

Recent data have expanded the phenotypic spectrum of Huntington’s Disease (HD) and the effects of pathogenic HTT repeat expansions on the human brain. Aside from neurodevelopmental abnormalities in human fetuses with pathogenic HTT gene expansion (PHGE) and a predilection for malformations in postmortem HD brains, recently, Dewan et al. demonstrated that 0.1% of individuals with frontotemporal dementia (FTD)/amyotrophic lateral sclerosis (ALS) have PHGE and the extent of expansion influences the age-of-onset of ALS suggesting a pathogenic link between HTT expansion and ALS [1, 3, 4].
To investigate the frequency of ALS in HD, neuropathology reports of 132 HD brains at the New York Brain Bank (NYBB- discovery cohort) and 619 HD brains from one of the author’s records (JPV- validation cohort) were reviewed. Both cohorts followed a standardized blocking series that included the motor cortex, various brainstem motor nuclei and corticospinal tracts. Neuropathologic criteria for the diagnosis of ALS were based on Brownell et al. [2].
Among 751 brains, 6 (0.8%) were diagnosed pathologically as HD and ALS (HD/ALS), exceeding the United States prevalence of ALS (0.0052%, P<0.0001, Fisher’s exact test) [5]. There was a broad range of age-at-death (44–90 years) without sex predisposition; CAG repeats ranged from 41 to 44 (Table 1). We compared the discovery cohort frequency with brains of essential tremor (ET) because many other neurodegenerative disorders have associations with FTD/ALS. While not statistically significant probably because of insufficient sample size, no ET brain had an ALS diagnosis (n=235), unlike 1.5% of HD brains (P=0.13, Fisher’s exact test).
Table 1:
Clinicopathologic summary of HD/ALS cohorts.
| Case # | Age at death (years) | Sex | CAG repeat size | Clinical details | Family history | Pathologic findings |
|---|---|---|---|---|---|---|
| Discovery cohort | ||||||
| 1 | 57 | Woman | 41 | ALS onset 24 months antemortem. She initially presented with reduced grip strength (left arm) that spread contralaterally. Electromyography showed scattered fasciculations 3 months before death, questionable chorea (non-specific signs- diagnostic confidence of HD: 1 indicating may be normal for age) | Paternal HD (CAG: 41) | ALS (TDP43+ aggregates) HD grade 1/4 (HTT+ aggregates) |
| 2 | 58 | Man | 44 | HD motor onset 6 years before death. Chorea, cognitive decline and behavioral changes 10 years prior to HD motor onset. ALS was not clinically diagnosed antemortem. | No known HD family history | HD grade 4/4 (HTT+ aggregates); ALS (TDP43+ aggregates) |
| Validation cohort | ||||||
| 3 | 55 | Not available | 41 | HD | Not available | HD, ALS |
| 4 | 44 | Not available | N/A | Not available | HD grade 1/4, ALS | |
| 5 | 61 | Man | 42 | HD and ALS | HD grade 2/4, ALS | |
| 6 | 67 | Woman | N/A | Not available | HD grade 3/4, ALS | |
Case #1 was a 57-year-old left-handed woman (CAG:41, C9orf72-mutation negative) with ALS onset 24 months before death. Examination 3 months antemortem revealed questionable facial chorea. On gross examination, the brain (1,362 grams) and spinal cord were unremarkable. Microscopically, there was corticospinal tract degeneration (fig. 1a) and motor neuron loss within the cortex (fig. 1b), facial and hypoglossal nuclei, and the anterior horns. Striatal degeneration was only detected in the tail of the caudate nucleus and was subtle (fig. 1e–h, Vonsattel grade 1). In addition to HTT (fig. 1d), p62 and ubiquitinated (fig. 1g) aggregates were TDP43 nucleocytoplasmic translocation and skeins (fig. 1c) confined to the motor nuclei. Altogether, she had ALS and low-grade HD neuropathologic changes.
