

Abstract
Advances in the understanding of lipid droplet biology have revealed essential roles for these organelles in mediating proper cellular homeostasis and stress response. Lipid droplets were initially thought to play a passive role in energy storage. However, recent studies demonstrate that they have substantially broader functions, including protection from reactive oxygen species, endoplasmic reticulum stress, and lipotoxicity. Dysregulation of lipid droplet homeostasis is associated with various pathologies spanning neurological, metabolic, cardiovascular, oncological, and renal diseases. This review provides an overview of the current understanding of lipid droplet biology in both health and disease.
Keywords: cell biology, cellular stress, lipid droplet, lipid dysregulation, lipophagy
PHYSIOLOGY OF LDs
Lipid droplets (LDs), initially considered mere subcellular cargo, are now recognized as cellular organelles with pivotal physiological functions. This shift was driven by the observation that LDs play a critical role in mediating normal cellular homeostasis and that dysregulation of this process is associated with various diseases (1). LDs store a variety of lipids, including fats, oils, and hormones. Lipids serve as energy-rich metabolic substrates, structural components of membranes, cell signal mediators, and posttranslational modifiers of proteins (2). Cells use multiple mechanisms to internalize, synthesize, store, and mobilize lipids within LDs to guarantee proper cellular homeostasis (3).
Structure, Biogenesis, and Composition of LDs
LDs are composed of a hydrophobic core consisting of primarily neutral lipids, such as triglycerides (TG) and cholesterol esters (CE), surrounded by an amphipathic phospholipid monolayer (4, 5). Phospholipids are synthesized at the endoplasmic reticulum (ER), and their presence in the phospholipid monolayer of LDs confers stability and regulates their size and numbers within cells (6). Other lipids, such as waxes, free cholesterol, and long-chain alcohols are also found in LDs but in significantly lesser quantities (7, 8). The amount and molecular composition of neutral lipids (NL) in LDs varies substantially among different cell types and may reflect cell-type specialization (9–11). LDs in adipocytes mostly contain TG (9), whereas LDs of nonadipocytes usually have a higher content of CE (10). Similarly, the CE content of LDs is higher in macrophages than in hepatocytes, whereas LDs of stellate cells are particularly enriched with retinoic acids (11). The NL composition of LDs also varies in response to pathological stressors. The NL, leukotriene, and prostaglandin content of LDs are elevated in neutrophils from patients with infectious and inflammatory diseases (12). The arrangement of these phospholipids in the phospholipid monolayer can vary in composition between different cell types and physiological states.
LDs originate from the ER; however, the exact mechanism of LD biogenesis is still a subject of active research. The current model of LD biogenesis involves resident ER proteins that synthesize NLs at the ER membrane (13–15). NLs aggregates develop between the membrane leaflets, leading to crescent or curved areas in the membrane, which ultimately bud into the cytoplasm with the help of specialized ER proteins to form the LD (16–20). In yeast, Pah1, which is involved in diacylglycerol (DAG) synthesis, and phospholipid:diacylglycerol acyltransferase (LRO1), a triacylglycerol (TAG) biosynthesis enzyme, were shown to localize to the ER-LD sites (20–22), suggesting DAG and TAG synthases are involved in LD formation. In mammalian cells, acyl-CoA synthetase long-chain family member 3 (ACSL3) and seipin coordinate the synthesis of LDs at the ER, with the recruitment of ACSL3 being regulated by seipin (23, 24). High-resolution structural data demonstrate that both human and Drosophila seipin form a helical ring-like structure with β-sheets oriented parallel to the membrane and α-helices situated perpendicular to the membrane (25, 26). These α-helices have been shown to interact with TAG, possibly ensuring efficient packaging of NLs into the LD.
Following the initiation of LD formation at the ER-LD biogenesis sites, LD expansion is the predominant process that determines LD size based on the number of NLs produced or sequestered (27–29). This process occurs upon relocation of ER-resident proteins to the LD via the ER-LD contact sites. Acetyltransferases, including glycerol-3-phosphate acyltransferase 3 (GPAT3), 1-acylglycerol-3-phosphate O-acyltransferase 3 (AGPAT3), and diacylglycerol O-acyltransferase 2 (DGAT2), move from the ER to the LD and induce LD expansion (27). As the LD matures, LD-associated proteins are trafficked to the LD, affecting its size, shape, and behavior. Approximately 200 different LD-associated proteins with essential functions in regulating LD lipid content and stability have been described (30). These proteins are critical for LD transport, intracellular distribution, and association with filaments and other organelles (2). LD-associated lipases, such as adipose triglyceride lipase (ATGL), degrade NLs, facilitate fatty acid synthesis, and contribute to the transport of sterols to other organelles (31, 32). Induction of lipase activity leads to a reduction in the size of LDs (33), whereas the activation of lipid synthases leads to the generation of LDs with more NLs or enlarged LDs (34). Perilipins (PLINs) are a group of proteins that influence LD catabolism by modulating the access of lipases to the LD surface (35) and have been implicated in chaperone-mediated autophagy (CMA) (36). Similarly, the loss of lysosomal-associated membrane protein 2 (LAMP2A) and PLIN3 activity leads to LD accumulation due to a decrease in CMA levels. Further evidence suggests that PLIN2, and possibly PLIN3, are needed on the surface of LDs to allow ATGL and other autophagic proteins access to LD contents (37). Within the cell, TG-rich LDs are separated from CE-rich LDs. Such separation of the two distinct LD populations was also observed using cell lines. For example, in rat McARH777 hepatocytes CE-rich LDs were found clustered in the cytoplasm centered around the nucleus whereas TG-rich LDs were localized in the periphery (38). More studies are needed to reveal the physiological significance and functional consequences of this spatial segregation.
After their initial formation, LDs undergo various fusion processes, ripening, or coalescence (39). Ripening defines the process by which one LD transfers its contents to another LD (39). A crucial component of ripening is cell death-inducing DNA fragmentation factor-α (DFFA)-like effector C/fat specific protein 27 (CIDEC/FSP27), a member of the cell death-inducing DNA fragmentation factor-α (DFFA)-like effector (CIDE) family of proteins, which potentially play a pivotal role in the formation of giant LDs (40–42). Conversely, coalescence is a much more rapid process by which LDs fuse. Conditions that favor coalescence include decreases in the phosphatidylcholine:phosphatidylethanolamine ratio of the LD membrane, whereby phosphatidylcholine prevents coalescence by acting as a surfactant (43, 44). Thus, adequate levels of phosphatidylcholine are necessary to prevent large LDs from accumulating through coalescence (45).
The Role of LDs in Lipid Metabolism
Metabolic responsiveness is critical for the ability of cells to cope with changing energetic demands. Cells store energy in the form of fatty acids (FAs) and NLs when the nutrient load is high, which can be released in instances of stress or low nutrient levels. The most abundant form of these NLs are TAG and sterol esters. LDs function as a site for the storage of these NLs and thus play a central role in energy and lipid metabolism, and the contact sites associated with LDs and organelles are instrumental for FA synthesis and breakdown (46, 47). FA metabolism utilizes multiple intracellular organelles in concert with LDs (46). Lipid transfer proteins and FA-modifying enzymes reside and function at LD-organelle contact sites and compartmentalize FA metabolism. FAs must be broken down into acyl-CoA via β oxidation, acyl-CoA can then enter the citric acid cycle. This process occurs in mitochondria and peroxisomes, the primary sites of lipid metabolism in the cell.
Lipolysis and Lipophagy
Lipolysis is the breakdown of LDs stored as TGs to free fatty acids (FFAs) to produce energy through the action of cytosolic lipases, including hormone-sensitive lipase (HSL), monoacylglycerol lipase, and ATGL (48–50). Traditionally, this was the predominant model of LD degradation until other regulatory mechanisms between lipolysis and autophagy were identified. Although insulin inhibits lipolysis and autophagy, glucagon acts in a stimulatory manner. A seminal paper by Singh et al. described the discovery of a novel mechanism of LD degradation by autophagy in hepatocytes. This observation led to the discovery of lipophagy, a process by which LDs are catabolized through autophagic pathways leading to lysosomal degradation of stored lipids into FFAs. This mechanism, termed lipophagy, was confirmed by others (51–53).
Lipophagy begins with the interaction of LDs with the autophagosome membrane through binding to microtubule-associated protein 1 light chain 3 (MAP1LC3) (54). This binding involves the cooperation between one or more proteins that function as cargo adapters (i.e., p62 and optineurin) that connect the LD and LC3 (55). The exact regulators of lipophagy are largely unknown. However, some Rab family proteins are thought to be involved in the induction or cessation of lipophagy (56–59). Rab proteins are readily detected on LDs in various disease states (56), and Rab7 (58), Rab10 (57), and Rab25 (59) are regulators of lipophagy. The cytosolic lipolysis-associated lipase ATGL, acting through the deacetylase SIRT1, functions as an initiator of lipophagy in mouse liver cells (60). Although there is tight coordination and connection between these two pathways, ATGL cannot initiate LD catabolism without lipophagy. LDs, depending on the size, can undergo macroautophagy or microautophagy. In macroautophagy, an entire small LD is incorporated into the autophagosome and processed. In microautophagy, small parts of larger LDs accumulate LC3 and bud off to be incorporated into the autophagosome for gradual processing (53). LDs are catabolized by special lipases known as lysosomal acid lipases (LALs) that have a broad range of lipid substrates (61–64). These lipases differ from their cytosolic counterparts in that they function in the acidic environment of the autophagosome (65).
Lipophagy is regulated by the nutrient status of the cell and proteins that detect changes in nutrient stores. Examples of these regulatory receptors include the nuclear receptors farnesoid X receptor (FXR), the transcriptional activator cAMP response element-binding protein (CREB), peroxisome proliferator-activated receptor alpha (PPARα) (66, 67), mammalian target of rapamycin (mTOR) (68–70), and AMP-activated protein kinase (AMPK) (71, 72). Increased intracellular nutrient stores inhibit lipophagy, whereas cellular starvation promotes it (73). Lipophagy also acts as a defense mechanism against lipotoxicity and is increased in a genetic mouse model of SOCE (store-operated calcium entry) deficiency (54). In this model there are reduced levels of cytosolic lipolysis, further demonstrating the importance of lipophagy as a mechanism for neutral lipid catabolism and in maintaining cellular homeostasis (54).
Although the primary function of each of these pathways lies in the catabolism of LD content, lipolysis functions mainly to release energy stores in the form of FFA β-oxidation. Lipophagy can also alleviate lipotoxic stressors through acidic degradation in the lysosome. Initially, it was thought that these two processes, lipolysis and lipophagy were independent. Recent studies propose that these two pathways may regulate each other directly (74). Evidence suggests that levels of autophagy are regulated by LDs and lipases (74). However, although both processes regulate FFA levels in the cell, lipolysis also functions in lipid signaling, which has implications in multiple downstream processes integral to homeostasis. Lipophagy has broader substrate specificity than lipolysis and can export hydrolyzed lipids out of the cell through a process called exophagy (63). Aberrant lipophagy may lead to a decreased ability of the cells to process a broad scope of lipid substrates and export excess FFA out of the cell, leading to persistent increases in LDs. Although lipolysis and lipophagy pathways and their interconnected regulation have been reviewed in detail elsewhere (62), this review will include a brief discussion on the lipophagy signaling cascades that may contribute to the accumulation of LDs in diseases including cancer, metabolic, and liver disease.
Lipid Export from LDs
The transport of lipids into LDs and subsequently outside of the cell counteracts the accumulation of lipids inside the cell and is necessary for proper protection from lipotoxicity. LDs can export toxic or excess lipid species through active and passive transport mechanisms to acceptor particles. One example of such a transport mechanism is reverse cholesterol transport (RCT), where cholesterol transport proteins such as ATP binding cassette subfamily A member 1 (ABCA1) and ATP binding cassette subfamily G member 1 (ABCG1) efflux cholesterol to the extracellular acceptor high-density lipid (HDL) particle in an apoprotein A1 (APOA1)-dependent mechanism (75, 76). Free cholesterol and phospholipids transported to HDL by ABCA1 are immediately esterified by lecithin-cholesterol acyltransferase (LCAT) (77). The interaction of ABCG1 and HDL further increases cholesterol efflux and the total capacity of HDL to uptake cholesterol (78). Although there are also passive mechanisms of lipid efflux, they account only for a minor fraction of the total efflux capacity of a cell (79). Therefore, the dysfunction of active efflux mechanisms mainly contributes to the accumulation of LDs and the pathophysiology of LD accumulation/lipotoxicity in disease. Reduced HDL levels and activity has been associated with many diseases resulting in LD accumulation and cell injury (80, 81). Although much is known about ABCA1- and ABCG1-mediated lipid efflux mechanisms, more studies are necessary to investigate the existence of other mechanisms that could be utilized as therapeutic targets for the treatment of disorders involving LD accumulation.
