

Abstract
Background
Chordomas are rare malignant bone cancers of the skull-base and spine. Patient survival is variable and not reliably predicted using clinical factors or molecular features. This study identifies prognostic epigenetic chordoma subtypes that are detected noninvasively using plasma methylomes.
Methods
Methylation profiles of 68 chordoma surgical samples were obtained between 1996 and 2018 across three international centers along with matched plasma methylomes where available.
Results
Consensus clustering identified two stable tissue clusters with a disease-specific survival difference that was independent of clinical factors in a multivariate Cox analysis (HR = 14.2, 95%CI: 2.1–94.8, P = 0.0063). Immune-related pathways with genes hypomethylated at promoters and increased immune cell abundance were observed in the poor-performing “Immune-infiltrated” subtype. Cell-to-cell interaction plus extracellular matrix pathway hypomethylation and higher tumor purity were observed in the better-performing “Cellular” subtype. The findings were validated in additional DNA methylation and RNA sequencing datasets as well as with immunohistochemical staining. Plasma methylomes distinguished chordomas from other clinical differential diagnoses by applying fifty chordoma-versus-other binomial generalized linear models in random 20% testing sets (mean AUROC = 0.84, 95%CI: 0.52–1.00). Tissue-based and plasma-based methylation signals were highly correlated in both prognostic clusters. Additionally, leave-one-out models accurately classified all tumors into their correct cluster based on plasma methylation data.
Conclusions
Here, we show the first identification of prognostic epigenetic chordoma subtypes and first use of plasma methylome-based biomarkers to noninvasively diagnose and subtype chordomas. These results may transform patient management by allowing treatment aggressiveness to be balanced with patient risk according to prognosis.
Keywords: bone cancer, central nervous system cancer, DNA methylation analysis, noninvasive diagnosis, prognostic biomarkers
Key Points.
Two prognostic DNA methylation-based chordoma subtypes were identified.
The Immune-infiltrated subtype has a poorer survival than the Cellular subtype.
It is possible to classify and subtype chordomas using plasma methylomes.
Importance of the Study.
We define, for the first time, two DNA methylation-based prognostic subgroups of chordomas; a tumor type with limited existing prognostic factors to stratify patient management currently. We utilize the largest cohort of chordomas with methylation data assembled to date, as well as published methylation and RNA sequencing data, to characterize these subgroups as an Immune-infiltrated subtype with a poorer disease-specific survival as well as a better-performing Cellular subtype. This works extends the concept of noninvasive tumor diagnosis to chordomas and shows feasibility for the classification and subtyping of chordomas using plasma cell-free tumor DNA methylation signatures. The noninvasive identification and prognostication of this rare but aggressive bone tumor that impacts the central nervous system can transform patient care by guiding treatment decisions based on prognostic methylation subtype. Preoperative noninvasive patient prognostication may individualize surgical planning by balancing aggressiveness in extent of surgical resection with the risk of treatment-induced neurological complications.
The identification of molecular tumor subtypes has changed the management approaches utilized for many cancer patients.1 Bone and central nervous system (CNS) tumors remain an exception, to a large extent, including chordomas. Chordomas are rare bone tumors of the skull-base and spine comprising 1–4% of all primary aggressive bone cancers.2 Patients with chordomas have a relatively high mortality rate, with the 10-year survival being 40%.3 They can cause also devastating neurological deficits resulting in a significant negative impact on patient quality of life and they have a relatively high propensity to become metastatic to other organs, which occurs in 30–40% of cases.4 Additionally, despite patients receiving standard of care treatment with surgery and radiotherapy according to global consensus guidelines,4,5 there is a very wide range of patient outcomes seen clinically with up to 10% surviving under 1 year and one-third living over 20 years.6
There are currently no robust prognostic histological features of chordoma as 97% of tumors fit one histological subtype and the World Health Organization (WHO) does not stratify chordomas into different tumor grades.7 A greater extent of surgical resection along with the use of high quality adjuvant radiation therapy both lead to better prognoses. However, there are no robust clinical factors that can identify high-risk patients in advance of treatment to guide decisions regarding treatment aggressiveness.4,5 Finally, genomic8–14 and transcriptomic9,15–17 studies of chordoma have identified chromosomal aberrations along with alterations in chromatin modeling, PI3K signaling, and cell cycle regulator genes within subsets of patients which warrants further assessment but none of which are used in routine clinical practice currently. Additionally, no prognostic tumor subtypes have been identified and, collectively, this limits the stratification of patient management based on prognosis.3,4
DNA methylation signatures can be used to accurately classify CNS tumors18–20 and our group has previously identified methylation signatures that can determine prognoses for patients with meningiomas.21 Collectively, this work leads to this study exploring chordoma methylation signatures. There are a few existing chordoma methylation studies, however, with very small sample sizes and they do not resolve prognostic chordoma subtypes.18,22,23 Here, using samples from multi-institutional sources to generate the largest cohort of chordomas reported on for DNA methylation analyses to date, we identified methylation-based prognostic tumor subtypes that have statistically significant differences in disease-specific survival.
Our group has shown that plasma methylated circulating cell-free tumor DNA (cfDNA) signatures obtained by immunoprecipitation and high-throughput sequencing (cfMeDIP-seq) can serve as a reliable biomarker for the diagnosis of many other cancer types.24–27 In this study we leveraged our group's established liquid biopsy approach to show that it is possible to diagnose chordomas noninvasively through plasma cfDNA methylomes, along with determining prognostic subtype. Identifying noninvasively obtained preoperative biomarkers for diagnosis and prognostication of chordomas will be transformative in their treatment. This patient prognostication would lead to the avoidance of the risks of invasive tumor biopsy, allow for diagnosis of unbiopsiable chordomas, and guide treatment aggressiveness based on patient risk. Tailoring of the extent of surgical resection and adjuvant radiotherapy provided based on patient risk may improve patient outcomes. In this way, high-risk patients may be considered for more aggressive treatment and lower risk patients may undergo less aggressive treatment in order to reduce the risk of therapy-induced morbidities associated with the loss of neurological function.
Materials and Methods
Patient Cohort
A multicenter surgical patient series was assembled across three international centers including the University Health Network (UHN) in Toronto, Canada (N = 41); Hôpital Lariboisière in Paris, France (N = 19); and Health Sciences Centre in Winnipeg, Canada (N = 9) with each institution having local research ethics board (REB) approval to utilize all stored tumor and plasma samples. From this cohort, a total of 68 tumor samples and 13 plasma samples were obtained based on tissue availability. Patients were operated on between 1996 and 2018. Hematoxylin and eosin slides from tumor samples were reviewed by experienced neuropathologists (SK/KDA/HD/NS) to confirm chordoma diagnoses and designate tumor tissue to be analyzed. Clinical data were collected locally at each institution in accordance with REB approvals for all clinical variables of interest including demographics, tumor location, treatment received, and patient outcomes. As described previously for chordomas, extent of resection was assessed based on surgeon's operative notes together with first postoperative magnetic resonance imaging of the brain and/or spine28 obtained within a month following surgery. Gross-totally resected lesions were defined as those with no evidence of residual tumor intraoperatively or on postoperative imaging. Subtotally-resected lesions had evidence of residual tumor. For the primary outcome of disease-specific survival, an event was defined as death due to the clinical impact of a patient's chordoma or its progression.
