

Abstract
BACKGROUND:
Newborn screening for lysosomal storage diseases (LSDs) has been gaining considerable interest owing to the availability of enzyme replacement therapies. We present a digital microfluidic platform to perform rapid, multiplexed enzymatic analysis of acid α-glucosidase (GAA) and acid α-galactosidase to screen for Pompe and Fabry disorders. The results were compared with those obtained using standard fluorometric methods.
METHODS:
We performed bench-based, fluorometric enzymatic analysis on 60 deidentified newborn dried blood spots (DBSs), plus 10 Pompe-affected and 11 Fabry-affected samples, at Duke Biochemical Genetics Laboratory using a 3-mm punch for each assay and an incubation time of 20 h. We used a digital microfluidic platform to automate fluorometric enzymatic assays at Advanced Liquid Logic Inc. using extract from a single punch for both assays, with an incubation time of 6 h. Assays were also performed with an incubation time of 1 h.
RESULTS:
Assay results were generally comparable, although mean enzymatic activity for GAA using microfluidics was approximately 3 times higher than that obtained using bench-based methods, which could be attributed to higher substrate concentration. Clear separation was observed between the normal and affected samples at both 6- and 1-h incubation times using digital microfluidics.
CONCLUSIONS:
A digital microfluidic platform compared favorably with a clinical reference laboratory to perform enzymatic analysis in DBSs for Pompe and Fabry disorders. This platform presents a new technology for a newborn screening laboratory to screen LSDs by fully automating all the liquid-handling operations in an inexpensive system, providing rapid results.
Newborn screening (NBS)3 was introduced in the US to screen for phenylketonuria, an inherited metabolic disorder that is best treated soon after birth (1), and has subsequently expanded globally to include many more conditions. The field of NBS has greatly benefited from the introduction of tandem mass spectrometry (MS/MS) as a multiplex technology to screen for >30 conditions from a single dried blood spot (DBS) specimen (2). Along with the development of corrective therapies for some lysosomal storage diseases (LSDs) such as enzyme replacement therapies (3), the general interest in screening for a panel of treatable LSDs has acquired impetus from the fact that methods to screen for these conditions in DBSs are also available (4). Effective enzyme replacement therapy is available for Pompe disease [also known as glycogen storage disease type II, caused by acid α-glucosidase (GAA) deficiency], Fabry disease [α-galactosidase (GLA) deficiency], Hurler syndrome (mucopolysaccharidosis type I,α-iduronidase deficiency), Hunter disease (mucopolysaccharidosis type II, iduronate-2-sulfatase deficiency), Maroteaux–Lamy disease (mucopolysaccharidosis type VI, N-acetylgalactosamine 4-sulfatase deficiency), and Gaucher disease (glucocerebrosidase deficiency) (5, 6). The disorders that will benefit the most from NBS are those that present with irreversible organ damage if not diagnosed and treated early in the course of the disease, which is true of several LSDs.
Newborn screening for Pompe and Fabry disorders can be performed by use of enzymatic functional assays based on either fluorometry (7, 8) or MS/MS (9–11) or by measurement of enzyme concentration based on immunoassay (12). New York state has been screening newborns for Krabbe disease by MS/MS since 2006 (13), and a research study of the application of the MS/MS-based enzyme assay for Pompe disease in the Austrian NBS program was recently published, with just over 10 000 newborn specimens (14). Other state programs (in Illinois, Missouri, and New Mexico) have recently mandated screening for certain LSDs. Using a modified version (15) of the fluorometric assay introduced by Chamoles et al. (8), the NBS program in Taiwan has screened 206 088 newborns for Pompe disease (16) and 171 977 for Fabry disease (17). A recently published review article provides a nice overview of the current state of NBS for LSDs (18).
Here, we present a digital microfluidic lab-on-a-chip (19) method as a novel platform for multiplex fluorometric enzymatic assays to screen for LSDs in <2 h using the extract from a single punch from a DBS sample. Essentially, the digital microfluidic platform automates all the liquid-handling steps involved in a standard benchtop fluorometric method and integrates all these steps onto a single disposable cartridge that is controlled by a desktop-sized instrument. The microfluidic method fully leverages the existing knowledge on enzymatic assays and provides complete automation, using a fraction of the sample and reagent volumes required by other methods (20). Assay times are also significantly reduced, so any newborn screening laboratory can take full advantage of this turnkey solution. In this study, we compared the GAA (EC 3.2.1.20) and GLA (EC 3.2.1.22) enzyme activities obtained using the digital microfluidic method with benchtop 96-well microplate fluorometric methods performed at Duke University Biochemical Genetics laboratory (BGL). By incorporation of a sensitive fluorometer on the digital microfluidic platform, incubation time was further reduced from 6 h to 1 h, thereby paving the way for rapid and multiplexed screening of LSDs.
Materials and Methods
DBS SAMPLES
Deidentified, presumed-normal DBS (NBS cards) samples were discarded specimens provided by the California Department of Public Health (CDPH) and the North Carolina Division of Public Health (NCDPH) NBS Laboratories, under individual material transfer agreements. DBS samples from CDPH were approximately 3 months old and stored at −20 °C; the samples from NCDPH were 1–2 days old. We obtained affected DBSs for Pompe (n = 10) and Fabry (n = 11) disease from Duke University Medical Center and the CDC. The affected spots, which were not from newborns, were anonymized and obtained under an institutional review board–approved protocol. QC DBS samples obtained from the CDC were stored at −20 °C.
REAGENTS
We obtained 4-methyumbelliferyl α-d-galactopyranoside (4-MU-α-gal), N-acetyl-d-galactosamine (GalNac), 4-MU-α-d-glucopyranoside (4-MU-α-gluc), acarbose, 4-methylumbelliferone sodium salt (4-MU), sodium acetate (99% pure), sodium bicarbonate, and Tween 20 from Sigma-Aldrich. Acetic acid (glacial) was from Fluka (Sigma-Aldrich), molecular-grade water was from Fisher Scientific, and 5cSt silicone oil was from Gelest Inc.
