

Abstract
There is an urgent need to understand how SARS-CoV-2 infects the airway epithelium and in a subset of individuals leads to severe illness or death. Induced pluripotent stem cells (iPSCs) provide a near limitless supply of human cells that can be differentiated into cell types of interest, including airway epithelium, for disease modeling. We present a human iPSC-derived airway epithelial platform, composed of the major airway epithelial cell types, that is permissive to SARS-CoV-2 infection. Subsets of iPSC-airway cells express the SARS-CoV-2 entry factors angiotensin-converting enzyme 2 (ACE2), and transmembrane protease serine 2 (TMPRSS2). Multiciliated cells are the primary initial target of SARS-CoV-2 infection. On infection with SARS-CoV-2, iPSC-airway cells generate robust interferon and inflammatory responses, and treatment with remdesivir or camostat mesylate causes a decrease in viral propagation and entry, respectively. In conclusion, iPSC-derived airway cells provide a physiologically relevant in vitro model system to interrogate the pathogenesis of, and develop treatment strategies for, COVID-19 pneumonia.
Keywords: airway epithelial cell, COVID-19, human induced pluripotent stem cells, lung, SARS-CoV-2
INTRODUCTION
Infection with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a zoonotic positive sense RNA virus, has been a major source of worldwide morbidity and mortality since its emergence, with current analyses indicating that it was the third leading cause of death in the United States in 2020 (1). The major morbidity and mortality from SARS-CoV-2 result from infection of the respiratory system causing COVID-19 pneumonia. Although respiratory failure and acute respiratory distress syndrome (ARDS) develop as consequences of infection involving the gas-exchanging alveolar compartment of the lung, the initial infection involves the nasal, and subsequently, airway epithelium (2, 3). Viral entry into cells is achieved by binding of the SARS-CoV-2 spike (S) glycoprotein to the human angiotensin-converting enzyme (ACE2) receptor and subsequent processing of the S protein by proteases, including transmembrane protease, serine 2 (TMPRSS2) (4, 5). ACE2 expression levels decrease along the respiratory tract with the highest expression in the nose and lowest expression in the distal lung (3). This gradient suggests a paradigm of initial infection in the susceptible nasal epithelium with subsequent propagation to the ACE2-expressing cells of the airways and alveoli (3). Although it is clear that the airway epithelium is a major target of SARS-CoV-2 (3, 5–7), the mechanism by which the airway responds to infection, triggers an immune response, and leads to a wide variation of disease severity is unclear and of major interest. A comprehensive understanding of the pathogenesis of SARS-CoV-2 in the airways is necessary to advance prognostic tools and therapeutics to combat COVID-19.
To enhance our understanding of SARS-CoV-2 infection in the airways, in vitro models that are permissive to SARS-CoV-2 infection and recapitulate the pathology of COVID-19 respiratory tract infection are required (8). SARS-CoV-2 can replicate in cell lines such as Calu-3 (6) and Caco-2 cells (7). However, these cancer cell lines have lost much of their tissue-specific cell programs (9). Vero and Vero E6 cells, immortalized kidney cell lines derived from an African green monkey, are widely used in SARS-CoV-2 research, since they are highly infectible by SARS-CoV-2 (10), amenable to high-throughput approaches, and were used in the original studies that identified ACE2 as the receptor for SARS-CoV-1 (11). However, the responses of these cell lines may not be representative of human or lung epithelial responses to infection (9). Human primary bronchial epithelial cells (HBECs) can be differentiated in well-established protocols into a mucociliary epithelium in an air-liquid interface (ALI) culture format that is considered the gold standard in vitro model of human airway epithelial biology (12). Multiple studies have demonstrated the permissiveness of primary HBECs to SARS-CoV-2 infection and documented an epithelial immune response (12–14). Relevant to future SARS-CoV-2 studies, there are aspects of HBECs that are limiting including 1) limited access to human tissues from diverse populations/diseases, 2) finite in vitro life span and differentiation capacity over time, and 3) limited success with gene-editing approaches to interrogate the role of specific genes or variants of interest (15, 16). Therefore, access to an additional physiologically relevant, renewable source of human airway epithelial cells that are permissive to SARS-CoV-2 infection has the potential to facilitate mechanistic studies into SARS-CoV-2 pathogenesis and drug development strategies.
Induced pluripotent stem cells (iPSCs) have several features relevant to COVID-19 disease modeling including 1) the near-limitless supply of cells, 2) their retention of the genetic profile of the individual from which they were generated, 3) the capacity to differentiate under appropriate conditions into tissue-specific cell types relevant to SARS-CoV-2 infection, including both proximal and distal lung epithelia, and 4) amenability to gene-editing (17–20). Through the in vitro recapitulation of developmental milestones via directed differentiation, we and others have generated iPSC-derived airway and alveolar epithelial cells (21–37). We recently demonstrated the feasibility of using iPSC-derived alveolar epithelial type 2 cells (iAT2s) to model SARS-CoV-2 infection (9, 38, 39). iAT2s were permissive to infection with SARS-CoV-2, which induced a rapid inflammatory response characterized by secretion of nuclear factor kappa B (NF-κB)-induced cytokines and moderate, delayed type I and III interferon responses. Treatment of infected iAT2s with remdesivir, an inhibitor of the SARS-CoV-2 RNA-dependent RNA polymerase, resulted in a decrease in viral replication, demonstrating the potential of the iPSC platform for drug testing (38). In terms of the initial target of SARS-CoV-2 infection, we recently developed methods to direct the differentiation of human iPSCs into airway basal cells (iBCs) (40). Basal cells are the major stem cells of the airway epithelium (41), and iBCs are molecularly similar to their endogenous counterparts and express canonical basal cell markers including the transcription factors tumor protein P63 (TP63) and NK2 homeobox 1 (NKX2-1) and the surface receptor nerve growth factor receptor (NGFR) (40). iBCs are functionally similar to primary basal cells based on their multilineage differentiation capacity in ALI cultures, forming pseudostratified epithelia composed of basal, secretory, and multiciliated cells. These iPSC-derived airway ALI cultures (iPSC airway) are similar in morphology and composition to primary HBEC-derived ALI cultures (40).
Here, we report the successful infection of iPSC airway by SARS-CoV-2 and characterization of the resulting epithelial response. We demonstrate that multiple cell types in our iPSC-airway model express the viral entry factors ACE2 and TMPRSS2, and we find that multiciliated cells are the initial primary targets of infection. Following infection, we identified robust type I and type III interferon responses that contrast with our findings in SARS-CoV-2 infection of iAT2 cells (38). In addition, we observed an inflammatory response implicating signaling via the NF-κB pathway. Finally, treatment with remdesivir and camostat mesylate caused a decrease in virus in our platform, demonstrating the feasibility of the platform for drug screening assays.
METHODS
Experimental Model and Subject Details
Maintenance of human iPSCs.
All experiments involving the differentiation of human pluripotent stem cell (PSC) lines were performed with the approval of the Institutional Review Board of Boston University (protocol H33122). The two iPSC lines, 1) BU3 NGPT, iPSC line carrying NKX2-1-GFP and TP63-tdTomato reporter and 2) 1566, a cystic fibrosis transmembrane conductance regulator (CFTR)-corrected line of a homozygous F508del CF iPSC, were previously described (40). BU3 NGPT was derived from the published single reporter, NKX2-1 GFP, iPSC line (BU3 NG), a normal donor iPSC carrying homozygous NKX2-1 GFP reporters (21). The BU3 NG line was then targeted and integrated with a TP63tdTomato fluorescent reporter using CRISPR/Cas9 technology. The homozygous F508del iPSC was generated from peripheral blood mononuclear cells at Boston Children’s Hospital Stem Cell Program, and monoallelic correction was performed by nucleofecting the cells with Cas9-GFP plasmid and plasmid containing the normal CFTR sequence (hereinafter abbreviated as wild-type, WT) that includes the exon encoding F508. Repaired clone 1566 (CFTR WT/F508del) was identified by polymerase chain reaction and Sanger sequencing. All iPSC lines used in this study displayed a normal karyotype when analyzed by G-banding (Cell Line Genetics). All iPSC lines were maintained in feeder-free conditions, on hESC-qualified Matrigel (Corning) in 6-well tissue culture dishes (Corning), in StemFlex (Gibco), or mTeSR1 medium (StemCell Technologies) using gentle cell dissociation reagent or ReLeSR (StemCell Technologies) for passaging.
Method Details
Directed differentiation of human into iPSCs airway epithelium via iBCs.
The protocol involves the stepwise-directed differentiation of iPSCs to airway as published previously (27, 40). In brief, it recapitulates the major stages of embryonic lung development as follows: Stage 1, definitive endoderm (D0–3) using STEMdiff Definitive Endoderm Kit. Stage 2, anterior foregut endoderm (D3–6) through transforming growth factor beta (TGFβ) and bone morphogenetic protein (BMP) inhibition. Stage 3, lung specification (D6–15) using a combination of CHIR 99021 (to activate WNT signaling), BMP4, and retinoic acid and evidenced by the expression of NKX2-1. We then purify these NKX2-1+ lung epithelial progenitors at D15 through either sorting on GFP (BU3 NGPT) or for nonreporter line, utilizing a CD47hi/CD26neg sorting strategy (21). Stage 4, early airway organoids (D15–30), where GFP+ (BU3 NGPT) or CD47hi/CD26neg-sorted cells (1566) are plated in growth-factor reduced Matrigel and cultured in media containing fibroblast growth factor 2 (FGF2) and fibroblast growth factor 10 (FGF10) for patterning toward proximal airway-like organoids composed of immature secretory and basal progenitors (21, 28). Stage 5, iBCs (D30–45), during which the iPSC-3D organoids are further expanded in dual-SMAD inhibition media with DMH-1 (inhibits BMP4-pSMAD 1/5/8) and A83-01 (inhibits TGFβ-pSMAD 2/3) for ∼12 days (42, 43). iBCs were either serially passaged or cryopreserved, thawed, and then serially passaged to generate cells for Stage 6. Stage 6, airway differentiation on ALI culture (>D45), where iBCs are identified and purified by sorting on NGFR+ cells, and purified cells are expanded and differentiated on Transwells in dual-SMAD inhibition media then transitioned to ALI media when >80% confluent. Apical chamber medium is removed to initiate ALI to recapitulate a physiologically relevant environment and stimulate differentiation. After 14 days, iBCs form a pseudostratified airway epithelium that displays morphologic, molecular, and functional similarities to primary human airway epithelial cells and comprises the major airway cell types of multiciliated, secretory, and basal cells (40).
Reanalysis of previously published single-cell RNA-seq data sets for viral entry factors.
Viral entry factors ACE2 and TMPRSS2 were assessed on scRNA-seq data of 1) BU3 NGPT and HBECs that were previously published (40) and 2) freshly isolated, uncultured lung epithelia (44).
For each of the three single cell data sets, previous analyses had performed dimensionality reduction using principal component analysis (PCA) and uniform manifold approximation and projection (UMAP) and clusters had been assigned using the Louvain algorithm. We annotated the clusters for each single-cell data set using known markers of secretory, ciliated, and basal cells. Following this annotation, the three data sets were merged. When the data sets were merged, in addition to regressing out mitochondrial content during our normalization procedure (SCTransform) (45), we also regressed out the “library” batch effect. We proceeded to compare the different cell types between the data sets using genes of interest (ACE2, TMPRSS2, etc.) and violin plots. Markers for each data set were computed using Motif Alignment and Search Tool (MAST) (46) and direct comparisons were made between each of the data sets to quantitatively assess the differences in expression between key genes (ACE2, TMPRSS2, etc). All single-cell visualizations were made with Seurat (heatmaps, UMAPs, violin plots) (47, 48). Positive cells were identified using a unique molecular identifier (UMI) threshold of 0 counts, so that any cell with 1 UMI or more for a specific gene was considered positive for that gene.
SARS-CoV-2 propagation.
