

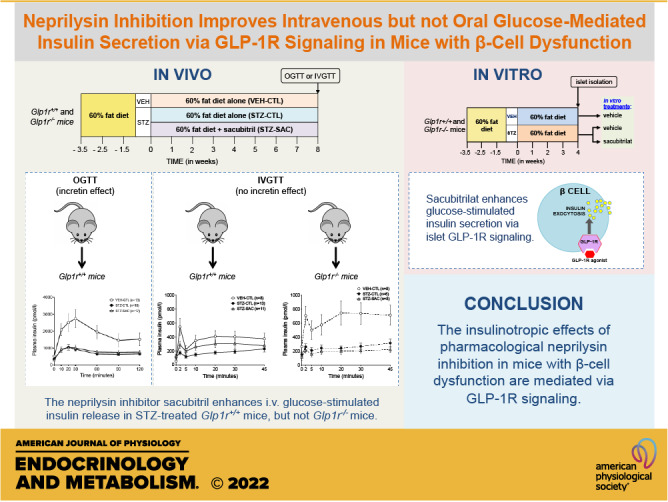
Keywords: GLP-1, insulin secretion, mouse, neprilysin, type 2 diabetes
Abstract
Type 2 diabetes is associated with the upregulation of neprilysin, a peptidase capable of cleaving glucoregulatory peptides such as glucagon-like peptide-1 (GLP-1). In humans, use of the neprilysin inhibitor sacubitril in combination with an angiotensin II receptor blocker was associated with increased plasma GLP-1 levels and improved glycemic control. Whether neprilysin inhibition per se is mediating these effects remains unknown. We sought to determine whether pharmacological neprilysin inhibition on its own confers beneficial effects on glycemic status and β-cell function in a mouse model of reduced insulin secretion, and whether any such effects are dependent on GLP-1 receptor (GLP-1R) signaling. High-fat-fed male wild-type (Glp1r+/+) and GLP-1R knockout (Glp1r−/−) mice were treated with low-dose streptozotocin (STZ) to recapitulate type 2 diabetes-associated β-cell dysfunction, or vehicle as control. Mice were continued on high-fat diet alone or supplemented with the neprilysin inhibitor sacubitril for 8 wk. At the end of the study period, β-cell function was assessed by oral or intravenous glucose-tolerance test. Fasting and fed glucose were significantly lower in wild-type mice treated with sacubitril, although active GLP-1 levels and insulin secretion during oral glucose challenge were unchanged. In contrast, insulin secretion in response to intravenous glucose was significantly enhanced in sacubitril-treated wild-type mice, and this effect was blunted in Glp1r−/− mice. Similarly, sacubitril enhanced insulin secretion in vitro in islets from STZ-treated Glp1r+/+ but not Glp1r−/− mice. Together, our data suggest the insulinotropic effects of pharmacological neprilysin inhibition in a mouse model of β-cell dysfunction are mediated via intra-islet GLP-1R signaling.
NEW & NOTEWORTHY The neprilysin inhibitor, sacubitril, improves glycemic status in a mouse model of reduced insulin secretion. Sacubitril enhances intravenous but not oral glucose-mediated insulin secretion. The increased glucose-mediated insulin secretion is GLP-1 receptor-dependent. Neprilysin inhibition does not raise postprandial circulating active GLP-1 levels.
INTRODUCTION
Neprilysin is a widely expressed peptidase upregulated in humans and rodents under conditions associated with impaired glucose tolerance and type 2 diabetes (1–3). Studies have shown that a combination drug approved for heart failure that comprises the neprilysin inhibitor sacubitril plus the angiotensin II receptor blocker valsartan improves insulin sensitivity (4) and glycemic control (5, 6) in obese individuals with or without type 2 diabetes. Although there are no reports of the metabolic effects of neprilysin inhibitors alone in humans, sacubitril/valsartan treatment was associated with greater reduction of HbA1c levels when compared with another blocker of the renin-angiotensin system (5). The latter suggests the benefits conferred by sacubitril/valsartan were due to neprilysin inhibition. In line with this, we previously reported that genetic ablation of neprilysin in high-fat-fed mice improves glycemic status and enhances insulin secretion and sensitivity (3). However, the mechanisms by which neprilysin inhibition improves glucose homeostasis are not fully understood.
Glucagon-like peptide-1 (GLP-1) is a gastrointestinal hormone that regulates glucose homeostasis in part via enhancing meal-stimulated insulin secretion, a process called the incretin effect (7, 8). Once secreted from the intestine, active GLP-1 [GLP-1(7–36)amide] is rapidly degraded by dipeptidyl peptidase-4 (DPP-4) via N-terminal cleavage between residues 8 and 9, generating GLP-1(9–36)amide (9, 10). Neprilysin can also cleave active GLP-1 at multiple sites, mainly in the C-terminal region (11–13). C-terminal amino acid residues of active GLP-1 are crucial for GLP-1 receptor (GLP-1R) binding, whereas N-terminal residues are essential for GLP-1R activation (14). Thus, neprilysin and DPP-4 can prevent active GLP-1 from GLP-1R binding and activation, respectively (15).
In high-fat-fed neprilysin-deficient mice, the improvement in glycemic status and β-cell function was associated with increased circulating levels of active GLP-1 (3). Elevated plasma GLP-1 has also been reported in humans treated with sacubitril/valsartan (6, 16, 17). However, in two of these clinical studies, measurements were made of total GLP-1 in the fasting state, rather than active GLP-1 in the postprandial state, where it exerts the greatest effects on insulin release and glucose tolerance (6, 16). In the third clinical study, an acute administration of sacubitril/valsartan in healthy young men increased circulating postprandial total GLP-1 (17). Furthermore, when sacubitril/valsartan was combined with a DPP-4 inhibitor, active GLP-1 levels were enhanced beyond that seen with DPP-4 inhibitor alone (17), suggesting neprilysin inhibition enhances levels of biologically active GLP-1. However, the effect of a more prolonged treatment or in conditions of type 2 diabetes was not evaluated. Finally, it could be possible that valsartan, rather than sacubitril, contributed to the increased levels of GLP-1. Indeed, in preclinical studies, the administration of an angiotensin receptor II blocker has been associated with increased pancreatic expression of GLP-1 immunoreactivity (18) and plasma GLP-1 levels (19).
Therefore, in this study, we sought to determine whether pharmacological inhibition of neprilysin on its own increases postprandial biologically active GLP-1 levels and improves β-cell function in a high-fat-fed mouse model of reduced insulin secretion. β-Cell function was assessed via both oral and intravenous glucose tolerance tests to stimulate (oral) or not [intravenous (iv)] the incretin effect. GLP-1R-deficient mice were also studied and in vitro islets studies were performed to determine whether any effects observed were dependent on GLP-1R signaling.
RESEARCH DESIGN AND METHODS
Animals and Diets/Drugs
Glp1r+/− mice were provided by Daniel Drucker (University of Toronto, Canada) to generate Glp1r+/+ and Glp1r−/− mice on a C57BL/6J background at VA Puget Sound Health Care System. Five-week-old male Glp1r+/+ and Glp1r−/− mice were fed a high-fat diet (HFD, 60% kcal fat, D12492; Research Diets Inc; New Brunswick, NJ) for 3 wk. Mice were then randomly assigned to receive intraperitoneal injections of either vehicle (VEH, citrate buffered saline, pH 4.5) or low-dose streptozotocin (STZ, 30 mg/kg; Sigma-Aldrich, St Louis, MO) for 3 consecutive days once daily, the latter to recapitulate β-cell dysfunction seen in human type 2 diabetes, as previously described (20, 21). Thereafter, mice were assigned via a block randomization method to receive 8 wk of HFD alone (CTL) or supplemented with sacubitril (SAC, 480 mg/kg diet; S080900, Toronto Research Chemicals Inc, Canada), with the goal of achieving a dose of 48 mg/kg body wt/day (based on a pilot study showing this dose was sufficient to significantly inhibit plasma neprilysin activity by at least 50% under conditions of HFD). Diet formulation was based on our prior experience and a pilot study in which each mouse consumed ∼0.1 g of food/g body wt/day.
Mice were housed two per cage, with a 12-h light/dark cycle, and had ad libitum access to diets and water for the duration of the study. Mice were weighed weekly and blood collected via lateral saphenous vein for measurement of nonfasting plasma glucose levels via glucose oxidase assay. Food and water intake were estimated weekly by weighing the remaining food and measuring water on a per-cage basis. The daily drug dose intake was calculated based on the food intake. At the end of the 8-wk treatment period, mice were subjected to an intraperitoneal insulin tolerance test (ITT) and either an oral glucose tolerance test (OGTT) or an intravenous glucose tolerance test (IVGTT) as described in Insulin and Glucose Tolerance Tests. In performing the in vivo tests and analytical assays, staff were blinded to treatment groups the mice were assigned to. Of a total of 159 mice enrolled in the study, 24 mice experienced complications during the surgery (carotid catheterization) for the IVGTT or OGTT procedures and were excluded from all analyses in the study.
For in vitro islet studies, another cohort of male Glp1r+/+ (n = 16) and Glp1r−/− (n = 13) mice initially on HFD for 3 wk then treated with STZ or VEH were subsequently continued on HFD alone for 4 wk. Blood was collected via lateral saphenous vein before the STZ and VEH injection and then after 3 wk on HFD for measurement of nonfasting plasma glucose levels via glucose oxidase assay. At the end of the 4-wk period on HFD alone, mice were euthanized and islets isolated for in vitro assessment of glucose-stimulated insulin secretion (GSIS) and islet insulin content, as described in In Vitro Assessment of GSIS.
Specific “n” for each end point is given in each figure or figure legend. Sample sizes were determined based on our own experience with the primary outcome being insulin secretion. All animal procedures were approved by the Institutional Animal Care and Use Committee of the VA Puget Sound Health Care System and adhered to the Animal Research: Reporting of In vivo Experiments (ARRIVE) guidelines.
