

Abstract
Hereditary pulmonary alveolar proteinosis (hPAP) is a rare disorder caused by recessive mutations in GM-CSF receptor subunit α/β genes (CSF2RA/CSF2RB, respectively) characterized by impaired GM-CSF-dependent surfactant clearance by alveolar macrophages (AMs) resulting in alveolar surfactant accumulation and hypoxemic respiratory failure. Because hPAP is caused by CSF2RA mutations in most patients, we created an animal model of hPAP caused by Csf2ra gene disruption (Csf2ra–/– mice) and evaluated the effects on AMs and lungs. Macrophages from Csf2ra–/– mice were unable to bind and clear GM-CSF, did not exhibit GM-CSF signaling, and had functional defects in phagocytosis, cholesterol clearance, and surfactant clearance. Csf2ra–/– mice developed a time-dependent, progressive lung disease similar to hPAP in children caused by CSF2RA mutations with respect to the clinical, physiological, histopathological, biochemical abnormalities, biomarkers of PAP lung disease, and clinical course. In contrast, Csf2ra+/– mice had functionally normal AMs and no lung disease. Pulmonary macrophage transplantation (PMT) without myeloablation resulted in long-term engraftment, restoration of GM-CSF responsiveness to AMs, and a safe and durable treatment effect that lasted for the duration of the experiment (6 mo). Results demonstrate that homozygous (but not heterozygous) Csf2ra gene ablation caused hPAP identical to hPAP in children with CSF2RA mutations, identified AMs as the cellular site of hPAP pathogenesis in Csf2ra–/– mice, and have implications for preclinical studies supporting the translation of PMT as therapy of hPAP in humans.
Keywords: alveolar macrophage, GM-CSF receptor, hereditary pulmonary alveolar proteinosis, surfactant homeostasis
INTRODUCTION
Hereditary pulmonary alveolar proteinosis (hPAP) is a recessive lung disorder characterized by surfactant accumulation caused by function-disrupting genetic mutations in either CSF2RA or CSF2RB, which encode the heterodimeric α or β subunits of the granulocyte/macrophage-colony stimulating factor (GM-CSF) receptor, respectively (1–5). Loss of GM-CSF stimulation impairs the maturation and functions of alveolar macrophages (AMs), which require GM-CSF to clear surfactant and perform numerous other functions (6). Although multiple disease-causing mutations have been identified in each subunit, recessive mutations in CSF2RA are the cause of the disease in ∼90% of patients with hPAP (BCT, unpublished observation). Currently, no specific pharmacologic therapy is approved for hPAP, and patients must be treated by whole lung lavage, an invasive procedure that requires general anesthesia and mechanical ventilation and targets the physical removal of surfactant by repeatedly washing the lungs with saline (7, 8). Bone marrow transplantation has been attempted in several children but has not had long-term success due to fatal complications arising from overwhelming infection (2) or the development of graft versus host disease (9).
Mice with homozygous disruption of the Csf2rb gene encoding the murine homologue of the human CSF2RB gene also develop hPAP identical to the disease in human patients with recessive CSF2RB mutations (4, 5, 10). We recently developed a novel, lung-targeted treatment approach for hPAP, pulmonary macrophage transplantation (PMT), and demonstrated in Csf2rb-deficient (Csf2rb−/−) mice that instillation of congenic wild type (WT) or Csf2rb gene-corrected Csf2rb−/− mouse-derived macrophages directly into the lungs—without myeloablation—was safe, well-tolerated, and resulted in an efficacious and durable treatment effect that increased the mouse’s lifespan by 20% (10). The extraordinary efficacy of PMT therapy of hPAP was attributable, in part, to a strong survival advantage of transplanted, GM-CSF-responsive cells compared with the dysfunctional native macrophages and driven by the markedly increased levels of pulmonary GM-CSF (10).
Here, we report the creation of a murine model of hPAP caused by homozygous disruption of the Csf2ra gene, its validation as an authentic model of human hPAP caused by recessive CSF2RA mutations and use of PMT to demonstrate that AMs are the cellular site of pathogenesis in hPAP lung disease. Csf2ra–/– mice created as part of this study were reported elsewhere in a preclinical, long-term safety and efficacy study of lentiviral-mediated gene therapy of hPAP (11). Mice heterozygous for Csf2ra gene ablation had a completely normal lung phenotype.
MATERIALS AND METHODS
Mice
Mice were housed, bred, and studied at the Cincinnati Children’s Hospital Research Foundation utilizing protocols approved by the Institutional Animal Care and Use Committee. C57BL/6-Ptprcb mice (Charles River) (wild type or WT mice hereafter) were used to create mice with homozygous disruption of Csf2ra alleles (Csf2ra−/− mice) using clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) nuclease 9 (CRISPR/Cas9) genome editing technology (12) as we previously reported (11). Briefly, two guide RNA molecules (sgRNA-1, 5′-CGC TAC TGC TTC CGC CAA CT-3′; sgRNA-2, 5′-CGT CCG AGT CCA GGG GTC GA-3′; each at 25 ng/μL and targeting Csf2ra gene exons 2 and 3, respectively (Fig. 1A); as well as Cas9 mRNA (at 100 ng/µL) were co-injected into the cytoplasm of C57Bl/6 zygotes by piezo-driven microinjection (13). This gene targeting strategy aimed to delete genomic DNA located between the two sgRNA target sites and smaller deletions within each of the individual guide RNA target sites (Fig. 1A). Deletions were confirmed by polymerase chain reaction (PCR) amplification analysis of mouse genomic DNA using a Csf2ra gene-specific oligonucleotide primer pair: forward (5'-CAT CAC ATG CCA TGA ACA TCA CC-3'), reverse (5'-ACC GGA AGT GAC ATC ATT GCG-3').
Figure 1.
