

Abstract
Solid tumor treatment relies heavily upon chemotherapies, radiation, surgical resection, and/or immunotherapies. Although many alternative non-invasive solid tumor therapies have been proposed through the years and continue to be tested in various contexts, tumor cell eradication remains a daunting task for the current cancer armamentarium. Indeed, solid tumors exhibit physically and biochemically heterogenous microenvironments, allowing them to easily acquire resistance mechanisms. Progress in sonodynamic therapy (SDT), a treatment modality capable of controlling tumor growth while limiting off-target effects and toxicities, has accelerated in recent years. SDT combines “sonosensitizing” agents with the non-invasive application of focused acoustic energy [i.e. focused ultrasound (FUS)] to drive highly localized formation of tumor cell-killing reactive oxygen species (ROS). Sonosensitizers selectively accumulate in tumor cells, after which FUS radiation eliminates the tumor by forcing the tumor cells to undergo cell death. In this article, we comprehensively review recent studies wherein SDT has been applied to treat primary and metastatic tumors. We discuss sonosensitizers, combination therapies with SDT, developments in defining the mechanism of SDT-induced cell cytotoxicity, and the promise SDT offers as a modulator of anti-tumor immunity.
Keywords: Sonosensitizers, Focused Ultrasound, Photodynamic therapy, Immunotherapy, Immunogenic cell death
1.0. Introduction
Anticancer therapies continue to progress, but highly effective treatments for many cancers remain elusive as cancer cells adapt and acquire resistance to therapy. Furthermore, solid tumors are often immunologically “cold”, or marked by an immunosuppressive environment preventing immune cell infiltration, expansion, and function. Thus, driving immunologically “cold” tumors into “hot” tumors while limiting off-target effects represents an attractive therapeutic approach.
Sonodynamic therapy (SDT) is a noninvasive anticancer modality derived from photodynamic therapy (PDT), which elicits tumor cell killing through activation of a photosensitive compound via localized light delivery [1]. Photosensitizers, which accumulate with high specificity in tumor cells, release energy upon activation and generate reactive oxygen species (ROS) in the presence of oxygen [2,3], resulting in cancer cell toxicity [3]. Cancer cells are especially reliant on antioxidants, making them particularly vulnerable to excessive ROS [3]. Nonetheless, one disadvantage of PDT is that light is significantly attenuated by intervening tissue, so the treatment success depends on tumor depth, composition, and position within the body.
SDT overcomes this limitation because the tumor is exposed to focused ultrasound (FUS), which offers improved tissue penetration, reducing potential off-target effects [1]. Compared to light, in some applications, FUS may provide a more diverse set of application options with regard to focusing and tumor coverage [1]. FUS activates “sonosensitizers”, which like photosensitizers, selectively accumulate in tumor cells and generate ROS. Additionally, SDT can be easily integrated with other FUS approaches. For example, FUS may be deployed with i.v.-injected contrast agent microbubbles (MBs) to permeabilize cell membranes and enhance drug delivery via transient opening of blood-brain and/or blood-tumor barriers [4]. Furthermore, sonosensitizer accumulation in tumors cells may be enriched by the enhanced permeability and retention (EPR) effect [5]. Mitochondria are especially vulnerable to ROS since they control activation of intrinsic apoptosis, autophagy, and ferroptosis, all of which can be triggered by excessive ROS [3]. However, tumor microenvironments (TMEs) are often devoid of oxygen (i.e. hypoxic) due to dysfunctional tumor vasculature [4], resulting in high glutathione levels (GSH) in the TME, which absorbs the ROS generated by SDT [6]. Therefore, alleviating tumor hypoxia is an important barrier for establishing the efficacy of SDT.
This review will examine SDT application to treat cancers including, but not limited to, breast, glioblastoma, pancreatic, lung, and leukemia. We first introduce sonosensitizers used over the last eight years, focusing on nanoparticle modifications that improve mitochondrial targeting, selective cancer cell accumulation, improved drug delivery, and alleviating tumor hypoxia. Details outlining tumor models and FUS parameters applied with the sonosensitizers discussed in this review are outlined in Table 1. We then discuss SDT combination therapies that may promote tumor control such as PDT-SDT, anti-tumor drug delivery, and immunotherapy-SDT. We conclude with an in-depth characterization of mechanisms driving SDT-induced tumor cell death and how SDT may stimulate an adaptive anti-tumor immune response. This information is summarized in Table 2.
Table 1.
Sonosensitizers with corresponding FUS parameters and models.
| Sonosensitizers | FUS Parameters f = frequency; dc = duty cycle; PRF = pulse repetition frequency; PNP = peak negative pressure; MI = mean Intensity; I = Intensity; ISATA = spatial average temporal average Intensity; ISPTA = spatial peak temporal average Intensity; t = time |
Cancer Model | Reference |
|---|---|---|---|
| Ce6 | f = 1.90 MHz; ISATA = 1.6 W/cm2; t = 3 min | 4T1 Breast | [1] |
| I = 1.5 W/cm2; t = 5 min | 4T1 Breast | [38] | |
| f = 1.56 MHz; I = 0, 2, 4 W/cm2; t = 3 min | H22 Liver | [13] | |
| f = 1.0 MHz; ISATA = 0.4, 0.8, 1.6 W/cm2; t = 2 min | SPCA1 Lung | [14] | |
| f = 1.0 MHz; duty cycle = 20%; Power = 1 W; burst interval = 1 s; t = 60 s | GL261 Glioma | [57] | |
| DVDMS | f = 1.1 MHz; Power = 1, 2, 4 W; t = 60 s | K562 & U937 Leukemia | [16] |
| f = 0.970 MHz; duty cycle = 30%; Power = 3.45 W; t = 3 min | HCT116 & RKO Colorectal | [17] | |
| f = 1.0 MHz; duty cycle = 30%; I = 0.4 W/cm2; t = 90 s | 4T1 Breast | [18] | |
| f = 1.9 MHz; ISATA = 2 W/cm2; t = 2 min (1 min on, 1 min off, 1 min on) | 4T1 Breast | [42] | |
| f = 1.0 MHz; I = 0.45 W/cm2; t = 1 min | U373 Glioma | [65] | |
| f = 1.1 MHz; duty cycle = 10%; PRF = 100 Hz; I = 1 W/cm2 | Hep-G2 Liver | [66] | |
| f = 1.1 MHz; duty cycle = 10%; PRF = 100 Hz; I = 0.5 W/cm2; t = 5 min | H446 Lung | [73] | |
| IR-780 | f = 1.0 MHz; duty cycle = 50%; PRF = 1 Hz; ISATA = 2 W/cm2; t = 4 min | 4T1 Breast | [19] |
| f = 1.0 MHz; duty cycle = 50%; I = 0.6 W/cm2; t = 3 min | MDA-MB-231 Breast | [20] | |
| f = 1.0 MHz; I = 2 W/cm2; t = 5 min | 4T1 Breast | [39] | |
| Rose Bengal | f = 1.0 MHz; duty cycle = 50%; I = 1.5 W/cm2; t = 5 min | HT-29 Colorectal | [4] |
| f = 1.0 MHz; duty cycle = 30%; PRF = 100 Hz; PNP = 0.48 MPa; MI = 0.48; I = 3.5 W/cm2; t = 3.5 min | MIA PaCa-2 Pancreatic | [31] | |
| f = 1.0 MHz; duty cycle = 30%; PRF = 100 Hz; PNP = 880 kPa; I = 3.5 W/cm2; t = 3.5 min | PSN-1 & BxPC-3 Pancreatic | [33] | |
| f = 1.17 MHz; duty cycle = 30%; PRF = 100 Hz; PNP = 0.7 MPa; ; t = 3.5 min | BxPC-3 Pancreatic | [36] | |
| f = 1.0 MHz; duty cycle = 30%; PRF = 100 Hz; PNP = 0.48 MPa; MI = 0.48; I = 3.5 W/cm2; t = 3.5 min | T110299 Pancreatic | [44] | |
| Metal-based Sensitizers | f = 1.0 MHz; Power = 2 W; t = 3 min | 4T1 Breast | [21] |
| f = 1.0 MHz; duty cycle = 50%; I = 2 W/cm2; t = 5 min | 4T1 Breast | [78] | |
| f = 1.0 MHz; duty cycle = 50%; I = 0.5 W/cm2; t = 2 min | 4T1 Breast | [79] | |
| f = 1.0 MHz; I = 1.5 W/cm2; t = 5 min | CT26 Colorectal | [5] | |
| f = 40 kHz; I = 2 W/cm2; t = 30 min | CT26 Colorectal | [77] | |
| f = 1.0 MHz; duty cycle = 100%; I = 0.5, 1 W/cm2; t = 1 min | B16/F10 Melanoma | [28] | |
| f = 1.0 MHz; duty cycle = 50%; I = 1 W/cm2; t = 10 min | B16-OVA Melanoma | [46] | |
| f = 1.0 MHz; duty cycle = 50%; I = 1 W/cm2; t = 5, 10 min | H22 Liver | [74] | |
| f = 1.0 MHz; I = 1 W/cm2; t = 0 – 24 hrs | C6 Glioma | [29] | |
| HMME | f = 0 – 1.0 MHz; I = 1 W/cm2; t = 60 s | C6 Glioma | [72] |
| f = 1.0 MHz; duty cycle = 30%; PRF = 100 Hz; I = 3.5 W/cm2; t = 3.5 min | LNCaP Prostate | [24] | |
| f = 1.0 MHz; duty cycle = 50%; I = 1.5 W/cm2; t = 5 min | 4T1 Breast & CT26 | [45] | |
| f = 1.34 MHz; I = 1 W/cm2; t = 15 – 60 s | Ehrlich ascites carcinoma (EAC) | [52] | |
| PpIX | f = 1.1 MHz; I = 1, 3, 5 W/cm2; t = 60 s | L1210 Leukemia | [50, 51] |
| f = 1.1 MHz; duty cycle = 50%; I = 2 W/cm2; t = 5 min | SAS Tongue | [60] | |
| ALA | f = 1.0 MHz; duty cycle = 60%; I = 2 W/cm2; t = 5 min | Capan-1 Pancreatic | [61] |
| f = 1.1 MHz; duty cycle = 10%; PNP = 500 kPa; ISPTA = 10 W/cm2; t = 3 min | C6 & U87 Glioma | [62] | |
| f = 1.06 MHz; PNP = 420 kPA; Power = 0.32 W; ISPTA = 2.8, 5.5, 11, 22 W/cm2; t = 20 min | C6 Glioma | [63] | |
| f = 1.0 MHz; d I = 2 W/cm2; t = 10 min | U251 Glioma | [64] | |
| f = 1.1 MHz; duty cycle = 10%; PRF = 100 Hz; I = 2 W/cm2; t = 5 min | B16/F10 Melanoma | [75] |
Table 2.
