

Abstract
Diabetes is a widespread disease characterized by high blood glucose levels due to abnormal insulin activity, production, or both. Chronic diabetes causes many secondary complications including cardiovascular disease: a life-threatening complication. Cerebral ischemia-related mortality, morbidity, and the extent of brain injury are high in diabetes. However, the mechanism of increase in ischemic brain injury during diabetes is not well understood. Multiple mechanisms mediate diabetic hyperglycemia and hypoglycemia-induced increase in ischemic brain injury. Endoplasmic reticulum (ER) stress mediates both brain injury as well as brain protection after ischemia-reperfusion injury. The pathways of ER stress are modulated during diabetes. Free radical generation and mitochondrial dysfunction, two of the prominent mechanisms that mediate diabetic increase in ischemic brain injury, are known to stimulate the pathways of ER stress. Increased ischemic brain injury in diabetes is accompanied by a further increase in the activation of ER stress. As there are many metabolic changes associated with diabetes, differential activation of the pathways of ER stress may mediate pronounced ischemic brain injury in subjects suffering from diabetes. We presently discuss the literature on the significance of ER stress in mediating increased ischemia-reperfusion injury in diabetes.
Keywords: Cell death, cerebral ischemia, hyperglycemia, hypoglycemia, unfolded protein response
Graphical Abstract
1. Introduction
As defined by the American Diabetes Association “Diabetes is a group of metabolic diseases characterized by hyperglycemia resulting from defects in insulin secretion, insulin action, or both” (American Diabetes Association, 2014). More than 34 million people in the USA are presently suffering from diabetes, and the estimated cost of diagnosed diabetes was $327 billion in 2017 (Kleindorfer et al., 2010). Long-term complications associated with chronic diabetes include nephropathy, retinopathy, neuropathy, and development of cardiovascular disease (CVD) along with other secondary complications (Tracey et al., 2016; Virani et al., 2021). It is an accepted fact that diabetes causes CVD in a large subset of patients (Spencer et al., 2008). Studies have shown that the prevalence of experiencing ischemic stroke and coronary heart disease is twice that of subjects suffering from diabetes (Emerging Risk Factors Collaboration et al., 2010). Increased hypercoagulability, dyslipidemia, inflammation, and endothelial dysfunction are some of the factors that may be mediating CVD (Plutzky J ZB, 2012). Individuals suffering from diabetes show increased macrophage infiltration, atheroma, atherosclerotic plaque, and thrombus formation (Cipollone et al., 2003; Moreno et al., 2000). These factors may cause a blockage in the cerebral vasculature and result in ischemia in subjects suffering from diabetes. CVD is considered an important cause of death in patients with diabetes (Virani et al., 2021). Diabetes is associated with increased cardiovascular and cerebrovascular mortality in male and female subjects (Liu et al., 2016; Virani et al., 2021). The estimated contribution of CVD toward the total direct costs of diabetes care is 20% to 49% (Einarson et al., 2018). Diabetes enhances the risk of cerebral ischemia, and cerebral ischemia-related mortality and morbidity are also greater in individuals suffering from diabetes (Centers for Disease Control and Prevention, 2003; Ottenbacher et al., 2004).
2. Diabetes and ischemia-reperfusion injury
Cerebral ischemia results from an interruption in cerebral blood flow (CBF) due to an occlusion in the blood vessel(s) in the brain (Smith, 2015). The level of drop in CBF depends on the structure of cerebral vessels, extent of collateral circulation, site and volume of blockage, and blood pressure. The main reasons for occlusion in cerebral vasculature that results in ischemia are an embolus formed at a different location in the body circulation, thrombosis inside the cerebral vasculature, and reduced CBF because of a stenosis in a prominent blood vessel. A long episode of cerebral ischemia induces cerebral infarction due to cell death in the area affected via the activation of many signal transduction cascades.
A meta-analysis study showed that chronic diabetes leads to poor clinical outcomes following stroke (Desilles et al., 2013). Cerebral infarction is observed more frequently in people with diabetes versus those without diabetes (Lithner et al., 1988). Focal cerebral ischemia in type‐2 diabetic db/db mice produces increased infarct size, neurological impairment, edema in the brain, inflammation, and mortality when compared to the non-diabetic control mice (Tureyen et al., 2011). A previous meta-analysis study showed that hyperglycemia is associated with an increased infarct size in an animal model of type 1 diabetes (T1D) (MacDougall and Muir, 2011). Our laboratory has shown that insulin treated diabetic rats experiencing previous episodes of recurrent hypoglycemia (RH) show enhanced ischemic brain injury (Dave et al., 2011; Rehni et al., 2019b; Shukla et al., 2019). However, the mechanism of increase in the ischemic brain injury during diabetes is not well understood. Studies have proposed the role of multiple mechanisms in mediating the diabetes-induced increase in ischemic brain injury. This article aims to review the scientific literature that shows the significance of endoplasmic reticulum (ER) stress in mediating increased ischemia-reperfusion injury in diabetes.
3. Endoplasmic reticulum stress
The ER is a membranous organelle that spans across the cell and serves the vital function of making new proteins. Besides, the ER facilitates proper protein folding and transport. In addition, ER plays a role in lipid biosynthesis, transfer and signaling. Further, the ER serves the function of calcium storage and release, and detoxification of compounds (Schwarz and Blower, 2016; Thomas D Pollard William C Earnshaw Jennifer Lippincott-Schwartz Graham Johnson, 2017). ER stress results when the burden of unfolded proteins in the ER lumen exceeds its ability to facilitate proper protein folding. Unfolded protein response (UPR) is a group of physiological signal transduction mechanisms that are responsible for sensing and responding to the protein folding capacity of the ER (Ron and Walter, 2007). The UPR regulates numerous genes that either maintain ER homeostasis or induce cell death (Ron and Walter, 2007). Increase in the levels of misfolded or unfolded proteins in the ER causes activation of various UPR pathways. First pathway is the activating transcription factor-6 (ATF6) pathway. The second pathway that is activated is the double-stranded RNA-dependent protein kinase (PKR)-like eukaryotic initiation factor 2α (eIF2α) kinase (PERK) pathway. And thirdly, inositol-requiring transmembrane kinase/endoribonuclease 1 (IRE1) pathway is another pathway activated by accumulation of improperly folded proteins. Activation of these pathways conveys the information through the membrane into the cytoplasm where several transcription factors then transmit information to the nucleus (Kadowaki and Nishitoh, 2013). Previous studies showed that ATF6, IRE1, and PERK are normally bound to the 78 kDa glucose-regulated protein or immunoglobulin heavy chain binding protein (GRP78 a.k.a BiP). BiP possesses a higher affinity for misfolded proteins in comparison to its affinity for ATF6, IRE1, and PERK (Wang and Kaufman, 2016). During conditions of stress in the ER, BiP detaches from the UPR proteins to bind to the misfolded proteins leading to the activation of the three UPR pathways (Bertolotti et al., 2000; Shen et al., 2002). Besides, the activation of IRE1 also occurs via direct binding to misfolded proteins (Gardner et al., 2013).
3.1. ATF6
ATF6 is a type II transmembrane protein with a luminal domain on the carboxy-terminal and a transcription factor domain on the amino terminal. During conditions of ER stress, ATF6 relocates from the network of the ER to the Golgi apparatus where site-1 and site-2 proteases act on them to detach its amino terminal transcription factor domain which moves to the nucleus and binds to ER stress response element (Haze et al., 1999; Shen et al., 2002). This activates the transcription of UPR target genes which corrects ER stress (Yoshida et al., 1998).
3.2. PERK
PERK is another type I transmembrane protein with a luminal stress-sensing domain and a cytosolic kinase domain. ER stress-induced-PERK trans-autophosphorylation causes phosphorylation of the eIF2α. eIF2α phosphorylation inhibits protein synthesis and thus reduces the ER protein load as discussed previously (Harding et al., 1999). However, certain mRNAs with inhibitory upstream open reading frames are preferentially translated (Jackson et al., 2010). Activating transcription factor-4 (ATF4) is upregulated resulting in increased transcription of factors like C/EBP homologous protein (CHOP) and Growth Arrest and DNA Damage-inducible 34 (GADD34) (Harding et al., 2000a; Scheuner et al., 2001). While the transcription factor GADD34 regulates a negative feedback mechanism that inhibits the PERK pathway, CHOP is known to mediate apoptotic cell death (Zinszner et al., 1998) (Wang et al., 1998; Zinszner et al., 1998). Therefore, the PERK pathway of ER stress exerts a beneficial effect on stressed cells. However, a concerted and prolonged activation of PERK pathway stimulates apoptotic cell death as reviewed previously (Walter and Ron, 2011).
3.3. IRE1
IRE1 is a type I transmembrane protein with an ER luminal domain on the amino-terminal, and cytoplasmic kinase and RNase domains on the carboxy terminal. During ER stress, IRE1 assembles into its oligomeric form by self-association of the ER luminal domains of its molecules (Kimata et al., 2007). This is followed by nucleotide binding which causes a conformational change, resulting in the activation of RNase activity of IRE1 (Papa et al., 2003). The RNase activity of IRE1 cleaves basic–leucine zipper transcription factor Hac1p mRNA (homologous to X-box binding protein 1: XBP1 in mammalian cells) (Cox and Walter, 1996) to remove introns. The resulting processed mRNA translates into an active transcription factor that up-regulates genes encoding ER quality control components (Yamamoto et al., 2007). Moreover, the nuclease activity of IRE1 relieves the cell from the load of unfolded proteins (Hollien et al., 2009).
4. Mechanisms of ER stress
ER stress is induced in several pathophysiological processes (Nakka et al., 2016; Sozen et al., 2015). Knowledge about the mechanisms activated in disease conditions is important in designing therapeutic strategies for them. To keep this manuscript succinct, we now provide two main mechanisms of ER stress.