SARS-CoV-2 stocks [isolate USA_WA1/2020, kindly provided by CDC’s Principal Investigator Natalie Thornburg and the World Reference Center for Emerging Viruses and Arboviruses (WRCEVA)] were grown in Vero E6 cells (ATCC CRL-1586) cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2% fetal calf serum (FCS), penicillin (50 U/mL), and streptomycin (50 mg/mL) and titrated as described previously (38). To remove confounding cytokines and other factors, viral stocks were purified by ultracentrifugation through a 20% sucrose cushion at 80,000 g for 2 h at 4°C as previously published (38). SARS-CoV-2 titer was determined in Vero E6 cells by tissue culture infectious dose 50 (TCID50) assay. All work with SARS-CoV-2 was performed in the biosafety level 4 (BSL-4) facility of the National Emerging Infectious Diseases Laboratories at Boston University following approved SOPs.
SARS-CoV-2 infection of iPSC airway in ALI culture.
Prior to infection, the apical side of iPSC airway plated in ALI culture was washed with 100 μL of PBS for 5–10 min at 37°C to remove the mucus. Then, purified SARS-CoV-2 stock (100 μL of inoculum prepared in PBS) or PBS without virus (mock infection) was added to the apical chambers of each Transwell at the indicated multiplicity of infection (MOI) and allowed to adsorb for 1 h at 37°C and 5% CO2. After adsorption, the inoculum was removed and cells were incubated at ALI at 37°C for 1–7 days. Basolateral chamber medium was changed every 2–3 days. To ensure that the levels of N-transcript were due to infected epithelial cells and not virions on the surface of the epithelium, we compared levels of N transcript at 1 day postinfection (dpi) to day 0 cultures, which had been exposed to SARS-CoV-2 for ∼10 min and saw more N in 1 dpi samples for both BU3 NGPT and 1566 (Fig. S2B). At the time of harvest, basolateral medium was collected for further analysis. Apical washes were performed by adding 100 μL medium to the apical chamber and incubating for 15 min at 37°C before collection for further analysis. Both the apical washes and basolateral media were used for viral titration and Luminex assays as described below. For immunofluorescent analysis or electron microscopy, cells were fixed in 10% formalin. For RT-qPCR and RNA-seq analysis, cells were harvested directly in TRIzol (ThermoFisher).
Cell viability assay.
For determining cell viability, iPSC airways cultured at ALI were detached by adding 0.2 mL accutase apically and 0.5 mL basolaterally and incubated at 37°C for 15 min. Detached cells were pelleted by low-speed centrifugation, resuspended in PBS, diluted 1:1 in trypan blue, and analyzed using a LUNA-II Automated Cell Counter (Logos Biosystems).
Immunofluorescence microscopy of iPSC airway.
SARS-CoV-2-infected or mock-infected cultures on Transwell inserts were fixed in 10% formalin for 6 h, washed twice in PBS (10 min each, room temperature), permeabilized with PBS containing 0.25% Triton X-100 and 2.5% normal donkey serum (30 min, room temperature), and blocked with PBS containing 2.5% normal donkey serum (20 min, room temperature). Subsequently, cells were incubated with primary antibody diluted in 4% normal donkey serum overnight at 4°C. The antibodies used were anti-SARS-CoV nucleoprotein (N) antibody (rabbit polyclonal, 1:2,500, Rockland Immunochemicals, Cat. No. 200-401-A50), anti-α-TUBULIN antibody (mouse monoclonal, 1:500 Sigma, Cat. No.T6199), anti-Mucin5B (MUC5B) antibody (mouse monoclonal, Santa Cruz Biotechnology, 1:500, Cat. No. SC-39395-2), and anti-secretoglobin family 1A member 1 (SCGB1A1) (goat polyclonal, 1:500 Millipore Sigma, Cat. No. ABS1673). Next, cells were washed with PBS three times (5 min, room temperature) and incubated with secondary antibody [AlexaFluor 546 AffiniPure Donkey Anti-mouse IgG (H + L), 1:500, and AlexFluor 647 donkey anti-mouse IgG (H + L) 1:500, Jackson ImmunoResearch] for 2 h at room temperature. Cells were washed with PBS three times (5 min, room temperature), incubated with DAPI (1:5,000, Life Technologies) or Hoescht 33342 (1:500, Life Technologies) for 5 min, and washed again. Transwell inserts were then excised with a razorblade and mounted with Prolong Diamond Mounting Reagent or Prolong Gold Anti-fade reagent (Life Technologies). Slides were imaged with either upright fluorescence microscope (Nikon Eclipse Ni-E) or confocal microscope (Leica SP5). Manual quantification of N+ cells were performed by distributing 10–15 images of iPSC airway infected with SARS-CoV-2 stained with DAPI and N (>200 cells/image) to 5 blinded scorers. Each image analyzed using ImageJ and DAPI+ or Hoescht 33342 nuclei and N+ cells were quantified with the multipoint tool. To determine cellular tropism, Z-stack images of the infected Transwells stained with either anti-SARS-CoV-2 N and α-TUBULIN antibodies or anti-SARS-CoV-2 N and anti-MUCIN 5B antibodies were taken on Nikon Eclipse NiE. For quantification of colocalization of N+ cells with airway cell types, at least 100 N+ cells were counted, and percentage of colocalization with α-TUBULIN, MUCIN 5B, and SCGB1A1 was determined.
ACE2 immunohistochemistry.
For ACE2 immunohistochemistry, iPSC airways on Transwells were fixed in 4% PFA for 30 min, washed three times with PBS (5 min each, room temperature), then dehydrated in 50% ethanol (15 min), 70% ethanol (15 min), 85% ethanol (15 min), 95% ethanol (15 min), 100% ethanol (three times, 15 min each), and Histoclear (Biocare Medical, 3 times 15 min each), and incubated in paraffin (3 changes 30 min each) at 60°C. The filter was excised from the Transwells, then cut in half, and embedded in paraffin. The embedded filters were sectioned on a Leica RM 2135 microtome at a thickness of 7 µM. Sections were deparaffinized, blocked with Inhibitor CM and antigen retrieval was performed with Cell Conditioner 1 (CC1). Anti-ACE2 (Abcam, Cat. No. ab108252) was applied and was visualized with anti-rabbit HQ and anti-HQ-HRP followed by ChromoMap DAB (Roche). Samples were counter stained with hematoxylin, rinsed with detergent, dehydrated, and coverslipped with permanent mounting media. For MUCIN costaining, before the incubation of the second primary ab, the samples were subjected to a heat denature step with Cell Conditioner 2 (CC2). Anti-Mucin 5AC (MUC5AC) (Abcam, Cat. No. ab198254) and anti-MUC5B (Abcam, Cat. No. ab87376) was applied and visualized with anti-rabbit NP (Roche) and anti-NP-AP (Roche) and detected with Discovery Yellow (Roche); counter stained with hematoxylin, rinsed with detergent, dehydrated, and cover-slipped with permanent mounting media. For tubulin costaining, sections were deparaffinized and blocked with Inhibitor CM and antigen retrieval was performed with CC1. Anti-ACE2 (Abcam, Cat. No. ab108252) was applied and was visualized with anti-rabbit HQ and anti-HQ-HRP followed by ChromoMap DAB (Roche). Prior to the incubation of the second primary ab, the samples were subjected to a heat denature step with CC2. Anti-α Tubulin (Abcam, Cat. No. ab24610) was applied and visualized with anti-mouse OmniMap-HRP (Roche) and detected with Discovery Purple (Roche) counter stained with hematoxylin, rinsed with detergent, dehydrated, and a coverslip placed with permanent mounting media.
Reverse transcriptase quantitative PCR.
RNA was extracted from TRIzol samples following the manufacturer’s protocol. Purified RNA was reverse transcribed into cDNA using the MultiScribe Reverse Transcriptase (Applied Biosystems). All qPCR was performed in 384-well plates and run for 40 cycles using an Applied Biosystems QuantStudio 384-well system. Predesigned TaqMan probes were from Applied Biosystems or Integrated DNA Technologies (IDT). Relative gene expression was calculated based on the average Ct value for technical triplicates and normalized to 18S control, and fold-change over mock-infected cells was calculated using 2−ΔΔCt. If probes were undetected, they were assigned a Ct value of 40 to allow for fold-change calculations, and replicates, as indicated in each figure legend, were run for statistical analyses. A replicate of the RT-qPCR is defined as an individual Transwell of airway epithelial cells generated from sorted NGFR+ cells from Stage 6 of the differentiation protocol.
Transmission electron microscopy.
iPSC airway (1566) on Transwell inserts were infected with SARS-CoV-2 at an MOI of 4 or mock-infected. At 1 dpi, cells were fixed in Karnovsky’s fixative (Tousimis) for 18 h at 4°C and washed with PBS. The membrane was excised from the Transwell, block stained in 1.5% uranyl acetate (Electron Microscopy Sciences, EMS) for 1 h at room temperature (RT). The samples were dehydrated quickly through acetone on ice, from 70% to 80% to 90%. The samples were then incubated 2 times in 100% acetone at RT for 10 min each, and in propylene oxide at RT for 15 min each. Finally, the samples were changed into EMbed 812 (EMS), left for 2 h at RT, changed into fresh EMbed 812, and left overnight at RT, after which they were embedded in fresh EMbed 812 and polymerized overnight at 60°C. Embedded samples were thin sectioned (70 nm) and grids were stained in 4% aqueous uranyl acetate for 10 min at RT followed by lead citrate for 10 min at RT. Electron microscopy was performed on a Philips CM12 EM operated at 100 kV, and images were recorded on a TVIPS F216 CMOS camera with a pixel size of 0.85–3.80 nm per pixel.
Drug efficacy testing in iPSC-airway cells.
iPSC airway plated in ALI culture were pretreated apically (100 μL) and basolaterally (600 μL) with the indicated concentrations of camostat mesylate (Tocris, Cat. No. 3193), remdesivir (Selleckchem, Cat. No. S8932), or DMSO control for 1 h at 37°C. After 1 h, all apical media were aspirated and SARS-CoV-2 (MOI 0.04) was added for 1 h without any drugs apically, after which the inoculum was removed, and samples were returned to 37°C in ALI culture format. iPSC airways were exposed to the compounds basolaterally for the entire duration of the experiment. Cells were harvested in TRIzol 2 dpi and processed for RT-qPCR.
Immune stimulation with poly(I:C) and IFNβ.
For immune stimulation treatments, iPSC airways cultured at ALI were treated with the TLR3 agonist poly(I:C) (InvivoGen) delivered with Oligofectamine Transfection Reagent (Invitrogen) or treated with recombinant human IFNβ (rhIFNβ; PeproTech). Prior to treatment, 1 μL poly(I:C) was mixed with 2.5 μL Oligofectamine and incubated at RT for 15 min. After the incubation period, the poly(I:C) and Oligofectamine mixture was added to 100 μL of ALI differentiation media. iPSC airways cultured at ALI were treated apically (100 μL) with poly(I:C) (10 μg/mL) and Oligofectamine, or apically and basolaterally (600 μL) with IFNβ (10 ng/mL) for 24 h at 37°C. Cells were subsequently harvested in Qiazol for RT-qPCR.
SARS-CoV-2 titration.
SARS-CoV-2 titers were determined in Vero E6 cells by tissue culture infectious dose 50 (TCID50) assay. Basolateral shedding of the virus was determined by directly assaying the basolateral media (600 μL). Apical shedding of virus was determined by assaying apical washes (100 μL) of the infected cells.
Luminex analysis.
Apical washes (100 μL) and basolateral media (600 μL) samples were clarified by centrifugation and analyzed using the Magnetic Luminex Human Discovery Assay (R&D Systems). Custom configured targets include CCL2/JE/MCP-1, CXCL-9/MIG, CXCL10/IP-10/CRG-2, GM-CSF, IFNβ, IL-6, TNFα, and TRAIL/TNFSF10. Mean fluorescence intensity was measured to calculate final concentration in pg/mL using Bioplex200 and Bioplex Manager 5 software (Bio-Rad).
RNA sequencing and bioinformatic analyses.