Sensitizers with known modes of action and immunomodulation.
| Sonosensitizers | Mode of Action | Immunomodulation | Reference |
|---|---|---|---|
| Ce6 | ROS-driven apoptosis ↓ VEGF & MMP |
Unknown | [1] |
| Necrosis S phase cell cycle arrest |
No change in white blood cell counts | [14] | |
| Apoptosis & necrosis ↑ intracellular ROS |
Unknown | [17] | |
| ↑ intracellular ROS, caspase-3, Bax, & cyt-c ↓ Bcl-2 & MMP-2 |
Unknown | [29] | |
| ROS-driven apoptosis | No change in white blood cell counts | [38] | |
| ROS-driven mitophagy promoting tumor cell death escape | Unknown | [58] | |
| Apoptosis ↑ cleaved caspase 3 & 9 ↓ MMP |
Unknown | [61] | |
| DVDMS | ROS-driven apoptosis & necrosis ↓ PCNA expression |
Unknown | [18] |
| ROS-driven apoptosis & necrosis ↑ cleaved caspase-3 ↓ ATP production, MMP, & PCNA |
Unknown | [42] | |
| Necrosis ROS-driven DNA double-strand breaks inducing apoptosis |
Unknown | [66] | |
| Apoptosis & necrosis ↑ cleaved caspase-3, 8, & 9 ↓ reduced Bcl-2, MMP, & VEGF ↓ RIP3 suggesting a lack of necroptotic cell death |
↑ TNFα | [74] | |
| IR-780 | Apoptosis & necrosis ↑ O2 & H2O2 |
Unknown | [19] |
| Apoptosis & necrosis ↑ O2, OH, & H2O2 ↓ MMP-2 & 9 |
Unknown | [20] | |
| ROS-driven apoptosis ↓ HIF1α & Ki67 |
Unknown | [39] | |
| Rose Bengal | ↓ cell viability ↑ singlet oxygen production |
Unknown | [4] |
| ↓ cellular metabolic activity ↓ angiogenic (VEGFC, IL-8, SMAD4), hypoxic (HIF1α, VHL, RUNX2), & cancer stem cell (NANOG, EPCAM) factors |
Unknown | [31] | |
| ↑ calreticulin indicative of ICD | ↑ tumor infiltrating CD4+ & CD8+ T cells | [44] | |
| Metal-based Sensitizers | ROS-driven apoptosis & necrosis | Unknown | [21] |
| ↑ O2 ↑ calreticulin & extracellular ATP indicating ICD ↓ GSH |
↑ DC maturation & CD8+ effector T cells | [46] | |
| Ferroptosis ↑ O2 ↓ HIF1α |
↓ M2 macrophages ↑ DC maturation & activated CD8+ T cells |
[75] | |
| ↑ calreticulin & extracellular ATP indicative of ICD | ↑ DC maturation & CD8+ T cells ↑ VCAM-1 expression |
[78] | |
| ↓ cell viability ↑ HMGB1, ATP, & calreticulin indicating ICD |
↑ DC maturation & NK cell activity ↑ CD4+ & CD8+ effector T cells ↓ Tregs & M2-macrophages |
[79] | |
| Apoptosis & necrosis ↑ HMGB1, HSP90, extracellular ATP, and calreticulin indicating ICD 𡤱 Ki67 expression |
↑ TNFα, IL-6, & IFN-γ | [80] | |
| PpIX | Apoptotic chromatin fragmentation Damaged F-actin driving apoptosis | Unknown | [52] |
| ROS-driven autophagy promoting tumor cell death escape | Unknown | [56] | |
| Apoptosis & necrosis G2/M phase cell cycle arrest ↓ CDK1 & Cyclin B1 ↑ p21 & p27 ↑ ROS-driven p53 expression |
Unknown | [67] | |
| HMME | Apoptosis | ↑ DC maturation & activated CD8+ T cells ↑ TNFα, IL-6, & IL-12p70 ↓ reduced Tregs |
[45] |
| Apoptosis ↑ intracellular Ca2+ ↑ cleaved caspase-3 and cytochrome-c |
Unknown | [72] | |
| ↑ Ca2+ induced apoptosis ↓ MMP |
Unknown | [73] | |
| Apoptosis & necrosis ↑ HMGB1, calreticulin, & HSP90 expression indicative of ICD ↓ Ki67 expression |
↑ DC maturation & effector CD8+ T cells ↑ pro-inflammatory cytokine production |
[81] | |
| ALA | ↓ tumor cell migration, invasion, & proliferation ↓ microvessel density |
Unknown | [60] |
| Apoptosis ↓ MMP & Bcl-2 ↑ caspase-9, cleaved caspase-3, & Bax |
Unknown | [62] | |
| ↑ caspase-3 & PARP-1 suggesting apoptotic cell death | Unknown | [63] | |
| Apoptosis ↓ Ki67 |
Unknown | [64] | |
| Apoptosis & necrosis ↓ MMP & Bcl-2 ↑ Bax & cleaved caspase 3 & 9 |
Unknown | [65] | |
| Necrosis | ↑ DC maturation ↓ M2 TAMs ↑ TNFα, IFN-γ, & IL-10 |
[76] |
2.0. Sonosensitizers
Sonosensitizers have unique chemical and physical properties that are critical for defining their utility in certain applications. Discussion of these chemical and physical features is beyond the scope of this review; however, excellent review articles focusing on these aspects of sonosensitizers are available [7–12].
2.1. Chlorin e6
Chlorin e6 (Ce6) is a chlorophyll derivative initially used as a photosensitizer. Ce6-based SDT of liver cancer showed potent sonosensitizer capacity with high tumor affinity and quick clearance from non-tumor tissues [13]. Ce6 has high affinity for non-small cell lung cancer (NSCLC) [14], as it accumulates specifically in SPCA1 lung tumor tissue, peaking at 18h and barely detectable by 72h. Furthermore, SDT-treated cells had a much higher rate of tumor cell necrosis relative to FUS and Ce6 alone. Interestingly, tumor cell apoptosis remained unaffected among the treatment groups, suggesting that Ce6 SDT predominately induces necrosis [14].
2.2. Sinoporphyrin sodium (DVDMS)
Porphyrin derivatives, including sinoporphyrin sodium (DVDMS), are effective photo- and sonosensitizers [15]. DVDMS accumulation in K562 leukemia cell mitochondria is seen as early as 1h post-SDT treatment, peaking at 2h [16]. Similar results were reported for human colorectal cancer [17], including increased intracellular ROS confirmation. Treating CT26 colorectal cancer with DVDMS and MBs decreased cell viability by 50% 4h after treatment compared to FUS+MBs without DVDMS. DVDMS+FUS+MBs increased apoptosis and necrosis rates by roughly 2-fold compared to FUS+MBs alone. These results corresponded with higher in vivo tumor growth control, leading the authors to conclude that MBs can improve DVDMS-mediated SDT’s antitumor effect. The investigators then hypothesized that DVDMS-loaded liposome-MB complexes (DLMBs) would further improve therapeutic efficacy [18]. DVDMS accumulation was indeed improved in human breast cancer MDA-MB-231 cells with FUS+DLMBs relative to DVDMS and DVDMS-liposome complexes alone. By 24h post-treatment, only 50% of the SDT-treated cells remained viable, compared to 30% with DVDMS treatment. In vivo, 4T1 mouse breast cancer treated with FUS+DLMB had significant growth inhibition (68%) relative to DVDMS-liposome and DVDMS alone groups (30% and 35%, respectively). In conclusion, a significant sonodynamic effect is observed in breast cancer models applying DVDMS-liposome-MB complexes combined with FUS.
2.3. IR-780
IR-780 is another PDT compound that may be repurposed for SDT. Mouse 4T1 breast cancer cells treated with IR-780 had reduced viability, corresponding to higher apoptosis and necrosis [19], and more O2 and H2O2 levels compared to controls. In vivo imaging of 4T1 tumor-bearing mice showed maximum intratumoral IR-780 concentration after 1h. Additionally, SDT-treated mice experienced tumor growth delay, suggesting that IR-780 may be a promising tumor-treating sonosensitizer.
One limitation of SDT is rapid metabolism of fat-soluble sonosensitizers, an obstacle avoided by integrating the compound into water-soluble carriers [20]. IR780 has been encapsulated in a polyethylene glycol (PEG) lipid-based carrier and delivered with Ce6 [20]. PEG-IR780@Ce6 produced more O2 and OH than free Ce6 and PEG-IR780 alone. Additionally, cancer cell invasion and migration after SDT treatment decreased, further supported by MMP-2 and MMP-9 metalloproteinase suppression which are critical for cell migration and invasion [20]. Thus, PEG-IR780@Ce6 offers better tumor control by generating multiple ROS compared to PEG-IR780 or Ce6 alone.
2.4. Rose Bengal
Sonosensitizer carriers often have low loading efficacy [20], previously addressed by using amphiphilic rose bengal (ARB) bound to MBs (RB-MBs) [4]. Singlet oxygen levels increased in response to RB-MBs+FUS treatment relative to controls, correlating with reduced HT-29 colorectal adenocarcinoma cell viability. Following in vivo treatment with RB-MBs+FUS, tumor growth was inhibited up to 76% compared to 24% growth inhibition by MBs+FUS, demonstrating that RB-MBs effectively induce tumor-specific cell death, thus improving tumor growth control.
2.5. Metal-based Sonosensitizers
While porphyrin and chlorophyll derivatives are well-studied for SDT, they often have low chemical and biological stability and poor tumor accumulation [21]. To overcome this, nanomaterials have been employed to prolong sonosensitizer circulation time, immunogenicity, and tissue penetration. Copper-cysteamine (Cu-Cy) metal-based nanoparticles have been applied to MCF-7, 4T1, and MDA-MB-231 breast cancer cells [15,16]. Following in vitro Cu-Cy+FUS treatment, cell viability decreased to 45%. Cu-Cy+FUS reduced 4T1 breast tumors to 74% of their starting volume, compared to Cu-Cy (26%) and FUS alone (37%), with reduced tumor burden corresponding with increased ROS levels [21].
A recent advance in metal-based sonosensitizers application employs Ag2S quantum dots (QD) encapsulated in red blood cell (RBC) vesicle membranes. RBCs possess catalase, an enzyme that oxidizes H2O2 into H2O and O2, thereby alleviating tumor hypoxia [23]. RBC membrane-coated Ag2S QDs were deployed to treat CT26 colorectal cancer cells [5], wherein they produced high amounts of O2 even under hypoxic conditions. In vivo analysis revealed high Ag2S QD accumulation in tumors between 6-9h post-injection, with dissipation after 24h. Polyglutamate (PGA), another common nanoparticle coating, is digested by cathepsin-B, a lysosomal protease often overexpressed in the TME [24]. This polymer coat has been used to generate PGA-tyrosine hematoporphyrin nanoparticles (HPNPs) for SDT treatment of prostate cancer [24]. When HPNPs were applied with FUS, LNCaP and PC3 prostate cancer cell viability was reduced by ~90%. LNCaP tumors were reduced in size by 36% at 24h post-SDT. These results suggest that cathepsin B-overexpressing tumors can be exploited to generate PGATyr-digested nanoparticles that can sonodynamically control prostate cancer.
3.0. Combination Treatments Employing SDT
3.1. PDT-SDT
It has been suggested that SDT with PDT (i.e. SPDT) may improve therapeutic effect. In SPDT, an agent with both photochemical and sonochemical activity is administered, then activated with light and sound [25]. This approach could allow for both lower sensitizer dosage and decreased total energy deposition, preventing potential negative side effects [26]. Applying Ce6 for SPDT to breast cancer cells reduced cell viability to 50-60% compared to a 30% viability reduction after PDT alone [1]. Peak Ce6 breast cancer cell uptake occurred at 2h, with a sustained signal until 6h. SPDT significantly increased ROS levels and inhibited breast cancer metastasis to the lungs. VEGF and MMP-9, which promote breast cancer invasion and metastases, exhibited reduced expression, suggesting that Ce6-mediated SPDT can inhibit cancer cell metastasis [1].
Metal nanoparticles are becoming more common sonosensitizers in SDT due to their improved biocompatibility [6]. For instance, SPDT with gold/MnO2 nanomaterials reduce intratumoral H2O2, alleviating tumor hypoxia [27]. Upon concurrent application of laser and FUS radiation with Au/MnO2 nanocomposites, B16/F10 melanoma cell viability declined more than with PDT alone. SPDT-treated melanoma tumors exhibit increased necrosis, supporting this photo- and sono-sensitizer for use in cancer treatment. Similar to Au/MnO2 nanocomposites, MnFe2O4/C nanocomposites have also been used to treat B16/F10 melanoma, eliciting reduced cell viability and increased tumor necrosis, further suggesting a synergistic effect of SDT-PDT combination treatment [28].