Additional sample sets were identified including N = 11 chordoma cell lines (Chordoma Foundation: MUG-Chor1, U-CH11, U-CH11R, U-CH12, U-CH14, U-CH17M, U-CH17P, U-CH17S, U-CH2, U-CH7, and UM-Chor1) for use in cell deconvolution plus N = 15 meningioma and N = 8 extradural spine prostate cancer metastasis patients with plasma samples available at UHN to serve as differential diagnoses for noninvasive chordoma diagnosis. Validation studies for the cell lines used can be found at https://www.chordomafoundation.org/research/disease-models/. Two external chordoma datasets were obtained from GSE14068629 (DNA methylation data from FFPE DNA, N = 61 samples) as well as dbGaP phs002301.v1.p114 (RNA sequencing, N = 27 patients with overlapping published copy number data).
Methylation Profiling of Tumor Tissue
DNA was extracted from formalin-fixed paraffin-embedded (FFPE) tissue (QIAamp DNA FFPE Tissue Kit, Qiagen) or fresh frozen surgical chordoma tissue samples or cell lines (DNeasy Blood & Tissue Kit, Qiagen). Between 200 and 500 ng of DNA was then bisulfite converted (EZ DNA methylation Kit, Zymo Research) and profiled on the Infinium MethylationEPIC BeadChip array (Illumina, San Diego, CA, USA) according to manufacturer instructions and after restoration of DNA obtained from FFPE tissue. Raw methylation data within.idat files, including those from external datasets, were processed in R v4.0.3 (R Foundation for Statistical Computing) using the minfi package and data was normalized using the single-sample Noob approach. CpG sites with low-quality data for one or more samples (CpG detection P > 0.01) were removed from further analysis as well as those located on X and Y chromosomes, overlapping with single-nucleotide polymorphisms, or designated as cross-reactive. All samples passed quality control assessment (sample detection P < 0.05). Batch correction was performed using the removeBatchEffect function of the limma package (v. 3.42.2). Postprocessed beta values were used for upstream analyses.
Generation and Validation of Prognostic Clusters
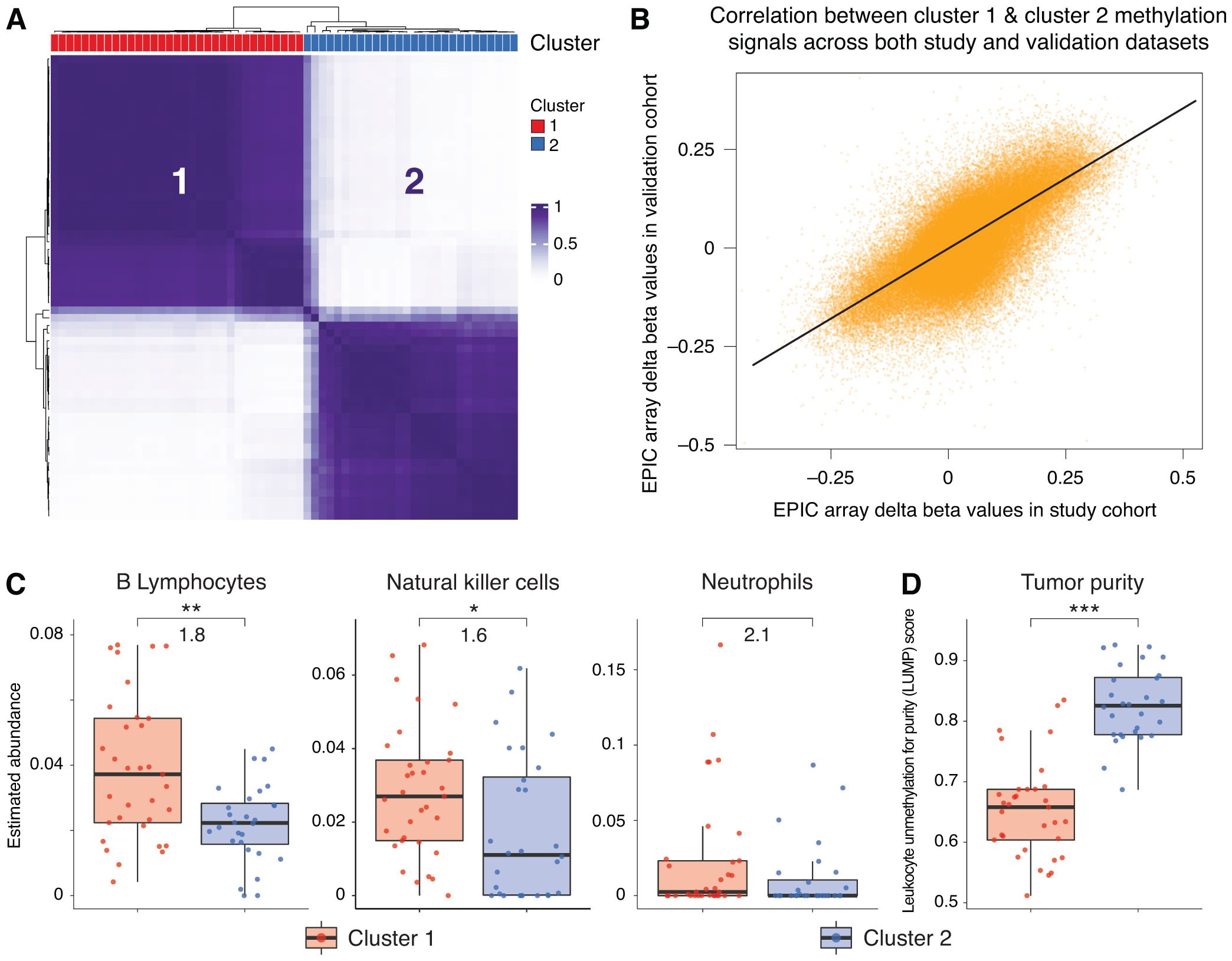
K-means consensus clustering was performed using the most variably methylated CpG sites across all tumor tissue samples and visualized in a heatmap with associated dendrograms. Silhouette scores and consensus cumulative distribution function plots were assessed to identify a k-value of two and 15,000 CpG sites as the optimal clustering parameters.30
Resulting clusters were compared in disease-specific survival times in Kaplan–Meier plots with associated log-rank test values. A multivariable Cox regression model was developed incorporating methylation-based cluster together with clinical factors as well as time from initial surgery to profiling (to control for sample age and changes in clinical practice over time). Hazard ratios (HR) of disease-specific survival along with associated 95% confidence intervals (CI) and P-values were determined for each variable in the multivariable Cox model.
The 7055 of these 15,000 CpGs that are represented on the Illumina Human Methylation 450K array, which many of the samples in the external dataset were profiled on, were used for k-means consensus clustering of the 61 external validation samples with k = 2. Delta beta values between cluster 1 and cluster 2 in the study cohort and validation cohort were compared in a scatterplot and the Pearson correlation coefficient was calculated. This analysis identified a positive correlation between cluster 1 tumors in the study dataset and cluster 1 tumors in the validation set as well as for the cluster 2 tumors in both datasets.