4.1. Oxidative stress
Reactive oxygen species (ROS) are highly reactive molecules produced in low quantities during physiological conditions and quenched by intrinsic antioxidant enzymes (Moro et al., 2005). However, during various disease conditions, increased production of ROS overpowers endogenous antioxidant systems leading to oxidative stress-induced cell death (Chong et al., 2005). NADPH oxidases and mitochondrial electron transport chain are important mechanisms responsible for superoxide production (Bedard and Krause, 2007; Murphy et al., 1999). Inhibition of NADPH oxidase decreases the levels of ER stress markers like GRP78, eIF2-α1, CHOP, and caspase 12 (Al-Saleh et al., 2020). Chemical chaperones that alleviate ER stress or inhibition of oxidative stress exert a protective effect on neurons undergoing cell death (Wei et al., 2008). The signaling mechanisms of ER stress and oxidative stress have been reported to be coordinated via PERK signaling by activating nuclear factor erythroid-2-related factor and ATF4 transcription factors (Cullinan and Diehl, 2006). Superoxide free radicals participate in ER stress-induced ischemic brain injury following focal cerebral ischemia in mice (Hayashi et al., 2005). Subjects with T2D show a positive correlation between ER stress and oxidative stress (Victor et al., 2021). Therefore, the literature implicates that oxidative stress plays a role in ER stress activation and limiting oxidative stress may help lower ER stress.
4.2. Mitochondrial dysfunction
Mitochondria are vital organelles responsible for maintaining respiration and energy production (Siekevitz, 1957). Both apoptosis and necrosis-based cell death essentially depend upon the participation of mitochondria (Kroemer et al., 1998), and mitochondrial dysfunction is a key mechanism that mediates ischemic brain injury as reviewed previously (Niizuma et al., 2010). Crosstalk among UPR, autophagy, and mitochondria underlies the development of ER stress as discussed previously (Senft and Ronai, 2015). Mitochondria and ER influence each other’s functions and thus mediate apoptotic cell death (Sanges and Marigo, 2006). As discussed previously, changes in the crosstalk between the mitochondria and ER participate in the development of T2D (Rieusset, 2011). Both mitochondrial dysfunction as well as ER stress are known to participate in the pathological progression of the T2D. However, the potential role of mitochondrial dysfunction-induced activation of ER stress in diabetic increase in ischemic brain injury is not well understood and merits future investigation.
5. ER stress and cell death pathways
Normal functioning of the ER is important for the cell and its survival. Sustained stimulation of the IRE1 pathway of UPR and/or increased transcription of CHOP can initiate apoptotic cell death (Szegezdi et al., 2006). ER stress-induced apoptotic cell death aims to eliminate cells in which UPR is unable to maintain normal ER physiology, and these cells may otherwise undergo necrosis and inflammation (Szegezdi et al., 2006). However, more severe and chronic ER stress can cause widespread cell death, as seen in several chronic disease conditions (Kaufman, 2002).
Studies have established the role of CHOP in ER stress-induced cell death in diseases like diabetes (Oyadomari et al., 2002), Parkinson’s disease (Silva et al., 2005), and atherosclerosis (Thorp et al., 2009). One of the mechanisms proposed to mediate the detrimental effect of CHOP include down-regulation of the anti-apoptotic protein Bcl-2 (McCullough et al., 2001). Moreover, an increase in the levels of Bim and Bax, ER oxidase 1α (ERO1α)-induced hyperoxidizing environment, and the ERO1α–inositol 1, 4, 5-trisphosphate receptor–Ca2+– Ca(2+)/calmodulin-dependent protein kinase II pathway are some of the other mechanisms proposed to mediate CHOP-induced cell death (Gotoh et al., 2004; Marciniak et al., 2004; Palomeque et al., 2009; Puthalakath et al., 2007). Death receptor-5 and Tribbles-related protein 3 are other mediators proposed to cause CHOP-induced cell death (Ohoka et al., 2005; Yamaguchi and Wang, 2004).
During ER stress, IRE1 causes adaptive degradation of membrane-associated mRNAs via regulated IRE1-dependent decay (RIDD) (Hollien et al., 2009). RIDD is known to contribute to cell death (Tam et al., 2014). Besides, prolonged ER stress activates a pro-apoptotic IRE1–TRAF2–JNK pathway (Tabas and Ron, 2011). Cells that lack IRE1 and its major downstream effectors display a better survival rate than their controls (Hetz et al., 2006). Therefore, ER stress pathways, if activated in a sustained manner and to a severe extent, do possess the potential to induce cell death and play a significant role in pathogenesis of diseases.
6. ER stress and diabetes
ER stress exerts an important role in onset of diabetes by causing pancreatic β-cell death and insulin resistance.
6.1. β-cell death
β-cells are specialized cells that produce insulin and their death or impaired functioning causes T1D and some aspects of later stages of type 2 diabetes (T2D) mellitus (Eizirik et al., 2008). Translation of proinsulin takes place on ribosomes and then transits into the ER for proper folding. As the synthesis of proinsulin varies largely according to physiological requirements, β-cells use the UPR pathways to balance the variable production of proinsulin and its folding in the ER (Dodson and Steiner, 1998). In particular, the initial activation of PERK and IRE1 pathways of the UPR supports the physiological functions of pancreatic β-cells such as the biosynthesis of proinsulin and total proteins (Elouil et al., 2007). However, chronic activation of UPR impairs the functioning of β-cells and contributes to their death leading to diabetes (Engin, 2016; Laybutt et al., 2007). Moreover, deficiency of the PERK pathway of ER stress causes β-cell death (Harding et al., 2001). Studies on human subjects demonstrated increased CHOP expression and increased ER size in the pancreatic β-cells of subjects suffering from diabetes (Marchetti et al., 2007). However, the mechanism of ER stress-mediated β-cell death is not well understood.
6.2. Insulin resistance
Insulin resistance is a situation when insulin is not able to mediate utilization of blood glucose in the body cells (https://www.niddk.nih.gov/health-information/diabetes/overview/what-is-diabetes/prediabetes-insulin-resistance (accessed April 15 2021)). Some of the subjects suffering from obesity have insulin resistance which makes the affected individuals vulnerable to T2D (Permutt et al., 2005). Studies have shown the role of ER stress in the pathogenesis of insulin resistance (Nakatani et al., 2005). The treatment of diabetic animals with chemical chaperones decreases ER stress, normalizes hyperglycemia and insulin sensitivity, and increases insulin activity in the liver, adipose tissues, and muscles (Ozcan et al., 2006). An in vitro study demonstrated that thapsigargin-induced ER stress in human hippocampal neurons leads to impaired insulin signaling (Sims-Robinson et al., 2016). The literature thus suggests a potential role for ER stress in insulin resistance.
6.3. Advanced glycation end products (AGEs)
AGEs are proteins, lipids and nucleic acids, which are progressively glycated because of chronic hyperglycemia in diabetes (Cho et al., 2007). They are a heterogeneous group of highly reactive molecules produced by a non-enzymatic reaction between reducing sugars and proteins, lipids, or nucleic acids (Cho et al., 2007). AGEs cause changes in the functions of extracellular proteins. Moreover, AGEs modulate certain signal transduction cascades by binding to their receptors (called receptor for AGEs) leading to free radical formation, cytokine secretion, and modulation of hormonal activity (Brownlee, 1995). A cell culture based in vitro study has shown that treatment with AGEs elicit ER stress (Adamopoulos et al., 2014). Moreover, chronic administration of AGEs causes ER stress activation in the brain, kidney, liver, and pancreas (Adamopoulos et al., 2016). An earlier study demonstrated that a precursor of AGEs causes ER stress-mediated activation of the caspase-3 cell death pathway potentially via NADPH oxidase 4 upregulation (Loughlin and Artlett, 2010). Additionally, ER stress inhibition decreases AGE-induced apoptotic cell death (Chen et al., 2008). These studies suggest the role of AGEs in the induction of ER stress in diabetes.
7. ER stress, diabetes, and the brain
Chronic hyperglycemia during diabetes affects the central nervous system leading to cognitive dysfunction, enhanced risk of dementia and cognitive impairment, and cerebrovascular diseases (Selvarajah and Tesfaye, 2006). ER stress mediates hippocampal neuronal apoptosis in diabetes and thus may be involved in cognitive impairment in diabetes (Zhang et al., 2013). Pharmacological modification of ER stress in animal models of T1D and T2D leads to alleviation of cognitive deficit (Ye et al., 2018; Zou et al., 2017). Metformin, an antidiabetic drug, inhibits ER stress in rat astrocytes (Wang et al., 2021). Moreover, hyperglycemia-induced ER stress activation and neuronal cell death occurs via the activation of transient receptor potential melastatin 7 channels (Huang et al., 2018). Hyperglycemia exacerbates oxygen-glucose deprivation-induced neuronal injury by activating the ER stress pathway (Lin et al., 2019). Therefore, the literature shows that chronic diabetes produces detrimental effects on the brain, possibly via activation of the ER stress pathway.
Studies have also evaluated ER stress in different brain cell types. Endothelial cells play an important role in various long-term complications of diabetes. Endothelial cells are continuously exposed to fluctuations in blood glucose levels during diabetes. Earlier studies have identified activation of all three pathways of ER stress in endothelial cells under hyperglycemic/diabetic conditions (Maamoun et al., 2019; Yao et al., 2021). Besides hyperglycemia, exposure to hypoglycemia also activates ER stress in endothelial cells (Soejima et al., 2018). Exposure to hyperglycemia is shown to activate ER stress in primary hippocampal neurons, NS20Y cells (cholinergic neuronal cell line obtained from mouse neuroblastoma), and dorsal root ganglion neurons (Huang et al., 2018; Liu et al., 2021; Sharma et al., 2016; Zhang et al., 2013). Moreover, glucose deprivation also activates ER stress in neurons (de la Cadena et al., 2014; Gomora-Garcia et al., 2021). Streptozotocin-induced diabetes is shown to activate ER stress in Müller cells: a type of retinal glial cells (Zhong et al., 2012). Exposure of rat primary astrocytes to high glucose results in increased phospho-PERK levels without any change in levels of total PERK (Wang et al., 2021). Moreover, primary human astrocytes exposed to an acute episode of hypoglycemia had increased levels of ER stress-related genes. However, ER stress was diminished in these astrocytes when they were exposed to recurrent low glucose conditions (Weightman Potter et al., 2021). On the other hand, recurrent low glucose exposure of mouse Schwann cells resulted in increased CHOP levels (Kato et al., 2019). The same study also reported that exposure to constant low, constant high, as well as intermittent high glucose increased CHOP levels in mouse Schwann cells. However, the role of various degrees of ER stress observed in different brain cell types, under hyperglycemic or hypoglycemic conditions, in brain-related long-term complications observed in diabetic patients requires further in-depth studies. Overall, the literature shows that chronic diabetes produces detrimental effects on the brain, possibly via activation of the ER stress pathway.
