

Abstract
The generation of chimeric antigen receptor (CAR) T cells requires the transfer of the CAR gene into primary T cells. Among various gene transfer strategies, gammaretroviral vectors have been widely used to generate CAR T cells for both preclinical and clinical settings. Here we describe the detailed method of generating CAR T cells utilizing gammaretroviral vectors. This approach consists of two parallel parts: 1) production of the gammaretroviral particles and 2) gammaretroviral transduction of activated T cells. The gammaretroviral particles are produced by co-transfecting the gammaretroviral vector with packaging plasmids into 293T cells. The manufactured viral particles then efficiently infect activated T cells where the CAR transgene is integrated into host genomic DNA, resulting in stable expression of the CAR molecule on the surface of T cells.
1. INTRODUCTION
Several gene transfer technologies have become readily available to incorporate transgenes encoding chimeric antigen receptors (CARs) into T cells, the most sophisticated of which have been developed preclinically and utilized in the clinical setting (Wang, & Rivière, 2016). These consist of both viral vectors and non-viral gene delivery methods including the transposon-transposase system (Sleeping Beauty, PiggyBac) (Nakazawa, et al., 2011,Singh, et al., 2008) and CAR mRNA transfection/electroporation (Beatty, et al., 2014,Yoon, et al., 2009). The application of transposon-mediated gene transfer leads to stable CAR expression on the T cell membrane while mRNA transfection produces transient CAR expression with the benefit of minimal potential toxicities as CAR T cell could damage healthy normal cells/tissues known as on-target/off-tumor toxicity (Brudno, & Kochenderfer, 2016). Non-viral gene transfer presents the advantages of i) simple manufacturing process and ii) relatively low-cost alternative compared to expensive high titer preparations required for viral vector mediated gene therapies. However, transgene introduction with these methods has yielded inferior expression compared to viral gene delivery methods (Yin, et al., 2014). Gammaretrovirus-derived gammaretroviral vectors and lentivirus-derived lentiviral vectors are most commonly used for viral gene transfer (Maude, et al., 2018,Park, et al., 2018). Both gammaretrovirus and lentivirus belong to the Retroviridae family which are characterized by a single stranded diploid RNA genome composed of coding and cis-regulatory sequences (Kay, Glorioso, & Naldini, 2001). These viruses are well apt for gene delivery as most of their genome can be replaced with the targeted transgene and once infected into host cells, their RNA genome is reverse transcribed into cDNA, which is then stably integrated into the host genomic DNA (Maetzig, et al., 2011,Morgan, & Boyerinas, 2016). These properties aid gammaretroviral or lentiviral vectors in proficient gene delivery and stable expression of CAR transgenes. In this chapter, we describe gene transfer methods specifically with gammaretroviral vectors for the expression of CARs on therapeutic T cells.
The RNA genome of gammaretrovirus coding sequences include gag (structural protein), pol (polymerase) and env (envelope) genes (Fig.1), that are essential for viral replication and formation of viral particles. In contrast to lentivirus, gammaretrovirus lacks a nuclear transporter element, preventing viral cDNA from passing through the nuclear membrane. Therefore, gammaretroviral genes cannot be integrated into host genome in non-dividing cells. To achieve stable gene expression with gammaretroviral vectors, target cells (e.g. T cells) must be actively undergoing mitosis which results in the breakdown of the nuclear membrane, allowing viral cDNA to be transported into nucleus (Miller, Adam, & Miller, 1990,Roe, et al., 1993). To generate CAR T cells with gammaretroviral vectors, stimulation of T cells with antibodies (CD3 - clone OKT3 and anti-CD28) is required to induce robust T cell proliferation to allow gammaretroviral cDNA transport into nucleus and subsequent integration into the host genomic DNA, leading to stable expression of transgenes.