Differential Methylation between Prognostic Subtypes
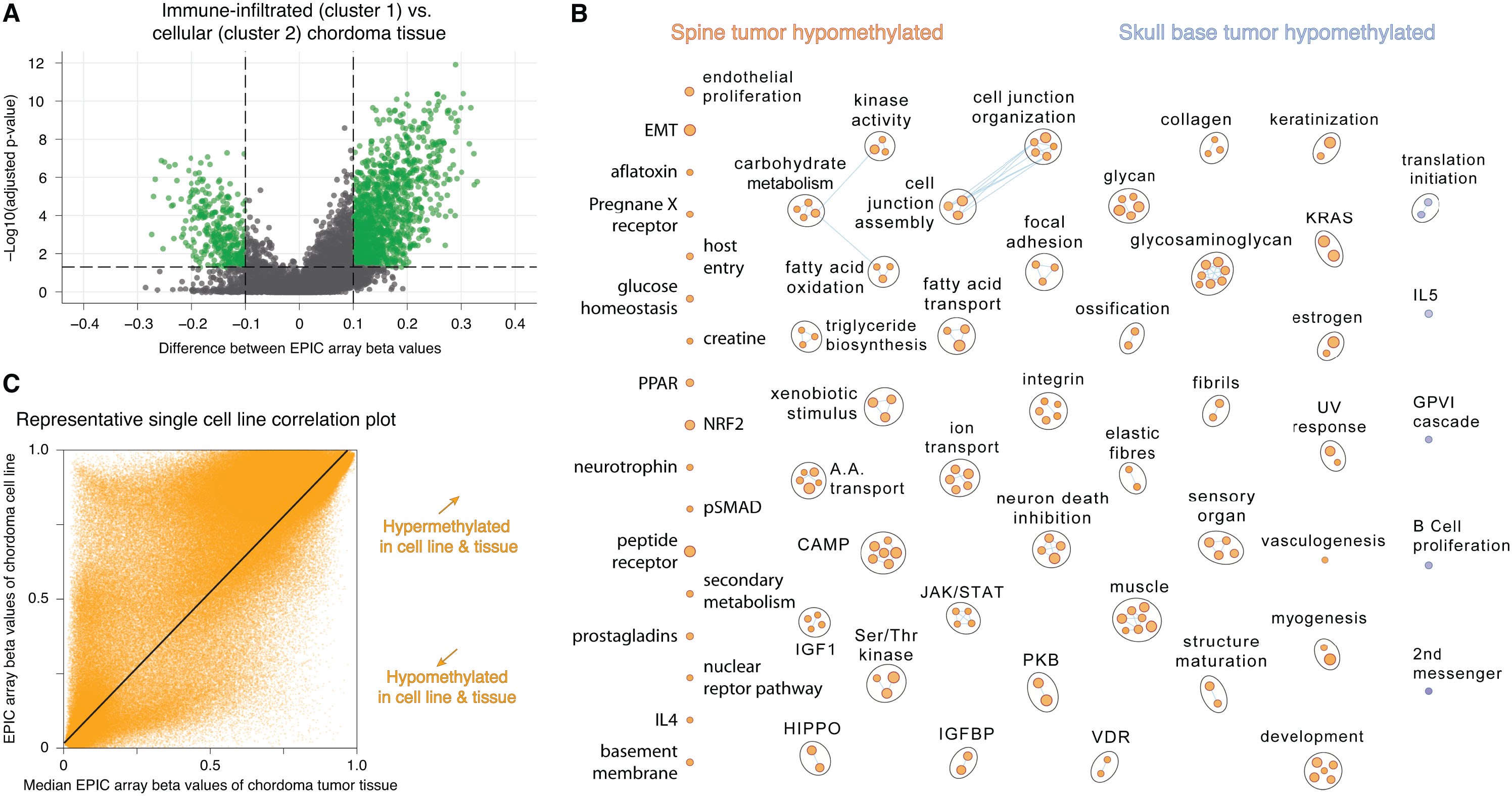
All CpGs were mapped to areas of the genome and those within gene promoters were selected. Median beta values between all CpGs within promoters of a given gene were calculated for all samples. Pathway analyses were pursued comparing prognostic clusters and tumor locations (skull-base versus spinal). GSEAs31,32 were performed using differentially methylated gene promoter data, identified using the limma package, ranked by scores that incorporate p-values and delta values with the gene set “Human_GOBP_AllPathways_no_GO_iea_November_01_2020_symbol.gmt” with 2000 permutations. Enrichment maps were generated with a Jaccard Coefficient of 0.25 and a p-value cutoff of 0.005. A volcano plot of differentially methylated gene promoters between prognostic clusters identified gene promoters with absolute delta beta >0.1 and false discovery rate (FDR) corrected P-value <0.05.
Cell Deconvolution and Leukocytes Unmethylation for Purity Analyses
We determined the composition of the microenvironment of the chordoma tissue samples in all tumor samples from both the study and external datasets using methylCIBERSORT.33 MethylCIBERSORT is an R package that adapts the support vector regression that is implemented in CIBERSORT34 to deconvolute samples into an estimated cancer cell fraction as well as fractions of neutrophils, B lymphocytes, natural killer cells, cytotoxic T lymphocytes, monocytes/macrophages, regulatory T lymphocytes, effector T lymphocytes, endothelial cells, fibroblasts, and eosinophils using DNA methylation data. We created the chordoma specific signature matrix by integrating methylation profiles of chordoma cell lines with methylation profiles of cells in their microenvironment within the methylCIBERSORT package using delta beta > 0.2, FDR < 0.01, and max number of features per pairwise comparison =100. In this way, the chordoma cell line signatures were used to identify the chordoma cell portion of the tumor tissue methylation data. MethylCIBERSORT has been validated on many CNS tumor including chordomas.18,35,36
The leukocytes unmethylation for purity (LUMP) estimate was calculated for all chordoma samples with DNA methylation data, as previously described, to estimate tumor purity/cellularity.37 In summary, the LUMP score is calculated by taking the average methylation levels of 44 CpG sites, which have been shown to be unmethylated in immune cells and methylated in tumors in a pan-cancer analysis, and dividing the number by 0.85. LUMP estimates were compared between prognostic subtypes. Wilcoxon's rank sum test was used to compare values between clusters for methylCIBERSORT and LUMP analyses.
Copy Number Alteration and Transcriptome Analyses
Chromosome level copy number alterations were inferred from raw methylation data after normalization using the conumee package (Bioconductor). Median intensity values for each chromosome arm were calculated and amplifications or deletions were identified with log2 copy number ratio > |0.3|.19 Composite copy number alteration signatures present in >50% of samples within one cluster and no samples within the other cluster were identified as genomic correlates in the study cohort. These genomic correlates were then identified in the external whole genome sequencing-based copy number data with >50% arm amplification or hemizygous deletion.
For external dataset RNA sequencing data, raw read quality assessment was performed using FastQC (v0.11.5). STAR aligner (v. 2.4.2a) was then used to align reads to the human reference genome (hg38) followed by HtSeq (v0.11.0) to count reads over gene exons. Genes were annotated using Gencode (v33) and data was normalized for differential expression analysis with DEseq2 (v1.26.0). A GSEA was performed using differential expression data identified with DEseq2 ranked by scores that incorporate adjusted P-values and log2 fold change with the gene set “Human_GOBP_AllPathways_no_GO_iea_November_01_2020_symbol.gmt” with 1000 permutations. Differentially expressed gene sets between tumors with and without the genomic correlate were identified using cutoffs of a normalized enrichment score >1.75 and FDR q-value <0.1 within the following databases: Hallmark, Pathways Integration Database, Reactome, WikiPathways, and Panther pathways. Significant pathways were portrayed in barplots with log10(FDR q-value). A volcano plot was used to depict differentially expressed genes between clusters with absolute log2 fold change >0.57 and false discovery rate (FDR) corrected P-value <0.05.
Immunohistochemical Validation of Immune Cells
FFPE tissue slides were analyzed by immunohistochemistry using myeloperoxidase and CD20 as markers of neutrophils and B lymphocytes, respectively. All stains were performed by the Pathology Research Program at the University Health Network. Three independent reviewers (JAZ, OS, and an experienced neuropathologist KDA) assessed slides for all stained cases.