For bulk RNA sequencing (RNA-Seq), biological triplicate (n = 3) samples of purified RNA extracts were harvested from each group of samples prepared as follows. After 87 days of total time in the directed differentiation protocol, iPSC airways cultured as serially passaged 3 D spheres were sorted and single-cell passaged onto Transwell inserts. Apical medium was removed on day 8 to initiate ALI culture. On day 24 (after removing the apical media), six replicate wells of iPSC airways were infected with SARS-CoV-2 from the apical surface and six replicate wells were mock-infected. Three wells per condition were harvested at each 1 and 3 dpi in TRIzol. Following total RNA isolation from the TRIzol samples, mRNA was isolated from each sample using magnetic bead-based poly(A) selection, followed by cDNA synthesis. The products were end-paired and PCR-amplified to create each final cDNA library. Sequencing of pooled libraries was done using a NextSeq 500 (Illumina). The quality of the raw data was assessed using FastQC v.0.11.7 (49).The sequence reads were aligned to a combination of the human genome reference (GRCh38) and the SARS-CoV-2 reference (NC_045512) using STAR v.2.6.0c (50). Counts per gene were summarized using the featureCounts function from the subread package v.1.6.2 (51). The edgeR package v.3.25.10 (52) was used to import, organize, filter, and normalize the counts. The matrix of log counts per million was then analyzed using the limma/voom normalization method (53). Genes were filtered based on the standard edgeR filtration method using the default parameters for the “filterByExpr” function. After exploratory data analysis with principal component analysis (PCA), contrasts for differential expression testing were done for each SARS-CoV-2-infected sample versus mock (controls) at each time point (days post infection). Differential expression testing was also conducted to compare the gene expression between the two infected time points and to investigate the time-specific effects in response to infection. The limma package v.3.42.2 (53) with its voom method, namely, linear modeling and empirical Bayes moderation was used to test differential expression (moderate t test). P values were adjusted for multiple testing using Benjamini-Hochberg correction [false-discovery rate (FDR)-adjusted P value]. Differentially expressed genes for each comparison were visualized using Glimma v. 1.11.1 (54) and FDR < 0.05 was set as the threshold for determining significant differential gene expression. Functional predictions were performed using the fgsea v.1.12.0 package for gene set analysis (55).
Quantification and Statistical Analysis
Statistical analyses were performed using GraphPad Prism 8, ggplot2 R package. Statistical significance was determined as P value of <0.05 using Student’s t test (*P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001).
RESULTS
Human iPSC Airway Expresses SARS-CoV-2 Entry Factors
We previously generated from a healthy donor a bifluorescent reporter iPSC line that carries a tdTomato coding sequence targeted to the TP63 locus and a GFP sequence targeted to the NKX2-1 locus (21, 40). When differentiated using a previously published six-stage protocol (Fig. 1A) (40), this BU3 NKX2-1GFP;TP63tdTomato dual reporter line (hereafter BU3 NGPT) allows the sequential identification, and purification via flow cytometry, of lung progenitors (NKX2-1GFP+), airway progenitors (NKX2-1GFP+/TP63tdTomato+), and finally basal cells (NKX2-1GFP+/TP63tdTomato+/NGFR+; Supplemental Fig. S1, A–C; see https://doi.org/10.6084/m9.figshare.16695133). As an alternative to the reporter-based approach, we also developed a surface marker-based strategy in which NKX2-1+ lung progenitors are identified using CD47hi/CD26neg sorting (21) followed by NGFR+ iBC purification. Following these established differentiation protocols, we differentiated BU3 NGPT and a previously described human iPSC line (hereafter “1566”) using reporter-based sorting or surface marker-based sorting, respectively (Supplemental Fig. S1, D–F) (40). Plating of purified iBCs from both iPSC lineages to a Transwell ALI culture format (removing media from the apical chamber and exposing the cells to air) resulted in their differentiation to a mucociliary airway epithelium (Fig. 1, B and C).
Figure 1.
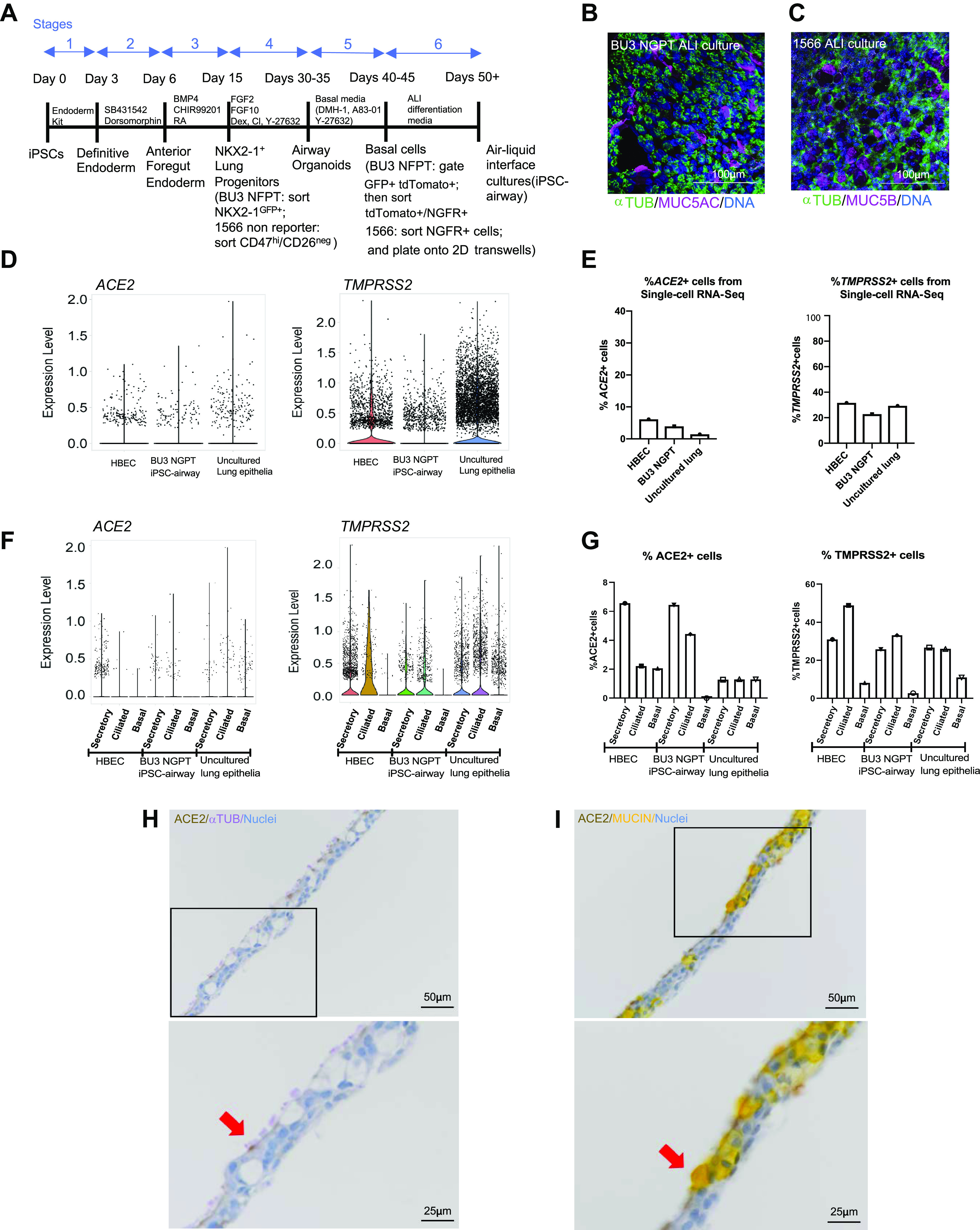
iPSC-derived airway cells express SARS-CoV-2 entry factors angiotensin converting enzyme 2 (ACE2), and transmembrane serine protease 2 (TMPRSS2). A: schematic of the six-stage differentiation protocol of generating iPSC airway. B: immunofluorescence analysis of BU3 NGPT iPSC airway stained with anti-α-TUBULIN and mucin 5AC (MUC5AC) (scale bar = 100 µm). Nuclei are stained with HOECHST (blue). C: immunofluorescence analysis of 1566 iPSC airway, stained with anti-α-TUBULIN and mucin 5B (MUC5B) (scale bar = 100 µm). Nuclei are stained with DAPI (blue). D–G: scRNA-seq analysis of human bronchial epithelial cells (HBEC)40, iPSC airway (BU3 NGPT) (40), and freshly isolated uncultured lung epithelia (42). D: violin plots of ACE2 and TMPRSS2 expression. E: percentage of ACE2- and TMPRSS2-positive cells in each data set (40). F: violin plots of ACE2 and TMPRSS2 expression by cellular type in each data set. G: comparison of the percentage of ACE2- and TMPRSS2-positive secretory, multiciliated, and basal cells in each data set. H and I: immunohistochemistry staining showing the localization of ACE2 (DAB), α-TUBULIN (purple, left) and MUCIN (yellow, right) in iPSC-derived airway (BU3 NGPT) counterstained with hematoxylin (×20, scale bar = 50 µm). Bottom: zoomed-in images of the black box in the upper panels. The red arrows indicated cells coexpressing ACE2/α-TUBULIN (left) and ACE2/MUCIN (right; scale bar = 25 µm). ACE2, human angiotensin-converting enzyme; ALI, air-liquid interface; HBECs, human primary bronchial epithelial cells; iPSC, induced pluripotent stem cell; TMPRSS2, transmembrane protease, serine 2.
To interrogate the feasibility of applying the iPSC-airway system to model SARS-CoV-2 infection of the airway, we first examined the expression levels of SARS-CoV-2 entry factors ACE2 and TMPRSS2. To do so, we compared previously published single-cell RNA-sequencing (scRNA-seq) data sets: airway epithelium derived from BU3 NGPT iBCs ALI cultures (BU3 NGPT iPSC airway) (40) and primary HBECs differentiated in ALI cultures from a healthy donor (40). We next integrated these data sets with a recently published scRNA-seq data set of freshly isolated (uncultured) lung epithelial tissue from six donors without underlying lung disease (44). Analyzing all airway epithelia in each of these samples, we identified subsets of cells expressing ACE2 and TMPRSS2 in BU3 NGPT iPSC airway, HBEC-derived ALI cultures (hereafter HBEC), and uncultured lung epithelia (Fig. 1D). We quantified the percentage of cells expressing ACE2 and TMPRSS2 transcripts: ACE2 was detected in 6.1% of HBEC, 3.9% of BU3 NGPT iPSC airway, and 1.4% of uncultured lung epithelia; TMPRSS2 was present in 31.6% of HBEC and 29.3% of uncultured lung epithelia, compared with 22.8% of BU3 NGPT iPSC airway (Fig. 1E).
Next, we examined the cell-type-specific expression of ACE2 and TMPRSS2 across these data sets (Fig. 1F). The annotated clusters and expression of canonical markers used to define secretory, multiciliated, and basal cells are shown in Supplemental Fig. S1G. The percentage of ACE2+ secretory cells were 6.6%, 6.5%, and 1.3% in HBEC, BU3 iPSC airway, and uncultured lung epithelia, respectively (Fig. 1G). The percentage of ACE2+ multiciliated cells were 2.2%, 4.4%, and 1.3%, and the percentage of ACE2+ basal cells were 2.1%, 0%, and 1.3% in HBEC, BU3 iPSC airway, and uncultured lung epithelia, respectively (Fig. 1G). For TMPRSS2, 31.0%, 25.8%, and 26.7% of secretory cells, 48.9%, 33.2%, and 26.0% of multiciliated cells, and 8.2%, 2.8%, and 11.0% of basal cells from HBEC, iPSC airway, and uncultured lung epithelia, respectively, were positive (Fig. 1G). Taken together, although there are apparent differences in ACE2 and TMPRSS2 expression when comparing in vitro platforms with in vivo, and iPSC airway to primary cells, in general, ACE2 is expressed in small subpopulations of cells across all three platforms. Furthermore, similar to a recent finding that cells that coexpress ACE2 and TMPRSS2 are enriched in pathways related to viral infection and immune response (56), we also found that the top enriched genes, ranked by Z-score, in iPSC-airway ACE2+ cells (vs. ACE2− cells) included interferon stimulated genes such as interferon alpha inducible protein 27 (IFI27), interferon induced protein with tetratricopeptide repeats 1 (IFIT1), radical S-adenosyl methionine domain containing 2 (RSAD2), interferon-stimulated gene 15 (ISG15), MX dynamin like GTPase 1 (MX1), interferon induced transmembrane protein 2 (IFITM2), interferon induced protein with tetratricopeptide repeats 3 (IFIT3) (Supplemental Table S1; see https://doi.org/10.6084/m9.figshare.16695130).