SAMPLE AND REAGENT PREPARATION
At the outset of the method comparison study, all the blood spots were punched out and stored at −20 °C before assay according to the following protocol. We obtained 3 3-mm punches from each of the 60 normal NBS cards and 2 from each of the 21 affected spots and placed them into separate microcentrifuge tubes. One punch from each normal and affected DBS was stored at Advanced Liquid Logic Inc. (ALL), and the remaining punches were stored at Duke BGL. Ten punches were obtained from each of the QC spots (low, medium, and high) and stored in separate microcentrifuge tubes at ALL. We prepared stock enzymatic substrate solutions in DMSO and stock inhibitor solutions in the respective assay buffers. Aliquots of the bulk substrate and inhibitor solutions were prepared and stored at −20 °C. Assay buffer, extraction buffer (EXT), and stop buffer (STB) solutions were stored in 15-mL tubes at room temperature. We diluted the substrate and inhibitor stock solutions using their respective assay buffers to result in the final reagent formulations that were used in the enzymatic analysis as presented in Table 1.
Table 1.
Comparison of microplate and microfluidics assay protocols for Pompe and Fabry disorders.
| Pompe assay (GAA) | Fabry assay (GLA) | |||
|---|---|---|---|---|
| Microplate (Duke) | Microfluidics (ALL) | Microplate (Duke) | Microfluidics (ALL) | |
| EXT | DI water | 0.1% (wt/vol) Tween20 in water | 1% sodium taurocholate in water | 0.1% (wt/vol) Tween20 in water |
| Extraction time and conditions | 360 μL EXT; incubated 1 h on ice | 100 μL EXT; incubated 30 min at room temperature | 250 μL EXT; incubated 1 h on ice | 100 μL EXT; incubated 30 min at room temperature |
| Substrate stock | 70 mmol/L in DMSO | 70 mmol/L in DMSO | 700 mmol/L in DMSO | 700 mmol/L in DMSO |
| Inhibitor stock | 80 μmol/L | 8 mmol/L | 0.5 mol/L in water | 750 μmol/L in water |
| Final reagent during incubation | 0.7 mmol/L 4-MU-α-gluc in sodium acetate, pH 3.8, with 8 μmol/L acarbose | 2.5 mmol/L 4-MU-α-gluc in sodium acetate, pH 3.8, with 6 μmol/L acarbose | 3.5 mmol/L 4-MU-α-gal in citrate-phosphate buffer, pH 4.5, with 75 mmol/L GalNac | 5 mmol/L 4-MU-α-gal in 40 mmol/L sodium acetate, pH 4.5, with 75 μmol/L GalNac |
| Incubation conditions | 20 h at 37 °C | 6 h at 37 °C | 20 h at 37 °C | 6 h at 37 °C |
| STB | 0.15 mol/L EDTA, pH 11.3 | 0.2 mol/L NaHCO3, pH 10.0, 0.01% (wt/vol) Tween20 | 0.15 mol/L EDTA, pH 11.3 | 0.2 mol/L NaHCO3, pH 10.0, 0.01% (wt/vol) Tween20 |
PROTOCOLS FOR THE BENCHTOP FLUOROMETRIC ENZYME ASSAYS
Individual 3-mm DBS punches obtained from NBS cards and DBS cards from the control and patient groups were assayed for Pompe and Fabry diseases using benchtop fluorometry methods previously described (7, 8, 15, 21). All procedures were carried out according to standard operating procedures in the Duke BGL, which is a College of American Pathologists (CAP) and Clinical Laboratory Improvement Amendment (CLIA) certified clinical laboratory.
Briefly, for Pompe disease, GAA activity in DBS extracts was determined fluorometrically using the artificial substrate 4-methylumbelliferyl-α-d-glucoside. Acarbose was used to inhibit maltase-glucoamylase, an enzyme present in polymorphonuclear neutrophils. The residual activity measured at acidic pH (3.8) in the presence of inhibitor is presented as pmol/punch/h. Similarly, for Fabry disease, extracts from 3-mm punches were tested for GLA activity using an artificial fluorogenic substrate (4-MU-α-gal) in the presence of an inhibitor, GalNac, for blocking the activity of α-N- acetylgalactosaminidase at acidic pH (4.5). Results are reported as pmol/punch/h of GLA activity. For comparison purposes, activity values were converted from the usually reported pmol/punch/h to μmol/L/h under the assumption that a 1/8-inch DBS punch contains 3.1 μL of whole blood (22).
PROTOCOLS FOR THE DIGITAL MICROFLUIDIC FLUOROMETRIC ENZYME ASSAYS
Over a period of 10 days, we analyzed 11 samples (6 normal NBS punches, 1 Pompe-affected DBS, 1 Fabry-affected DBS, and 1 each of low, medium, and high QC DBS samples) each day on a single digital microfluidic cartridge. Each set of 11 NBS/DBS punches in their respective microcentrifuge tubes was removed from the freezer, and extraction was performed by adding 100 μL EXT and incubating for 30 min on a shaker at room temperature. After extraction, the tubes were centrifuged briefly (2152g for 30 s), and the supernatant fluids were transferred within a few minutes to the digital microfluidic cartridge sample reservoirs for assay. The digital microfluidic cartridge used to perform the enzymatic assays for this study is depicted in Fig. 1. It is similar to one used for performing immunoassays and PCR, the details of which are published elsewhere (23). Briefly, the cartridge consists of 12 sample reservoirs and 8 reagent reservoirs. Sample reservoirs were loaded with 3.4 μL from each of the 11 DBS extracts. Two reagent reservoirs were loaded with 6.4 μL GAA or GLA reagents. Other reagents (STB, EXT) required for the analysis were loaded into other available reservoirs as depicted in Fig. 1. The entire space between the cover plate and circuit board (approximately 3 by 5 cm by 0.3 mm) was filled with approximately 2 mL 5cSt silicone oil with 0.1% Triton X-15 to minimize evaporation and facilitate droplet transport. In the following procedure, each unit droplet has a volume of 0.3 μL.
Fig. 1. Disposable digital microfluidic cartridge used for multiplexed enzyme analyses.