Sequencing of Methylated Cell-free DNA
Plasma samples from chordoma, meningioma, and spinal metastasis patients were obtained at the time of first presentation or surgery at UHN from peripheral blood that underwent centrifugation at 20˚C (2500g for 15 min) and subsequent cryopreservation. cfDNA was extracted from 0.5-3mL of plasma (QIAamp Circulating Nucleic Acid Kit, Qiagen). cfDNA from each sample was processed using the cfMeDIP-seq protocol as has been described and used by our group.25,27 In summary, 1–10 ng of cfDNA quantified by a Qubit fluorometer (Qubit 4, Thermo Fisher Scientific) underwent library preparation (Kapa HyperPrep Kit, Roche), methylated DNA immunoprecipitation (MagMeDIP Kit, Diagenode), and DNA purification (IPure Kit v2 (Diagenode)) followed by library amplification and cleanup. Final libraries were sequenced using the Illumina Novaseq6000 (100 bp paired end reads, median 60.7 million reads/sample) after confirmation of optimal fragment size selection on Bioanalyzer traces (2100 Bioanalyzer, Agilent).
Plasma-based Diagnosis and Subtyping
Plasma cfMeDIP-seq data was processed as described previously.24 Briefly, reads were aligned to the human genome with Bowtie2, deduplicated, and indexed with SAMtools. Reads per kilobase million (RPKM) counts were then calculated for 300 bp genomic windows which cover regulatory features (CpG islands, shores, shelves, and FANTOM5 enhancers) using the MEDIPS package. RPKM values were converted to count per million (CPM) values based on the 300 kb window size.
To assess whether chordomas are distinguishable from meningiomas and spine metastases, as representative typical clinical differential diagnoses, the combined 36 tumors were divided into 50 random training and testing cohorts including 80% and 20% of tumors, respectively, balanced by each of these 3 tumor classes. In each training set, negative-binomial generalized linear models for each tumor class were developed using the top 300 DMRs for each pairwise comparison with the other two tumor classes combined, as identified by moderated t-statistic using limma-trend. One-class-versus-other models were optimized with three iterations of 10-fold cross validation and evaluated in the testing sets not used to build the models with AUROC calculations. Representative clinical chordoma cases are displayed where preoperative magnetic resonance imaging and clinical factors suggested diagnoses of a skull-base meningioma and two spinal metastases after review by two expert neuroradiologists and an expert neurosurgeon, but neuropathological diagnoses of surgical tumor tissue were consistent with chordoma. Boxplots of the distribution of class probabilities from all one-class-versus-other models, where the tumor of interest was in the testing set, allow for the assessment of the accuracy of plasma cfDNA methylomes to make the correct diagnosis of chordoma. Additional cases show the correct identification of a meningioma and a spine metastasis using testing set class probabilities.
To determine whether methylation signatures of prognostic chordoma clusters may be detectable in plasma cfDNA, the correlation between tissue methylation data and cfMeDIP-seq data was performed at a single-sample level for all chordoma patients with matched/paired tissue/plasma samples (N = 12). For this analysis, Pearson's correlation was utilized to assess for associations between normalized read counts (CPM values) at each window with averaged beta values for CpGs within a given window. Correlation coefficients were calculated for each sample. Additionally, a leave-one-out analysis was performed by developing a set of 12 random forest models, one model for each sample that is left out, which are each trained on the remaining 11 samples using the top 500 DMRs between cluster 1 and 2 patients. These models provided output probabilities of the held out sample being in cluster 1 or in cluster 2.
Statistical Analysis
Statistically significant P-values were defined as those with two-tailed P < 0.05, after false discovery rate correction for multiple comparisons where necessary, unless otherwise specified. Chi-squared and t-tests were used to compare clusters by clinical factors. Boxplot central bars indicate medians, boxes show upper and lower distribution quartiles, and whiskers extend across the 1.5× interquartile range. GSEA nodes represent gene sets, with number of genes in the set proportional to node size and connections between nodes signifying gene set overlap.
Data Availability
The DNA methylation and cfMeDIP-seq data for patient samples that support the findings of this study are available upon request from the corresponding authors (G.Z.) to comply with institutional ethics regulation.
Results
Chordoma Tumor Cohort
Tumor methylation profiles were obtained on 68 chordoma samples collected over a 22-year period with all variables of interest included in the clinical annotation. Baseline clinical features of the cohort are shown in Supplementary Table S1 along with time from initial surgery to the time of sample profiling. All craniospinal tumor locations are represented with both subtotally (STR) and gross totally resected (GTR) lesions as well as tumors that did and did not undergo radiotherapy.
Identification of Prognostic DNA Methylation-based Chordoma Subtypes
Consensus clustering of all 68 tumor samples using the top 15,000 most variably methylated CpG sites across all samples identified two stable tumor clusters that are shown in Figure 1A. Both clusters have representation from a range of relevant clinical factors shown (Figure 1A and Supplementary Table S1) with cluster 2 having a higher proportion of spinal chordomas (49% v. 16%, P = 0.0166) and a higher mean age (56 v. 47 years, P = 0.0184) than cluster 1. In contrast, cluster 1 had a higher proportion of skull-base chordomas (84% v. 51%, P = 0.0166) than cluster 2. Cluster 1 also had a statistically significant poorer disease-specific survival than cluster 2 as portrayed in Figure 1B (log-rank P = 0.0062). The median survival was 6.0 years in cluster 1 compared to 17.3 years in cluster 2.
Fig. 1.

Tumor DNA methylation signatures identify two prognostic chordoma subtypes. A, Unsupervised K-means consensus clustering of 68 chordoma tissue samples using the top 15,000 most variably methylated CpG sites identifies two stable tumor clusters. Color annotations show the distribution of clinical factors across the dataset. B, Kaplan–Meier plot showing a statistically significant poorer disease-specific survival in cluster 1 compared to cluster 2. C, Gene-set enrichment analysis revealing main pathways with genes having hypomethylated promoters within each prognostic cluster. Cluster 1 shows enrichment of immune- and transcription/translation-related pathways (“Immune-infiltrated subtype”) compared to those of cell-to-cell interaction, extracellular matrix, angiogenesis, and metabolism in cluster 2 (“Cellular subtype”). *** P < 0.00005, ** P < 0.005, * P < 0.05. EOR, extent of resection; aRT, adjuvant radiotherapy; N/A, not available.
Table 1 displays a multivariable analysis combining prognostic and nonprognostic clinical factors with methylation-based clusters. Independently statistically significant prognostic variables include methylation-based cluster (Cluster 1 vs. 2; HR = 14.2, 95%CI: 2.1–94.8, P = 0.0063) along with the receipt of adjuvant radiotherapy treatment (Unradiated vs. radiated; HR = 13.9, 95%CI: 2.2–89.3, P = 0.0056) after controlling for age, location, extent of resection, and time from initial surgery to sample profiling.
Table 1.