Insulin and Glucose Tolerance Tests
In the initial cohort of Glp1r+/+ mice (n = 48), ITTs (0.75 IU insulin/kg) were performed following the 8-wk treatment period in conscious mice fasted for 3.5 h. Tail vein blood was collected at baseline and at 15, 30, 45, and 60 min after insulin administration for glucose measurement. Two to five days later, OGTTs (2 g glucose/kg) were performed, as previously described (3), in pentobarbital (80 mg/kg) anesthetized mice fasted for 6 h, wherein blood was collected via carotid catheter for measurement of glucose. Incremental areas under the curve (iAUC) for glucose and insulin from 0 to 30 min and from 0 to 120 min were calculated.
To determine whether the use of anesthesia affected the action of sacubitril on OGTT measures, OGTTs were performed in a subgroup of conscious Glp1r+/+ mice (n = 33) fasted for 6 h. Tail vein blood was collected at baseline and at 10, 20, 30, 60, 90, and 120 min after glucose bolus for measuring glucose, and then iAUC for glucose from 0 to 30 min and 0 to 120 min were calculated. Retro-orbital blood was collected at baseline and at 30 min after glucose bolus for insulin measurement. GSIS was calculated as the ratio of the change in insulin to the change in glucose from 0 to 30 min.
In another cohort of both Glp1r+/+ (n = 32) and Glp1r−/− mice (n = 22), IVGTTs (1 g glucose/kg) were performed, as previously described (20), in pentobarbital (80 mg/kg) anesthetized mice fasted for 16 h, wherein blood was collected via carotid catheter for the measurement of glucose and insulin as above. Glucose elimination rate (Kg) was computed as the negative slope of the natural log of glucose versus time from 10 to 45 min. First- and second-phase insulin secretion were calculated as the ratio of iAUC for insulin over glucose for 0–5 min and 5–45 min, respectively.
Glucose, Insulin, Active GLP-1, and Glucagon Measurements
For weekly bleeds and IVGTTs, plasma glucose levels were determined using the glucose oxidase method. For ITTs and OGTTs, blood glucose levels were measured using an Accu-Chek Aviva Plus glucometer (Roche, Basel, Switzerland). Insulin levels in plasma and pancreas extracts were determined using a Mouse Ultrasensitive Insulin ELISA (Alpco, Salem, NH). For glucagon measurement, retro-orbital blood was collected at the end of the study period from overnight fasted conscious mice. To avoid GLP-1 and glucagon degradation, aprotinin and diprotin A were added to the blood as previously described (3), and plasma samples were stored at −80°C before assay. Plasma concentrations of active GLP-1 and glucagon were measured using the active GLP-1 assay (Meso Scale Discovery, Rockville, MD) and glucagon ELISA kit (Mercodia, Upsala, Sweden), respectively.
Plasma Neprilysin and DPP-4 Activity Assays
Neprilysin and DPP-4 activities were determined as previously described (3) in plasma samples taken from conscious fed mice via lateral saphenous vein bleed at week 0 (after STZ or VEH injections) and at the end of the 8-wk treatment period.
Tissue Collections and Histological Assessments
Following glucose tolerance tests, mice were euthanized, and epididymal fat pad mass and inguinal fat pad mass were recorded. A small portion of the pancreas (tail) was weighed and homogenized in 0.18 M HCl/95% ethanol for evaluation of insulin content (expressed per total protein content). The same pancreatic region from each mouse was extracted to avoid variability that may arise due to random sampling. The remaining pancreas was excised, weighed, and fixed in 10% neutral-buffered formalin overnight, paraffin-embedded, and sectioned at 4 µm thickness. Sections representing three different areas of the pancreas per mouse were deparaffinized and stained with anti-insulin (1:2,000; clone K36AC10, Sigma-Aldrich, St Louis, MO) and anti-glucagon (1:2,000; EP3070, Abcam, Cambridge, MA) antibodies followed by Alexa Fluor 488-conjugated anti-mouse and Alexa Fluor 546-conjugated anti-rabbit to visualize islet β cells and α cells, respectively. The sections were then counterstained with Hoechst 33258 (2 μg/mL) to visualize cell nuclei. Morphometric analyses were performed using NIS-Elements HC/JOBS software on a Nikon Ni-E microscope system (Nikon USA, Melville, NY), as previously described (22). The data collector was blinded to the identity of each specimen. Histological assessments were made for all islets visible on each section (Glp1r+/+: 20 ± 9 and Glp1r−/−: 23 ± 2 islets, respectively). Section area, islet area, insulin, and glucagon areas were quantified, and the following measures were calculated: β-cell and α-cell area expressed relative to islet area (Σ insulin area/Σ islet area × 100), and β-cell and α-cell mass (β-cell or α-cell area/pancreas area × total pancreas weight).
In Vitro Assessment of GSIS
Pancreatic islets were isolated from a cohort of VEH- or STZ-treated Glp1r+/+ and Glp1r−/− mice fed an HFD alone for 4 wk. Islets were isolated by collagenase digestion and purified by gradient separation, as previously described (23). After an overnight recovery in RPMI 1640 media, islets were preincubated for 90 min in Krebs buffer containing 2.8 mmol/L glucose, Then, basal or glucose-stimulated insulin release were determined in triplicate batches of islets incubated in Krebs buffer containing 2.8 or 20 mmol/L glucose, respectively. To match the treatment groups from the in vivo study, islets from VEH-treated mice were coincubated with vehicle, and islets from STZ-treated mice were coincubated with vehicle or 1 μmol/L sacubitrilat (the active metabolite of sacubitril; Sigma-Aldrich, St. Louis, MO). After 60 min, supernatant was collected and islets extracted for insulin content using acid-ethanol. Insulin in supernatant and islet extracts was assayed using the Mouse Ultrasensitive Insulin ELISA (Alpco, Salem, NH).
Statistical Analyses
Data are presented as means ± SE. Bar graphs also show individual values. Time-course data were analyzed via repeated-measures two-way ANOVA/mixed-effect model and mean data were compared among groups by one-way ANOVA or Brown–Forsythe ANOVA when variances were not equal between groups. Dunnet or Sidak correction for multiple comparisons was used for post hoc analyses. A Student’s t test was used when two groups were compared. A P value ≤0.05 was considered statistically significant. Statistical calculations and graphs were made using GraphPad Prism (v. 9.01 for Mac; GraphPad Software, La Jolla, CA).
RESULTS
Sacubitril Decreases Plasma Neprilysin Activity but Does Not Alter DPP-4 Activity
In an initial cohort of Glp1r+/+ mice, plasma neprilysin (Fig. 1A) and DPP-4 (Fig. 1B) activities at baseline were comparable among all groups of mice. After 8 wk of treatment with sacubitril, STZ-injected mice (STZ-SAC) displayed a reduction of plasma neprilysin activity by 54.6% compared with baseline and by 70.5% compared with STZ mice receiving control diet (STZ-CTL; Fig. 1A). Plasma DPP-4 activity remained unchanged (Fig. 1B). Food and water intake were overall similar between groups (Supplemental Fig. S1; https://doi.org/10.6084/m9.figshare.14815689). Over the 8-wk period, the sacubitril-treated mice consumed 82.3 ± 2.2% of the drug target dose.
Figure 1.
