

Abstract
Mitochondrial defects result in dysregulation of metabolomics and energy homeostasis that are detected in upper motor neurons (UMNs) with TDP-43 pathology, a pathology that is predominantly present in both familial and sporadic cases of amyotrophic lateral sclerosis (ALS). While same mitochondrial problems are present in the UMNs of ALS patients with TDP-43 pathology and UMNs of TDP-43 mouse models, and since pathologies are shared at a cellular level, regardless of species, we first analyzed the metabolite profile of both healthy and diseased motor cortex to investigate whether metabolomic changes occur with respect to TDP-43 pathology. High-performance liquid chromatography, high-resolution mass spectrometry and tandem mass spectrometry (HPLC–MS/MS) for metabolite profiling began to suggest that reduced levels of NAD+ is one of the underlying causes of metabolomic problems. Since nicotinamide mononucleotide (NMN) was reported to restore NAD+ levels, we next investigated whether NMN treatment would improve the health of diseased corticospinal motor neurons (CSMN, a.k.a. UMN in mice). prpTDP-43A315T-UeGFP mice, the CSMN reporter line with TDP-43 pathology, allowed cell-type specific responses of CSMN to NMN treatment to be assessed in vitro. Our results show that metabolomic defects occur early in ALS motor cortex and establishing NAD+ balance could offer therapeutic benefit to UMNs with TDP-43 pathology.
Subject terms: Cellular neuroscience, Amyotrophic lateral sclerosis
Introduction
Mitochondria are best studied for their ability to generate energy, but they are also essential for lipid homeostasis, ensuring proper Ca+2 storage inside the cells, and maintaining metabolomic balance1–5. With their multiple important functions, they serve as one of the key organelles for cellular health. Therefore, it is no surprise that mitochondrial dysfunction emerges as one of the common causes of neurodegeneration in many different diseases, and ALS is no exception.
TDP-43 pathology is one of the common proteinopathies observed in a large spectrum of ALS patients, including familial, sporadic and ALS with frontotemporal lobar dementia (ALS/FTLD)6–9. Even though some patients have mutations in TARDBP, the gene that codes for TDP-43 protein, not all TDP-43 pathologies are associated with the mutation10, and not all protein accumulations occur in the cytoplasm, some are detected in the nucleus—11,12. In fact, most of the patients with TDP-43 pathology do not have mutations in their TARDP gene, and yet have protein accumulations containing phosphorylated TDP-4313, and mostly ubiquitinated protein aggregates10,14,15. One of the interesting findings related to the neurons with TDP-43 pathology is the mitochondrial defects observed at a cellular level16–18. Such mitochondrial defects are also present in the upper motor neurons (UMNs) that are diseased in ALS. Regardless of species, the UMNs of a broad spectrum of ALS patients and corticospinal motor neurons (CSMN; a.k.a UMN in mice) of well characterized mouse models of TDP-43 pathology, both display prominent mitochondrial defects in a cell-type specific manner16.
There is an immense effort to reveal the underlying causes of TDP-43 pathology and the downstream cellular events that are perturbed in diseased neurons. Since mitochondrial defects have been broadly observed in many different diseases in which TDP-43 pathology is commonly detected, and because mitochondria are crucially important for maintaining metabolomic homeostasis, one of the key questions that remains unanswered is: “What are the metabolomic changes that occur with respect to TDP-43 pathology?” We investigated the metabolomic perturbations that occur in the motor cortex of prp-TDP-43A315T mice, a mouse model which recapitulates many aspects of TDP-43 pathology observed in ALS and ALS/FTLD patients19. An early symptomatic stage of the disease was selected to investigate whether metabolomic perturbations begin to occur early in the motor cortex with TDP-43 pathology.
Our findings began to reveal reduced ATP production as well as perturbations in the balance of key metabolites, such as NAD+, GSH (glutathione), and PEP (phosphoenol pyruvate). We thus tested the hypothesis that increasing NAD+ levels would help restore balance and improve the health of UMNs diseased due to TDP-43 pathology. NAD+ plays a crucial role in several physiological conditions such as neurological disorders, diabetes, aging, oxidative stress and neurodegenerative diseases20–26. Since NMN (nicotinamide mononucleotide) was previously reported to increase the levels of NAD+ by acting as the precursor for NAD+27, we investigated whether NMN treatment would have therapeutic impact on diseased UMNs.
In an effort to bring cellular clarity to our analyses, and to investigate UMNs direct response to treatment, we took advantage of the prpTDP-43A315T-UeGFP mice, a CSMN reporter line in which diseased UMNs in mice that have TDP-43 pathology retain their fluorescence both in vivo and in vitro. Our results not only reveal early metabolomic problems in the motor cortex, but also suggest that maintaining NAD+ balance could offer a therapeutic treatment strategy for diseased UMNs with TDP-43 pathology.
Results
Metabolite profiling in the motor cortex with TDP-43 pathology reveals energy deficiency and oxidative stress
We previously identified major mitochondrial problems, especially within the corticospinal motor neurons (CSMN) of prpTDP-43A315T mice16 (Fig. 1a). Mitochondrial problems were present as early as P15 (postnatal day 15)17, and they were also observed in the upper motor neurons (UMNs) of a broad spectrum of ALS patients with TDP-43 pathology16. Since mitochondrial problems begin to occur early and because they are shared among species, we first investigated potential changes in metabolite dynamics, especially in the motor cortex of prpTDP-43A315T mice, which closely recapitulates ALS pathology in patients16,17,19. Primary motor cortices of 90 days old (P90) prpTDP-43A315T and healthy wildtype (WT) control mice were dissected out and metabolites were isolated by methanol extraction method followed by high performance mass spectrometry profiling of metabolites (Fig. 1b). We chose P90 as the time of investigation, because this is a symptomatic stage with profound CSMN loss, and it coincides to the disease stage, when patients show symptoms and TDP-43 pathology begins to occur. Analyses of metabolites resulted in the identification of distinct set of metabolites (Supplementary Table 1), some of which are upregulated (Fig. 1c), and some downregulated (Fig. 1d) with respect to healthy WT control motor cortex.
Figure 1.

Metabolite profiling of motor cortex revealed dysregulated metabolite due to TDP-43 pathology. (a) representative electron microscopy images of mitochondria in WT healthy CSMN and in CSMN that are diseased due to TDP-43 pathology (b) schematic representation of workflow of study design. (c) list of selected upregulated metabolites. (d) List of selected downregulated metabolites. Scale bar = 200 nm.