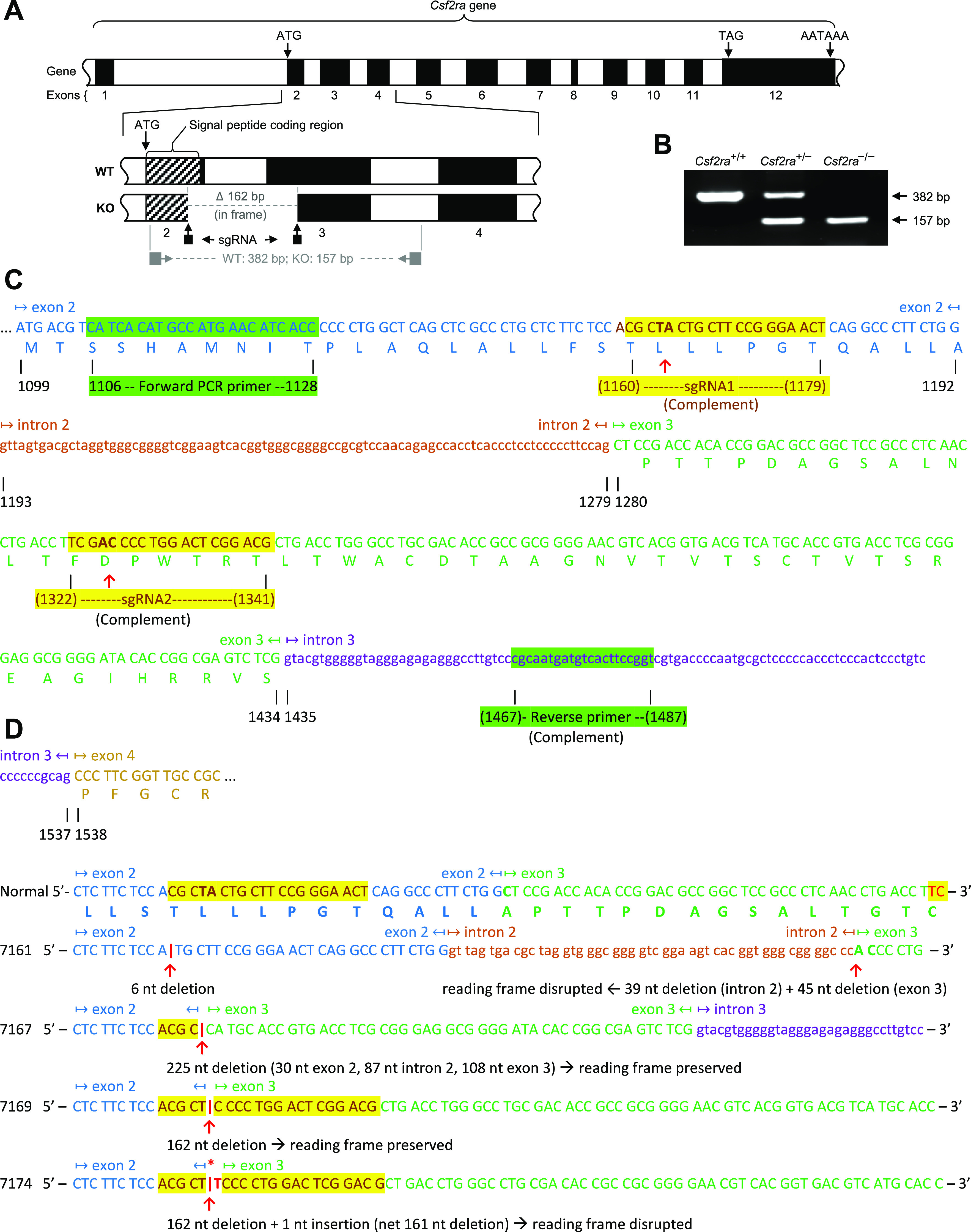
Disruption of Csf2ra gene expression in mice by specific exon-targeted gene editing. A: molecular strategy used to disrupt the Csf2ra gene using CRISPR-Cas9 genome-editing. The normal structure of the Csf2ra gene is shown with the Csf2ra deletion present in mouse line 7169. The location of the single-strand guide ribonucleic acid (sgRNA) and the oligonucleotide primers used for polymerase chain reaction (PCR) amplification-based genotyping of mice are shown. B: PCR-based Csf2ra genotyping analysis showing mice homozygous for wild-type (WT) alleles (Csf2ra+/+), heterozygous for disrupted and WT Csf2ra alleles (Csf2ra+/–), and homozygous for disrupted Csf2ra alleles (Csf2ra–/–). The sizes of normal [382 base pairs (bp)] and disrupted (157 bp) Csf2ra alleles are indicated. C: molecular strategy used to create Csf2ra gene disruption. The sequence of the two sgRNA and oligonucleotide primers used for PCR-based genotyping are shown in alignment to the sequence of mouse chromosome 19 showing a portion of the WT Csf2ra gene [Mus muscularis (C57BL/6J) Chromosome 19, nucleotides 61217191–61228463, mRNA coding strand shown, Genbank accession number NM_009970.2]. D: sequence of the WT Csf2ra gene and the region encompassing exon-2 to exon-3 in each of the four mouse lines (7161, 7167, 7169, and 7174) with disruption of the Csf2ra gene (Csf2ra–/–). The sequences of exons (capital letters) and introns (lower case letters) are indicated, alternating colors are used to improve readability, yellow highlighting identifies the regions affected by gene editing, and the specific deletion in each mouse line is indicated.
All experiments utilized age-matched male and female mice that were housed together, underwent PCR genotyping, and were assigned to groups without randomization. WT B6.SJL-Ptprca (BoyJ) mice were used to generate Csf2ra+/+/CD45.1+ donor cells for the pulmonary macrophage transplantation (PMT) experiments.
Characterization of the Lung Disease in Csf2ra−/− Mice
Lung epithelial lining fluid and nonadherent cells were collected by bronchoalveolar lavage (BAL), processed immediately, and evaluated for BAL turbidity (optical absorption at 600 nm) and cytology. Cholesterol was measured in total BAL using the Amplex Red Assay (Thermo Fisher Scientific) according to the manufacturer’s protocol as previously described (11, 14). The levels of proinflammatory cytokines (IL-1β, IL-6, and TNF-α) and BAL cytokine biomarkers of PAP [GM-CSF, M-CSF, and monocyte chemoattractant protein (MCP)-1] in BAL fluid were measured by enzyme-linked immunosorbent assay (ELISA) (R&D Systems). Lung histopathology was evaluated after removal of heart and lungs—en bloc, inflation-fixation at 25 cm of pressure, and sectioning (5 µm), staining, and microscopic examination as described previously (10).
Characterization of Macrophages
Lung epithelial lining fluid and nonadherent cells were collected by BAL (five 1 mL aliquots of PBS plus 0.5 mM EDTA) and processed immediately as described (10). AMs in BAL were evaluated directly or immediately after isolation by brief adherence. Bone marrow-derived macrophages (BMDMs) were isolated by flushing the tibias and femurs of 6- to 8-wk-old WT mice with culture medium [Dulbecco’s modified Eagle’s medium (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS), 50 U/mL penicillin, and 50 µg/mL streptomycin], cultured in the presence of GM-CSF and M-CSF for 5 days from seeding, and utilized in PMT experiments (10). Expression of the GM-CSF receptor α subunit (CD116) on BAL cells was evaluated after sedimentation and immunostaining with a monoclonal mouse GM-CSF receptor α PE-conjugated antibody (Rat IgG2A Clone No. 698423, R and D systems) by fluorescence microscopy [ZeissAxioplan2 microscope (Zeiss)] or by flow cytometry as described (11). A rat IgG2A PE-conjugated antibody (monoclonal rat IgG2A Clone No. 54447, R and D systems) was used as the isotype control antibody for GM-CSF receptor α staining. GM-CSF receptor function was measured as the capacity of macrophages to remove GM-CSF from the culture media as a function of time (1) by quantifying GM-CSF levels remaining in the culture by ELISA (R&D Systems). GM-CSF receptor signal transduction was evaluated by measuring GM-CSF-stimulated phosphorylation of signal transducer and activator of transcription (STAT5) by using flow cytometry as described (3). Phagocytic capacity was determined by measuring the uptake of opsonized Nile red beads using flow cytometry (11). Surfactant clearance was determined by measuring the elimination of internalized surfactant-related cholesterol using the Amplex Red Cholesterol Assay as described (14).
Pulmonary Macrophage Transplantation
Pulmonary macrophage transplantation (PMT) was performed by instillation of cultured WT BMDMs (2 × 106 cells/recipient mouse) and mice were evaluated as described (10). Hematologic indices were measured using an automated Hemavet 850 (Drew Scientific). Transcriptomic analysis was done using total mRNA from AMs after isolation, reverse transcription, barcoding, and high-throughput sequencing (Illumina HiSeq2500, Rapid Mode) (10).
Statistical Analysis
Numeric data were evaluated for a normal distribution using the Shapiro–Wilk test and are presented as means ± SE. Two-group comparisons were made with Student’s t test (parametric data) or Mann–Whitney test (nonparametric data) as appropriate. Multiple group comparisons were done using one-way analysis of variance (ANOVA) with post hoc pairwise comparisons using the Student–Newman–Keuls or Dunn’s method as appropriate. P values of <0.05 were considered to indicate statistical significance. All experiments were repeated at least twice with similar results.