Finally, we examined ACE2 protein expression and localization in iPSC-airway cells (Fig. 1, H and I). In concordance with our scRNA-seq data, ACE2 was detected in subsets of α-TUBULIN+ multiciliated and MUCIN+ secretory cells and was apically localized (Fig. 1, H and I). Taken together, we demonstrate that SARS-CoV-2 entry factors ACE2 and TMPRSS2 are expressed in multiple lineages of iPSC-airway epithelial cells.
iPSC Airway Is Permissive to SARS-CoV-2 Infection
To determine whether iPSC airway is permissive to SARS-CoV-2 infection, iPSC-airway cultures from BU3 NGPT and 1566 were differentiated following the airway differentiation protocol as described above. After ∼21 days in ALI culture, the cells were infected from the apical surface with purified SARS-CoV-2 particles (Fig. 2A). Viral infection was detected by immunofluorescence analysis (IFA) using an antibody directed against the SARS-CoV nucleoprotein (N) with cross-reactivity against SARS-CoV-2 N protein (Fig. 2, B–G) and by RT-qPCR of the N transcript (Fig. 2H and Supplemental Fig. S2, A–D; see https://doi.org/10.6084/m9.figshare.17100023). We observed a dose-dependent increase in viral N transcript with increasing multiplicity of infection (Supplemental Fig. S2A). MOI of 4 was selected for the subsequent studies. At 1 day postinfection (dpi), an average of 6.87% (SE 0.548%) of BU3 NGPT iPSC-airway cells were N+ by IFA (Fig. 2, B and C). In 1566 iPSC airway infected with SARS-CoV-2, 11.21% (SE 1.075) of cells were N+ by IFA at 1 dpi (Fig. 2, D and E). RT-qPCR of both BU3 NGPT and 1566 iPSC airway confirmed the presence of N-transcript at 1 and 3 dpi (Fig. 2H and Supplemental Fig. S2D). In five separate infections of BU3 NGPT iPSC airway and three separate infections of 1566 iPSC airway, we detected an average N transcript at 1 dpi of 2.64 × 107 (fold-change over mock) and 2.44 × 107 (fold-change over mock), respectively (data not shown). We then followed the course of SARS-CoV-2 infection of iPSC airway over time. In both iPSC lines, peak infection was detected a 1 dpi and decreased ∼10-fold by 3 dpi (Fig. 2H and Supplemental Fig. S2, C and D). Cellular toxicity was suggested at 3 dpi by lower cell density, patches devoid of nuclei, an increase in trypan-blue-labeled nonviable cells, and an increase in fragmented nuclei (Fig. 2, G and I and Supplemental Fig. S2, E and H). To determine whether infectious virus was released from the infected iPSC airway epithelium, viral titers were performed on apical washes and basolateral medium from 1 and 3 dpi samples. Viral particles were mainly released form the apical surface (Fig. 2J), which is in line with the transmission route of SARS-CoV-2 and similar to findings reported using primary HBECs (57). We also observed increased shedding of infectious virus from both the apical and basolateral compartments from 1 to 3 dpi (Fig. 2J), whereas intracellular viral transcripts remained steady or decreased, similar to what has been observed during infection of primary HBEC ALI cultures (58).
Figure 2.
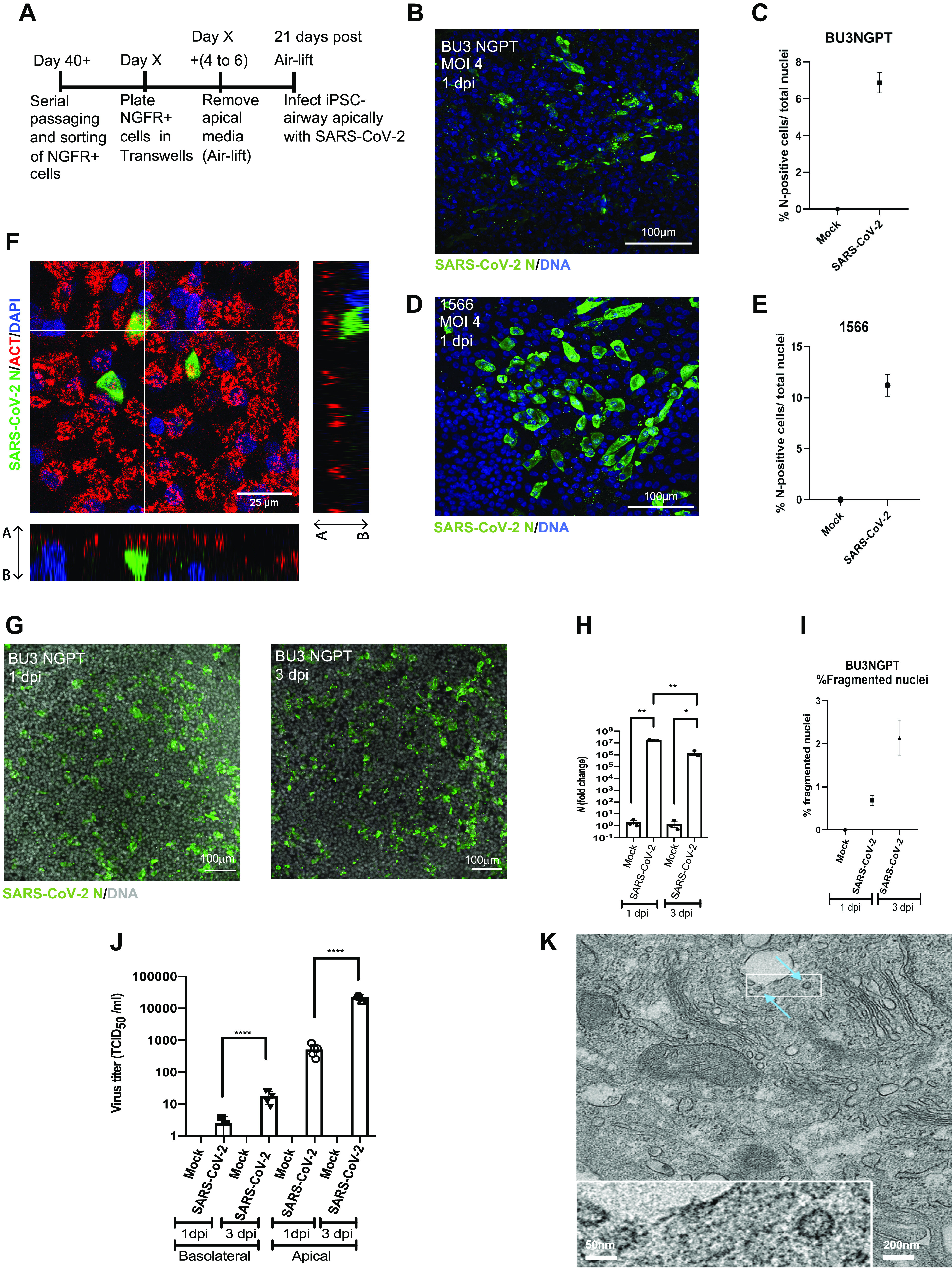
iPSC-derived airway is permissive to SARS-CoV-2 infection with time-dependent restriction in viral growth. A: schematic of the protocol to iPSC airway with SARS-CoV-2. B–E: immunofluorescence and quantification of viral nucleoprotein+ (SARS-CoV-2 N, green) cells in BU3 NGPT (B and C) and 1566 (D and E) cells at 1 dpi (×40, scale bar = 100 µm). BU3 NGPT mean nucleoprotein+ cells = 6.87 ± 0.548% (SE). 1566 mean nucleoprotein+ cells = 11.21 ± 1.075% (SE). F: confocal immunofluorescence microscopy of BU3 NGPT with antibodies against SARS-CoV-2 N positive (green) cells and α-TUBULIN (red). Nuclei are stained with DAPI (blue). G: immunofluorescence of infected iPSC airway (BU3 NGPT) at 1 and 3 dpi, labeled with anti-SARS-CoV-2 N antibody and nuclei labeled with DAPI (×20, scale bar = 100 µm). H: RT-qPCR of viral N gene expression of iPSC airway (BU3 NGPT) at 1 and 3 dpi (n = 3 Transwells per sample). Fold-change expression compared with mock (2−ddCt) after 18S normalization is shown. I: percentage of fragmented nuclei detected at 1 and 3 dpi in iPSC airway (BU3 NGPT) [1 dpi = mean 0.69 ± 0.12% (SE) and 3 dpi = mean 2.14 ± 0.32% (SE)]. J: viral titers from apical washes and basolateral media at 1 and 3 dpi from iPSC airway (BU3 NGPT) compared with mock. K: transmission electron micrograph of SARS-CoV-2-infected iPSC airway (1566) at 1 dpi. Blue arrows indicate viral particles (scale bar = 200 nm, 50 nm in enclosed box). Statistical significance was determined as P value of <0.05 using Student’s t test (*P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001). iPSC, induced pluripotent stem cell.
To study the cellular tropism of SARS-CoV-2 in our iPSC-airway system, we performed IFA with 1 dpi samples using antibodies directed against SARS-CoV nucleoprotein and canonical markers of multiciliated (alpha-tubulin, ACT), basal (TP63), or secretory cells (MUC5AC or SCGB1A1). By confocal microscopy, all cells that coexpressed a lineage marker and viral nucleoprotein were ACT+ multiciliated cells (Fig. 2F). We did not identify colocalization of viral nucleoprotein with MUC5AC+ (Supplemental Fig. S2F) or SCGB1A1+ (Supplemental Fig. S2G) secretory cells, or TP63+ basal cells (not shown). The observation that multiciliated cells are the predominant initial airway target cells is in agreement with previous studies in SARS-CoV-2 infected primary airway cultures (3, 59). Ultrastructural analysis of SARS-CoV-2-infected iPSC airway by transmission electron microscopy (TEM) revealed the presence of intracellular viral particles, confirming productive viral infection (Fig. 2K). Taken together, our findings indicate iPSC-airway cells from two different individuals are permissive to SARS-CoV-2 infection and multiciliated cells are the initial target cells. Though there may be increased virion release or potential extracellular accumulation on the apical surface of the infected cells at 3 dpi, the infection of the epithelium peaks before 3 dpi with declining intracellular viral presence over time, associated with cytotoxicity.
SARS-CoV-2 Infection Stimulates an Epithelial-Intrinsic Interferon and Inflammatory Response in iPSC Airway
Having demonstrated the permissiveness of the iPSC-airway system to SARS-CoV-2 infection, we next sought to assess the global transcriptomic response to infection. To do so, we performed bulk RNA-sequencing (RNA-Seq) of SARS-CoV-2-infected iPSC airway (BU3 NGPT) at 1 and 3 dpi, compared with time-matched mock-infected controls (n = 3 replicates at each time point; Fig. 3A). Principal component analysis (PCA) indicated that the predominant variance in global gene expression (PC1; 42.6% variance) was explained by the infection state of the cells, with less variance (PC2; 20.2% variance) explained by time in culture (Fig. 3B and Supplemental Table S2; see https://doi.org/10.6084/m9.figshare.16679614). For example, 529 genes and 5134 genes were differentially expressed at 1 and 3 dpi, respectively, compared with time-matched, mock-infected samples (FDR) <0.05). Comparing 3 to 1 dpi, 6,136 genes were differentially expressed, with 2,853 upregulated and 3,283 downregulated genes (Fig. 3B and Supplemental Table S2).