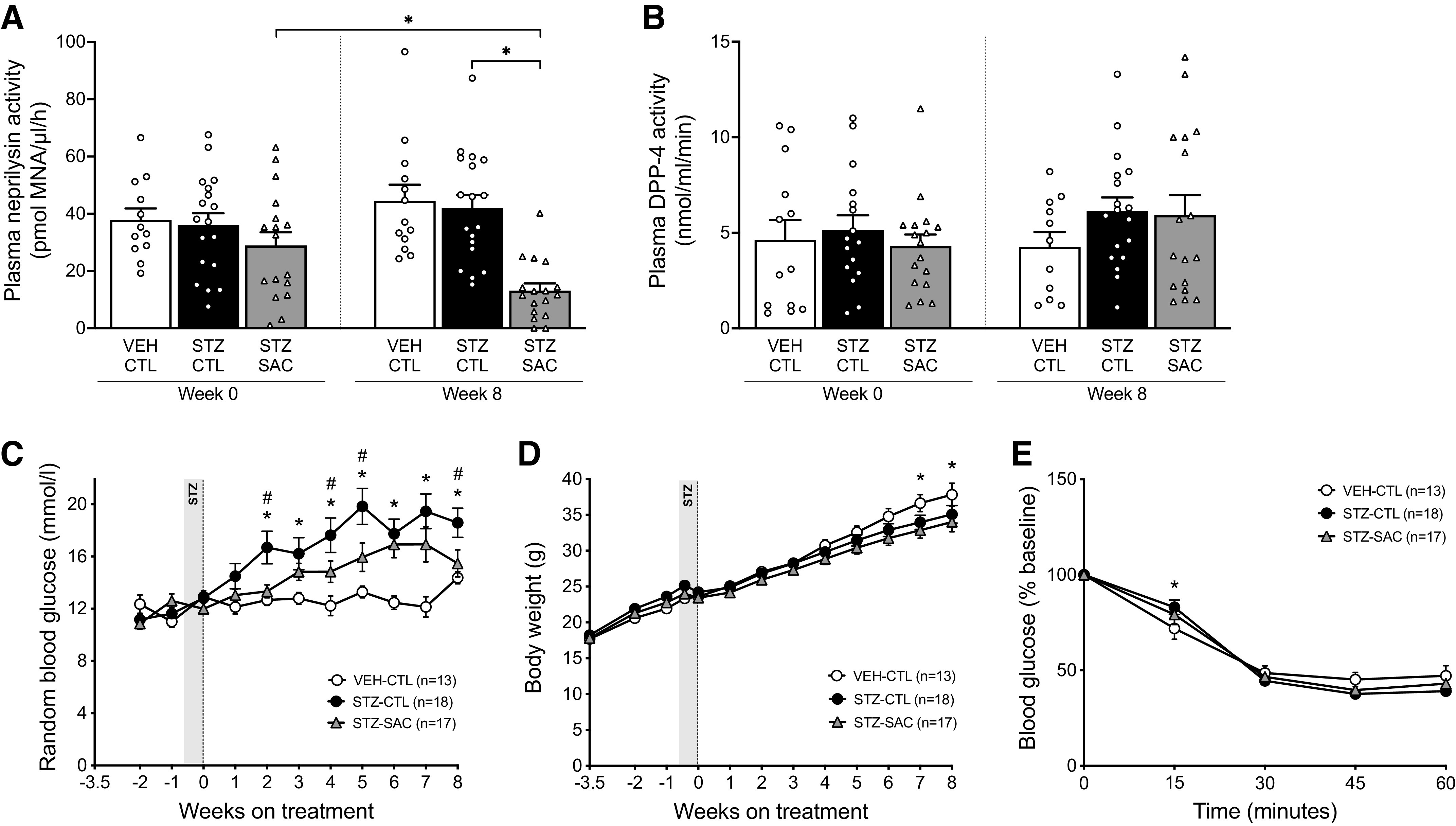
Pharmacological inhibition of neprilysin with sacubitril reduces streptozotocin (STZ)-induced hyperglycemia without altering body weight and insulin sensitivity. Plasma neprilysin (A) and DPP-4 (B) activities at baseline (week 0) and at the end of the 8-week study period in vehicle (VEH)- or streptozotocin (STZ)-injected male Glp1r+/+ mice fed a high-fat diet (HFD) alone or supplemented with sacubitril (SAC). n = 13–18/group, *P ≤ 0.05 (analyzed using two-way ANOVA followed by Dunnet or Sidak’s multiple comparisons test). Random fed blood glucose levels (C) and body weight (D) over time in VEH-CTL (open circles, n = 13), STZ-CTL (black circles, n = 18) and STZ-SAC (gray triangles, n = 17) groups of male Glp1r+/+ mice. *P ≤ 0.05 STZ-CTL vs. VEH-CTL; #P ≤ 0.05 STZ-SAC vs. STZ-CTL (analyzed using repeated-measures two-way ANOVA/mixed effects followed by Dunnet’s multiple comparisons test). E: blood glucose levels as percentage of baseline during an IPITT at the end of the 8-wk treatment period in VEH-CTL (open circles, n = 13), STZ-CTL (black circles, n = 18), and STZ-SAC (gray triangles, n = 17) male Glp1r+/+ mice. *P ≤ 0.05 STZ-CTL vs. VEH-CTL (analyzed using repeated-measures two-way ANOVA followed by Dunnet’s multiple comparisons test). Data are means ± SE.
Neprilysin Inhibition is Associated with Decreased Fed and Fasting Glucose without Change in Body Weight and Insulin Sensitivity
At baseline, fed glucose levels were comparable in all groups of Glp1r+/+ mice (Fig. 1C). Throughout the 8-wk study period, STZ-CTL mice displayed increased fed glucose compared with mice that received VEH injections and control diet (VEH-CTL) (Fig. 1C). Similarly, fasting glucose levels were increased in STZ-CTL mice at the end of the study period (Table 1). In STZ mice, sacubitril lowered fed glucose over time (Fig. 1C) and fasting glucose levels at 8 wk post-treatment (Table 1).
Table 1.
Fasting plasma glucose and insulin, body weight gain, fat pad mass, and pancreas weight in Glp1r+/+ mice injected with vehicle (VEH) or streptozotocin (STZ) and fed HFD alone (control, CTL) or HFD supplemented with sacubitril (SAC) for 8 wk
| VEH-CTL | STZ-CTL | STZ-SAC | ANOVA P Value | |
|---|---|---|---|---|
| Fasting plasma glucose, mg/dL | 258.7 ± 8.43 | 345.6 ± 22.01* | 298.8 ± 22.1# | 0.0007 |
| Fasting plasma insulin, pmol/L | 509.4 ± 71.13 | 349.1 ± 51.06 | 305.8 ± 35.40 | 0.0291 |
| Body weight gain, g | 14.45 ± 1.25 | 10.92 ± 0.85* | 10.46 ± 0.91 | 0.0181 |
| Inguinal fat pad mass, g | 1.91 ± 0.19 | 1.59 ± 0.17 | 1.40 ± 1.17 | 0.1581 |
| Epididymal fat pad mass, g | 0.236 ± 0.027 | 0.207 ± 0.020 | 0.175 ± 0.018 | 0.1598 |
| Pancreas weight, g | 0.298 ± 0.015 | 0.303 ± 0.015 | 0.319 ± 0.024 | 0.7173 |
Data are expressed as means ± SE. One-way ANOVA was performed to determine whether the means were significantly different across the three groups of mice and the overall P value is displayed in the far right column. If ANOVA P < 0.05, Dunnet’s multiple comparisons were performed and are displayed as followed: *P < 0.05 vs. STZ-CTL vs. VEH-CTL; #STZ-SAC vs. STZ-CTL. CTL, control; HFD: high-fat diet; SAC, sacubitril; STZ, streptozotocin; VEH, vehicle.
Although body weight gain was reduced at the end of the study period in STZ versus VEH mice receiving control diet (Fig. 1D and Table 1), treatment of STZ mice with sacubitril did not further impact body weight (Fig. 1D and Table 1). Fat pad mass was not different among groups (Table 1).
Sacubitril did not affect insulin sensitivity, as assessed by ITT at the end of the 8-wk treatment period (Fig. 1E).
Neprilysin Inhibition Does Not Improve Oral GSIS and Does Not Increase Circulating Active GLP-1 Levels
At the end of the 8-wk treatment period, OGTTs were performed in anesthetized Glp1r+/+ mice. Although fasting glucose was reduced with sacubitril (Fig. 2A, Table 1), glucose tolerance during the OGTT was similar between groups of mice, as quantified by iAUC for glucose (Fig. 2B). Insulin release in response to oral glucose was significantly reduced in STZ-CTL mice and was similarly reduced in STZ-SAC mice (Fig. 2C, D), even after accounting for the prevailing glucose concentration (Fig. 2E). Similar results were obtained when OGTTs were performed in conscious mice (Supplemental Fig. S2; https://doi.org/10.6084/m9.figshare.14815704). Circulating active GLP-1 levels measured during the OGTT (10 min after oral glucose administration) did not increase with neprilysin inhibition (Fig. 2F). Since glucagon is also a substrate of neprilysin (11, 24, 25), fasting plasma glucagon levels were also measured at the end of the study period and were not significantly different among the three groups of Glp1r+/+ mice (8.48 ± 1.92 vs. 4.85 ± 0.51 vs. 6.07 ± 0.99 pmol/L in VEH-CTL, STZ-CTL, and STZ-SAC respectively, ANOVA P value = 0.11).
Figure 2.

Sacubitril does not increase oral GSIS or circulating active GLP-1 levels in anesthetized mice. Plasma glucose (A) and insulin (C) levels during an OGTT at the end of the 8-wk treatment period in VEH-CTL (open circles, n = 13), STZ-CTL (black circles, n = 18), and STZ-SAC (gray triangles, n = 17) groups of anesthetized male Glp1r+/+ mice. Calculated incremental areas under the curve (iAUC) for glucose (B), insulin (D), and insulin/glucose (E) from 0–30’ and 0–120’ during the OGTT. F: active GLP-1 levels measured during the OGTT, 10 min after glucose administration. *P ≤ 0.05 (analyzed using one-way ANOVA followed by Dunnet’s multiple comparisons test). Data are means ± SE. CTL, control; GLP-1, glucagon-like peptide-1; HFD: high-fat diet; OGTT, oral glucose tolerance test; SAC, sacubitril; STZ, streptozotocin; VEH, vehicle.
Neprilysin Inhibition Enhances Intravenous GSIS in a GLP-1R-Dependent Manner
To evaluate the effect of neprilysin inhibition on β-cell function independent of an incretin effect, IVGTTs were performed at the end of the 8-wk treatment period in another cohort of Glp1r+/+ mice. In this cohort, the sacubitril intake reached 73.4% of the target dose with plasma neprilysin activity reduced by 86.6% in STZ-injected mice that received sacubitril compared with STZ-CTL mice (Fig. 3A). Plasma glucose levels during the IVGTT are shown in Fig. 3B. The rate of glucose disappearance (Kg) was reduced in STZ-CTL versus VEH-CTL mice, whereas neprilysin inhibition in STZ-SAC mice resulted in increased Kg compared with STZ-CTL mice (inset, Fig. 3B). Consistent with our previous data (20) and OGTTs in the present study, STZ-CTL mice displayed a significant reduction in first- and second-phase insulin secretion during the IVGTT when compared with VEH-CTL mice (Fig. 3, C and D). Importantly, STZ-SAC mice had significantly increased insulin release when compared with STZ-CTL mice (Fig. 3, C and D). To determine whether this improvement in β-cell function was dependent on GLP-1R signaling, IVGTTs were also performed in Glp1r−/− mice. In STZ-injected Glp1r−/− mice treated with sacubitril for 8 wk, the drug intake reached 75.6% of the target dose and plasma neprilysin activity was reduced by 82.8% compared with STZ-CTL Glp1r−/− mice (Fig. 3E). During the IVGTT, plasma glucose levels and Kg were similar among groups (Fig. 3F). Insulin secretion was significantly reduced in STZ-CTL Glp1r−/− mice (Fig. 3, G and H). In contrast to what was observed in Glp1r+/+ mice, sacubitril did not enhance GSIS in STZ-injected Glp1r−/− mice (Fig. 3, G and H).