Enrichment analysis was performed using Metaboanalysis software28. Heatmap of hierarchical clustering analysis of top 50 highly enriched metabolites revealed differential presence of key metabolites (Fig. 2a). Pyruvate metabolism, tyrosine and nicotinamide metabolism, glycine and serine metabolism, citric acid cycle, glutathione metabolism, and carnitine synthesis were among important metabolite pathways that were different between healthy and diseased motor cortex (Fig. 2b,c). Glutamate is precursor for glutathione biosynthesis. Purine and glutamate metabolism were the top hits, suggesting perhaps ATP and glutathione biology are impacted. (Fig. 2a–c).
Figure 2.

Enrichment analysis of metabolites. (a) Hierarchical clustering heat map of top 50 differentially present metabolites, (b) overview of enriched metabolites, and (c) analysis of enriched metabolites.
Detailed analyses revealed striking imbalance especially in the ratio of metabolites that play a key role in energy homeostasis and oxidative stress, such as ATP, NAD+ and phosphoenol pyruvate. The ATP/ADP ratio was significantly lower in prpTDP-43A315T motor cortex (WT: 0.17 ± 0.01, n = 4; prpTDP-43A315T: 0.12 ± 0.006, n = 4, p = 0.044, Fig. 3a), and so was NAD+/NADH ratio (WT: 42.94 ± 11.73, n = 4; prpTDP-43A315T: 4.94 ± 0.77, n = 4, p = 0.017, Fig. 3b).
Figure 3.
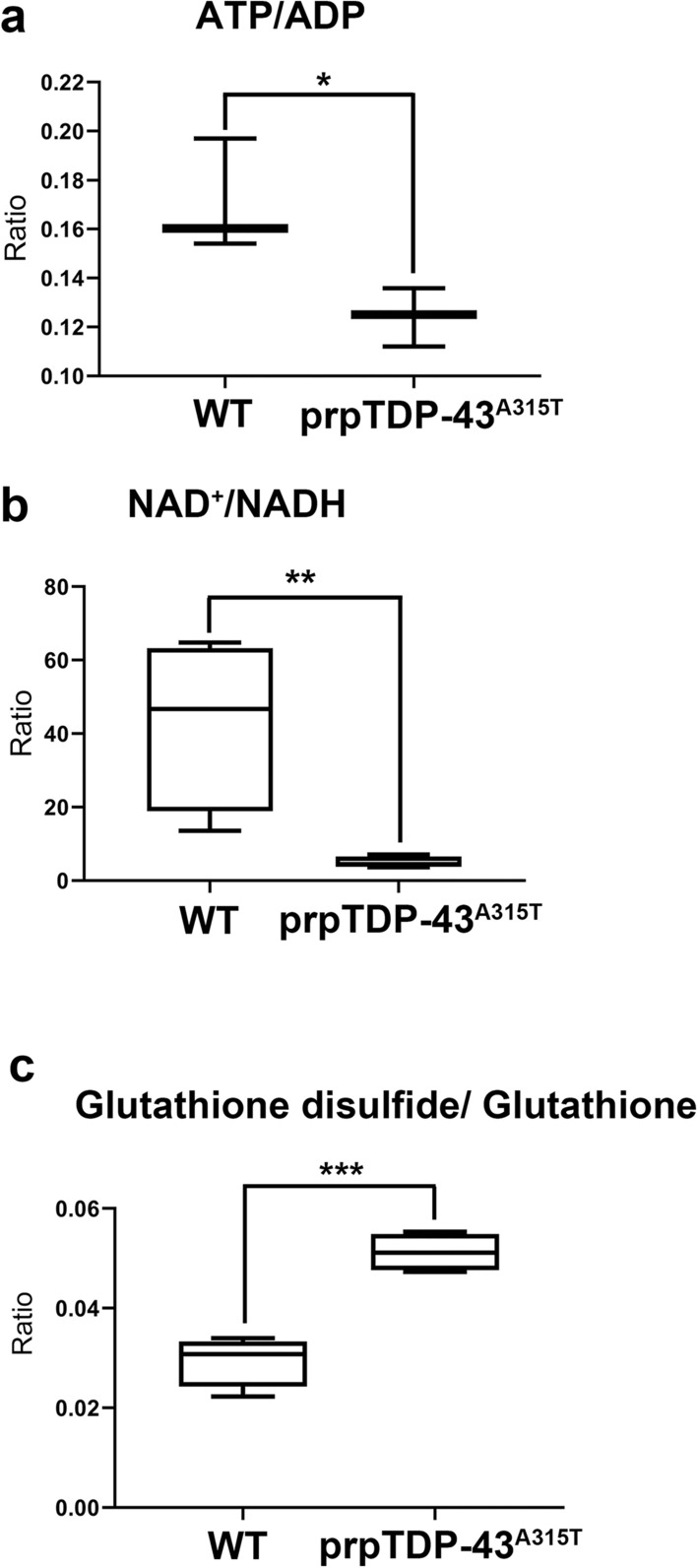
The ratio of key metabolites that are involved in energy homeostasis and oxidative stress are dysregulated in the motor cortex that is diseased due to TDP-43 pathology. (a) ratio of ATP/ADP; (b) ratio of NAD+/NADH and (c) ratio of GSSG/GSH.
The ratio of GSH and its oxidative derivative GSSG directly corelates with cellular oxidative stress29. GSH scavange reactive oxygen species (ROS) and converts into GSSH30. Thus, higher GSSG/GSH ratio indicates increased oxidative stress. We find that GSSG/GSH ratio is significantly higher in the motor cortex of prpTDP-43A315T mice even at P90 (WT: 0.02 ± 0.002, n = 4; prpTDP-43A315T: 0.051 ± 0.001, n = 4, p = 0.0005, Fig. 3c). Thus, these altered metabolomic changes and perturbed balance in the motor cortex of prpTDP-43A315T mice suggest a deficit in energy homeostasis and increased oxidative stress, events that are associated with problems with mitochondrial integrity and function.
UMN culture is an important tool to investigate UMN responses to treatment in vitro
UCHL-eGFP is a reporter mouse line for corticospinal motor neurons (CSMN, a.k.a the UMNs in mice), in which CSMN express eGFP, and CSMN identity of eGFP+ neurons in the layer 5 of the motor cortex is confirmed by retrograde labeling, molecular marker expression and electrophysiological properties31. Interestingly, CSMN also retain their eGFP expression in culture. Therefore, UCHL1-eGFP mice allow precise visualization and cellular assessment of UMNs in vitro31.