LDs IN RESPONSE TO CELLULAR STRESS
The essential role of LDs in maintaining homeostasis includes functions pertaining to lipid storage and trafficking, lipid metabolism, innate immunity, and cellular signaling, which have been extensively reviewed elsewhere (2, 13, 82–89). In addition to these functions, LDs mediate cellular stress responses common to multiple disease states. Recent studies have demonstrated that LDs may mediate ER stress, reactive oxygen species (ROS), and lipotoxicity, and maladaptive processing of LDs can have deleterious effects (Fig. 1).
Figure 1.
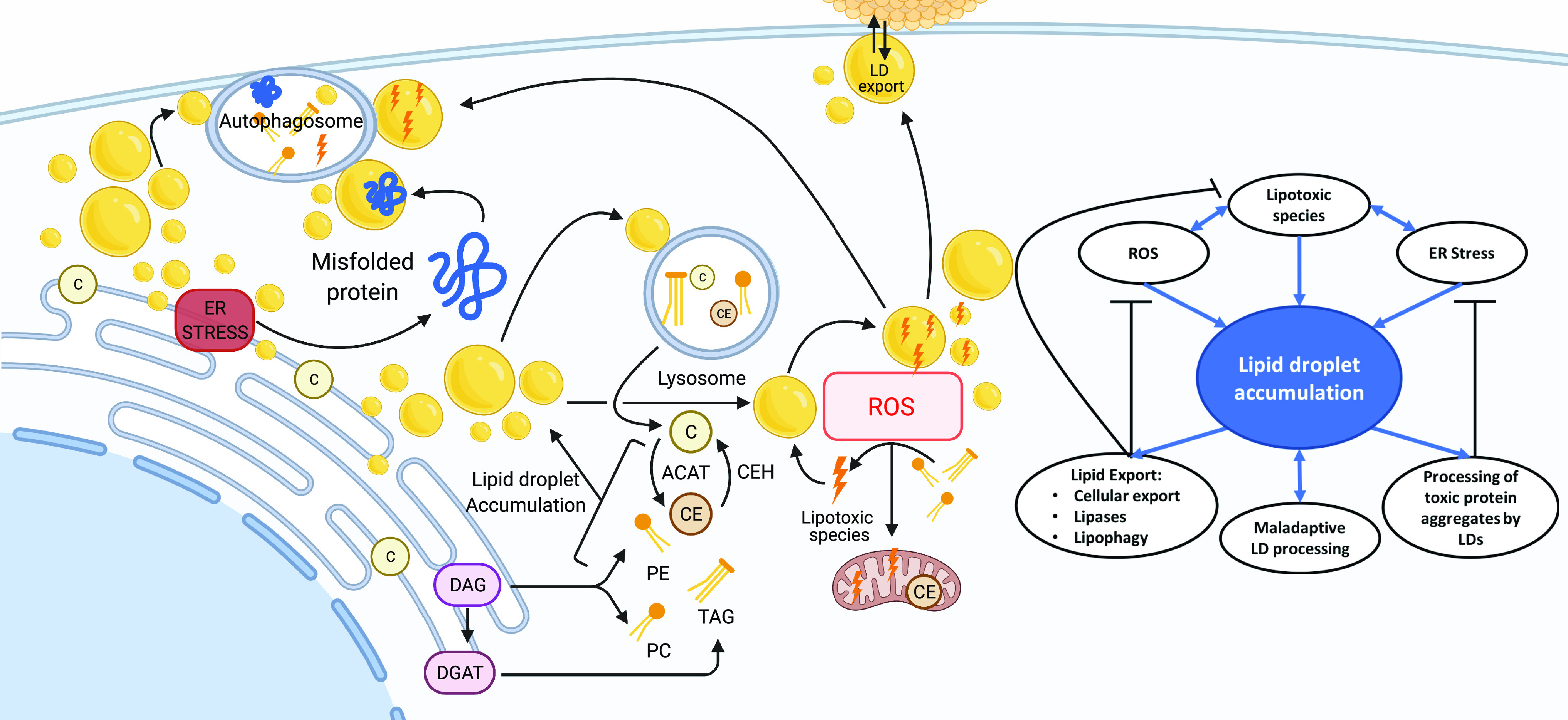
Recent studies demonstrating lipid droplets (LDs) may mediate endoplasmic reticulum (ER) stress, reactive oxygen species (ROS), and lipotoxicity, and maladaptive processing of LDs can have deleterious effects. Figure created with Biorender.com.
ER Stress
ER stress and unfolded protein response (UPR) signaling are central in the pathogenesis of many diseases, including metabolic diseases, neurodegenerative diseases, and cancer (90–98). ER stress is a general term that refers to aberrant calcium uptake and lipid composition, which leads to dysfunction of ER protein folding capacity (99). In response to ER stress, cells initiate UPR, which re-establishes proper ER homeostasis (99). Accumulation of unfolded proteins initiates the activity of three independent UPR transducers, inositol-requiring protein 1 (IRE1), protein kinase RNA (PKR)-like ER kinase (PERK; also known as EIF2AK3), and activating transcription factor 6 (ATF6) (2). This initiates signaling cascades to slow protein translation and to increase the expression of genes involved in protein folding, protein degradation, and lipid biosynthesis (2). In yeast, any perturbation in LD biogenesis results in the upregulation of UPR (100). However, the mechanisms connecting UPR activation to LD biogenesis remain unclear. It is possible that alterations in the ER composition directly activate the UPR, independent of complications in protein folding (97, 101–103).
Polyubiquitylated proteins marked for degradation are observed in LD-enriched cellular fractions during ER stress, suggesting that LDs aid in the removal of defective proteins (100). However, the mechanism of these faulty proteins localizing to LDs is unclear and warrants further investigation. Research suggests that lipid export or trafficking of ubiquitinated proteins leads to the removal of proteins and decreased ER stress. In yeast and hepatocytes, impaired ER protein degradation and UPR inducers (e.g., tunicamycin and dithiothreitol) lead to LD accumulation, indicating that LDs are induced in response to ER stress to help restore cellular homeostasis (104–106). LD accumulation may also counteract ER stress through the direct redistribution of specific phospholipids and the restoration of proper function to the ER membrane. Increased UPR activation is observed in yeast strains that lack the enzymes required for the biosynthesis of LDs (107–109). LDs accumulate near ER aggregates in yeast models of ER stress (100). The absence of LDs leads to an increase in phosphatidylinositol, resulting in atypical ER morphology (109, 110). In mammalian adipocytes, LDs also play a role in preventing ER stress and UPR activation. Recent studies demonstrate that noradrenergic stimulation of lipolysis results in the release of fatty acids and the generation of LDs, which inhibitors of acyl-CoA synthetases and DGAT1 can block, indicating a role for fatty acid re-esterification and packaging of lipids into DGAT1-dependent LDs (111–113). These data strongly suggest an essential role for LDs in ER homeostasis and as a critical organelle that mediates intracellular lipid trafficking in response to ER stress.
Reactive Oxygen Species
LDs play an essential role in protecting cells from ROS-induced injury (114). High levels of ROS are produced during mitochondrial dysfunction, hypoxia, and aberrant lipid catabolism. Studies demonstrate a positive correlation between ROS levels and LD numbers and indicate an ability for LDs to ameliorate ROS. Increases in ROS lead to the expression of LD-associated proteins that may increase LDs (108, 109) directly. LDs appear to provide a buffer against damage caused by ROS by sequestering fatty acids that would otherwise become oxidative species (115, 116). LDs function to export neutral and peroxidized lipids that are not substrates for cytoplasmic lipases, and which would otherwise contribute to cell damage and activate cell death processes. Although nuclear and mitochondrial DNA, proteins, lipids, mitochondrial enzymes, calcium signaling, receptor trafficking, and metabolic enzymes are all cell components affected by ROS, LDs play a crucial role in reducing ROS by preventing the further oxidation of lipids, which could lead to mitochondrial dysfunction.
Lipotoxic Stress
In models of cancer, LDs can function as a buffer to prevent toxic lipid buildup and lipotoxicity by redistributing lipids within and outside the cell (117). This protective mechanism mediated by LDs inside the cell involves lipolysis via autophagy pathways (59, 62, 118–124). LDs can also accumulate inert TGs, CEs, and acyl ceramides and mitigate their potentially harmful impact on cell structures by processing, neutralizing, and exporting lipid species (125–129). Free fatty acids at high levels can be incorporated into cytotoxic lipid species or act as detergents that disrupt cellular membranes. In cancer cell models, LDs limit peroxidation by actively accumulating polyunsaturated FAs, a lipid species prone to oxidation, in their lipid core (130). This sequestration of oxidation-prone lipids maintains cellular membrane composition and cellular homeostasis during periods of lipotoxic stress (115, 125, 131). LDs also protect against increases in intracellular lipids, a hallmark of many metabolic diseases (132, 133).
LDs IN DISEASE
LDs decrease the amount of toxic lipids, accumulated proteins, and oxidative stressors freely available within the cell, limiting the damage from exogenous and endogenous stressors. Once these toxic species are incorporated into LDs, there is a need to further process and ultimately export or neutralize the contents. Dysfunction of export mechanisms associated with LD trafficking can lead to cellular insult and inability to recover from cellular stress. The emerging evidence indicates that in various diseases, LD accumulation can initially act to remedy lipotoxicity mediated by ER stress and ROS, whereby LDs aid in the sequestration, trafficking, and ultimate export of cytotoxic components. However, disruptions in these dynamic processes that control LD distribution, composition, and transport may overwhelm and destabilize this system, resulting in a failed stress response and contributing to disease pathology.
The roles of LDs in pathologies including cancer, atherosclerosis, obesity, nonalcoholic fatty liver disease, renal disease, and neurological disorders provide the option for the discovery of new therapeutic targets with a potential for novel clinical interventions.
Metabolic Disease
Obesity and diabetes.
Adipose tissue functions as the body’s energy reservoir by storing TGs during energy surplus and converting these to FFA during energy deficit. There are two types of adipose tissue, white adipose tissue (WAT) and brown adipose tissue (BAT). In vertebrates, around 90% of TGs are stored in WAT. WAT can expand its capacity to buffer energy stores by fat cell hypertrophy and function predominantly as an energy storage mechanism. On the other hand, BAT is a highly oxidative tissue with high mitochondrial density to generate heat. These two types of adipose tissue function very differently, and the role of LDs predominantly involves the storage and release of lipids in WAT. These cells are capable of de novo synthesis of lipids from intermediates of carbohydrate metabolism, storing dietary lipids, and regulating their distribution. Excessive lipids in adipocytes induce enlargement of the adipose tissue and eventually cause the release of stress signals that induce macrophage migration and infiltration. Macrophage migration and inflammation within adipose tissue can modulate the endocrine response in this tissue. Oversecretion of cytokines by adipocytes, such as PAI-1, tumor necrosis factor-α (TNF), or interleukin 6 (IL-6) in conjunction with hyposecretion of adipocytokines, such as adiponectin, may contribute to the pathogenesis of lifestyle-related diseases including diabetes mellitus, hyperlipidemia, hypertension, and atherosclerosis, comprising metabolic syndrome (134).
Elevated levels of circulating FAs and altered endocrine function results in ectopic lipid accumulation in peripheral tissues, including the liver and skeletal muscle. Accumulation of intramyocellular lipids is associated with the loss of insulin sensitivity and altered systemic metabolism (30). The number of LDs in the subsarcolemmal space of patients with type 2 diabetes was larger than in athletes and nondiabetic patients with obesity. Exercise intervention in these patients with type 2 diabetes reduced the number of LDs to almost the same as a control group of nondiabetic and nonathlete subjects (135). Therefore, exercise may play a role in cycling LDs and initiating LD clearance from muscle cells and potentially other cells in the body.
Atherosclerosis.
