

Abstract
Purpose of Review
Scavenger receptor class B type I (SR-BI) serves a key role in the reverse cholesterol transport in the liver as the high-affinity receptor for HDL. SR-BI is abundantly expressed in endothelium, and earlier works indicate that the receptor mediates anti-atherogenic actions of HDL. However, more recent studies uncovered novel functions of endothelial SR-BI as a lipoprotein transporter, which regulates transcellular transport process of both LDL and HDL. This brief review focuses on the unique functions of endothelial SR-BI and how they influence atherogenesis.
Recent Findings
Earlier studies indicate that SR-BI facilitates anti-atherogenic actions of HDL through modulation of intracellular signaling to stimulate endothelial nitric oxide synthase. In vivo studies in global SR-BI knockout mice also showed a strong atheroprotective role of the receptor; however, a contribution of endothelial SR-BI to atherosclerosis process in vivo has not been fully appreciated. Recent studies using cultured endothelial cells and in mice with endothelial-specific deletion of the receptor revealed previously unappreciated pro-atherogenic actions of SR-BI, which relates to its ability to deliver LDL into arteries. On the other hand, SR-BI has also been implicated in transport of HDL to the sub-intimal space as a part of reverse cholesterol transport.
Summary
SR-BI mediates internalization and transcellular transport of both HDL and LDL, and the cellular and molecular mechanism of the process has just begun to emerge. Harnessing these dual transport functions of the endothelial SR-BI may provide a novel, effective intervention to atherosclerosis.
Keywords: Endothelium, Scavenger receptor type B class I, Transcytosis, Atherosclerosis
Introduction
Scavenger receptor class B type I (SR-BI, SR-B1, or SCARB1) was initially discovered as a scavenger protein that binds to a series of native and modified lipoproteins [1]. The receptor was subsequently identified as the high-affinity receptor for high-density lipoprotein (HDL), which mediates the selective uptake of HDL-associated cholesterol esters in the hepatocytes [2]. The receptor is composed of two cytoplasmic domains and a large highly glycosylated extracellular domain, where the ligands interact [3, 4]. Global deletion of SR-BI in mice results in abnormal lipid profiles and severe atherosclerotic phenotypes [5–9]. This strong atheroprotective role of SR-BI has been mainly assigned to its function in the liver, in which SR-BI mediates HDL-cholesterol uptake as a crucial part of reverse cholesterol transport (RCT) [5–8, 10]. In addition to the liver, SR-BI is abundantly expressed in macrophages and endothelial cells, and accumulating evidence indicates that the receptor has extrahepatic functions to influence the progression of atherosclerosis [10–14]. Although atheroprotective function of macrophage SR-BI was demonstrated in vivo through bone marrow transplantation experiments [12, 15, 16], an in vivo role of endothelial SR-BI in atherogenesis has not been well understood until recently [14••]. A number of studies in cultured endothelial cells have shown that SR-BI mediates upregulation of nitric oxide (NO) production induced by HDL through activation of endothelial NO synthase, which plays a pivotal role in maintaining atheroprotective endothelial integrity [17–19]. In addition to being a key mediator of anti-atherogenic actions of HDL, more recent studies in cultured cells and in vivo have revealed that SR-BI is involved in both HDL and low-density lipoprotein (LDL) transport across the endothelium [20–22]. During the initial step of the RCT pathway, HDL need to cross the endothelial barrier to subendothelial spaces, where HDL removes excess cholesterol deposited in macrophages in atherosclerotic plaques [23]. Multiple studies in cultured endothelial cells indicate SR-BI is involved in the HDL transport process. In contrast, the subendothelial accumulation of pro-atherogenic LDL particles represents a pivotal step in the early stage of atherosclerosis [24, 25]. Most recently, using a hyperlipidemic mouse model lacking the receptor selectively in endothelium, Huang and his colleagues discovered a novel pro-atherogenic role of endothelial SR-BI, which relates to its function as an LDL transporter [14••]. In this review, we summarize the function of the endothelial SR-BI in the regulation of HDL and LDL transport, and how it influences the progression of atherosclerosis. More comprehensive recent reviews on endothelial transcytosis are available [20, 26].