Figure 3.

Transcriptomic analysis of SARS-CoV-2-infected iPSC airway shows robust interferon response. A: schematic of the experimental design to compare SARS-CoV-2-infected iPSC airway samples (BU3 NGPT) at 1 and 3 dpi to mock controls by RNA-seq (n = 3 Transwells). B–I: analyses of this experiment. B: PCA comparing PC1 vs. PC2 of the samples described in A. C: the top 50 differentially expressed genes (DEGs) ranked by fold-change (FDR < 0.05) of 1 dpi vs. mock. D: the top 50 DEGs ranked by fold-change (FDR < 0.05) of 3 dpi vs. 1 dpi. E: volcano plots of differentially expressed genes in 1 dpi versus mock for iPSC airway. F: volcano plots of differentially expressed genes in 3 dpi versus 1 dpi for iPSC airway. G: local regression (LOESS) plots of viral, interferon and ISG, and inflammatory gene expression levels quantified by RNA-seq normalized expression (counts per million reads). H: gene set enrichment analysis (GSEA) of the top 10 upregulated gene sets in 1 dpi versus mock for iPSC airway (black color indicates statistical significance; Padj < 0.05). I: gene set enrichment analysis (GSEA) of the top 10 upregulated gene sets in 1 dpi versus 3 dpi iPSC airway (black color indicates statistical significance; Padj < 0.05). FDR, false discovery rate; iPSC, induced pluripotent stem cell; PCA, principal component analysis.
Given the large number of genes changing after infection, we analyzed the most robust transcriptomic changes by focusing on the top 50 differentially expressed genes (DEGs) ranked by log fold change (logFC) (and filtered by FDR < 0.05) at 1 dpi (vs. mock; Fig. 3C), 3 dpi (vs. mock; Supplemental Fig. 3S3A), and 3 dpi versus 1 dpi (Fig. 3D). We found that 9/50 (18%) of the top DEGs at 1 dpi were viral transcripts as expected (logFC; P < 0.05; Fig. 3, C and E and Supplemental Fig. S3A). At 1 dpi, genes associated with type I [including interferon beta 1 (IFNB1)], type III interferon [including interferon lambda 1 (IFNL1), interferon lambda 3 (IFNL3)], and downstream interferon-stimulated genes [ISGs; interferon induced protein with tetratricopeptide repeats 2 (IFIT2), MX dynamin-like GTPase 2 (MX2), MX dynamin-like GTPase 1 (MX1)] were upregulated (Fig. 3, C and E). Furthermore, genes encoding cytokines involved in the recruitment and differentiation of immune cells, including C-X-C motif chemokine ligand 9 (CXCL9), C-X-C motif chemokine ligand 10 (CXCL10), C-X-C motif chemokine ligand 11 (CXCL11) were also upregulated at 1 dpi compared with mock-infected samples (Fig. 3, C and E). From 1 to 3 dpi, there was a decrease in viral gene expression, including transcripts from N, M, E, and ORF3a, consistent with RT-qPCR N transcript profiles (Supplemental Table S1, FDR < 0.05). This was accompanied by further upregulation of type I and type III interferon genes (IFNB, IFNL1, interferon lambda 2 (IFNL2) and ISGs (interferon induced protein with tetratricopeptide repeats1B (IFIT1B), MX1), suggesting increasing interferon response over time (Fig. 3, D and F). Furthermore, there was an upregulation of inflammatory mediators (CXCL9, CXCL10, and CXCL11) and a modest increase in interleukin 6 (IL6) expression from 1 to 3 dpi (Fig. 3F).
The expression kinetics of viral transcripts, receptors, interferon genes, ISGs, and genes involved in the inflammatory response (including NF-κB and IL-6) from 1 to 3 dpi were also visualized using local regression (LOESS) plots (Fig. 3G and Supplemental Fig. S3E). In addition to upregulation of type I/III interferons and ISGs as described above, there was a time-dependent increase in viral sensors [DExD/H-Box Helicase 58 (DDX58), Toll-like receptor 3 (TLR3)] and adaptor molecule (myeloid differentiation primary response 88 (MYD88); Supplemental Fig. S3E). It was shown previously that ACE2 is an interferon-inducible gene (60), and we also found an increase in ACE2 at 3 dpi compared with 1 dpi (Supplemental Fig. S3E). Furthermore, we observed an increase in chemokines important for T- and NK-cell recruitment, including CXCL9, CXCL10, and CXCL11 from 1 to 3 dpi. Similarly, we also observed an increase in IL-23, which is implicated in the IL-17 pathway, and C-X-C motif chemokine ligand 8 (CXCL8), which is important for neutrophil migration (Supplemental Fig. S3E). C-C motif chemokine ligand 2 (CCL2), which is important for macrophage recruitment, was not upregulated (Supplemental Fig. S3E). Consistent with the findings that inflammation is a hallmark of SARS-CoV-2 infection (61–63), gene set enrichment analysis (GSEA) based on the DEGs at 1 dpi (vs. mock) suggested enrichment of the following pathways: “TNFA signaling via NF-κB”; “IL-2-STAT5 signaling”; “Inflammatory response”; and “Complement” (Fig. 3H). Furthermore, GSEA also showed enrichment of “interferon-γ” and “interferon-α” pathways, consistent with an interferon response. GSEA of DEGs between 3 dpi versus 1 dpi suggested enrichment for the following signaling pathways: “TNFA signaling via NF-κB”; “Complement”; “Interferon-γ”; and “Inflammatory response” (Fig. 3I).
To confirm the secretion of interferon and inflammatory mediators from iPSC airway infected with SARS-CoV-2, we performed Luminex assays using the basolateral supernatants and apical washes of mock-infected and SARS-CoV-2-infected iPSC airway (Fig. 4A). Consistent with the transcriptomic analysis, IFNβ secretion was increased modestly at 1 dpi in both the apical and basolateral chambers, and increased more drastically by 3 dpi in both chambers. The time-dependent increases in IL6, CXCL9, CXCL10, and TNFα transcripts were also validated by levels of secreted protein in basolateral media and apical washes (Fig. 4A). Though the lack of significant upregulation of CCL2 from 1 to 3 dpi was also confirmed by Luminex assay, there was significant upregulation of granulocyte-macrophage colony-stimulating factor (GM-CSF) in both apical and basolateral chambers. In addition, we observed an increase in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) secretion, suggesting the initiation of apoptosis (Fig. 4A). We used RT-qPCR to further validate the key RNA-seq transcriptomic changes in IFNB1, IFNL1, IFNL2, IFIT1, MX1, and IL6, confirming an initial modest but significant increase in expression in infected iPSC airway compared with mock at 1 dpi and a more robust increase by 3 dpi (Fig. 4B). Of note, the induction of interferon signaling was considerably lower in SARS-CoV-2-infected iPSC airway compared with recombinant IFNβ-treated iPSC airway (Supplemental Fig. S3F), and induction of IL-6 in infected iPSC airway was lower compared with poly(I:C)-treated iPSC airway (Supplemental Fig. S3F). This result is in line with multiple reports showing that SARS-CoV-2 blocks innate immune signaling (64, 65). In contrast to iPSC-airway cells, SARS-CoV-2-infected iPSC-derived alveolar type 2 cells (iAT2s) exhibited a delayed and dampened interferon response in our previously published studies (38).
Figure 4.

iPSC airway infected with SARS-CoV-2 secrete inflammatory cytokines and chemokines and detects antiviral drug effects. A: luminex analysis of apical washes and basolateral media collected from iPSC airway (BU3 NGPT, cultures; n = 3 Transwells). B: RT-qPCR of select genes iPSC airway (BU3 NGPT) infected with SARS-CoV-2 (MOI 4) and harvested at 1 and 3 dpi with their respective mock-infected samples (n = 3 Transwells). Fold-change expression compared with mock (2−ddCt) after 18S normalization is shown. C and D: RT-qPCR of N, IL6, IFNL2 gene expression of mock-infected and SARS-CoV-2-infected (MOI 0.04) iPSC airway (BU3 NGPT) at 2 dpi pretreated with vehicle control (DMSO) or remdesivir (10 μM; C) or vehicle control (DMSO) or camostat mesylate (TMPRSS2 inhibitor, 1, 10, 100 μM; D), as indicated (n = 3 Transwells for both C and D). Statistical significance was determined as P value of <0.05 using Student’s t test (*P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001). iPSC, induced pluripotent stem cell; MOI, multiplicity of infection.
We next analyzed the expression of markers of airway epithelial cells, apoptosis, and cell death in our RNA-seq data set. Markers of multiciliated cells [including forkhead box J1 (FOXJ1), tubulin alpha 1a (TUBA1A), dynein light chain LC8-type 1 (DYNLL1)] decreased significantly in the SARS-CoV-2-infected samples by 3 dpi, suggesting loss or perturbation of multiciliated cells. Analysis of secretory cell markers demonstrated stable expression of SCGB1A1 but a relative increase in MUC5B at 3 dpi (Supplemental Fig. S3E). Although some markers for cell death were not elevated [BLC2 associated agonist of cell death (BAD)], apoptosis markers [caspase 3 (CASP3), phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1), B-cell lymphoma 2 (BLC2)] and necrosis/necroptosis markers [receptor interacting serine/threonine kinase 3 (RIPK3)] were upregulated at 3 dpi compared with 1 dpi (Supplemental Fig. S3E).
To determine whether the transcriptional response of iPSC airway was recapitulated in a genetically distinct iPSC line, we performed bulk-sequencing of SARS-CoV-2-infected iPSC airway at 1 dpi versus mock-infected samples using the 1566 iPSC line. Similar to findings with BU3 NGPT iPSCs, there was evidence of type I and type III interferon response to viral infection by 1 dpi (Supplemental Fig. S3, C and D). GSEA of 1566-infected iPSC airway revealed almost identical pathway enrichment, including TNF-NF-κB, IFN-γ, IFN-α, IL-6/JAK/STAT3, and complement (Supplemental Fig. S3B).
Taken together, our results indicate that SARS-CoV-2-infected iPSC airways exhibit transcriptomic changes characterized by significant type I and type III interferon responses, loss of multiciliated cell markers, and a proinflammatory phenotype characterized by progressively increasing levels of NF-κB signaling. These data are in agreement with other studies in airway and alveolar cultures showing that SARS-CoV-2 infection induces a proinflammatory response (14, 38).
iPSC Airway Can Be Used as a Platform for Antiviral Drug Screening
Finally, to assess the feasibility of iPSC-airway platform to screen for COVID-19 therapeutic compounds, we tested the effect of the FDA-approved antiviral drug remdesivir and found that viral N transcripts were reduced ∼100-fold in remdesivir-treated cells (Fig. 4C), with a concurrent decrease in IL6 and IFNL2 (Fig. 4C). We also tested the effect of the TMPRSS2 inhibitor camostat mesylate, which significantly reduced the amount of viral N transcripts in a dose-dependent manner (Fig. 4D). Similarly, IL6 and IFNL2 were also decreased with increasing doses of camostat mesylate (Fig. 4D). This indicates that SARS-CoV-2 infection of iPSC airway relies on priming by the protease TMPRSS2, similar to infection in iAT2 cells (38).
DISCUSSION
We show here that human iPSC-airway cells derived from multiple individuals express viral entry factors ACE2 and TMPRSS2 are permissive to SARS-CoV-2 infection, and on infection, generate an interferon and inflammatory response. Furthermore, treatment with remdesivir or camostat mesylate causes a decrease in viral replication, demonstrating the feasibility of using this iPSC-airway platform for antiviral drug screening assays. To our knowledge, this is the most detailed assessment of the expression of profile of SARS-CoV-2 entry factors and epithelial response to SARS-CoV-2 infection using iPSC-derived airway cells.