Multivariable Cox Analysis of Methylation Cluster and Known Prognostic Variables
| Covariate | Univariable Cox | Multivariable Cox | |||
|---|---|---|---|---|---|
| HR (95% CI) | P-value | HR (95% CI) | P-value | ||
| Methylation cluster | 1 vs. 2 | 4.3 (1.4–13.4) | 0.0119 | 14.2 (2.1–94.8) | 0.0063 |
| Age | Years | 0.6 (0.3–1.2) | 0.1413 | 0.7 (0.3–1.5) | 0.3352 |
| Location | Skull-base vs. spinal | 2.5 (0.7–8.6) | 0.1593 | 5.3 (0.6–45.2) | 0.1292 |
| Extent of resection | Subtotal vs. gross-total | 1.8 (0.5–6.7) | 0.3484 | 3.2 (0.4–24.0) | 0.2674 |
| Adjuvant radiotherapy | Not received vs. received | 2.0 (0.7–5.9) | 0.2303 | 13.9 (2.2–89.3) | 0.0056 |
| Time from initial treatment | Years | 0.8 (0.4–1.3) | 0.3177 | 0.8 (0.4–1.6) | 0.5181 |
Abbreviations: CI, confidence interval; HR, hazard ratio.
Characterization of Prognostic Chordoma Subtypes
A gene-set enrichment analysis (GSEA) was performed to characterize clusters by assessing pathways with genes that are differentially methylated at promoters within each cluster (Supplementary Figure S1A). Significant pathways with hypomethylated gene promoters, typically resulting in transcription, are depicted for each cluster in Figure 1C. Cluster 1 pathways were mainly immune- and transcription/translation-related while those in cluster 2 included cell-to-cell interaction, extracellular matrix, angiogenesis, and metabolic pathways. Accordingly, cluster 1 was termed the Immune-infiltrated subtype and cluster 2 the Cellular subtype.
Although tumor location (skull-base vs. spine) was not predictive of disease-specific survival (univariable Cox P = 0.1593), a GSEA of pathways with genes that were differentially methylated at promoters (Supplementary Figure S1B) revealed many hypomethylated pathways in spinal chordomas. It was assessed whether any currently studied chordoma drug targets38 were identified in this GSEA and it was observed that both kinase activity (encompassing PDGFR & KIT genes) as well as vascular proliferation hypomethylated pathways were enriched in spinal chordomas.
To assess tumor immune cell compositions, a cell deconvolution analysis of the tumor methylation data was pursued using methylCIBERSORT.36 MethylCIBERSORT adapts CIBERSORT models that deconvolute gene expression data to identify immune and nonimmune cell compositions from methylation profiles.34 After removing cell fractions pertaining to nonimmune and chordoma cells, the fractions of immune cells were compared between methylation-based clusters. The chordoma cell fraction was determined using chordoma cell line methylation data and the chordoma cell line methylation data was strongly correlated to that of chordoma tumors, for each individual cell line used in the study, with a median Pearson correlation coefficient of 0.84 (95%CI: 0.72–0.95, P < 2.2 × 10−16 for all cell lines). Supplementary Figure S1C shows a representative cell line to chordoma tumor tissue correlation plot. Figure 2A shows a greater abundance of neutrophils (7.0 fold, P < 0.0001), B lymphocytes (2.5 fold, P = 0.002), and natural killer cells (1.6 fold, P = 0.045) in cluster 1 chordomas that have been termed the Immune-infiltrated subtype. Cytotoxic T lymphocytes and macrophages/monocytes were not differentially abundant between subtypes. Furthermore, the leukocytes unmethylation for purity (LUMP) estimates in Figure 2B, which assess tumor purity indirectly through measuring immune/stromal tissue components, show higher tumor purity in cluster 2 (Cellular subtype) than cluster 1 (median 0.66 vs. 0.45, P < 0.0001).
Fig. 2.

Immune cell infiltration in cluster 1 chordomas and higher tumor purity in cluster 2 chordomas. A, Boxplots of deconvoluted tumor cell compositions showing greater immune cell abundance in Immune-infiltrated (cluster 1) tumors compared to Cellular (cluster 2) chordomas. Numerical values displayed between boxes represent fold changes between clusters, cluster 1 versus cluster 2. B, Boxplots of leukocyte unmethylation for purity (LUMP) estimates are consistent with higher tumor purity and cellularity in cellular chordomas (cluster 2). C, Immunohistochemical (IHC) staining for immune cell marker validation. Representative images are shown of myeloperoxidase (left panels) and CD20 (right panels) stained tissue showing more frequent neutrophil and B lymphocytes presence, respectively, in cluster 1 (upper panels) than cluster 2 (lower panels). Boxplot central bars indicate medians, boxes show upper and lower distribution quartiles, and whiskers extend across the 1.5× interquartile range. *** P < 0.00005, ** P < 0.005, * P < 0.05.
Immunohistochemical Validation of Immune Cells
Immunohistochemical staining for myeloperoxidase and CD20 identified the presence of neutrophils and B lymphocytes, respectively. Qualitatively, cluster 1 tumors had more frequent immune cells than cluster 2 tumors, based on assessment by three reviewers including an independent experienced neuropathologist, which is depicted in Figure 2C representative images.
External Validation of Chordoma Subgroups
We next analyzed an independent cohort of chordomas that is a publicly available DNA methylation dataset. Consensus clustering of these 61 chordomas using our prognostic CpG sites showed two stable clusters as shown in Supplementary Figure S2A. Delta beta values between cluster 1 and cluster 2 chordomas were well correlated between tumors in the study cohort and those in the external dataset (Supplementary Figure S2B; r = 0.68, P < 0.0001), showing alignment between cluster 1s as well as alignment between cluster 2s across both datasets. After controlling for other cell fractions in a methylCIBERSORT analysis, the immune cells with increased abundance in cluster 1 from the study cohort had similar increased fractions in the external dataset cluster 1 subgroup (Supplementary Figure S2C). Cluster 2 similarly had higher tumor purity in the external dataset as was seen in the study cohort cluster 2 tumors (Supplementary Figure S2D).