Fig 1. Schema of three plasmids for transient gammaretroviral particle production:
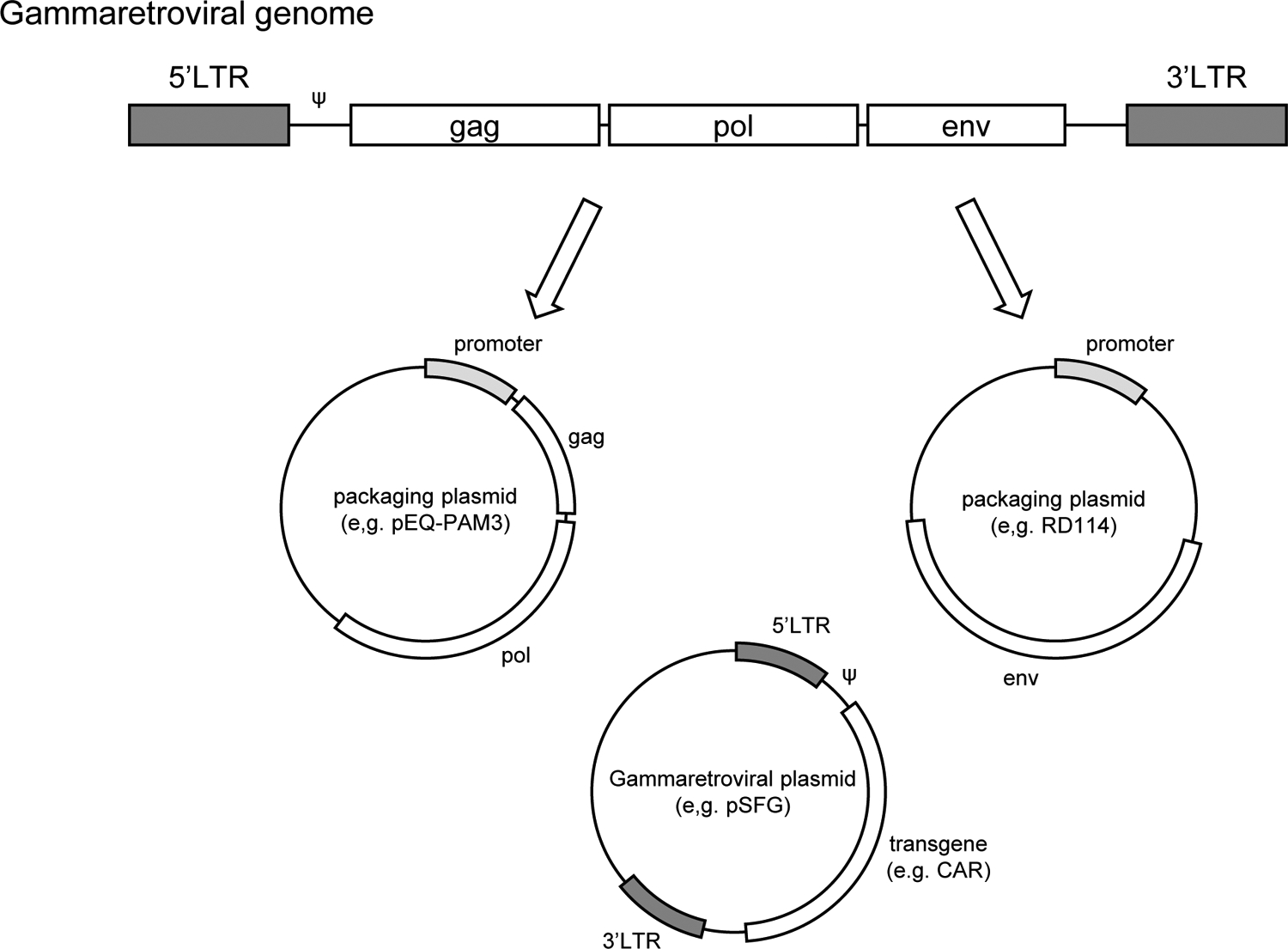
Gammaretroviral genome encoding gag, pol and env are divided into separate vectors(packaging plasmids). Long-terminal repeat (LTR) with packaging signal (ψ) from gammaretroviral genome is used to package transgene into gammaretroviral particles.
