightcap CM, Kari G, Arias-Romero LE, Chernoff J, Rodeck U & Williams JC (2009). Interaction with LC8 is required for Pak1 nuclear import and is indispensable for zebrafish development. PLoS One 4(6), e6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightcap CM, Sun S, Lear JD, Rodeck U, Polenova T & Williams JC (2008). Biochemical and structural characterization of the Pak1-LC8 interaction. Journal of Biological Chemistry 283(40), 27314–27324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Katz S, Gupta R & Mayer BJ (1997). Activation of Pak by membrane localization mediated by an SH3 domain from the adaptor protein Nck. Current Biology 7(2), 85–94. [DOI] [PubMed] [Google Scholar]
- Lu W & Mayer BJ (1999). Mechanism of activation of Pak1 kinase by membrane localization. Oncogene 18(3), 797–806. [DOI] [PubMed] [Google Scholar]
- Maglorius Renkilaraj MRL, Baudouin L, Wells CM, Doulazmi M, Wehrle R, Cannaya V, Bachelin C, Barnier JV, Jia Z, Nait Oumesmar B, Dusart I & Bouslama-Oueghlani L (2017). The intellectual disability protein PAK3 regulates oligodendrocyte precursor cell differentiation. Neurobiology of Disease 98, 137–148. [DOI] [PubMed] [Google Scholar]
- Manser E, Loo TH, Koh CG, Zhao ZS, Chen XQ, Tan L, Tan I, Leung T & Lim L (1998). PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Molecular Cell 1(2), 183–192. [DOI] [PubMed] [Google Scholar]
- Mao K, Kobayashi S, Jaffer ZM, Huang Y, Volden P, Chernoff J & Liang Q (2008). Regulation of Akt/PKB activity by P21-activated kinase in cardiomyocytes. Journal of Molecular and Cellular Cardiology 44(2), 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques S, Zeisel A, Codeluppi S, van Bruggen D, Mendanha Falcao A, Xiao L, Li H, Haring M, Hochgerner H, Romanov RA, Gyllborg D, Munoz Manchado A, La Manno G, Lonnerberg P, et al. (2016). Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 352(6291), 1326–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei F, Lehmann-Horn K, Shen YA, Rankin KA, Stebbins KJ, Lorrain DS, Pekarek K, S AS, Xiao L, Teuscher C, von Budingen HC, Wess J, Lawrence JJ, Green AJ, Fancy SP, Zamvil SS, et al. (2016). Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard RE & Mattingly RR (2004). Gbetagamma subunits stimulate p21-activated kinase 1 (PAK1) through activation of PI3-kinase and Akt but act independently of Rac1/Cdc42. FEBS Letters 556(1–3), 187–192. [DOI] [PubMed] [Google Scholar]
- Meng J, Meng Y, Hanna A, Janus C & Jia Z (2005). Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. Journal of Neuroscience 25(28), 6641–6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino JG (2019). White matter hyperintensities on magnetic resonance imaging: what is a clinician to do? Mayo Clinic Proceedings 94(3), 380–382. [DOI] [PubMed] [Google Scholar]
- Miglietta A, Bozzo F, Gabriel L, Bocca C & Canuto RA (2006). Extracellular signal-regulated kinase 1/2 and protein phosphatase 2A are involved in the antiproliferative activity of conjugated linoleic acid in MCF-7 cells. British Journal of Nutrition 96(1), 22–27. [DOI] [PubMed] [Google Scholar]
- Mo JS, Kim MY, Han SO, Kim IS, Ann EJ, Lee KS, Seo MS, Kim JY, Lee SC, Park JW, Choi EJ, Seong JY, Joe CO, Faessler R & Park HS (2007). Integrin-linked kinase controls Notch1 signaling by down-regulation of protein stability through Fbw7 ubiquitin ligase. Molecular and Cellular Biology 27(15), 5565–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott HR, Nietlispach D, Evetts KA & Owen D (2005). Structural analysis of the SH3 domain of beta-PIX and its interaction with alpha-p21 activated kinase (PAK). Biochemistry 44(33), 10977–10983. [DOI] [PubMed] [Google Scholar]