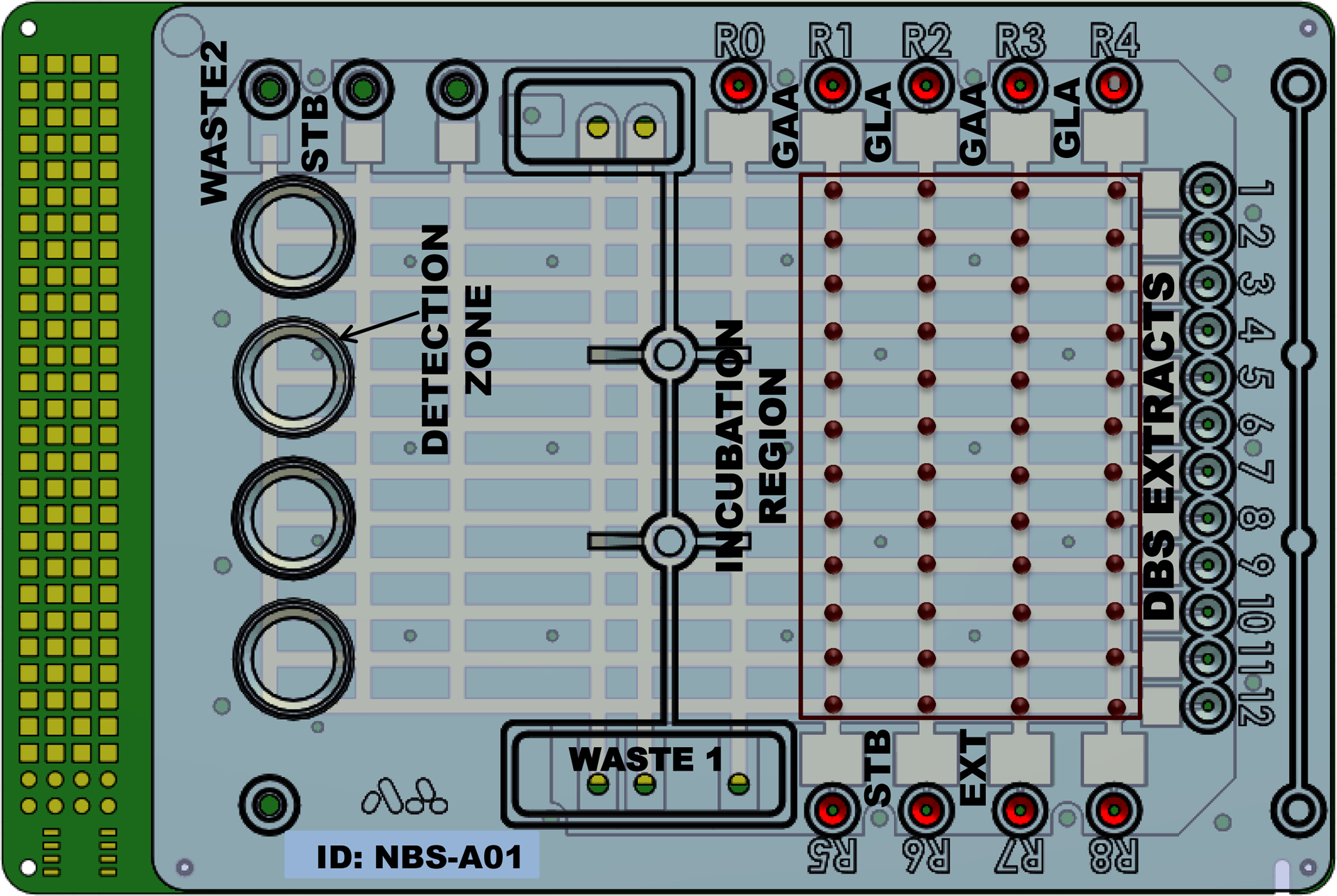
DBS extracts are loaded into reservoirs 1–12 and reagents in reservoirs R1–R8 as needed. All liquid handling is digitally programmed and automated using droplets of 0.3 μL volume.
We measured fluorescence (excitation 365 nm/emission 450 nm) at a specific location on the cartridge (the detection zone, see Fig. 1) using a custom-built microfluorometer with a limit of detection of 4-MU at pH 10.0 of 5.5 nmol/L. We calibrated each cartridge by measuring a known concentration of 4-MU as described below. Two unit droplets of STB were dispensed from the STB reservoir (labeled R5 in Fig. 1) and merged to form a double-sized droplet. Background fluorescence of the stop buffer droplet was recorded as a low calibrant value. Two unit droplets of the calibrant solution were dispensed from 1 of the 12 sample reservoirs and merged to form a double-sized droplet. Fluorescence from this droplet was measured and recorded as high calibrant value. We used a 2-point calibration curve from these 2 values to convert absolute fluorescence values into moles of 4-MU produced. Before starting the enzymatic reaction, we obtained background fluorescence readings of the substrate formulations (GAA and GLA) from the fluorescence signal of a substrate droplet mixed with EXT and STB (R5 in Fig. 1), simulating the assay protocol without the DBS sample. Eleven unit droplets of GAA reagent were dispensed, and each merged with 1 droplet dispensed from each of the sample reservoirs, resulting in 11 double-sized droplets. We repeated this process with the other 3 reagent reservoirs to form a total of 44 double-sized reaction droplets, which were split, resulting in 2 sets of unit-sized droplets for each reaction. Each of the first set of unit-sized droplets was merged with 1 droplet of STB, and the fluorescence was recorded as the t = 0 measurement for all the reaction droplets. The second set of unit-sized droplets was incubated for 6 h at 37 °C. After incubation, each reaction droplet was mixed with 1 droplet of STB, and endpoint fluorescence was measured. Enzymatic activities were expressed as micromoles of substrate hydrolyzed per hour of incubation per liter of blood in the sample.
PROTOCOLS TO DETERMINE VARIABILITY IN ENZYME ACTIVITY
We determined intercartridge variation in enzymatic activity by use of the CDC QC samples (QCL, QCM, and QCH, corresponding to low, medium, and high concentrations) that were analyzed over a period of 10 days as described above. We determined intracartridge variability from a single QCH sample that was loaded in all 11 sample reservoirs on a single cartridge and analyzed for GAA and GLA assays using the protocols described above. Intracartridge, punch-to-punch variability was determined using DBS extracts that were obtained from 10 different punches from a single QCH DBS sample, which were then analyzed for GAA and GLA activities on a single cartridge.
DIGITAL MICROFLUIDIC PROTOCOL FOR ENZYME ANALYSIS WITH REDUCED INCUBATION TIME
We performed a further set of experiments unrelated to the comparison study using an improved fluorometer (limit of detection 0.55 nmol/L of 4-MU at pH 10) on a different set of 105 normal DBSs. The assay incubation times were reduced from 6 h to 1 h at 37 °C, with all the other steps of the protocol remaining the same except for a few minor changes. The high calibrant was a 0.15 μmol/L 4-MU solution prepared in STB instead of 3.75 μmol/L. Extraction was performed in 96-well plates instead of microcentrifuge tubes to make the method compatible with high-throughput methods. The sub-sequent centrifugation step was also removed for the same reason.