Moloney murine leukemia virus (MMLV)-derived gammaretroviral vector has been widely used for transgene expression in gammaretroviral transduction. To encode for the gene of interest (transgene), the coding sequences for gag, pol and env are removed from the retroviral genome, leaving the MMLV long terminal repeat (LTR) regulatory sequence which promotes/enhances transgene expression (Fig.1). Therefore, to generate competent viral particles encoding the gene of interest, the transgene expressing gammaretroviral vector needs to be either co-transfected with helper plasmids (packaging plasmids) encoding gag, pol and env into producer cell line (e.g. 293T cells), or transfected into a packaging cell line which stably expresses gag, pol and env. Providing structural proteins in trans with the gammaretroviral vector expressing the gene of interest reduces the recombination of the vector into a replication competent retrovirus, thus providing increased safety making the gammaretroviral particles replication deficient (Fig. 1). There are several packaging plasmids available for co-transfection. The animal source of the env gene regulates the efficiency of transduction in different cell types creating a broad tropism of retroviral infections. The envelope protein is classified into either ecotropic (mouse or rat cells), amphotropic (majority of mammalian cells) or pantropic (all species). In this chapter, we describe a successful co-transfection method to generate gammaretroviral particles using i) pSFG vector (gammaretroviral vector - transfer plasmid containing the CAR gene of interest), ii) pEQ-PAM (packaging plasmid encoding gag and pol) (Persons, et al., 1998) and iii) RD114 (packaging plasmid encoding env derived from feline endogenous retrovirus, an amphotropic protein reported to be highly efficient in transducing human T cells) (Kelly, et al., 2000). After co-transfection of those plasmids into 293T cells, viral particles containing the CAR gene are assembled and released into culture supernatant (Fig. 2A). The resulting infectious viral particles are used for ensuing transduction into T cells, resulting in the generation of CAR-expressing T cells (Fig. 2B).
Fig 2. Gammaretroviral gene transfer overview:
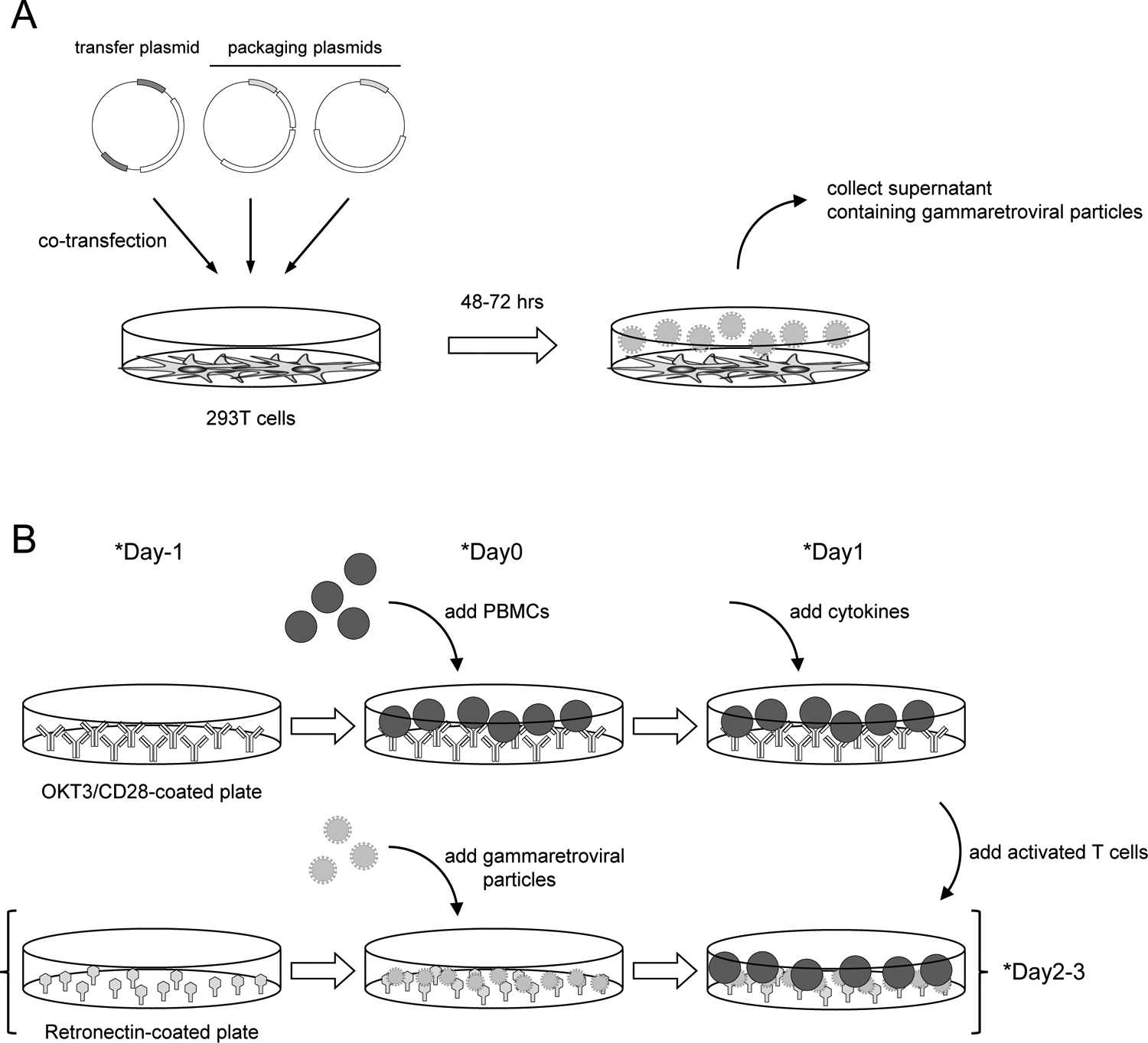
(A) Outline of the production of gammaretroviral particles. Transfection of transfer plasmid and packaging plasmids into 293T cells results in the production of gammaretroviral particles in the culture superatant. (B) Outline of CAR T cell generation process. PBMCs are stimulated with CD3/CD28 to promote their proliferation (topline). Culture supernatant containing gammaretroviral particles is added to Retronectin-coated plate and then centrifuged (bottom line). Activated T cells are added onto gammaretroviral particle-bound Retronectin plate for transduction. *Timeline can be modified.