In atherosclerosis, arterial injury causes endothelial dysfunction and the retention and oxidation of low-density lipoproteins (LDL) and other apolipoprotein B-containing lipoproteins rich in cholesterol in the arteries. Oxidized LDL promotes the infiltration of macrophages in the subendothelial space. Scavenger receptors expressed in macrophages recognize and internalize oxidized LDL. Internalization of oxidized LDL causes remarkable lipid accumulation and enlargement of LDs in macrophages, earning them the name “foam cells.” Foam cells comprise the majority of cells found in the earliest lesions during atherogenesis, called the “fatty streak” (136). The actual chemical composition of oxidized LDL varies and may depend on the extent of oxidation of the LDL molecule. Oxidation may affect the phospholipid monolayer, the apolipoprotein, or the lipids in the internal core. Thus, oxidized LDL can contain various oxidation products like lysophosphatidic choline in the phospholipid monolayer, peroxidized FAs, aldehydes, and cross-linked proteins (137). LDL oxidation is catalyzed by transition metal ions, several free radicals, and some oxidizing enzymes. Exacerbated LD accumulation in foamy macrophages leads to perturbations in lipid signaling, resulting in an inflammatory phenotype.
Macrophage inflammation results in increased oxidative stress and cytokine/chemokine secretion, causing a vicious cycle of additional endothelial cell recruitment, LDL oxidation, macrophage infiltration, and foam cell formation. Macrophage inflammatory chemokines promote infiltration of other cell types, including lymphocytes and dendritic cells, which also participate in the modulation of cytokine production within the lesion and smooth muscle cells. Smooth muscle cells become activated, increase, and overexpress extracellular matrix components, forming a fibrous cap over the core of foam cells. Smooth muscle cells in the vicinity of foamy macrophages also accumulate CEs in LDs and become foamy (138). Foamy macrophages and smooth muscle cells show dysfunctional lysosomal activity and cholesterol efflux. The resulting atherosclerotic plaque can remain stable until exacerbated inflammation, and oxidation triggers the formation of a vulnerable plaque, with a more necrotic macrophage core, thinning of the fibrous cap, and enrichment in 18:0 lysophosphatidylcholine, that is more prone to rupture. Rupture of atherosclerotic plaque releases prothrombotic factors and results in thrombus formation at the site of plaque rupture (138). Foamy macrophages have also been found in other pathologies, including multiple sclerosis and spinal cord injury (139). Therefore, it is possible that strategies initially designed to alleviate foamy macrophage formation in atherosclerosis could have utility in the treatment of other neurological diseases as well (140).
Neurological Disease
Nervous tissue contains the highest relative amount of lipids and the greatest variety in lipid species, suggesting a complex role of lipid trafficking and signaling involved in neuronal homeostasis and function compared with other tissue types (141). LD accumulation occurs in both neurons and glial cells (141–143), with the latter being the primary target of LD accumulation in Parkinson’s disease and Alzheimer’s disease (AD), multiple sclerosis (139), and aging of the brain (144). Lipids and nutrients are transferred between glial cells and neurons, which results in preferential accumulation of LDs in glial cells (145). This may reflect a critical interplay between these two cell types whereby glial cells protect from neuronal dysfunction by preventing an excessive LD accumulation in neurons. In support of this concept, in the Drosophila larvae model of neuronal damage, mutations in mitochondrial genes introduced in neurons lead to neuronal damage via ROS accumulation, accompanied by a transient accumulation of LDs in glial cells but not in neurons. In contrast, when the same mutations were introduced in glial cells, they did not cause an increase in LDs (146). These data support the concept that glial cells assist in the export of LDs from neuronal cells. Moreover, it was found that glial cells supply lactate to neurons, which neurons utilize to synthesize lipids (147). Neuronal damage via ROS accumulation induces the synthesis of lipids, which are then transferred to glial cells via apolipoproteins, ApoD, or ApoE (116). In the presence of neuronal stressors, including ROS and ER stress, LD accumulation worsens neurodegeneration (146, 148). Reduction of LDs via lipase overexpression or downregulation of TG synthesis in the absence of neuronal damage induced neurodegeneration (149), indicating that dysregulation of LD composition and dynamics, not the mere presence of LDs, is detrimental to the cell. Additional studies are required to elucidate the role of LDs in normal and neurological disease states and identify therapeutic targets of pathological LD accumulation in neurological diseases.
Alzheimer’s disease.
Alois Alzheimer first reported lipid inclusions in glial cells in AD. Increases in lipid species, such as lysophosphatidic acid, sphingomyelin, ganglioside GM3, and CEs, are present in the entorhinal cortex of patients with AD, suggesting a pathogenic mechanism associated with endo-lysosomal storage disorders (150). In patients, APOE4, an isoform of the APOE apolipoprotein, is the strongest genetic risk factor for the development of late-onset AD. APOE has a multifaceted function in cholesterol efflux and the clearance of amyloid-β plaques (151). Fibroblasts treated with cultured media from APOE4 expressing astrocytes show an increased number of LDs and increased phospholipid synthesis and CE accumulation compared with those exposed to the media from APOE3 expressing astrocytes (116, 146). Liu et al. (116) demonstrated that APOE and APOD mediate the transport of lipids from neurons to glial cells in a Drosophila larvae model of AD. When APOE4 Drosophila larvae were exposed to rotenone, which increases ROS production, APOE4 harboring neurons failed to transport lipids to glia, leading to neurodegeneration (116). Astrocytes harboring APOE4 contained an increased number of LDs of reduced size than those harboring APOE3, possibly indicating that LD fission occurs and suggesting incomplete fatty acid oxidation (152). These findings support a role for LD accumulation in the pathogenesis of AD, caused by increases in ROS or ER stress, hallmarks of the disease (145).
Age-related macular degeneration.
Age-related macular (AMD) degeneration is one of the significant causes of blindness worldwide. Genome-wide association studies have identified lipid metabolism and inflammation as AMD-associated pathogenic pathways. Cultured retinal epithelial cells from AMD donors exhibit accumulation of LDs, impaired autophagy, increased ROS, susceptibility to oxidative stress, and decreased mitochondrial activity compared with regular donors (153). The role of lipid efflux in the homeostasis of retinal epithelial cells was recently demonstrated in a study showing that mice with retinal pigment epithelium-specific ABCA1 and ABCG1 deletions were characterized by increased accumulation of LDs, reduced retinal function, retinal inflammation, and retinal pigment epithelium/photoreceptor degeneration (154). LDs in the retinal pigment epithelium were enriched with CEs (154), which have been shown to increase under conditions of ROS stress (155). LD accumulation in macular degeneration may be a response to increased ROS levels, and failure to export lipids, such as cholesterol, via ABCA1 and ABCG1 transporters could promote the disease phenotype.
Parkinson’s disease.
Parkinson’s disease is characterized by the formation of fibrillary lesions containing α-synuclein (αS) aggregates that are neurotoxic. Changes in membrane lipids drive the formation of toxic pathological S aggregates. Under normal conditions, αS undergoes transient cyclic conformations, binds to membranes, is assembled in homo-tetramers, and then disassembles into the soluble form. Under certain pathological conditions where the membrane composition is altered, αS self-aggregates into toxic intermediates resulting in progressive neurotoxicity (156, 157). Overexpression of A53T αS, a mutant that preferentially adopts the toxic conformation, induces accumulation of LDs and TGs in neurons (158). In vitro, αS binds to LDs with low affinity to phospholipids, likely due to a more exposed hydrophobic core (39). The presence of free monounsaturated fatty acid, particularly oleic acid, favors the formation of toxic αS aggregates in neurons, and the addition of oleic acid worsens the neurotoxicity of αS (159). Reduction of stearoyl-CoA, a key enzyme in the synthesis of oleic acid, via pharmacological inhibition or gene deletion, reduces the formation of toxic aggregates, thus preventing the neurotoxic effect (159). Interestingly, the accumulation of LDs in response to αS is associated with a beneficial effect, whereas the neurons with lower LD numbers had worsened neurotoxicity (157). This effect highlights the need for LDs in preventing the damaging effects associated with the accumulation of toxic lipid species. The accumulation of LD in Parkinson’s disease that coincides with the increase in TGs may therefore sequester the toxic αS aggregates and embed them in the core of high TG-containing LDs.
Liver Disease
Steatosis occurs when hepatocytes accumulate TGs due to aberrant lipid metabolism (160). Excess TG is stored in LDs, and the formation of very large LDs is a hallmark of liver steatosis (161). Phosphatidylcholine is required for very low-density lipoprotein (VLDL) production and secretion in hepatocytes, which lysophosphatidylcholine acyltransferase 3 (LPCAT3) contributes to a critical step in VLDL production (162, 163). Mice lacking LPCAT3 accumulate LDs in the liver when fed a high-sucrose diet and show altered TG secretion, which are hallmarks of steatosis (163).
Increased LD accumulation in hepatic steatosis can act to remedy lipotoxicity, ER stress, and ROS stress (160). In mouse models of obesity, hepatocytes demonstrate high levels of ER stress and concurrent accumulation of LDs (92, 164). ER stress increases the expression of lipogenic genes in hepatocytes and initiates inflammatory cascades that further promote insulin resistance and lipogenesis (165). These events can contribute to the progression of other diseases, including atherosclerosis, diabetes, obesity, and renal disease, all of which are characterized by additional local tissue increases in LDs (164, 166). Lipotoxicity-induced LD accumulation in hepatic steatosis is also driven by dysregulation of lipophagy (103, 104). Studies have shown decreased levels of transcription factor EB (TFEB), a transcription regulator involved in lipophagy, and glycine N-methyltransferase (GNMT), an enzyme involved in autophagy initiation, are due to LD accumulation in hepatic steatosis (103, 104). People with naturally occurring mutations in immunity-related GTPase family M (IRGM), a protein involved in autophagic clearance of bacteria, demonstrate increased levels of hepatic lipid accumulation, which is also observed in IRGM knockout mice (106–108). This indicates that dysregulation of lipophagy-related pathways contributes to the progression of hepatic steatosis. Several therapeutics that upregulate autophagy are currently under investigation as potential treatments for liver disease (86, 109, 110). Future studies are needed to investigate the contribution of LD lipophagy, including exophagy-mediated clearance of LD contents, and lipolysis in liver diseases to better understand the role of LD accumulation and lipotoxicity, and to enable the development of novel therapeutics.
Cancer
The solid tumor microenvironment is characterized by abnormal vascularization and decreased blood supply (167). Hypoxia, the condition of low oxygen levels in tissue, occurs in most solid and more significant tumor types (168). Cancer cells have increased levels of ROS due to internal oncogenic signaling and immune cell infiltration into the tumor microenvironment, both of which lead to cytokine release, inflammation, and activation of cell proliferation pathways in human cancer cells (169). High levels of ROS lead to the oxidation of other molecules in the cell, which is typically associated with cell damage and death. However, cancer cells can upregulate redox pathways and activate mechanisms to counteract prolonged exposure to elevated ROS. NADH production in cancer cells is essential in protecting these cells from ROS damage (170). Increased FA accumulation in response to impaired lipolysis occurs in cancer cells, which induces the formation of LDs (171, 172). LD accumulation in response to hypoxia has been observed in various cancer types, including brain, breast, renal, and prostate cancers. It may protect cancer cells by embedding potentially toxic lipid species into the core of the LD (117). Studies in a rat glioma model demonstrate that LDs accumulate mainly in the necrotic areas of the tumor core and function to decrease the ROS build-up induced by the hypoxic conditions associated with necrotic tissue (173, 174). Hypoxia-inducing factors, HIF-1α and HIF-2α, mediate the production of DAG and stimulate LD accumulation in cancer cells through Lipin 1 and PLIN2, respectively (175, 176). In addition, hypoxia leads to decreases in endogenous production of FAs through the inhibition of stearoyl-coenzyme A desaturase 1, SCD1, thus increasing FA uptake to compensate for hypoxia-induced ER stress (171, 177, 178). LD accumulation in breast cancer and glioblastoma cells exposed to hypoxia promoted cell survival (172). Knockdown of FABP3, FABP7, or PLIN2 inhibited LD formation during hypoxia resulting in reduced NADPH levels and increased ROS accumulation (172). LDs aggregate lipotoxic species generated by increased ROS levels, which need to be processed or exported for the survival of cancer cells in the hypoxic tumor microenvironment. Lipophagy is a significant contributor to the catalytic breakdown of LDs, and recent studies indicate a role for this pathway in cancer progression (179–184). Different experimental models of cancer and cancer cell types exhibit different modulations on lipophilic signaling proteins. Future work to clarify the role of LD accumulation and catabolism, lipophagy, and the specialized export of autophagosomal contents by exophagy in various cancer types and tumor microenvironments may unveil new therapeutic targets for the treatment of cancer.