- Narayanan SP, Flores AI, Wang F & Macklin WB (2009). Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. Journal of Neuroscience 29(21), 6860–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naska S, Park KJ, Hannigan GE, Dedhar S, Miller FD & Kaplan DR (2006). An essential role for the integrin-linked kinase-glycogen synthase kinase-3 beta pathway during dendrite initiation and growth. Journal of Neuroscience 26(51), 13344–13356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawaz S, Sanchez P, Schmitt S, Snaidero N, Mitkovski M, Velte C, Bruckner BR, Alexopoulos I, Czopka T, Jung SY, Rhee JS, Janshoff A, Witke W, Schaap IAT, Lyons DA, et al. (2015). Actin filament turnover drives leading edge growth during myelin sheath formation in the central nervous system. Developmental Cell 34(2), 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndubaku CO, Crawford JJ, Drobnick J, Aliagas I, Campbell D, Dong P, Dornan LM, Duron S, Epler J, Gazzard L, Heise CE, Hoeflich KP, Jakubiak D, La H, Lee W, et al. (2015). Design of selective PAK1 inhibitor G-5555: improving properties by employing an unorthodox low-pK a polar moiety. ACS Medicinal Chemistry Letters 6(12), 1241–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbern JM, Li X, Shoemaker SE, Zhou J, Zhong J, Wu Y, Bonder D, Hollenback S, Coppola G, Geschwind DH, Landreth GE & Snider WD (2011). Specific functions for ERK/MAPK signaling during PNS development. Neuron 69(1), 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic M (2008). The Pak1 kinase: an important regulator of neuronal morphology and function in the developing forebrain. Molecular Neurobiology 37(2–3), 187–202. [DOI] [PubMed] [Google Scholar]
- Nikolic M, Chou MM, Lu W, Mayer BJ & Tsai LH (1998). The p35/Cdk5 kinase is a neuron-specific Rac effector that inhibits Pak1 activity. Nature 395(6698), 194–198. [DOI] [PubMed] [Google Scholar]
- Oladimeji P & Diakonova M (2016). PAK1 translocates into nucleus in response to prolactin but not to estrogen. Biochemical and Biophysical Research Communications 473(1), 206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oladimeji P, Skerl R, Rusch C & Diakonova M (2016). Synergistic activation of ERalpha by estrogen and prolactin in breast cancer cells requires tyrosyl phosphorylation of PAK1. Cancer Research 76(9), 2600–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald F, Kostezka U, Astrahantseff K, Bourteele S, Dillinger K, Zechner U, Ludwig L, Wilda M, Hameister H, Knochel W, Liptay S & Schmid RM (2002). SHARP is a novel component of the Notch/RBP-Jkappa signalling pathway. EMBO Journal 21(20), 5417–5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacey LK, Xuan IC, Guan S, Sussman D, Henkelman RM, Chen Y, Thomsen C & Hampson DR (2013). Delayed myelination in a mouse model of fragile X syndrome. Human Molecular Genetics 22(19), 3920–3930. [DOI] [PubMed] [Google Scholar]
- Pan X, Chang X, Leung C, Zhou Z, Cao F, Xie W & Jia Z (2015). PAK1 regulates cortical development via promoting neuronal migration and progenitor cell proliferation. Molecular Brain 8, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HC & Appel B (2003). Delta-Notch signaling regulates oligodendrocyte specification. Development 130(16), 3747–3755. [DOI] [PubMed] [Google Scholar]
- Park MH, Kim DJ, You ST, Lee CS, Kim HK, Park SM, Shin EY & Kim EG (2012). Phosphorylation of beta-catenin at serine 663 regulates its transcriptional activity. Biochemical and Biophysical Research Communications 419(3), 543–549. [DOI] [PubMed] [Google Scholar]