2. CELL CULTURE AND MAINTENANCE OF 293T CELLS
All procedures should be performed in a tissue culture hood with aseptic technique. 293T cells (ATCC CRL-3216) are routinely maintained at 37°C, 5% CO2, in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% Fetal bovine serum (FBS) and 1X GlutaMAX in T75 (15 mL) or T175 flask (30 mL). Culture medium should be pre-warmed (37°C) before use (see Note 1).
Recover frozen 293T cells by thawing cryovial in a 37°C water bath. DO NOT completely thaw the cells. There should be a small frozen ice pellet in the vial when removing from the water bath.
Spray the cryovial with 70% EtOH and wipe before bringing it into the tissue culture hood.
Add 9ml of pre-warmed IMDM media to a 15ml conical tube.
Using a p1000 pipette, transfer 1ml of prewarmed media from the 15ml conical tube in step 3 to the cryovial. Then transfer media and cells back to the 15ml tube drop by drop.
Centrifuge the tube at 150 xg for 5 min (see Note 2).
Aspirate the supernatant and break the cell pellet by finger flicking. Resuspend cells with appropriate volume of culture medium and transfer them to tissue culture flask. Place the culture flask in a 37°C incubator.
Monitor the health of 293T cells regularly under the microscope by observing cell morphology and viability. 293T cells should attach to the bottom of the culture flask and exhibit some dendrites. Passaging of 293T cells should be done before reaching 90% confluency as follows (see Note 3).
To passage 293T cells, aspirate culture medium, add PBS slowly (5 mL for T75 flask, 10 mL for T175 flask) without disturbing attached 293T cells and rinse by rocking the flask 3–5 times.
Aspirate PBS and add 0.25% trypsin/EDTA (3 mL for T75 flask, 5 mL for T175 flask). Rock the flask to make sure it covers entire surface and place the flask in 37°C incubator to incubate 3–5 min.
Remove the flask from the incubator and rock 3–5 more times. Verify that the 293T cells have detached from the culture by looking under the microscope. If not, place the flask back in the incubator and incubate another 3–5 min (see Note 4).
Add serum containing culture medium (10%FBS IMDM) to inactivate trypsin (7 mL for T75 flask, 15 mL for T175 flask) and thoroughly mix by pipetting up and down with a serological pipette.