Results and Discussion
POMPE AND FABRY ENZYME ANALYSIS COMPARISON BETWEEN MICROPLATE AND MICROFLUIDICS METHODS
One of the objectives of this study was to develop a method that would use a single punch for performing both Pompe and Fabry assays. Current bench-based, fluorometric enzyme assay protocols require 2 separate punches for measuring the activity of GAA and GLA, because the punch for Pompe assay is extracted in deionized water, whereas the punch for Fabry assay requires 1% sodium taurocholate. We formulated a single DBS extraction buffer for both assays and substantially modified the assays to accommodate this change. The main details are summarized in Table 1. Substrate concentration for both the assays on cartridge is greater than that used in conventional microplate assays. (See Fig. 2 and Supplemental Fig. 1, which accompanies the online version of this article at http://www.clinchem.org/content/vol57/issue10, for Michaelis–Menten kinetics and Lineweaver–Burk plots for Pompe and Fabry assays.) To verify that the modified protocol on a digital microfluidic platform gives results equivalent to those obtained using the standard benchtop assay, we performed a method comparison between the digital microfluidic assay protocol at ALL and fluorescence plate-reader protocol at Duke BGL, comparing GAA and GLA activities in the same set of specimens. Fig. 2 shows the distribution of GAA and GLA activities obtained for the normal controls and affected samples with the ALL and Duke BGL assay protocols. The mean, maximum, and minimum activity values for all the data are presented in Table 2.
Fig. 2.
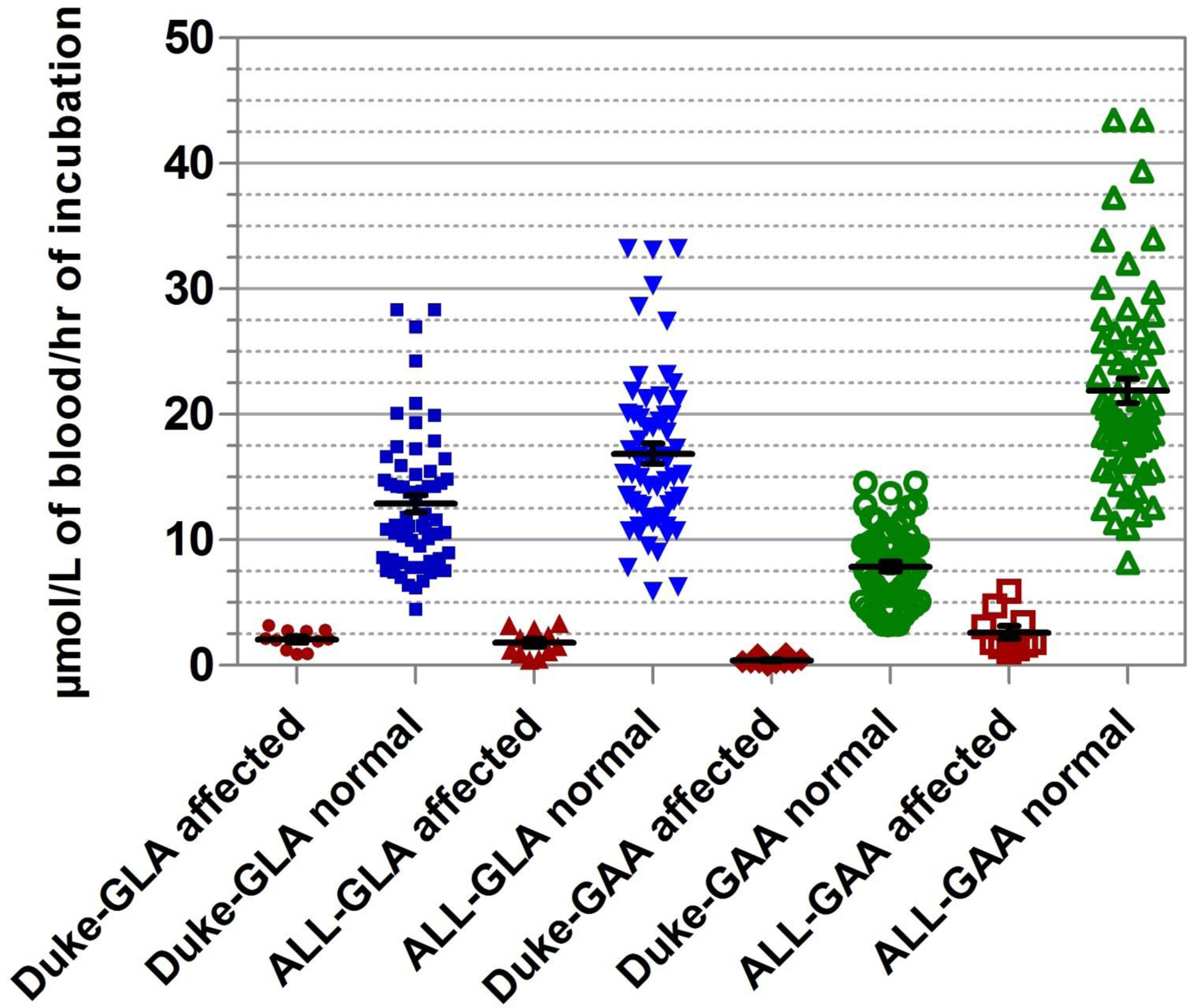
Comparison of enzyme assay results for GLA and GAA between benchtop (Duke) and digital microfluidic (ALL) fluorometric assay methods, using the same set of normal (n = 60) and affected (GLA: n = 11; GAA: n = 12) DBS specimens.
Table 2.
Activity values (μmol/L/h) between microplate and microfluidics methods.a
| Pompe (GAA) | Fabry (GLA) | |||||||
|---|---|---|---|---|---|---|---|---|
| Enzyme activity | Microplate (Duke) | Microfluidics (ALL) | Microplate (Duke) | Microfluidics (ALL) | ||||
| Normal | Affected | Normal | Affected | Normal | Affected | Normal | Affected | |
| Mean | 7.9 | 0.4 | 21.9 | 2.3 | 12.9 | 2.1 | 16.9 | 1.8 |
| Maximum | 14.6 | 0.9 | 43.5 | 4.7 | 28.3 | 3.2 | 33.2 | 3.4 |
| Minimum | 3.2 | 0 | 10.9 | 1.1 | 4.5 | 0.9 | 5.9 | 0.5 |
Normal sample size is 60 DBSs. Affected sample size is 11 DBSs for Fabry and 10 DBSs for Pompe.
Variation in enzymatic activityof CDC QC samples.