Renal Disease
LD accumulation has been strongly implicated in the progression and pathology of renal disease. Here, we will describe mechanisms in which LDs accumulate, the responses of different renal cells to LD accumulation, and the contribution of LD accumulation to renal disease.
Tubular epithelial disease.
Renal lipid accumulation has been extensively documented in acute and chronic kidney injury (185). In acute kidney injury, most damage is typically to kidney tubular epithelial cells. These cells consume and store fatty acids to accommodate the high energy demand required for the active transport of ions and molecules and are highly susceptible to hypoxia. Proximal tubules, which account for ∼ 70% of kidney cortex mass, are rich in TGs compared with glomeruli, accounting for most of the TGs present in the kidney cortex (186). Acute kidney injury in the left kidney is induced within 15 min after ischemia in angiotensin II-infused mice with nephrectomy of the right kidney and persists for hours after reperfusion (187) or starvation (188). In mice, CEs and TGs accumulated in proximal tubules after acute kidney injury is induced by different insults, including LPS injection, glycerol-induced rhabdomyolysis, and ischemia/reperfusion (187, 189). This phenomenon occurs independently from cholesterol and TG blood levels (187, 189), suggesting an important role for cellular rather than systemic lipids in acute kidney injury. LD accumulation in tubular cells may be mediated by increased TG synthesis, increased fatty acid uptake, or decreased fatty acid oxidation. Increased phospholipase A2 (PLA2) activity releases FFAs from phospholipids of the cell membrane, supplying FFA for LD biogenesis. Decreased expression of genes involved in cholesterol efflux, such as ABCA1 and ABCG1, can also account for LD accumulation (190). Whether lipid accumulation in tubular cells is beneficial or detrimental in renal disease depends on the duration of the injury and/or the extent of the damage. For example, LD accumulation after LPS injection protects tubular cells by limiting further exposure to hypoxia or oxidative stress (187). On the other hand, oleate-loaded tubular cells are more sensitive to oxidants and ATP depletion insults (187). Moreover, genetic or drug activation of PPARα receptors, which induces FA oxidation and consumption of LD cargo, protected mice from tubulointerstitial fibrosis (191, 192). LD accumulation forms part of a cytoprotective response but can become part of a maladaptive response that worsens the disease if left unresolved.
Glomerular disease.
Glomerular LD accumulation has been documented in many renal diseases of metabolic and nonmetabolic origin by us and others (190, 193–202). LD accumulation in podocytes, endothelial cells, and mesangial cells has been described in kidney biopsies from patients with diabetic kidney disease (203). Decreased expression of genes participating in cholesterol efflux and fatty acid oxidation along with upregulation of LDL and oxidized LDL receptors was found in kidney biopsies of these patients (203). Studies from our research group demonstrated that cultured human podocytes exposed to sera from patients with diabetic kidney disease are characterized by an increased number of LDs and downregulation of ABCA1 (190). In this context, renal lipid accumulation occurs in a manner independent of serum lipid levels (194), suggesting that this is an active cytopathological process and not the mere passive accumulation of circulating lipids in podocytes.
Glomerular diseases, such as focal segmental glomerulosclerosis (FSGS) and Alport syndrome (195), are other notable examples in which glomerular lipid accumulation was described (197–200). We previously described the accumulation of LDs in glomeruli of Col4A3 knockout (KO) mice, a mouse model of renal disease associated with Alport syndrome. These mice present with proteinuria and exhibit substantial glomerular damage and renal failure (195). Similarly, glomerular LD accumulation can be observed in mice with adriamycin-induced nephropathy, a mouse model for FSGS. More importantly, in both mouse models, compounds that induce ABCA1 expression, and ABCA1-mediated cholesterol efflux, significantly reduce the number of LDs and proteinuria. This suggests that promoting the resolution of the LD stress response in podocytes by preventing or remedying excessive LD accumulation and lipotoxicity improves renal function in glomerular disease.
TRANSLATIONAL APPLICATIONS
One strategy to accelerate moving drugs that target lipid metabolism forward into the clinical setting is repurposing existing FDA-approved drugs. For example, Verapamil, a calcium channel blocker commonly used to treat high blood pressure, was also shown to reduce hepatic lipid droplet accumulation, insulin resistance, and steatohepatitis, indicating a potential therapeutic advantage of calcium channel blockers that could be utilized in the treatment of general nonalcoholic fatty liver disease pathologies (204). Furthermore, several small molecules that target lipid metabolic pathways are currently being investigated as potential cancer therapeutics, which have been reviewed extensively elsewhere (205). The role of LDs in multiple disease states represents a significant opportunity for therapeutic intervention. LDs are ubiquitous among all cellular life forms, and thus the mechanisms that regulate their biosynthesis and activity are highly conserved. This allows for translational study of LDs in various organisms, including Drosophila, Caenorhabditis elegans, yeast, mammals, and fish. Efforts are underway to identify small molecule modulators of LD accumulation using phenotypic drug screens, which will provide further insight into the mechanisms involved in regulating LD formation and function. Phenotypic drug screens using these model organisms that investigate the potential to remedy LD accumulation will provide further insight into the mechanisms involved in regulating LD formation and function. These phenotypic screens could identify new compounds that can promote the resolution of the LD stress response by preventing excessive lipid buildup and broaden the pharmacological tools available to treat diseases characterized by uncontrolled LD dysregulation.
CONCLUSION
LD dysregulation occurs in multiple diseases including metabolic disease, neurological disease, liver disease, renal disease, and cancer. Targeting dysregulated cellular lipid metabolism may represent a potential therapeutic strategy for treating many of these diseases. The emerging evidence summarized in this review indicates that in various diseases, LD accumulation can initially act to remedy lipotoxicity mediated by ER stress and ROS, whereby LDs aid in the sequestration, trafficking, and ultimately the export of cytotoxic components. Disruptions in these dynamic processes that control LD distribution, composition, and transport may overwhelm and destabilize this system, resulting in a failed stress response and contributing to disease pathology. Depending on the pathophysiology and progression of known diseases involving LD accumulation, drugs that modulate cellular lipid metabolism may prove to represent a robust tool in the clinical setting.
In future studies, insights into the time scale of lipid accumulation in disease development and progression will provide important mechanistic data to enhance our understanding of LD dysfunction. Although LDs exercise an initial beneficial role in maintaining proper cellular homeostasis, the excessive accumulation of LDs is pathological and therefore represents an attractive new target for drug discovery. Live imaging of LDs in disease will provide valuable information on the behavior of LDs in disease states and is currently gaining momentum with the increase in lipid-specific dyes and advanced techniques. Determining the signaling cascades that enable the switch between various LD behaviors, size, and composition will be instrumental in treating metabolic, liver, renal, and neurological diseases. Molecular interventions that switch the behavior of LDs may provide novel clinical tools for the treatment of many diseases, including cancer.
This review underscores the critical need for further research into mechanisms of intercellular lipid trafficking, lipid metabolism, and the behavior of LDs. The knowledge obtained from further research will benefit the development of future therapeutic strategies aimed at adjusting maladaptive lipid accumulation, which can have a substantial clinical impact on an extensive range of diseases.
GRANTS
This work was supported by Boeheringer Ingelheim (to A.F. and S. M.), NIH Grants R01CA227493, R01DK117599, and R01DK104753 (to J.D.P., M.Z.G, and J.T.V.); Grants UL1TR000460, U54DK083912, UM1DK100846, and U01DK116101 (to A.F.); Grants DK076169 and DK115255 (to H.A-A.); and US Department of Defense Grant W81XWH2110940 (to H.A-A.).
DISCLOSURES
A. Fornoni and S. Merscher are inventors on pending (PCT/US2019/032215; PCT/US2019/041730; PCT/US2013/036484; US17/057,247; US17/259,883; Japan No. 501309/2021; Europe No. 19834217.2; China No. 201980060078.3; Canada Nos. 2,852,904, 2,930,119, 3,012,773) and issued patents (US10,183,038; US10,052,345) aimed at preventing and treating renal disease. They stand to gain royalties from their future commercialization. A. Fornoni and S. Merscher hold indirect equity interest in and potential royalty from ZyVersa Therapeutics Inc. by virtue of assignment and licensure of a patent estate. H. Al-Ali is an inventor on pending patent application PCT/US2018/058411 (China CN111801322A, Europe EP3704103A1, South Korea KR20200080273A, Japan JP2021503442A, Australia AU2018359413A1, and Israel IL274067D0). He is also a cofounder of Truvitech LLC, a university of Miami-based startup focused on developing and commercializing idTRAX, a biotechnology platform for drug target identification. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
J.D.P. prepared figures; J.D.P., M.Z.G., and J.T.V.S. drafted manuscript; J.D.P., M.Z.G., J.T.V.S., A.F., S.M., and H.A-A. edited and revised manuscript; J.D.P., J.T.V.S., A.F., S.M., and H.A-A. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Alexis Sloan for help in editing this review.