RESULTS
Csf2ra Gene Disruption
Eleven distinct lines of mice harboring homozygous disruption of the Csf2ra genes were created in C57BL/6 mice using CRISPR/Cas9 gene editing to delete a region of the Csf2ra gene encompassing the 3′ end of exon 2, all of intron 2, and the 5′ end of exon 3 (Fig. 1A). Csf2ra deletions were confirmed by PCR-based genotype analysis (Fig. 1B) and mice homozygous or heterozygous for Csf2ra deletion or harboring WT Csf2ra alleles (Csf2ra–/–, Csf2ra+/–, or Csf2ra+/+ mice, respectively) were created by breeding together male and female Csf2ra+/– mice. The sequences of the two guide RNA molecules and PCR primers pairs used are indicated (Fig. 1C). Of the eleven mouse lines created, four (7161, 7167, 7169, and 7174) underwent detailed genomic sequencing and other analyses (Fig. 1D); results for two lines (7167 and 7169) are described in detail and reported here. A prior report on the long-term safety and efficacy of lentiviral-mediated gene therapy includes data on the lung phenotype in mice homozygous for ablation of the Csf2ra gene (11).
Macrophage Dysfunction in Csf2ra–/– Mice
The Csf2ra gene product, the GM-CSF receptor α subunit (CD116), was readily detected by flow cytometry on AMs and BMDMs from mice harboring at least one normal Csf2ra allele (Csf2ra+/– and Csf2ra+/+) but not from mice with homozygous Csf2ra gene disruption (Csf2ra–/–) (Fig. 2, A, B, H, and I). BMDMs from Csf2ra+/– or Csf2ra+/+ but not Csf2ra–/– mice rapidly cleared GM-CSF from the culture media in a time-dependent fashion (Fig. 2C) and underwent GM-CSF-stimulated phosphorylation of signal transducer and activator of transcription 5 (STAT5)—a signaling molecule immediately downstream from the GM-CSF receptor that becomes phosphorylated upon ligand binding (15) (Fig. 2D). Phagocytosis was impaired in Csf2ra–/– BMDMs compared with WT BMDMs as demonstrated by a reduced percentage of cells internalizing opsonized fluorescent beads and reduced number of beads internalized per cell (Fig. 2E) and by reduced total mean fluorescence intensity (MFI) (Fig. 2F). Cholesterol clearance was markedly reduced in Csf2ra–/– mice compared with Csf2ra+/– and Csf2ra+/+ mice (Fig. 2G). Results indicate macrophages homozygous for Csf2ra disruption did not bind GM-CSF, exhibit GM-CSF receptor signaling, and had reduced GM-CSF-dependent functions whereas macrophages heterozygous for Csf2ra disruption did bind GM-CSF, exhibited GM-CSF signaling, and had GM-CSF-dependent functions similar to macrophages from WT mice.
Figure 2.
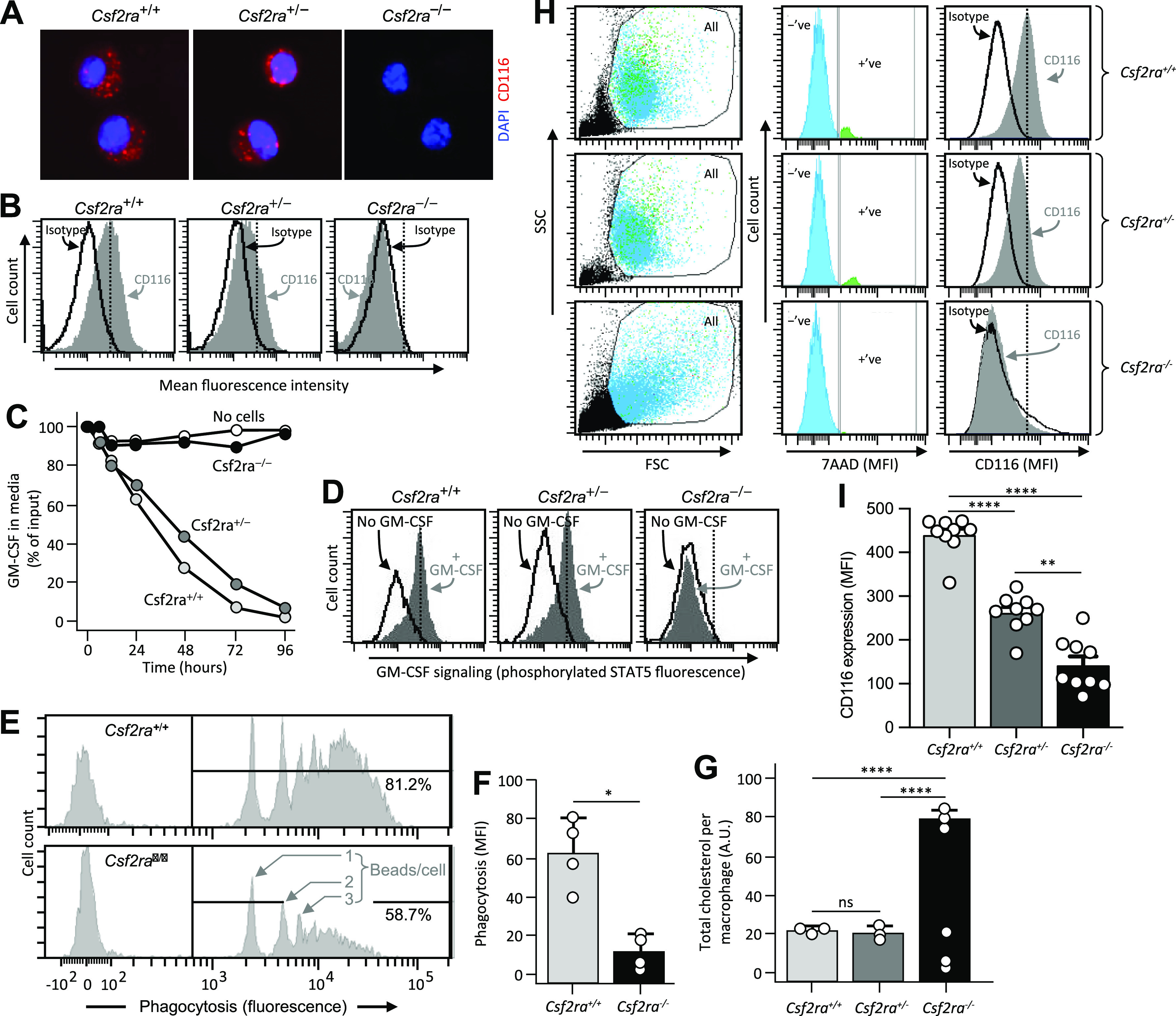
Effects of Csf2ra gene disruption on macrophage functions. A: photomicrographs showing immunofluorescence indicating the presence of the granulocyte/macrophage-colony stimulating factor (GM-CSF) receptor α subunit (CD116) on alveolar macrophages (AMs) from age-matched Csf2ra+/+ and Csf2ra+/− but not Csf2ra–/– mice. B: detection of CD116 by flow cytometry on bone marrow-derived macrophages (BMDMs) from age-matched Csf2ra+/+ and Csf2ra+/− but not Csf2ra–/– mice after staining with isotype control antibody or anti-CD116 antibody (indicated). C: macrophage-mediated binding and clearance of GM-CSF. BMDMs from the indicated mouse lines were cultured in medium to which GM-CSF had been added without further media changes and at the indicated times media aliquots were obtained and the concentration of GM-CSF was measured by ELISA. Three separate plates were evaluated at each condition and time point. D: GM-CSF receptor signaling. BMDMs from the indicated mouse lines were evaluated for GM-CSF receptor signaling by incubation without or with GM-CSF (10 ng/mL) to stimulate phosphorylation of signal transducer and activator of transcription (STAT5) measured by flow cytometry as described (10). E and F: Fc-receptor mediated phagocytosis. BMDMs were incubated with opsonized beads (2-μm diameter fluorescent microspheres coated with bovine serum albumin (BSA) bound by anti-BSA antibodies) for 1 h and internalization was measured by flow cytometry. E: histograms showing peaks of fluorescence corresponding to 1, 2, or 3, … beads/cell. The percentage of cells internalizing fluorescent beads is shown numerically. F: phagocytosis was quantified as the means ± SE mean fluorescence intensity from four measurements per condition. *P < 0.05. G: surfactant clearance. BMDMs in culture were incubated for 24 h with pulmonary alveolar proteinosis (PAP) patient-derived surfactant sediment (from whole lung lavage), then for 24 h in media without added surfactant to allow cells to clear the internalized surfactant phospholipids, cholesterol, and proteins, and then the retained intracellular cholesterol ester was measured by Amplex Red assay and expressed after normalization to cellular protein from three measurements per condition. H and I: evaluation of CD116 expression on AMs from age-matched Csf2ra+/+, Csf2ra+/–, and Csf2ra–/– mice by flow cytometry. H, Left: scatterplots showing side scatter (SSC) versus forward scatter (FSC) and the gating strategy used to identify cells (All) and exclude noncell debris. H, Middle: histogram plots and gating strategy used to identify cells with positive (+ve) or negative (−ve) 7AAD-antibody staining. H, Right: histogram plots showing 7AAD-negative cells (determined from the gate shown in central panels) that were stained with isotype control antibody or anti-CD116 antibody (indicated). I: CD116 expression on AMs was quantified as means ± SE mean fluorescence intensity from 8 to 9 mice/group. **Significant P < 0.01, ****P < 0.0001.