- Parrini MC, Lei M, Harrison SC & Mayer BJ (2002). Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Molecular Cell 9(1), 73–83. [DOI] [PubMed] [Google Scholar]
- Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Bruck W, Lucchinetti C & Lassmann H (2006). Remyelination is extensive in a subset of multiple sclerosis patients. Brain 129(Pt 12), 3165–3172. [DOI] [PubMed] [Google Scholar]
- Peippo M, Koivisto AM, Sarkamo T, Sipponen M, von Koskull H, Ylisaukko-oja T, Rehnstrom K, Froyen G, Ignatius J & Jarvela I (2007). PAK3 related mental disability: further characterization of the phenotype. American Journal of Medical Genetics A 143A(20), 2406–2416. [DOI] [PubMed] [Google Scholar]
- Podbielska M, Krotkiewski H & Hogan EL (2012). Signaling and regulatory functions of bioactive sphingolipids as therapeutic targets in multiple sclerosis. Neurochemical Research 37(6), 1154–1169. [DOI] [PubMed] [Google Scholar]
- Popko B (2003). Notch signaling: a rheostat regulating oligodendrocyte differentiation? Developmental Cell 5(5), 668–669. [DOI] [PubMed] [Google Scholar]
- Puto LA, Pestonjamasp K, King CC & Bokoch GM (2003). p21-activated kinase 1 (PAK1) interacts with the Grb2 adapter protein to couple to growth factor signaling. Journal of Biological Chemistry 278(11), 9388–9393. [DOI] [PubMed] [Google Scholar]
- Qian Y, Wu B, Lu Y, Zhou W, Wang S & Wang H (2020). Novel PAK3 gene missense variant associated with two Chinese siblings with intellectual disability: a case report. BMC Medical Genetics 21(1), 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radu M, Semenova G, Kosoff R & Chernoff J (2014). PAK signalling during the development and progression of cancer. Nature Reviews Cancer 14(1), 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid T, Banerjee M & Nikolic M (2001). Phosphorylation of Pak1 by the p35/Cdk5 kinase affects neuronal morphology. Journal of Biological Chemistry 276(52), 49043–4952. [DOI] [PubMed] [Google Scholar]
- Rayala SK, Molli PR & Kumar R (2006a). Nuclear p21-activated kinase 1 in breast cancer packs off tamoxifen sensitivity. Cancer Research 66(12), 5985–5988. [DOI] [PubMed] [Google Scholar]
- Rayala SK, Talukder AH, Balasenthil S, Tharakan R, Barnes CJ, Wang RA, Aldaz CM, Khan S & Kumar R (2006b). P21-activated kinase 1 regulation of estrogen receptor-alpha activation involves serine 305 activation linked with serine 118 phosphorylation. Cancer Research 66(3), 1694–1701. [DOI] [PubMed] [Google Scholar]
- Roll-Mecak A (2019). A Microtubule-Myelination Connection. Cell 179(1), 54–56. [DOI] [PubMed] [Google Scholar]
- Rousseau V, Goupille O, Morin N & Barnier JV (2003). A new constitutively active brain PAK3 isoform displays modified specificities toward Rac and Cdc42 GTPases. Journal of Biological Chemistry 278(6), 3912–3920. [DOI] [PubMed] [Google Scholar]
- Rudel T & Bokoch GM (1997). Membrane and morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science 276(5318), 1571–1574. [DOI] [PubMed] [Google Scholar]
- Rudolph J, Murray LJ, Ndubaku CO, O’Brien T, Blackwood E, Wang W, Aliagas I, Gazzard L, Crawford JJ, Drobnick J, Lee W, Zhao X, Hoeflich KP, Favor DA, Dong P, et al. (2016). Chemically diverse group I p21-activated kinase (PAK) inhibitors impart acute cardiovascular toxicity with a narrow therapeutic window. Journal of Medicinal Chemistry 59(11), 5520–5541. [DOI] [PubMed] [Google Scholar]
- Sanders LC, Matsumura F, Bokoch GM & de Lanerolle P (1999). Inhibition of myosin light chain kinase by p21-activated kinase. Science 283(5410), 2083–2085. [DOI] [PubMed] [Google Scholar]
- Say E, Tay HG, Zhao ZS, Baskaran Y, Li R, Lim L & Manser E (2010). A functional requirement for PAK1 binding to the KH(2) domain of the fragile X protein-related FXR1. Molecular Cell 38(2), 236–249. [DOI] [PubMed] [Google Scholar]