Intracartridge CV of the QCH sample was 3% (n = 11) for both GLA and GAA assays, when extract from a single punch was loaded in all the sample reservoirs. Similarly, punch-to-punch, intracartridge CV of 15% was observed in the enzymatic activity of GAA and GLA assays, which were performed on extracts from 10 different punches from a single spot and run on the same cartridge. Overall intercartridge CV values of 13%, 25%, and 23% were obtained for QCL, QCM, and QCH samples, respectively, for Pompe assay and 24%, 23%, and 16% for Fabry assay over the 10-day study period. These numbers are in general agreement with the published data on QC DBS samples (24). (See online Supplemental Table 1 for more data on CDC QC spots.) Some potential sources of variability are variability in reagent preparation between days, distribution of enzyme concentration over the DBS sample, and variation from cartridges fabricated in different batches.
Pompe GAA activity comparison.
The GAA assay activity measured by the microfluidic method was almost twice that of the benchtop microplate method for normal controls, and approximately 3 times greater for the affected samples. This difference can be accounted for, at least partly, by the higher concentrations of the substrate in the reagent formulation (about 3.5-fold) and of the enzyme in the DBS extract (3.6-fold) using the digital microfluidic method. The separation between the highest activity in the Pompe-affected spot and the lowest activity in the normal spot is 6.2μmol/L/h using the microfluidics protocol, compared with 2.3 μmol/L/h using the microplate protocol. As one would expect, the separation boundary between the lowest normal spot and the highest affected spot also scales linearly with the activity, thus increasing the tolerance for discrimination between normal and affected spots on the digital microfluidic cartridge, despite the reduced incubation time of 6 h and DBS extract sample volume of only 0.3 μL.
Fabry GLA activity comparisons.
For the GLA assay, the measured activities using the 2 methods were similar for both normal controls and affected samples, although the concentration of the substrate was higher with the digital microfluidic assay (1.4-fold). We reasoned that any gains in activity from the increased concentration of substrate and lower DBS extraction volume are more than likely offset by reduced extraction efficiency of GLA due to the lack of sodium taurocholate in the extraction buffer in the microfluidic protocol. Besides this key difference, other differences between the assay protocols outlined in Table 1 also might have an effect on the analytical sensitivity of the assay. The inhibitor concentration (GalNac) in the microfluidic method is 3 orders of magnitude lower than the bench method. Such change was found to have no effect on decreased inhibition of the assay, as was recently independently observed (11). The separation boundary between the highest activity in the affected spot and the lowest activity in the normal spot was 2.5 μmol/L/h using the microfluidic protocol compared with 1.3 μmol/L/h using the microplate protocol. Again, clear separation between the normal controls and the disease samples on the digital microfluidic cartridge was achieved using a much lower incubation time and sample volume.
EFFECT OF HEMATOCRIT ON ASSAYS PERFORMED ON THE MICROFLUIDIC PLATFORM
Another objective of this study was to determine the impact of varying amounts of hematocrit between different DBS samples on the measured fluorescence signal. It is known that hemoglobin inhibits the fluorescence of 4-MU (25). Time 0 fluorescence data obtained from all the QCL DBSs (from CDC) and Pompe- and Fabry-affected DBS samples mixed with the respective fluorescence substrates during the entire study are compared with the substrate background measurements for both GAA and GLA assays in Fig. 3. Substrate background measurements were obtained by measuring the endpoint fluorescence of a droplet mixture containing 1 droplet of EXT, 1 droplet of the substrate, and 1 droplet of STB. It is essentially same as t = 0 fluorescence measurements without DBS extract. All the QCL samples from CDC are known to have a hematocrit of 50%. The t = 0 signal for the GAA assay on all the QCL samples (mean 3.3 μmol/L) is almost the same as that of the GAA substrate (mean 3.1 μmol/L), which of course has 0% hematocrit, and that of the Pompe-affected spots (mean 3.1 μmol/L). Similarly, the t = 0 signal for GLA assay on all the QCL samples (mean 5.3 μmol/L) is virtually the same as for the GLA substrate-only (mean 5.5 μmol/L) and Fabry-affected (mean 5.5 μmol/L) spots. Although hematocrit levels in the affected spots were not measured, it is presumed that they would vary. Because these are t = 0 measurements, all the relative fluorescence unit (RFU) counts were converted to moles of 4-MU per liter of blood using the calibration curve. From Fig. 3, it can be seen that the average concentration of 4-MU with and without the DBS extract is essentially the same in both assays, suggesting that there is no significant quenching effect on 4-MU by hemoglobin on the digital microfluidics platform. In another experiment, varying amounts of QC base pool (50.5% hematocrit) DBS extract were added to a fixed concentration of 4-MU to simulate hematocrit levels ranging from 30% to 70% in the droplet, and fluorescence of 4-MU was measured. The variability in the fluorescence signal was <10% (see online Supplemental Fig. 4). This is readily explained by the much-reduced pathlength of a droplet (<0.3 mm) compared with the column of liquid in a microplate. On the basis of Beer’s law (A = E ⋆c ⋆ l, where A is absorbance, E is absorptivity, c is concentration, and l is pathlength), this reduced pathlength would make the droplets less susceptible to changes in absorbance due, for example, to differences in hemoglobin concentration between samples. Thus, it is deemed unnecessary to require a separate time 0 measurement for each sample. Instead, single fluorescence background measurements for the fluorogenic substrates can be used as surrogate t = 0 values for each assay. This offers a significant advantage for the digital microfluidic platform: a reduction in the total number of droplet operations that enables more efficient use of the available space on each cartridge.
Fig. 3.