Passaging of 293T cells for maintenance in culture can be completed at a 1:3 to 1:8 split according to ATCC but could be further diluted to a 1:20 split observed from personal experience.
3. PRODUCTION OF RETROVIRAL SUPERNATANT (TRANSFECTION OF RETROVIRAL CONSTRUCT INTO 293T CELLS)
The following procedure is for transfecting one 10 cm tissue culture dish. This will produce 20 mL of viral supernatant (see Note 5).
Day −1: Plating of 293T cells for transfection
Harvest 293T cells as described above (Section 2. step 8–11).
Transfer single cell suspension to 15 or 50 mL conical tube (pending T75 or T175 culture flask).
Centrifuge at 400 xg for 5 min.
Aspirate supernatant carefully and break cell pellet by finger flicking.
Resuspend the cells with appropriate volume of culture medium and acquire the cell count.
Adjust the cell concentration to 1.5–3.0×106 cells / 10 mL by adding culture medium and plate 10 mL of cell suspension into 10 cm tissue culture dish (see Note 6).
Rock the plate in various directions to ensure that 293T cells distribute equally across the entire surface. DO NOT swirl the plate, otherwise the cells will be concentrated at the center of the tissue culture dish, which will impair 293T cell growth and transfection efficiency.
Place the plate in a 37°C incubator and incubate 18–24hrs.
Day 0: Transfection
The following procedure is for a 10 cm dish with GeneJuice reagent (see Note 7). The confluency of 293T cells should be around 70% at the time of transfection to maximize virus production.
Add 470 μL of pure IMDM (no serum, no anti-biotics) to an appropriate tube (1.5mL Eppendorf tube or 50 mL conical tube) followed by 30 μL of GeneJuice reagent per construct. Mix gently by pipetting 3–4 times (label tube #1) (see Note 8).
Incubate at room temperature for 5 min (see Note 9).
- Meanwhile, prepare DNA plasmid mixture in a separate 1.5 mL tube as follows (label tube #2) (see Note 10);
a. Gammaretroviral vector (pSFG) 3.75 μg b. pEQ-PAM 3.75 μg c. RD114 2.5 μg Transfer 500 μL from tube #1 to tube #2. Mix gently by pipetting 3–4 times.
Incubate at room temperature for 15 min. This incubation is required for the GeneJuice reagent and DNA(s) to form a lipophilic complex in order to enter the 293T cells.
Remove 293T tissue culture dish from the incubator. Mix the GeneJuice + DNA mixture gently and slowly by pipetting 2–3 times (see Note 11) and then transfer the complete volume onto 293T cells drop-by-drop in a spiral direction to cover entire plate.
Rock the plate several times in different directions to ensure the DNA complexes are evenly distributed across the entire plate.
Place the plate in a 37°C incubator and incubate for 48 hrs (see Note 12).
Day 2: Collection of viral supernatant (48hrs post transfection)
Prewarm complete IMDM culture media in 37°C water bath.
Remove transfected plate from the incubator.
Retroviral particles are being produced by 293T cells and secreted in the supernatant. Tilt the plate toward you, collect supernatant with a 10 mL serological pipette and transfer to a 15 or 50 mL conical tube.
Carefully replenish the media by adding 10ml pre-warmed culture medium into 10 cm dish along the plate wall to avoid disrupting attached 293T cells (see Note 13).
Place the transfected 293T cells back into the incubator for another collection of viral supernatant the following day.
If you need to use viral supernatant right away for transduction, filter viral supernatant with 45 μm syringe disk filter. To store the filtered viral supernatant, aliquot the volume into several conical tubes to avoid multiple freeze-thaw cycles. Place conical tubes into ethanol (or isopropanol) + dry ice and wait 10 min to perform snap freezing. Transfer and store the frozen viral supernatant in a −80 °C freezer. Alternatively, store the viral supernatant at 4°C overnight and mix with the subsequent 72hr viral supernatant (see Note 14).
Day 3: Collection of viral supernatants (72hrs) and checking transfection efficiency
Remove the transfected plate from the incubator (see Note 15).
Tilt the plate towards you, collect and transfer supernatant by using 10 mL serological pipette into 15 or 50 mL conical tube (see Note 16).