Crossbreeding of UCHL-eGFP with prpTDP-43A315T mouse model of ALS, generated prpTDP-43A315T-UeGFP mice (diseased) and WT-UeGFP mice (healthy control). When dissociated cortical cultures are established from these mice, CSMN are distinguished among many different cortical cells and neurons, as they retain their eGFP expression (Fig. 4a,b). Motor cortex is isolated from P3 pups of WT-eGFP and and prpTDP-43A315T-UeGFP mice, and dissociated cells are plated on glass coverslips to establish motor cortex cultures in vitro32. CSMN of WT-UeGFP mice express eGFP and were healthy with large pyramidal cell bodies, prominent apical dendrites and long axons (Fig. 4a). In contrast, CSMN of prpTDP-43A315T-UeGFP mice failed to extend long axons and lacked branching and arborization, even though they retained eGFP expression (Fig. 4b). There was significant reduction in diseased CSMN axon length (WT: 371.7 ± 10.96 μm, n = 3 mice; prpTDP-43A315T: 203.9 ± 13.9 μm, n = 3 mice, p = 0.0007, Fig. 4c). Percent distribution of axon length also confirmed that majority of CSMN in prpTDP-43A315T-UeGFP mice were shorter when compared to the CSMN of WT-UeGFP mice (Fig. 4d). Diseased CSMN had significantly reduced branching and arborization, as revealed by Sholl analyses (Fig. 4e). Since average CSMN axon length and the extent of neuronal branching and arborization enabled quantitative assessment of differences between healthy and diseased CSMN, these outcome measures were utilized to assess CSMN response to treatment with a cellular precision that was not possible before (Fig. 4c–e).
Figure 4.

CSMN that express mutated from of human TDP-43A315T extend shorter neurites in vitro. (a) representative images of CSMN from WT-UeGFP mouse; (b) representative images of CSMN from prpTDP-43A315T-UeGFP mouse model of ALS; (c) bar graph representation of average neurite length; (d) percent distribution of axon length; and (e) Sholl analysis of arborization of neurites. Scale bar = 20 µm.
Nicotinamide mononucleotide (NMN) improved CSMN health
CSMN display early vulnerability and progressive degeneration in ALS motor cortex and are particularly vulnerable to oxidative stress and energy deficiency16,17. Metabolomics analyses revealed NAD+ deficiency in the motor cortex. After having established a robust in vitro model for evaluating CSMN health, we next investigated whether increasing NAD+ levels could improve their health.
Since NMN was previously reported to be an NAD+ precursor27 and NMN led to the production of NAD+26, we tested the hypothesis that NMN treatment would help maintain the NAD+ balance in diseased CSMN, and thus improve their health. NMN was added to the culture medium (1 µM) of both healthy (WT-UeGFP) and diseased (prpTDP-43A315T-UeGFP ) motor cultures in vitro, and CSMN responses to treatment was quantitatively assessed by measuring changes in average axon length and the extent of branching and arborization (Fig. 5). CSMN of prpTDP-43A315T-UeGFP mice had short axons and did not display branching and arborization (Fig. 5a). However, upon NMN treatment (1 µM), there were significant improvements (Fig. 5b). For example, the average axon length increased in diseased CSMN (prpTDP-43A315T-UeGFP: 203.9 ± 13.9 μm, n = 3 mice, prpTDP-43A315T-UeGFP + NMN: 372.8 ± 15.8 μm, n = 3 mice; adjusted p value = 0.0002). In addition, the percent distribution of CSMN based on their axon length also revealed an overall and significant shift towards longer axons, such that about 80% of treated CSMN had axons longer than 300 μm, whereas about 90% of untreated CSMN had maximum axon lenth of 200 μm (Fig. 5f).
Figure 5.

Nicotinamide mononucleotide (NMN) improves the health of CSMN that become diseased due to TDP-43 pathology. (a) a representative image of CSMN from prpTDP-43A315T-UeGFP mouse; (b) a representative image of CSMN from prpTDP-43A315T-UeGFP mouse treated with 1 µM NMN; (c) a representative image of CSMN from WT-UeGFP mouse; (d) a representative image of CSMN from WT-UeGFP mouse treated with NMN. (e) Bar graph representation of average neurite length; (f) Percent distribution of axon length; and (g) Sholl analysis of arborization of neurites. Scale bar = 20 µm.
NIMN treatment was not toxic to WT-UeGFP CSMN (Fig. 5c,d). Regardless of their NMN treatment, they had similar axon lenth and arborization, and were comparable (WT-eGFP: 371.7 ± 10.96 μm, n = 3 mice; WT-eGFP + NMN: 350.3 ± 18.7 μm, n = 3 mice, adjusted p value = 0.753, Fig. 5e).
In addition to axon length, diseased CSMN treated with NMN also displayed improved branching and arborization especially within the 50–200 μm radius, as revealed by Sholl analyses (Fig. 5g, Supplementary Table 1).
NMN treatment improved mitochondrial stability
We used dissociated motor cortex cultures isolated from prpTDP-43A315T-UeGFP mice to investigate the basis of NMN-mediated improvement in CSMN health. To visualize the precise ultrastructural changes that occur at an organnelle level, we utilized correlative light electron microscopy (CLEM), on a total of 4 CSMN (untreated (n = 2) and NMN treated (n = 2)). CSMN were evident with their eGFP expression in vitro (Fig. 6a) and they were prepared for single cell EM analyses to reveal potential changes, especially at the site of mitochondrion. Healthy CSMN from WT-UeGFP mice were used as internal controls with similar treatment paradigms. Most of mitochondria in CSMN of prpTDP-43A315T-UeGFP mice had defects especially in their inner mitochondrial membrane; cristae structures were disintegrated and were not detectable (Fig. 6b; CSMN#1: 59%, n = 46 mitochondria; CSMN #2: 60%, n = 40 mitochondria). However, upon NMN treatment, there were significant improvements in the overall architecture of mitochondria and they had a more intact inner membrane with visible cristae structures. CSMN of prpTDP-43A315T-UeGFP mice had fewer diseased mitochondria (Fig. 6c; CSMN #3: 27%, n = 55 mitochondria; CSMN # 4: 45%, n = 49 mitochondria). The CLEM results were obtained from 4 independent CSMN isolated from four different mice, revealed the impact of NMN treatment on mitochondrial structure and integrity (Fig. 6b,c).
Figure 6.

Correlative light electron microscopy shows improvement of mitochondrial ultrastructure upon NMN treatment. (a) a representative image of motor cortex neurons cultured on a gridded glass bottom 35 mm dish; boxed area enlarged to highlight presence of GFP expressing CSMN; (b) representative electron micrograph of an individual CSMN from prpTDP-43A315T-UeGFP mouse showing mitochondrial ultrastructural defects, images of mitochondrion are enlarged to right; and (c) representative electron micrograph of an individual CSMN from prpTDP-43A315T-UeGFP treated with 1 µM NMN showing improved mitochondrial ultrastructure, images of mitochondrion are enlarged to right. Scale bar = 1 µm.