3.2. Antitumor Drug Delivery and SDT
Chemotherapies are the standard of care for most cancers. A quintessential example is glioblastoma treatment with Temozolomide (TMZ). TMZ has many known forms of intrinsic and acquired resistance, so it has often been a candidate for combination therapies [29]. Overexpression of sodium-hydrogen exchanger isoform 1 (NHE1) is one such mode of resistance which can enhance tumor cells’ invasive capacity [23,24]. Hematoporphyrin monomethyl ether (HMME)-mediated SDT alters NHE1 expression in TMZ-resistant glioma cells, preventing cancer cell proliferation and invasion, as well as potentially re-sensitizing the cells to TMZ. Following SDT and TMZ, C6 glioma cells showed reduced capacity to migrate and invade. Furthermore, caspase-3, cleaved caspase-3, Bcl-2, and Cyt-c were increased following TMZ+SDT treatment, suggesting mitochondrial apoptotic pathway induction.
Gemcitabine (Gem), the most widely used chemotherapeutic to treat pancreatic cancer, is an antimetabolite drug that can synergize with SDT- and FUS-targeted microbubble destruction (UTMD) [31]. UTMD occurs when FUS facilitates MB cavitation and collapse, driving increased uptake of chemotherapeutics at the target site and improved treatment efficacy [32]. Chemo-sonodynamic treatment of pancreatic cancer using Gem-O2MB+RB-O2MB decreased pancreatic cancer cell metabolic activity [31]. Treatment also reduced expression of pro-angiogenic factors, VEGF-C and IL-8, and HIF1α, VHL, and RUNX2 expression, suggesting a less hypoxic tumor microenvironment. Thus, chemo-SDT using Gem-O2MB and RB-O2MB successfully increases drug delivery to the tumor cells, limiting tumor growth.
Gem is also radiosensitive and has been used in combination with RB-O2MBs for chemoradiation therapy (CRT) [33]. While CRT has been combined with PDT, CRT-SDT efficacy had not been evaluated [28,29]. Using BxPC-3 and PSN-1 pancreatic tumors, CRT-SDT treatment significantly delayed tumor growth in PSN-1 tumor bearing mice, but not BxPC-3 tumor bearing mice. This differential treatment success could stem from poor vascularization in PSN-1 tumors. As a result, RB-O2MBs may be unable to extravasate through the vascular endothelium, leading to increased ROS generation in the vasculature itself and SDT-mediated vascular damage.
While the CRT-SDT treatment of BxPC-3 pancreatic tumors proved unsuccessful, BxPC-3 tumor growth inhibition does occur when using magnetically-responsive oxygen MBs (MagO2MBs) bound to RB, in the presence of co-applied magnetic and ultrasound fields using a magnetic-acoustic-device (MAD) [36]. It was hypothesized that the field alignment generated by the MAD is critical to maintain the MBs in the ultrasound beam [36]. Though a strong argument is made for the necessity of field alignment, without assessing tumor vascularity prior to treatment, it is unknown whether the tumor response was due to a differing ability of the MBs to enter the vasculature or the field alignment specifically. Regardless, co-alignment and application of ultrasound and magnetic fields resulted in greater MagO2MBs-RB-Gem treatment efficacy in controlling tumor growth.
Another chemotherapeutic, doxorubicin (DOX), is used for treating breast cancer by forcing apoptosis-inducing DNA strand breaks [37]. Recently, a biodegradable polydopamine nanoparticle embedded with platinum and co-loaded with DOX and Ce6-modified with TPP, a mitochondrial-targeting molecule (CDP@HP-T) (Figure 1), has been deployed [38]. The CDP@HP-T nanoprobe accumulates in mitochondria and subsequently produces ROS in 4T1 breast tumor cells. In vivo, peak intratumoral CDP@HP-T accumulation occurred ~3h post-injection. Tumor growth rates were also diminished in the CDP@HP-T+FUS group, supporting this nanoprobe’s application in the chemo-SDT of breast cancer.
Figure 1.
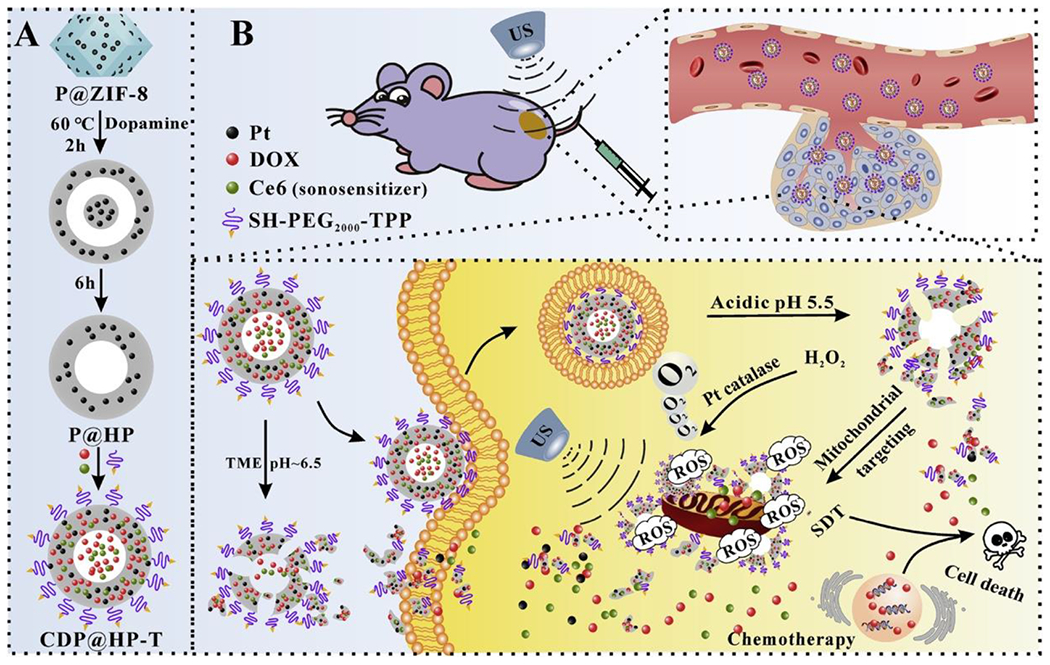
Schematic illustration of the synthesis route of CDP@HP-T and chemo-sonodynamic combined therapy. (a) Generation of CDP@HP-T nanoparticles. (b) I.V. injection of nanoparticles and Ce6 sonosensitizer followed by FUS treatment of the tumor. Nanoparticles in the blood stream extravasate into the tumor where the acidic environment prompts destruction of the nanoparticle coat releasing DOX and platinum. FUS-driven Ce6 accumulation in the tumor cells leads to ROS generation, whereby the mitochondrial cell death pathway is induced [38]. Reproduced with permission.
To overcome tumor hypoxia, an oxygen nanoplatform with a perfluorocarbon (PFC) compound, which can readily dissolve high amount of oxygen [39], has been developed. Here, a polymer shell and PFC core shelter IR780, DOX, and oxygen (PPID-NPs) [39]. PPID-NPs+FUS decreased 4T1 and MCF-7 breast cancer cell viability under normoxic and hypoxic conditions, and co-treatment produced significantly more ROS. 4T1 tumor staining showed decreased Ki67 expression and increased TUNEL staining following PPID-NPs+FUS treatment, suggesting lower cancer cell proliferation and higher levels of tumor cell death, respectively. Additionally, chemo-SDT reduced tumor burden, leading the authors to conclude that the treatment regimen may successfully treat hypoxic tumors.
While we have focused primarily on chemo-sonodynamic therapies, metabolic inhibitors have also been combined with SDT. Cancer cells rely on elevated glycolytic capacity [34,35], which facilitates cancer cell invasion, metastasis, and apoptosis. One method to prevent tumor growth is to sensitize cancer cells to apoptosis using the anti-glycolytic compound 2-deoxyglucose (2-DG) [42]. 2-DG is phosphorylated by the enzyme hexokinase II, often overexpressed in tumors [42]. It has been hypothesized that by targeting glycolysis with 2-DG and mitochondria with DVDMS-mediated SDT, breast cancer progression would be significantly reduced [42]. Indeed, SDT-2DG induced breast cancer cell death and decreased PCNA expression, implying inhibited proliferation of viable cells. Tumor growth was inhibited up to 70% after co-treatment, supporting the idea that blocking glycolysis can promote cell death upon SDT treatment. A follow-up study showed that SDT-2DG treatment can also reduce breast cancer’s metastatic capacity [43].
3.3. Immunotherapy-SDT
More recently, immunotherapy-SDT combination approaches have been used to control tumor growth and initiate antitumor immune responses. Many cancer immunotherapies strive to improve the immune system’s capacity to control cancer by enhancing T cell effector function [44]. For example, anti-PD-L1 blocks PD-L1 protein expressed on tumor cells, which binds its cognate receptor PD-1 on tumor-specific T cells. Blocking anti-PD-L1 allows tumor-specific T cells to be properly activated and drive necessary antitumor immunity. A PD-L1 inhibitor has been recently combined with HMME-directed SDT [45], where it augmented activated dendritic cells (DCs) and the production of proinflammatory IL-6 and TNF-α cytokines in 4T1 breast tumors. Anti-PD-L1+SDT reduced tumor volume and metastatic pulmonary nodules and increased the number of CD8+ T effector cells, suggesting that anti-PD-L1 in combination with SDT can effectively drive antitumor immune responses.
Similarly, a SDT-nanovaccine approach has been hypothesized to improve anti-PD-1 antibody efficacy against malignant melanoma [46]. The investigators constructed a manganese porphyrin-based metal-organic frameworks (Mn-MOF) nanoplatform bound to CpG (a TLR9 agonist) coated in the cell membrane of murine B16 melanoma cells expressing ovalbumin (OVA), an antigen used to track tumor-specific immune responses. This cMn-MOF@CM nanovaccine platform aimed to induce DC maturation and subsequent T cell activation by displaying OVA via MHCII. Anti-PD-1+SDT significantly reduced tumor growth and increased survival. The number of mature DCs and effector CD8+ T cells within the tumor were also increased. In a bilateral B16-OVA melanoma model, both primary and distant tumors decreased in size, suggesting that the combined anti-PD-1 and SDT may control breast cancer tumor growth by inducing an adaptive antitumor immune response.
Anti-PD-L1 therapy has also been applied with MB-RB-SDT to treat T110299 bilateral pancreatic tumors [44]. The investigators showed upregulated expression of calreticulin (CRT), an ‘eat me’ signal and marker of immunogenic cell death (ICD) expressed on the surface of dying cells. SDT alone inhibited treated and distal tumor growth, which was enhanced when combined with PD-L1 therapy. Treated tumor CD4+ T cells increased only in the anti-PD-L1 treatment group. However, at the distal tumor site, CD4+ and CD8+ T cells increased following SDT+PD-L1 therapy, suggesting that this treatment regimen may be a strong candidate for control of metastatic disease.
While anti-PD1/PD-L1 therapies are widely used in the clinic, they often lack efficacy due to similarly functioning negative regulatory molecules. One of these molecules, indoleamine 2,3-dioxygenase (IDO), is frequently overexpressed in tumor cells and antigen presenting cells (APCs) [47]. IDO enhances immune tolerance due to increased regulatory T cells (Tregs), which promote immune suppression in the TME [45, 46]. The IDO inhibitor NLG919 has been employed in combination with Au-BMSN nanoparticles to treat 4T1 breast cancer. Au-BMSN nanoparticles contain CO-releasing molecules and are coated in a macrophage cell membrane (N@CAu-BMSNs) [47]. Anti-IDO and SDT treatment yielded decreased tumor volume and lung metastases and increased maturation and antigen-presenting capacity of DCs in tumor draining lymph nodes. CD8+ T effector cells also increased and levels of FOXP3+ cells declined, suggesting less immune suppression conferred by Tregs. In conclusion, anti-IDO-SDT combination shows promise in promoting an antitumor immune response against breast cancer.