REFERENCES
- 1.Coleman RA. The “discovery” of lipid droplets: a brief history of organelles hidden in plain sight. Biochim Biophys Acta Mol Cell Biol Lipids 1865: 158762, 2020. doi: 10.1016/j.bbalip.2020.158762. [DOI] [PubMed] [Google Scholar]
- 2.Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol 20: 137–155, 2019. doi: 10.1038/s41580-018-0085-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosch M, Parton RG, Pol A. Lipid droplets, bioenergetic fluxes, and metabolic flexibility. Semin Cell Dev Biol 108: 33–46, 2020. doi: 10.1016/j.semcdb.2020.02.010. [DOI] [PubMed] [Google Scholar]
- 4.Fujimoto T, Parton RG. Not just fat: the structure and function of the lipid droplet. Cold Spring Harb Perspect Biol 3: a004838, 2011. doi: 10.1101/cshperspect.a004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo Y, Cordes KR, Farese RV Jr, Walther TC. Lipid droplets at a glance. J Cell Sci 122: 749–752, 2009. doi: 10.1242/jcs.037630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fagone P, Jackowski S. Membrane phospholipid synthesis and endoplasmic reticulum function. J Lipid Res 50, Suppl: S311–S316, 2009. doi: 10.1194/jlr.R800049-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walther TC, Farese RV Jr.. The life of lipid droplets. Biochim Biophys Acta 1791: 459–466, 2009. doi: 10.1016/j.bbalip.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McVicker BL, Rasineni K, Tuma DJ, McNiven MA, Casey CA. Lipid droplet accumulation and impaired fat efflux in polarized hepatic cells: consequences of ethanol metabolism. Int J Hepatol 2012: 978136, 2012. doi: 10.1155/2012/978136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Onal G, Kutlu O, Gozuacik D, Dokmeci Emre S. Lipid droplets in health and disease. Lipids Health Dis 16: 128, 2017. doi: 10.1186/s12944-017-0521-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujimoto T, Ohsaki Y, Cheng J, Suzuki M, Shinohara Y. Lipid droplets: a classic organelle with new outfits. Histochem Cell Biol 130: 263–279, 2008. doi: 10.1007/s00418-008-0449-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blaner WS, O'Byrne SM, Wongsiriroj N, Kluwe J, D'Ambrosio DM, Jiang H, Schwabe RF, Hillman EM, Piantedosi R, Libien J. Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage. Biochim Biophys Acta 1791: 467–473, 2009. doi: 10.1016/j.bbalip.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melo RC, Weller PF. Lipid droplets in leukocytes: organelles linked to inflammatory responses. Exp Cell Res 340: 193–197, 2016. doi: 10.1016/j.yexcr.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walther TC, Chung J, Farese RV Jr.. Lipid droplet biogenesis. Annu Rev Cell Dev Biol 33: 491–510, 2017. doi: 10.1146/annurev-cellbio-100616-060608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chorlay A, Monticelli L, Ferreira JV, M’barek KB, Ajjaji D, Wang S, Johnson E, Beck R, Omrane M, Beller MJ. Membrane asymmetry imposes directionality on lipid droplet emergence from the ER. Dev Cell 50: 25–42.e27, 2019. doi: 10.1016/j.devcel.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 15.Renne MF, Emerling BM. ORP5 regulates PI (4) P on the lipid droplet: novel players on the monolayer. J Cell Biol 219: e201912010, 2020. doi: 10.1083/jcb.201912010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiam AR, Ikonen EJ. Lipid droplet nucleation. Trends Cell Biol 31: 108–118, 2021. doi: 10.1016/j.tcb.2020.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Joshi AS, Nebenfuehr B, Choudhary V, Satpute-Krishnan P, Levine TP, Golden A, Prinz WA. Lipid droplet and peroxisome biogenesis occur at the same ER subdomains. Nat Commun 9: 2940, 2018.doi: 10.1038/s41467-018-05277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang S, Idrissi F-Z, Hermansson M, Grippa A, Ejsing CS, Carvalho P. Seipin and the membrane-shaping protein Pex30 cooperate in organelle budding from the endoplasmic reticulum. Nat Commun 9: 2939, 2018. doi: 10.1038/s41467-018-05278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salo VT, Li S, Vihinen H, Hölttä-Vuori M, Szkalisity A, Horvath P, Belevich I, Peränen J, Thiele C, Somerharju P, Zhao H, Santinho A, Thiam AR, Jokitalo E, Ikonen E. Seipin facilitates triglyceride flow to lipid droplet and counteracts droplet ripening via endoplasmic reticulum contact. Dev Cell 50: 478–493.e479, 2019. doi: 10.1016/j.devcel.2019.05.016. [DOI] [PubMed] [Google Scholar]
- 20.Choudhary V, El Atab O, Mizzon G, Prinz WA, Schneiter RJ. Seipin and Nem1 establish discrete ER subdomains to initiate yeast lipid droplet biogenesis. J Cell Biol 219: e201910177, 2020. doi: 10.1083/jcb.201910177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adeyo O, Horn PJ, Lee S, Binns DD, Chandrahas A, Chapman KD, Goodman JM. The yeast lipin orthologue Pah1p is important for biogenesis of lipid droplets. J Cell Biol 192: 1043–1055, 2011. doi: 10.1083/jcb.201010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barbosa AD, Sembongi H, Su W-M, Abreu S, Reggiori F, Carman GM, Siniossoglou SJ. Lipid partitioning at the nuclear envelope controls membrane biogenesis. Mol Biol Cell 26: 3641–3657, 2015. doi: 10.1091/mbc.E15-03-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salo VT, Belevich I, Li S, Karhinen L, Vihinen H, Vigouroux C, Magré J, Thiele C, Hölttä-Vuori M, Jokitalo E, Ikonen E. Seipin regulates ER–lipid droplet contacts and cargo delivery. EMBO J 35: 2699–2716, 2016. doi: 10.15252/embj.201695170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kassan A, Herms A, Fernández-Vidal A, Bosch M, Schieber NL, Reddy BJN, Fajardo A, Gelabert-Baldrich M, Tebar F, Enrich C, Gross SP, Parton RG, Pol A. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J Cell Biol 203: 985–1001, 2013. doi: 10.1083/jcb.201305142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sui X, Arlt H, Brock KP, Lai ZW, DiMaio F, Marks DS, Liao M, Farese RV Jr, Walther TC. Cryo-electron microscopy structure of the lipid droplet-formation protein seipin. J Cell Biol 217: 4080–4091, 2018. doi: 10.1083/jcb.201809067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan R, Qian H, Lukmantara I, Gao M, Du X, Yan N, Yang H. Human SEIPIN binds anionic phospholipids. Dev Cell 47: 248–256.e4, 2018. doi: 10.1016/j.devcel.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 27.Wilfling F, Wang H, Haas JT, Krahmer N, Gould TJ, Uchida A, Cheng J-X, Graham M, Christiano R, Fröhlich F, Liu X, Buhman KK, Coleman RA, Bewersdorf J, Farese RV, Walther TC. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev Cell 24: 384–399, 2013. doi: 10.1016/j.devcel.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Currie E, Guo X, Christiano R, Chitraju C, Kory N, Harrison K, Haas J, Walther TC, Farese RV Jr.. High confidence proteomic analysis of yeast LDs identifies additional droplet proteins and reveals connections to dolichol synthesis and sterol acetylation [S]. J Lipid Res 55: 1465–1477, 2014. doi: 10.1194/jlr.M050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bersuker K, Peterson CWH, To M, Sahl SJ, Savikhin V, Grossman EA, Nomura DK, Olzmann JA. A proximity labeling strategy provides insights into the composition and dynamics of lipid droplet proteomes. Dev Cell 44: 97–112.e7, 2018. doi: 10.1016/j.devcel.2017.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu S, Zhang X, Liu P. Lipid droplet proteins and metabolic diseases. Biochim Biophys Acta Mol Basis Dis 1864: 1968–1983, 2018. doi: 10.1016/j.bbadis.2017.07.019. [DOI] [PubMed] [Google Scholar]
- 31.Smirnova E, Goldberg EB, Makarova KS, Lin L, Brown WJ, Jackson CL. ATGL has a key role in lipid droplet/adiposome degradation in mammalian cells. EMBO Rep 7: 106–113, 2006. doi: 10.1038/sj.embor.7400559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X, Heckmann BL, Zhang X, Smas CM, Liu J. Distinct mechanisms regulate ATGL-mediated adipocyte lipolysis by lipid droplet coat proteins. Mol Endocrinol 27: 116–126, 2013. doi: 10.1210/me.2012-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schott MB, Weller SG, Schulze RJ, Krueger EW, Drizyte-Miller K, Casey CA, McNiven MA. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J Cell Biol 218: 3320–3335, 2019. doi: 10.1083/jcb.201803153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arrese EL, Saudale FZ, Soulages JL. Lipid droplets as signaling platforms linking metabolic and cellular functions. Lipid Insights 7: 7–16, 2014. doi: 10.4137/LPI.S11128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimmel AR, Sztalryd C. The perilipins: major cytosolic lipid droplet-associated proteins and their roles in cellular lipid storage, mobilization, and systemic homeostasis. Annu Rev Nutr 36: 471–509, 2016. doi: 10.1146/annurev-nutr-071813-105410. [DOI] [PubMed] [Google Scholar]
- 36.Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol 17: 759–770, 2015. doi: 10.1038/ncb3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaushik S, Cuervo AM. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 12: 432–438, 2016. doi: 10.1080/15548627.2015.1124226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsieh K, Lee YK, Londos C, Raaka BM, Dalen KT, Kimmel AR. Perilipin family members preferentially sequester to either triacylglycerol-specific or cholesteryl-ester-specific intracellular lipid storage droplets. J Cell Sci 125: 4067–4076, 2012. doi: 10.1242/jcs.104943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thiam AR, Farese RV Jr, Walther TC. The biophysics and cell biology of lipid droplets. Nat Rev Mol Cell Biol 14: 775–786, 2013. doi: 10.1038/nrm3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gong J, Sun Z, Wu L, Xu W, Schieber N, Xu D, Shui G, Yang H, Parton RG, Li P. Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J Cell Biol 195: 953–963, 2011. doi: 10.1083/jcb.201104142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu X, Park JG, So JS, Lee AH. Transcriptional activation of Fsp27 by the liver-enriched transcription factor CREBH promotes lipid droplet growth and hepatic steatosis. Hepatology 61: 857–869, 2015. doi: 10.1002/hep.27371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langhi C, Baldán Á. CIDEC/FSP27 is regulated by peroxisome proliferator-activated receptor alpha and plays a critical role in fasting- and diet-induced hepatosteatosis. Hepatology 61: 1227–1238, 2015. doi: 10.1002/hep.27607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ling J, Chaba T, Zhu LF, Jacobs RL, Vance DE. Hepatic ratio of phosphatidylcholine to phosphatidylethanolamine predicts survival after partial hepatectomy in mice. Hepatology 55: 1094–1102, 2012. doi: 10.1002/hep.24782. [DOI] [PubMed] [Google Scholar]
- 44.Krahmer N, Guo Y, Wilfling F, Hilger M, Lingrell S, Heger K, Newman HW, Schmidt-Supprian M, Vance DE, Mann M, Farese RV Jr, Walther TC. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab 14: 504–515, 2011. doi: 10.1016/j.cmet.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee HS, Nam Y, Chung YH, Kim HR, Park ES, Chung SJ, Kim JH, Sohn UD, Kim HC, Oh KW, Jeong JH. Beneficial effects of phosphatidylcholine on high-fat diet-induced obesity, hyperlipidemia and fatty liver in mice. Life Sci 118: 7–14, 2014. doi: 10.1016/j.lfs.2014.09.027. [DOI] [PubMed] [Google Scholar]