Discussion
Mitochondrial problems are mainly studied within the context of energy depletion. However, since mitochondria are also a major site for metabolomic regulation, problems or dysregulation of metabolites are now gaining attention, especially for motor neuron diseases, which display mitochondrial problems as one of the early cellular pathologies. We previously showed the presence of mitochondrial dysfunction as early as P15 in the motor cortex, but more precisely in the CSMN of TDP-43A315T mice17, one of the best characterized mouse models of TDP-43 pathology16,19,33,34. Numerous other studies, utilizing different animal models35–37 or cell lines38, all agreed on the presence of mitochondrial problems especially with respect to TDP-43 pathology.
Therefore, we investigated potential metabolomic dysregulation that may occur in the motor cortex of ALS with TDP-43 pathology. We have chosen prpTDP-43A315T mice for our analyses, because this mouse model of TDP-43 pathology was generated based on the A315T mutation detected in the TARDP gene of ALS patients, who had TDP-43 pathology in their cortex19, and because the mouse model strictly recapitulated many aspects of the disease, including progressive CSMN degeneration16. Numerous other studies have also chosen this mouse model as one of the best characterized and most informative mouse models for TDP-43 pathology39–43. More importantly, when detailed cellular analyses were performed with electron microscopy (EM), exact same cellular problems were detected both in the UMNs of ALS patients with TDP-43 pathology and the CSMN of this mouse model of TDP-43 pathology16.
TDP-43 pathology was originally defined with the accumulation of proteins that include phosphorylated TDP-43 (pTDP-43) in the cytoplasm of neurons that undergo degeneration10, and this pathology was broadly observed in a wide spectrum of ALS cases44. However, recent evidence also reveals TDP-43 accumulations in the nucleus, not only in the cytoplasm11,12. Especially in the case of some mutated forms of TDP-43, the protein aggregations are vastly ubiquitinated, but the presence of pTDP-43 is not dominant14,15,45. Regardless, patients with TDP-43 mutations have TDP-43 pathology, and the mouse model that have A315T mutation has TDP-43 accumulations mostly in the nucleus, have extensive ubiquitinated protein accumulations in the cytoplasm, and based on the antibody used, pTDP-43 are also detected in the cytoplasm19,46. Therefore, in line with the current definition of TDP-43 pathology, this mouse model has been considered one of the most reliable models for TDP-43 pathology, especially in the cortex16,19,39–43.
Metabolomic dysregulations in the plasma samples isolated from ALS patients were previously reported47–54. These studies showed significant alterations in ceramide levels as well as benzoate, creatine and fatty acid metabolisms and sphingomyelins pathways, with respect to ALS progression49, and these changes in metabolite levels correlated with patient’s ALS Functional Rating Scale (ALSFRS-R) scores55. Likewise, alteration in glucose and lipid metabolisms were reported to contribute to neuronal vulnerability in diseased motor neurons50. Components of lipid metabolism such as β-hydroxybutyric acid and medium-chain fatty acids were also found to be dysregulated56.
Metabolomic changes begin to reveal the some of the underlying causes of vulnerability in the motor cortex. In this study, we report that a select set of metabolites, such as phosphoenol pyruvate (PEP), glutathione (GSH), 2-hydroxyglutarate (2-HG), and NAD+, display significant differences between the healthy and diseased motor cortex with TDP-43 pathology at a symptomatic stage.
Oxidative stress is a prominent physiological feature of ALS, and mechanisms responsible for reducing oxidative stress are compromised22. In the motor cortex of ALS patients, glutathione (GSH) levels were reported to be reduced when compared to healthy controls57. Likewise, in hSOD1G93A mouse models of ALS, depletion of GSH resulted in degeneration of spinal motor neurons, dysfunctional mitochondria, and neuron loss58. GSH is critically important for scavenging reactive oxygen species (ROS) generated by mitochondria30, and is also a substrate for antioxidant enzymes that detoxify hydrogen peroxide and lipid peroxides59. Importantly, early studies demonstrate a correlation between GSH deficiency and mitochondrial damage60. GSH normally exists in its reduced from and is converted into its oxidized form, glutathione disulfide (GSSG), due to oxidative stress. To diminish the effects of oxidative stress, the enzyme glutathione reductase reverts GSSG into GSH. Thus, the ratio of GSH/GSSG is a marker of neuronal oxidative stress and is often used as a marker of cellular toxicity30. In resting state, GSSG/GSH ratio were maintained at lower levels, however, in a cell that is experiencing oxidative stress this ratio shifts toward more GSSG29. In our study, the motor cortex with TDP-43 pathology displayed increased GSSG/GSH ratio indicating ongoing oxidative stress.
Perturbation of NAD+/NADH ratio indicates energy deficiency and is associated with many diseases, including ALS22. NAD+ accepts H+ released from various substrates and converts into NADH, releasing ATP. A balanced NAD+/NADH ratio is critical for continuous generation of ATP. Metabolic dysregulation leads into depletion of NAD+ and that in turn affects energy homeostasis. NAD+ levels can be restored by compounds such as NR (nicotinamide riboside), NAM (nicotinamide), NA (niacin), and NMN (nicotinamide mononucleotide)61. These precursors convert into NAD+ that is ready to be used in energy generation pathways. Restoration of NAD+ levels had been beneficial and neuroprotective in Alzheimer’s disease and intracerebral hemorrhage62–65. Moreover, NAD+ enhancement has shown attenuation of astrocyte mediated neurotoxicity in vitro66,67. Treatment of hSOD1G93A mouse model of ALS with NR slowed down degeneration of spinal motor neurons and reduced neuroinflammation68. In an in vitro model of TDP-43 deficiency in spinal motor neurons, loss of TDP-43 is accompanied by axonal defects. Lack of TDP-43 leads to perturbed transcriptome in axon and dendrites and dysregulated transcripts code for genes that are key component of mRNA translation, cytoskeleton, and mitochondria mediated energy production and metabolism35.
Building evidence, including the current study, suggests that mitochondria mediated metabolite disturbances are emerging as prevalent feature in ALS, and restoration of NAD+ levels by NAD+ precursor compounds could in fact be a potential therapeutic strategy for improving neuron health. Therefore, we tested whether NMN treatment would help improve the health of CSMN that are diseased due to TDP-43 pathology.