Lung Disease in Csf2ra–/– Mice
BAL cytology showed homozygous disruption of Csf2ra resulted in accumulation of amorphous debris in pulmonary alveoli whereas heterozygous disruption did not (Fig. 3A). Compared with WT, AMs from mice with homozygous Csf2ra gene disruption were enlarged, foamy-appearing, and had intense oil-red-O staining whereas those from mice with heterozygous disruption appeared normal (Fig. 3A). Histologic examination of the lungs revealed that homozygous (but not heterozygous) disruption of Csf2ra was associated with the accumulation of amorphous material in alveoli that stained heavily with hematoxylin and eosin (Fig. 3B) and periodic acid-Schiff reagent (Fig. 3C). Homozygous (but not heterozygous) disruption of Csf2ra was also associated with marked peribronchiolar lymphocytosis (Fig. 3B, white/black arrows). Pulmonary fibrosis was not observed in mice with homozygous or heterozygous Csf2ra gene disruption (Fig. 3D). These results demonstrate that mice with homozygous disruption of Csf2ra (but not heterozygous disruption) spontaneously develop a lung disease with cytologic and histopathologic features identical to that of children with hPAP caused by recessive CSF2RA mutations (1–3).
Figure 3.

Effects of Csf2ra gene disruption on bronchoalveolar lavage (BAL) cytology and lung histology. A: examination of BAL after Oil-red-O staining revealed normal cytology in Csf2ra+/+ and Csf2ra+/– mice and abnormal cytology, numerous large, red macrophages (indicating cholesterol ester-rich inclusions) and “dirty”-appearing extracellular debris in Csf2ra–/– mice. Scale bar is 100 µm in main panel and 20 µm in inset. B: lung histology was normal in Csf2ra+/+ and Csf2ra+/– mice and abnormal in Csf2ra–/– mice; hematoxylin and eosin staining revealed well-preserved alveoli filled with homogeneous eosinophilic sediment (second row, right) and lymphocytosis (arrows) in perivascular distribution (third row, right). Scale bar is 100 µm. C: periodic acid-Schiff staining was normal in Csf2ra+/+ and Csf2ra+/– mice and abnormal histology in Csf2ra–/– showing a pattern typical of hereditary pulmonary alveolar proteinosis (hPAP) (fourth row, right). Scale bar is 100 µm. D: Masson trichrome staining did not reveal any significant fibrosis in either Csf2ra+/+, Csf2ra+/–, or Csf2ra–/– mice (bottom row). Scale bar is 100 µm.
To evaluate the severity of lung disease in Csf2ra–/– mice, we measured BAL turbidity (an excellent global measure of hPAP lung disease severity in mice (10)). BAL turbidity was markedly increased in Csf2ra–/– mice compared with WT mice and Csf2ra+/– mice (Fig. 4A). BAL turbidity was abnormally increased to a similar degree in all four Csf2ra–/– mouse lines evaluated (data not shown). Furthermore, BAL cholesterol—another excellent global measure of hPAP lung disease severity in mice (10)—was also increased in Csf2ra–/– mice compared with WT and normal in Csf2ra+/– mice (Fig. 4B). Consistent with these observations, the BAL from Csf2ra–/– mice had a markedly more opaque and milky gross appearance than BAL of Csf2ra+/+ mice (see Fig. 6C) or Csf2ra+/− mice (not shown), which were normal in appearance.
Figure 4.

Effects of Csf2ra gene disruption on lung surfactant accumulation. Mice of the indicated genotypes (20/group) were evaluated at 10 wk of age by bronchoalveolar lavage (BAL) followed by measurement of BAL turbidity (A) and cholesterol level (B). ns, not significant. ****P < 0.0001.
Next, we evaluated levels of BAL cytokine biomarkers of hPAP (increased GM-CSF, M-CSF, and MCP-1) observed in humans with hPAP caused by CSF2RA mutations (1–3); all were elevated in Csf2ra–/– mice compared with Csf2ra+/+ mice and normal in Csf2ra+/– mice (Fig. 5).
Figure 5.
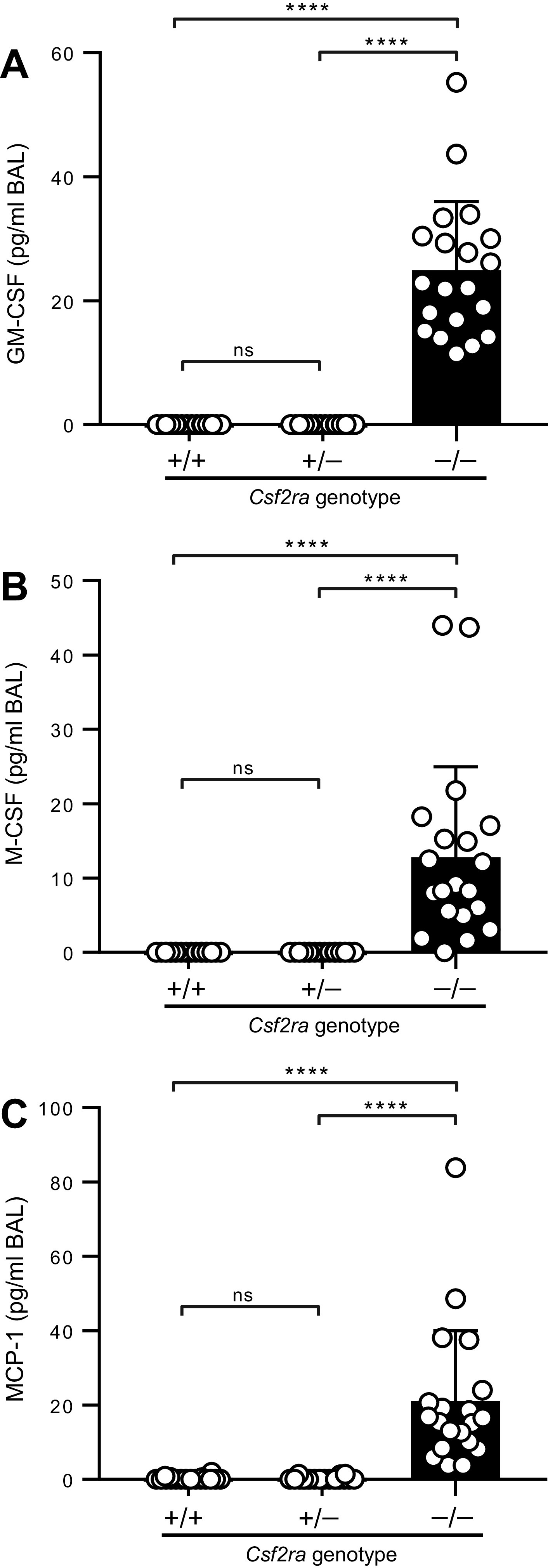
Effects of Csf2ra gene disruption on lung cytokine biomarkers of hereditary pulmonary alveolar proteinosis (hPAP). Mice of the indicated genotypes (20/group) were evaluated at 10 wk of age by bronchoalveolar lavage (BAL) followed by ELISA-based measurement of granulocyte/macrophage-colony stimulating factor (GM-CSF) (A), M-CSF (B), and monocyte chemoattractant protein (MCP)-1 (C). ns, not significant. ****P < 0.0001.