Genomic Correlates of Subgroups and Corresponding Gene Expression Profiles
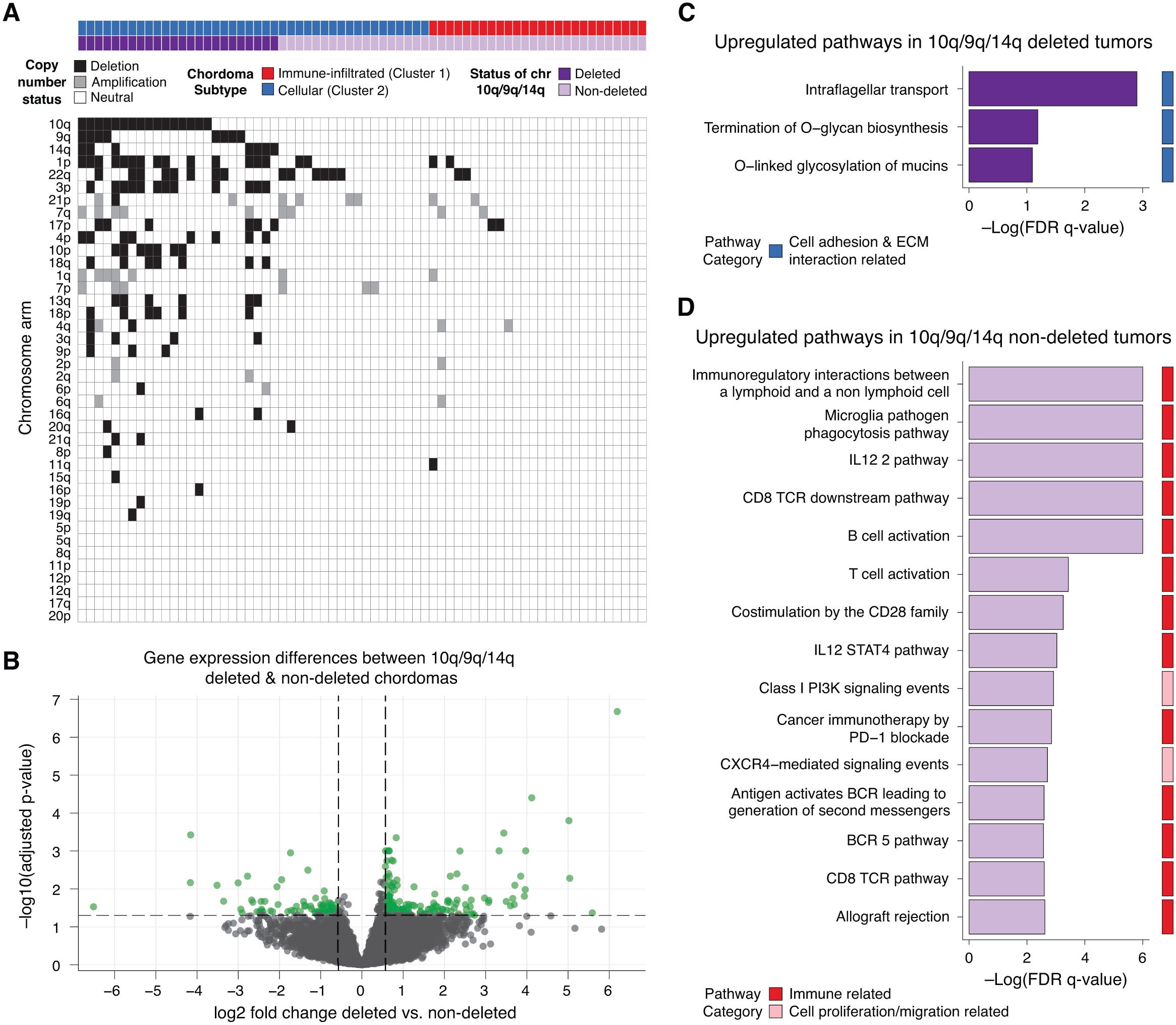
Copy number deletions and amplifications across the study dataset are shown in Supplementary Figure S3A with the majority of alterations occurring in cluster 2 tumors. The presence of a 10q deletion, 9q deletion, or 14q deletion was seen exclusively in cluster 2 but not cluster 1 chordomas. In an external cohort with available WGS-derived copy number and RNAseq data, gene expression differences between chordomas with (16/27) and without (11/27) 10q/9q/14q deletions were identified and assessed (Supplementary Figure S3B). Supplementary Figure S3C and D shows that 10q/9q/14q deleted tumors exclusive to cluster 2 had upregulated cell adhesion and ECM related pathways (similar to those hypomethylated at promoters in study cohort cluster 2 in Figure 1C), while chordomas without these deletions showed relative overexpression of immune pathways similar to those which were hypomethylated at promoters in study cohort cluster 1 tumors.
Noninvasive Diagnosis and Subtyping of Chordoma
Circulating cfDNA methylomes were obtained, using the cfMeDIP-seq technique, from matched plasma samples in patients with tumor DNA methylation data already obtained in this study. Additionally, cfDNA methylomes were obtained from patients with meningiomas as well as spine metastases which are representative clinical entities that are commonly included in the differential diagnoses of chordomas. Our established machine-learning algorithms were applied to distinguish these tumor types within this representative clinical differential diagnosis by randomly splitting all tumors, in an 80%:20% manner, into fifty training and testing sets. The top 300 differentially methylated regions (DMRs) in each one-class-versus-others comparison for each tumor type in training sets were identified.
Fifty negative-binomial generalized linear models for each tumor type were built using DMRs derived from training sets and these models were evaluated in corresponding independent randomly selected testing sets with area under the receiver operating characteristic curve (AUROC) values. The ensemble of fifty models differentiated chordomas from meningiomas and spinal metastases with a high discriminative capacity (mean AUROC = 0.84, 95%CI: 0.52–1.00) as shown in Figure 3A. Model ensembles also distinguished meningiomas from other diagnoses (mean AUROC = 0.92, 95%CI: 0.66–1.00) and spinal metastases from other classes (mean AUROC = 0.88, 95%CI: 0.62–1.00) with similarly high discriminative capacities.
Fig. 3.

Plasma cfDNA methylomes distinguish chordomas from other clinical differential diagnoses of meningiomas and spine metastases while also detecting chordoma subtype signatures. A, Boxplots of AUROC values across 50 iterations of one-class-versus-other models show accurate discrimination of chordomas from differential diagnoses. Mean AUROCs (95% CIs) are 0.84 (0.52–1.00), 0.92 (0.66–1.00), and 0.88 (0.62–1.00) for chordomas, meningiomas, and spinal metastases, respectively. B, Representative scatterplot showing the correlation between tumor tissue methylation beta values and plasma cfMeDIP-seq window CPM values within paired samples from a single chordoma patient. C–E, Representative clinical cases of neuropathologically-diagnosed chordomas where preoperative neuroimaging and clinical factors suggested diagnoses of (C) a skull-base meningioma or (D–E) spine metastases. The top cfMeDIP-seq model class was that of chordoma for all three cases, based on class probabilities in model testing sets, and consistent with the neuropathological diagnosis. F–G, Representative cases of clinical differential diagnoses, including both meningioma (F) and spine metastasis (G) patients, confirmed by histopathology and accurately identified by plasma methylomes according to testing set class probability data. Boxplot central bars indicate medians, boxes show upper and lower distribution quartiles, and whiskers extend across the 1.5× interquartile range. AUROC, area under the receiver operating characteristic curve; cfMeDIP-seq, cell-free methylated DNA immunoprecipitation and high-throughput sequencing; CPM, count per million; MRI, magnetic resonance imaging.
A high correlation between chordoma tumor tissue methylation values and plasma cfMeDIP-seq signals was observed in matched/paired patient samples. Figure 3B displays a representative plot of the correlation between tissue and plasma methylation signals for one patient. Pearson coefficients from all such correlation plots for all patients demonstrate high tumor-to-plasma correlations for both Immune-infiltrated (cluster 1) chordomas (median r = 0.69, 95%CI: 0.66–0.72, P < 2.2 × 10−16 for all) and Cellular (cluster 2) chordomas (median r = 0.67, 95%CI: 0.62–0.72, P < 2.2 × 10−16 for all). In a leave-one-out analysis, models were applied to each held out sample, after being trained on the remaining 11 chordomas, to determine the probabilities of it being in cluster 1 or cluster 2 based on plasma-derived DMRs. Probabilities for each held out sample being in the correct cluster were assessed. The probability of being in the correct cluster was higher than that of the incorrect cluster for all samples. For cluster 1 samples, the median cluster 1 probability was 0.82 (range: 0.81–0.98) and the median cluster 2 probability for cluster 2 samples was 0.92 (range: 0.80–0.95).
Illustrative Clinical Cases Showing Utility of Noninvasive Chordoma Diagnosis
Three representative clinical cases are illustrated, in Figure 3C–E magnetic resonance images, where two expert neuroradiologists provided top differential diagnoses of a skull-base meningioma (C) or a spinal metastasis (D–E) prior to both surgery. In all three cases, histopathology confirmed chordoma diagnoses after surgery and definitively ruled out other diagnoses, including meningioma or metastasis. Figure 3C–E boxplots depict probability values for each tumor type in the differential diagnosis for each of our cfMeDIP-seq based models where the patient shown was randomly assigned into the testing set and not used to build the model. In case C, the median testing set probability of a chordoma diagnosis was 0.54 (range = 0.40–0.67) and significantly higher than 0.10 for meningioma (range = 0.07–0.15). Likewise, in case D, the median testing set chordoma probability of 0.65 (range = 0.60–0.73) was higher than that of a spinal metastasis (median = 0.07, range = 0.05–0.08). Finally, median test set chordoma probability of 0.62 (range = 0.52–0.76) was higher than that of a spinal metastasis (median = 0.09, range = 0.07–0.12) for case E.