Transcytosis of Lipoproteins Across Endothelium
The endothelium forms a cellular monolayer which selectively regulates the passage of molecules between the bloodstream and tissues [27]. The transport of macromolecules, including lipoproteins, across the endothelium is actively controlled by endothelial cells via the transcellular pathway, termed transcytosis [28]. The process of transcytosis involves ligand uptake by receptor-mediated endocytosis or fluid-phase pinocytosis, trafficking of the cargo through the cytoplasm, and exocytotic release of the cargo at the sub-intimal side [26]. The transcytosis is initiated by the interaction of molecules with their receptors or transporters on the luminal surface of endothelium. During the initial stages of atherosclerosis, LDL particles are transported across the endothelial barrier and accumulate in the subendothelial space. These LDL particles are then oxidized to form oxidized LDL (oxLDL), which promotes the formation of macrophage foam cells within atherosclerotic lesions [24, 25, 29]. Early studies examining LDL transport across the endothelium in vivo in rats using transmission electron microscopy clearly visualized the process of LDL transcytosis via a vesicular trafficking pathway [30]. Despite the relevance of LDL transport across the endothelium during atherogenesis, the molecular mechanism that controls this process has not been fully understood. Recent studies have identified several key molecules that participate in transcytosis, which include SR-BI, caveolin-1 and activin receptor-like kinase 1 (ALK1) [14, 20, 31, 32]. In contrast to atherogenic LDL, the atheroprotective effect of HDL is attributed to its ability to facilitate RCT through which cholesterol is delivered from the peripheral macrophages to the liver for biliary excretion [24, 33–36]. To achieve the removal of excess cholesterol deposited in the atherosclerotic lesions, circulating HDL need to cross the endothelial barrier to get access to lipid-laden macrophages in atherosclerotic plaques [23]. Previous studies found that the endothelial transcytosis of mature HDL requires SR-BI and an ATP-binding cassette transporter, ABCG1 [37, 38]. Thus, these studies imply that SR-BI may have a dual role in endothelial transport of both HDL and LDL. Here, we will highlight recent findings on the role of SR-BI in endothelial lipoprotein transcytosis.
Endothelial SR-BI and HDL Transport
Blood Endothelial SR-BI
Earlier studies established that the transport of lipids from HDL to SR-BI occurs through a selective cholesteryl ester uptake, in which the lipid content of HDL is transferred without concomitant internalization of the HDL particle itself [39–41]. On the other hand, the study by Silver et al. first showed in polarized hepatocytes in culture that SR-BI mediates HDL internalization and transport to the endosomal recycling compartment and apical membrane regions [42]. Pagler et al. also demonstrated concomitant endocytosis of SR-BI and HDL, which was followed by HDL particle resecretion [43, 44]. Although an exact itinerary of HDL-SR-BI in hepatocytes is yet to be determined, these studies and others suggest that HDL is likely routed to two possible intracellular pools after internalization, which include endosomal recycling compartment and multivesicular bodies [42, 43, 45–48]. In contrast to HDL internalization and secretion process in hepatocytes, HDL transport pathway in endothelium is less defined. HDL is considered to be actively transported through endothelial cells by transcytosis, which involves apical endocytosis, intracellular trafficking, and basolateral exocytosis. Previous studies indicate that in arteries, HDL transcytosis only occurs from the luminal surface to the basolateral intima and not in the reverse direction [20••], and that HDL leaves the intima mainly via the lymphatic system [49]. Endothelial SR-BI has been implicated in transport of HDL from circulation to the intima, as well as the removal of HDL from intimal plaques via lymphatics [50, 51]. The study by von Eckardstein’s group first revealed that both SR-BI and ABCG1 mediate HDL transcytosis in polarized aortic endothelial cells [37]. Using cell surface biotinylation experiments, they showed that approximately 30% of the total cell-associated [125I]-HDL was recovered in intracellular compartments. Internalization of HDL by endothelial cells was further investigated by confocal fluorescence microscopy in endothelial cells incubated with FITC-conjugated HDL and Alexa 594-transferrin. After 10 min of incubation, vesicles containing HDL were partially co-localized with transferrin, indicating that HDL is internalized into early endosomes. In the polarized endothelial cells, HDL translocated from the apical to the basolateral compartment, but no HDL transport was detected from basolateral to apical compartment. Furthermore, HDL binding was partially reduced by