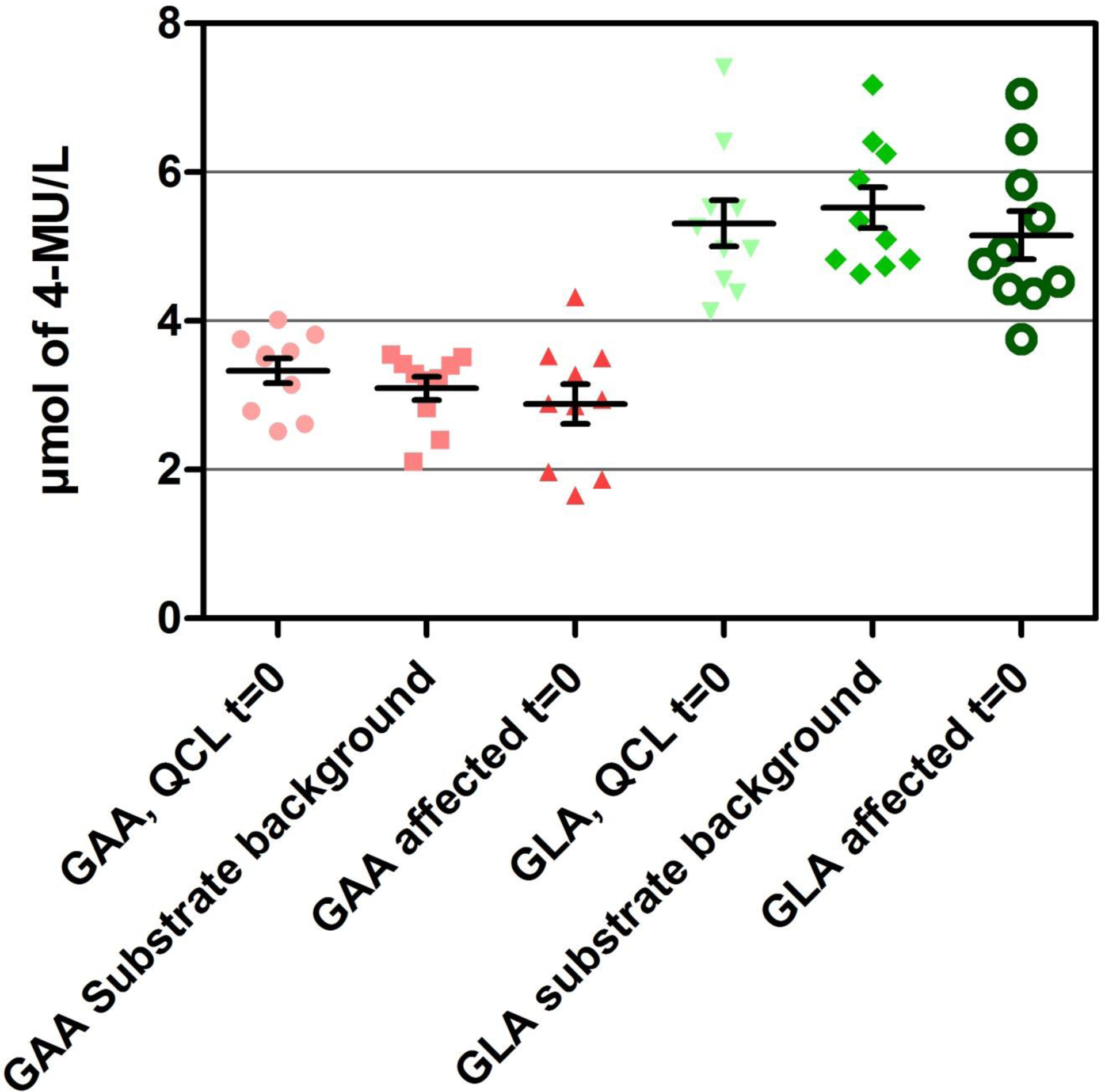
Comparison of fluorescence signals at base-line (t = 0) for all assays performed on the digital microfluidics platform in this study with substrate blanks, demonstrating lack of interference from hemoglobin in the DBS extracts.
POMPE AND FABRY ENZYME ANALYSIS ON DIGITAL MICROFLUIDIC CARTRIDGE WITH REDUCED INCUBATION TIME
Experiments were performed using a fresh set of 105 presumptive normal DBS samples to evaluate the impact of reducing the assay incubation time from 6 h to 1 h. Both the assays had linear response over an incubation period of 2 h (see online Supplemental Fig. 3 for the kinetic data). The distribution of GAA and GLA activities obtained from the control newborn DBSs compared with 6 Pompe-affected and 6 Fabry-affected DBS samples are depicted in Fig. 4. The mean GAA and GLA enzyme activities using 1-h incubation time (16.5 and 14.8 μmol/L/h, respectively) were comparable with those obtained using 6-h incubation time on the cartridge (see Table 2). The separation boundaries between the highest activity in the affected spot and the lowest activity in the normal spot for GAA and GLA assays were 4.7 and 1.3 μmol/L/h, respectively. Differences between the 1- and 6-h data sets are relatively minor, and may be ascribed to the different sources and storage conditions of the DBSs and that a more sensitive fluorometer was used to generate the 1-h data set.
Fig. 4. GLA and GAA assays with 1-h incubation.
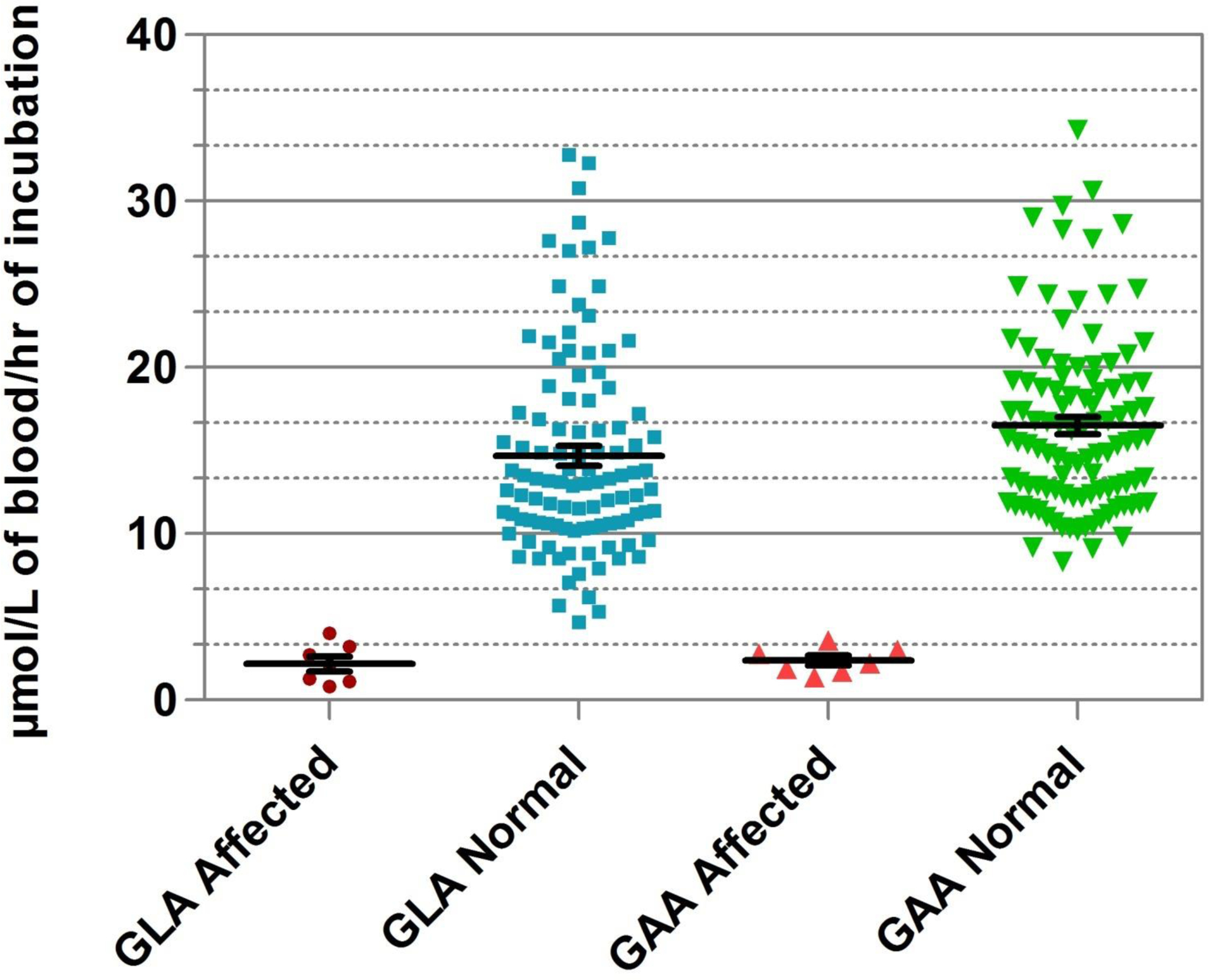
GLA activity for normal control samples (n = 105), mean 14.8 μmol/L/h (min–max, 5.3–32.8). GLA activity for affected samples (n = 6), mean 2.2 μmol/L/h (min–max, 0.8–4.0). GAA activity for normal control samples, mean 16.5 μmol/L/h (min–max, 8.3–34.3). GAA activity for affected samples (n = 7), mean 2.4 μmol/L/h (min–max, 1.4–3.6).