In an effort to investigate whether NMN treatment would have an impact on the health of UMNs diseased with TDP-43 pathology, we utilized dissociated motor cortex cultures established from prpTDP-43A315T-UeGFP mice, in which CSMN are genetically labeled with eGFP expression that is stable and long-lasting, and when in culture retain their CSMN identity and eGFP expression. These diseased reporter mice were exceptionally valuable for directly assessing the cellular responses of healthy and diseased CSMN to treatments in vitro and in vivo69–71. Here, we utilized them to investigate their response to NMN treatment. We find that acute and short-term treatment of diseased CSMN with NMN not only improve their neuronal health, but also help mitochondria to regain the integrity of their inner membrane. Our results reveal early metabolomic dysfunction in the motor cortex of prpTDP-43A315T mice and begin to suggest that reestablishing the perturbed balance may help UMNs regain their health and stability.
Even though long-term exposure to NMN may not be favorable for axonal stability or neuron health due to its interaction with Sterile alpha and Toll/interleukin-1 receptor motif-containing 1 (SARM1), and by modulating its activity72,73, maintaining NAD+/ NADH balance appears to be an important contributor to UMN health, especially the ones that are diseased due to TDP-43 pathology. Therefore, our results suggest the unique importance of understanding the metabolomic problems that occur early in the diseased motor cortex, and encourages for the development of novel therapeutic strategies by reestablishing the perturbed NAD+ levels in the brain.
Conclusions
Metabolomic defects begin to occur early in the motor cortex with TDP-43 pathology. There is altered energy balance and reduced levels of ATP. Since metabolomic analyses reveal reduced levels of NAD+ as a potential converging outcome of altered metabolomic imbalance, NMN treatment is applied to upregulate NAD+ levels and to investigate the relationship between increased NAD+ and CSMN health. Our findings, which utilize a novel CSMN reporter line with TDP-43 pathology, not only reveal the presence of metabolomic defects at an early stage of the disease, but also suggest that proper modulation of metabolomic imbalance could offer therapeutic strategies to improve the integrity of mitochondria, and thus the health of diseased UMNs.
Materials and methods
Animals
All animal experiments were performed in compliance with the standards set by National Institutes of Health (NIH) and were reviewed and approved by the Northwestern University Institutional Animal Care and Use committee (IACUC approval number # IS00004844). All experiments were conducted in compliance with the ARRIVE guidelines. The following mouse strains were used in this study: prp-TDP-43A315T (procured from Jackson Laboratory, stock no. 010700), and UCHL1-eGFP (generated by the Ozdinler Laboratory at Northwestern Targeted Mutagenesis Core Facility, now also available at Jackson Laboratory, stock no. 022476). Gastrointestinal obstruction have been reported in the prp-TDP-43A315T mouse line74. However, feeding these mice with high fat jelly diet prevented their gastrointestinal problems and their death was reported to be due to TDP-43 pathology mediated degeneration, and not due to gastrointestinal problems75,76. In this study, all TDP-43 mouse models were fed with the jelly diet. Hemizygous UCHL1-eGFP females were bred to hemizygous prp-TDP-43A315T males to generate prp-TDP-43A315T-UeGFP mice. All the mice used in this study were on C57/BL6 background and were P90 at the time of analyses. Animals were housed at a controlled temperature (23 °C) and controlled light–dark cycle (12–12 h), with free access to water and food16,17.
Tissue preparation and metabolite extraction
Mice were deeply anesthetized with intraperitoneal injection of Ketamine (90 mg/kg, and Xylazine (10 mg/kg; Fort Dodge Animal Health, Fort Dodge, IA, USA)16. Intact brain was removed and motor cortex was dissected out, weighed and immediately frozen in liquid nitrogen. The tissue was homogenized in 1 ml chilled 80% chromatography grade methanol and vigorously vortexed three times. 200 µL tissue homogenate was mixed into a tube pre-added with 800 µL of ice-cold methanol/water 80% (vol/vol) followed by vortexing rigorously for 1 min, and then centrifuge at ~ 20,160g for 15 min in a refrigerated centrifuge. The metabolite-containing supernatant was separated into a fresh tube and stored at − 80 °C until metabolite profiling was performed77.
Method for sample reconstitution after extraction
Extraction solution was dried using SpeedVac. 50% acetonitrile was added to the tube for reconstitution following by vigorously shaking for 30 s. Samples solution was then centrifuged for 15 min @ 20,000g, 4 °C. Supernatant was collected for LCMS analysis77.
Hydrophilic metabolites profiling
Comprehensive metabolomics was performed as described earlier77. Briefly, samples were analyzed by High-Performance Liquid Chromatography and High-Resolution Mass Spectrometry and Tandem Mass Spectrometry (HPLC–MS/MS). Specifically, system consisted of a Thermo Q-Exactive in line with an electrospray source and an Ultimate3000 (Thermo) series HPLC consisting of a binary pump, degasser, and auto-sampler outfitted with a Xbridge Amide column (Waters; dimensions of 4.6 mm × 100 mm and a 3.5 µm particle size). The mobile phase A contained 95% (vol/vol) water, 5% (vol/vol) acetonitrile, 20 mM ammonium hydroxide, 20 mM ammonium acetate, pH = 9.0; B was 100% Acetonitrile. The gradient was as following: 0 min, 15% A; 2.5 min, 30% A; 7 min, 43% A; 16 min, 62% A; 16.1–18 min, 75% A; 18–25 min, 15% A with a flow rate of 400 μL/min. The capillary of the ESI source was set to 275 °C, with sheath gas at 45 arbitrary units, auxiliary gas at 5 arbitrary units and the spray voltage at 4.0 kV. In positive/negative polarity switching mode, an m/z scan range from 70 to 850 was chosen and MS1 data was collected at a resolution of 70,000. The automatic gain control (AGC) target was set at 1 × 106 and the maximum injection time was 200 ms. The top 5 precursor ions were subsequently fragmented, in a data-dependent manner, using the higher energy collisional dissociation (HCD) cell set to 30% normalized collision energy in MS2 at a resolution power of 17,500. Data acquisition and analysis were carried out by Xcalibur 4.1 software and Tracefinder 4.1 software, respectively (both from Thermo Fisher Scientific).