4.0. Mechanisms of SDT
Broadly speaking, mechanisms of sonosensitizer activation with FUS include mechanical stress, sonoporation, cavitation, and sonocytotoxicity. Some studies have suggested that PDT drives changes in the F-actin structure of tumor cells, which subsequently affects cell shape and adhesion characteristics [54–56]. To test whether SDT acts through similar mechanisms, Ehrlich ascites carcinoma (EAC) cells were treated with SDT-PpIX [52]. F-Actin staining indicated lost plasma membrane integrity due to actin cytoskeletal damage and apoptotic chromatin fragmentation. Thus, one potential mechanism by which SDT works is by damaging tumor cell F-actin organization, which correlates with apoptosis induction.
Before 2013, apoptosis and necrosis were well-established pathways driving SDT-induced tumor cell death. Concurrently, the autophagy survival pathway was identified as a mechanism by which cells can survive without sufficient nutrients by eliminating damaged proteins and organelles [53]. Other publications also suggested cross-talk between autophagy and apoptosis [58,59]. Against this backdrop, the role of PpIX-mediated SDT-induced autophagy was explored in L1210 leukemia cells [50,51]. Autophagosome formation increased following SDT and ~35% of the leukemia cells underwent apoptosis. When the cells were treated with an autophagy inhibitor, the percentage of apoptotic cells increased to 50-60%, further supported by the inability of caspase inhibitors to prevent cell cytotoxicity. Additionally, ROS levels increased following SDT, but were abrogated in the presence of the ROS inhibitor N-acetyl-L-cysteine (NAC), suggesting that ROS may be driving autophagosome formation. Collectively, these results suggest that combining autophagy inhibitors and SDT may be more effective than SDT alone for initiating tumor cell death.
It has been suggested that autophagy functions as a cell survival pathway, which could lead to a resistant phenotype preventing SDT-mediated apoptosis [53]. To investigate this hypothesis, hydroxychloroquine sulfate (HCQ), an autophagy inhibitor that prevents autophagosome fusion with lysosomes and facilitates vessel normalization, was deployed. This reduced tumor hypoxia and further enhanced the SDT-mediated antitumor effect (Figure 2). A cancer cell membrane-coated HMTNP was loaded with HCQ to treat MCF-7 breast cancer cells. Autophagy was highest in the CCM-HMTNPs/HCQ+FUS group. Additionally, the CCM-HMTNPs/HCQ+FUS treatment significantly reduced tumor volume. Based on these results, blocking the autophagic flux following SDT treatment is a promising strategy to sensitize breast cancer cells to SDT-mediated apoptosis.
Figure 2.
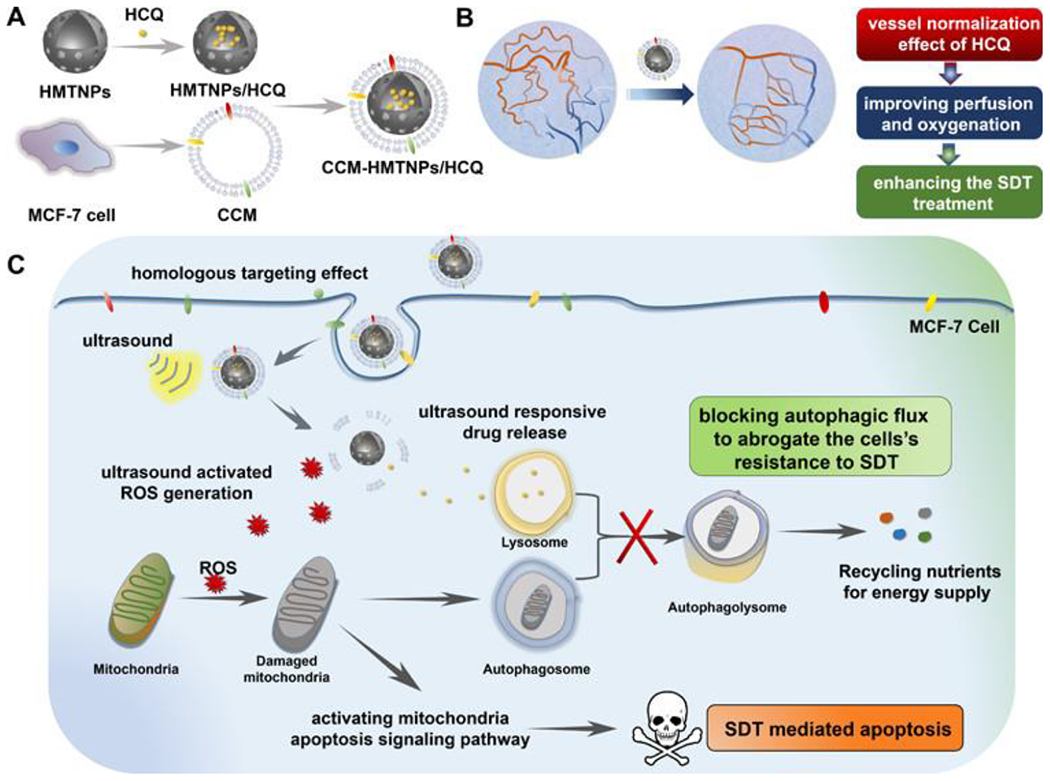
Mechanism of autophagy inhibition on SDT-mediated apoptosis of MCF-7 breast cancer cells. (a) Nanoparticle development. (b) Nanoparticle employment with HCQ leads to vessel normalization and improved perfusion and oxygenation of the tumor, potentiating enhanced SDT therapeutic efficacy. (c) CD44 MCF-7 surface marker leads to directed targeting of MCF-7 tumor cells. Application of ultrasound induces ROS generation and release of HCQ. ROS damages tumor cell mitochondria initiating apoptosis. HCQ prevents fusion of the autophagosome and lysosome blocking the autophagic flux and preventing the tumor cell’s resistance to SDT-mediated apoptosis [53]. Reproduced with permission.
Once ROS have been generated by sonosensitizer activation, mitochondria are stimulated and begin generating a secondary form of ROS, driving apoptosis and clearance of damaged mitochondria through mitophagy [58]. To understand the role of mitophagy following SDT in GL261 glioma cells, a nanoparticle using Ce6 and the autophagy inhibitor HCQ encapsulated in angiopep-2 peptide-modified liposomes was developed. Ce6 co-localized with mitochondria. PINK1, a marker of depolarization, accumulated on the outer mitochondrial membrane by 30 minutes post-SDT treatment [59]. In the presence of NAC, this accumulation was abrogated, suggesting that ROS drives mitophagy induction. In the future, combining SDT and autophagy inhibition may prove effective in preventing tumor cell death escape, inhibiting mitophagy, and forcing increased tumor cell mitochondrial damage.
The balance between sufficient tumor oxygenation for effective SDT-mediated ROS production while limiting pro-tumorigenic angiogenesis is challenging, as more blood vessels infiltrating a tumor provides more oxygen and nutrients to the tumor, making tumor growth and metastasis more likely [60]. The anti-angiogenic capacity of ALA-mediated SDT in human tongue cancer has been examined, with results showing that ALA+FUS significantly reduces microvessel density (MVD) [60]. Decreased MVD correlated with lower VEGF expression. These results suggest that ALA-mediated SDT drives angiogenesis inhibition, which contributes to SDT-mediated tumor growth control.
The role of apoptosis in SDT-induced cytotoxicity after FUS has been studied in detail [61]. Following SDT, ~18% of the leukemia cells underwent apoptosis, further supported by increased cleaved caspase-3. Cleaved caspase-9, which functions just upstream of caspase-3 in the intrinsic apoptosis signaling cascade, peaked at 0.5h post-SDT treatment. Similar upregulation of apoptosis-related genes were seen in Capan-1 pancreatic cells, including the apoptosis promoting gene BAX, cleaved caspase-3, and caspase-9 [62]. Additionally, expression of the anti-apoptotic gene Bcl-2 decreased, further supporting apoptosis induction. Treatment of C6 and U87 glioma cells with 5-ALA+FUS significantly increased caspase-positive cells [63]. Furthermore, staining for PARP-1, a mitochondrial protein activated downstream of caspase-3 pathway inhibiting DNA repair and driving apoptosis, was also significantly increased in the SDT treated cells. 5-ALA+FUS has also been shown to significantly diminish Ki67 expression in C6 glioma tumors [64]. These data further support that 5-ALA-mediated SDT drives apoptosis induction through a mitochondrial-mediated pathway.
“Hyperthermotherapy” (HT) is also known to drive SDT [65]. HT destroys cell membranes, activates lysosomes, and prevents DNA/RNA/protein synthesis [65]. 5-ALA+HT significantly reduced glioma cell viability relative to HT alone. 5-ALA+HT also increased rates of apoptosis, corresponding with decreased tumor volume. Furthermore, ROS-positive glioma cells increased in the SDT-HT group. Bcl-2 protein expression decreased, while expression of Bax and cleaved caspase-3, −8, and −9, all increased. Thus, HT-mediated SDT successfully reduced tumor cell viability, generated ROS, and promoted apoptosis, though further studies are necessary to establish if HT provides an increased sonodynamic effect relative to traditional FUS.
Though many of the papers herein focus on ROS from SDT promoting cell death, excessive ROS can exert other effects on tumor cells. DNA fragmentation is induced by DVDMS-mediated SDT in U373 glioblastoma cells [66]. Histone γH2A.X, which marks double-strand breaks in the DNA, was significantly increased following SDT. Expression of γH2A.X was reduced with NAC, suggesting that ROS drives double-strand breaks glioma cell DNA, further supporting SDT-driven apoptosis induction.
Evidence of SDT mechanisms has been shown in studies treating hepatocellular carcinoma with DVDMS [67]. Following SDT, Hep-G2 cells had higher rates of apoptosis than wild-type hepatocytes. SDT augmented cell number in the G2/M phase, while decreasing cells stalled in G0/G1. Furthermore, p21 and p27 levels were elevated, suggesting decelerated cell cycling. Meanwhile, cell cycle-promoters cyclin B1 and CDK1 were decreased [67]. The investigators also observed increased apoptosis-related pathway genes like p53, cleaved caspase-3, and Bax. In conclusion, upregulation of p21 and p27 by SDT drives an arrest in the G2/M phase of hepatocellular carcinoma cells, limiting proliferation and facilitating caspase-mediated apoptosis.
Aside from ROS, mitochondrial damage and calcium overload can also initiate apoptosis. Increases in intracellular calcium cause mitochondrial damage, release of apoptotic promoters, and subsequent activation of caspases [71–73]. Both FUS alone and HMME-mediated SDT can increase intracellular calcium, though underlying mechanisms have not yet been investigated [74,75]. It has been hypothesized that release of internal calcium stores drives ROS production, loss of mitochondrial membrane potential, and release of cytochrome c (cyt-c) [73]. SDT of C6 glioblastoma tumor cells significantly increased intracellular calcium, and upon treatment with nimodipine, which blocks the L-type extracellular calcium channel [73], the SDT-mediated calcium increase was not reduced, suggesting that internal calcium stores drive the calcium increase. Additionally, following SDT, mitochondrial membrane permeability (MMP) significantly decreased and cyt-c protein levels increased, indicating mitochondria-driven apoptosis. Furthermore, cells with the highest calcium burden also corresponded with the highest rate of apoptosis. In summary, HMME-SDT increases intracellular calcium levels, driving ROS production and ultimately leading to MMP loss and induction of apoptosis.