8. ER stress modulation and brain injury
To understand the role of ER stress in mediating pathological changes in the brain in many neurological conditions, studies have evaluated the effect of ER stress inhibition on such conditions. Inhibiting ER stress using salubrinal protects neurons from excitotoxic injury (Sokka et al., 2007). ER stress inhibition using either salubrinal or guanabenz exerts an ameliorative effect on trauma-induced brain injury (Hood et al., 2018; Tan et al., 2018). Similarly, treatment with a chemical chaperone that reduces unfolded protein loss also produces a beneficial effect on the brain during Alzheimer’s disease by preventing aging-associated deficits in memory (Ricobaraza et al., 2011; Ricobaraza et al., 2012). Lowering unfolded protein response in Parkinson’s disease using various pharmacological strategies may be prosurvival for dopaminergic neurons as reviewed previously (Kovaleva and Saarma, 2021). Moreover, ER stress inhibition improves learning and memory and synaptic plasticity damage in aged rats (Carvajal-Flores et al., 2020). Treatment with 4-phenylbutyric acid (4-PBA), an ER stress inhibitor, lowers the detrimental effect of diabetes on spinal cord injury in rats (He et al., 2017). Therefore, there is a sizable amount of published data showing the significance of ER stress activation on brain injury.
9. ER stress and ischemic brain injury
Cerebral ischemia is one of the pathological events that activates ER stress. A severe episode of cerebral ischemia causes a sudden increase in misfolded proteins. Brain cells tend to correct the overload of misfolded proteins by decreasing protein synthesis, enhancing the expression of chaperones that facilitate proper folding of misfolded proteins, and activating protein degradation mechanisms for eliminating damaged organelles (Kristian and Hu, 2018). Recovery of protein synthesis in vulnerable neurons is not complete even after a brief period of ischemia (Thilmann et al., 1986), showing that their failure to revive protein synthesis is associated with cell death. Inhibition of protein translation initiated by the activation of eIF2α kinase causes cell death (Srivastava et al., 1998). A 10 min period of global cerebral ischemia activates the PERK pathway of ER stress along with an increase in eIF2α phosphorylation, leading to the inhibition of protein translation in the cerebral cortex, brain stem, and hippocampus when quantified after 4 h of reperfusion in rats (Kumar et al., 2003). Other studies confirmed that global cerebral ischemia and reperfusion inhibit protein synthesis (Krause and Tiffany, 1993) via a PERK-dependent acute increase in the levels eIF2α phosphorylation (Kumar et al., 2001). A study by Hayashi et al. (Hayashi et al., 2003) showed that the cerebral ischemia-induced increased phosphorylation of eIF2α and PERK is lower in transgenic rats overexpressing copper/zinc superoxide dismutase when compared to their wild-type counterparts. This study implicates the role of oxidative stress in ischemic activation of PERK and further proposes that ischemia reduces PERK-GRP78 binding resulting in PERK activation.
Aspirin is shown to limit cerebral infarction along with downregulation of toll-like receptor 4/nuclear factor kappa light chain enhancer of activated B cells-mediated ER stress measured in terms of PERK, eIF2α, and CHOP levels (Wang et al., 2018). Moreover, diazoxide exerts a marked neuroprotective effect on ER-stress-induced apoptotic cell death in an in vitro and in vivo model of cerebral ischemia (Lei et al., 2018). Another study showed that dexmedetomidine decreases ischemia-reperfusion injury in the brain by inhibiting ER stress-dependent apoptosis via modulation of the PERK pathway (Liu et al., 2018). Melatonin treatment exerts a protective effect on ischemic brain injury. The protective effect of melatonin occurs via decrease in autophagy. Activation of PERK- and IRE1-dependent pathways of UPR mediate the effect of melatonin on ischemic brain injury (Feng et al., 2017). Although a study showed that global cerebral ischemia did not activate IRE1 and ATF6 pathways, another study showed that xbp1 mRNA splicing is facilitated after transient focal as well as global cerebral ischemia, suggesting that ischemia may activate the IRE1-pathway of ER stress (Kumar et al., 2003; Paschen et al., 2003).
Treatment with salubrinal, a potent inhibitor of dephosphorylation of phosphorylated-eIF2α, has been shown to restore the ischemia-induced increase in phosphorylated eIF2α in these mice and improve functional recovery after focal (Wang et al., 2020) and global cerebral ischemia (Font-Belmonte et al., 2019). Concomitant treatment with robenacoxib and salubrinal post-ischemia reduces ischemic neuronal damage (Anuncibay-Soto et al., 2018). A recent study showed that neuron-specific inducible PERK knockout mice display larger infarcts and worse neurological outcomes when compared to control mice, showing that PERK activation-induced eIF2α phosphorylation and resulting suppression of translation exerts a protective effect on ischemic neurons (Wang et al., 2020). Yu et al demonstrated that the activation of the ATF6 pathway of ER stress in an inducible sATF6 knock-in mice reduced infarct size and improved functional outcome 24 h after stroke (Yu et al., 2017b). While the deletion of XBP1 exerted a detrimental effect on outcomes following experimental stroke, pharmacological activation of O-GlcNAcylation exerted a beneficial effect on stroke outcomes (Jiang et al., 2017).
The literature cited presents conflicting views that both strategies that lower activation of ER stress pathways as well as preservation of ER stress pathways results in protection against ischemic brain injury. It is plausible that suppressing activation of ER stress during the early period after ischemia, when cellular energy levels are not yet normalized, may have beneficial effects. On the contrary, during the late/recovery stage, when protein synthesis is required for recovery, preservation of ER stress pathways can have protective effects against cerebral ischemic damage.
10. ER stress and ischemic brain injury in diabetes
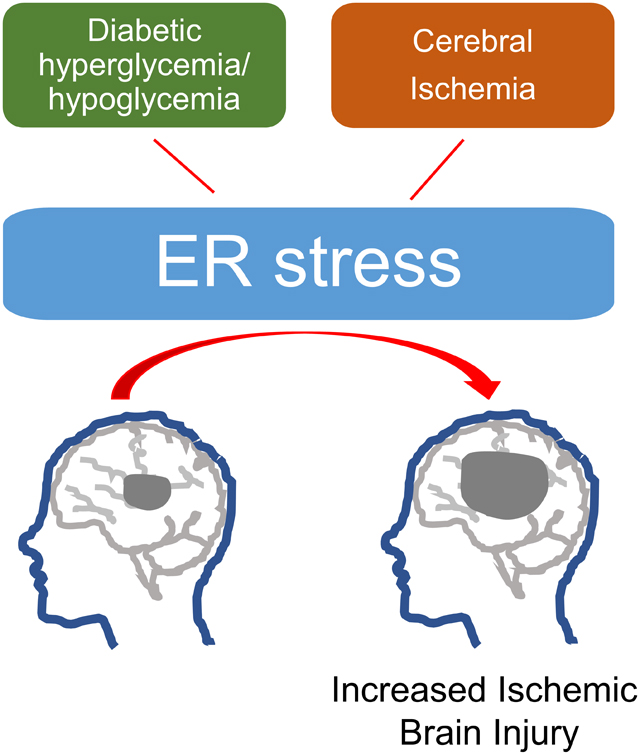
Diabetes is another pathological event that activates ER stress (Fig. 1). ER stress plays an important role in ischemia/reperfusion injury in various organ systems including brain in diabetes. For example, ischemia-reperfusion injury in the liver causes excessive inflammation and exacerbated ischemic injury in diabetic and hyperglycemic animals, possibly via increased CHOP levels (Rao et al., 2017). However, treatment with diallyl trisulfide, an inhibitor of ER stress, exerts an ameliorative effect on ischemic myocardium in T1D rats possibly via inhibition of PERK pathway of ER stress (Yu et al., 2017a). Further, tauroursodeoxycholic acid-induced in vivo inhibition of ER stress exerts a significant protective effect against ischemic damage in the myocardium of db−/db− mice with T2D (Mali et al., 2018). Moreover, chronic treatment with tauroursodeoxycholic acid for 4 weeks attenuated ER stress, inflammation, and neovascularization and resulted in recovered blood flow following hind-limb ischemia in db/db mouse model of T2D (Amin et al., 2012). Preconditioning with tunicamycin, a selective ER stress inducer, exerts an ameliorative effect on ischemic injury in the myocardium in streptozotocin-diabetic rats (Yan et al., 2019). ER stress decreases erythropoietin-induced cardioprotective effect on ischemic myocardium in a rat model of T2D (Miki et al., 2009).