- 46.Shai N, Yifrach E, van Roermund CW, Cohen N, Bibi C, IJlst L, Cavellini L, Meurisse J, Schuster R, Zada L, Mari MC, Reggiori FM, Hughes AL, Escobar-Henriques M, Cohen MM, Waterham HR, Wanders RJA, Schuldiner M, Zalckvar E. Systematic mapping of contact sites reveals tethers and a function for the peroxisome-mitochondria contact. Nat Commun 9: 1761, 2018. doi: 10.1038/s41467-018-03957-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Valm AM, Cohen S, Legant WR, Melunis J, Hershberg U, Wait E, Cohen AR, Davidson MW, Betzig E, Lippincott-Schwartz J. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546: 162–167, 2017. doi: 10.1038/nature22369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306: 1383–1386, 2004.doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 49.Ong KT, Mashek MT, Bu SY, Greenberg AS, Mashek DG. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology 53: 116–126, 2011. doi: 10.1002/hep.24006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Obrowsky S, Chandak PG, Patankar JV, Povoden S, Schlager S, Kershaw EE, Bogner-Strauss JG, Hoefler G, Levak-Frank S, Kratky D. Adipose triglyceride lipase is a TG hydrolase of the small intestine and regulates intestinal PPARα signaling. J Lipid Res 54: 425–435, 2013. doi: 10.1194/jlr.M031716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ashford TP, Porter KR. Cytoplasmic components in hepatic cell lysosomes. J Cell Biol 12: 198–202, 1962. doi: 10.1083/jcb.12.1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Novikoff AB, Essner E. Cytolysomes and mitochondrial degeneration. J Cell Biol 15: 140–146, 1962. doi: 10.1083/jcb.15.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature 458: 1131–1135, 2009. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maus M, Cuk M, Patel B, Lian J, Ouimet M, Kaufmann U, Yang J, Horvath R, Hornig-Do H-T, Chrzanowska-Lightowlers ZM, Moore KJ, Cuervo AM, Feske S. Store-operated Ca2+ entry controls induction of lipolysis and the transcriptional reprogramming to lipid metabolism. Cell Metab 25: 698–712, 2017. doi: 10.1016/j.cmet.2016.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rogov V, Dötsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell 53: 167–178, 2014. doi: 10.1016/j.molcel.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 56.Kiss RS, Nilsson TJ. Rab proteins implicated in lipid storage and mobilization. J Biomed Res 28: 169, 2014. doi: 10.7555/JBR.28.20140029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Z, Schulze RJ, Weller SG, Krueger EW, Schott MB, Zhang X, Casey CA, Liu J, Stöckli J, James DE, McNiven MA. A novel Rab10-EHBP1-EHD2 complex essential for the autophagic engulfment of lipid droplets. Sci Adv 2: e1601470, 2016. doi: 10.1126/sciadv.1601470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, McNiven MA. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 61: 1896–1907, 2015. doi: 10.1002/hep.27667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Z, Yao Z, Chen Y, Qian L, Jiang S, Zhou J, Shao J, Chen A, Zhang F, Zheng S. Lipophagy and liver disease: new perspectives to better understanding and therapy. Biomed Pharmacother 97: 339–348, 2018. doi: 10.1016/j.biopha.2017.07.168. [DOI] [PubMed] [Google Scholar]
- 60.Sathyanarayan A, Mashek MT, Mashek DG. ATGL promotes autophagy/lipophagy via SIRT1 to control hepatic lipid droplet catabolism. Cell Rep 19: 1–9, 2017. doi: 10.1016/j.celrep.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grumet L, Eichmann TO, Taschler U, Zierler KA, Leopold C, Moustafa T, Radovic B, Romauch M, Yan C, Du H, Haemmerle G, Zechner R, Fickert P, Kratky D, Zimmermann R, Lass A. Lysosomal acid lipase hydrolyzes retinyl ester and affects retinoid turnover. J Biol Chem 291: 17977–17987, 2016. doi: 10.1074/jbc.M116.724054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schulze RJ, Sathyanarayan A, Mashek DG. Breaking fat: the regulation and mechanisms of lipophagy. Biochim Biophys Acta Mol Cell Biol Lipids 1862: 1178–1187, 2017. doi: 10.1016/j.bbalip.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sheriff S, Du H, Grabowski GA. Characterization of lysosomal acid lipase by site-directed mutagenesis and heterologous expression. J Biol Chem 270: 27766–27772, 1995. doi: 10.1074/jbc.270.46.27766. [DOI] [PubMed] [Google Scholar]
- 64.Warner TG, Dambach LM, Shin JH, O'Brien JS. Purification of the lysosomal acid lipase from human liver and its role in lysosomal lipid hydrolysis. J Biol Chem 256: 2952–2957, 1981. doi: 10.1016/S0021-9258(19)69707-3. [DOI] [PubMed] [Google Scholar]
- 65.Zechner R, Madeo F, Kratky D. Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat Rev Mol Cell Biol 18: 671–684, 2017. doi: 10.1038/nrm.2017.76. [DOI] [PubMed] [Google Scholar]
- 66.Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 516: 112–115, 2014. doi: 10.1038/nature13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seok S, Fu T, Choi S-E, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, Ma J, Kemper B, Kemper JK. Transcriptional regulation of autophagy by an FXR–CREB axis. Nature 516: 108–111, 2014. doi: 10.1038/nature13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lapierre LR, Gelino S, Meléndez A, Hansen M. Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr Biol 21: 1507–1514, 2011. doi: 10.1016/j.cub.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin C-W, Zhang H, Li M, Xiong X, Chen X, Chen X, Dong XC, Yin X-M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol 58: 993–999, 2013. doi: 10.1016/j.jhep.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang H, Yan S, Khambu B, Ma F, Li Y, Chen X, Martina JA, Puertollano R, Li Y, Chalasani N, Yin X-M. Dynamic MTORC1-TFEB feedback signaling regulates hepatic autophagy, steatosis and liver injury in long-term nutrient oversupply. Autophagy 14: 1779–1795, 2018. doi: 10.1080/15548627.2018.1490850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seo AY, Lau P-W, Feliciano D, Sengupta P, Le Gros MA, Cinquin B, Larabell CA, Lippincott-Schwartz J. AMPK and vacuole-associated Atg14p orchestrate μ-lipophagy for energy production and long-term survival under glucose starvation. eLife 6: e21690, 2017. doi: 10.7554/eLife.21690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Y, Yang P, Zhao L, Chen Y, Zhang X, Zeng S, Wei L, Varghese Z, Moorhead JF, Chen Y, Ruan XZ. CD36 plays a negative role in the regulation of lipophagy in hepatocytes through an AMPK-dependent pathway [S]. J Lipid Res 60: 844–855, 2019. doi: 10.1194/jlr.M090969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mony VK, Benjamin S, O'Rourke EJ. A lysosome-centered view of nutrient homeostasis. Autophagy 12: 619–631, 2016. doi: 10.1080/15548627.2016.1147671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cingolani F, Czaja MJ. Regulation and functions of autophagic lipolysis. Trends Endocrinol Metab 27: 696–705, 2016. doi: 10.1016/j.tem.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marques LR, Diniz TA, Antunes BM, Rossi FE, Caperuto EC, Lira FS, Gonçalves DC. Reverse cholesterol transport: molecular mechanisms and the non-medical approach to enhance HDL cholesterol. Front Physiol 9: 526, 2018. doi: 10.3389/fphys.2018.00526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ouimet M, Barrett TJ, Fisher EA. HDL and reverse cholesterol transport: basic mechanisms and their roles in vascular health and disease. Circ Res 124: 1505–1518, 2019. doi: 10.1161/CIRCRESAHA.119.312617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tall AR. An overview of reverse cholesterol transport. Eur Heart J 19: A31–A35, 1998. [PubMed] [Google Scholar]
- 78.Yvan-Charvet L, Wang N, Tall AR. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol 30: 139–143, 2010. doi: 10.1161/ATVBAHA.108.179283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem 289: 24020–24029, 2014. doi: 10.1074/jbc.R114.583658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Holzer M, Birner-Gruenberger R, Stojakovic T, El-Gamal D, Binder V, Wadsack C, Heinemann A, Marsche G. Uremia alters HDL composition and function. J Am Soc Nephrol 22: 1631–1641, 2011. doi: 10.1681/ASN.2010111144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kon V, Ikizler TA, Fazio S. Importance of high-density lipoprotein quality: evidence from chronic kidney disease. Curr Opin Nephrol Hypertens 22: 259–265, 2013. doi: 10.1097/MNH.0b013e32835fe47f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang C, Liu P. The lipid droplet: a conserved cellular organelle. Protein Cell 8: 796–800, 2017. doi: 10.1007/s13238-017-0467-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thiam AR, Beller M. The why, when and how of lipid droplet diversity. J Cell Sci 130: 315–324, 2017. doi: 10.1242/jcs.192021. [DOI] [PubMed] [Google Scholar]
- 84.Ohsaki Y, Sołtysik K, Fujimoto T. The lipid droplet and the endoplasmic reticulum. Adv Exp Med Biol 997: 111–120, 2017. doi: 10.1007/978-981-10-4567-7_8. [DOI] [PubMed] [Google Scholar]
- 85.Jackson CL. Lipid droplet biogenesis. Curr Opin Cell Biol 59: 88–96, 2019. doi: 10.1016/j.ceb.2019.03.018. [DOI] [PubMed] [Google Scholar]
- 86.Bersuker K, Olzmann JA. Establishing the lipid droplet proteome: mechanisms of lipid droplet protein targeting and degradation. Biochim Biophys Acta Mol Cell Biol Lipids 1862: 1166–1177, 2017. doi: 10.1016/j.bbalip.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barneda D, Christian M. Lipid droplet growth: regulation of a dynamic organelle. Curr Opin Cell Biol 47: 9–15, 2017. doi: 10.1016/j.ceb.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 88.Barbosa AD, Siniossoglou S. Function of lipid droplet-organelle interactions in lipid homeostasis. Biochim Biophys Acta Mol Cell Res 1864: 1459–1468, 2017. doi: 10.1016/j.bbamcr.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 89.Bosch M, Sánchez-Álvarez M, Fajardo A, Kapetanovic R, Steiner B, Dutra F, Moreira L, López JA, Campo R, Marí M, Morales-Paytuví F, Tort O, Gubern A, Templin RM, Curson JEB, Martel N, Català C, Lozano F, Tebar F, Enrich C, Vázquez J, Del Pozo MA, Sweet MJ, Bozza PT, Gross SP, Parton RG, Pol A. Mammalian lipid droplets are innate immune hubs integrating cell metabolism and host defense. Science 370: eaay8085, 2020. doi: 10.1126/science.aay8085. [DOI] [PubMed] [Google Scholar]
- 90.Yoshida H. ER stress and diseases. FEBS J 274: 630–658, 2007. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 91.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 16: 1345–1355, 2002. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306: 457–461, 2004. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 93.Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, Mori K, Glimcher LH, Denko NC, Giaccia AJ, Le QT, Koong AC. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res 64: 5943–5947, 2004. doi: 10.1158/0008-5472.CAN-04-1606. [DOI] [PubMed] [Google Scholar]
- 94.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 403: 98–103, 2000. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 95.Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441: 513–517, 2006. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 96.Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 109: 525–532, 2002. doi: 10.1172/JCI0214550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Görgün CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313: 1137–1140, 2006. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Blais JD, Addison CL, Edge R, Falls T, Zhao H, Wary K, Koumenis C, Harding HP, Ron D, Holcik M, Bell JC. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol Cell Biol 26: 9517–9532, 2006. doi: 10.1128/MCB.01145-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086, 2011. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 100.Vevea JD, Garcia EJ, Chan RB, Zhou B, Schultz M, Di Paolo G, McCaffery JM, Pon LA. Role for lipid droplet biogenesis and microlipophagy in adaptation to lipid imbalance in yeast. Dev Cell 35: 584–599, 2015. doi: 10.1016/j.devcel.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci USA 110: 4628–4633, 2013. doi: 10.1073/pnas.1217611110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Promlek T, Ishiwata-Kimata Y, Shido M, Sakuramoto M, Kohno K, Kimata Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol Biol Cell 22: 3520–3532, 2011. doi: 10.1091/mbc.E11-04-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473: 528–531, 2011. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fei W, Wang H, Fu X, Bielby C, Yang H. Conditions of endoplasmic reticulum stress stimulate lipid droplet formation in Saccharomyces cerevisiae. Biochem J 424: 61–67, 2009. doi: 10.1042/BJ20090785. [DOI] [PubMed] [Google Scholar]
- 105.Lee JS, Mendez R, Heng HH, Yang ZQ, Zhang K. Pharmacological ER stress promotes hepatic lipogenesis and lipid droplet formation. Am J Transl Res 4: 102–113, 2012. [PMC free article] [PubMed] [Google Scholar]