Chamoles and colleagues have reported performance of Pompe (8) and Fabry (7) assays individually by use of microplate fluorometric technology with an incubation time of 20 h. The aforementioned characteristics of digital microfluidics facilitate kinetic enzyme assays that have an incubation time of 1 h. This standard will significantly help the flow of work in newborn screening laboratories. Generally, there is no significant benefit to a public health laboratory if the time-to-result is reduced to about 7 h (6 h incubation + 1 h extraction and processing), because only 1 assay run can be accomplished in an 8-h shift. However, a reduction of the total time to 2 h (incubation time of 1 h) makes it possible to perform multiple runs in a single 8-h shift and thus reduces the time to report results, which is a major advantage in a newborn screening laboratory setting.
Conclusions
We successfully demonstrated and validated the performance of multiplexed enzymatic assays to screen for Pompe and Fabry diseases on the digital microfluidic platform using deidentified newborn DBS samples and affected DBS samples. The results were in close agreement with those obtained using standard bench-based, fluorometric methods in a clinical reference laboratory. The mean activity for GAA enzyme obtained using the digital microfluidic method with 6-h incubation time was almost 3 times higher than that obtained using the bench-based method with 20-h incubation time, whereas the mean enzymatic activity for GLA was essentially the same on both platforms. There was clear separation between the normal samples and the confirmed affected samples for both Pompe and Fabry despite the much lower incubation time on the microfluidic platform. Fluorescence of 4-MU on the microfluidic platform was unaffected by hemoglobin coextracted from the DBS, removing the requirement for an additional measurement for each sample. A more sensitive fluorometer facilitated a further significant reduction in incubation time from 6 h to only 1 h, thereby reducing the time from sample extraction to results to <2 h. This method was further validated by performing GAA and GLA assays on 105 normal samples plus 6 affected samples for each disorder. These experiments are essential precursors to the deployment of digital microfluidics for pilot-phase LSD assays in a newborn screening laboratory. The results shown here are highly encouraging, and the method will be extended to encompass assays for other treatable LSDs. Digital microfluidics therefore promises a low-cost alternative to enzymatic methods currently used for the recognition of LSDs.
Supplementary Material
Acknowledgments:
We acknowledge Drs. Fred Lorey and Ajit Bhandal (CDPH), Shu Chaing (NCDPH) for deidentified normal DBS, Hui Zhou and Robert Vogt (CDC) for QC spots, and Gwen Dicker-son of Duke BGL for technical assistance. The content is solely the responsibility of the authors.
Authors’ Disclosures or Potential Conflicts of Interest:
Upon manuscript submission, all authors completed the Disclosures of Potential Conflict of Interest form. Potential conflicts of interest:
Employment or Leadership: R.S. Sista, Advanced Liquid Logic Inc.; A.E. Eckhardt, Advanced Liquid Logic Inc.; T. Wang, Advanced Liquid Logic Inc.; C. Graham, Advanced Liquid Logic Inc.; J.L. Rouse, Advanced Liquid Logic Inc.; S.M. Norton, Advanced Liquid Logic Inc.; V. Srinivasan, Advanced Liquid Logic Inc.; M.G. Pollack, Advanced Liquid Logic Inc.; V.K. Pamula, Advanced Liquid Logic Inc.
Consultant or Advisory Role: S.M. Norton, Advanced Liquid Logic Inc., D.S. Millington, Advanced Liquid Logic Inc.
Stock Ownership: R.S. Sista, Advanced Liquid Logic Inc.; A.E. Eckhardt, Advanced Liquid Logic Inc.; T. Wang, Advanced Liquid Logic Inc.; C. Graham, Advanced Liquid Logic Inc.; J.L. Rouse, Advanced Liquid Logic Inc.; S.M. Norton, Advanced Liquid Logic Inc.; V. Srinivasan, Advanced Liquid Logic Inc.; M.G. Pollack, Advanced Liquid Logic Inc.; D.S. Millington, Advanced Liquid Logic Inc.; V.K. Pamula, Advanced Liquid Logic Inc.
Honoraria: None declared.
Research Funding: R44HD057713 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development to Advanced Liquid Logic Inc.
Expert Testimony: None declared.
Role of Sponsor:
The funding organizations played no role in the design of study, choice of enrolled patients, review and interpretation of data, or preparation or approval of manuscript.