As discussed previously, SDT drives both programmed cell death via apoptosis and unprogrammed cell death via necrosis. However, other classes of cell death exist, including programmed necrotic cell death [i.e. necroptosis]. While apoptosis is driven by caspase activation, resulting in membrane permeability, necroptosis does not rely on caspases and is characterized by cell membrane rupture, which can release cellular components and thus drive immune cell recruitment [74]. Necroptosis is initiated when RIP3 and RIP1 complex and are phosphorylated. Whether DVDMS-mediated SDT leads to necroptosis in small-cell lung cancer has been studied [74]. In this study, RIP3 mRNA and protein levels decreased, suggesting that SDT does not drive necroptosis induction. RIP3 levels returned to normal after treatment with NAC, suggesting that ROS production drives decreased RIP3 expression. After investigating the expression of cleaved caspase-3, −8, and −9, and caspase-10, apoptosis exerted the largest impact on SDT-mediated cell death. In the presence of the Z-VAD-FMK pan-caspase inhibitor, increased caspase activity was abrogated. In all, these results suggest that DVDMS-mediated SDT does not drive tumor cells to undergo necroptosis.
Ferroptosis is yet another form of regulated cell death. It is an iron-dependent regulated form of cell death which promotes ROS accumulation and lipid peroxidation (LPO), both of which are lethal to the tumor cell [75]. Glutathione peroxidase 4 (GPX4) acts downstream of ferroptosis to inhibit LPO accumulation by using GSH to reduce the toxic hyperoxides. Furthermore, GSH levels are notably high in hypoxic tumors [75]. It has been hypothesized that Mn-MOFs, which deliver oxygen to the tumor, could alleviate hypoxia and facilitate increased ferroptosis [75]. Reduced (i.e. up to 2-fold) GSH levels are observed in 4T1 and H22 hepatocellular tumor cells in the presence of Mn-MOF nanoparticles, suggesting alleviated tumor hypoxia through oxygen deposition. Additionally, GPX4 activity decreased by ~80% after Mn-MOF+FUS relative to Mn-MOF alone. These results suggest that ferroptosis is a possible mechanism of SDT-mediated cell death. In addition, the role of both ROS and increased ferroptosis in inducing an antitumor immune response has been studied. Here, the investigators observed an increase in CD4+ and CD8+ T cells, as well as CD69+ recently activated T cells, which returned to levels comparable to the controls in the presence of UAMC-3203, a ferroptosis inhibitor. CD8+ T cells secreting IFN-γ and granzyme B were also increased, as were mature DCs. In conclusion, these data suggest that Mn-MOF-mediated SDT can alleviate ferroptosis inhibition and can reshape the tumor immune landscape, showing promise for eliciting an effective antitumor response.
5.0. Modulating the Tumor Immune Landscape
This final section explores SDT’s ability to modulate the TME to increase immune cell infiltration. Figure 3 illustrates mechanisms through which SDT may elicit ICD and damage associated molecular pattern (DAMP) expression, leading to APC (i.e. DCs and macrophages) activation, T cell priming, and potentially triggering an adaptive immune response. The effect of 5-ALA-mediated SDT on macrophages and DCs has been studied in B16/F10 melanoma tumors [76], leading to the finding that both FUS alone and 5-ALA+FUS can increase intratumoral macrophages. There was a decrease in the number of CD163+ M2-type macrophages, which are traditionally thought of as pro-tumorigenic, in the presence of FUS alone and SDT, suggesting that 5-ALA-mediated SDT may drive the re-education of pro-tumorigenic macrophages into M1-type pro-inflammatory and anti-tumorigenic macrophages. In addition, the authors saw an increase in CD80 and CD86 expression following FUS alone and SDT, indicating that FUS may drive DC activation and subsequent antigen presentation to T cells. Furthermore, IL-10, IFN-γ, and TNF-α expression also increased in the FUS and SDT groups. These cytokines facilitate immune recruitment and are typically associated with M1 macrophages, further supporting that SDT drives re-education of macrophages to an antitumor phenotype.
Figure 3.
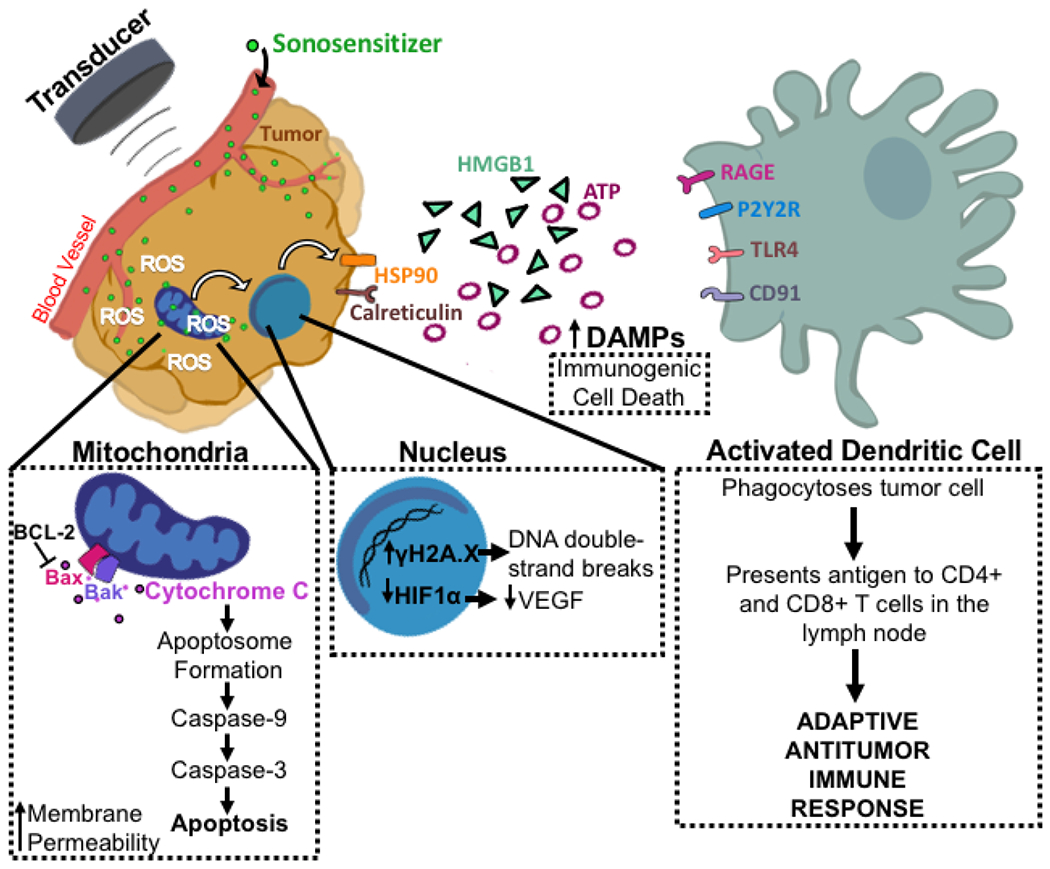
Proposed mechanism of SDT inducing ICD and release of DAMPs. The administered sonosensitizer accumulates specifically in the tumor. FUS administration activates the sensitizer to generate ROS and induce mitochondrial-mediated apoptosis. Bax and Bak form a pore on the outer mitochondrial membrane for release of cyt-c. Cyt-c complexes with adaptor proteins to form the apoptosome which cleaves caspase-9. Caspase-3 is then cleaved and functions as the executioner caspase to induce apoptotic cell death. In addition, ROS generation leads to increased γH2A.X [66], which marks DNA double-strand breaks, and decreased HIF1α, which reduces VEGF expression [77], in the nucleus. Finally, DAMPs including HSP90, calreticulin, HMGB1, and ATP are upregulated [78]–[81]. These DAMPs are then recognized by DCs which drives their activation leading to phagocytosis of the tumor cell and an adaptive antitumor immune response.
Like standard chemotherapeutic treatments and radiation, SDT may release tumor antigens, facilitating better immune cell tumor recognition. Studies have employed Zn-TCPP nanosheets loaded with CpG (a TLR9 agonist) to treat CT26 colorectal cancer [78]. The investigators evaluated whether SDT induced ICD, characterized by increased cell membrane CRT expression on dying cancer cells, adenosine triphosphate (ATP) release, heat shock proteins (HSPs), and high mobility group box 1 (HMBG1), all of which facilitate immune cell recognition of dying cells. Surface CRT and ATP levels increased following Zn-TCPP+FUS treatment, suggesting that SDT initiated ICD. DCs were more mature following treatment with Zn-TCPP as evidenced by augmented CD80 and CD86 expression, and a significant increase in the CD8+ T cells infiltration into the tumor was reported. Furthermore, VCAM-1 expression, which facilitates tethering of circulating T cells to endothelial cells, was increased following treatment with both Zn-TCPP+FUS and Zn-TCPP/CpG+FUS. Finally, generation of immunological memory was confirmed as T effector cells increased in the re-challenged tumor bearing mice that received SDT treatment 40 days prior. Thus, SDT has the potential to reverse the immunosuppressive environment often associated with tumors and perhaps drive an immunological memory response that could prevent disease recurrence.
The potential for SDT to mediate ICD was also tested by designing a liposome-coated manganese-protoporphyrin complex (MnP) with folate (Figure 4), as many folate receptors are overexpressed on the surface of cancers [79]. Stimulation of 4T1 breast cancer cells with FA-MnPs+FUS significantly increased CRT, HMGB1, and ATP expression, suggesting that FA-MnP-SDT induced DAMP release and ICD. SDT treatment also led to the upregulation of DC activation markers. Further analysis of tumor-draining lymph node (TDLN) and tumor immune infiltrate also showed increased CRT expression and mature DCs. While the percent of activated CD8+ T cells were not increased in the TDLN, they were increased in the tumor. In addition, activated CD4+ T cells and NK cells increased, while the number of Tregs decreased in both the tumor and TDLN. Interestingly, analysis of macrophage polarization revealed a decrease in M2-type macrophages, supporting a previous study [76]. In all, these data suggest that SDT can drive ICD leading to DC maturation and subsequent activation of T cells and NK cells, while also re-educating immunosuppressive macrophages in the TME.
Figure 4.
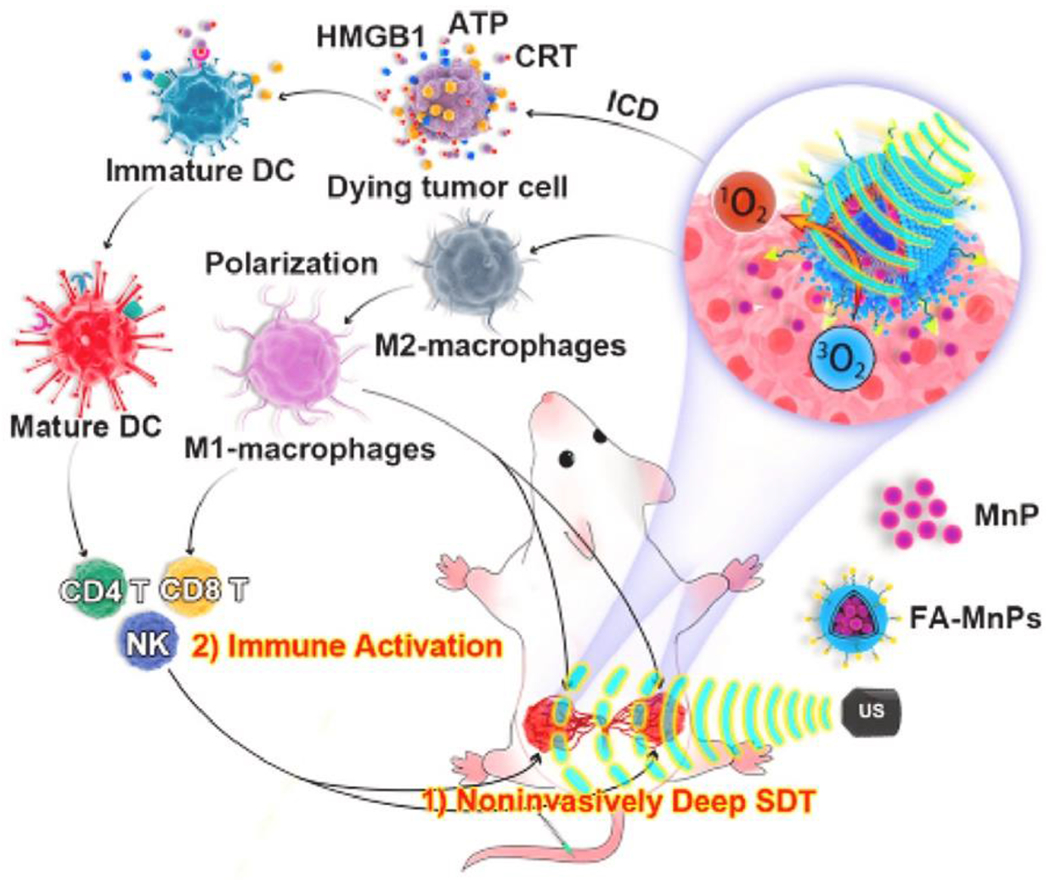
Schematic of immune activation by FA-MnPs for tumor suppression. FA-MnPs accumulate in bilateral tumors in mice which are activated by FUS. The activation generates O2 triggering ICD, DAMP release, M1 macrophage polarization, DC maturation, and subsequent activation of an adaptive antitumor immunity inhibiting tumor growth [79]. Reproduced with permission.