Together, these results demonstrate that mice homozygous for Csf2ra gene disruption develop a lung disease that is cytologically, histologically, and biochemically identical to hPAP in children caused by CSF2RA mutations (1–3) and that mice heterozygous for Csf2ra disruption do not develop lung disease.
Identifying the Cellular Site of hPAP Pathogenesis in Csf2ra–/– Mice
Since instillation of WT macrophages into the lungs of Csf2rb–/– mice resulted in the long-term pulmonary engraftment and time-dependent replacement of the dysfunctional, less-viable endogenous (Csf2rb–/–) macrophages (10), we utilized PMT to test the hypothesis that AMs were the cellular site of pathogenesis of the lung disease observed in Csf2ra–/– mice. Instillation of Csf2ra+/+ BMDMs directly into the lungs of Csf2ra–/– mice without preparatory radiologic or chemical myeloablation resulted in excellent engraftment of CD116-expressing macrophages in the lungs as shown by flow cytometry (Fig. 6A) and immunofluorescence (Fig. 6B). Within 2 mo, PMT normalized the milky appearance of Csf2ra–/– BAL fluid (Fig. 6C), abnormal BAL cytology and AM morphology (Fig. 6D), BAL turbidity (Fig. 6F), BAL cholesterol levels (Fig. 6G), BAL cytokine biomarkers of hPAP (Fig. 6H), and alveolar PAP sediment accumulation (Fig. 6E) and resolved the lung disease (Fig. 6E). In untreated Csf2ra–/– mice, BAL turbidity increased progressively over time; in contrast, BAL turbidity remained low in Csf2ra+/– and Csf2ra+/+ mice (Fig. 6L). Importantly, one PMT of WT (Csf2ra+/+) macrophages caused a time-dependent reduction of BAL turbidity (Fig. 6L). Furthermore, the reduction of turbidity (Fig. 6I), cholesterol (Fig. 6J), and cytokine biomarkers of hPAP (Fig. 6K) in Csf2ra–/– mice persisted for at least 6 mo after PMT. PMT was not associated with significant changes in gross changes in hematologic indices (Table 1), or lung levels of proinflammatory cytokines [interleukin (IL)-1β; IL-6, tumor necrosis factor-α] (Table 2).
Figure 6.
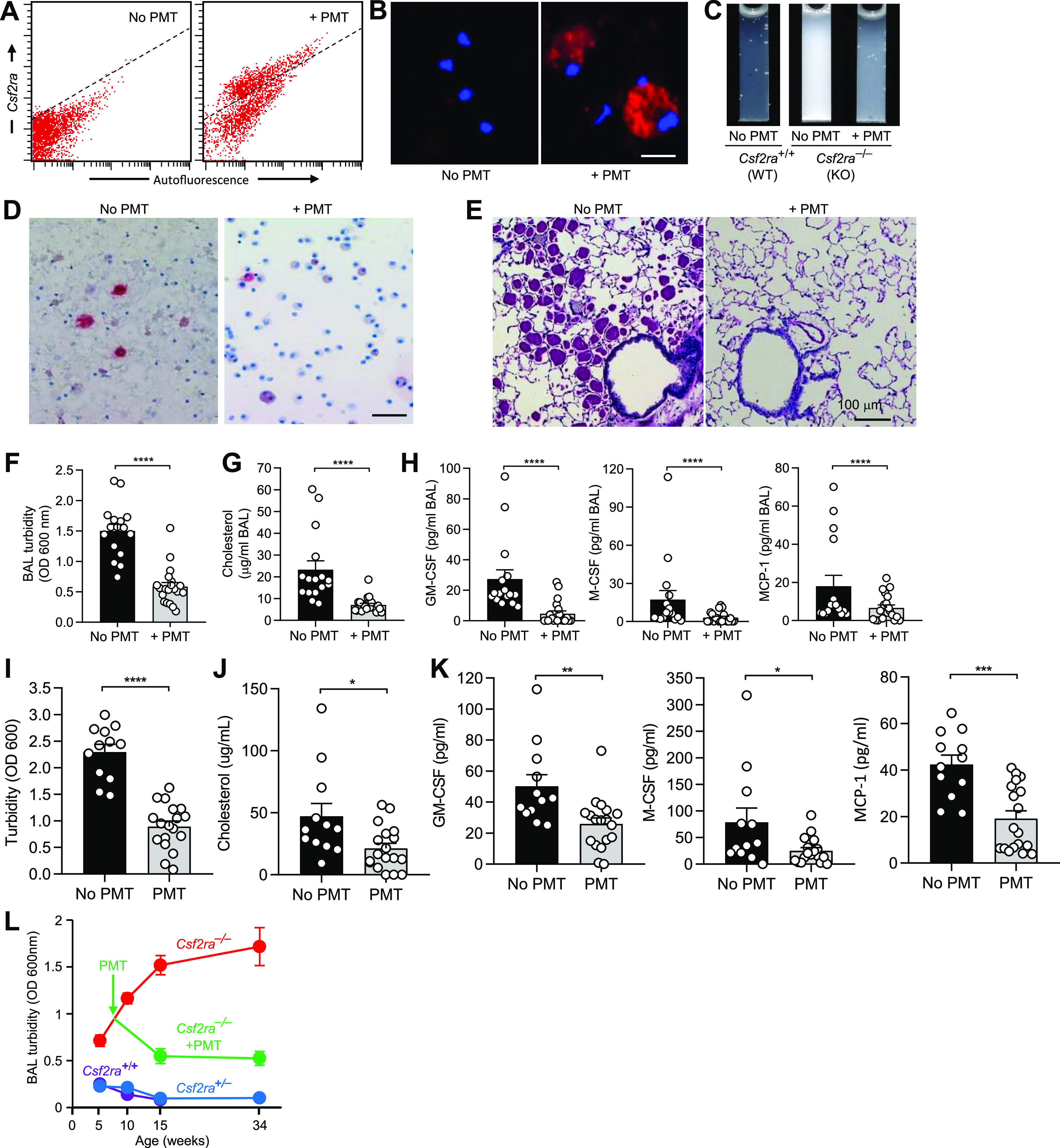
Effects of pulmonary macrophage transplantation (PMT) on granulocyte/macrophage-colony stimulating factor (GM-CSF) signaling and functions of the alveolar macrophage (AM) population in Csf2ra–/– mice to identify the cellular site of disease in Csf2ra–/– mice. Age-matched Csf2ra–/– mice without/with PMT Csf2ra+/+ donor cells were evaluated at subsequent times to characterize the lung phenotype. A: engraftment of transplanted macrophages measured 2 mo after PMT by flow cytometric detection of CD116-positive cells (above dotted line) in PMT-treated mice (right); only autofluorescence was detected in mice without PMT (below dotted line). B: evaluation for expression of CD116 (red color) and counterstaining to detect DAPI nuclear counterstaining (blue color) on AMs in age-matched Csf2ra–/– mice without/with PMT after 2 mo. Scale bar = 20 µm. C: gross appearance of BAL fluid 2 mo after PMT in age-matched, untreated (No PMT) Csf2ra+/+ or Csf2ra–/– mice and PMT-treated (+PMT) Csf2ra–/– mice. D: bronchoalveolar lavage (BAL) cytology after 2 mo in untreated (No PMT) and PMT-treated (+PMT) Csf2ra–/– mice. Oil-red-O stain. Scale bar = 100 µm. E and F: effect of PMT on lung disease severity. E: representative lung histology after 2 mo in Csf2ra–/– mice that were untreated or PMT-treated. Periodic acid-Schiff stain. F: BAL turbidity after 2 mo in age-matched Csf2ra–/– mice without (16 mice/group) or with PMT (20 mice/group). G: BAL cholesterol after 2 mo in age-matched Csf2ra–/– mice without (16 mice/group) or with PMT (20 mice/group). H: hereditary pulmonary alveolar proteinosis (PAP) BAL biomarkers [granulocyte/macrophage-colony stimulating factor (GM-CSF), M-CSF, and monocyte chemoattractant protein (MCP)-1] after 2 mo in age-matched Csf2ra–/– mice without (16 mice/group) or with (20 mice/group) PMT. I–K: BAL turbidity, cholesterol, or PAP cytokine biomarkers, respectively, after 6 mo in age-matched Csf2ra–/– mice without (12–16 mice/group) or with (18–20 mice/group) PMT. L: BAL turbidity at various times in age-matched Csf2ra–/– mice without or with PMT. The numbers of mice at risk at each time point were [genotype, treatment; age (wk)—number of mice]: Csf2ra–/–, untreated; 5 wk—16, 10 wk—22, 16 wk—23, 34 wk—12; Csf2ra–/–, PMT-treated; 16 wk—16, 34 wk—16; Csf2ra+/+, untreated; 5 wk—15, 10 wk—16, 16 wk—7; Csf2ra+/−, untreated; 5 wk—23, 10 wk—16, 16 wk—15, 34 wk—8. Scale bar = 100 µm. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Table 1.