Two additional cases are displayed in Figure 3F–G. In these cases, neuropathological diagnoses of a skull base meningioma (F) and a spinal metastasis (G) matched the classification identified by comparing testing set model probabilities using our noninvasive cfMeDIP-seq based models. In case F, the meningioma median test set probability was 0.60 (range = 0.52–0.65) and this was higher than the chordoma probability of 0.14 (range = 0.11–0.22). Similarly, in case G the median spine metastasis probability was 0.47 (range = 0.40–0.54) and greater than that of chordomas (0.17, range = 0.11–0.23).
Discussion
In this study we demonstrate, for the first time, two distinct DNA methylation-based subtypes of chordoma that have prognostic value independent of clinical factors. These methylation-based subtypes contribute to resolving the range of chordoma outcomes that are seen clinically despite standard of care treatment.6 We further characterize these prognostic chordoma subtypes and show that the subtype with a poorer disease-specific survival has higher fractions of immune cells within tumors, including neutrophils and B lymphocytes, which was validated with immunohistochemical staining. This pattern is similar to that of other poorly prognostic immune-related tumors.39 This cluster also shows enrichment of immune-related pathways with genes hypomethylated at promoters. Therefore, we termed this subtype Immune-infiltrated. Comparatively, the better-performing Cellular chordoma subtype shows higher tumor cellularity as well as an enrichment of extracellular matrix and cellular interaction pathways involved in chordoma tissue architecture. Chromosome 10q, 9q, and 14q deletions were identified as genomic correlates in cluster 2 chordomas and tumors with these deletions showed expression of cell adhesion and extracellular matrix related gene sets,40,41 while those without this cluster 2 correlate showed upregulation of immune-related pathways similar to those pathways with hypomethylated promoters in cluster 1. The limited overall efficacy of targeted therapies currently being trialed for chordoma may relate to tumor heterogeneity between methylation-based subtypes, which may potentially respond differently to treatments, although further work to assess this is required.38
Here we show that circulating cell-free DNA methylomes can be used for the diagnosis of chordomas from meningiomas and spinal metastases as representative clinical differential diagnoses. We show that this discrimination can be done with good and comparable accuracy to what we have shown previously for noninvasive identification of other systemic and CNS cancers.24,25 Knowing the preoperative diagnosis of chordoma in patients where it is a potential differential diagnosis will have a considerable impact on how these tumors are managed. Current practice is to obtain preoperative needle biopsies for patients with bone tumors where chordoma is in the clinical differential diagnosis and where a biopsy can be safely performed, as maximal resection and minimal tumor violation during surgery improves outcomes in chordoma and requires upfront operative planning. However, there is a risk of tumor seeding and spreading due to the biopsy procedure, biopsies are typically unfeasible for skull-base or cervical tumors, and many patients with chordoma are not identified for biopsy due to chordoma rarity and so are instead diagnosed at the time of surgery.4,42 Accordingly, noninvasive preoperative biopsies of chordomas may allow for optimal surgical planning while eliminating seeding risk and allowing safe diagnosis of unbiopsiable lesions or those where chordoma is not considered in the differential diagnosis. In these patients, liquid biopsy may either rule out chordoma in favor of potentially more benign tumors amenable to more conservative treatment or, alternatively, identify chordomas preoperatively and ensure that their maximal safe surgical resection with limited tumor violation is planned for in advance to optimize patient outcomes.4,5
In addition to determining the diagnosis preoperatively, we show that plasma methylome signatures are highly consistent with tumor tissue methylation signatures for both prognostic subtypes, suggesting feasibility for liquid biopsies to identify chordoma subtype. We also show a leave-one-out analysis that accurately identifies chordoma subtype. Such noninvasive subtyping has the potential to transform clinical care of patients with chordomas as the aggressiveness of treatment may be appropriately tailored based on whether a chordoma is Immune-infiltrated (cluster 1) with a poorer prognosis or Cellular (cluster 2) with a good prognosis. This information may also guide patient counseling to provide more accurate depictions of patient prognoses based on the methylation subtype of their tumor. In patients with a poorer prognosis, a greater extent of resection may be pursued along with potentially more frequent monitoring for tumor progression using neuroimaging to most optimally delay progression and allow for early treatment of tumor recurrence. Additionally, surgery and radiation for chordoma have significant risks due to proximity to and involvement of critical neurovascular structures and so avoidance of overtreatment in less aggressive chordomas holds promise for improving patient care by reducing the risk for treatment-induced neurological morbidities. A lower risk patient may be considered for nonsurgical treatment with close neuroimaging follow-up to identify tumor progression unless there is evidence of compression of critical structures and/or neurological compromise.
This study presents new knowledge on chordomas, however is limited in sample size due to the relative rarity of this tumor type, in particular for our plasma methylation analyses where our leave-one-out analysis suggested feasibility for identifying chordoma subtype noninvasively. It is important to establish consortiums that can assemble significantly larger cohorts with both tissue and plasma samples for validation of the findings in this study. Further work assessing whether immune infiltration is a driver or marker of poor prognosis, along with whether immune cells may be potential therapeutic targets, is warranted in future studies.19,20
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The GSE14068629 dataset was made publicly available by the German Cancer Research Centre. The phs002301.v1.p114 data was available through the dbGaP and was provided by Beijing Neurosurgical Institute. This research was supported by Beijing Municipal Science and Technology Commission [grant number Z171100000117002], Research Special Fund for Public Welfare Industry of Health [grant number 201402008], and the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Division of Cancer Epidemiology and Genetics.
Conflict of interest statement. D.D.D.C. is co-founder and shareholder of DNAMx, Inc. D.D.D.C. and A.C. are inventors in patents related to cfMeDIP-seq.
Authorship statement. Study design: J.A.Z., V.P., S.M., K.D.A., D.D.D.C., G.Z. Laboratory work and sample processing: J.A.Z., S.M., F.N., S.Ka., O.S., N.S., H.A., K.D.A. Clinical data collection: J.A.Z., F.N., J.P.A., A.B., M.H., H.A. Data analysis: J.A.Z., V.P., S.M., O.S., J.C.L., Y.M. Study supervision: GZ. Data interpretation: J.A.Z., V.P, S.M., F.N., A.C., O.S., K.D.A., D.D.D.C., G.Z. First manuscript draft preparation: J.A.Z., V.P, S.M., G.Z. Final data interpretation, manuscript revision, manuscript approval: all authors.
Funding
This work was supported by the following sources: Canadian Institute of Health Research Canada Graduate Scholarship Doctoral Award (to J.A.Z); Strategic Training in Transdisciplinary Radiation Science for the 21st Century Program Scholarship (to J.A.Z.); Princess Margaret Cancer Foundation Post-Doctoral Fellowship Award (to V.P.); Canadian Institute of Health Research New Investigator salary award [grant number 201512MSH360794-228629 to D.D.D.C.]; Helen M Cooke professorship from Princess Margaret Cancer Foundation (to D.D.D.C.); Canada Research Chair (to D.D.D.C.); Canadian Institute of Health Research Foundation Grant [grant number FDN 148430 to D.D.D.C.]; Canadian Institute of Health Research Project Grant [grant number PJT 165986 to D.D.D.C.]; Natural Sciences and Engineering Research Council of Canada [grant number 489073 to D.D.D.C.]; Terry Fox Research Institute Program Projects Grant [grant number 1106 to D.D.D.C.]; Ontario Institute for Cancer Research (Province of Ontario to D.D.D.C.); Canadian Cancer Society Operating Grant [grant number 706135 to G.Z.].