Figure 1: Schematic representation of the possible ER stress mechanisms that mediate diabetes-induced increase in ischemic cell death:
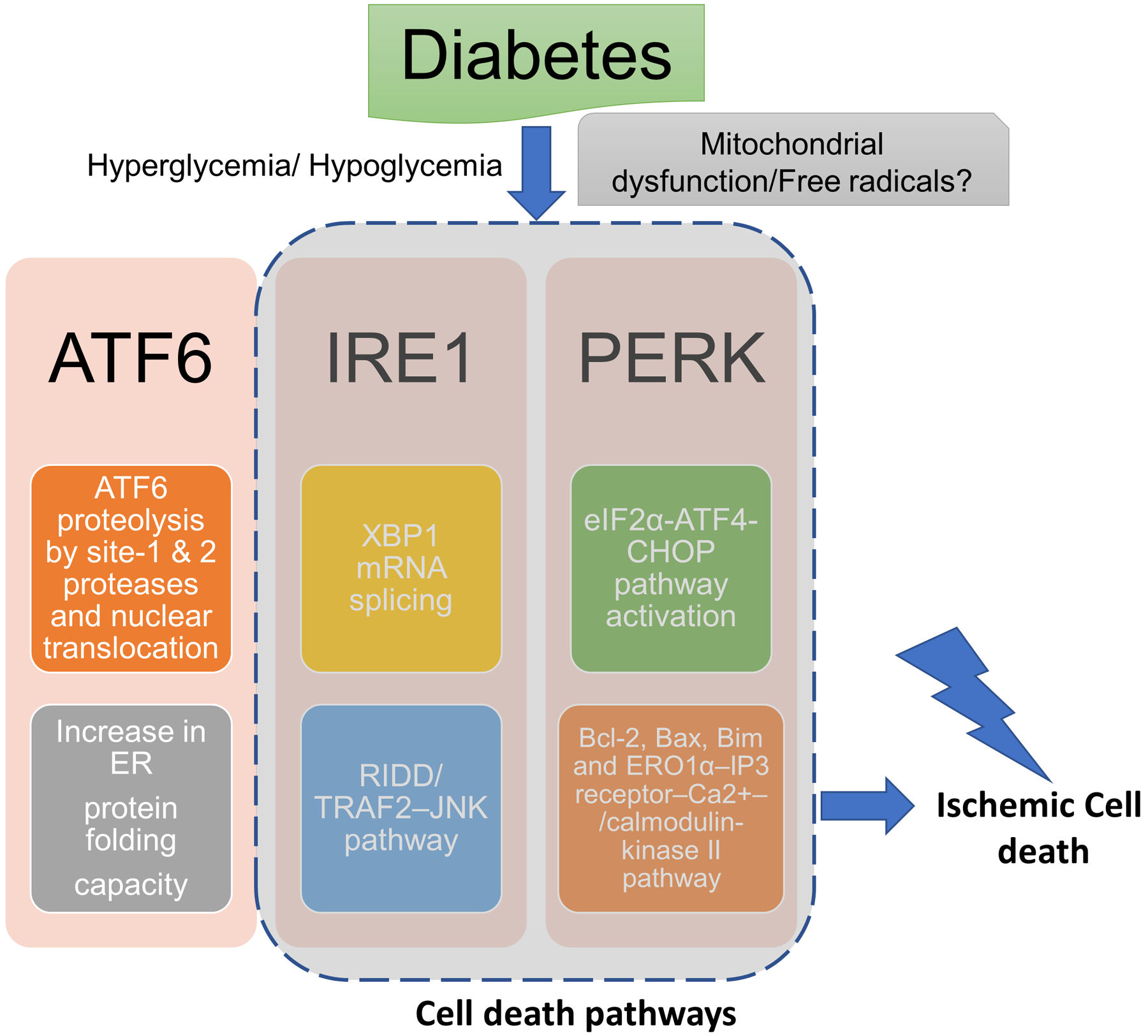
Diabetic hypoglycemia/ hyperglycemia may activate endoplasmic reticulum (ER) stress via mitochondrial dysfunction-induced free radical generation. The major possible pathways of ER stress that may mediate pronounced ischemic brain injury in diabetes: (1) activating transcription factor-6 (ATF6) translocates from the ER compartment of the cell to the Golgi apparatus where it is activated by site-1 and site-2 proteases via proteolysis. The activated ATF6 then moves to the nucleus and stimulates the production of UPR proteins via ER stress response element dependent changes in gene transcription. This binds to the ER stress response element and activates the transcription of UPR target genes which corrects ER stress. (2) inositol-requiring transmembrane kinase/endoribonuclease 1 (IRE1α) splices X-box protein 1 mRNA resulting in the activation of regulated IRE1-dependent decay (RIDD) and the IRE1α cytosolic domain activates tumor necrosis factor receptor-associated factor (TRAF)-TRAF2–JUN N-terminal kinase (JNK) signaling. (2) PERK causes phosphorylation of eukaryotic translation initiation factor 2 subunit-α (eIF2α) resulting in inhibition of protein synthesis by reducing mRNA translation initiation. Nevertheless, PERK-eIF2α pathway causes increase in the levels of ATF4 and C/EBP homologous protein (CHOP) and associated cell death pathways.
The role of ER stress has also been explored in models of cerebral ischemia. An earlier study using streptozotocin-induced diabetes showed that infarct volume as well as the levels of GRP78 and CHOP were significantly higher in diabetic animals subjected to focal cerebral ischemia (Srinivasan and Sharma, 2011a). Using transient focal cerebral ischemia in diabetic rats as a model system the group further showed the beneficial effect of sodium 4-phenylbutyrate, a chemical chaperone, in terms of a decrease in cerebral infarct size and improvement in functional outcomes (Srinivasan and Sharma, 2011b). Therefore, some studies have implicated the role of ER stress in diabetic exacerbation of ischemic brain injury (Srinivasan and Sharma, 2011a, b, 2012; Su et al., 2017). A recent study showed that glucagon-like peptide-1 / glucose-dependent insulinotropic polypeptide dual agonist DA3-CH reduces ER stress as well as reduces both the infarct size and the neurological deficit score in streptozotocin-diabetic rats exposed to cerebral ischemia-reperfusion injury (Bai et al., 2021). A neuroprotective agent monosialotetrahexosy-1 ganglioside (GM1) is observed to reduce cerebral infarct size, and neuronal apoptosis and improve the neurological behavior in streptozotocin-diabetic rats (Su et al., 2017). The beneficial effect of GM1 on ischemic brain is accompanied by an increase in the level of GRP78 and a decrease in the level of CHOP. Nonetheless, there are a limited number of studies that show the role of ER stress pathways in cerebral ischemia-induced injury in animal models of diabetes. Therefore, the literature indicates that increased ER stress seen during diabetes may cause an exacerbated activation of ER stress following cerebral ischemia in diabetes, and a corresponding increase in ischemic damage (Fig. 2).
Figure 2: Representation of the tested ER stress interventions that exerts a protective effect on ischemic damage during diabetes:
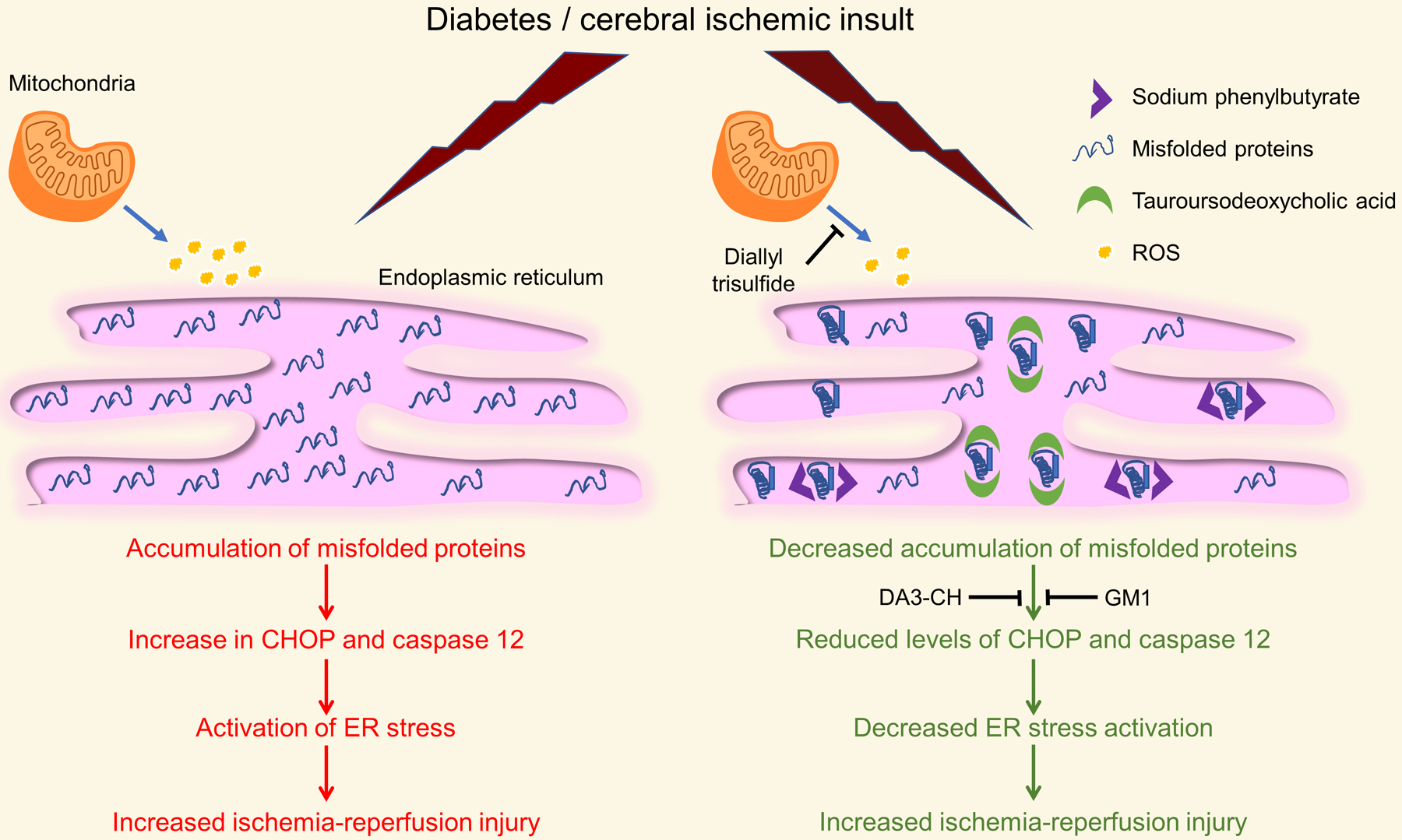
Treatment with chemical chaperones like sodium phenylbutyrate and tauroursodeoxycholic acid and modulators of ER stress like DA3-CH (glucagon-like peptide-1/ glucose-dependent insulinotropic polypeptide dual agonist), diallyl trisulfide, and monosialotetrahexosy-1 ganglioside (GM1) decreases diabetic exacerbation of ischemic injury. CHOP: C/EBP homologous protein; ROS: Reactive oxygen species.