In another study, ZrO2–xNPs (ZPR NPs) were coated with cyclic-Arg-Gly-Asp peptide, which interacts with overexpressed αvβ3 integrin on tumor cells to help facilitate targeted drug delivery [80]. While CRT and HSP90 expression increased in treated 4T1 breast cancer cells, HMGB1 levels were somewhat decreased. HSP90, another marker of dying cells, interacts with CD91 on DCs leading to maturation and antigen presentation on MHCI, driving cytotoxic T lymphocyte activation (Figure 5) [80]. Furthermore, extracellular ATP levels, which drive inflammasome activation and recruitment of APCs, were increased following SDT treatment [80], suggesting that ZPR-NPs promote ICD. Moreover, this corresponded to 4T1 tumor growth control. Of note, TNF-α and IL-6 proinflammatory cytokines were upregulated, and MHCI expression was increased on APCs, leading to better T cell differentiation. These data support the induction of ICD by SDT and the subsequent release of DAMPs leading to an antitumor immune response.
Figure 5.
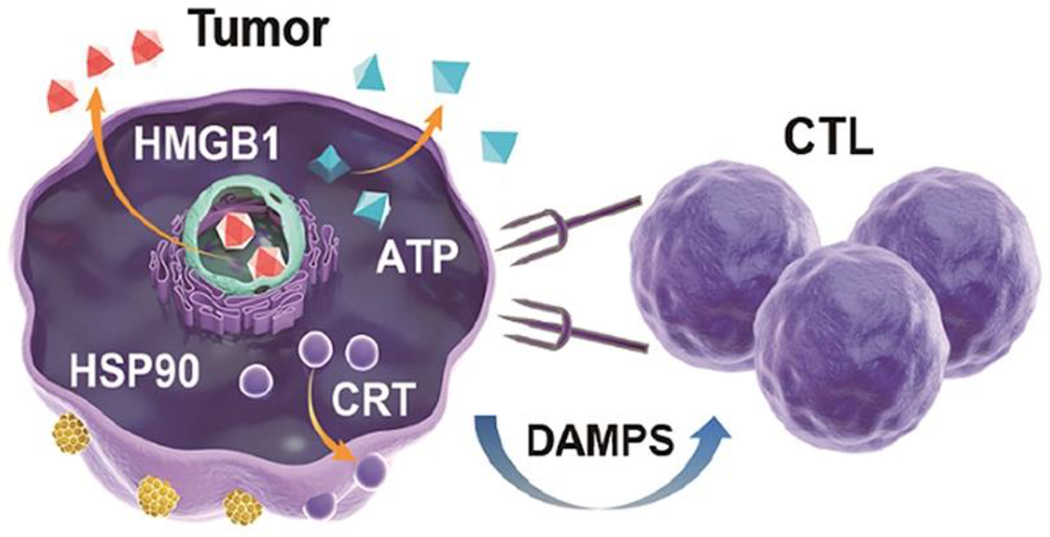
Diagram of ICD promoting cytotoxic T lymphocyte (CTL)-directed tumor targeting. Release of HMGB1 and ATP and surface expression of CRT and HSP90 signal to DCs and CTLs for tumor destruction [80]. Reproduced with permission.
Though ICD is a natural oncological immunotherapy, it is often inhibited by the immunosuppressive TME [81]. To address this issue, a continuous ultrasound-triggered inertial cavitation (UIC) SDT platform has been employed to continuously release carbon dioxide, allowing for the production of enough ROS to overcome the immunosuppressive TME and induce ICD [81]. In the presence of this treatment approach, 4T1 breast cancer tumors showed increased CRT, HMGB1, and HSP70 expression. The number of mature DCs and CD8+ cytotoxic T cells increased. Levels of proinflammatory cytokines IFN-γ, IL-12, TNF-α, and IL-6 all increased as well. These data suggest that this UIC-SDT platform can successfully overcome the immunosuppressive tumor environment and facilitate ICD and subsequent immune cell infiltration into an otherwise immunologically “cold” tumor.
6.0. Future Directions
Throughout this review we have outlined promising SDT approaches for controlling tumor growth. Moving forward, amelioration of SDT-immunotherapy approaches for treating solid tumors is essential, likely facilitated by understanding the tumor immune landscape to assess immunotherapy targets. Furthermore, it is important to keep in mind that tumor cells can acquire resistance to immune checkpoint inhibitors (ICIs), much like chemotherapeutics. Therefore, targeting multiple non-redundant immune checkpoint pathways in combination with SDT may provide better therapeutic responses, and may also be necessary for long-term tumor-specific immunological control [82].
After discussing the combination therapy approaches, we discussed mechanisms through which SDT elicits tumor cytotoxicity. Mechanisms of apoptosis induction from SDT have been well characterized, and some data suggests that autophagy plays a role in resisting the cytotoxic effects of SDT through cross-talk with the apoptosis pathway [47–50], leaving room for investigation. Additionally, ferroptosis may mediate an additional form of SDT-induced tumor cell death [75]. More research needs to be conducted to establish if other cell death pathways may contribute to the sonocytotoxicity of tumor cells.
Regarding SDT’s immunomodulatory capacity, it is clear that SDT can initiate ICD, facilitating tumor cell recognition and phagocytosis [70,73]. ICD post-SDT spurs phagocytosis of dying cells, antigen processing, and presentation of tumor antigens, critical steps for T cell recognition and antigen-specific targeting of tumor cells. This suggests that SDT can not only improve target cancer cells for destruction but can also elicit a more effective antitumor immune response. Additional research is necessary to improve ICD’s effects and determine if ICIs administered concurrently with ICD elicits enhanced adaptive antitumor immunity. Furthermore, future research should continue to assess and optimize the ability of SDT-mediated ICD to convert immunologically “cold” tumors into immunologically “hot” tumors.
Sonodynamic therapy (SDT) elicits tumor cell death through ROS production
ROS generation is the result of the activation of sonosensitizers with ultrasound
SDT modulates the tumor immune landscape
Solid tumor treatments may combine SDT with chemo- and/or immunotherapies
Progress to date in sonodynamic therapy is comprehensively reviewed
Acknowledgements
Supported by NIH R01 CA204968, R01EB030409, R01EB030744, R01EB030007, R21CA230088, and R21NS118278 to RJP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Wang P et al. “Anti-metastatic and pro-apoptotic effects elicited by combination photodynamic therapy with sonodynamic therapy on breast cancer both in vitro and in vivo,” Ultrason. Sonochem, vol. 23, pp. 116–127, Mar. 2015, doi: 10.1016/j.ultsonch.2014.10.027. [DOI] [PubMed] [Google Scholar]
- [2].Dolmans DEJGJ, Fukumura D, and Jain RK, “Photodynamic therapy for cancer,” Nat. Rev. Cancer, vol. 3, no. 5, pp. 380–387, May 2003, doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]
- [3].An Y-W et al. “Sinoporphyrin sodium is a promising sensitizer for photodynamic and sonodynamic therapy in glioma,” Oncol. Rep, vol. 44, no. 4, pp. 1596–1604, Oct. 2020, doi: 10.3892/or.2020.7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hou R, Liang X, Li X, Zhang X, Ma X, and Wang F, “In situ conversion of rose bengal microbubbles into nanoparticles for ultrasound imaging guided sonodynamic therapy with enhanced antitumor efficacy,” Biomater. Sci, vol. 8, no. 9, pp. 2526–2536, 2020, doi: 10.1039/C9BM02046B. [DOI] [PubMed] [Google Scholar]
- [5].Li C et al. “Red blood cell membrane-enveloped O2 self-supplementing biomimetic nanoparticles for tumor imaging-guided enhanced sonodynamic therapy,” Theranostics, vol. 10, no. 2, pp. 867–879, Jan. 2020, doi: 10.7150/thno.37930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Soratijahromi E, Mohammadi S, Dehdari Vais R, Azarpira N, and Sattarahmady N, “Photothermal/sonodynamic therapy of melanoma tumor by a gold/manganese dioxide nanocomposite: In vitro and in vivo studies,” Photodiagnosis Photodyn. Ther, vol. 31, p. 101846, Sep. 2020, doi: 10.1016/j.pdpdt.2020.101846. [DOI] [PubMed] [Google Scholar]
- [7].Gong Z and Dai Z, “Design and Challenges of Sonodynamic Therapy System for Cancer Theranostics: From Equipment to Sensitizers,” Adv. Sci, vol. 8, no. 10, p. 2002178, 2021, doi: 10.1002/advs.202002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li L, Lin H, Li D, Zeng Y, and Liu G, “Ultrasound activated nanosensitizers for sonodynamic therapy and theranostics,” Biomed. Mater, vol. 16, no. 2, p. 022008, Feb. 2021, doi: 10.1088/1748-605X/abd382. [DOI] [PubMed] [Google Scholar]
- [9].Pan X et al. “Sonodynamic therapy (SDT): a novel strategy for cancer nanotheranostics,” Sci. China Life Sci, vol. 61, no. 4, pp. 415–426, Apr. 2018, doi: 10.1007/s11427-017-9262-x. [DOI] [PubMed] [Google Scholar]
- [10].Son S et al. “Multifunctional sonosensitizers in sonodynamic cancer therapy,” Chem. Soc. Rev, vol. 49, no. 11, pp. 3244–3261, 2020, doi: 10.1039/C9CS00648F. [DOI] [PubMed] [Google Scholar]
- [11].Xu T, Zhao S, Lin C, Zheng X, and Lan M, “Recent advances in nanomaterials for sonodynamic therapy,” Nano Res, vol. 13, no. 11, pp. 2898–2908, Nov. 2020, doi: 10.1007/s12274-020-2992-5. [DOI] [Google Scholar]
- [12].Zhou Y, Wang M, and Dai Z, “The molecular design of and challenges relating to sensitizers for cancer sonodynamic therapy,” Mater. Chem. Front, vol. 4, no. 8, pp. 2223–2234, 2020, doi: 10.1039/D0QM00232A. [DOI] [Google Scholar]
- [13].Shi H, Liu Q, Qin X, Wang P, and Wang X, “Pharmacokinetic study of a novel sonosensitizer chlorin-e6 and its sonodynamic anti-cancer activity in hepatoma-22 tumor-bearing mice,” Biopharm. Drug Dispos, vol. 32, no. 6, pp. 319–332, 2011, doi: 10.1002/bdd.761. [DOI] [PubMed] [Google Scholar]
- [14].Chen B, Zheng R, Liu D, Li B, Lin J, and Zhang W, “The tumor affinity of chlorin e6 and its sonodynamic effects on non-small cell lung cancer,” Ultrason. Sonochem, vol. 20, no. 2, pp. 667–673, Mar. 2013, doi: 10.1016/j.ultsonch.2012.09.008. [DOI] [PubMed] [Google Scholar]
- [15].Yumita N, Nishigaki R, Umemura K, and Umemura S, “Hematoporphyrin as a Sensitizer of Cell-damaging Effect of Ultrasound,” Jpn. J. Cancer Res, vol. 80, no. 3, pp. 219–222, 1989, doi: 10.1111/j.1349-7006.1989.tb02295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hu J et al. “Sinoporphyrin sodium: a novel sensitizer in sonodynamic therapy,” Anticancer. Drugs, vol. 25, no. 2, pp. 174–182, Feb. 2014, doi: 10.1097/CAD.0000000000000031. [DOI] [PubMed] [Google Scholar]