- 106.Rutkowski DT, Wu J, Back SH, Callaghan MU, Ferris SP, Iqbal J, Clark R, Miao H, Hassler JR, Fornek J, Katze MG, Hussain MM, Song B, Swathirajan J, Wang J, Yau GD, Kaufman RJ. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell 15: 829–840, 2008. doi: 10.1016/j.devcel.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Olzmann JA, Kopito RR. Lipid droplet formation is dispensable for endoplasmic reticulum-associated degradation. J Biol Chem 286: 27872–27874, 2011. doi: 10.1074/jbc.C111.266452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Petschnigg J, Wolinski H, Kolb D, Zellnig G, Kurat CF, Natter K, Kohlwein SD. Good fat, essential cellular requirements for triacylglycerol synthesis to maintain membrane homeostasis in yeast. J Biol Chem 284: 30981–30993, 2009. doi: 10.1074/jbc.M109.024752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Velázquez AP, Tatsuta T, Ghillebert R, Drescher I, Graef M. Lipid droplet-mediated ER homeostasis regulates autophagy and cell survival during starvation. J Cell Biol 212: 621–631, 2016. doi: 10.1083/jcb.201508102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gaspar ML, Hofbauer HF, Kohlwein SD, Henry SA. Coordination of storage lipid synthesis and membrane biogenesis: evidence for cross-talk between triacylglycerol metabolism and phosphatidylinositol synthesis. J Biol Chem 286: 1696–1708, 2011. doi: 10.1074/jbc.M110.172296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chitraju C, Mejhert N, Haas JT, Diaz-Ramirez LG, Grueter CA, Imbriglio JE, Pinto S, Koliwad SK, Walther TC, Farese RV Jr.. Triglyceride synthesis by DGAT1 protects adipocytes from lipid-induced ER stress during lipolysis. Cell Metab 26: 407–418.e3, 2017. doi: 10.1016/j.cmet.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Paar M, Jüngst C, Steiner NA, Magnes C, Sinner F, Kolb D, Lass A, Zimmermann R, Zumbusch A, Kohlwein SD, Wolinski H. Remodeling of lipid droplets during lipolysis and growth in adipocytes. J Biol Chem 287: 11164–11173, 2012. doi: 10.1074/jbc.M111.316794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hashimoto T, Segawa H, Okuno M, Kano H, Hamaguchi HO, Haraguchi T, Hiraoka Y, Hasui S, Yamaguchi T, Hirose F, Osumi T. Active involvement of micro-lipid droplets and lipid-droplet-associated proteins in hormone-stimulated lipolysis in adipocytes. J Cell Sci 125: 6127–6136, 2012. doi: 10.1242/jcs.113084. [DOI] [PubMed] [Google Scholar]
- 114.Islam A, Kagawa Y, Miyazaki H, Shil SK, Umaru BA, Yasumoto Y, Yamamoto Y, Owada Y. FABP7 protects astrocytes against ROS toxicity via lipid droplet formation. Mol Neurobiol 56: 5763–5779, 2019. doi: 10.1007/s12035-019-1489-2. [DOI] [PubMed] [Google Scholar]
- 115.Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, Lechene CP, Postle AD, Gould AP. Antioxidant role for lipid droplets in a stem cell niche of Drosophila. Cell 163: 340–353, 2015. doi: 10.1016/j.cell.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liu L, MacKenzie KR, Putluri N, Maletić-Savatić M, Bellen HJ. The glia-neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab 26: 719–737.e6, 2017. doi: 10.1016/j.cmet.2017.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Petan T, Jarc E, Jusović M. Lipid Droplets in cancer: guardians of fat in a stressful world. Molecules 23: 1941, 2018. doi: 10.3390/molecules23081941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shi L, Wang K, Deng Y, Wang Y, Zhu S, Yang X, Liao W. Role of lipophagy in the regulation of lipid metabolism and the molecular mechanism. Nan Fang Yi Ke Da Xue Xue Bao 39: 867–874, 2019. doi: 10.12122/j.issn.1673-4254.2019.07.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang CW. Lipid droplets, lipophagy, and beyond. Biochim Biophys Acta 1861: 793–805, 2016. doi: 10.1016/j.bbalip.2015.12.010. [DOI] [PubMed] [Google Scholar]
- 120.Sztalryd C, Brasaemle DL. The perilipin family of lipid droplet proteins: gatekeepers of intracellular lipolysis. Biochim Biophys Acta Mol Cell Biol Lipids 1862: 1221–1232, 2017. doi: 10.1016/j.bbalip.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schulze RJ, McNiven MA. Lipid droplet formation and lipophagy in fatty liver disease. Semin Liver Dis 39: 283–290, 2019. doi: 10.1055/s-0039-1685524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Martinez-Lopez N, Singh R. Autophagy and lipid droplets in the liver. Annu Rev Nutr 35: 215–237, 2015. doi: 10.1146/annurev-nutr-071813-105336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Maan M, Peters JM, Dutta M, Patterson AD. Lipid metabolism and lipophagy in cancer. Biochem Biophys Res Commun 504: 582–589, 2018. doi: 10.1016/j.bbrc.2018.02.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Khawar MB, Gao H, Li W. Autophagy and lipid metabolism. Adv Exp Med Biol 1206: 359–374, 2019. doi: 10.1007/978-981-15-0602-4_17. [DOI] [PubMed] [Google Scholar]
- 125.Jarc E, Kump A, Malavašič P, Eichmann TO, Zimmermann R, Petan T. Lipid droplets induced by secreted phospholipase A2 and unsaturated fatty acids protect breast cancer cells from nutrient and lipotoxic stress. Biochim Biophys Acta Mol Cell Biol Lipids 1863: 247–265, 2018. doi: 10.1016/j.bbalip.2017.12.006. [DOI] [PubMed] [Google Scholar]
- 126.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr. Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA 100: 3077–3082, 2003. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Senkal CE, Salama MF, Snider AJ, Allopenna JJ, Rana NA, Koller A, Hannun YA, Obeid LM. Ceramide is metabolized to acylceramide and stored in lipid droplets. Cell Metab 25: 686–697, 2017. doi: 10.1016/j.cmet.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bosma M, Dapito DH, Drosatos-Tampakaki Z, Huiping-Son N, Huang LS, Kersten S, Drosatos K, Goldberg IJ. Sequestration of fatty acids in triglycerides prevents endoplasmic reticulum stress in an in vitro model of cardiomyocyte lipotoxicity. Biochim Biophys Acta 1841: 1648–1655, 2014. doi: 10.1016/j.bbalip.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Herms A, Bosch M, Reddy BJ, Schieber NL, Fajardo A, Rupérez C, Fernández-Vidal A, Ferguson C, Rentero C, Tebar F, Enrich C, Parton RG, Gross SP, Pol A. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat Commun 6: 7176, 2015. doi: 10.1038/ncomms8176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pan X, Wilson M, McConville C, Arvanitis TN, Griffin JL, Kauppinen RA, Peet AC. Increased unsaturation of lipids in cytoplasmic lipid droplets in DAOY cancer cells in response to cisplatin treatment. Metabolomics 9: 722–729, 2013. doi: 10.1007/s11306-012-0483-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li N, Lizardo DY, Atilla-Gokcumen GE. Specific triacylglycerols accumulate via increased lipogenesis during 5-FU-induced apoptosis. ACS Chem Biol 11: 2583–2587, 2016. doi: 10.1021/acschembio.6b00410. [DOI] [PubMed] [Google Scholar]
- 132.Nguyen TB, Louie SM, Daniele JR, Tran Q, Dillin A, Zoncu R, Nomura DK, Olzmann JA. DGAT1-dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy. Dev Cell 42: 9–21.e5, 2017. doi: 10.1016/j.devcel.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nguyen TB, Olzmann JA. Lipid droplets and lipotoxicity during autophagy. Autophagy 13: 2002–2003, 2017. doi: 10.1080/15548627.2017.1359451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Coelho M, Oliveira T, Fernandes R. Biochemistry of adipose tissue: an endocrine organ. Arch Med Sci 9: 191–200, 2013. doi: 10.5114/aoms.2013.33181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nielsen J, Mogensen M, Vind BF, Sahlin K, Højlund K, Schrøder HD, Ortenblad N. Increased subsarcolemmal lipids in type 2 diabetes: effect of training on localization of lipids, mitochondria, and glycogen in sedentary human skeletal muscle. Am J Physiol Endocrinol Physiol 298: E706–E713, 2010. doi: 10.1152/ajpendo.00692.2009. [DOI] [PubMed] [Google Scholar]
- 136.Itabe H, Obama T, Kato R. The dynamics of oxidized LDL during atherogenesis. J Lipids 2011: 418313, 2011. doi: 10.1155/2011/418313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Parthasarathy S, Raghavamenon A, Garelnabi MO, Santanam N. Oxidized low-density lipoprotein. Methods Mol Biol 610: 403–417, 2010. doi: 10.1007/978-1-60327-029-8_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Linton MF, Yancey PG, Davies SS, Jerome WG, Linton EF, Song WL, Doran AC, Vickers KC. The role of lipids and lipoproteins in atherosclerosis. In Endotext, edited by Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, Grossman A, Hershman JM, Hofland HJ, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Purnell J, Singer F, Stratakis CA, Trence DL, Wilson DP.. South Dartmouth, MA: MDText.com, Inc., 2000. [Google Scholar]
- 139.Grajchen E, Hendriks JJA, Bogie JFJ. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol Commun 6: 124, 2018. doi: 10.1186/s40478-018-0628-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Milich LM, Ryan CB, Lee JK. The origin, fate, and contribution of macrophages to spinal cord injury pathology. Acta Neuropathol 137: 785–797, 2019. [Erratum in Acta Neuropathol 137: 799–800, 2019] doi: 10.1007/s00401-019-01992-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kis V, Barti B, Lippai M, Sass M. Specialized cortex glial cells accumulate lipid droplets in Drosophila melanogaster. PLoS One 10: e0131250, 2015. doi: 10.1371/journal.pone.0131250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci 13: 567–576, 2010. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Papadopoulos C, Orso G, Mancuso G, Herholz M, Gumeni S, Tadepalle N, Jüngst C, Tzschichholz A, Schauss A, Höning S, Trifunovic A, Daga A, Rugarli EI. Spastin binds to lipid droplets and affects lipid metabolism. PLoS Genet 11: e1005149, 2015. doi: 10.1371/journal.pgen.1005149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Marschallinger J, Iram T, Zardeneta M, Lee SE, Lehallier B, Haney MS, Pluvinage JV, Mathur V, Hahn O, Morgens DW, Kim J, Tevini J, Felder TK, Wolinski H, Bertozzi CR, Bassik MC, Aigner L, Wyss-Coray T. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci 23: 194–208, 2020. [Erratum in Nat Neurosci 23: 294, 2020 and in Nat Neurosci 23: 1308, 2020] doi: 10.1038/s41593-019-0566-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Farmer BC, Walsh AE, Kluemper JC, Johnson LA. Lipid droplets in neurodegenerative disorders. Front Neurosci 14: 742–742, 2020. doi: 10.3389/fnins.2020.00742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A, Bellen HJ. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160: 177–190, 2015. doi: 10.1016/j.cell.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Philips T, Rothstein JD. Oligodendroglia: metabolic supporters of neurons. J Clin Invest 127: 3271–3280, 2017. doi: 10.1172/JCI90610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dourlen P, Sujkowski A, Wessells R, Mollereau B. Fatty acid transport proteins in disease: new insights from invertebrate models. Prog Lipid Res 60: 30–40, 2015. doi: 10.1016/j.plipres.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 149.Van Den Brink DM, Cubizolle A, Chatelain G, Davoust N, Girard V, Johansen S, Napoletano F, Dourlen P, Guillou L, Angebault-Prouteau C, Bernoud-Hubac N, Guichardant M, Brabet P, Mollereau B. Physiological and pathological roles of FATP-mediated lipid droplets in Drosophila and mice retina. PLoS Genet 14: e1007627, 2018. doi: 10.1371/journal.pgen.1007627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chan RB, Oliveira TG, Cortes EP, Honig LS, Duff KE, Small SA, Wenk MR, Shui G, Di Paolo G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J Biol Chem 287: 2678–2688, 2012. doi: 10.1074/jbc.M111.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dafnis I, Raftopoulou C, Mountaki C, Megalou E, Zannis VI, Chroni A. ApoE isoforms and carboxyl-terminal-truncated apoE4 forms affect neuronal BACE1 levels and Aβ production independently of their cholesterol efflux capacity. Biochem J 475: 1839–1859, 2018. doi: 10.1042/BCJ20180068. [DOI] [PubMed] [Google Scholar]
- 152.Farmer BC, Kluemper J, Johnson LA. Apolipoprotein E4 alters astrocyte fatty acid metabolism and lipid droplet formation. Cells 8: 182, 2019. doi: 10.3390/cells8020182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Golestaneh N, Chu Y, Xiao YY, Stoleru GL, Theos AC. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis 8: e2537, 2017. doi: 10.1038/cddis.2016.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Storti F, Klee K, Todorova V, Steiner R, Othman A, van der Velde-Visser S, Samardzija M, Meneau I, Barben M, Karademir D, Pauzuolyte V, Boye SL, Blaser F, Ullmer C, Dunaief JL, Hornemann T, Rohrer L, den Hollander A, von Eckardstein A, Fingerle J, Maugeais C, Grimm C. Impaired ABCA1/ABCG1-mediated lipid efflux in the mouse retinal pigment epithelium (RPE) leads to retinal degeneration. eLife 8: e45100, 2019. doi: 10.7554/eLife.45100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Choi S-H, Sviridov D, Miller YI. Oxidized cholesteryl esters and inflammation. Biochim Biophys Acta Mol Cell Biol Lipids 1862: 393–397, 2017. doi: 10.1016/j.bbalip.2016.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL. Lipid droplet binding and oligomerization properties of the Parkinson's disease protein α-synuclein. J Biol Chem 277: 6344–6352, 2002. doi: 10.1074/jbc.M108414200. [DOI] [PubMed] [Google Scholar]
- 157.Fanning S, Haque A, Imberdis T, Baru V, Barrasa MI, Nuber S, Termine D, Ramalingam N, Ho GPH, Noble T, Sandoe J, Lou Y, Landgraf D, Freyzon Y, Newby G, Soldner F, Terry-Kantor E, Kim TE, Hofbauer HF, Becuwe M, Jaenisch R, Pincus D, Clish CB, Walther TC, Farese RV Jr, Srinivasan S, Welte MA, Kohlwein SD, Dettmer U, Lindquist S, Selkoe D. Lipidomic analysis of α-synuclein neurotoxicity identifies stearoyl CoA desaturase as a target for Parkinson treatment. Mol Cell 73: 1001–1014.e8, 2019. doi: 10.1016/j.molcel.2018.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sánchez Campos S, Alza NP, Salvador GA. Lipid metabolism alterations in the neuronal response to A53T α-synuclein and Fe-induced injury. Arch Biochem Biophys 655: 43–54, 2018. doi: 10.1016/j.abb.2018.08.007. [DOI] [PubMed] [Google Scholar]