Figure 3.
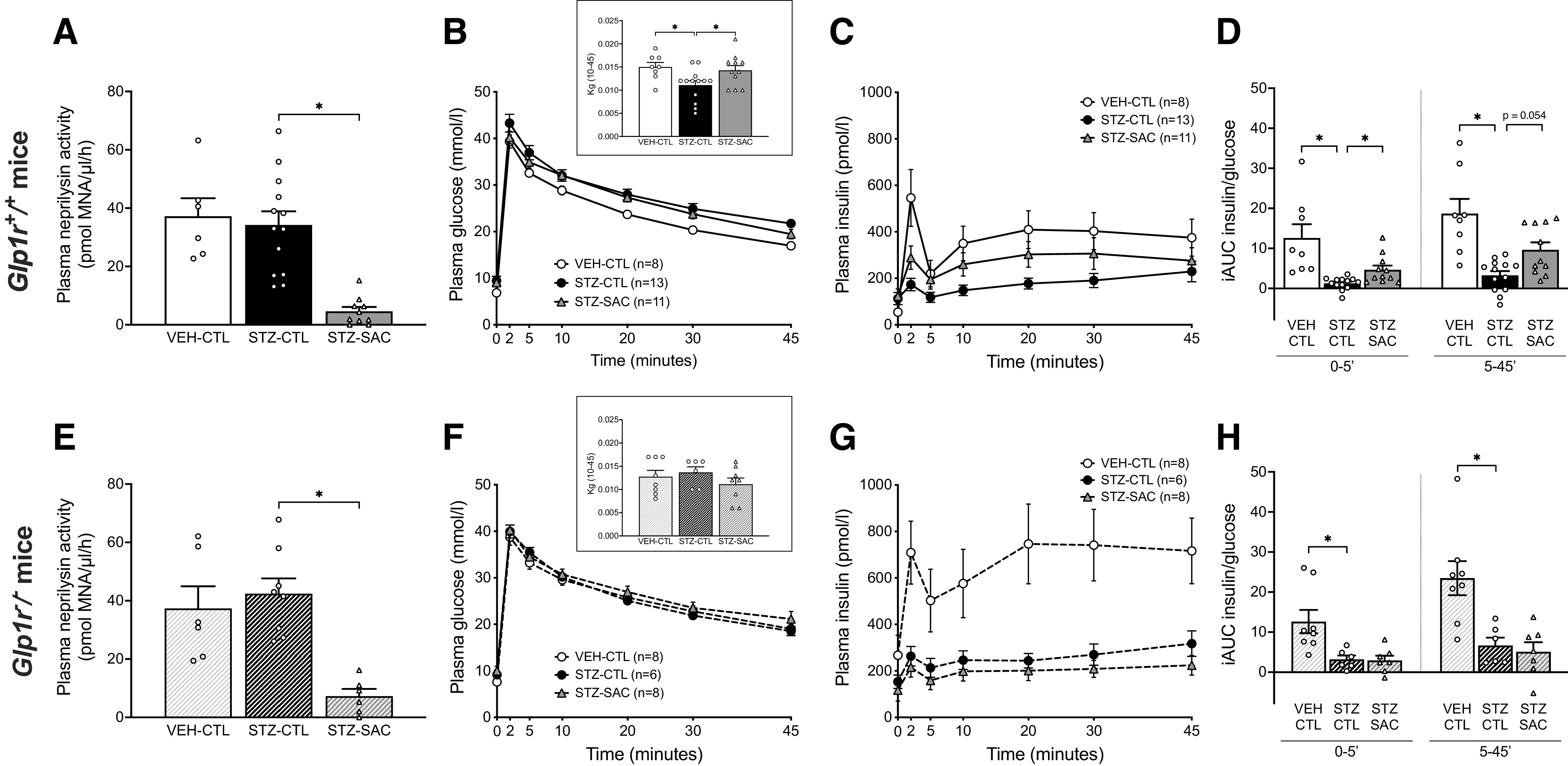
Sacubitril increases intravenous GSIS in a GLP-1R-dependent manner. Plasma neprilysin activity at the end of the 8-wk treatment period in VEH-CTL, STZ-CTL, and STZ-SAC groups of male Glp1r+/+ (A) and Glp1r−/− mice (E). Plasma glucose (B and F) and insulin (C and G) levels during an IVGTT in VEH-CTL (open circles), STZ-CTL (black circles), and STZ-SAC (gray triangles) Glp1r+/+ (B and C) and Glp1r−/− (F and G) mice at the end of the 8-wk study period. The insets in B and F represent the glucose disposal (Kg) computed from 10 to 45 min during the IVGTT. First-phase (0–5’) and second-phase (5–45’) GSIS during the IVGTT in VEH-CTL, STZ-CTL, and STZ-SAC groups of male Glp1r+/+ (D) and Glp1r−/− (H) mice. n = 6–13/group, *P ≤ 0.05 (analyzed using one-way or Brown–Forsythe ANOVA followed by Dunnet’s multiple comparisons test). Data are means ± SE. CTL, control; GLP-1, glucagon-like peptide-1; GLP-1R, glucagon-like peptide-1 receptor; GSIS, glucose-stimulated insulin secretion; IVGTT, intravenous glucose tolerance test; SAC, sacubitril; STZ, streptozotocin; VEH, vehicle.
Neprilysin Inhibition Increases Pancreatic Insulin Content in a GLP-1R Dependent-Manner but Is Not Associated with Changes in β- and α-Cell Mass
At the end of the study period, pancreatic insulin content in Glp1r+/+ mice was decreased by 75% in the STZ-CTL versus VEH-CTL group. Neprilysin inhibition in STZ-SAC mice resulted in pancreatic insulin content that was increased by 50% when compared with the STZ-CTL group (Fig. 4A). In Glp1r−/− mice, pancreatic insulin content was similarly decreased in the STZ-CTL versus VEH-CTL group; however, no increase in pancreatic insulin content was observed in STZ-SAC versus STZ-CTL mice (Fig. 4B). β-Cell mass in both Glp1r+/+ and Glp1r−/− mice was decreased in STZ-CTL versus VEH-CTL groups, but did not change in the STZ-SAC group (Fig. 5, A and C). α-Cell mass remained comparable among all groups in both genotypes (Fig. 5, B and D). Representative images of insulin and glucagon staining are shown in Fig. 5E.
Figure 4.
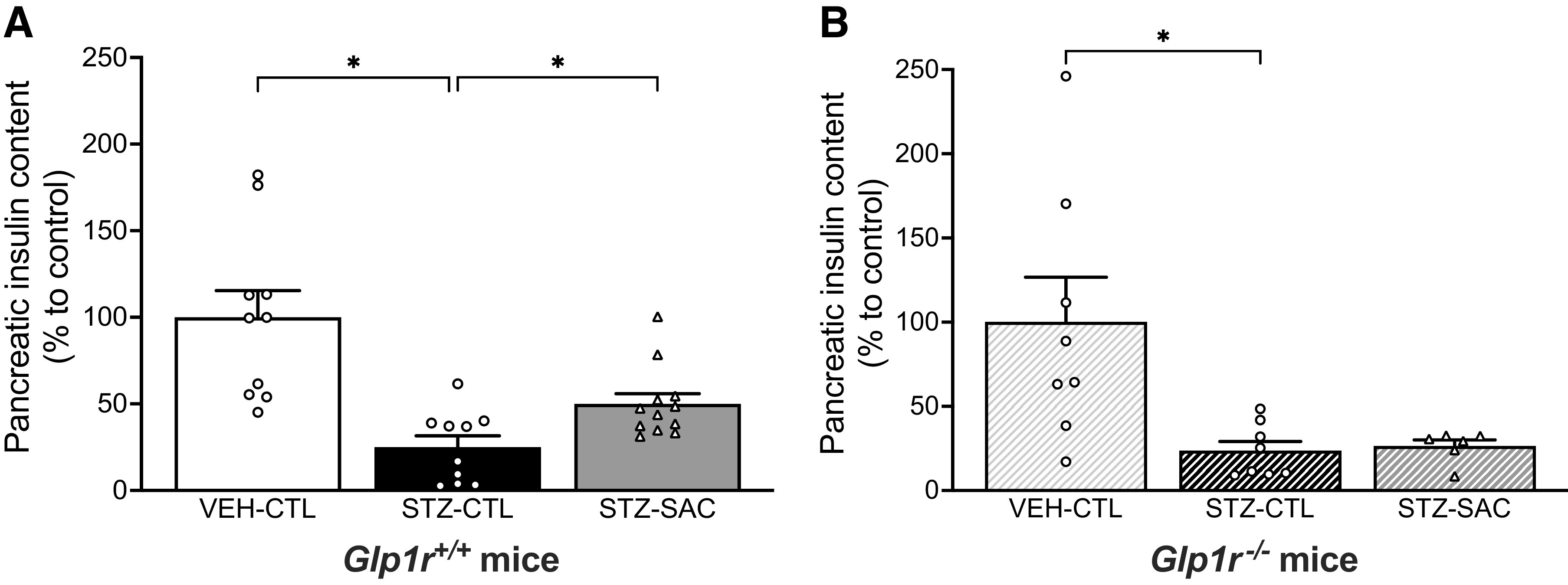
Sacubitril increases insulin content in a GLP-1R-dependent manner. Insulin content at the end of the 8-wk treatment period in VEH-CTL (n = 10), STZ-CTL (n = 10), and STZ-SAC (n = 12) groups of male Glp1r+/+ mice (A) and VEH-CTL (n = 8), STZ-CTL (n = 8), and STZ-SAC (n = 6) groups of male Glp1r−/− mice (B). *P ≤ 0.05 (analyzed using one-way or Brown–Forsythe ANOVA followed by Dunnet’s multiple comparisons test). Data are means ± SE. GLP-1R, glucagon-like peptide-1 receptor; SAC, sacubitril; STZ, streptozotocin; VEH, vehicle.