- Semenova G & Chernoff J (2017). Targeting PAK1. Biochem Soc Trans 45(1), 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin YJ, Kim YB & Kim JH (2013). Protein kinase CK2 phosphorylates and activates p21-activated kinase 1. Molecular Biology of the Cell 24(18), 2990–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha Y, Schafer EJ, Boehm JS, Thomas SR, He F, Du J, Wang S, Barretina J, Weir BA, Zhao JJ, Polyak K, Golub TR, Beroukhim R & Hahn WC (2012). PAK1 is a breast cancer oncogene that coordinately activates MAPK and MET signaling. Oncogene 31(29), 3397–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RR, Song C, Yang Z & Kumar R (2005). Nuclear localization and chromatin targets of p21-activated kinase 1. Journal of Biological Chemistry 280(18), 18130–18137. [DOI] [PubMed] [Google Scholar]
- Staser K, Shew MA, Michels EG, Mwanthi MM, Yang FC, Clapp DW & Park SJ (2013). A Pak1-PP2A-ERM signaling axis mediates F-actin rearrangement and degranulation in mast cells. Experimental Hematology 41(1), 56–66 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenweg ME, Vanderver A, Blaser S, Bizzi A, de Koning TJ, Mancini GM, van Wieringen WN, Barkhof F, Wolf NI & van der Knaap MS (2010). Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 133(10), 2971–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stidworthy MF, Genoud S, Li WW, Leone DP, Mantei N, Suter U & Franklin RJ (2004). Notch1 and Jagged1 are expressed after CNS demyelination, but are not a major rate-determining factor during remyelination. Brain 127(Pt 9), 1928–1941. [DOI] [PubMed] [Google Scholar]
- Sun J, Khalid S, Rozakis-Adcock M, Fantus IG & Jin T (2009). P-21-activated protein kinase-1 functions as a linker between insulin and Wnt signaling pathways in the intestine. Oncogene 28(35), 3132–3144. [DOI] [PubMed] [Google Scholar]
- Suo N, Guo YE, He B, Gu H & Xie X (2019). Inhibition of MAPK/ERK pathway promotes oligodendrocytes generation and recovery of demyelinating diseases. Glia 67(7), 1320–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglieri DM, Monasky MM, Knezevic I, Sheehan KA, Lei M, Wang X, Chernoff J, Wolska BM, Ke Y & Solaro RJ (2011). Ablation of p21-activated kinase-1 in mice promotes isoproterenol-induced cardiac hypertrophy in association with activation of Erk1/2 and inhibition of protein phosphatase 2A. Journal of Molecular and Cellular Cardiology 51(6), 988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Zhou H, Chen A, Pittman RN & Field J (2000). The Akt proto-oncogene links Ras to Pak and cell survival signals. Journal of Biological Chemistry 275(13), 9106–9109. [DOI] [PubMed] [Google Scholar]
- Thiel DA, Reeder MK, Pfaff A, Coleman TR, Sells MA & Chernoff J (2002). Cell cycle-regulated phosphorylation of p21-activated kinase 1. Current Biology 12(14), 1227–1232. [DOI] [PubMed] [Google Scholar]
- Thullberg M, Gad A, Beeser A, Chernoff J & Stromblad S (2007). The kinase-inhibitory domain of p21-activated kinase 1 (PAK1) inhibits cell cycle progression independent of PAK1 kinase activity. Oncogene 26(12), 1820–1828. [DOI] [PubMed] [Google Scholar]
- Thurnherr T, Benninger Y, Wu X, Chrostek A, Krause SM, Nave KA, Franklin RJ, Brakebusch C, Suter U & Relvas JB (2006). Cdc42 and Rac1 signaling are both required for and act synergistically in the correct formation of myelin sheaths in the CNS. Journal of Neuroscience 26(40), 10110–10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadlamudi RK, Bagheri-Yarmand R, Yang Z, Balasenthil S, Nguyen D, Sahin AA, den Hollander P & Kumar R (2004a). Dynein light chain 1, a p21-activated kinase 1-interacting substrate, promotes cancerous phenotypes. Cancer Cell 5(6), 575–585. [DOI] [PubMed] [Google Scholar]