Footnotes
Nonstandard abbreviations: NBS, newborn screening; MS/MS, tandem mass spectrometry; DBS, dried blood spot; LSD, lysosomal storage disease; GAA, acid α-glucosidase; GLA, acid α-galactosidase; BGL, Duke Biochemical Genetics Laboratory; CDPH, California Department of Public Health; NCDPH, North Carolina Division of Public Health; 4-MU-α-gal, 4-methyumbelliferyl α-d-galactopyranoside; GalNac, N-acetyl-d-galactosamine; 4-MU-α-gluc, 4-methyumbelliferyl α-d-glucopyranoside; 4-MU, 4-methylumbelliferone sodium salt; ALL, Advanced Liquid Logic Inc.; EXT, extraction buffer; STB, stop buffer; CAP, College of American Pathologists; CLIA, Clinical Laboratory Improvement Amendment; RFU, relative fluorescence units.
References
- 1.Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 1963;32: 338–43. [PubMed] [Google Scholar]
- 2.Millington DS, Kodo N, Norwood DL, Roe CR. Tandem mass spectrometry: a new method for acylcarnitine profiling with potential for neonatal screening for inborn errors of metabolism. J Inherit Metab Dis 1990;13:321–4. [DOI] [PubMed] [Google Scholar]
- 3.Lim-Melia ER, Kronn DF. Current enzyme replacement therapy for the treatment of lysosomal storage diseases. Pediatr Ann 2009;38:448–55. [DOI] [PubMed] [Google Scholar]
- 4.Millington DS. Rapid and effective screening for lysosomal storage disease: how close are we? Clin Chem 2008;54:1592–4. [DOI] [PubMed] [Google Scholar]
- 5.Hwu WL, Chien YH, Lee NC. Newborn screening for neuropathic lysosomal storage disorders. J Inherit Metab Dis 2010;33:381–6. [DOI] [PubMed] [Google Scholar]
- 6.Marsden D, Levy H. Newborn screening of lysosomal storage disorders. Clin Chem 2010;56: 1071–9. [DOI] [PubMed] [Google Scholar]
- 7.Chamoles NA, Blanco M, Gaggioli D. Fabry disease: enzymatic diagnosis in dried blood spots on filter paper. Clin Chim Acta 2001;308:195–6. [DOI] [PubMed] [Google Scholar]
- 8.Chamoles NA, Niizawa G, Blanco M, Gaggioli D, Casentini C. Glycogen storage disease type II: enzymatic screening in dried blood spots on filter paper. Clin Chim Acta 2004;347:97–102. [DOI] [PubMed] [Google Scholar]
- 9.Li Y, Scott RC, Chamoles NA, Ghavami A, Pinto BM, Turecek F, Gelb MH. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin Chem 2004;50:1785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang XK, Elbin CS, Chuang WL, Cooper SK, Marashio CA, Beauregard C, et al. Multiplex enzyme assay screening of dried blood spots for lysosomal storage disorders by using tandem mass spectrometry. Clin Chem 2008;54:1725–8. [DOI] [PubMed] [Google Scholar]
- 11.Duffey TA, Bellamy G, Elliott S, Fox AC, Glass M, Turecek F, et al. A tandem mass spectrometry triplex assay for the detection of Fabry, Pompe and mucopolysaccharidosis-I (Hurler). Clin Chem 2010;56:1854–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meikle PJ, Grasby DJ, Dean CJ, Lang DL, Bockmann M, Whittle AM, et al. Newborn screening for lysosomal storage disorders. Mol Genet Metab 2006;88:307–14. [DOI] [PubMed] [Google Scholar]
- 13.Duffner PK, Caggana M, Orsini JJ, Wenger DA, Patterson MC, Crosley CJ, et al. Newborn screening for Krabbe disease: the New York State model. Pediatr Neurol 2009;40:245–52. [DOI] [PubMed] [Google Scholar]
- 14.Dajnoki A, Mühl A, Fekete G, Keutzer J, Orsini J, DeJesus V, et al. Newborn screening for Pompe disease by measuring acid alpha-glucosidase activity using tandem mass spectrometry. Clin Chem 2008;54:1624–9. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, Kallwass H, Young SP, Carr C, Dai J, Kishnani PS, et al. A comparison of maltose and acarbose as inhibitors of maltase-glucoamylase activity in assaying acid α-glucosidase activity in dried blood spots for the diagnosis of infantile Pompe disease. Genet Med 2006;8:302–6. [DOI] [PubMed] [Google Scholar]
- 16.Chien YH, Lee NC, Thurberg BL, Chiang SC, Zhang XK, Keutzer J, et al. Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics 2009;124:e1116–25. [DOI] [PubMed] [Google Scholar]
- 17.Hwu WL, Chien YH, Lee NC, Chiang SC, Dobro-volny R, Huang AC, et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c. 936+ 919G>A (IVS4+919G>A). Hum Mutat 2009;30: 1397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou H, Fernhoff P, Vogt RF. Newborn bloodspot screening for lysosomal storage disorders. J Pediatr 2011;159:7–13e1. [DOI] [PubMed] [Google Scholar]
- 19.Srinivasan V, Pamula VK, Fair RB. An integrated digital microfluidic lab-on-a-chip for clinical diagnostics on human physiological fluids. Lab Chip 2004;4:310–5. [DOI] [PubMed] [Google Scholar]
- 20.Millington DS, Sista RS, Eckhardt A, Rouse J, Bali D, Goldberg R, et al. Digital microfluidics: a future technology in the newborn screening laboratory? Semin Perinatol 2010;34:163–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Desnick RJ, Ioannou YA, Eng CM. α-Galactosidase A deficiency: Fabry disease. In: Wonsiewicz M, Noujaim S, Boyle P, eds. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001. p 3733–74. [Google Scholar]
- 22.Adam B, Alexander J, Smith S, Chace D, Loeber J, Elvers L, et al. Recoveries of phenylalanine from two sets of dried-blood-spot reference materials: prediction from hematocrit, spot volume and paper matrix. Clin Chem 2000;46:126–8. [PubMed] [Google Scholar]
- 23.Sista RS, Hua Z, Thwar P, Sudarsan A, Srinivasan V, Eckhardt A, et al. Development of a digital microfluidic platform for point of care testing. Lab Chip 2008;8:2091–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Jesus VR, Zhang XK, Keutzer J, Bodamer OA, Muhl A, Orsini JJ et al. Development and evaluation of quality control dried blood spot materials in newborn screening for lysosomal storage disorders. Clin Chem 2009;55:158–64. [DOI] [PubMed] [Google Scholar]
- 25.Oemardien LF, Boer AM, Ruijter GJG, Ploeg ATV, De Klerk JBC, Reuser AJJ, et al. Hemoglobin precipitation greatly improves 4-methylumbelliferone-based diagnostic assays for lysosomal storage diseases in dried blood spots. Mol Genet Metab 2011;102:44–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.