Figure 5.
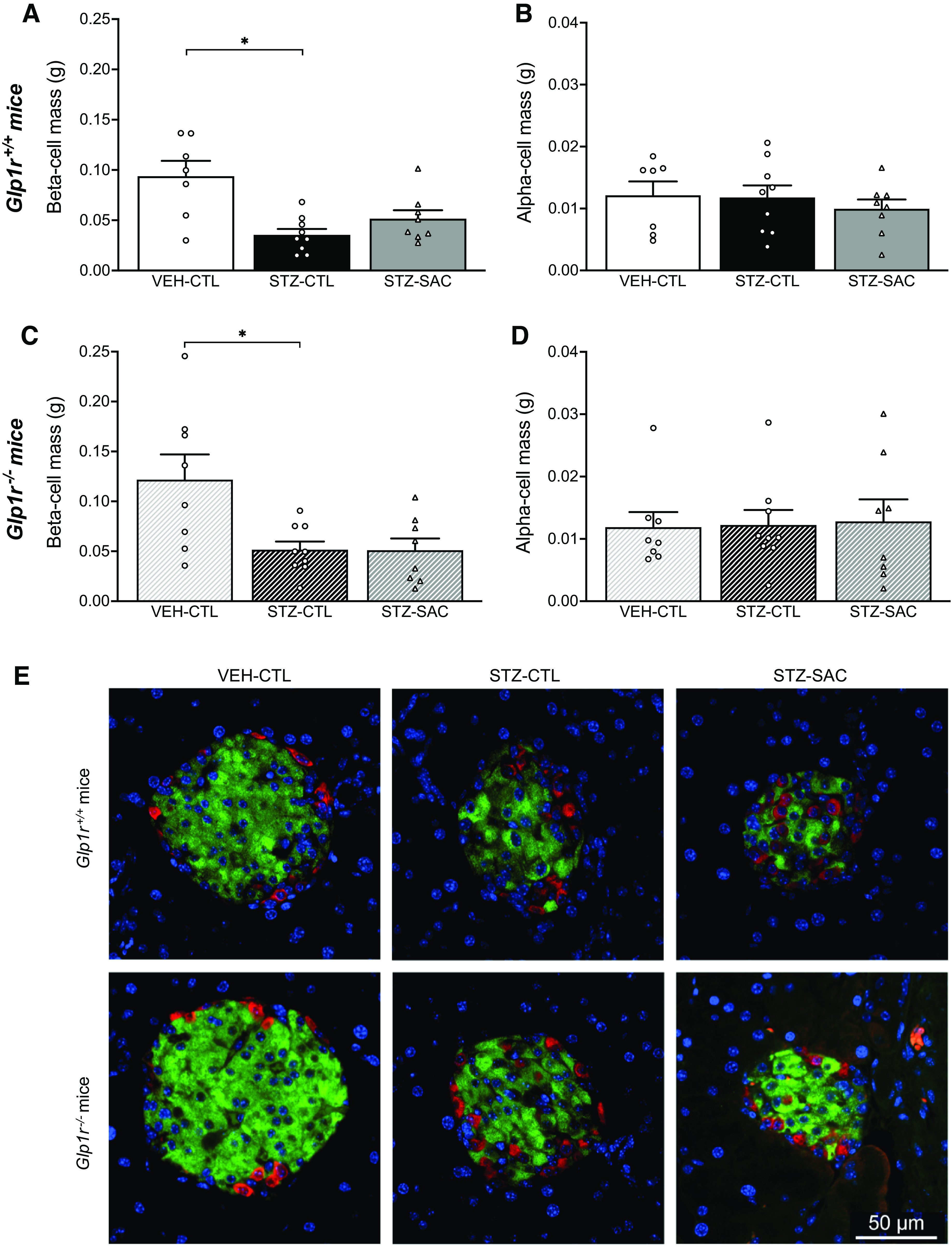
Sacubitril does not alter β-cell and α-cell mass in Glp1r+/+ or Glp1r−/− mice. β-Cell mass (A and C) and α-cell mass (B and D) at the end of the 8-wk treatment period in VEH-CTL (n = 6), STZ-CTL (n = 9), and STZ-SAC (n = 8) groups of male Glp1r+/+ mice (A and B) and VEH-CTL (n = 8), STZ-CTL (n = 9), and STZ-SAC (n = 8) groups of male Glp1r−/− mice (C and D). E: representative images showing β cell (insulin; green), α cell (glucagon; red), and nuclei (blue) staining in pancreata from VEH-CTL (left), STZ-CTL (middle), and STZ-SAC (right) groups of male Glp1r+/+ (top) and male Glp1r−/− mice (bottom); scale bar = 50 μm. *P ≤ 0.05 (analyzed using one-way or Brown–Forsythe ANOVA followed by Dunnet’s multiple comparisons test). Data are means ± SE. GLP-1R, glucagon-like peptide-1 receptor; SAC, sacubitril; STZ, streptozotocin; VEH, vehicle.
Neprilysin Inhibition Enhances GSIS via Intra-Islet GLP-1R Signaling
Since our IVGTT insulin data in STZ-SAC mice suggest a role for GLP-1R signaling that is independent of the incretin effect, we sought to determine the direct effect of neprilysin inhibition on intra-islet GLP-1R signaling and GSIS. Islets were isolated from a cohort of STZ- or VEH-treated Glp1r+/+ and Glp1r−/− mice fed an HFD alone for 4 wk. GSIS was then assessed in vitro in the absence or presence of sacubitrilat, the active metabolite of sacubitril. Before STZ or VEH injections, all mice had comparable nonfasting plasma glucose levels [Glp1r+/+ mice before STZ (n = 11) or VEH (n = 5): 12.0 ± 0.3 vs. 12.8 ± 0.6 mmol/L, P = 0.14; Glp1r−/− mice before STZ (n = 8) or VEH (n = 5): 14.8 ± 1.4 vs. 13.8 ± 0.8 mmol/L, P = 0.60). After 3 wk on HFD, both Glp1r+/+ and Glp1r−/− mice that received STZ injection had increased plasma glucose levels when compared with mice injected with VEH (Fig. 6, A and B). As expected, insulin content and GSIS were significantly reduced in islets from mice treated with STZ compared with VEH in both genotypes (Fig. 6, C–F). Acute treatment with sacubitrilat significantly enhanced GSIS in islets from STZ-treated Glp1r+/+ mice (1.6-fold; Fig. 6E), but not in islets from STZ-treated Glp1r−/− mice (0.89-fold; Fig. 6F).
Figure 6.
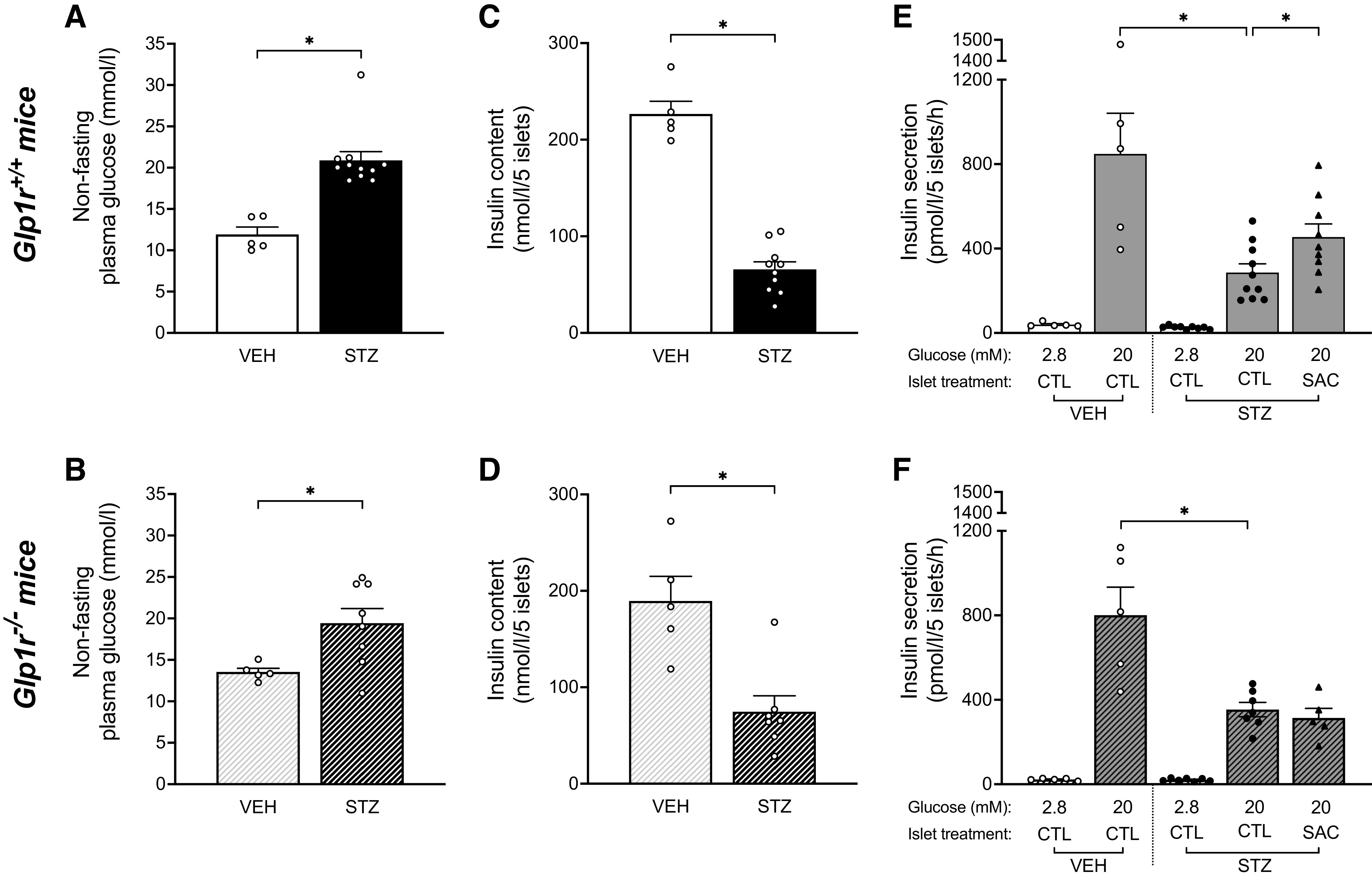
Sacubitrilat increases GSIS via GLP-1R pathway in islets isolated from STZ-treated high fat-fed mice. Nonfasting plasma glucose after 3 wk on HFD alone in vehicle (VEH)- or streptozotocin (STZ)-treated male Glp1r+/+ (n = 16; A) and Glp1r−/− (n = 13; B) mice. *P ≤ 0.05 (analyzed using Student’s t test). At the end of the 4-wk period on HFD, mice were euthanized and islets isolated for assessment of insulin content and GSIS. Insulin content in islets isolated from VEH- or STZ-treated Glp1r+/+ (C) and Glp1r−/− (D) mice. *P ≤ 0.05 (analyzed using Student’s t test). Insulin secretion in response to 2.8 or 20 mmol/glucose with vehicle (CTL) or 1 μmol/L sacubitrilat (SAC) in islets isolated from VEH- or STZ-treated Glp1r+/+ (E) and Glp1r−/− (F) mice. n = 5–10/group, *P ≤ 0.05 (analyzed using ANOVA followed by Sidak’s multiple comparison). Data are means ± SE. CTL, control; GLP-1R, glucagon-like peptide-1 receptor; GSIS, glucose-stimulated insulin secretion; HFD: high-fat diet; STZ, streptozotocin; VEH, vehicle.