- Vadlamudi RK, Barnes CJ, Rayala S, Li F, Balasenthil S, Marcus S, Goodson HV, Sahin AA & Kumar R (2005a). p21-activated kinase 1 regulates microtubule dynamics by phosphorylating tubulin cofactor B. Molecular and Cellular Biology 25(9), 3726–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadlamudi RK, Li F, Barnes CJ, Bagheri-Yarmand R & Kumar R (2004b). p41-Arc subunit of human Arp2/3 complex is a p21-activated kinase-1-interacting substrate. EMBO Reports 5(2), 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadlamudi RK, Manavathi B, Singh RR, Nguyen D, Li F & Kumar R (2005b). An essential role of Pak1 phosphorylation of SHARP in Notch signaling. Oncogene 24(28), 4591–4596. [DOI] [PubMed] [Google Scholar]
- Van Kanegan MJ, Adams DG, Wadzinski BE & Strack S (2005). Distinct protein phosphatase 2A heterotrimers modulate growth factor signaling to extracellular signal-regulated kinases and Akt. Journal of Biological Chemistry 280(43), 36029–36036. [DOI] [PubMed] [Google Scholar]
- Viaud J & Peterson JR (2009). An allosteric kinase inhibitor binds the p21-activated kinase autoregulatory domain covalently. Molecular Cancer Therapies 8(9), 2559–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl SE, McLane LE, Bercury KK, Macklin WB & Wood TL (2014). Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. Journal of Neuroscience 34(13), 4453–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Rusielewicz T, Tewari A, Leitman EM, Einheber S & Melendez-Vasquez CV (2012). Myosin II is a negative regulator of oligodendrocyte morphological differentiation. Journal of Neuroscience Research 90(8), 1547–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J & Wang Z (2008). Negative regulation of caspase 3-cleaved PAK2 activity by protein phosphatase 1. Science in China Series C - Life Sciences 51(1), 1–11. [DOI] [PubMed] [Google Scholar]
- Wang S, Sdrulla AD, diSibio G, Bush G, Nofziger D, Hicks C, Weinmaster G & Barres BA (1998). Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 21(1), 63–75. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zeng C, Li J, Zhou Z, Ju X, Xia S, Li Y, Liu A, Teng H, Zhang K, Shi L, Bi C, Xie W, He X, Jia Z, et al. (2018). PAK2 Haploinsufficiency results in synaptic cytoskeleton impairment and autism-related behavior. Cell Reports 24(8), 2029–2041. [DOI] [PubMed] [Google Scholar]
- Wang Z, Fu M, Wang L, Liu J, Li Y, Brakebusch C & Mei Q (2013). p21-activated kinase 1 (PAK1) can promote ERK activation in a kinase-independent manner. Journal of Biological Chemistry 288(27), 20093–20099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner HL (2008). A shift from adaptive to innate immunity: a potential mechanism of disease progression in multiple sclerosis. Journal of Neurology 255 Suppl 1, 3–11. [DOI] [PubMed] [Google Scholar]
- Weisz Hubsman M, Volinsky N, Manser E, Yablonski D & Aronheim A (2007). Autophosphorylation-dependent degradation of Pak1, triggered by the Rho-family GTPase, Chp. Biochemical Journal 404(3), 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willatt L, Cox J, Barber J, Cabanas ED, Collins A, Donnai D, FitzPatrick DR, Maher E, Martin H, Parnau J, Pindar L, Ramsay J, Shaw-Smith C, Sistermans EA, Tettenborn M, et al. (2005). 3q29 microdeletion syndrome: clinical and molecular characterization of a new syndrome. American Journal of Human Genetics 77(1), 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann T, Bokoch GM & Waterman-Storer CM (2004). Regulation of microtubule destabilizing activity of Op18/stathmin downstream of Rac1. Journal of Biological Chemistry 279(7), 6196–6203. [DOI] [PubMed] [Google Scholar]