The global burden of respiratory viruses is significant, suggesting the need for in vitro platforms that accurately recapitulate the biology of the human lung. The application of iPSC technology to study viral infections is expanding (66), with a recent focus on organs infected by SARS-CoV-2 (67). We previously described the application of iAT2s to study infection of the distal lung epithelium by SARS-CoV-2 (38). Interestingly, iAT2s were highly permissive to infection, with 20% of cells infected (MOI 5) at 1 dpi and 60% at 4 dpi (38). Despite similar culture and infection conditions, iPSC-airway cells were less permissive to infection with ∼6–12% infected cells at 1 dpi (MOI of 4), and a decrease in viral transcripts by 3 dpi. Consistent with prior studies in primary cells, we detected peak virus release from iPSC airway, preferentially from the apical surface, at 3 dpi (13, 57). However, by 3 dpi viral transcripts were decreasing within the epithelium, suggesting either a restriction of viral replication, death of infected cells, or both.
Our transcriptomic analysis revealed that decrease in SARS-CoV-2 transcripts in iPSC airways between 1 and 3 dpi was accompanied by a significant and progressive induction of both type I and III interferon responses, raising the question of whether the antiviral response in part accounts for the restriction of epithelial infection in our system. Though these results mirror a recent scRNA-seq analysis of SARS-CoV-2-infected primary airways (59), they contrast with other studies of SARS-CoV-2-infected primary airway cultures that showed minimal type I and type III interferon response (14, 68). These differences may be due to the observation that iPSC-derived tissues tend to be less mature than primary comparators; whether the robust antiviral response observed here reflects the biology of young/fetal epithelial tissue will require further study. The robust interferon response in iPSC airway also differs from the delayed and dampened interferon response of SARS-CoV-2-infected iAT2 cells (38). These observations give rise to the intriguing speculation that airway epithelial cells may respond differently to SARS-CoV-2 infection than alveolar epithelial cells. In SARS-CoV-1 and MERS-CoV infections, a delayed IFN response in infected human airway epithelial cells was a determinant of disease severity (69–71). Does the late onset of ARDS in a subset of individuals infected with SARS-CoV-2 develop as a consequence of a failure of the airways to restrict the infection? Future studies aimed at interrogating differential responses of airway and alveolar lung epithelia to SARS-CoV-2 infection may help to address these questions.
Given that hyperinflammation is a hallmark of SARS-CoV-2 infection and is associated with morbidity/mortality, we also focused on pathways associated with proinflammatory cytokine/chemokine production in SARS-CoV-2-infected iPSC airway. Indeed, SARS-CoV-2 infection was associated with activation of the NF-κB pathway as well as chemokines implicated in the recruitment of downstream immune mediators and leukocytes. Specifically, there was a significant upregulation and secretion of IL6 and TNFα, and T/NK-cell chemokines CXCL9 and CXCL10. There was also an increase in neutrophil-associated chemokines CXCL2 and CXCL8, consistent with the observation that neutrophils have a role in the biology of SARS-CoV-2 infection (72, 73). IL23, part of TH17 response axis important for mucosal immunity, was modestly upregulated. Though we did not detect a time-dependent increase in monocyte-associated chemokines CCL2 and CCL8 as has been reported in primary airways (59, 68), we detected an increase in GM-CSF expression from 1 to 3 dpi, suggesting monocyte-macrophage activation may result from epithelial secretion of these cytokines. Our results suggest that iPSC-airway epithelium, on SARS-CoV-2 infection, is primed to orchestrate downstream immune responses.
In agreement with previous reports on the tropism of SARS-CoV-2 in airway epithelial cells (3, 59, 74) and in keeping with the tropisms of coronaviruses HCoV-NL63 and SARS-CoV-1 (75–77), we find that multiciliated cells are the primary initial airway target cells of SARS-CoV-2. Given lack of convincing colocalization of N+ cells with MUC5AC and TP63, we suspect the majority of N+ cells represent multiciliated cells that had apical acetylated-tubulin on a separate plane as N+ staining. Consistent with this possibility, we observed a time-dependent downregulation of multiciliated cell markers during SARS-CoV-2 infection, which likewise mirrors observations made in primary cell infection (74). Significant injury to multiciliated cells in this context could impair mucociliary clearance, promoting viral spread to the distal airway as well as promoting secondary bacterial infection, both of which are associated with worse outcomes for COVID-19 patients (74, 78). Primary cell studies have shown that goblet cells eventually become infected with SARS-CoV-2, whereas basal cells are not permissive to infection, possibly due to inaccessibility to the virus due to their basolateral location, and minimal expression of TMPRSS2 (2). We similarly find that iPSC-airway basal cells have minimal expression of TMPRSS2 and ACE2 and are not detectably infected in our model. In individuals with more severe or prolonged infection with SARS-CoV-2, it will be important to determine if basal cells are ultimately infected and understand potential consequences on airway regeneration.
Lastly, we found that treatment with remdesivir or camostat mesylate led to a decrease of viral N transcripts, IL-6, and IFNL2 in our platform. There remains an urgent need for effective therapies targeting pulmonary viral infections. The iPSC airway offers an additional physiologically relevant drug-validating platform to accelerate their development.
Our data are in congruence with similar publications analyzing SARS-CoV-2 infection of various airway epithelium models. Our transcriptomic analyses extend on the findings of a previously published iPSC-derived airway model, which was infectable with SARS-CoV-2 and also showed decrease in infection from 24 to 48 h, as well as induction of an interferon response (79). Another recently published study of iPSC-derived airway epithelium observed similar but more muted induction of inflammatory cytokines and interferons and did not describe a role for IFNL (80). The observed differences in cellular responses between this study and ours may, in large part, be due to differences in MOIs used for infections. Alternatively, different directed differentiation protocols may result in differences in the frequencies and maturity of airway cell types. We utilized higher MOIs to ensure the highest possible infection rate to facilitate the generation of meaningful transcriptional responses in our cultures where not all cells are capable of being infected.
Studies using primary and human embryonic stem cell-derived (hESC) airway epithelial SARS-CoV-2 infection models have shown similar induction of inflammatory cytokines, interferons, and ISGs as observed in our model when similar MOIs were used (58, 81, 82), highlighting the accuracy and usefulness of our model system. However, although SARS-CoV-2 infections with a low MOI of 0.2 and a high MOI of 2 in an A549-ACE2 cell culture model resulted in a similar moderate cytokine signature, many more genes were upregulated in the high-MOI infection, including moderate induction of Type I and III IFNs (68). In conclusion, both high and low MOI infections have their limitations, and single-cell sequencing approaches will be instrumental to get a better understanding of the similarities and differences of both infection models. Note that for common human coronaviruses such as HCoV 119E and OC43, which have the capacity to infect all or nearly all cells within a culture, lower MOIs may be more appropriate for bulk analyses (83). Similarly, because of differences in infection rates between common human coronaviruses and SARS-CoV-2 within airway epithelial cultures, comparisons of host cell responses by single-cell approaches may be more appropriate than bulk transcriptomic analyses.
This study is not without limitations. The iPSC-airway epithelium is transcriptionally similar to primary airway epithelium and can be used to functionally model diseases such as cystic fibrosis (CF) in vitro (40). However, iPSC-derived cells differentiated into many cell types of different organs tend to be more fetal or immature than their endogenous counterparts in adults (84–86), and there may be differences including receptor expression and cell type distributions. Whether our model reflects a more fetal or pediatric response to SARS-CoV-2 will require further investigation. Another limitation of this model is that the epithelial response is studied in isolation, and the complex interplay between epithelial, immune, interstitial, and endothelial cells that leads to COVID-19 pneumonia is not captured in our model. However, the iPSC system offers a reductionist, physiologically relevant model system to study the intrinsic epithelial response and provides key insights into the initial stages of COVID-19. Furthermore, given the clinical spectrum of disease severity caused by SARS-CoV-2 infection, there is pressing need to further understand the mechanisms that lead to serve disease. Numerous genes, variants, and pathways are implicated in modulating the response to infection and require further investigation (15, 16). This iPSC-based platform, coupled with gene-editing technology, opens up future directions to evaluate the mechanisms of the airway response to SARS-CoV-2 response.
DATA AVAILABILITY
Data will be made available upon reasonable request. The RNA-Seq data have been deposited at NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE196464 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE196464).
SUPPLEMENTAL DATA
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.16695130.
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.16679614.
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.16695133.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.17100023.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.17100047.
Supplemental figure legends: https://doi.org/10.6084/m9.figshare.17100110.
GRANTS
The current work was supported by Harry Shwachman Cystic Fibrosis Clinical Investigator Award, Gilead Sciences Research Scholars, Alfred and Gilda Slifka Fund, and CF/MS fund to R.W.; T32 HL007035 to J.L.S.; F30HL147426 to K.M.A.; R01HL122442, R01HL095993, U01HL134745, U01HL134766 to D.N.K.; U01HL148692 to D.N.K. and F.J.H.; Evergrande MassCPR awards to D.N.K. and E.M.; Fast Grants and NIH NCATS grant UL1TR001430 to E.M.; R01HL139799 to F.J.H. iPSC distribution and disease modeling is supported by NIH grants U01TR001810 and NO1 75N92020C00005.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.W., S.R., K.M.A., A.A.W., E.M., D.N.K., F.J.H., and A.J.H. conceived and designed research; R.W., E.B., A.H., M.G., S.R., R.B.W., K.M.A., F.J.H., A.J.H., M.L.B., C.S.-R., J.L.S., J.H., and J.O. performed experiments; R.W., E.B., A.H., S.R., R.B.W., K.M.A., A.A.W., E.M., D.N.K., F.J.H., A.J.H., M.L.B., C.S.-R., J.L.-V., J.L.S., J.H., J.O., and C.V.-M. analyzed data; R.W., E.B., A.H., S.R., R.B.W., K.M.A., A.A.W., E.M., D.N.K., F.J.H., A.J.H., M.L.B., J.L.-V., J.L.S., and C.V.-M. interpreted results of experiments; R.W., E.B., S.R., F.J.H., M.L.B., C.S.-R., and J.L.S. prepared figures; R.W. drafted manuscript; R.W., E.B., S.R., R.B.W., K.M.A., A.A.W., E.M., D.N.K., F.J.H., A.J.H., M.L.B., C.S.-R., J.L.-V., J.L.S., J.H., J.O., and C.V.-M. edited and revised manuscript; R.W., E.B., A.H., M.G., S.R., R.B.W., K.M.A., A.A.W., E.M., D.N.K., F.J.H., A.J.H., M.L.B., C.S.-R., J.L.-V., J.L.S., J.H., J.O., and C.V.-M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Brian R. Tilton of the BUSM Flow Cytometry Core and Yuriy Alekseyev of the Boston University School of Medicine (BUSM) Sequencing Core (supported by NIH Grant 1UL1TR001430), and Thorsten Schlaeger and Yang Tang of Stem Cell Core Facility, Stem Cell Program of Boston Children’s Hospital (supported by Alfred and Gilda Slifka Fund and CF/MS Fund). For facilities management, we are indebted to Greg Miller (CReM Laboratory Manager) and Marianne James (CReM iPSC Core Manager).
A preprint is available at https://www.biorxiv.org/content/10.1101/2021.07.06.451340v1.