Correlative light electron microscopy (CLEM)
Mixed cortical cultures from prpTDP-43A315T mice were plated on a gridded glass coverslip bottom petridishes (MetTek) and cultured for 3 DIV either in SFM alone or treated with 1 µM NMN (Sigma). Correlative Light Electron microscopy (CLEM) was performed as describes previously78. After fixing with 2% PFA and 0.5% glutaraldehyde for 10 min at room temperature, GFP+ CSMN were identified under epifluorescent microscope, and their location on the grid marked before processing for EM analysis. Cells were further post-fixed in 2% PFA and 2% glutaraldehyde for 10 min at room temperature and 50 min at 4 °C. Cells were washed with 0.12 M phosphate buffer pH 7.4 and treated with 1% osmium tetroxide (EMS) and 1.5% potassium ferrocyanide (Sigma) dissolved in 0.12 M phosphate buffer. Cells were dehydrated by subjecting them to ascending series of alcohol 50%, 70%, 80%, 90% and 100% followed by treatment with Epoxy resin for 24 h. The grids were mounted on resin blocks and cured at 65 °C for 3 days. The blocks were trimmed to contain only CSMN, before proceeding to ultra-thin sectioning. Resin blocks were ultra thin sectioned on a Leica Ultracut UC6 ultramicrotome (Leica Inc., Nussloch, Germany). 70 nm thin sections were collected on 200-mesh copper–palladium grids. Ultra-thin sections were counterstained on a drop of UranyLess solution (Electron Microscopy Sciences, Hatfield, PA, USA) and 0.2% lead citrate. Grids were examined on FEI Tecnai Spirit G2 TEM (FEI company, Hillsboro, OR, USA), and digital images were captured on a FEI Eagle camera.
Motor cortex cultures
P3 motor cortices isolated from WT-UeGFP, and prpTDP-43A315T–UeGFP mice, dissected, dissociated, and cultured on glass coverslips (4 × 104 cells per 18 mm diameter coverslip, Fisherbrand) coated with poly-L-lysine (10 mg/mL, Sigma). Neurons were cultured in serum-free medium [SFM; 0.034 mg/L BSA, 1 mM L-glutamine, 25 U/mL penicillin, 0.025 mg/mL streptomycin, 35 mM glucose, and 0.5% B27 in Neurobasal-A medium (Life Technologies)] in a humidified tissue culture incubator in the presence of 5% CO2 at 37 °C as previously described28. NMN (1 µM) was added at the start of the culture. Cultures were fixed after 3 days in vitro.
Immunocytochemistry
Anti-GFP (1:1000, Abcam) antibody was applied in blocking solution (PBS, 0.05% BSA, 2% FBS, 1% Triton X-100, and 0.1% saponin) for 2 h at room temperature. After incubation, and washes with PBS, appropriate secondary fluorescent antibodies (1:500, AlexaFluor-conjugated, Invitrogen) were added to the blocking solution at room temperature for 2 h in the dark. Nuclei were counterstained with DAPI.
Data analysis
Measurement of axon length
CSMN from WT-UeGFP, and prp-TDP-43A315T-UeGFP mice (n = 3) were culture in vitro for 3 days. Immunocytochemistry was performed with eGFP to enhance visualization of GFP+ neurons. Images were acquired using epifluorescent microscope. Neurite length was measure using FIJI ImageJ software. CSMN processes were traced using the Simple Neurite Tracer plugin from FIJI (NIH), which enables semi-autonomous tracing of neuron morphology. The longest neurite was selected to measure the length of the axon in µm.
Sholl analyses
The aggregation of the neurite tracings centered at the soma generates a profile available for Sholl analysis to quantify number of intersections at 5 µm radius intervals for each neuron included in analysis. Minimum 15 neurons per treatment from 3 independent experiments were used for analysis.
Statistics
Statistical analyses were performed using Prism software (GraphPad Software Inc., La Jolla, CA, USA)16. For each quantification at least n = 3 mice were used for each genotype and group. D’Agostino and Pearson normality tests were performed on all data sets. Student’s t test was used to determine statistical differences between two experimental groups depending on the genotype, experimental conditions and the disease group. One way ANOVA was used for analysis of more than two groups. Data are shown as mean ± SEM of at least three replicates and are representative of three independent experiments unless otherwise stated and statistically significant differences were taken at p < 0.05, and p values are reported in the text.
Supplementary Information
Acknowledgements
We thank A Long Swim for Ellen McConnell Blakeman Fellowship support to M.G. Metabolomic profiling was performed at Metabolomics Core, Lurie Cancer Center, Northwestern University, Chicago.
Author contributions
Conceptualization, M.G., N.C., and P.H.O.; methodology, M.G.; data curation, M.G., A.G., and P.H.O.; writing, M.G. N.C., and P.H.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by NIA/NINDS RO1 AG061708 (P.H.O) and was made possible with the generous philanthropic support the Ozdinler Lab received from many different donors. We thank the Estaban Bullrich Foundation for covering publication costs.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-08068-5.
References
- 1.Benard G, et al. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007;120:838–848. doi: 10.1242/jcs.03381. [DOI] [PubMed] [Google Scholar]
- 2.Dimmer KS, Scorrano L. (De)constructing mitochondria: What for? Physiology (Bethesda) 2006;21:233–241. doi: 10.1152/physiol.00010.2006. [DOI] [PubMed] [Google Scholar]
- 3.Esterhuizen K, van der Westhuizen FH, Louw R. Metabolomics of mitochondrial disease. Mitochondrion. 2017;35:97–110. doi: 10.1016/j.mito.2017.05.012. [DOI] [PubMed] [Google Scholar]
- 4.Steinert EM, Vasan K, Chandel NS. Mitochondrial metabolism regulation of T cell-mediated immunity. Annu. Rev. Immunol. 2021;39:395–416. doi: 10.1146/annurev-immunol-101819-082015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandel NS. Lipid metabolism. Cold Spring Harb. Perspect. Biol. 2021;13:a040576. doi: 10.1101/cshperspect.a040576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bigio EH, et al. TDP-43 pathology in primary progressive aphasia and frontotemporal dementia with pathologic Alzheimer disease. Acta Neuropathol. 2010;120:43–54. doi: 10.1007/s00401-010-0681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao J, Wang L, Huntley ML, Perry G, Wang X. Pathomechanisms of TDP-43 in neurodegeneration. J. Neurochem. 2018 doi: 10.1111/jnc.14327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nana AL, et al. Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol. 2019;137:27–46. doi: 10.1007/s00401-018-1942-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shenouda M, Zhang AB, Weichert A, Robertson J. Mechanisms associated with TDP-43 neurotoxicity in ALS/FTLD. Adv. Neurobiol. 2018;20:239–263. doi: 10.1007/978-3-319-89689-2_9. [DOI] [PubMed] [Google Scholar]
- 10.Neumann M, Kwong LK, Sampathu DM, Trojanowski JQ, Lee VM. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch. Neurol. 2007;64:1388–1394. doi: 10.1001/archneur.64.10.1388. [DOI] [PubMed] [Google Scholar]