To check transfection efficiency, carefully add 3–5 mL of PBS along the plate wall without disrupting the attached 293T cells. Rock 3–5 times to rinse (see Note 17).
Store the viral supernatant as described before (Day2 - step 5), or use filtered viral supernatant for transduction.
Remove PBS from 293T cell plate and carefully add 3 mL of 0.25% trypsin/EDTA. Rock the plate to make sure it covers entire surface of the plate. Place the plate into 37°C incubator and incubate 3–5 min.
Remove the plate from the incubator and rock 3–5 times. Confirm 293T cells detachment under the microscope. If not, place the plate back to incubator and incubate for another 3–5 min.
Add 7 mL of serum containing culture medium to inactivate trypsin and mix by pipetting up and down.
Take 0.5–1 mL of cell suspension to check transfection efficiency by staining them with appropriate CAR detection antibody and measure expression by flow cytometry. In general, you can expect that more than 50% of 293T cells express the CAR.
4. RETROVIRUS TRANSDUCTION OF T CELLS
All procedures should be performed in a tissue culture hood with aseptic technique. T cells are cultured and expanded in a T cell culture medium consisting of 45% RPMI-1640, 45% Click’s medium, 10% FBS and 1X GlutaMax (see Note 18). The following procedure is for the use of a multi antibody-coated plate to activate T cells (see Note 19).
Day 0: Stimulation of PBMCs with CD3/CD28 antibodies
To prepare the antibody-coated plate: dilute anti-CD3 (clone: OKT3) and anti-CD28 antibodies in sterile water to a final concentration of 1 μg/ml and add 0.5 mL per well in a 24-well non-tissue culture treated plate.
Place the plate into 37°C incubator and incubate 2–3 hours. Alternatively, seal the plate with parafilm and leave at 4°C overnight.
After the incubation, aspirate CD3/CD28 solution from each well and add 1 mL of T cell culture medium along the well wall to rinse.
Resuspend and adjust the concentration of PBMCs at 1×106 cells in 2 mL with T cell culture medium (see Note 20).
Aspirate culture medium from the antibody-coated plate and add 2 mL of PBMCs suspension per well (1×106 cells / well).
Place the plate in a 37°C incubator.
Day 1: Supplementation of cytokines
Remove the plate from the incubator and add IL7 and IL15 to reach a final concentration of 10 ng/mL (see Note 21).
Place the plate into 37°C incubator.
Day 2 or Day3: Transduction (see Note 22)
Calculate how many wells are required for transduction in non-tissue culture treated 24-well plate.
Dilute Retronectin in sterlie PBS at 7 μg/mL and add 1 mL per well.
Place the plate in a 37°C incubator and incubate 2–3 hrs. Alternatively, seal the plate with parafilm and leave at 4°C overnight.
After the incubation, aspirate Retronectin solution from the wells and add 1 mL of T cell culture medium along the well wall to rinse and place in the incubator for 30 min (see Note 23).
Thaw the viral supernatant in a 37°C water bath (or use fresh viral supernatant).
Aspirate the culture medium from the Retronectin-coated plate and add 1 mL of viral supernatant along the well wall (see Note 24).
Centrifuge the plate at 2,000 xg for 2 hrs at 32°C (see Note 25).
Meanwhile, collect activated T cells from CD3/CD28 plate and transfer to 15 or 50 mL conical tubes.
Centrifuge T cells at 400 xg for 5 min.
Aspirate supernatant and break the cell pellet by finger flicking. Resuspend T cells with cytokine-containing T cell medium and count.
Adjust the cell concentration to 0.2×106 cells / 2 mL (see Note 26).
After centrifugation of the viral supernatant, aspirate viral supernatant from the wells and add T cell culture medium along the well wall to rinse (see Note 27).
Aspirate culture medium from the wells and add 2 mL of T cell suspension along the well wall (0.2×106 / well) (see Note 28).
Centrifuge the plate at 1,000 xg for 10 min at room temperature.
Place the plate into the incubator and leave cells on transduced plate for at least 4 hrs for efficient viral entry.
Post transduction: Maintenance of T cell culture
Transfer transduced T cells to a tissue culture treated plate 24–48hrs post transduction.
Monitor cell density and medium color. Split cells into multiple wells and / or replenish T cell culture medium with cytokines as needed.