Hematological indices in PMT-treated and age-matched, untreated Csf2ra–/–mice*
| Parameter | Normal Range | PMT-Treated (n = 12) |
No PMT (n = 11) |
|---|---|---|---|
| Evaluation 2 mo after PMT | |||
| Hemoglobin, g/dL | 11.0–15.1 | 11.7 [10.9–12.2] | 10.5 [9.7–11.8] |
| Hematocrit, % | 35.1–45.4 | 41.2 [38.9–41.7] | 38.0 [34.0–39.6] |
| WBC, ×103/µL | 1.8–10.7 | 4.1 [3.3–5.09] | 3.8 [2.5–4.8] |
| Neutrophils, ×103/µL | 0.1–2.4 | 1.3 [1.2–1.50] | 0.5 [0.4–0.6] |
| Lymphocytes, ×103/µL | 0.9–9.3 | 2.4 [1.8–2.66] | 2.7 [1.9–3.7] |
| Monocytes, ×103/µL | 0.0–0.4 | 0.2 [0.2–0.38] | 0.47 [0.4–0.60] |
| Eosinophils, ×103/µL | 0.0–0.2 | 0.0 [0.0–0.02] | 0.02 [0.0–0.08] |
| Basophils, ×103/µL | 0.0–0.2 | 0.00 [0.0–0.00] | 0.01 [0.0–0.02] |
| Platelets, ×103/µL | 592–2972 | 904 [749–959] | 829 [624–849] |
| Parameter | Normal Range | PMT (n = 18) |
No PMT (n = 12) |
|---|---|---|---|
| Evaluation 6 mo after PMT | |||
| Hemoglobin, g/dL | 11.0–15.1 | 12.1 [11.6–12.50] | 12.4 [12.2–12.8] |
| Hematocrit, % | 35.1–45.4 | 38.2 [35.6–39.6] | 40.3 [39.6–41.8] |
| White blood cell count, ×103/µL | 1.8–10.7 | 3.1 [2.9–4.01] | 4.0 [3.8–4.4] |
| Neutrophils, ×103/µL | 0.1–2.4 | 0.3 [0.2–0.65] | 0.6 [0.5–0.82] |
| Lymphocytes, ×103/µL | 0.9–9.3 | 2.3 [2.0–2.96] | 2.79 [2.4–3.01] |
| Monocytes, ×103/µL | 0.0–0.4 | 0.3 [0.3–0.47] | 0.48 [0.3–0.62] |
| Eosinophils, ×103/µL | 0.0–0.2 | 0.03 [0.0–0.47] | 0.06 [0.0–0.09] |
| Basophils, ×103/µL | 0.0–0.2 | 0.01 [0.0–0.02] | 0.01 [0.0–0.04] |
| Platelets, ×103/µL | 592–2,972 | 573 [492–709] | 680 [580–798] |
Data are median and interquartile range.
PMT, pulmonary macrophage transplantation (direct intrapulmonary instillation of congenic, wild-type, bone-marrow-derived macrophages).
Table 2.
Levels of proinflammatory cytokines in bronchoalveolar lavage fluid from PMT-treated and age-matched, untreated, Csf2ra–/–mice*
| Cytokine | PMT-Treated (n = 20) |
No PMT (n = 16) |
|---|---|---|
| Evaluation 2 mo after PMT | ||
| Interleukin-6, pg/mL | 0 [0.0–0.0] | 0 [0.0–26] |
| Interleukin-1β, pg/mL | 0 [0.0–0.3] | 0 [0.0–1.0] |
| Tumor necrosis factor-α, pg/mL | 1.5 [0.1–3.8] | 1.4 [0.3–3.4] |
| Cytokine | PMT-treated (n = 20) |
No PMT (n = 16) |
|---|---|---|
| Evaluation 6 mo after PMT | ||
| Interleukin-6, pg/mL | 3.1 [0.0–10.7] | 11.5 [4.5–46] |
| Interleukin-1β, pg/mL | 0 [0.0–10.7] | 7.1 [0.0–39] |
| Tumor necrosis factor-α, pg/mL | 0.5 [0.0–3.9] | 4.7 [1.8–6.9] |
Data are median and interquartile range.
PMT, pulmonary macrophage transplantation (direct intrapulmonary instillation of congenic, wild-type, bone-marrow-derived macrophages).
Having demonstrated PMT restored GM-CSF signaling in AMs, we evaluated the effects on the transcriptome by performing genome-wide expression profiling on AMs isolated from age-matched untreated Csf2ra+/+ and Csf2ra–/– mice and Csf2ra–/– mice 6 mo after PMT of Csf2ra+/+ BMDMs. Unsupervised hierarchical analysis of RNA-seq data indicated marked co-clustering between PMT-treated Csf2ra–/– and Csf2ra+/+ mice whereas untreated Csf2ra–/– mice clustered separately (Fig. 7A). Supervised gene ontology analysis revealed that genes regulated by GM-CSF and peroxisome proliferator-activated receptor gamma (PPARγ), and genes in multiple pathways involved in macrophage survival, cellular proliferation, lipid metabolism, and immune functions were coordinately dysregulated in Csf2ra–/– mice and normalized by PMT (Fig. 7B).