11. ER stress and hypoglycemia in diabetes
Chronic drug therapy of diabetes is known to elicit episodes of iatrogenic hypoglycemia (Dagogo-Jack, 2004). Repeated episodes of such transient hypoglycemia inhibit the mechanisms of glucose counter-regulation (Dagogo-Jack et al., 1993), increase hypoglycemia unawareness (Cryer, 2004), and thus enhance the risk of hypoglycemia (Cryer, 2013). Continuous glucose monitoring studies have shown that patients suffering from T1D experience hypoglycemia for 30 to 90 mins every day (Battelino et al., 2011; Heinemann et al., 2018; Oliver et al., 2020). Patients with T2D experience approximately 2 episodes of hypoglycemia every 5 days (McNally et al., 2007).
PERK pathway of ER stress is an adaptive mechanism of the cell (Harding et al., 2000b; Harding et al., 1999), but a prolonged severe activation of this pathway activates cell death mechanisms (Rutkowski et al., 2006; Wang and Kaufman, 2016). ER stress mediates cell death in glucose-deprived neurons via PERK activation (de la Cadena et al., 2014) and acute hypoglycemia increases CHOP levels (Gonzales et al., 2008). The upstream mechanism by which hypoglycemia causes ER stress is however not well understood. As discussed above, cerebral ischemia stimulates PERK activation and increases ATF4 and CHOP levels (Hadley et al., 2018; Paschen et al., 1998), and CHOP mediates ischemic neuronal death in the brain (Tajiri et al., 2004). We have reported that prior RH exposure in treated diabetic rats causes increased cerebral ischemic damage in comparison to the normoglycemic treated diabetic control rats (Dave et al., 2011; Rehni et al., 2019b; Shukla et al., 2019). Our previous data showed that the exposure to RH alone or cerebral ischemia in animals exposed to prior RH causes an increase in free radical production (Dave et al., 2011; Rehni et al., 2019a). Oxidative stress activates ER calcium release channels like ryanodine receptors (Bull et al., 2003) and IP3 receptors (Bansaghi et al., 2014). ER calcium depletion/increased calcium release causes ER stress (Doutheil et al., 1997; Farrukh et al., 2014; Luciani et al., 2009; Paschen and Doutheil, 1999). Cerebral ischemia-induced oxidative stress activates the PERK pathway (Hayashi et al., 2005; Hayashi et al., 2003). Dantrolene, a ryanodine receptor inhibitor (Zhao et al., 2001), inhibits ischemic activation of the PERK pathway of ER stress (Li et al., 2005). Therefore, it may be hypothesized that RH-induced activation of the PERK pathway of ER stress is dependent on free radical production and renders the brain of RH-exposed insulin-treated diabetic rats susceptible to increased ischemic injury. However, future studies are warranted to test such a hypothesis.
A recent study showed that acute glucose deprivation activates PERK and IRE1α pathways of ER stress (Gomora-Garcia et al., 2021), a pathway also known to mediate ischemic brain injury (Sanderson et al., 2015). Although direct studies conclusively showing the role of ER stress in diabetic exaggeration of ischemic brain injury are still required, the above literature shows that hypoglycemia-induced ER stress activation may play an important role in diabetic increase in ischemic brain injury.
12. Summary and Future Directions
Studies have shown that the PERK, ATF6, and IRE1 pathways of ER stress play an adaptive role in cerebral ischemia injury. The literature shows that ER stress plays a central role in diabetes by mediating pancreatic β-cell death and insulin resistance (Eizirik et al., 2008). The literature reviewed above demonstrates that diabetes activates ER stress pathways as well as increases ischemic brain injury. Because several metabolic changes are associated with diabetes, the effect of ER stress on ischemic brain injury in diabetic subjects is expected to produce a differential effect in comparison to normal subjects. However, experimental evaluation is required to characterize the effect of diabetes on ischemic activation of the signal transduction cascades of ER stress. Additionally, studies aimed to better understand the upstream pathways that cause the ischemic activation of ER stress pathways in diabetes (both hyperglycemia and hypoglycemia) are also needed. Moreover, studies are also required to identify the downstream mechanisms that potentially cause ER stress-induced changes in ischemic brain injury in diabetes. Given the multifarious pathogenesis of cerebral ischemia and diabetes, it is important to conduct studies using an experimental design that allows evaluating the impact of both cerebral ischemia and diabetes separately as well as together. Such detailed studies may also help confirm, if any, the therapeutic relevance of the role of ER stress in diabetes-induced increase in ischemic brain injury.
13. Conclusion
In conclusion, only a few studies have demonstrated the role of activation of the ER stress pathway in ischemia-reperfusion injury in the brain of diabetic animals. We expect that diabetes-induced ER stress activation plays a major role in mediating diabetes-induced exacerbation of ischemic brain injury, possibly via modulating multiple cell death mechanisms associated with the pathway. A complete understanding of these pathways in diabetes-induced exacerbation of ischemic brain injury may help develop new therapies to treat ischemic brain damage in patients with diabetes.
Acknowledgement
We would like to thank Dr. Brant Watson for critical reading of this manuscript.
Funding:
This work was supported by the National Institutes of Health [grants number R01NS122808, and R01NS073779]; and the American Heart Association [grants number 20TPA35490411, and 18POST34070061]. The sponsors did not have any role in study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Footnotes
Consent for publication and submission declaration and verification:
The work described has not been published previously, that it is not under consideration for publication elsewhere, that its publication is approved by all authors, and that, if accepted, it will not be published elsewhere in the same form, in English or in any other language, including electronically without the written consent of the copyright-holder.
References
- Adamopoulos C, Farmaki E, Spilioti E, et al. , 2014. Advanced glycation end-products induce endoplasmic reticulum stress in human aortic endothelial cells. Clin. Chem. Lab. Med, 52, 151–160. [DOI] [PubMed] [Google Scholar]
- Adamopoulos C, Mihailidou C, Grivaki C, et al. , 2016. Systemic effects of AGEs in ER stress induction in vivo. Glycoconj. J, 33, 537–544. [DOI] [PubMed] [Google Scholar]
- Al-Saleh F, Khashab F, Fadel F, et al. , 2020. Inhibition of NADPH oxidase alleviates germ cell apoptosis and ER stress during testicular ischemia reperfusion injury. Saudi. J. Biol. Sci, 27, 2174–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Diabetes Association, 2014. Diagnosis and classification of diabetes mellitus. Diabetes Care, 37, S81–90. [DOI] [PubMed] [Google Scholar]
- Amin A, Choi SK, Galan M, et al. , 2012. Chronic inhibition of endoplasmic reticulum stress and inflammation prevents ischaemia-induced vascular pathology in type II diabetic mice. J. Pathol, 227, 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anuncibay-Soto B, Perez-Rodriguez D, Santos-Galdiano M, et al. , 2018. Salubrinal and robenacoxib treatment after global cerebral ischemia. Exploring the interactions between ER stress and inflammation. Biochem. Pharmacol, 151, 26–37. [DOI] [PubMed] [Google Scholar]
- Bai B, Li D, Xue G, et al. , 2021. The novel GLP-1/GIP dual agonist DA3-CH is more effective than liraglutide in reducing endoplasmic reticulum stress in diabetic rats with cerebral ischemia-reperfusion injury. Nutr. Metab. Cardiovasc. Dis, 31, 333–343. [DOI] [PubMed] [Google Scholar]
- Bansaghi S, Golenar T, Madesh M, et al. , 2014. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem, 289, 8170–8181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battelino T, Phillip M, Bratina N, et al. , 2011. Effect of continuous glucose monitoring on hypoglycemia in type 1 diabetes. Diabetes Care, 34, 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH, 2007. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev, 87, 245–313. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, et al. , 2000. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol, 2, 326–332. [DOI] [PubMed] [Google Scholar]
- Brownlee M, 1995. Advanced protein glycosylation in diabetes and aging. Annu. Rev. Med, 46, 223–234. [DOI] [PubMed] [Google Scholar]
- Bull R, Marengo JJ, Finkelstein JP, et al. , 2003. SH oxidation coordinates subunits of rat brain ryanodine receptor channels activated by calcium and ATP. Am. J. Physiol. Cell. Physiol, 285, C119–128. [DOI] [PubMed] [Google Scholar]