- Xia S, Zhou Z & Jia Z (2018). PAK1 regulates inhibitory synaptic function via a novel mechanism mediated by endocannabinoids. Small GTPases 9(4), 322–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D, Li C, Rajoka MSR, He Z, Huang J, Wang J & Zhang J (2020). P21-activated kinase 1: emerging biological functions and potential therapeutic targets in cancer. Theranostics 10(21), 9741–9766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye DZ & Field J (2012). PAK signaling in cancer. Cellular Logistics 2(2), 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, Hu T, Taketo MM, van Es JH, Clevers H, Hsieh J, Bassel-Duby R, Olson EN & Lu QR (2009). HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nature Neuroscience 12(7), 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Mo JS, Ann EJ, Ahn JS, Jo EH, Lee HJ, Hong SH, Kim MY, Kim EG, Lee K & Park HS (2016). NOTCH1 intracellular domain negatively regulates PAK1 signaling pathway through direct interaction. Biochimica et Biophysica Acta 1863(2), 179–188. [DOI] [PubMed] [Google Scholar]
- Zang M, Hayne C & Luo Z (2002). Interaction between active Pak1 and Raf-1 is necessary for phosphorylation and activation of Raf-1. Journal of Biological Chemistry 277(6), 4395–4405. [DOI] [PubMed] [Google Scholar]
- Zhang S, Zhu X, Gui X, Croteau C, Song L, Xu J, Wang A, Bannerman P & Guo F (2018). Sox2 is essential for oligodendroglial proliferation and differentiation during postnatal brain myelination and CNS remyelination. Journal of Neuroscience 38(7), 1802–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Argaw AT, Gurfein BT, Zameer A, Snyder BJ, Ge C, Lu QR, Rowitch DH, Raine CS, Brosnan CF & John GR (2009). Notch1 signaling plays a role in regulating precursor differentiation during CNS remyelination. Proceedings of the National Academy of Sciences USA 106(45), 19162–19167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of Neuroscience 34(36), 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZS, Manser E, Chen XQ, Chong C, Leung T & Lim L (1998). A conserved negative regulatory region in alphaPAK: inhibition of PAK kinases reveals their morphological roles downstream of Cdc42 and Rac1. Molecular and Cellular Biology 18(4), 2153–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZS, Manser E & Lim L (2000). Interaction between PAK and nck: a template for Nck targets and role of PAK autophosphorylation. Molecular and Cellular Biology 20(11), 3906–3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong JL, Banerjee MD & Nikolic M (2003). Pak1 and its T212 phosphorylated form accumulate in neurones and epithelial cells of the developing rodent. Developmental Dynamics 228(1), 121–127. [DOI] [PubMed] [Google Scholar]
- Zhou GL, Zhuo Y, King CC, Fryer BH, Bokoch GM & Field J (2003). Akt phosphorylation of serine 21 on Pak1 modulates Nck binding and cell migration. Molecular and Cellular Biology 23(22), 8058–8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Wang Y, Huang B, Liang J, Ding Y, Xu A & Wu W (2012). A Rac1/PAK1 cascade controls beta-catenin activation in colon cancer cells. Oncogene 31(8), 1001–1012. [DOI] [PubMed] [Google Scholar]
- Zuchero JB, Fu MM, Sloan SA, Ibrahim A, Olson A, Zaremba A, Dugas JC, Wienbar S, Caprariello AV, Kantor C, Leonoudakis D, Lariosa-Willingham K, Kronenberg G, Gertz K, Soderling SH, et al. (2015). CNS myelin wrapping is driven by actin disassembly. Developmental Cell 34(2), 152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]