Figure 7.

Effects of pulmonary macrophage transplantation (PMT) on genome-wide gene expression of the alveolar macrophage (AM) population in Csf2ra–/– mice. A: unsupervised hierarchical clustering dendrogram for AMs obtained from age-matched mice (3 mice/group) including untreated Csf2ra–/– mice (knockout, KO), PMT-treated Csf2ra–/– mice (KO + PMT), and untreated Csf2ra+/+ mice (wild type, WT). B: heat maps showing differentially expressed granulocyte/macrophage-colony stimulating factor (GM-CSF)-regulated genes peroxisome proliferator-activated receptor gamma (PPARγ)-regulated genes, and genes in KEGG pathways related to cell cycle control, apoptosis, peroxisome function, glycophospholipid metabolism, and immune host defense. Genes with increased or decreased transcript levels are shown by red and blue colors, respectively, as indicated on the color scale.
These results show that a single intrapulmonary instillation of WT macrophages into Csf2ra–/– mice without myeloablative “niche-making” resulted in long-term pulmonary engraftment, normalization of AM gene expression and function, and correction of hPAP lung disease. These results identify AMs as the cellular site (and GM-CSF receptors on AMs as the molecular site) of hPAP pathogenesis in Csf2ra–/– mice.
DISCUSSION
In this report, we demonstrate that homozygous disruption of the Csf2ra gene in mice results in a lung disease identical to hPAP in children caused by recessive or compound heterozygous CSF2RA mutations, characterized by the absence of receptor-mediated GM-CSF clearance and GM-CSF signaling, functional impairment of macrophages including reduced phagocytic clearance of particles, cholesterol and surfactant, a clinical course comprising insidious, progressive filling of alveoli with surfactant sediment and parenchymal lymphocytosis. Results also demonstrate that AMs are the cellular site of hPAP pathogenesis and that GM-CSF receptors on AMs are the molecular site of hPAP pathogenesis in Csf2ra–/– mice. These results extend previous observations (11) by demonstrating that mice heterozygous for Csf2ra gene ablation are phenotypically normal and also provide a proof-of-concept supporting the translation of PMT therapy to human patients with hPAP.
The observation that Csf2ra–/– mice have the same clinical, physiological, histopathological, and biochemical abnormalities, disease biomarkers, and natural history as children with hPAP caused by recessive or compound heterozygous CSF2RA gene mutations (1–3) indicates that Csf2ra–/– mice comprise a valid model of the human disease. As is the case for human carriers of a function-disruption CSF2RA mutation, mice heterozygous for Csf2ra gene disruption had no detectable abnormalities of AM function or any evidence of lung disease, which confirms the recessive nature of hPAP caused by Csf2ra gene disruption. It is notable that all the abnormalities identified as affecting AMs or the lungs in Csf2ra–/– mice have similar corresponding abnormalities in Csf2rb–/– mice (10) and, where evaluations have been done, also with human patients with hPAP caused by recessive CSF2RA or CSF2RB mutations (4, 5). Except for the increased levels of GM-CSF, all the abnormalities we observed in Csf2ra–/– mice are also present in Csf2-deficient mice (16–18). Thus, disruption of GM-CSF signaling causes a similar disease phenotype in mice and humans whether caused by disruption of the gene encoding GM-CSF itself or either one of its receptor subunits or by autoantibodies targeting GM-CSF. This has important implications for their use in preclinical toxicology and efficacy studies (11) supporting the development of novel therapies for individuals affected by this devastating disease.
The observation that PMT of Csf2ra+/+ macrophages into Csf2ra–/– mice resulted in long-term engraftment, restored a GM-CSF-responsive AM population, normalized all the cellular and lung disease-specific abnormalities identified in Csf2ra–/– mice, and restored a gene expression profile of AMs in Csf2ra–/– mice that was similar to Csf2ra+/+ (and not Csf2ra–/–) mice strongly supports the concept that loss of GM-CSF signaling in AMs is the primary driver of hPAP pathogenesis in Csf2ra–/– mice. This is important since GM-CSF receptors are also expressed on other cell types, notably respiratory epithelial cells, and overexpression of GM-CSF in the lungs of Csf2–/– mice causes hyperplasia and increased proliferation of alveolar epithelial type 2 cells (19).
The feasibility of translating PMT to humans as therapy of hPAP is supported by our safety and efficacy results for PMT in Csf2ra–/– mice (this report and Ref. 11) and by the striking similarity of hPAP in mice and humans homozygous for disruption of their respective homologous Csf2ra/CSF2RA genes. Transplanted macrophages quickly engrafted in the lungs and persisted long-term, replacing the less-viable, dysfunctional endogenous AMs, resulting in near complete elimination of the accumulated alveolar sediment by 2 mo after PMT, and a persistent treatment effect lasting the duration of the study (6 mo). These observations are supported by similar results for PMT of Csf2ra gene-corrected macrophages into Csf2ra–/– mice (11) and by PMT of WT (Csf2ra+/+/Csf2rb+/+) or Csf2rb gene-corrected Csf2rb–/– macrophages into Csf2rb–/– mice (10). The observation that Csf2ra+/– mice had absolutely no detectable clinical, cytological, histopathological, or biochemical manifestations of hPAP has implications for the level of gene expression targeted in translation of PMT as therapy of hPAP. A level of therapeutic gene expression equivalent to one Csf2ra gene copy is expected to be sufficient to achieve clinical benefit. Furthermore, since ∼40% engraftment/replacement of endogenous AMs by PMT-derived, GM-CSF-responsive macrophages was associated with normalization of the BAL turbidity, cytology, and lung histopathology after PMT, the cellular engraftment target is also less than 50%. Although we did not observe any inflammatory response to PMT in this study, further studies performed under the auspices of regulatory oversight by the US Food and Drug Administration will be needed to formally establish nonclinical safety of PMT as therapy of hPAP.
The limitations of this study include that it did not evaluate or establish a dose-response relationship, minimum effective dose, maximum tolerated dose, or evaluate the effects of repeat administration of PMT. Notwithstanding, the durability of the treatment benefit from the current results and the results of prior studies (10, 11) suggest that a single administration of PMT may be very durable and repeat dosing may not be required. Longer duration studies will be needed to evaluate the effect of PMT in Csf2ra–/– recipients on overall survival as was shown for PMT in Csf2rb–/– mice (10).