DISCUSSION
While previous preclinical studies have shown that genetic neprilysin deficiency (3) or acute pharmacological reduction in neprilysin activity (26–29) are associated with improved glucose tolerance and insulin secretion, our study is the first to assess β-cell function and active GLP-1 levels following a more prolonged pharmacological inhibition of neprilysin, as would occur in humans treated with the sacubitril/valsartan combination drug. Importantly, we show that in a mouse model of reduced insulin secretion, sacubitril alone reduced STZ-induced hyperglycemia and enhanced intravenous but not oral GSIS in a GLP-1R-dependent manner. Furthermore, acute treatment of islets in vitro with sacubitrilat partially rescued STZ-induced deficits in GSIS in islets from Glp1r+/+ mice but not from Glp1r−/− mice. These findings suggest that in obese or type 2 diabetic humans treated with sacubitril/valsartan (5, 6), the observed improvements in glycemic control may be mediated, at least in part, by neprilysin inhibition and islet GLP-1R signaling.
Neprilysin is known to cleave and inactivate up to 50% of GLP-1 entering the circulation (11, 13, 29). Previous studies have suggested a glucose-lowering effect of neprilysin deficiency/inhibition via increased circulating levels of GLP-1 (3, 6, 16), although none have reported increases in the active form of GLP-1 in the postprandial state after a more prolonged pharmacological neprilysin inhibition. Importantly, our study showed that following oral glucose administration, neprilysin inhibition did not improve insulin secretion and did not increase plasma active GLP-1 levels. This is in contrast with our previous data from high-fat-fed neprilysin-deficient mice where elevated plasma active GLP-1 levels were associated with enhanced β-cell function during an OGTT (3). However, in the latter, neprilysin deficiency was also associated with a decrease in plasma DPP-4 activity, which may have contributed to preservation of gut-derived active GLP-1 and thus, an improved insulin response to oral glucose. In the present study, sacubitril did not decrease plasma DPP-4 activity, and thereby active GLP-1 would still be rapidly degraded by DPP-4. This is in accordance with a recent clinical study in which acute administration of sacubitril/valsartan alone did not increase plasma concentrations of active GLP-1, whereas it did when combined with a DPP-4 inhibitor (17). The lack of increase in plasma active GLP-1 levels during the OGTT might therefore explain why we did not observe an enhancement of GSIS during the test.
A major mechanism of action by which intestinal L-cell-derived GLP-1 controls GSIS is via neuronal activation of an enteroinsular or gut-to-brain-to-pancreas axis (30). In fact, because of its rapid degradation by DPP-4 and neprilysin, only 10%–15% of intact secreted GLP-1 reaches the β cells via the circulation (31). This has led to the suggestion that GLP-1 can induce its metabolic effects by interacting locally with its receptors located on afferent neurons within submucosal and myenteric nervous plexi, which then transmit the signal to the brain and the pancreas through the vagus nerve (32, 33). Since pentobarbital anesthesia has been shown to exert a suppressive action on pancreatic parasympathetic nerves (34), OGTTs were also performed in conscious mice. Under these conditions, neprilysin inhibition also did not improve GSIS, suggesting pentobarbital anesthesia had not masked a GLP-1-mediated local activation of the enteroinsular axis.
In contrast with our OGTT findings, insulin secretion in response to intravenous glucose was significantly enhanced with neprilysin inhibition, and this beneficial effect was dependent on GLP-1R signaling. When we performed in vitro studies of islets from STZ-treated mice, we found that neprilysin inhibition enhanced GSIS in a GLP-1R-dependent manner. Our observation that GLP-1R signaling within the islet can mediate the insulinotropic effect of neprilysin inhibition is corroborated by our previous in vitro study (35). Also, there is now compelling evidence demonstrating a role for α-cell-derived GLP-1 in modulating β-cell function (36–38), and that GLP-1 production is increased under metabolic stress, as well as STZ-induced diabetes (38, 39). Of note, α-cell-derived active GLP-1 does not seem to contribute to the circulating concentrations of GLP-1, as circulating active GLP-1 levels were almost undetectable in mice with intestine-specific inactivation of the proglucagon gene (40). Although we did not measure intra-islet GLP-1 levels per se, our data raise the possibility that neprilysin inhibition could improve β-cell function by preserving islet-derived active GLP-1. In contrast to the OGTT, upon assessing insulin secretion by IVGTT, any incretin effect of circulating GLP-1 is likely to be minimal due to the intravenous route of glucose administration. Rather, the paracrine action of intra-islet GLP-1 is intact, allowing for sacubitril to enhance levels of α-cell-derived GLP-1 and thereby its insulinotropic effect. Of note, we previously observed that genetic ablation of neprilysin also improves insulin responses during an IVGTT (23), which would be in line with our current findings and may be similarly attributed to enhanced islet-derived active GLP-1 levels and/or islet GLP-1R signaling.
In addition to the well-known effect of GLP-1 to potentiate GSIS, it can also enhance β-cell insulin stores by increasing insulin gene transcription and content, and stabilizing insulin mRNA (41, 42). Our data demonstrated a GLP-1R-dependent increase in pancreatic insulin content in STZ-SAC mice, which may further support a role for intra-islet GLP-1 in sacubitril’s beneficial effects, especially since circulating levels of active GLP-1 were unchanged. Glucagon, another substrate of neprilysin (11, 24, 25), is also able to act on the GLP-1R to modulate GSIS (43–46). Although circulating glucagon levels were not increased with neprilysin inhibition, it cannot be definitively excluded that the GLP-1R-dependent increase in insulin secretion was mediated by α-cell derived glucagon. On the other hand, glucagon is not known to alter insulin content.
In interpreting our data, there are some considerations. First, the GLP-1 degradation product GLP-1(9–36), which may be generated in the setting of decreased neprilysin activity but preserved DPP-4 activity, has been reported to exhibit glucoregulatory actions (47, 48). Although GLP-1(9–36) does not appear to increase insulin secretion (47, 48), it cannot be ruled out that increased levels of this peptide could have contributed to the beneficial effects of neprilysin inhibition in our study. Second, since neprilysin has broad substrate specificity and can cleave other substrates with glucoregulatory properties (15), it is possible that other GLP-1-independent mechanisms, such as increased levels of intra-islet gastric inhibitory polypeptide (11) or peptide YY (49), may have also contributed to the beneficial effect of sacubitril alone on β-cell function.
Last, it is worth noting that while the rate of glucose disappearance (Kg) during the IVGTT was improved in STZ-SAC versus STZ-CTL Glp1r+/+ mice, it is possible that this beneficial effect of neprilysin inhibition was partly mediated by insulin-independent mechanisms. Indeed, insulin-independent glucose disposal is a major mechanism driving glucose tolerance in mice (50). In Glp1r−/− mice, we did not observe an increase in Kg in STZ-SAC versus STZ-CTL mice. The lack of an effect of neprilysin inhibition on Kg in Glp1r−/− mice is consistent with recent studies suggesting that the GLP-1R is required for insulin-independent glucose disposal (51).
In conclusion, our study shows that pharmacological neprilysin inhibition on its own does not increase circulating active GLP-1 levels in a mouse model of reduced insulin secretion, but raises the interesting possibility that it could improve β-cell function by enhancing intra-islet GLP-1R signaling. These results confirm that neprilysin inhibitors have beneficial glycemic and insulinotropic effects and may be useful in treatment of type 2 diabetes.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.14815689.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.14815704.
GRANTS
This work was supported by the research service of the Seattle VA Puget Sound Health Care System and Seattle Institute for Biomedical and Clinical Research, along with funding from the National Institutes of Health (NIH) Grants DK-098506 (to S.Z.) and P30 DK-017047 (University of Washington Diabetes Research Center, Cell Function Analysis Core and Cellular and Molecular Imaging Core). N.E. was supported by the Dick and Julia McAbee Endowed Fellowship from the University of Washington, the French Society of Diabetes, the Baillet-Latour Found and the Belgian American Educational Foundation, the Belgian Association of Diabetes, the Horlait-Dapsens Foundation, and the Leon Fredericq Foundation. J.J.C. was supported by NIH Grant T32 HL-007028 and a Cystic Fibrosis Foundation Fellowship (CASTIL20F0). R.A. was supported by NIH Grant T32 DK-007247.