REFERENCES
- 1.Ahmad FB, Cisewski JA, Miniño A, Anderson RN. Provisional mortality data—United States, 2020. MMWR Morb Mortal Wkly Rep 70: 519–522, 2021. [Erratum in MMWR Morb Mortal Wkly Rep 70: 900, 2021]. doi: 10.15585/mmwr.mm7014e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, Talavera-López C, Maatz H, Reichart D, Sampaziotis F, Worlock KB, Yoshida M, Barnes JL; HCA Lung Biological Network. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med 26: 681–687, 2020. doi: 10.1038/s41591-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hou YJ, Okuda K, Edwards CE, Martinez DR, Asakura T, Dinnon KH 3rd, et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 182: 429–446.e14, 2020. doi: 10.1016/j.cell.2020.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Müller MA, Drosten C, Pöhlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181: 271–280.e8, 2020. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 183: 1735, 2020. doi: 10.1016/j.cell.2020.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu H, Chan JF, Yuen TT, Shuai H, Yuan S, Wang Y, Hu B, Yip CC, Tsang JO, Huang X, Chai Y, Yang D, Hou Y, Chik KK, Zhang X, Fung AY, Tsoi HW, Cai JP, Chan WM, Ip JD, Chu AW, Zhou J, Lung DC, Kok KH, To KK, Tsang OT, Chan KH, Yuen KY. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: an observational study. Lancet Microbe 1: e14–e23, 2020. doi: 10.1016/S2666-5247(20)30004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim JM, Chung YS, Jo HJ, Lee NJ, Kim MS, Woo SH, Park S, Kim JW, Kim HM, Han MG. Identification of coronavirus isolated from a patient in Korea with COVID-19. Osong Public Health Res Perspect 11: 3–7, 2020. doi: 10.24171/j.phrp.2020.11.1.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takayama K. In vitro and animal models for SARS-CoV-2 research. Trends Pharmacol Sci 41: 513–517, 2020. doi: 10.1016/j.tips.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hekman RM, Hume AJ, Goel RK, Abo KM, Huang J, Blum BC, et al. Actionable cytopathogenic host responses of human alveolar type 2 cells to SARS-CoV-2. Mol Cell 81: 212, 2021. doi: 10.1016/j.molcel.2020.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harcourt J, Tamin A, Lu X, Kamili S, Sakthivel SK, Murray J, et al. Isolation and characterization of SARS-CoV-2 from the first US COVID-19 patient. bioRxiv, 2020. doi: 10.1101/2020.03.02.972935. [DOI] [PMC free article] [PubMed]
- 11.Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426: 450–454, 2003. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mulay A, Konda B, Garcia G Jr, Yao C, Beil S, Villalba JM, Koziol C, Sen C, Purkayastha A, Kolls JK, Pociask DA, Pessina P, de Aja JS, Garcia-de-Alba C, Kim CF, Gomperts B, Arumugaswami V, Stripp BR. SARS-CoV-2 infection of primary human lung epithelium for COVID-19 modeling and drug discovery. Cell Rep 35: 109055, 2021. doi: 10.1016/j.celrep.2021.109055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu N, Wang W, Liu Z, Liang C, Wang W, Ye F, Huang B, Zhao L, Wang H, Zhou W, Deng Y, Mao L, Su C, Qiang G, Jiang T, Zhao J, Wu G, Song J, Tan W. Morphogenesis and cytopathic effect of SARS-CoV-2 infection in human airway epithelial cells. Nat Commun 11: 3910, 2020. doi: 10.1038/s41467-020-17796-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vanderheiden A, Ralfs P, Chirkova T, Upadhyay AA, Zimmerman MG, Bedoya S, Aoued H, Tharp GM, Pellegrini KL, Manfredi C, Sorscher E, Mainou B, Lobby JL, Kohlmeier JE, Lowen AC, Shi PY, Menachery VD, Anderson LJ, Grakoui A, Bosinger SE, Suthar MS. Type I and Type III interferons restrict SARS-CoV-2 infection of human airway epithelial cultures. J Virol 94: e00985-20, 2020. doi: 10.1128/JVI.00985-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kachuri L, Francis SS, Morrison ML, Wendt GA, Bosse Y, Cavazos TB, Rashkin SR, Ziv E, Witte JS. The landscape of host genetic factors involved in immune response to common viral infections. Genome Med 12: 93, 2020. doi: 10.1186/s13073-020-00790-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang R, Simoneau CR, Kulsuptrakul J, Bouhaddou M, Travisano KA, Hayashi JM, Carlson-Stevermer J, Zengel JR, Richards CM, Fozouni P, Oki J, Rodriguez L, Joehnk B, Walcott K, Holden K, Sil A, Carette JE, Krogan NJ, Ott M, Puschnik AS. Genetic screens identify host factors for SARS-CoV-2 and common cold coronaviruses. Cell 184: 106–119.e14, 2021. doi: 10.1016/j.cell.2020.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: 663–676, 2006. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 18.Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov 16: 115–130, 2017. doi: 10.1038/nrd.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861–872, 2007. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 20.Crane AM, Kramer P, Bui JH, Chung WJ, Li XS, Gonzalez-Garay ML, Hawkins F, Liao W, Mora D, Choi S, Wang J, Sun HC, Paschon DE, Guschin DY, Gregory PD, Kotton DN, Holmes MC, Sorscher EJ, Davis BR. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Reports 4: 569–577, 2015. doi: 10.1016/j.stemcr.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hawkins F, Kramer P, Jacob A, Driver I, Thomas DC, McCauley KB, Skvir N, Crane AM, Kurmann AA, Hollenberg AN, Nguyen S, Wong BG, Khalil AS, Huang SX, Guttentag S, Rock JR, Shannon JM, Davis BR, Kotton DN. Prospective isolation of NKX2-1-expressing human lung progenitors derived from pluripotent stem cells. J Clin Invest 127: 2277–2294, 2017. doi: 10.1172/JCI89950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang SX, Islam MN, O'Neill J, Hu Z, Yang YG, Chen YW, Mumau M, Green MD, Vunjak-Novakovic G, Bhattacharya J, Snoeck HW. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat Biotechnol 32: 84–91, 2014. doi: 10.1038/nbt.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hurley K, Ding J, Villacorta-Martin C, Herriges MJ, Jacob A, Vedaie M, Alysandratos KD, Sun YL, Lin C, Werder RB, Huang J, Wilson AA, Mithal A, Mostoslavsky G, Oglesby I, Caballero IS, Guttentag SH, Ahangari F, Kaminski N, Rodriguez-Fraticelli A, Camargo F, Bar-Joseph Z, Kotton DN. Reconstructed single-cell fate trajectories define lineage plasticity windows during differentiation of human PSC-derived distal lung progenitors. Cell Stem Cell 26: 593–608.e8, 2020. doi: 10.1016/j.stem.2019.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacob A, Morley M, Hawkins F, McCauley KB, Jean JC, Heins H, Na CL, Weaver TE, Vedaie M, Hurley K, Hinds A, Russo SJ, Kook S, Zacharias W, Ochs M, Traber K, Quinton LJ, Crane A, Davis BR, White FV, Wambach J, Whitsett JA, Cole FS, Morrisey EE, Guttentag SH, Beers MF, Kotton DN. Differentiation of human pluripotent stem cells into functional lung alveolar epithelial cells. Cell Stem Cell 21: 472–488.e10, 2017. doi: 10.1016/j.stem.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Longmire TA, Ikonomou L, Hawkins F, Christodoulou C, Cao Y, Jean JC, Kwok LW, Mou H, Rajagopal J, Shen SS, Dowton AA, Serra M, Weiss DJ, Green MD, Snoeck HW, Ramirez MI, Kotton DN. Efficient derivation of purified lung and thyroid progenitors from embryonic stem cells. Cell Stem Cell 10: 398–411, 2012. doi: 10.1016/j.stem.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCauley KB, Alysandratos KD, Jacob A, Hawkins F, Caballero IS, Vedaie M, Yang W, Slovik KJ, Morley M, Carraro G, Kook S, Guttentag SH, Stripp BR, Morrisey EE, Kotton DN. Single-cell transcriptomic profiling of pluripotent stem cell-derived SCGB3A2 airway epithelium. Stem Cell Reports 10: 1579–1595, 2018. doi: 10.1016/j.stemcr.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCauley KB, Hawkins F, Kotton DN. Derivation of epithelial-only airway organoids from human pluripotent stem cells. Curr Protoc Stem Cell Biol 45: e51, 2018. doi: 10.1002/cpsc.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCauley KB, Hawkins F, Serra M, Thomas DC, Jacob A, Kotton DN. Efficient derivation of functional human airway epithelium from pluripotent stem cells via temporal regulation of Wnt signaling. Cell Stem Cell 20: 844–857.e6, 2017. doi: 10.1016/j.stem.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Serra M, Alysandratos KD, Hawkins F, McCauley KB, Jacob A, Choi J, Caballero IS, Vedaie M, Kurmann AA, Ikonomou L, Hollenberg AN, Shannon JM, Kotton DN. Pluripotent stem cell differentiation reveals distinct developmental pathways regulating lung- versus thyroid-lineage specification. Development 144: 3879–3893, 2017. doi: 10.1242/dev.150193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gotoh S, Ito I, Nagasaki T, Yamamoto Y, Konishi S, Korogi Y, Matsumoto H, Muro S, Hirai T, Funato M, Mae S, Toyoda T, Sato-Otsubo A, Ogawa S, Osafune K, Mishima M. Generation of alveolar epithelial spheroids via isolated progenitor cells from human pluripotent stem cells. Stem Cell Reports 3: 394–403, 2014. doi: 10.1016/j.stemcr.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Konishi S, Gotoh S, Tateishi K, Yamamoto Y, Korogi Y, Nagasaki T, Matsumoto H, Muro S, Hirai T, Ito I, Tsukita S, Mishima M. Directed induction of functional multi-ciliated cells in proximal airway epithelial spheroids from human pluripotent stem cells. Stem Cell Reports 6: 18–25, 2016. doi: 10.1016/j.stemcr.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamamoto Y, Gotoh S, Korogi Y, Seki M, Konishi S, Ikeo S, Sone N, Nagasaki T, Matsumoto H, Muro S, Ito I, Hirai T, Kohno T, Suzuki Y, Mishima M. Long-term expansion of alveolar stem cells derived from human iPS cells in organoids. Nat Methods 14: 1097–1106, 2017. doi: 10.1038/nmeth.4448. [DOI] [PubMed] [Google Scholar]
- 33.Dye BR, Hill DR, Ferguson MA, Tsai YH, Nagy MS, Dyal R, Wells JM, Mayhew CN, Nattiv R, Klein OD, White ES, Deutsch GH, Spence JR. In vitro generation of human pluripotent stem cell derived lung organoids. eLife 4: e05098, 2015. doi: 10.7554/eLife.05098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller AJ, Dye BR, Ferrer-Torres D, Hill DR, Overeem AW, Shea LD, Spence JR. Generation of lung organoids from human pluripotent stem cells in vitro. Nat Protoc 14: 518–540, 2019. doi: 10.1038/s41596-018-0104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Firth AL, Dargitz CT, Qualls SJ, Menon T, Wright R, Singer O, Gage FH, Khanna A, Verma IM. Generation of multiciliated cells in functional airway epithelia from human induced pluripotent stem cells. Proc Natl Acad Sci USA 111: E1723–E1730, 2014. doi: 10.1073/pnas.1403470111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang SX, Green MD, de Carvalho AT, Mumau M, Chen YW, D'Souza SL, Snoeck HW. The in vitro generation of lung and airway progenitor cells from human pluripotent stem cells. Nat Protoc 10: 413–425, 2015. doi: 10.1038/nprot.2015.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Green MD, Chen A, Nostro MC, d'Souza SL, Schaniel C, Lemischka IR, Gouon-Evans V, Keller G, Snoeck HW. Generation of anterior foregut endoderm from human embryonic and induced pluripotent stem cells. Nat Biotechnol 29: 267–272, 2011. doi: 10.1038/nbt.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang J, Hume AJ, Abo KM, Werder RB, Villacorta-Martin C, Alysandratos KD, Beermann ML, Simone-Roach C, Lindstrom-Vautrin J, Olejnik J, Suder EL, Bullitt E, Hinds A, Sharma A, Bosmann M, Wang R, Hawkins F, Burks EJ, Saeed M, Wilson AA, Mühlberger E, Kotton DN. SARS-CoV-2 infection of pluripotent stem cell-derived human lung alveolar type 2 cells elicits a rapid epithelial-intrinsic inflammatory response. Cell Stem Cell 27: 962–973.e7, 2020. doi: 10.1016/j.stem.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abo KM, Ma L, Matte T, Huang J, Alysandratos KD, Werder RB, Mithal A, Beermann ML, Lindstrom-Vautrin J, Mostoslavsky G, Ikonomou L, Kotton DN, Hawkins F, Wilson A, Villacorta-Martin C. Human iPSC-derived alveolar and airway epithelial cells can be cultured at air-liquid interface and express SARS-CoV-2 host factors. bioRxiv, 2020. doi: 10.1101/2020.06.03.132639. [DOI] [PMC free article] [PubMed]