- 11.Droppelmann CA, Campos-Melo D, Moszczynski AJ, Amzil H, Strong MJ. TDP-43 aggregation inside micronuclei reveals a potential mechanism for protein inclusion formation in ALS. Sci. Rep. 2019;9:19928. doi: 10.1038/s41598-019-56483-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miguel L, Frebourg T, Campion D, Lecourtois M. Both cytoplasmic and nuclear accumulations of the protein are neurotoxic in Drosophila models of TDP-43 proteinopathies. Neurobiol. Dis. 2011;41:398–406. doi: 10.1016/j.nbd.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Mackenzie IR, Rademakers R. The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr. Opin. Neurol. 2008;21:693–700. doi: 10.1097/WCO.0b013e3283168d1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanelli T, et al. Evidence that TDP-43 is not the major ubiquitinated target within the pathological inclusions of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007;66:1147–1153. doi: 10.1097/nen.0b013e31815c5edd. [DOI] [PubMed] [Google Scholar]
- 15.Swarup V, et al. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain. 2011;134:2610–2626. doi: 10.1093/brain/awr159. [DOI] [PubMed] [Google Scholar]
- 16.Gautam M, et al. Mitochondria, ER, and nuclear membrane defects reveal early mechanisms for upper motor neuron vulnerability with respect to TDP-43 pathology. Acta Neuropathol. 2019;137:47–69. doi: 10.1007/s00401-018-1934-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gautam M, Xie EF, Kocak N, Ozdinler PH. Mitoautophagy: A unique self-destructive path mitochondria of upper motor neurons with TDP-43 pathology take, very early in ALS. Front. Cell. Neurosci. 2019;13:489. doi: 10.3389/fncel.2019.00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang P, et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019;15:e1007947. doi: 10.1371/journal.pgen.1007947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA. 2009;106:18809–18814. doi: 10.1073/pnas.0908767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mithal DS, Chandel NS. The Gro3p factor: Restoring NAD+/NADH homeostasis to ameliorate mitochondrial disease. Cell Metab. 2021;33:1905–1907. doi: 10.1016/j.cmet.2021.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Obrador E, Salvador-Palmer R, Lopez-Blanch R, Dellinger RW, Estrela JM. NAD(+) precursors and antioxidants for the treatment of amyotrophic lateral sclerosis. Biomedicines. 2021;9:1000. doi: 10.3390/biomedicines9081000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J. Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Srivastava S. Emerging therapeutic roles for NAD(+) metabolism in mitochondrial and age-related disorders. Clin. Transl. Med. 2016;5:25. doi: 10.1186/s40169-016-0104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- 26.Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee CF, Caudal A, Abell L, Nagana Gowda GA, Tian R. Targeting NAD(+) metabolism as interventions for mitochondrial disease. Sci. Rep. 2019;9:3073. doi: 10.1038/s41598-019-39419-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pang Z, et al. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021;49:W388–W396. doi: 10.1093/nar/gkab382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zitka O, et al. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012;4:1247–1253. doi: 10.3892/ol.2012.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forman HJ, Zhang H, Rinna A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 2009;30:1–12. doi: 10.1016/j.mam.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yasvoina MV, et al. eGFP expression under UCHL1 promoter genetically labels corticospinal motor neurons and a subpopulation of degeneration-resistant spinal motor neurons in an ALS mouse model. J. Neurosci. 2013;33:7890–7904. doi: 10.1523/JNEUROSCI.2787-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozdinler PH, Macklis JD. IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat. Neurosci. 2006;9:1371–1381. doi: 10.1038/nn1789. [DOI] [PubMed] [Google Scholar]
- 33.Jiang T, et al. Amyotrophic lateral sclerosis mutant TDP-43 may cause synaptic dysfunction through altered dendritic spine function. Dis. Model Mech. 2019 doi: 10.1242/dmm.038109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez-Gonzalez L, et al. Motor neuron preservation and decrease of in vivo TDP-43 phosphorylation by protein CK-1delta kinase inhibitor treatment. Sci. Rep. 2020;10:4449. doi: 10.1038/s41598-020-61265-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Briese M, et al. Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol. Commun. 2020;8:116. doi: 10.1186/s40478-020-00987-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salvatori I, et al. Differential toxicity of TAR DNA-binding protein 43 isoforms depends on their submitochondrial localization in neuronal cells. J. Neurochem. 2018;146:585–597. doi: 10.1111/jnc.14465. [DOI] [PubMed] [Google Scholar]
- 37.Wang W, et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016;22:869–878. doi: 10.1038/nm.4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis SA, et al. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018;678:8–15. doi: 10.1016/j.neulet.2018.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garofalo S, et al. Natural killer cells modulate motor neuron-immune cell cross talk in models of Amyotrophic Lateral Sclerosis. Nat. Commun. 2020;11:1773. doi: 10.1038/s41467-020-15644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marques RF, et al. Motor neuron translatome reveals deregulation of SYNGR4 and PLEKHB1 in mutant TDP-43 amyotrophic lateral sclerosis models. Hum. Mol. Genet. 2020;29:2647–2661. doi: 10.1093/hmg/ddaa140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orr BO, et al. Presynaptic homeostasis opposes disease progression in mouse models of ALS-like degeneration: Evidence for homeostatic neuroprotection. Neuron. 2020;107:95–111.e116. doi: 10.1016/j.neuron.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodriguez-Cueto C, et al. Inactivation of the CB2 receptor accelerated the neuropathological deterioration in TDP-43 transgenic mice, a model of amyotrophic lateral sclerosis. Brain Pathol. 2021;31:e12972. doi: 10.1111/bpa.12972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu CH, et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell. 2020;183:636–649.e618. doi: 10.1016/j.cell.2020.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arnold ES, et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc. Natl. Acad. Sci. USA. 2013;110:E736–745. doi: 10.1073/pnas.1222809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goossens J, et al. TDP-43 as a possible biomarker for frontotemporal lobar degeneration: A systematic review of existing antibodies. Acta Neuropathol. Commun. 2015;3:15. doi: 10.1186/s40478-015-0195-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blasco H, et al. Understanding and managing metabolic dysfunction in amyotrophic lateral sclerosis. Expert Rev. Neurother. 2020;20:907–919. doi: 10.1080/14737175.2020.1788389. [DOI] [PubMed] [Google Scholar]