DISCLAIMERS
S.Z. is the guarantor of this work and as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
DISCLOSURES
S.Z. has previously received research support from Novartis Pharmaceuticals Corporation. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
N.E., S.M.M., J.P., B.M.B., T.O.M., M.J.L., R.L.H., and S.Z. conceived and designed research; N.E., S.M.M., J.P., B.M.B., T.O.M., B.S.F., M.J.L., and S.Z. performed experiments; N.E., S.M.M., J.P., B.M.B., T.O.M., B.S.F., M.J.L., R.L.H., and S.Z. analyzed data; N.E., S.M.M., J.P., B.M.B., T.O.M., M.J.L., J.J.C., R.A., R.L.H., and S.Z. interpreted results of experiments; N.E. prepared figures; N.E. drafted manuscript; N.E., S.M.M., J.P., B.M.B., T.O.M., B.S.F., M.J.L., J.J.C., R.A., R.L.H., and S.Z. edited and revised manuscript; N.E., S.M.M., J.P., B.M.B., T.O.M., B.S.F., J.J.C., R.A., R.L.H., and S.Z. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Daniel Drucker (University of Toronto, Canada) for providing Glp1r+/− mice to generate the wild-type and knockout mice used in this study. We thank A. Aplin, E. Giering, D. Hackney, and C. Schmidt (Seattle Institute for Biomedical and Clinical Research,) for excellent technical support. We thank Steven Kahn (VA Puget Sound Health Care System and University of Washington, Seattle, WA) for valuable discussions during the writing of this manuscript.
REFERENCES
- 1.Rice GI, Jones AL, Grant PJ, Carter AM, Turner AJ, Hooper NM. Circulating activities of angiotensin-converting enzyme, its homolog, angiotensin-converting enzyme 2, and neprilysin in a family study. Hypertension 48: 914–920, 2006. doi: 10.1161/01.HYP.0000244543.91937.79. [DOI] [PubMed] [Google Scholar]
- 2.Standeven KF, Hess K, Carter AM, Rice GI, Cordell PA, Balmforth AJ, Lu B, Scott DJ, Turner AJ, Hooper NM, Grant PJ. Neprilysin, obesity and the metabolic syndrome. Int J Obes (Lond) 35: 1031–1040, 2011. doi: 10.1038/ijo.2010.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willard JR, Barrow BM, Zraika S. Improved glycaemia in high-fat-fed neprilysin-deficient mice is associated with reduced DPP-4 activity and increased active GLP-1 levels. Diabetologia 60: 701–708, 2017. doi: 10.1007/s00125-016-4172-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jordan J, Stinkens R, Jax T, Engeli S, Blaak EE, May M, Havekes B, Schindler C, Albrecht D, Pal P, Heise T, Goossens GH, Langenickel TH. Improved insulin sensitivity with angiotensin receptor neprilysin inhibition in individuals with obesity and hypertension. Clin Pharmacol Ther 101: 254–263, 2017. doi: 10.1002/cpt.455. [DOI] [PubMed] [Google Scholar]
- 5.Seferovic JP, Claggett B, Seidelmann SB, Seely EW, Packer M, Zile MR, Rouleau JL, Swedberg K, Lefkowitz M, Shi VC, Desai AS, McMurray JJV, Solomon SD. Effect of sacubitril/valsartan versus enalapril on glycaemic control in patients with heart failure and diabetes: a post-hoc analysis from the PARADIGM-HF trial. Lancet Diabetes Endocrinol 5: 333–340, 2017. doi: 10.1016/S2213-8587(17)30087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nougué H, Pezel T, Picard F, Sadoune M, Arrigo M, Beauvais F, Launay JM, Cohen-Solal A, Vodovar N, Logeart D. Effects of sacubitril/valsartan on neprilysin targets and the metabolism of natriuretic peptides in chronic heart failure: a mechanistic clinical study. Eur J Heart Fail 21: 598–605, 2019. doi: 10.1002/ejhf.1342. [DOI] [PubMed] [Google Scholar]
- 7.Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet 2: 1300–1304, 1987. doi: 10.1016/s0140-6736(87)91194-9. [DOI] [PubMed] [Google Scholar]
- 8.Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest 79: 616–619, 1987. doi: 10.1172/JCI112855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kieffer TJ, McIntosh CH, Pederson RA. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 136: 3585–3596, 1995. doi: 10.1210/endo.136.8.7628397. [DOI] [PubMed] [Google Scholar]
- 10.Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab 80: 952–957, 1995. doi: 10.1210/jcem.80.3.7883856. [DOI] [PubMed] [Google Scholar]
- 11.Hupe-Sodmann K, McGregor GP, Bridenbaugh R, Göke R, Göke B, Thole H, Zimmermann B, Voigt K. Characterisation of the processing by human neutral endopeptidase 24.11 of GLP-1(7-36) amide and comparison of the substrate specificity of the enzyme for other glucagon-like peptides. Regul Pept 58: 149–156, 1995. doi: 10.1016/0167-0115(95)00063-H. [DOI] [PubMed] [Google Scholar]
- 12.Hupe-Sodmann K, Göke R, Göke B, Thole HH, Zimmermann B, Voigt K, McGregor GP. Endoproteolysis of glucagon-like peptide (GLP)-1 (7-36) amide by ectopeptidases in RINm5F cells. Peptides 18: 625–632, 1997. doi: 10.1016/S0196-9781(97)00123-X. [DOI] [PubMed] [Google Scholar]
- 13.Windeløv JA, Wewer Albrechtsen NJ, Kuhre RE, Jepsen SL, Hornburg D, Pedersen J, Jensen EP, Galsgaard KD, Winther-Sørensen M, Ørgaard A, Deacon CF, Mann M, Kissow H, Hartmann B, Holst JJ. Why is it so difficult to measure glucagon-like peptide-1 in a mouse? Diabetologia 60: 2066–2075, 2017. doi: 10.1007/s00125-017-4347-7. [DOI] [PubMed] [Google Scholar]
- 14.Adelhorst K, Hedegaard BB, Knudsen LB, Kirk O. Structure-activity studies of glucagon-like peptide-1. J Biol Chem 269: 6275–6278, 1994. [PubMed] [Google Scholar]
- 15.Esser N, Zraika S. Neprilysin inhibition: a new therapeutic option for type 2 diabetes? Diabetologia 62: 1113–1122, 2019. doi: 10.1007/s00125-019-4889-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vodovar N, Nougué H, Launay JM, Solal AC, Logeart D. Sacubitril/valsartan in PARADIGM-HF. Lancet Diabetes Endocrinol 5: 495–496, 2017. [Erratum in Lancet Diabetes Endocrinol 7: e19, 2019]. doi: 10.1016/S2213-8587(17)30177-8. [DOI] [PubMed] [Google Scholar]
- 17.Wewer Albrechtsen NJ, Mark PD, Terzic D, Hansen LH, Andersen UØ, Hartmann B, Carr RD, Gustafsson F, Deacon CF, Holst JJ, Goetze JP, Plomgaard P. Sacubitril/valsartan augments postprandial plasma concentrations of active GLP-1 when combined with sitagliptin in men. J Clin Endocrinol Metab 104: 3868–3876, 2019. doi: 10.1210/jc.2019-00515. [DOI] [PubMed] [Google Scholar]
- 18.Graus-Nunes F, Marinho TS, Barbosa-da-Silva S, Aguila MB, Mandarim-de-Lacerda CA, Souza-Mello V. Differential effects of angiotensin receptor blockers on pancreatic islet remodelling and glucose homeostasis in diet-induced obese mice. Mol Cell Endocrinol 439: 54–64, 2017. doi: 10.1016/j.mce.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 19.Shen M, Sun D, Li W, Liu B, Wang S, Zhang Z, Cao F. The synergistic effect of valsartan and LAF237 [(S)-1-[(3-hydroxy-1-adamantyl)ammo]acetyl-2-cyanopyrrolidine] on vascular oxidative stress and inflammation in type 2 diabetic mice. Exp Diabetes Res 2012: 146194, 2012. doi: 10.1155/2012/146194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parilla JH, Willard JR, Barrow BM, Zraika S. A mouse model of beta-cell dysfunction as seen in human type 2 diabetes. J Diabetes Res 2018: 6106051, 2018. doi: 10.1155/2018/6106051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mu J, Woods J, Zhou YP, Roy RS, Li Z, Zycband E, Feng Y, Zhu L, Li C, Howard AD, Moller DE, Thornberry NA, Zhang BB. Chronic inhibition of dipeptidyl peptidase-4 with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes 55: 1695–1704, 2006. doi: 10.2337/db05-1602. [DOI] [PubMed] [Google Scholar]
- 22.Hogan MF, Hackney DJ, Aplin AC, Mundinger TO, Larmore MJ, Castillo JJ, Esser N, Zraika S, Hull RL. SGLT2-i improves markers of islet endothelial cell function in db/db diabetic mice. J Endocrinol 248: 95–106, 2021. doi: 10.1530/JOE-20-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zraika S, Koh DS, Barrow BM, Lu B, Kahn SE, Andrikopoulos S. Neprilysin deficiency protects against fat-induced insulin secretory dysfunction by maintaining calcium influx. Diabetes 62: 1593–1601, 2013. doi: 10.2337/db11-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trebbien R, Klarskov L, Olesen M, Holst JJ, Carr RD, Deacon CF. Neutral endopeptidase 24.11 is important for the degradation of both endogenous and exogenous glucagon in anesthetized pigs. Am J Physiol Endocrinol Physiol 287: E431–E438, 2004. doi: 10.1152/ajpendo.00353.2003. [DOI] [PubMed] [Google Scholar]