- 159.Vincent BM, Tardiff DF, Piotrowski JS, Aron R, Lucas MC, Chung CY, Bacherman H, Chen Y, Pires M, Subramaniam R, Doshi DB, Sadlish H, Raja WK, Solís EJ, Khurana V, Le Bourdonnec B, Scannevin RH, Rhodes KJ. Inhibiting stearoyl-CoA desaturase ameliorates α-synuclein cytotoxicity. Cell Rep 25: 2742–2754.e31, 2018. doi: 10.1016/j.celrep.2018.11.028. [DOI] [PubMed] [Google Scholar]
- 160.Gluchowski NL, Becuwe M, Walther TC, Farese RV Jr.. Lipid droplets and liver disease: from basic biology to clinical implications. Nat Rev Gastroenterol Hepatol 14: 343–355, 2017. doi: 10.1038/nrgastro.2017.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sahini N, Borlak J. Recent insights into the molecular pathophysiology of lipid droplet formation in hepatocytes. Prog Lipid Res 54: 86–112, 2014. doi: 10.1016/j.plipres.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 162.Yao ZM, Vance DE. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J Biol Chem 263: 2998–3004, 1988. doi: 10.1016/S0021-9258(18)69166-5. [DOI] [PubMed] [Google Scholar]
- 163.Rong X, Wang B, Dunham MM, Hedde PN, Wong JS, Gratton E, Young SG, Ford DA, Tontonoz P. Lpcat3-dependent production of arachidonoyl phospholipids is a key determinant of triglyceride secretion. eLife 4: e06557, 2015. doi: 10.7554/eLife.06557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol 29: 415–445, 2011. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 165.Zhang X-Q, Xu C-F, Yu C-H, Chen W-X, Li Y-M. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol 20: 1768–1776, 2014. doi: 10.3748/wjg.v20.i7.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320: 1492–1496, 2008. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Pouysségur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441: 437–443, 2006. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 168.Vaupel P, Höckel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal 9: 1221–1235, 2007. doi: 10.1089/ars.2007.1628. [DOI] [PubMed] [Google Scholar]
- 169.Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab 2: 17, 2014. doi: 10.1186/2049-3002-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ralph SJ, Moreno-Sánchez R, Neuzil J, Rodríguez-Enríquez S. Inhibitors of succinate: quinone reductase/complex II regulate production of mitochondrial reactive oxygen species and protect normal cells from ischemic damage but induce specific cancer cell death. Pharm Res 28: 2695–2730, 2011. [Erratum in Pharm Res 28: 3274, 2011] doi: 10.1007/s11095-011-0566-7. [DOI] [PubMed] [Google Scholar]
- 171.Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB, Rabinowitz JD. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci USA 110: 8882–8887, 2013. doi: 10.1073/pnas.1307237110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, Zhang Q, Wakelam MJO, Karpe F, Schulze A, Harris AL. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep 9: 349–365, 2014. doi: 10.1016/j.celrep.2014.08.056. [DOI] [PubMed] [Google Scholar]
- 173.Zoula S, Rijken PF, Peters JP, Farion R, Van der Sanden BP, Van der Kogel AJ, Décorps M, Rémy C. Pimonidazole binding in C6 rat brain glioma: relation with lipid droplet detection. Br J Cancer 88: 1439–1444, 2003. doi: 10.1038/sj.bjc.6600837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rémy C, Fouilhé N, Barba I, Sam-Laï E, Lahrech H, Cucurella MG, Izquierdo M, Moreno A, Ziegler A, Massarelli R, Décorps M, Arús C. Evidence that mobile lipids detected in rat brain glioma by 1H nuclear magnetic resonance correspond to lipid droplets. Cancer Res 57: 407–414, 1997. [PubMed] [Google Scholar]
- 175.Mylonis I, Sembongi H, Befani C, Liakos P, Siniossoglou S, Simos G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J Cell Sci 125: 3485–3493, 2012. doi: 10.1242/jcs.106682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Qiu B, Ackerman D, Sanchez DJ, Li B, Ochocki JD, Grazioli A, Bobrovnikova-Marjon E, Diehl JA, Keith B, Simon MC. HIF2α-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov 5: 652–667, 2015. doi: 10.1158/2159-8290.CD-14-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ackerman D, Simon MC. Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell Biol 24: 472–478, 2014. doi: 10.1016/j.tcb.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Young RM, Ackerman D, Quinn ZL, Mancuso A, Gruber M, Liu L, Giannoukos DN, Bobrovnikova-Marjon E, Diehl JA, Keith B, Simon MC. Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev 27: 1115–1131, 2013. doi: 10.1101/gad.198630.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, Attardi LD. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev 27: 1016–1031, 2013. doi: 10.1101/gad.212282.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Goldstein I, Rotter V. Regulation of lipid metabolism by p53—fighting two villains with one sword. Trends Endocrinol Metab 23: 567–575, 2012. doi: 10.1016/j.tem.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 181.Kaini RR, Sillerud LO, Zhaorigetu S, Hu CAA. Autophagy regulates lipolysis and cell survival through lipid droplet degradation in androgen‐sensitive prostate cancer cells. Prostate 72: 1412–1422, 2012. doi: 10.1002/pros.22489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Mukhopadhyay S, Schlaepfer IR, Bergman BC, Panda PK, Praharaj PP, Naik PP, Agarwal R, Bhutia SK. ATG14 facilitated lipophagy in cancer cells induce ER stress mediated mitoptosis through a ROS dependent pathway. Free Radic Biol Med 104: 199–213, 2017. doi: 10.1016/j.freeradbiomed.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 183.Park E-J, Lee AY, Chang S-H, Yu K-N, Kim J-H, Cho M-H. Role of p53 in the cellular response following oleic acid accumulation in Chang liver cells. Toxicol Lett 224: 114–120, 2014. doi: 10.1016/j.toxlet.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 184.Xu G, Jiang Y, Xiao Y, Liu XD, Yue F, Li W, Li X, He Y, Jiang X, Huang H, Chen Q, Jonasch E, Liu L. Fast clearance of lipid droplets through MAP1S-activated autophagy suppresses clear cell renal cell carcinomas and promotes patient survival. Oncotarget 7: 6255–6265, 2016. doi: 10.18632/oncotarget.6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Weinberg J. Lipotoxicity. Kidney Int 70: 1560–1566, 2006. doi: 10.1038/sj.ki.5001834. [DOI] [PubMed] [Google Scholar]
- 186.Bobulescu IA, Lotan Y, Zhang J, Rosenthal TR, Rogers JT, Adams-Huet B, Sakhaee K, Moe OW. Triglycerides in the human kidney cortex: relationship with body size. PLoS One 9: e101285, 2014. doi: 10.1371/journal.pone.0101285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zager RA, Johnson AC, Hanson SY. Renal tubular triglyercide accumulation following endotoxic, toxic, and ischemic injury. Kidney Int 67: 111–121, 2005. doi: 10.1111/j.1523-1755.2005.00061.x. [DOI] [PubMed] [Google Scholar]
- 188.Minami S, Yamamoto T, Takabatake Y, Takahashi A, Namba T, Matsuda J, Kimura T, Kaimori JY, Matsui I, Hamano T, Takeda H, Takahashi M, Izumi Y, Bamba T, Matsusaka T, Niimura F, Isaka Y. Lipophagy maintains energy homeostasis in the kidney proximal tubule during prolonged starvation. Autophagy 13: 1629–1647, 2017. doi: 10.1080/15548627.2017.1341464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zager RA, Johnson A, Anderson K, Wright S. Cholesterol ester accumulation: an immediate consequence of acute in vivo ischemic renal injury. Kidney Int 59: 1750–1761, 2001. doi: 10.1046/j.1523-1755.2001.0590051750.x. [DOI] [PubMed] [Google Scholar]
- 190.Ducasa GM, Mitrofanova A, Mallela SK, Liu X, Molina J, Sloan A, Pedigo CE, Ge M, Santos JV, Hernandez Y, Kim JJ, Maugeais C, Mendez AJ, Nair V, Kretzler M, Burke GW, Nelson RG, Ishimoto Y, Inagi R, Banerjee S, Liu S, Szeto HH, Merscher S, Fontanesi F, Fornoni A. ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J Clin Invest 129: 3387–3400, 2019. doi: 10.1172/JCI125316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21: 37–46, 2015. doi: 10.1038/nm.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Jao TM, Nangaku M, Wu CH, Sugahara M, Saito H, Maekawa H, Ishimoto Y, Aoe M, Inoue T, Tanaka T, Staels B, Mori K, Inagi R. ATF6α downregulation of PPARα promotes lipotoxicity-induced tubulointerstitial fibrosis. Kidney Int 95: 577–589, 2019. doi: 10.1016/j.kint.2018.09.023. [DOI] [PubMed] [Google Scholar]
- 193.Ge M, Fontanesi F, Merscher S, Fornoni A. The vicious cycle of renal lipotoxicity and mitochondrial dysfunction. Front Physiol 11: 732, 2020. doi: 10.3389/fphys.2020.00732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Merscher-Gomez S, Guzman J, Pedigo CE, Lehto M, Aguillon-Prada R, Mendez A, Lassenius MI, Forsblom C, Yoo T, Villarreal R, Maiguel D, Johnson K, Goldberg R, Nair V, Randolph A, Kretzler M, Nelson RG, Burke GW 3rd, Groop PH, Fornoni A; FinnDiane Study Group. Cyclodextrin protects podocytes in diabetic kidney disease. Diabetes 62: 3817–3827, 2013. doi: 10.2337/db13-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mitrofanova A, Molina J, Varona Santos J, Guzman J, Morales XA, Ducasa GM, Bryn J, Sloan A, Volosenco I, Kim JJ, Ge M, Mallela SK, Kretzler M, Eddy S, Martini S, Wahl P, Pastori S, Mendez AJ, Burke GW, Merscher S, Fornoni A. Hydroxypropyl-β-cyclodextrin protects from kidney disease in experimental Alport syndrome and focal segmental glomerulosclerosis. Kidney Int 94: 1151–1159, 2018. doi: 10.1016/j.kint.2018.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Pedigo CE, Ducasa GM, Leclercq F, Sloan A, Mitrofanova A, Hashmi T, Molina-David J, Ge M, Lassenius MI, Forsblom C, Lehto M, Groop PH, Kretzler M, Eddy S, Martini S, Reich H, Wahl P, Ghiggeri G, Faul C, Burke GW 3rd, Kretz O, Huber TB, Mendez AJ, Merscher S, Fornoni A. Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J Clin Invest 126: 3336–3350, 2016. doi: 10.1172/JCI85939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hara S, Kobayashi N, Sakamoto K, Ueno T, Manabe S, Takashima Y, Hamada J, Pastan I, Fukamizu A, Matsusaka T, Nagata M. Podocyte injury-driven lipid peroxidation accelerates the infiltration of glomerular foam cells in focal segmental glomerulosclerosis. Am J Pathol 185: 2118–2131, 2015. doi: 10.1016/j.ajpath.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lee HS, Kruth HS. Accumulation of cholesterol in the lesions of focal segmental glomerulosclerosis. Nephrology (Carlton) 8: 224–223, 2003. doi: 10.1046/j.1440-1797.2003.00160.x. [DOI] [PubMed] [Google Scholar]
- 199.Gassler N, Elger M, Kränzlin B, Kriz W, Gretz N, Hähnel B, Hosser H, Hartmann I. Podocyte injury underlies the progression of focal segmental glomerulosclerosis in the fa/fa Zucker rat. Kidney Int 60: 106–116, 2001. doi: 10.1046/j.1523-1755.2001.00777.x. [DOI] [PubMed] [Google Scholar]
- 200.Meyrier A, Dairou F, Callard P, Mougenot B. Lipoprotein glomerulopathy: first case in a white European. Nephrol Dial Transplant 10: 546–549, 1995. [Erratum in Nephrol Dial Transplant 10: 1273, 1995] doi: 10.1093/ndt/10.4.546. [DOI] [PubMed] [Google Scholar]
- 201.Chun J, Zhang JY, Wilkins MS, Subramanian B, Riella C, Magraner JM, Alper SL, Friedman DJ, Pollak MR. Recruitment of APOL1 kidney disease risk variants to lipid droplets attenuates cell toxicity. Proc Natl Acad Sci USA 116: 3712–3721, 2019. doi: 10.1073/pnas.1820414116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ge M, Molina J, Ducasa GM, Mallela SK, Varona Santos J, Mitrofanova A, Kim JJ, Liu X, Sloan A, Mendez AJ, Banerjee S, Liu S, Szeto HH, Shin MK, Hoek M, Kopp JB, Fontanesi F, Merscher S, Fornoni A. APOL1 risk variants affect podocyte lipid homeostasis and energy production in focal segmental glomerulosclerosis. Hum Mol Genet 30: 182–197, 2021. doi: 10.1093/hmg/ddab022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Herman-Edelstein M, Scherzer P, Tobar A, Levi M, Gafter U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J Lipid Res 55: 561–572, 2014. doi: 10.1194/jlr.P040501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Park H-W, Lee JH. Calcium channel blockers as potential therapeutics for obesity-associated autophagy defects and fatty liver pathologies. Autophagy 10: 2385–2386, 2014. doi: 10.4161/15548627.2014.984268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Cha J-Y, Lee H-J. Targeting lipid metabolic reprogramming as anticancer therapeutics. J Cancer Prev 21: 209–215, 2016. doi: 10.15430/JCP.2016.21.4.209. [DOI] [PMC free article] [PubMed] [Google Scholar]