- 48.Cistaro A, et al. The metabolic signature of C9ORF72-related ALS: FDG PET comparison with nonmutated patients. Eur. J. Nucl. Med. Mol. Imaging. 2014;41:844–852. doi: 10.1007/s00259-013-2667-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goutman SA, et al. Untargeted metabolomics yields insight into ALS disease mechanisms. J. Neurol. Neurosurg. Psychiatry. 2020 doi: 10.1136/jnnp-2020-323611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jawaid A, Khan R, Polymenidou M, Schulz PE. Disease-modifying effects of metabolic perturbations in ALS/FTLD. Mol Neurodegener. 2018;13:63. doi: 10.1186/s13024-018-0294-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar A, Ghosh D, Singh RL. Amyotrophic lateral sclerosis and metabolomics: Clinical implication and therapeutic approach. J. Biomark. 2013;2013:538765. doi: 10.1155/2013/538765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palamiuc L, et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 2015;7:526–546. doi: 10.15252/emmm.201404433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steyn FJ, et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry. 2018;89:1016–1023. doi: 10.1136/jnnp-2017-317887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vandoorne T, De Bock K, Van Den Bosch L. Energy metabolism in ALS: An underappreciated opportunity? Acta Neuropathol. 2018;135:489–509. doi: 10.1007/s00401-018-1835-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lawton KA, et al. Plasma metabolomic biomarker panel to distinguish patients with amyotrophic lateral sclerosis from disease mimics. Amyotroph. Lateral Scler. Frontotemporal Degener. 2014;15:362–370. doi: 10.3109/21678421.2014.908311. [DOI] [PubMed] [Google Scholar]
- 56.Manzo E, et al. Medium-chain fatty acids, beta-hydroxybutyric acid and genetic modulation of the carnitine shuttle are protective in a Drosophila model of ALS based on TDP-43. Front. Mol. Neurosci. 2018;11:182. doi: 10.3389/fnmol.2018.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weiduschat N, et al. Motor cortex glutathione deficit in ALS measured in vivo with the J-editing technique. Neurosci. Lett. 2014;570:102–107. doi: 10.1016/j.neulet.2014.04.020. [DOI] [PubMed] [Google Scholar]
- 58.Chi L, Ke Y, Luo C, Gozal D, Liu R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience. 2007;144:991–1003. doi: 10.1016/j.neuroscience.2006.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Poli G, Albano E, Dianzani MU. The role of lipid peroxidation in liver damage. Chem. Phys. Lipids. 1987;45:117–142. doi: 10.1016/0009-3084(87)90063-6. [DOI] [PubMed] [Google Scholar]
- 60.Jain A, Martensson J, Stole E, Auld PA, Meister A. Glutathione deficiency leads to mitochondrial damage in brain. Proc. Natl. Acad. Sci. USA. 1991;88:1913–1917. doi: 10.1073/pnas.88.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rajman L, Chwalek K, Sinclair DA. Therapeutic potential of NAD-boosting molecules: The in vivo evidence. Cell Metab. 2018;27:529–547. doi: 10.1016/j.cmet.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gong B, et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-gamma coactivator 1alpha regulated beta-secretase 1 degradation and mitochondrial gene expression in Alzheimer's mouse models. Neurobiol. Aging. 2013;34:1581–1588. doi: 10.1016/j.neurobiolaging.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hou Y, et al. NAD(+) supplementation normalizes key Alzheimer's features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA. 2018;115:E1876–E1885. doi: 10.1073/pnas.1718819115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sorrentino V, et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature. 2017;552:187–193. doi: 10.1038/nature25143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wei CC, et al. Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Sci. Rep. 2017;7:717. doi: 10.1038/s41598-017-00851-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harlan BA, Pehar M, Killoy KM, Vargas MR. Enhanced SIRT6 activity abrogates the neurotoxic phenotype of astrocytes expressing ALS-linked mutant SOD1. FASEB J. 2019;33:7084–7091. doi: 10.1096/fj.201802752R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harlan BA, et al. Enhancing NAD+ salvage pathway reverts the toxicity of primary astrocytes expressing amyotrophic lateral sclerosis-linked mutant superoxide dismutase 1 (SOD1) J. Biol. Chem. 2016;291:10836–10846. doi: 10.1074/jbc.M115.698779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Harlan BA, et al. Evaluation of the NAD(+) biosynthetic pathway in ALS patients and effect of modulating NAD(+) levels in hSOD1-linked ALS mouse models. Exp. Neurol. 2020;327:113219. doi: 10.1016/j.expneurol.2020.113219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Genc B, et al. Improving mitochondria and ER stability helps eliminate upper motor neuron degeneration that occurs due to mSOD1 toxicity and TDP-43 pathology. Clin. Transl. Med. 2021;11:e336. doi: 10.1002/ctm2.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dervishi I, Ozdinler PH. Incorporating upper motor neuron health in ALS drug discovery. Drug Discov. Today. 2018;23:696–703. doi: 10.1016/j.drudis.2018.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Genc B, Ozdinler PH. Moving forward in clinical trials for ALS: Motor neurons lead the way please. Drug Discov. Today. 2014;19:441–449. doi: 10.1016/j.drudis.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bloom AJ, et al. Constitutively active SARM1 variants that induce neuropathy are enriched in ALS patients. Mol. Neurodegener. 2022;17:1. doi: 10.1186/s13024-021-00511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Figley MD, et al. SARM1 is a metabolic sensor activated by an increased NMN/NAD(+) ratio to trigger axon degeneration. Neuron. 2021;109:1118–1136.e1111. doi: 10.1016/j.neuron.2021.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Esmaeili MA, Panahi M, Yadav S, Hennings L, Kiaei M. Premature death of TDP-43 (A315T) transgenic mice due to gastrointestinal complications prior to development of full neurological symptoms of amyotrophic lateral sclerosis. Int. J. Exp. Pathol. 2013;94:56–64. doi: 10.1111/iep.12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Coughlan KS, Halang L, Woods I, Prehn JH. A high-fat jelly diet restores bioenergetic balance and extends lifespan in the presence of motor dysfunction and lumbar spinal cord motor neuron loss in TDP-43A315T mutant C57BL6/J mice. Dis. Model Mech. 2016;9:1029–1037. doi: 10.1242/dmm.024786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Herdewyn S, et al. Prevention of intestinal obstruction reveals progressive neurodegeneration in mutant TDP-43 (A315T) mice. Mol. Neurodegener. 2014;9:24. doi: 10.1186/1750-1326-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kong H, et al. Metabolic determinants of cellular fitness dependent on mitochondrial reactive oxygen species. Sci. Adv. 2020 doi: 10.1126/sciadv.abb7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arai R, Waguri S. Improved electron microscopy fixation methods for tracking autophagy-associated membranes in cultured mammalian cells. Methods Mol. Biol. 2019;1880:211–221. doi: 10.1007/978-1-4939-8873-0_13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.