- 25.Kjeldsen SAS, Hansen LH, Esser N, Mongovin S, Winther-Sørensen M, Galsgaard KD, Hunt JE, Kissow H, Ceutz FR, Terzic D, Mark PD, Plomgaard P, Goetze JP, Goossens GH, Blaak EE, Deacon CF, Rosenkilde MM, Zraika S, Holst JJ, Wewer Albrechtsen NJ. Neprilysin inhibition increases glucagon levels in humans and mice with potential effects on amino acid metabolism. J Endocr Soc 5: bvab084, 2021. doi: 10.1210/jendso/bvab084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chipkin RE, Kreutner W, Billard W. Potentiation of the hypoglycemic effect of insulin by thiorphan, an enkephalinase inhibitor. Eur J Pharmacol 102: 151–154, 1984. doi: 10.1016/0014-2999(84)90349-2. [DOI] [PubMed] [Google Scholar]
- 27.Arbin V, Claperon N, Fournié-Zaluski MC, Roques BP, Peyroux J. Acute effect of the dual angiotensin-converting enzyme and neutral endopeptidase 24-11 inhibitor mixanpril on insulin sensitivity in obese Zucker rat. Br J Pharmacol 133: 495–502, 2001. doi: 10.1038/sj.bjp.0704098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu HT, Chang CK, Cheng KC, Chang CH, Yeh CH, Cheng JT. Increase of plasma insulin by racecadotril, an inhibitor of enkephalinase, in Wistar rats. Horm Metab Res 42: 261–267, 2010. doi: 10.1055/s-0029-1246190. [DOI] [PubMed] [Google Scholar]
- 29.Plamboeck A, Holst JJ, Carr RD, Deacon CF. Neutral endopeptidase 24.11 and dipeptidyl peptidase IV are both mediators of the degradation of glucagon-like peptide 1 in the anaesthetised pig. Diabetologia 48: 1882–1890, 2005. doi: 10.1007/s00125-005-1847-7. [DOI] [PubMed] [Google Scholar]
- 30.Donath MY, Burcelin R. GLP-1 effects on islets: hormonal, neuronal, or paracrine? Diabetes Care 36, Suppl 2: S145–S148, 2013. doi: 10.2337/dcS13-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-like peptide-1-(7-36)amide is transformed to glucagon-like peptide-1-(9-36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 140: 5356–5363, 1999. doi: 10.1210/endo.140.11.7143. [DOI] [PubMed] [Google Scholar]
- 32.Washington MC, Raboin SJ, Thompson W, Larsen CJ, Sayegh AI. Exenatide reduces food intake and activates the enteric nervous system of the gastrointestinal tract and the dorsal vagal complex of the hindbrain in the rat by a GLP-1 receptor. Brain Res 1344: 124–133, 2010. doi: 10.1016/j.brainres.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 33.Waget A, Cabou C, Masseboeuf M, Cattan P, Armanet M, Karaca M, Castel J, Garret C, Payros G, Maida A, Sulpice T, Holst JJ, Drucker DJ, Magnan C, Burcelin R. Physiological and pharmacological mechanisms through which the DPP-4 inhibitor sitagliptin regulates glycemia in mice. Endocrinology 152: 3018–3029, 2011. doi: 10.1210/en.2011-0286. [DOI] [PubMed] [Google Scholar]
- 34.Havel PJ, Paquette TL, Taborsky GJ Jr.. Halothane is less suppressive than pentobarbital on reflex and neural activation of pancreatic F-cell. Am J Physiol Endocrinol Physiol 251: E111–E116, 1986. doi: 10.1152/ajpendo.1986.251.1.E111. [DOI] [PubMed] [Google Scholar]
- 35.Esser N, Barrow BM, Choung E, Shen NJ, Zraika S. Neprilysin inhibition in mouse islets enhances insulin secretion in a GLP-1 receptor dependent manner. Islets 10: 175–180, 2018. doi: 10.1080/19382014.2018.1502521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wideman RD, Covey SD, Webb GC, Drucker DJ, Kieffer TJ. A switch from prohormone convertase (PC)-2 to PC1/3 expression in transplanted alpha-cells is accompanied by differential processing of proglucagon and improved glucose homeostasis in mice. Diabetes 56: 2744–2752, 2007. doi: 10.2337/db07-0563. [DOI] [PubMed] [Google Scholar]
- 37.Chambers AP, Sorrell JE, Haller A, Roelofs K, Hutch CR, Kim KS, Gutierrez-Aguilar R, Li B, Drucker DJ, D'Alessio DA, Seeley RJ, Sandoval DA. The role of pancreatic preproglucagon in glucose homeostasis in mice. Cell Metab 25: 927–934.e3, 2017. doi: 10.1016/j.cmet.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Traub S, Meier DT, Schulze F, Dror E, Nordmann TM, Goetz N, Koch N, Dalmas E, Stawiski M, Makshana V, Thorel F, Herrera PL, Boni-Schnetzler M, Donath MY. Pancreatic α cell-derived glucagon-related peptides are required for β cell adaptation and glucose homeostasis. Cell Rep 18: 3192–3203, 2017. doi: 10.1016/j.celrep.2017.03.005. [DOI] [PubMed] [Google Scholar]
- 39.Nie Y, Nakashima M, Brubaker PL, Li QL, Perfetti R, Jansen E, Zambre Y, Pipeleers D, Friedman TC. Regulation of pancreatic PC1 and PC2 associated with increased glucagon-like peptide 1 in diabetic rats. J Clin Invest 105: 955–965, 2000. doi: 10.1172/JCI7456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song Y, Koehler JA, Baggio LL, Powers AC, Sandoval DA, Drucker DJ. Gut-proglucagon-derived peptides are essential for regulating glucose homeostasis in mice. Cell Metab 30: 976–986.e3, 2019. doi: 10.1016/j.cmet.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fehmann HC, Habener JF. Insulinotropic hormone glucagon-like peptide-I(7-37) stimulation of proinsulin gene expression and proinsulin biosynthesis in insulinoma beta TC-1 cells. Endocrinology 130: 159–166, 1992. doi: 10.1210/endo.130.1.1309325. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Egan JM, Raygada M, Nadiv O, Roth J, Montrose-Rafizadeh C. Glucagon-like peptide-1 affects gene transcription and messenger ribonucleic acid stability of components of the insulin secretory system in RIN 1046-38 cells. Endocrinology 136: 4910–4917, 1995. doi: 10.1210/endo.136.11.7588224. [DOI] [PubMed] [Google Scholar]
- 43.Moens K, Flamez D, Van Schravendijk C, Ling Z, Pipeleers D, Schuit F. Dual glucagon recognition by pancreatic beta-cells via glucagon and glucagon-like peptide 1 receptors. Diabetes 47: 66–72, 1998. doi: 10.2337/diab.47.1.66. [DOI] [PubMed] [Google Scholar]
- 44.Svendsen B, Larsen O, Gabe MBN, Christiansen CB, Rosenkilde MM, Drucker DJ, Holst JJ. Insulin secretion depends on intra-islet glucagon signaling. Cell Rep 25: 1127–1134.e2, 2018. doi: 10.1016/j.celrep.2018.10.018. [DOI] [PubMed] [Google Scholar]
- 45.Capozzi ME, Wait JB, Koech J, Gordon AN, Coch RW, Svendsen B, Finan B, D'Alessio DA, Campbell JE. Glucagon lowers glycemia when β-cells are active. JCI Insight 4: e129954, 2019. doi: 10.1172/jci.insight.129954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu L, Dattaroy D, Pham J, Wang L, Barella LF, Cui Y, Wilkins KJ, Roth BL, Hochgeschwender U, Matschinsky FM, Kaestner KH, Doliba NM, Wess J. Intra-islet glucagon signaling is critical for maintaining glucose homeostasis. JCI Insight 5: e127994, 2019. doi: 10.1172/jci.insight.127994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deacon CF, Plamboeck A, Møller S, Holst JJ. GLP-1-(9-36) amide reduces blood glucose in anesthetized pigs by a mechanism that does not involve insulin secretion. Am J Physiol Endocrinol Physiol 282: E873–E879, 2002. doi: 10.1152/ajpendo.00452.2001. [DOI] [PubMed] [Google Scholar]
- 48.Meier JJ, Gethmann A, Nauck MA, Götze O, Schmitz F, Deacon CF, Gallwitz B, Schmidt WE, Holst JJ. The glucagon-like peptide-1 metabolite GLP-1-(9-36) amide reduces postprandial glycemia independently of gastric emptying and insulin secretion in humans. Am J Physiol Endocrinol Physiol 290: E1118–E1123, 2006. doi: 10.1152/ajpendo.00576.2005. [DOI] [PubMed] [Google Scholar]
- 49.Medeiros MD, Turner AJ. Processing and metabolism of peptide-YY: pivotal roles of dipeptidylpeptidase-IV, aminopeptidase-P, and endopeptidase-24.11. Endocrinology 134: 2088–2094, 1994. doi: 10.1210/endo.134.5.7908871. [DOI] [PubMed] [Google Scholar]
- 50.Pacini G, Thomaseth K, Ahrén B. Contribution to glucose tolerance of insulin-independent vs. insulin-dependent mechanisms in mice. Am J Physiol Endocrinol Physiol 281: E693–E703, 2001. doi: 10.1152/ajpendo.2001.281.4.E693. [DOI] [PubMed] [Google Scholar]
- 51.Ovlund T, Pacini G, Ahrén B. Impact of incretin hormone receptors on insulin-independent glucose disposal in model experiments in mice. Front Endocrinol (Lausanne) 12: 680153, 2021. doi: 10.3389/fendo.2021.680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.14815689.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.14815704.