- [17].Shen Y et al. “An in vitro study on sonodynamic treatment of human colon cancer cells using sinoporphyrin sodium as sonosensitizer,” Biomed. Eng. OnLine, vol. 19, no. 1, p. 52, Jun. 2020, doi: 10.1186/s12938-020-00797-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Li Y et al. “Ultrasound-triggered release of sinoporphyrin sodium from liposome-microbubble complexes and its enhanced sonodynamic toxicity in breast cancer,” Nano Res, vol. 11, no. 2, pp. 1038–1056, Feb. 2018, doi: 10.1007/s12274-017-1719-8. [DOI] [Google Scholar]
- [19].Li Y et al. “IR-780 Dye as a Sonosensitizer for Sonodynamic Therapy of Breast Tumor,” Sci. Rep, vol. 6, no. 1, Art. no. 1, May 2016, doi: 10.1038/srep25968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Han X et al. “Mitochondria-targeted high-load sound-sensitive micelles for sonodynamic therapy to treat triple-negative breast cancer and inhibit metastasis,” Mater. Sci. Eng. C, vol. 124, p. 112054, May 2021, doi: 10.1016/j.msec.2021.112054. [DOI] [PubMed] [Google Scholar]
- [21].Wang P et al. “Nanosonosensitization by Using Copper—Cysteamine Nanoparticles Augmented Sonodynamic Cancer Treatment - Wang - 2018 - Particle & Particle Systems Characterization - Wiley Online Library,” Part. Part. Syst. Charact, vol. 35, no. 4, p. 1700378, Feb. 2018, doi: 10.1002/ppsc.201700378. [DOI] [Google Scholar]
- [22].Ma L et al. “A new Cu—cysteamine complex: structure and optical properties,” J. Mater. Chem. C, vol. 2, no. 21, pp. 4239–4246, May 2014, doi: 10.1039/C4TC00114A. [DOI] [Google Scholar]
- [23].Rao L et al. “Red Blood Cell Membrane as a Biomimetic Nanocoating for Prolonged Circulation Time and Reduced Accelerated Blood Clearance,” Small Weinh. Bergstr. Ger, vol. 11, no. 46, pp. 6225–6236, Dec. 2015, doi: 10.1002/smll.201502388. [DOI] [PubMed] [Google Scholar]
- [24].Hadi MM et al. “Investigating the performance of a novel pH and cathepsin B sensitive, stimulus-responsive nanoparticle for optimised sonodynamic therapy in prostate cancer,” J. Controlled Release, vol. 329, pp. 76–86, Jan. 2021, doi: 10.1016/j.jconrel.2020.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sadanala KC et al. “Sono-photodynamic combination therapy: a review on sensitizers,” Anticancer Res, vol. 34, no. 9, pp. 4657–4664, Sep. 2014. [PubMed] [Google Scholar]
- [26].Su X, Li L, and Wang P, “[Research progress of the anti-tumor effect of sonodynamic and photodynamic therapy],” Sheng Wu Yi Xue Gong Cheng Xue Za Zhi J. Biomed. Eng. Shengwu Yixue Gongchengxue Zazhi, vol. 29, no. 3, pp. 583–587, Jun. 2012. [PubMed] [Google Scholar]
- [27].Wang S, Lu W, Tovmachenko O, Rai US, Yu H, and Ray PC, “Challenge in understanding size and shape dependent toxicity of gold nanomaterials in human skin keratinocytes,” Chem. Phys. Lett, vol. 463, no. 1, pp. 145–149, Sep. 2008, doi: 10.1016/j.cplett.2008.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gorgizadeh M et al. “A MnFe2O4/C nanocomposite as a novel theranostic agent in MRI, sonodynamic therapy and photothermal therapy of a melanoma cancer model,” J. Alloys Compd, vol. 816, p. 152597, Mar. 2020, doi: 10.1016/j.jallcom.2019.152597. [DOI] [Google Scholar]
- [29].Chen L, Cong D, Li Y, Wang D, Li Q, and Hu S, “Combination of sonodynamic with temozolomide inhibits C6 glioma migration and promotes mitochondrial pathway apoptosis via suppressing NHE-1 expression,” Ultrason. Sonochem, vol. 39, pp. 654–661, Nov. 2017, doi: 10.1016/j.ultsonch.2017.05.013. [DOI] [PubMed] [Google Scholar]
- [30].Bower M et al. “Multicentre CRC phase II trial of temozolomide in recurrent or progressive high-grade glioma,” Cancer Chemother. Pharmacol, vol. 40, no. 6, pp. 484–488, Sep. 1997, doi: 10.1007/S002800050691. [DOI] [PubMed] [Google Scholar]
- [31].Nesbitt H et al. “Gemcitabine loaded microbubbles for targeted chemo-sonodynamic therapy of pancreatic cancer,” J. Controlled Release, vol. 279, pp. 8–16, Jun. 2018, doi: 10.1016/j.jconrel.2018.04.018. [DOI] [PubMed] [Google Scholar]
- [32].Xing L et al. “Ultrasound-Mediated Microbubble Destruction (UMMD) Facilitates the Delivery of CA19-9 Targeted and Paclitaxel Loaded mPEG-PLGA-PLL Nanoparticles in Pancreatic Cancer,” Theranostics, vol. 6, no. 10, pp. 1573–1587, Jan. 2016, doi: 10.7150/thno.15164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Browning RJ et al. “Combining sonodynamic therapy with chemoradiation for the treatment of pancreatic cancer,” J. Controlled Release, vol. 337, pp. 371–377, Sep. 2021, doi: 10.1016/j.jconrel.2021.07.020. [DOI] [PubMed] [Google Scholar]
- [34].Kusuzaki K et al. “Clinical trial of photodynamic therapy using acridine orange with/without low dose radiation as new limb salvage modality in musculoskeletal sarcomas,” Anticancer Res, vol. 25, no. 2B, pp. 1225–1235, Mar. 2005. [PubMed] [Google Scholar]
- [35].Freitag L, Ernst A, Thomas M, Prenzel R, Wahlers B, and Macha H-N, “Sequential photodynamic therapy (PDT) and high dose brachytherapy for endobronchial tumour control in patients with limited bronchogenic carcinoma,” Thorax, vol. 59, no. 9, pp. 790–793, Sep. 2004, doi: 10.1136/thx.2003.013599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Beguin E et al. “Magnetic microbubble mediated chemo-sonodynamic therapy using a combined magnetic-acoustic device,” J. Controlled Release, vol. 317, pp. 23–33, Jan. 2020, doi: 10.1016/j.jconrel.2019.11.013. [DOI] [PubMed] [Google Scholar]
- [37].Bardal SK, Waechter JE, and Martin DS, “Chapter 20 - Neoplasia,” in Applied Pharmacology, Bardal SK, Waechter JE, and Martin DS, Eds. Philadelphia: W.B. Saunders, 2011, pp. 305–324. doi: 10.1016/B978-1-4377-0310-8.00020-8. [DOI] [Google Scholar]
- [38].An J et al. “ROS-augmented and tumor-microenvironment responsive biodegradable nanoplatform for enhancing chemo-sonodynamic therapy,” Biomaterials, vol. 234, p. 119761, Mar. 2020, doi: 10.1016/j.biomaterials.2020.119761. [DOI] [PubMed] [Google Scholar]
- [39].Huang B et al. “Oxygen-Sufficient Nanoplatform for Chemo-Sonodynamic Therapy of Hypoxic Tumors,” Front. Chem, vol. 8, Apr. 2020, doi: 10.3389/fchem.2020.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Warburg O, “On the Origin of Cancer Cells,” Sci. New Ser, vol. 123, no. 3191, pp. 309–314, 1956, [Online], Available: http://www.jstor.org/stable/1750066 [DOI] [PubMed] [Google Scholar]
- [41].Vander Heiden MG, Cantley LC, and Thompson CB, “Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation,” Science, vol. 324, no. 5930, pp. 1029–1033, May 2009, doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Xie L et al. “Blocking the Glycolytic Pathway Sensitizes Breast Cancer to Sonodynamic Therapy,” Ultrasound Med. Biol, vol. 44, no. 6, pp. 1233–1243, Jun. 2018, doi: 10.1016/j.ultrasmedbio.2018.01.020. [DOI] [PubMed] [Google Scholar]
- [43].Xie L, Feng X, Huang M, Zhang K, and Liu Q, “Sonodynamic Therapy Combined to 2-Deoxyglucose Potentiate Cell Metastasis Inhibition of Breast Cancer,” Ultrasound Med. Biol, vol. 45, no. 11, pp. 2984–2992, Nov. 2019, doi: 10.1016/j.ultrasmedbio.2019.07.008. [DOI] [PubMed] [Google Scholar]
- [44].Nesbitt H et al. “Sonodynamic therapy complements PD-L1 immune checkpoint inhibition in a murine model of pancreatic cancer,” Cancer Lett, vol. 517, pp. 88–95, Oct. 2021, doi: 10.1016/j.canlet.2021.06.003. [DOI] [PubMed] [Google Scholar]
- [45].Yue W et al. “Checkpoint blockade and nanosonosensitizer-augmented noninvasive sonodynamic therapy combination reduces tumour growth and metastases in mice,” Nat. Commun, vol. 10, no. 1, Art. no. 1, May 2019, doi: 10.1038/s41467-019-09760-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhan G et al. “Biomimetic sonodynamic therapy-nanovaccine integration platform potentiates Anti-PD-1 therapy in hypoxic tumors,” Nano Today, vol. 38, p. 101195, Jun. 2021, doi: 10.1016/j.nantod.2021.101195. [DOI] [Google Scholar]
- [47].Zhang D et al. “Ultrasound-Driven Biomimetic Nanosystem Suppresses Tumor Growth and Metastasis through Sonodynamic Therapy, CO Therapy, and Indoleamine 2,3-Dioxygenase Inhibition,” ACS Nano, vol. 14, no. 7, pp. 8985–8999, Jul. 2020, doi: 10.1021/acsnano.0c03833. [DOI] [PubMed] [Google Scholar]
- [48].Cheng K et al. “Sequentially Responsive Therapeutic Peptide Assembling Nanoparticles for Dual-Targeted Cancer Immunotherapy,” Nano Lett, vol. 18, no. 5, pp. 3250–3258, May 2018, doi: 10.1021/acs.nanolett.8b01071. [DOI] [PubMed] [Google Scholar]
- [49].Juarranz A et al. “Photodamage Induced by Zinc(II)-phthalocyanine to Microtubules, Actin, α-Actinin and Keratin of HeLa Cells¶,” Photochem. Photobiol, vol. 73, no. 3, pp. 283–289, 2001, doi: 10.1562/0031-8655(2001)0730283PIBZIP2.0.CO2. [DOI] [PubMed] [Google Scholar]
- [50].Uzdensky A, Kolpakova E, Juzeniene A, Juzenas P, and Moan J, “The effect of sub-lethal ALA-PDT on the cytoskeleton and adhesion of cultured human cancer cells,” Biochim. Biophys. Acta BBA - Gen. Subj, vol. 1722, no. 1, pp. 43–50, Feb. 2005, doi: 10.1016/j.bbagen.2004.11.011. [DOI] [PubMed] [Google Scholar]