- 40.Hawkins FJ, Suzuki S, Beermann ML, Barillà C, Wang R, Villacorta-Martin C, Berical A, Jean JC, Le Suer J, Matte T, Simone-Roach C, Tang Y, Schlaeger TM, Crane AM, Matthias N, Huang SXL, Randell SH, Wu J, Spence JR, Carraro G, Stripp BR, Rab A, Sorsher EJ, Horani A, Brody SL, Davis BR, Kotton DN. Derivation of airway basal stem cells from human pluripotent stem cells. Cell Stem Cell 28: 79–95.e8, 2020. doi: 10.1016/j.stem.2020.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rock JR, Onaitis MW, Rawlins EL, Lu Y, Clark CP, Xue Y, Randell SH, Hogan BL. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci USA 106: 12771–12775, 2009. doi: 10.1073/pnas.0906850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mou H, Vinarsky V, Tata PR, Brazauskas K, Choi SH, Crooke AK, Zhang B, Solomon GM, Turner B, Bihler H, Harrington J, Lapey A, Channick C, Keyes C, Freund A, Artandi S, Mense M, Rowe S, Engelhardt JF, Hsu YC, Rajagopal J. Dual SMAD signaling inhibition enables long-term expansion of diverse epithelial basal cells. Cell Stem Cell 19: 217–231, 2016. doi: 10.1016/j.stem.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rock JR, Randell SH, Hogan BL. Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis Model Mech 3: 545–556, 2010. doi: 10.1242/dmm.006031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carraro G, Mulay A, Yao C, Mizuno T, Konda B, Petrov M, Lafkas D, Arron JR, Hogaboam CM, Chen P, Jiang D, Noble PW, Randell SH, McQualter JL, Stripp BR. Single-cell reconstruction of human basal cell diversity in normal and idiopathic pulmonary fibrosis lungs. Am J Respir Crit Care Med 202: 1540–1550, 2020. doi: 10.1164/rccm.201904-0792OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20: 296, 2019. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, Linsley PS, Gottardo R. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 16: 278, 2015. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36: 411–420, 2018. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R. Comprehensive integration of single-cell data. Cell 177: 1888–1902.e21, 2019. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Babraham Bioinformatics. FastQC. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 50.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21, 2013. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930, 2014. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 52.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140, 2010. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47, 2015. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Su S, Law CW, Ah-Cann C, Asselin-Labat ML, Blewitt ME, Ritchie ME. Glimma: interactive graphics for gene expression analysis. Bioinformatics 33: 2050–2052, 2017. doi: 10.1093/bioinformatics/btx094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sergushichev A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv, 2016. doi: 10.1101/060012. [DOI]
- 56.Muus C, Luecken MD, Eraslan G, Sikkema L, Waghray A, Heimberg G, et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat Med 27: 546–559, 2021. doi: 10.1038/s41591-020-01227-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hao S, Ning K, Kuz CA, Vorhies K, Yan Z, Qiu J. Long-term modeling of SARS-CoV-2 infection of in vitro cultured polarized human airway epithelium. mBio 11: e02852-20, 2020. doi: 10.1128/mBio.02852-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.V'Kovski P, Gultom M, Kelly JN, Steiner S, Russeil J, Mangeat B, Cora E, Pezoldt J, Holwerda M, Kratzel A, Laloli L, Wider M, Portmann J, Tran T, Ebert N, Stalder H, Hartmann R, Gardeux V, Alpern D, Deplancke B, Thiel V, Dijkman R. Disparate temperature-dependent virus-host dynamics for SARS-CoV-2 and SARS-CoV in the human respiratory epithelium. PLoS Biol 19: e3001158, 2021. doi: 10.1371/journal.pbio.3001158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ravindra NG, Alfajaro MM, Gasque V, Huston NC, Wan H, Szigeti-Buck K, Yasumoto Y, Greaney AM, Habet V, Chow RD, Chen JS, Wei J, Filler RB, Wang B, Wang G, Niklason LE, Montgomery RR, Eisenbarth SC, Chen S, Williams A, Iwasaki A, Horvath TL, Foxman EF, Pierce RW, Pyle AM, van Dijk D, Wilen CB. Single-cell longitudinal analysis of SARS-CoV-2 infection in human airway epithelium identifies target cells, alterations in gene expression, and cell state changes. PLoS Biol 19: e3001143, 2021. doi: 10.1371/journal.pbio.3001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181: 1016–1035.e19, 2020. doi: 10.1016/j.cell.2020.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395: 497–506, 2020. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ; HLH Across Speciality Collaboration, UK. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395: 1033–1034, 2020. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, Guan L, Wei Y, Li H, Wu X, Xu J, Tu S, Zhang Y, Chen H, Cao B. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395: 1054–1062, 2020. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, Wang C, Wang Y, Li L, Ren L, Guo F, Zhao Z, Zhou Z, Xiang Z, Wang J. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun 11: 3810, 2020. doi: 10.1038/s41467-020-17665-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sa Ribero M, Jouvenet N, Dreux M, Nisole S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog 16: e1008737, 2020. doi: 10.1371/journal.ppat.1008737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen YW, Huang SX, de Carvalho ALRT, Ho SH, Islam MN, Volpi S, Notarangelo LD, Ciancanelli M, Casanova JL, Bhattacharya J, Liang AF, Palermo LM, Porotto M, Moscona A, Snoeck HW. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat Cell Biol 19: 542–549, 2017. doi: 10.1038/ncb3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Simoneau CR, Ott M. Modeling multi-organ infection by SARS-CoV-2 using stem cell technology. Cell Stem Cell 27: 859–868, 2020. doi: 10.1016/j.stem.2020.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D, Wang TT, Schwartz RE, Lim JK, Albrecht RA, tenOever BR. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181: 1036–1045.e9, 2020. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, Perlman S. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 19: 181–193, 2016. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Menachery VD, Eisfeld AJ, Schäfer A, Josset L, Sims AC, Proll S, Fan S, Li C, Neumann G, Tilton SC, Chang J, Gralinski LE, Long C, Green R, Williams CM, Weiss J, Matzke MM, Webb-Robertson BJ, Schepmoes AA, Shukla AK, Metz TO, Smith RD, Waters KM, Katze MG, Kawaoka Y, Baric RS. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. mBio 5: e01174-14, 2014. doi: 10.1128/mBio.01174-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Spiegel M, Pichlmair A, Martínez-Sobrido L, Cros J, García-Sastre A, Haller O, Weber F. Inhibition of beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J Virol 79: 2079–2086, 2005. doi: 10.1128/JVI.79.4.2079-2086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, Xie C, Ma K, Shang K, Wang W, Tian DS. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis 71: 762–768, 2020. doi: 10.1093/cid/ciaa248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y, Xia J, Yu T, Zhang X, Zhang L. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395: 507–513, 2020. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Robinot R, Hubert M, de Melo GD, Lazarini F, Bruel T, Smith N, Levallois S, Larrous F, Fernandes J, Gellenoncourt S, Rigaud S, Gorgette O, Thouvenot C, Trébeau C, Mallet A, Duménil G, Gobaa S, Etournay R, Lledo PM, Lecuit M, Bourhy H, Duffy D, Michel V, Schwartz O, Chakrabarti LA. SARS-CoV-2 infection induces the dedifferentiation of multiciliated cells and impairs mucociliary clearance. Nat Commun 12: 4354, 2021. doi: 10.1038/s41467-021-24521-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.S Banach B, Orenstein JM, Fox LM, Randell SH, Rowley AH, Baker SC. Human airway epithelial cell culture to identify new respiratory viruses: coronavirus NL63 as a model. J Virol Methods 156: 19–26, 2009. doi: 10.1016/j.jviromet.2008.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dijkman R, Jebbink MF, Koekkoek SM, Deijs M, Jónsdóttir HR, Molenkamp R, Ieven M, Goossens H, Thiel V, van der Hoek L. Isolation and characterization of current human coronavirus strains in primary human epithelial cell cultures reveal differences in target cell tropism. J Virol 87: 6081–6090, 2013. doi: 10.1128/JVI.03368-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sims AC, Baric RS, Yount B, Burkett SE, Collins PL, Pickles RJ. Severe acute respiratory syndrome coronavirus infection of human ciliated airway epithelia: role of ciliated cells in viral spread in the conducting airways of the lungs. J Virol 79: 15511–15524, 2005. doi: 10.1128/JVI.79.24.15511-15524.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Garcia-Vidal C, Sanjuan G, Moreno-García E, Puerta-Alcalde P, Garcia-Pouton N, Chumbita M, Fernandez-Pittol M, Pitart C, Inciarte A, Bodro M, Morata L, Ambrosioni J, Grafia I, Meira F, Macaya I, Cardozo C, Casals C, Tellez A, Castro P, Marco F, García F, Mensa J, Martínez JA, Soriano A; COVID-19 Researchers Group. Incidence of co-infections and superinfections in hospitalized patients with COVID-19: a retrospective cohort study. Clin Microbiol Infect 27: 83–88, 2021. doi: 10.1016/j.cmi.2020.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yin X, Riva L, Pu Y, Martin-Sancho L, Kanamune J, Yamamoto Y, Sakai K, Gotoh S, Miorin L, De Jesus PD, Yang CC, Herbert KM, Yoh S, Hultquist JF, García-Sastre A, Chanda SK. MDA5 governs the innate immune response to SARS-CoV-2 in lung epithelial cells. Cell Rep 34: 108628, 2021. doi: 10.1016/j.celrep.2020.108628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Djidrovski I, Georgiou M, Hughes GL, Patterson EI, Casas-Sanchez A, Pennington SH, Biagini GA, Moya-Molina M, van den Bor J, Smit MJ, Chung G, Lako M, Armstrong L. SARS-CoV-2 infects an upper airway model derived from induced pluripotent stem cells. Stem Cells 39: 1310–1321, 2021. doi: 10.1002/stem.3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fiege JK, Thiede JM, Nanda HA, Matchett WE, Moore PJ, Montanari NR, Thielen BK, Daniel J, Stanley E, Hunter RC, Menachery VD, Shen SS, Bold TD, Langlois RA. Single cell resolution of SARS-CoV-2 tropism, antiviral responses, and susceptibility to therapies in primary human airway epithelium. PLoS Pathog 17: e1009292, 2021. doi: 10.1371/journal.ppat.1009292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pei R, Feng J, Zhang Y, Sun H, Li L, Yang X, He J, Xiao S, Xiong J, Lin Y, Wen K, Zhou H, Chen J, Rong Z, Chen X. Host metabolism dysregulation and cell tropism identification in human airway and alveolar organoids upon SARS-CoV-2 infection. Protein Cell 12: 717–733, 2021. doi: 10.1007/s13238-020-00811-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Loo SL, Wark PAB, Esneau C, Nichol KS, Hsu AC, Bartlett NW. Human coronaviruses 229E and OC43 replicate and induce distinct antiviral responses in differentiated primary human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 319: L926–L931, 2020. doi: 10.1152/ajplung.00374.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Volpato V, Smith J, Sandor C, Ried JS, Baud A, Handel A, et al. Reproducibility of molecular phenotypes after long-term differentiation to human iPSC-derived neurons: a multi-site omics study. Stem Cell Reports 11: 897–911, 2018. doi: 10.1016/j.stemcr.2018.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Goversen B, van der Heyden MAG, van Veen TAB, de Boer TP. The immature electrophysiological phenotype of iPSC-CMs still hampers in vitro drug screening: special focus on I. Pharmacol Ther 183: 127–136, 2018. doi: 10.1016/j.pharmthera.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 86.Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res 117: 80–88, 2015. doi: 10.1161/CIRCRESAHA.117.305365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.16695130.
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.16679614.
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.16695133.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.17100023.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.17100047.
Supplemental figure legends: https://doi.org/10.6084/m9.figshare.17100110.
Data Availability Statement
Data will be made available upon reasonable request. The RNA-Seq data have been deposited at NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE196464 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE196464).