- Carvajal-Flores FN, Diaz A, Flores-Gomez GD, et al. , 2020. Phenylbutyrate ameliorates prefrontal cortex, hippocampus, and nucleus accumbens neural atrophy as well as synaptophysin and GFAP stress in aging mice. Synapse, 74, e22177. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention, 2003. Self-reported heart disease and stroke among adults with and without diabetes--United States, 1999–2001. MMWR Morb. Mortal. Wkly. Rep, 52, 1065–1070. [PubMed] [Google Scholar]
- Chen Y, Liu CP, Xu KF, et al. , 2008. Effect of taurine-conjugated ursodeoxycholic acid on endoplasmic reticulum stress and apoptosis induced by advanced glycation end products in cultured mouse podocytes. Am. J. Nephrol, 28, 1014–1022. [DOI] [PubMed] [Google Scholar]
- Cho SJ, Roman G, Yeboah F, et al. , 2007. The road to advanced glycation end products: a mechanistic perspective. Curr. Med. Chem, 14, 1653–1671. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K, 2005. Oxidative stress in the brain: novel cellular targets that govern survival during neurodegenerative disease. Prog. Neurobiol, 75, 207–246. [DOI] [PubMed] [Google Scholar]
- Cipollone F, Iezzi A, Fazia M, et al. , 2003. The receptor RAGE as a progression factor amplifying arachidonate-dependent inflammatory and proteolytic response in human atherosclerotic plaques: role of glycemic control. Circulation, 108, 1070–1077. [DOI] [PubMed] [Google Scholar]
- Cox JS, Walter P, 1996. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell, 87, 391–404. [DOI] [PubMed] [Google Scholar]
- Cryer PE, 2004. Diverse causes of hypoglycemia-associated autonomic failure in diabetes. N. Engl. J. Med, 350, 2272–2279. [DOI] [PubMed] [Google Scholar]
- Cryer PE, 2013. Hypoglycemia-associated autonomic failure in diabetes. Handb. Clin. Neurol, 117, 295–307. [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Diehl JA, 2006. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol, 38, 317–332. [DOI] [PubMed] [Google Scholar]
- Dagogo-Jack S, 2004. Hypoglycemia in type 1 diabetes mellitus: pathophysiology and prevention. Treat. Endocrinol, 3, 91–103. [DOI] [PubMed] [Google Scholar]
- Dagogo-Jack SE, Craft S, Cryer PE, 1993. Hypoglycemia-associated autonomic failure in insulin-dependent diabetes mellitus. Recent antecedent hypoglycemia reduces autonomic responses to, symptoms of, and defense against subsequent hypoglycemia. J. Clin. Invest, 91, 819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave KR, Tamariz J, Desai KM, et al. , 2011. Recurrent hypoglycemia exacerbates cerebral ischemic damage in streptozotocin-induced diabetic rats. Stroke, 42, 1404–1411. [DOI] [PubMed] [Google Scholar]
- de la Cadena SG, Hernandez-Fonseca K, Camacho-Arroyo I, et al. , 2014. Glucose deprivation induces reticulum stress by the PERK pathway and caspase-7- and calpain-mediated caspase-12 activation. Apoptosis, 19, 414–427. [DOI] [PubMed] [Google Scholar]
- Desilles JP, Meseguer E, Labreuche J, et al. , 2013. Diabetes mellitus, admission glucose, and outcomes after stroke thrombolysis: a registry and systematic review. Stroke, 44, 1915–1923. [DOI] [PubMed] [Google Scholar]
- Dodson G, Steiner D, 1998. The role of assembly in insulin’s biosynthesis. Curr. Opin. Struct. Biol, 8, 189–194. [DOI] [PubMed] [Google Scholar]
- Doutheil J, Gissel C, Oschlies U, et al. , 1997. Relation of neuronal endoplasmic reticulum calcium homeostasis to ribosomal aggregation and protein synthesis: implications for stress-induced suppression of protein synthesis. Brain Res, 775, 43–51. [DOI] [PubMed] [Google Scholar]
- Einarson TR, Acs A, Ludwig C, et al. , 2018. Economic Burden of Cardiovascular Disease in Type 2 Diabetes: A Systematic Review. Value Health, 21, 881–890. [DOI] [PubMed] [Google Scholar]
- Eizirik DL, Cardozo AK, Cnop M, 2008. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev, 29, 42–61. [DOI] [PubMed] [Google Scholar]
- Elouil H, Bensellam M, Guiot Y, et al. , 2007. Acute nutrient regulation of the unfolded protein response and integrated stress response in cultured rat pancreatic islets. Diabetologia, 50, 1442–1452. [DOI] [PubMed] [Google Scholar]
- Emerging Risk Factors Collaboration, Sarwar N, Gao P, et al. , 2010. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet, 375, 2215–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin F, 2016. ER stress and development of type 1 diabetes. J. Investig. Med, 64, 2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrukh MR, Nissar UA, Afnan Q, et al. , 2014. Oxidative stress mediated Ca(2+) release manifests endoplasmic reticulum stress leading to unfolded protein response in UV-B irradiated human skin cells. J. Dermatol. Sci, 75, 24–35. [DOI] [PubMed] [Google Scholar]
- Feng D, Wang B, Wang L, et al. , 2017. Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via PERK and IRE1 signalings. J. Pineal Res, 62. [DOI] [PubMed] [Google Scholar]
- Font-Belmonte E, Ugidos IF, Santos-Galdiano M, et al. , 2019. Post-ischemic salubrinal administration reduces necroptosis in a rat model of global cerebral ischemia. J. Neurochem, 151, 777–794. [DOI] [PubMed] [Google Scholar]
- Gardner BM, Pincus D, Gotthardt K, et al. , 2013. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol, 5, a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomora-Garcia JC, Geronimo-Olvera C, Perez-Martinez X, et al. , 2021. IRE1alpha RIDD activity induced under ER stress drives neuronal death by the degradation of 14-3-3 theta mRNA in cortical neurons during glucose deprivation. Cell. Death. Discov, 7, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales JC, Gentile CL, Pfaffenbach KT, et al. , 2008. Chemical induction of the unfolded protein response in the liver increases glucose production and is activated during insulin-induced hypoglycaemia in rats. Diabetologia, 51, 1920–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh T, Terada K, Oyadomari S, et al. , 2004. hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ, 11, 390–402. [DOI] [PubMed] [Google Scholar]
- Hadley G, Neuhaus AA, Couch Y, et al. , 2018. The role of the endoplasmic reticulum stress response following cerebral ischemia. Int. J. Stroke, 13, 379–390. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, et al. , 2000a. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell, 6, 1099–1108. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, et al. , 2001. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol. Cell, 7, 1153–1163. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, et al. , 2000b. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell, 5, 897–904. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D, 1999. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature, 397, 271–274. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Saito A, Okuno S, et al. , 2005. Damage to the endoplasmic reticulum and activation of apoptotic machinery by oxidative stress in ischemic neurons. J. Cereb. Blood Flow Metab, 25, 41–53. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Saito A, Okuno S, et al. , 2003. Oxidative damage to the endoplasmic reticulum is implicated in ischemic neuronal cell death. J. Cereb. Blood Flow Metab, 23, 1117–1128. [DOI] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, et al. , 1999. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell, 10, 3787–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Zou S, Yin J, et al. , 2017. Inhibition of Endoplasmic Reticulum Stress Preserves the Integrity of Blood-Spinal Cord Barrier in Diabetic Rats Subjected to Spinal Cord Injury. Sci. Rep, 7, 7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann L, Freckmann G, Ehrmann D, et al. , 2018. Real-time continuous glucose monitoring in adults with type 1 diabetes and impaired hypoglycaemia awareness or severe hypoglycaemia treated with multiple daily insulin injections (HypoDE): a multicentre, randomised controlled trial. Lancet, 391, 1367–1377. [DOI] [PubMed] [Google Scholar]
- Hetz C, Bernasconi P, Fisher J, et al. , 2006. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science, 312, 572–576. [DOI] [PubMed] [Google Scholar]
- Hollien J, Lin JH, Li H, et al. , 2009. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol, 186, 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood KN, Zhao J, Redell JB, et al. , 2018. Endoplasmic Reticulum Stress Contributes to the Loss of Newborn Hippocampal Neurons after Traumatic Brain Injury. J. Neurosci, 38, 2372–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.niddk.nih.gov/health-information/diabetes/overview/what-is-diabetes/prediabetes-insulin-resistance (accessed 15 April 2021).