Figure 1:
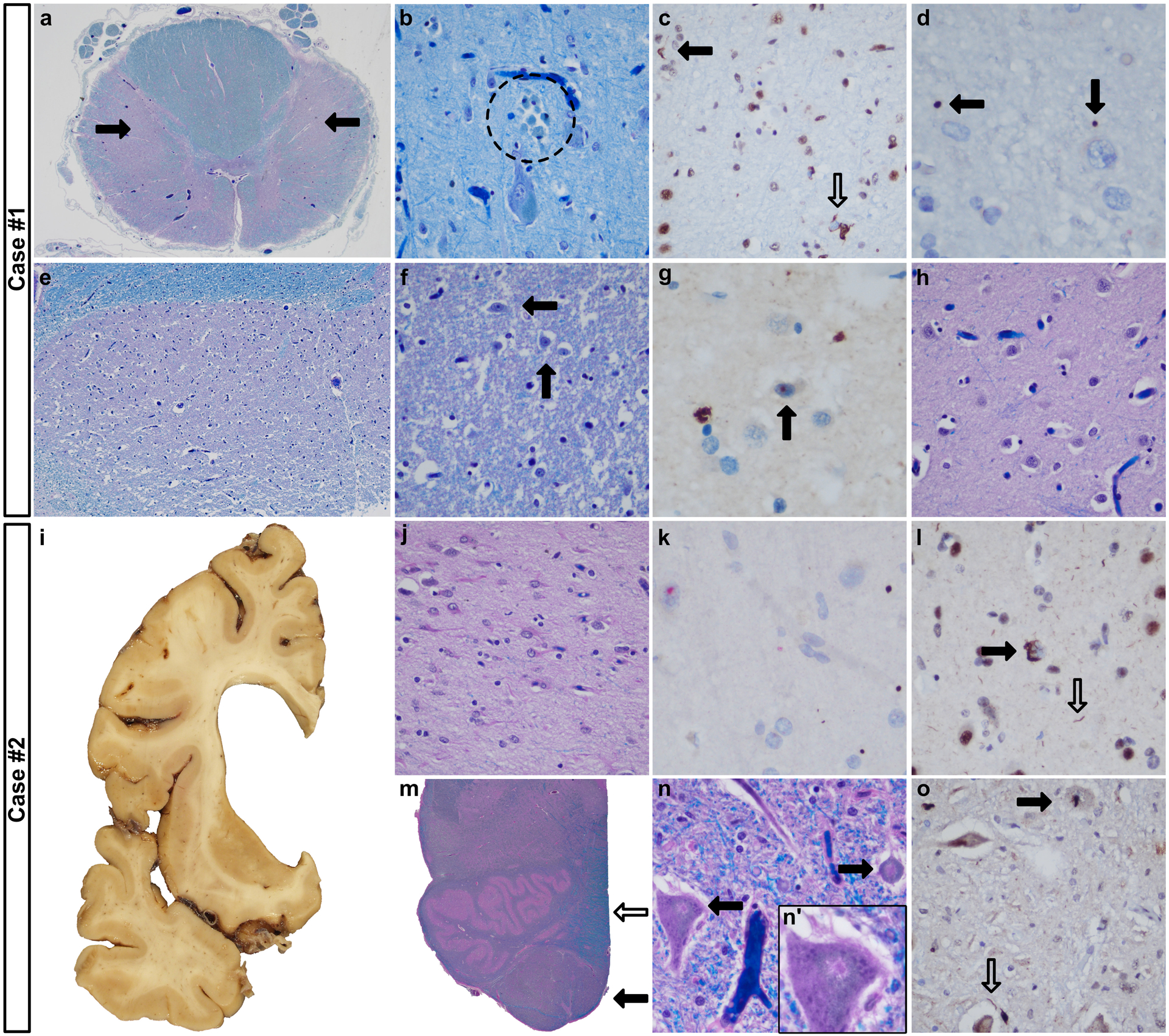
Selected findings in cases 1 (a-h) and 2 (i-o). a: Severe myelin loss of the lateral corticospinal tracts (closed arrows). b: Clusters of macrophages suggested Betz cell neuronophagia (dashed circle, motor cortex, layer V). c-d: Translocation of TDP43 (open arrow) and TDP43-labeled threads (closed arrow) (motor cortex, layer V, (c)) and HTT aggregates (d). e-g: Subtle neuronal loss (e-f, arrows indicating neurons) and an intranuclear ubiquitinated inclusion (g) in the tail of the caudate nucleus. h: The dorsal caudate nucleus showed no apparent neurodegeneration (level of nucleus accumbens). i-j: Severe striatal degeneration; dorsal head of the caudate nucleus (level of nucleus accumbens, (j)). k: HTT aggregates (brown) and intranuclear p62 inclusions (red) (motor cortex, layer V). l: TDP43 translocation (closed arrow) and TDP43-labeled threads (motor cortex). m: Pyramidal myelin pallor (closed arrow) contrasts with the medial lemniscus (open arrow). n: Eosinophilic aggregates in the hypoglossal (n, n’) and facial nuclei. o: TDP43 translocation (closed arrow) and threads (open arrow) (hypoglossal nucleus). Original magnifications: a, m: 12.5x, b-d, e: 20x, f-h, j-l, n-o: 400x.