Culture T cells at least 72 hrs before checking CAR expression (see Note 29).
5. CONCLUDING REMARKS
The procedure outlined here should induce high expression of CAR on the surface of T cells. As we described, two separate steps are required to generate CAR T cells; i) production of gammaretroviral particles and ii) transduction of gammaretroviral particles into T cells. Depending on the choice of reagent, vectors and the method to activate T cells, each step will need to be carefully optimized. However, in general you can expect to achieve more than 60% of CAR-expressing T cells by utilizing gammaretroviral vectors. Since there are two separate steps, a low transduction efficiency could be due to the failure of production of retroviral particles, transduction technique, or both. Failure of CAR expression can also be attributed to a mutation in the sequence encoding the CAR gene that may lead to an incorrect open reading frame or misfolding of the CAR protein after the translation which leads to proteosome degradation. In contrast, too high expression of the CAR can cause toxicity to T cells as described elsewhere (Gomes-Silva, et al., 2017). The expression levels of the CAR can be fine-tuned by using i) varying amounts of retroviral supernatant (i.e., less amount of retroviral supernatant will reduce CAR expression level), and ii) different number of T cells used for transduction (i.e., more cells will reduce the CAR expression level). Based on the described protocol, it is best to optimize each step depending on the CAR construct of choice in order to have optimal CAR expression on T cells.
6. NOTE
293T cells can be maintained in Dulbecco’s Modified Eagle Medium (DMEM), IMDM or RPMI-1640 media supplemented with 10 % FBS and 2 mM L-glutamine.
It is recommended to centrifuge thawed cells at lower speed to reduce the damage.
Leaving 293T cells at full confluency may result in a change their morphology and impact their growth rate after passaging. It is very important to maintain 293T cells at optimal confluency for consistent results.
Do not leave trypsin on the cells for too long as over-trypsinization will reduce the viability.
Other types of tissue culture plate/dish/flask can be used for plating 293T cells for transfection. The amount of transfection reagents and medium should be scaled up or down proportionally based on the surface area of the tissue culture vessels.
The target cell concentration is dependent on the growth rate of 293T cells which is affected by the number of passages and confluency of 293T cells at the time of harvest. You want to achieve 70% cell confluency at the time of transfection the day after plating.
Transfection can be done with other transfection reagents such as Lipofectamine and FuGene. The transfection protocol should be optimized according to your transfection reagent of choice.
You can adjust the amount of reagent and medium depending on the number of constructs you intend to transfect. In this case, it is recommended to make a mixture for the number of constructs +1 with consideration for pipette error.
If you transfect multiple constructs, it is better to prepare tube #2 first, then make tube #1 since incubation time of tube #1 is critical.
The amount of gammaretroviral vector and packaging plasmids may need to be adjusted based on the vector of choice. All the plasmids used for transfection should be sufficiently concentrated in water to avoid excessive dilution of the transfection reagent.
DO NOT mix vigorously otherwise you will disrupt the complex.
If you observe toxicity due to the transfection reagent, you can replace the culture medium at 18–24 hrs post transfection.
To avoid drying up the plate, steps 3–4 should be completed on a per plate basis.
You can either freeze the 48hrs viral supernatant separately from 72hrs viral supernatant to maximize the titer, or store 48hrs viral supernatant at 4°C overnight and mix with 72hrs viral supernatant and freeze to equalize the titer.
Take 48hr viral supernatant stored in the 4°C if you plan to combine with 72hr supernatant.
If you have 48hrs viral supernatant, you can transfer 72hrs viral supernatant into those tubes and mix.
This step needs to be done before proceeding to the cryopreservation of viral supernatant to prevent drying up of 293T cells.
Medium components are not restricted to this formula. You can use other medias to support T cell expansion.
T cells can be activated by other methods such as antibody-coated beads or phytohemagglutinin (PHA) + IL2. Details of transduction protocol including cell number and timing of the transduction needs to be determined based on the activation method of choice.
Both freshly isolated PBMCs and frozen PBMCs can be used. If you use frozen PBMCs, refer to section 2, steps 1–6.
Other cytokines can be used here including IL2, IL18, IL21 or a combination.