References
- 1. Carbone A. Cancer classification at the crossroads. Cancers (Basel). 2020; 12(4):980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chugh R, Tawbi H, Lucas DR, Biermann JS, Schuetze SM, Baker LH. Chordoma: the nonsarcoma primary bone tumor. Oncologist. 2007; 12(11):1344–1350. [DOI] [PubMed] [Google Scholar]
- 3. George B, Bresson D, Herman P, Froelich S. Chordomas: a review. Neurosurg Clin N Am. 2015; 26(3):437–452. [DOI] [PubMed] [Google Scholar]
- 4. Stacchiotti S, Sommer J; Chordoma Global Consensus Group. Building a global consensus approach to chordoma: a position paper from the medical and patient community. Lancet Oncol. 2015; 16(2):e71–e83. [DOI] [PubMed] [Google Scholar]
- 5. Stacchiotti S, Gronchi A, Fossati P, et al. Best practices for the management of local-regional recurrent chordoma: a position paper by the chordoma global consensus group. Ann Oncol. 2017; 28(6):1230–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smoll NR, Gautschi OP, Radovanovic I, Schaller K, Weber DC. Incidence and relative survival of chordomas: the standardized mortality ratio and the impact of chordomas on a population. Cancer. 2013; 119(11):2029–2037. [DOI] [PubMed] [Google Scholar]
- 7. Frezza AM, Botta L, Trama A, Dei Tos AP, Stacchiotti S. Chordoma: update on disease, epidemiology, biology and medical therapies. Curr Opin Oncol. 2019; 31(2):114–120. [DOI] [PubMed] [Google Scholar]
- 8. Tarpey PS, Behjati S, Young MD, et al. The driver landscape of sporadic chordoma. Nat Commun. 2017; 8(1):890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sa JK, Lee IH, Hong SD, Kong DS, Nam DH. Genomic and transcriptomic characterization of skull base chordoma. Oncotarget. 2017; 8(1):1321–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Le LP, Nielsen GP, Rosenberg AE, et al. Recurrent chromosomal copy number alterations in sporadic chordomas. PLoS One. 2011; 6(5):e18846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Choy E, MacConaill LE, Cote GM, et al. Genotyping cancer-associated genes in chordoma identifies mutations in oncogenes and areas of chromosomal loss involving CDKN2A, PTEN, and SMARCB1. PLoS One. 2014; 9(7):e101283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Diaz RJ, Guduk M, Romagnuolo R, et al. High-resolution whole-genome analysis of skull base chordomas implicates FHIT loss in chordoma pathogenesis. Neoplasia. 2012; 14(9):788–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang L, Zehir A, Nafa K, et al. Genomic aberrations frequently alter chromatin regulatory genes in chordoma. Genes Chromosomes Cancer. 2016; 55(7):591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bai J, Shi J, Li C, et al. Whole genome sequencing of skull-base chordoma reveals genomic alterations associated with recurrence and chordoma-specific survival. Nat Commun. 2021; 12(1):757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bell AH, DeMonte F, Raza SM, et al. Transcriptome comparison identifies potential biomarkers of spine and skull base chordomas. Virchows Arch. 2018; 472(3):489–497. [DOI] [PubMed] [Google Scholar]
- 16. Bell D, Raza SM, Bell AH, Fuller GN, DeMonte F. Whole-transcriptome analysis of chordoma of the skull base. Virchows Arch. 2016; 469(4):439–449. [DOI] [PubMed] [Google Scholar]
- 17. Jäger D, Barth TFE, Brüderlein S, et al. HOXA7, HOXA9, and HOXA10 are differentially expressed in clival and sacral chordomas. Sci Rep. 2017; 7(1):2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018; 555(7697):469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Capper D, Stichel D, Sahm F, et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol. 2018; 136(2):181–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karimi S, Zuccato JA, Mamatjan Y, et al. The central nervous system tumor methylation classifier changes neuro-oncology practice for challenging brain tumor diagnoses and directly impacts patient care. Clin Epigenetics. 2019; 11(1):185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Olar A, Wani KM, Wilson CD, et al. Global epigenetic profiling identifies methylation subgroups associated with recurrence-free survival in meningioma. Acta Neuropathol. 2017; 133(3):431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alholle A, Brini AT, Bauer J, et al. Genome-wide DNA methylation profiling of recurrent and non-recurrent chordomas. Epigenetics. 2015; 10(3):213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rinner B, Weinhaeusel A, Lohberger B, et al. Chordoma characterization of significant changes of the DNA methylation pattern. PLoS One. 2013; 8(3):e56609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shen SY, Singhania R, Fehringer G, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018; 563(7732):579–583. [DOI] [PubMed] [Google Scholar]
- 25. Nassiri F, Chakravarthy A, Feng S, et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat Med. 2020; 26(7):1044–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nuzzo PV, Berchuck JE, Korthauer K, et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat Med. 2020; 26(7):1041–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shen SY, Burgener JM, Bratman SV, De Carvalho DD. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat Protoc. 2019; 14(10):2749–2780. [DOI] [PubMed] [Google Scholar]
- 28. Sanusi O, Arnaout O, Rahme RJ, Horbinski C, Chandler JP. Surgical resection and adjuvant radiation therapy in the treatment of skull base chordomas. World Neurosurg. 2018; 115:e13–e21. [DOI] [PubMed] [Google Scholar]
- 29. Koelsche C, Schrimpf D, Stichel D, et al. Sarcoma classification by DNA methylation profiling. Nat Commun. 2021; 12(1):498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010; 26(12):1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003; 34(3):267–273. [DOI] [PubMed] [Google Scholar]
- 33. Baghban R, Roshangar L, Jahanban-Esfahlan R, et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal. 2020; 18(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015; 12(5):453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grabovska Y, Mackay A, O’Hare P, et al. Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat Commun. 2020; 11(1):4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chakravarthy A, Furness A, Joshi K, et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat Commun. 2018; 9(1):3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nat Commun. 2015; 6:8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meng T, Jin J, Jiang C, et al. Molecular targeted therapy in the treatment of chordoma: a systematic review. Front Oncol. 2019; 9:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim S, Kim A, Shin JY, Seo JS. The tumor immune microenvironmental analysis of 2,033 transcriptomes across 7 cancer types. Sci Rep. 2020; 10(1):9536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Collins I, Wann AKT. Regulation of the extracellular matrix by ciliary machinery. Cells. 2020; 9(2):278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gupta R, Leon F, Rauth S, Batra SK, Ponnusamy MP. A systematic review on the implications of O-linked glycan branching and truncating enzymes on cancer progression and metastasis. Cells. 2020; 9(2):446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zuccato JA, Witiw CD, Keith J, Dyer E, Saghal A, da Costa L. The importance of preoperative tissue sampling for mobile spine chordomas: literature review and report of two cases. Spinal Cord Ser Cases. 2018; 4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The DNA methylation and cfMeDIP-seq data for patient samples that support the findings of this study are available upon request from the corresponding authors (G.Z.) to comply with institutional ethics regulation.