- Huang Y, Leng TD, Inoue K, et al. , 2018. TRPM7 channels play a role in high glucose-induced endoplasmic reticulum stress and neuronal cell apoptosis. J. Biol. Chem, 293, 14393–14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CU, Pestova TV, 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol, 11, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Yu S, Yu Z, et al. , 2017. XBP1 (X-Box-Binding Protein-1)-Dependent O-GlcNAcylation Is Neuroprotective in Ischemic Stroke in Young Mice and Its Impairment in Aged Mice Is Rescued by Thiamet-G. Stroke, 48, 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki H, Nishitoh H, 2013. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes (Basel), 4, 306–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Tatsumi Y, Yako H, et al. , 2019. Recurrent short-term hypoglycemia and hyperglycemia induce apoptosis and oxidative stress via the ER stress response in immortalized adult mouse Schwann (IMS32) cells. Neurosci. Res, 147, 26–32. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ, 2002. Orchestrating the unfolded protein response in health and disease. J. Clin. Invest, 110, 1389–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y, Ishiwata-Kimata Y, Ito T, et al. , 2007. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J. Cell Biol, 179, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleindorfer DO, Khoury J, Moomaw CJ, et al. , 2010. Stroke incidence is decreasing in whites but not in blacks: a population-based estimate of temporal trends in stroke incidence from the Greater Cincinnati/Northern Kentucky Stroke Study. Stroke, 41, 1326–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaleva V, Saarma M, 2021. Endoplasmic Reticulum Stress Regulators: New Drug Targets for Parkinson’s Disease. J. Parkinsons. Dis, 11 (s2), S219–S228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause GS, Tiffany BR, 1993. Suppression of protein synthesis in the reperfused brain. Stroke, 24, 747–755. [DOI] [PubMed] [Google Scholar]
- Kristian T, Hu B, 2018. The Protein Modification and Degradation Pathways after Brain Ischemia. Transl Stroke Res, 9, 199–200. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Dallaporta B, Resche-Rigon M, 1998. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol, 60, 619–642. [DOI] [PubMed] [Google Scholar]
- Kumar R, Azam S, Sullivan JM, et al. , 2001. Brain ischemia and reperfusion activates the eukaryotic initiation factor 2alpha kinase, PERK. J. Neurochem, 77, 1418–1421. [DOI] [PubMed] [Google Scholar]
- Kumar R, Krause GS, Yoshida H, et al. , 2003. Dysfunction of the unfolded protein response during global brain ischemia and reperfusion. J. Cereb. Blood Flow Metab, 23, 462–471. [DOI] [PubMed] [Google Scholar]
- Laybutt DR, Preston AM, Akerfeldt MC, et al. , 2007. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia, 50, 752–763. [DOI] [PubMed] [Google Scholar]
- Lei X, Lei L, Zhang Z, et al. , 2018. Diazoxide inhibits of ER stressmediated apoptosis during oxygenglucose deprivation in vitro and cerebral ischemiareperfusion in vivo. Mol Med Rep, 17, 8039–8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Hayashi T, Jin G, et al. , 2005. The protective effect of dantrolene on ischemic neuronal cell death is associated with reduced expression of endoplasmic reticulum stress markers. Brain Res, 1048, 59–68. [DOI] [PubMed] [Google Scholar]
- Lin M, Ling J, Geng X, et al. , 2019. RTN1-C is involved in high glucose-aggravated neuronal cell subjected to oxygen-glucose deprivation and reoxygenation injury via endoplasmic reticulum stress. Brain Res. Bull, 149, 129–136. [DOI] [PubMed] [Google Scholar]
- Lithner F, Asplund K, Eriksson S, et al. , 1988. Clinical characteristics in diabetic stroke patients. Diabete Metab, 14, 15–19. [PubMed] [Google Scholar]
- Liu C, Fu Q, Mu R, et al. , 2018. Dexmedetomidine alleviates cerebral ischemia-reperfusion injury by inhibiting endoplasmic reticulum stress dependent apoptosis through the PERK-CHOP-Caspase-11 pathway. Brain Res, 1701, 246–254. [DOI] [PubMed] [Google Scholar]
- Liu L, Simon B, Shi J, et al. , 2016. Impact of diabetes mellitus on risk of cardiovascular disease and all-cause mortality: Evidence on health outcomes and antidiabetic treatment in United States adults. World J. Diabetes, 7, 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Shang Y, Yan Z, et al. , 2021. Pim1 kinase provides protection against high glucose-induced stress and apoptosis in cultured dorsal root ganglion neurons. Neurosci. Res, 169, 9–16. [DOI] [PubMed] [Google Scholar]
- Loughlin DT, Artlett CM, 2010. Precursor of advanced glycation end products mediates ER-stress-induced caspase-3 activation of human dermal fibroblasts through NAD(P)H oxidase 4. PLoS One, 5, e11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciani DS, Gwiazda KS, Yang TL, et al. , 2009. Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes, 58, 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maamoun H, Abdelsalam SS, Zeidan A, et al. , 2019. Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes. Int J Mol Sci, 20, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougall NJ, Muir KW, 2011. Hyperglycaemia and infarct size in animal models of middle cerebral artery occlusion: systematic review and meta-analysis. J. Cereb. Blood Flow Metab, 31, 807–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali V, Haddox S, Hornersmith C, et al. , 2018. Essential role for EGFR tyrosine kinase and ER stress in myocardial infarction in type 2 diabetes. Pflugers Arch, 470, 471–480. [DOI] [PubMed] [Google Scholar]
- Marchetti P, Bugliani M, Lupi R, et al. , 2007. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia, 50, 2486–2494. [DOI] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, et al. , 2004. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev, 18, 3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz LO, et al. , 2001. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol, 21, 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally PG, Dean JD, Morris AD, et al. , 2007. Using continuous glucose monitoring to measure the frequency of low glucose values when using biphasic insulin aspart 30 compared with biphasic human insulin 30: a double-blind crossover study in individuals with type 2 diabetes. Diabetes Care, 30, 1044–1048. [DOI] [PubMed] [Google Scholar]
- Miki T, Miura T, Hotta H, et al. , 2009. Endoplasmic reticulum stress in diabetic hearts abolishes erythropoietin-induced myocardial protection by impairment of phospho-glycogen synthase kinase-3beta-mediated suppression of mitochondrial permeability transition. Diabetes, 58, 2863–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno PR, Murcia AM, Palacios IF, et al. , 2000. Coronary composition and macrophage infiltration in atherectomy specimens from patients with diabetes mellitus. Circulation, 102, 2180–2184. [DOI] [PubMed] [Google Scholar]
- Moro MA, Almeida A, Bolanos JP, et al. , 2005. Mitochondrial respiratory chain and free radical generation in stroke. Free Radic. Biol. Med, 39, 1291–1304. [DOI] [PubMed] [Google Scholar]
- Murphy AN, Fiskum G, Beal MF, 1999. Mitochondria in neurodegeneration: bioenergetic function in cell life and death. J. Cereb. Blood Flow Metab, 19, 231–245. [DOI] [PubMed] [Google Scholar]
- Nakatani Y, Kaneto H, Kawamori D, et al. , 2005. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J. Biol. Chem, 280, 847–851. [DOI] [PubMed] [Google Scholar]
- Nakka VP, Prakash-Babu P, Vemuganti R, 2016. Crosstalk Between Endoplasmic Reticulum Stress, Oxidative Stress, and Autophagy: Potential Therapeutic Targets for Acute CNS Injuries. Mol. Neurobiol, 53, 532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niizuma K, Yoshioka H, Chen H, et al. , 2010. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta, 1802, 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N, Yoshii S, Hattori T, et al. , 2005. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J, 24, 1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver N, Gimenez M, Calhoun P, et al. , 2020. Continuous Glucose Monitoring in People With Type 1 Diabetes on Multiple-Dose Injection Therapy: The Relationship Between Glycemic Control and Hypoglycemia. Diabetes Care, 43, 53–58. [DOI] [PubMed] [Google Scholar]
- Ottenbacher KJ, Ostir GV, Peek MK, et al. , 2004. Diabetes mellitus as a risk factor for stroke incidence and mortality in Mexican American older adults. J. Gerontol. A Biol. Sci. Med. Sci, 59, M640–645. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Koizumi A, Takeda K, et al. , 2002. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Invest, 109, 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, et al. , 2006. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science, 313, 1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomeque J, Rueda OV, Sapia L, et al. , 2009. Angiotensin II-induced oxidative stress resets the Ca2+ dependence of Ca2+-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ. Res, 105, 1204–1212. [DOI] [PubMed] [Google Scholar]
- Papa FR, Zhang C, Shokat K, et al. , 2003. Bypassing a kinase activity with an ATP-competitive drug. Science, 302, 1533–1537. [DOI] [PubMed] [Google Scholar]
- Paschen W, Aufenberg C, Hotop S, et al. , 2003. Transient cerebral ischemia activates processing of xbp1 messenger RNA indicative of endoplasmic reticulum stress. J. Cereb. Blood Flow Metab, 23, 449–461. [DOI] [PubMed] [Google Scholar]
- Paschen W, Doutheil J, 1999. Disturbances of the functioning of endoplasmic reticulum: a key mechanism underlying neuronal cell injury? J. Cereb. Blood Flow Metab, 19, 1–18. [DOI] [PubMed] [Google Scholar]
- Paschen W, Gissel C, Linden T, et al. , 1998. Activation of gadd153 expression through transient cerebral ischemia: evidence that ischemia causes endoplasmic reticulum dysfunction. Brain Res. Mol. Brain Res, 60, 115–122. [DOI] [PubMed] [Google Scholar]
- Permutt MA, Wasson J, Cox N, 2005. Genetic epidemiology of diabetes. J. Clin. Invest, 115, 1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plutzky J ZB BJ, 2012. Vascular Biology of Atherosclerosis in Patients with Diabetes: Dyslipidemia, Hypercoagulability, Endothelial Dysfunction and Inflammation, Diabetes in Cardiovascular Disease: A Companion to Braunwald’s Heart Disease, 9th Edition (McGuire DK MN, ed) ed,ELSEVIER Saunders, pp. 111–126. [Google Scholar]
- Puthalakath H, O’Reilly LA, Gunn P, et al. , 2007. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell, 129, 1337–1349. [DOI] [PubMed] [Google Scholar]
- Rao Z, Sun J, Pan X, et al. , 2017. Hyperglycemia Aggravates Hepatic Ischemia and Reperfusion Injury by Inhibiting Liver-Resident Macrophage M2 Polarization via C/EBP Homologous Protein-Mediated Endoplasmic Reticulum Stress. Front. Immunol, 8, 1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehni AK, Cho S, Dave KR, 2019a. Prior exposure to recurrent hypoglycemia causes post-ischemic ER stress via increased free radical production in treated diabetic rats, https://www.abstractsonline.com/pp8/#!/7883/presentation/43842 (Accessed 13 September 2021).