CD3/CD28 blasts can be transduced on day 2 or 3 post stimulation.
This step will i) ensure the removal of excess soluble Retronectin which may block the binding sites on the virus and cells and ii) block the virus from binding to exposed hydrophobic plastic surfaces that may result in loss of the virus.
Depending on the titer of your viral supernatant and the expression level you want, the amount of viral supernatant to add to the well may vary. You can also use mixed-viral supernatant for co-transduction if you expect similar retroviral titers for both viral supernatants. Otherwise, high titer viruses will be integrated into most of the available integration site and impair the integration of other viruses. In this case, it is better to perform serial transductions on days 2 and 3 for co-expression.
In our personal experience, centrifugation for 1.5–2 hrs at room temperature yields similar results.
Cell numbers may vary. Increasing the cell number might result in better cell viability after transduction but lower the transduction efficiency.
Perform this step quickly to avoid drying up the wells. If you have multiple wells, it is better to split into multiple times.
Perform this step quickly to avoid drying up the wells. If you have multiple wells, it is better to split them into sections to complete.
This will allow for viral integration into genome and transgene expression to take place.
ACKNOWLEDGMENTS
This work was supported by the Lymphoma SPORE DRP award and by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number 5T32HL092332-17 supervised by Dr. Helen Heslop.
REFERENCES
- Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G et al. (2014). Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer immunology research, 2, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno JN & Kochenderfer JN (2016). Toxicities of chimeric antigen receptor T cells: recognition and management. Blood, 127, 3321–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes-Silva D, Mukherjee M, Srinivasan M, Krenciute G, Dakhova O, Zheng Y et al. (2017). Tonic 4–1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell reports, 21, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay MA, Glorioso JC & Naldini L (2001). Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nature medicine, 7, 33–40. [DOI] [PubMed] [Google Scholar]
- Kelly PF, Vandergriff J, Nathwani A, Nienhuis AW & Vanin EF (2000). Highly efficient gene transfer into cord blood nonobese diabetic/severe combined immunodeficiency repopulating cells by oncoretroviral vector particles pseudotyped with the feline endogenous retrovirus (RD114) envelope protein. Blood, 96, 1206–1214. [PubMed] [Google Scholar]
- Maetzig T, Galla M, Baum C & Schambach A (2011). Gammaretroviral vectors: biology, technology and application. Viruses, 3, 677–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H et al. (2018). Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. The New England journal of medicine, 378, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DG, Adam MA & Miller AD (1990). Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Molecular and cellular biology, 10, 4239–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA & Boyerinas B (2016). Genetic Modification of T Cells. Biomedicines, 4, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa Y, Huye LE, Salsman VS, Leen AM, Ahmed N, Rollins L et al. (2011). PiggyBac-mediated cancer immunotherapy using EBV-specific cytotoxic T-cells expressing HER2-specific chimeric antigen receptor. Molecular therapy : the journal of the American Society of Gene Therapy, 19, 2133–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ et al. (2018). Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. The New England journal of medicine, 378, 449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persons DA, Mehaffey MG, Kaleko M, Nienhuis AW & Vanin EF (1998). An improved method for generating retroviral producer clones for vectors lacking a selectable marker gene. Blood cells, molecules & diseases, 24, 167–182. [DOI] [PubMed] [Google Scholar]
- Roe T, Reynolds TC, Yu G & Brown PO (1993). Integration of murine leukemia virus DNA depends on mitosis. The EMBO journal, 12, 2099–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H et al. (2008). Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer research, 68, 2961–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X & Rivière I (2016). Clinical manufacturing of CAR T cells: foundation of a promising therapy. Molecular therapy oncolytics, 3, 16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR & Anderson DG (2014). Non-viral vectors for gene-based therapy. Nature reviews. Genetics, 15, 541–555. [DOI] [PubMed] [Google Scholar]
- Yoon SH, Lee JM, Cho HI, Kim EK, Kim HS, Park MY et al. (2009). Adoptive immunotherapy using human peripheral blood lymphocytes transferred with RNA encoding Her-2/neu-specific chimeric immune receptor in ovarian cancer xenograft model. Cancer gene therapy, 16, 489–497. [DOI] [PubMed] [Google Scholar]