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
This work was supported by NIH Grants R01 HL085453 and HL118342 (to B. C. Trapnell), R01HL136721 (to T. Suzuki), Cincinnati Children’s Hospital Medical Center (CCHMC) Foundation Trustee Grant (to T. Suzuki), JSPS KAKENHI Grantsw 16K21750 and 19H03669 (to T. Suzuki), Astellas Foundation for Research on Metabolic Disorders (to T. Suzuki), and the Center for Rheumatic Disease Research (NIH AR070549) P30 grant.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.C.T. and T.S. conceived and designed research; K.S., P.A., A.S., Y.H., Y.M., C.T., M.W., C.C., B.C.C., and T.S. performed experiments; K.S., P.A., A.S., Y.H., Y.M., C.T., M.W., C.C., C.M., B.C.C., B.C.T., and T.S. analyzed data; K.S., P.A., A.S., Y.H., Y.M., C.T., M.W., C.C., C.M., B.C.C., B.C.T., and T.S. interpreted results of experiments; K.S., C.T., B.C.T., and T.S. prepared figures; C.M. and B.C.T. drafted manuscript; K.S., P.A., A.S., Y.H., Y.M., C.T., M.W., C.C., C.M., B.C.C., B.C.T., and T.S. edited and revised manuscript; K.S., P.A., A.S., Y.H., Y.M., C.T., M.W., C.C., C.M., B.C.C., B.C.T., and T.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank D. Black for excellent technical help and the CCRF Transgenic Animal and Genome Editing Core at Cincinnati Children’s Hospital Medical Center for assistance in generation and genotyping of Csf2ra−/− mice. We also acknowledge the assistance of the Research Flow Cytometry Core in the Division of Rheumatology at Cincinnati Children’s Hospital Medical Center. All flow cytometric data were acquired using equipment maintained by the Research Flow Cytometry Core in the Division of Rheumatology at Cincinnati Children’s Hospital Medical Center.
REFERENCES
- 1.Suzuki T, Sakagami T, Rubin BK, Nogee LM, Wood RE, Zimmerman SL, Smolarek T, Dishop MK, Wert SE, Whitsett JA, Grabowski G, Carey BC, Stevens C, van der Loo JC, Trapnell BC. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med 205: 2703–2710, 2008. doi: 10.1084/jem.20080990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinez-Moczygemba M, Doan ML, Elidemir O, Fan LL, Cheung SW, Lei JT, Moore JP, Tavana G, Lewis LR, Zhu Y, Muzny DM, Gibbs RA, Huston DP. Pulmonary alveolar proteinosis caused by deletion of the GM-CSFRalpha gene in the X chromosome pseudoautosomal region 1. J Exp Med 205: 2711–2716, 2008. doi: 10.1084/jem.20080759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suzuki T, Sakagami T, Young LR, Carey BC, Wood RE, Luisetti M, Wert SE, Rubin BK, Kevill K, Chalk C, Whitsett JA, Stevens C, Nogee LM, Campo I, Trapnell BC. Hereditary pulmonary alveolar proteinosis: pathogenesis, presentation, diagnosis, and therapy. Am J Respir Crit Care Med 182: 1292–1304, 2010. doi: 10.1164/rccm.201002-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki T, Maranda B, Sakagami T, Catellier P, Couture CY, Carey BC, Chalk C, Trapnell BC. Hereditary pulmonary alveolar proteinosis caused by recessive CSF2RB mutations. Eur Respir J 37: 201–204, 2011. doi: 10.1183/09031936.00090610. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka T, Motoi N, Tsuchihashi Y, Tazawa R, Kaneko C, Nei T, Yamamoto T, Hayashi T, Tagawa T, Nagayasu T, Kuribayashi F, Ariyoshi K, Nakata K, Morimoto K. Adult-onset hereditary pulmonary alveolar proteinosis caused by a single-base deletion in CSF2RB. J Med Genet 48: 205–209, 2011. doi: 10.1136/jmg.2010.082586. [DOI] [PubMed] [Google Scholar]
- 6.Trapnell BC. Granulocyte macrophage-colony stimulating factor augmentation therapy in sepsis: is there a role? Am J Respir Crit Care Med 2002 166: 129–130, 2002. doi: 10.1164/rccm.2205017. [DOI] [PubMed] [Google Scholar]
- 7.Griese M. Pulmonary alveolar proteinosis: a comprehensive clinical perspective. Pediatrics 140: e20170610, 2017. doi: 10.1542/peds.2017-0610. [DOI] [PubMed] [Google Scholar]
- 8.Trapnell BC, Nakata K, Bonella F, Campo I, Griese M, Hamilton J, Wang T, Morgan C, Cottin V, McCarthy C. Pulmonary alveolar proteinosis. Nat Rev Dis Primers 5: 16, 2019. doi: 10.1038/s41572-019-0066-3. [DOI] [PubMed] [Google Scholar]
- 9.Frémond ML, Hadchouel A, Schweitzer C, Berteloot L, Bruneau J, Bonnet C, Cros G, Briand C, Magnani A, Pochon C, Delacourt C, Cavazzana M, Moshous D, Fischer A, Blanche S, Blic J, Neven B. Successful haematopoietic stem cell transplantation in a case of pulmonary alveolar proteinosis due to GM-CSF receptor deficiency. Thorax 73: 590–592, 2018. doi: 10.1136/thoraxjnl-2017-211076. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki T, Arumugam P, Sakagami T, Lachmann N, Chalk C, Sallese A, Abe S, Trapnell C, Carey B, Moritz T, Malik P, Lutzko C, Wood RE, Trapnell BC. Pulmonary macrophage transplantation therapy. Nature 514: 450–454, 2014. doi: 10.1038/nature13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arumugam P, Suzuki T, Shima K, McCarthy C, Sallese A, Wessendarp M, Ma Y, Meyer J, Black D, Chalk C, Carey BC, Lachmann N, Moritz T, Trapnell BC. Long-term safety and efficacy of gene-pulmonary macrophage transplantation therapy of PAP in Csf2ra−/− mice. Mol Ther 27: 1597–1611, 2019. doi: 10.1016/j.ymthe.2019.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiu CY, Su SC, Fan WL, Lai SH, Tsai MH, Chen SH, Wong KS, Chung WH. Whole-genome sequencing of a family with hereditary pulmonary alveolar proteinosis identifies a rare structural variant involving CSF2RA/CRLF2/IL3RA gene disruption. Sci Rep 7: 43469, 2017. doi: 10.1038/srep43469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scott MA, Hu YC. Generation of CRISPR-edited rodents using a piezo-driven zygote injection technique. Methods Mol Biol 1874: 169–178, 2019. doi: 10.1007/978-1-4939-8831-0_9. [DOI] [PubMed] [Google Scholar]
- 14.Sallese A, Suzuki T, McCarthy C, Bridges J, Filuta A, Arumugam P, Shima K, Ma Y, Wessendarp M, Black D, Chalk C, Carey B, Trapnell BC. Targeting cholesterol homeostasis in lung diseases. Sci Rep 7: 10211, 2017. doi: 10.1038/s41598-017-10879-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mui AL, Wakao H, O'Farrell AM, Harada N, Miyajima A. Interleukin-3, granulocyte-macrophage colony stimulating factor and interleukin-5 transduce signals through two STAT5 homologs. EMBO J 14: 1166–1175, 1995. doi: 10.1002/j.1460-2075.1995.tb07100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, Dickersin GR, Bachurski CJ, Mark EL, Whitsett JA, Mulligan RC. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science 264: 713–716, 1994. doi: 10.1126/science.8171324. [DOI] [PubMed] [Google Scholar]
- 17.Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 15: 557–567, 2001. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- 18.Berclaz PY, Zsengellér Z, Shibata Y, Otake K, Strasbaugh S, Whitsett JA, Trapnell BC. Endocytic internalization of adenovirus, nonspecific phagocytosis, and cytoskeletal organization are coordinately regulated in alveolar macrophages by GM-CSF and PU.1. J Immunol 169: 6332–6342, 2002. doi: 10.4049/jimmunol.169.11.6332. [DOI] [PubMed] [Google Scholar]
- 19.Huffman Reed JA, Rice WR, Zsengellér ZK, Wert SE, Dranoff G, Whitsett JA. GM-CSF enhances lung growth and causes alveolar type II epithelial cell hyperplasia in transgenic mice. Am J Physiol Lung Cell Mol Physiol 273: L715–L725, 1997. doi: 10.1152/ajplung.1997.273.4.L715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.