Case #2 was a 58-year-old man who was diagnosed with HD at age 52 years (CAG: 44). His chorea and depression required hospitalizations, however no features of ALS were recognized 21 months before death. His formalin-fixed left half brain (648 grams) showed severe HD (Vonsattel grade 4, fig. 1i–j) and degeneration of the motor cortex, corticospinal tract (fig. 1m), facial and hypoglossal nuclei (fig. 1n), consistent with ALS. In addition to HTT and p62 aggregates (fig. 1k), TDP43 nucleocytoplasmic translocation and threads within the motor cortex and motor nuclei were present (fig 1l, 1o).
We demonstrate that ALS is over-represented in individuals with HD neuropathologic changes. Cases #1 and #2 differed in their clinical onset and these diseases may therefore have asynchronous presentations. Bias from common ancestry between HD/ALS individuals cannot be excluded and analysis for ALS-causing genes was unattainable. Earlier reports have documented HD/ALS, although only recently has a large-scale study shown that ALS with PHGE may occur without chorea [3, 6, 7]. Further investigation of the potential biologic mechanism that is driving motor neuron degeneration in PHGE is warranted to establish causality. If demonstrated, such ALS patients may benefit from future HTT-related gene therapies.
Acknowledgements
RAH was supported by grant funding from the Huntington Disease Society of America and Hereditary Disease Foundation and was a Columbia University Irving Medical Center ADRC Research Education Component trainee (P30 AG066462-01 PI: Small, Scott, MD). The New York Brain Bank is supported by P50 AG008702 (PI Scott Small, MD). We extend our gratitude to the patients, families, and caregivers who were critical for accomplishing this study and the Essential Tremor Centralized Brain Repository (PI Elan Louis, MD). This work was supported in part by the Intramural Research Programs of the NIH, National Institute on Aging (Z01-AG000949-02).
Footnotes
The original publication is available at https://link.springer.com/article/10.1007%2Fs00401-021-02385-1
References
- 1.Barnat M, Capizzi M, Aparicio E, Boluda S, Wennagel D, Kacher R, Kassem R, Lenoir S, Agasse F, Braz BY et al. (2020) Huntington’s disease alters human neurodevelopment. Science (New York, NY) 369: 787–793 Doi 10.1126/science.aax3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brownell B, Oppenheimer DR, Hughes JT (1970) The central nervous system in motor neurone disease. J Neurol Neurosurg Psychiatry 33: 338–357 Doi 10.1136/jnnp.33.3.338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dewan R, Chia R, Ding J, Hickman RA, Stein TD, Abramzon Y, Ahmed S, Sabir MS, Portley MK, Tucci A et al. (2021) Pathogenic Huntingtin Repeat Expansions in Patients with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neuron 109: 448–460.e444 Doi 10.1016/j.neuron.2020.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hickman RA, Faust PL, Rosenblum MK, Marder K, Mehler MF, Vonsattel JP (2021) Developmental malformations in Huntington disease: neuropathologic evidence of focal neuronal migration defects in a subset of adult brains. Acta Neuropathol 141: 399–413 Doi 10.1007/s00401-021-02269-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mehta P, Kaye W, Raymond J, Punjani R, Larson T, Cohen J, Muravov O, Horton K (2018) Prevalence of Amyotrophic Lateral Sclerosis - United States, 2015. MMWR Morb Mortal Wkly Rep 67: 1285–1289 Doi 10.15585/mmwr.mm6746a1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papageorgiou SG, Antelli A, Bonakis A, Vassos E, Zalonis I, Kalfakis N, Panas M (2006) Association of genetically proven Huntington’s disease and sporadic amyotrophic lateral sclerosis in a 72-year-old woman. J Neurol 253: 1649–1650 Doi 10.1007/s00415-006-0267-z [DOI] [PubMed] [Google Scholar]
- 7.Tada M, Coon EA, Osmand AP, Kirby PA, Martin W, Wieler M, Shiga A, Shirasaki H, Tada M, Makifuchi T et al. (2012) Coexistence of Huntington’s disease and amyotrophic lateral sclerosis: a clinicopathologic study. Acta Neuropathol 124: 749–760 Doi 10.1007/s00401-012-1005-5 [DOI] [PMC free article] [PubMed] [Google Scholar]