- [51].Pluskalová M, Pešlová G, Grebeňová D, Halada P, and Hrkal Z, “Photodynamic treatment (ALA-PDT) suppresses the expression of the oncogenic Bcr-Abl kinase and affects the cytoskeleton organization in K562 cells,” J. Photochem. Photobiol. B, vol. 83, no. 3, pp. 205–212, Jun. 2006, doi: 10.1016/j.jphotobiol.2006.01.003. [DOI] [PubMed] [Google Scholar]
- [52].Zhao X et al. “Damage effects of protoporphyrin IX — Sonodynamic therapy on the cytoskeletal F-actin of Ehrlich ascites carcinoma cells,” Ultrason. Sonochem, vol. 16, no. 1, pp. 50–56, Jan. 2009, doi: 10.1016/j.ultsonch.2008.05.005. [DOI] [PubMed] [Google Scholar]
- [53].Feng Q et al. “Cancer Cell Membrane-Biomimetic Nanoplatform for Enhanced Sonodynamic Therapy on Breast Cancer via Autophagy Regulation Strategy | ACS Applied Materials & Interfaces,” ACS Appl Mater. Interfaces, vol. 11, no. 36, pp. 32729–32738, Aug. 2019, doi: 10.1021/acsami.9b10948. [DOI] [PubMed] [Google Scholar]
- [54].Maiuri MC, Zalckvar E, Kimchi A, and Kroemer G, “Self-eating and self-killing: crosstalk between autophagy and apoptosis,” Nat. Rev. Mol. Cell Biol, vol. 8, no. 9, pp. 741–752, Sep. 2007, doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- [55].Boya P et al. “Inhibition of macroautophagy triggers apoptosis,” Mol. Cell. Biol, vol. 25, no. 3, pp. 1025–1040, Feb. 2005, doi: 10.1128/mcb.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang X, Wang P, Zhang K, Su X, Hou J, and Liu Q, “Initiation of autophagy and apoptosis by sonodynamic therapy in murine leukemia L1210 cells,” Toxicol. In Vitro, vol. 27, no. 4, pp. 1247–1259, Jun. 2013, doi: 10.1016/j.tiv.2012.12.023. [DOI] [PubMed] [Google Scholar]
- [57].Wang X et al. “Role of Autophagy in Sonodynamic Therapy-Induced Cytotoxicity in S180 Cells,” Ultrasound Med. Biol, vol. 36, no. 11, pp. 1933–1946, Nov. 2010, doi: 10.1016/j.ultrasmedbio.2010.06.022. [DOI] [PubMed] [Google Scholar]
- [58].Qu F et al. “Full article: Manipulation of Mitophagy by ‘All-in-One’ nanosensitizer augments sonodynamic glioma therapy,” Autophagy, vol. 16, no. 8, pp. 1413–1435, Nov. 2019, doi: 10.1080/15548627.2019.1687210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].King L and Plun-Favreau H, “Chapter 5 - Mitophagy,” in Parkinson’s Disease, Verstreken P, Ed. San Diego: Academic Press, 2017, pp. 139–177. doi: 10.1016/B978-0-12-803783-6.00005-5. [DOI] [Google Scholar]
- [60].Gao Z et al. “Sonodynamic therapy inhibits angiogenesis and tumor growth in a xenograft mouse model,” Cancer Lett, vol. 335, no. 1, pp. 93–99, Jul. 2013, doi: 10.1016/j.canlet.2013.02.006. [DOI] [PubMed] [Google Scholar]
- [61].Li Y, Wang P, Wang X, Su X, and Liu Q, “Involvement of Mitochondrial and Reactive Oxygen Species in the Sonodynamic Toxicity of Chlorin e6 in Human Leukemia K562 Cells,” Ultrasound Med. Biol, vol. 40, no. 5, pp. 990–1000, May 2014, doi: 10.1016/j.ultrasmedbio.2013.11.022. [DOI] [PubMed] [Google Scholar]
- [62].Li YJ et al. “Sonodynamically Induced Anti-tumor Effect of 5-Aminolevulinic Acid on Pancreatic Cancer Cells,” Ultrasound Med. Biol, vol. 40, no. 11, pp. 2671–2679, Nov. 2014, doi: 10.1016/j.ultrasmedbio.2014.07.003. [DOI] [PubMed] [Google Scholar]
- [63].Sheehan K et al. “Investigation of the tumoricidal effects of sonodynamic therapy in malignant glioblastoma brain tumors | SpringerLink,” J. Neurooncol, vol. 148, pp. 9–16, May 2020, doi: 10.1007/s11060-020-03504-w. [DOI] [PubMed] [Google Scholar]
- [64].Wu S-K, Santos M, Marcus S, and Hynynen K, “MR-guided Focused Ultrasound Facilitates Sonodynamic Therapy with 5-Aminolevulinic Acid in a Rat Glioma Model | Scientific Reports,” Sci. Rep, vol. 9, p. 10465, Jul. 2019, doi: 10.1038/s41598-019-46832-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ju D et al. “Hyperthermotherapy enhances antitumor effect of 5-aminolevulinic acid-mediated sonodynamic therapy with activation of caspase-dependent apoptotic pathway in human glioma | SpringerLink,” Tumor Biol, vol. 37, pp. 10415–10426, Feb. 2016, doi: 10.1007/s13277-016-4931-3. [DOI] [PubMed] [Google Scholar]
- [66].Sun Y et al. “Sonodynamic therapy induces oxidative stress, DNA damage and apoptosis in glioma cells,” RSC Adv, vol. 8, no. 63, pp. 36245–36256, 2018, doi: 10.1039/C8RA07099G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Li E et al. “Sinoporphyrin sodium based sonodynamic therapy induces anti-tumor effects in hepatocellular carcinoma and activates p53/caspase 3 axis,” Int. J. Biochem. Cell Biol, vol. 113, pp. 104–114, Aug. 2019, doi: 10.1016/j.biocel.2019.01.009. [DOI] [PubMed] [Google Scholar]
- [68].Adachi S et al. “Cyclin A/cdk2 activation is involved in hypoxia-induced apoptosis in cardiomyocytes,” Circ. Res, vol. 88, no. 4, pp. 408–414, Mar. 2001, doi: 10.1161/01.res.88.4.408. [DOI] [PubMed] [Google Scholar]
- [69].Green DR and Reed JC, “Mitochondria and apoptosis,” Science, vol. 281, no. 5381, pp. 1309–1312,Aug. 1998, doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- [70].Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, and Vandenabeele P, “Toxic proteins released from mitochondria in cell death,” Oncogene, vol. 23, no. 16, pp. 2861–2874, Apr. 2004, doi: 10.1038/sj.onc.1207523. [DOI] [PubMed] [Google Scholar]
- [71].Honda H, Kondo T, Zhao G-L, Feril LB, and Kitagawa H, “Role of intracellular calcium ions and reactive oxygen species in apoptosis induced by ultrasound,” Ultrasound Med. Biol, vol. 30, no. 5, pp. 683–692, May 2004, doi: 10.1016/j.ultrasmedbio.2004.02.008. [DOI] [PubMed] [Google Scholar]
- [72].Li J-H et al. “Calcium overload induces C6 rat glioma cell apoptosis in sonodynamic therapy,” Int. J. Radiat. Biol, vol. 87, no. 10, pp. 1061–1066, Oct. 2011, doi: 10.3109/09553002.2011.584938. [DOI] [PubMed] [Google Scholar]
- [73].Hao D, Song Y, Che Z, and Liu O, “Calcium Overload and in vitro Apoptosis of the C6 Glioma Cells Mediated by Sonodynamic Therapy (Hematoporphyrin monomethyl ether and ultrasound),” Cell Biochem. Biophys, vol. 70, no. 2, pp. 1445–1452, 2014, doi: 10.1007/s12013-014-0081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Shen J et al. “Sinoporphyrin Sodium-Mediated Sonodynamic Therapy Inhibits RIP3 Expression and Induces Apoptosis in the H446 Small Cell Lung Cancer Cell Line - Abstract - Cellular Physiology and Biochemistry 2018, Vol. 51, No. 6 - Karger Publishers,” Cell. Physiol. Biochem, vol. 51, no. 6, pp. 2938–2954, Jan. 2019, doi: 10.1159/000496045. [DOI] [PubMed] [Google Scholar]
- [75].Xu Q, Zhan G, Zhang Z, Yong T, Yang X, and Gan L, “Manganese porphyrin-based metal-organic framework for synergistic sonodynamic therapy and ferroptosis in hypoxic tumors,” Theranostics, vol. 11, no. 4, pp. 1937–1952, Jan. 2021, doi: 10.7150/thno.45511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang S et al. “5-Aminolevulinic acid-mediated sonodynamic therapy reverses macrophage and dendritic cell passivity in murine melanoma xenografts,” Ultrasound Med. Biol, vol. 40, no. 9, pp. 2125–2133, Sep. 2014, doi: 10.1016/j.ultrasmedbio.2014.05.007. [DOI] [PubMed] [Google Scholar]
- [77].Mizukami Y, Li J, Zhang X, Zimmer MA, Iliopoulos O, and Chung DC, “Hypoxia-inducible factor-1-independent regulation of vascular endothelial growth factor by hypoxia in colon cancer,” Cancer Res, vol. 64, no. 5, pp. 1765–1772, Mar. 2004, doi: 10.1158/0008-5472.can-03-3017. [DOI] [PubMed] [Google Scholar]
- [78].Zhu W et al. “Sonodynamic therapy with immune modulatable two-dimensional coordination nanosheets for enhanced anti-tumor immunotherapy,” Nano Res, vol. 14, no. 1, pp. 212–221, Jan. 2021, doi: 10.1007/s12274-020-3070-8. [DOI] [Google Scholar]
- [79].Chen H et al. “Noninvasively immunogenic sonodynamic therapy with manganese protoporphyrin liposomes against triple-negative breast cancer,” Biomaterials, vol. 269, p. 120639, Feb. 2021, doi: 10.1016/j.biomaterials.2020.120639. [DOI] [PubMed] [Google Scholar]
- [80].Jiao X et al. “Engineering oxygen-deficient ZrO2-x nanoplatform as therapy-activated ‘immunogenic cell death (ICD)’ inducer to synergize photothermal-augmented sonodynamic tumor elimination in NIR-II biological window,” Biomaterials, vol. 272, p. 120787, May 2021, doi: 10.1016/j.biomaterials.2021.120787. [DOI] [PubMed] [Google Scholar]
- [81].Yin Y et al. “Continuous inertial cavitation evokes massive ROS for reinforcing sonodynamic therapy and immunogenic cell death against breast carcinoma,” Nano Today, vol. 36, p. 101009, Feb. 2021, doi: 10.1016/j.nantod.2020.101009. [DOI] [Google Scholar]
- [82].O’Donnell JS, Teng MWL, and Smyth MJ, “Cancer immunoediting and resistance to T cell-based immunotherapy,” Nat. Rev. Clin. Oncol, vol. 16, no. 3, pp. 151–167, Mar. 2019, doi: 10.1038/s41571-018-0142-8. [DOI] [PubMed] [Google Scholar]