- Rehni AK, Shukla V, Perez-Pinzon MA, et al. , 2019b. Blockade of Acid-Sensing Ion Channels Attenuates Recurrent Hypoglycemia-Induced Potentiation of Ischemic Brain Damage in Treated Diabetic Rats. Neuromolecular Med., 21, 454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricobaraza A, Cuadrado-Tejedor M, Garcia-Osta A, 2011. Long-term phenylbutyrate administration prevents memory deficits in Tg2576 mice by decreasing Abeta. Front. Biosci (Elite Ed), 3, 1375–1384. [DOI] [PubMed] [Google Scholar]
- Ricobaraza A, Cuadrado-Tejedor M, Marco S, et al. , 2012. Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus, 22, 1040–1050. [DOI] [PubMed] [Google Scholar]
- Rieusset J, 2011. Mitochondria and endoplasmic reticulum: mitochondria-endoplasmic reticulum interplay in type 2 diabetes pathophysiology. Int. J. Biochem. Cell Biol, 43, 1257–1262. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P, 2007. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol, 8, 519–529. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Arnold SM, Miller CN, et al. , 2006. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol, 4, 2024–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson TH, Gallaway M, Kumar R, 2015. Unfolding the unfolded protein response: unique insights into brain ischemia. Int. J. Mol. Sci, 16, 7133–7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanges D, Marigo V, 2006. Cross-talk between two apoptotic pathways activated by endoplasmic reticulum stress: differential contribution of caspase-12 and AIF. Apoptosis, 11, 1629–1641. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, et al. , 2001. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell, 7, 1165–1176. [DOI] [PubMed] [Google Scholar]
- Schwarz DS, Blower MD, 2016. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell. Mol. Life Sci, 73, 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvarajah D, Tesfaye S, 2006. Central nervous system involvement in diabetes mellitus. Curr. Diab. Rep, 6, 431–438. [DOI] [PubMed] [Google Scholar]
- Senft D, Ronai ZA, 2015. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci, 40, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D, Singh JN, Sharma SS, 2016. Effects of 4-phenyl butyric acid on high glucose-induced alterations in dorsal root ganglion neurons. Neurosci. Lett, 635, 83–89. [DOI] [PubMed] [Google Scholar]
- Shen J, Chen X, Hendershot L, et al. , 2002. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell, 3, 99–111. [DOI] [PubMed] [Google Scholar]
- Shukla V, Fuchs P, Liu A, et al. , 2019. Recurrent Hypoglycemia Exacerbates Cerebral Ischemic Damage in Diabetic Rats via Enhanced Post-Ischemic Mitochondrial Dysfunction. Transl Stroke Res, 10, 78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siekevitz P, 1957. Powerhouse of the cell. Sci. Am, 197, 131–140. [Google Scholar]
- Silva RM, Ries V, Oo TF, et al. , 2005. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J. Neurochem, 95, 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims-Robinson C, Bakeman A, Glasser R, et al. , 2016. The role of endoplasmic reticulum stress in hippocampal insulin resistance. Exp. Neurol, 277, 261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WS, Johnston SC, Hemphill JC,, 2015. Cerebrovascular Diseases, In: Kasper D, Fauci A, Hauser S, Longo D, Jameson JL, Loscalzo J (Ed.), Harrison’s Principles of Internal Medicine, 19th Edition ed,19e. McGraw-Hill Education, New York, NY. [Google Scholar]
- Soejima E, Ohki T, Kurita Y, et al. , 2018. Protective effect of 3-hydroxybutyrate against endoplasmic reticulum stress-associated vascular endothelial cell damage induced by low glucose exposure. PLoS One, 13, e0191147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokka AL, Putkonen N, Mudo G, et al. , 2007. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J. Neurosci, 27, 901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozen E, Karademir B, Ozer NK, 2015. Basic mechanisms in endoplasmic reticulum stress and relation to cardiovascular diseases. Free Radic. Biol. Med, 78, 30–41. [DOI] [PubMed] [Google Scholar]
- Spencer EA, Pirie KL, Stevens RJ, et al. , 2008. Diabetes and modifiable risk factors for cardiovascular disease: the prospective Million Women Study. Eur. J. Epidemiol, 23, 793–799. [DOI] [PubMed] [Google Scholar]
- Srinivasan K, Sharma SS, 2011a. Augmentation of endoplasmic reticulum stress in cerebral ischemia/reperfusion injury associated with comorbid type 2 diabetes. Neurol. Res, 33, 858–865. [DOI] [PubMed] [Google Scholar]
- Srinivasan K, Sharma SS, 2011b. Sodium phenylbutyrate ameliorates focal cerebral ischemic/reperfusion injury associated with comorbid type 2 diabetes by reducing endoplasmic reticulum stress and DNA fragmentation. Behav. Brain Res, 225, 110–116. [DOI] [PubMed] [Google Scholar]
- Srinivasan K, Sharma SS, 2012. 3-Bromo-7-nitroindazole attenuates brain ischemic injury in diabetic stroke via inhibition of endoplasmic reticulum stress pathway involving CHOP. Life Sci., 90, 154–160. [DOI] [PubMed] [Google Scholar]
- Srivastava SP, Kumar KU, Kaufman RJ, 1998. Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J. Biol. Chem, 273, 2416–2423. [DOI] [PubMed] [Google Scholar]
- Su D, Ma J, Yang J, et al. , 2017. Monosialotetrahexosy-1 ganglioside attenuates diabetes-associated cerebral ischemia/reperfusion injury through suppression of the endoplasmic reticulum stress-induced apoptosis. J. Clin. Neurosci, 41, 54–59. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Logue SE, Gorman AM, et al. , 2006. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep, 7, 880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D, 2011. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol, 13, 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajiri S, Oyadomari S, Yano S, et al. , 2004. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ, 11, 403–415. [DOI] [PubMed] [Google Scholar]
- Tam AB, Koong AC, Niwa M, 2014. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep, 9, 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HP, Guo Q, Hua G, et al. , 2018. Inhibition of endoplasmic reticulum stress alleviates secondary injury after traumatic brain injury. Neural. Regen. Res, 13, 827–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thilmann R, Xie Y, Kleihues P, et al. , 1986. Persistent inhibition of protein synthesis precedes delayed neuronal death in postischemic gerbil hippocampus. Acta Neuropathol, 71, 88–93. [DOI] [PubMed] [Google Scholar]
- Pollard Thomas D Earnshaw William C Lippincott-Schwartz Jennifer Johnson Graham, 2017. Endoplasmic Reticulum, In: Pollard Thomas D Earnshaw William C Lippincott-Schwartz Jennifer Johnson Graham (Ed.), Cell Biology, 3rd Edition ed. Vol. 2021,ELSEVIER, pp. 331–350. [Google Scholar]
- Thorp E, Li G, Seimon TA, et al. , 2009. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metab, 9, 474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey ML, Gilmartin M, O’Neill K, et al. , 2016. Epidemiology of diabetes and complications among adults in the Republic of Ireland 1998–2015: a systematic review and meta-analysis. BMC Public Health, 16, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tureyen K, Bowen K, Liang J, et al. , 2011. Exacerbated brain damage, edema and inflammation in type-2 diabetic mice subjected to focal ischemia. J. Neurochem, 116, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor P, Umapathy D, George L, et al. , 2021. Crosstalk between endoplasmic reticulum stress and oxidative stress in the progression of diabetic nephropathy. Cell Stress Chaperones, 26, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virani SS, Alonso A, Aparicio HJ, et al. , 2021. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation, 143, e254–e743. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D, 2011. The unfolded protein response: from stress pathway to homeostatic regulation. Science, 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Wang G, Cui W, Chen S, et al. , 2021. Metformin alleviates high glucose-induced ER stress and inflammation by inhibiting the interaction between caveolin1 and AMPKalpha in rat astrocytes. Biochem. Biophys. Res. Commun, 534, 908–913. [DOI] [PubMed] [Google Scholar]
- Wang M, Kaufman RJ, 2016. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature, 529, 326–335. [DOI] [PubMed] [Google Scholar]
- Wang X, Shen B, Sun D, et al. , 2018. Aspirin ameliorates cerebral infarction through regulation of TLR4/NFkappaBmediated endoplasmic reticulum stress in mouse model. Mol Med Rep, 17, 479–487. [DOI] [PubMed] [Google Scholar]
- Wang XZ, Kuroda M, Sok J, et al. , 1998. Identification of novel stress-induced genes downstream of chop. EMBO J, 17, 3619–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YC, Li X, Shen Y, et al. , 2020. PERK (Protein Kinase RNA-Like ER Kinase) Branch of the Unfolded Protein Response Confers Neuroprotection in Ischemic Stroke by Suppressing Protein Synthesis. Stroke, 51, 1570–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Kim SJ, Zhang Z, et al. , 2008. ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum. Mol. Genet, 17, 469–477. [DOI] [PubMed] [Google Scholar]
- Weightman Potter PG, Washer SJ, Jeffries AR, et al. , 2021. Attenuated Induction of the Unfolded Protein Response in Adult Human Primary Astrocytes in Response to Recurrent Low Glucose. Front. Endocrinol. (Lausanne), 12, 671724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Wang HG, 2004. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem, 279, 45495–45502. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sato T, Matsui T, et al. , 2007. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell, 13, 365–376. [DOI] [PubMed] [Google Scholar]
- Yan B, Liu S, Li X, et al. , 2019. Preconditioning with endoplasmic reticulum stress alleviated heart ischemia/reperfusion injury via modulating IRE1/ATF6/RACK1/PERK and PGC-1alpha in diabetes mellitus. Biomed. Pharmacother, 118, 109407. [DOI] [PubMed] [Google Scholar]
- Yao W, Wang K, Wang X, et al. , 2021. Icariin ameliorates endothelial dysfunction in type 1 diabetic rats by suppressing ER stress via the PPARalpha/Sirt1/AMPKalpha pathway. J. Cell. Physiol, 236, 1889–1902. [DOI] [PubMed] [Google Scholar]
- Ye T, Meng X, Wang R, et al. , 2018. Gastrodin Alleviates Cognitive Dysfunction and Depressive-Like Behaviors by Inhibiting ER Stress and NLRP3 Inflammasome Activation in db/db Mice. Int. J. Mol. Sci, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Haze K, Yanagi H, et al. , 1998. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem, 273, 33741–33749. [DOI] [PubMed] [Google Scholar]
- Yu L, Li S, Tang X, et al. , 2017a. Diallyl trisulfide ameliorates myocardial ischemia-reperfusion injury by reducing oxidative stress and endoplasmic reticulum stress-mediated apoptosis in type 1 diabetic rats: role of SIRT1 activation. Apoptosis, 22, 942–954. [DOI] [PubMed] [Google Scholar]
- Yu Z, Sheng H, Liu S, et al. , 2017b. Activation of the ATF6 branch of the unfolded protein response in neurons improves stroke outcome. J. Cereb. Blood Flow Metab, 37, 1069–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Xu L, He D, et al. , 2013. Endoplasmic reticulum stress-mediated hippocampal neuron apoptosis involved in diabetic cognitive impairment. Biomed. Res. Int, 2013, 924327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, Li P, Chen SR, et al. , 2001. Dantrolene inhibition of ryanodine receptor Ca2+ release channels. Molecular mechanism and isoform selectivity. J. Biol. Chem, 276, 13810–13816. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Li J, Chen Y, et al. , 2012. Activation of endoplasmic reticulum stress by hyperglycemia is essential for Muller cell-derived inflammatory cytokine production in diabetes. Diabetes, 61, 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, et al. , 1998. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev, 12, 982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Yuan J, Tang ZJ, et al. , 2017. Hydrogen sulfide ameliorates cognitive dysfunction in streptozotocin-induced diabetic rats: involving suppression in hippocampal endoplasmic reticulum stress. Oncotarget, 8, 64203–64216. [DOI] [PMC free article] [PubMed] [Google Scholar]