silencing of SR-BI or ABCG1, but not after reducing ABCA1 expression. However, co-silencing of SR-BI and ABCG1 did not further decrease HDL transcytosis, indicating existence of additional pathways. More recently, using a combination of spinning-disc confocal and total internal reflection fluorescence microscopy, Fung et al. examined the uptake and transcytosis of HDL by human primary brain microvascular endothelial cell monolayers [52•]. They found that internalized HDL partially co-localized with SR-BI and that knockdown of SR-BI attenuated HDL internalization. Interestingly, Velagapudi et al. reported that vascular endothelial growth factor (VEGF)-A regulates cellular localization of SR-BI to influence transendothelial transport of HDL [38•]. In search of kinases that may be required for HDL uptake in endothelial cells, they performed a microscopy-based high-throughput screening by incubating human aortic endothelial cells with 141 kinase inhibitors and fluorescent-labeled LDL or HDL. They identified the inhibitors of VEGFR as suppressors of HDL uptake, but not LDL, and further confirmed that silencing of VEGFR2 decreased cellular binding, association, and transendothelial transport of HDL. HDL transport was reduced without VEGF-A in cell culture medium, and it was restored by an addition of VEGF-A. The effect of VEGF-A on HDL transport was dependent on activation of phosphoinositide 3-kinase-Akt pathway or p38 mitogen-activated protein kinase, as well as the presence of SR-BI. They further found that VEGF-A is required for the localization of SR-BI in the plasma membrane of endothelial cells. The identification of VEGF as a regulatory factor of transendothelial transport of HDL but not LDL supports the concept that the endothelium is a highly specific barrier for the entry of lipoproteins into the vascular wall, and that this unique function can be a target of pharmaceutical interventions. Although HDL transport across endothelium is largely considered unidirectional from plasma to the intima, additional role for SR-BI in free cholesterol transport from basolateral to apical side of endothelial layer has been reported [53]. Using cultured mouse aortic endothelial cells, Miao et al. observed that unesterified cholesterol can be transported across the endothelial cell monolayer from the basolateral to the apical compartment. Administration of HDL or apolipoprotein AI (apoAI) to the apical compartment enhanced transendothelial cholesterol transport in a concentration-dependent manner. Knockdown of ABCG1 or SR-BI, or inhibition of SR-BI by BLT-1 diminished HDL-induced transendothelial cholesterol transport. SR-BI-mediated transport of cholesterol from the subendothelial intima back to the circulating blood may contribute to atheroprotection, but whether the process occurs in vivo is yet to be determined.
Lymphatic Endothelial SR-BI
Delivery of excess cholesterol from peripheral tissues and macrophages to the bloodstream by HDL is one of the early steps in the RCT process [33, 34]. The lymphatic system is considered to be the primary location for the return of lipoproteins from the interstitial space to circulation [50, 54–56]. It has been shown that lymphatic transport of cholesterol by HDL is mediated by SR-BI expressed on lymphatic endothelium [50]. Lim et al. [50] demonstrated for the first time that SR-BI is expressed in lymphatic endothelial cells both in culture and in vivo, and that treatment of wild-type mice with SR-BI blocking antibody suppressed the transport of HDL via lymphatic vessels. They further confirmed that in vivo reverse cholesterol transport was also impaired in wild-type mice treated with the SR-BI neutralizing antibody or in global SR-BI−/− mice. Their findings indicate that lymphatic vessels play a key role in HDL-mediated RCT through a mechanism that involves the ability of lymphatic endothelial SR-BI to facilitate HDL transcytosis. Using a mouse strain in which endothelial SR-BI is overexpressed by the Tie2 promoter-driven SR-BI transgene, Vaisman et al. have shown that endothelial cell-specific overexpression of SR-BI in high-fat, high-cholesterol diet-fed C57BL/6 mice results in an increase in plasma HDL-cholesterol levels and a decrease in total cholesterol levels, thereby contributing to a reduction in atherosclerosis [57]. The mechanism by which endothelial SR-BI overexpression alters plasma lipid profiles is not clear; however, because Tie2 is expressed both in blood and lymphatic endothelium [58], the changes in plasma lipid profiles observed in these mice may be consistent with the finding that lymphatic endothelial SR-BI participates in the removal of cholesterol from peripheral tissues by lymphatic vessels through the uptake and transcytosis of HDL [56].
Collectively, accumulating evidence suggests that HDL transport across endothelial cells via transcytosis plays a major role in atheroprotective RCT and that both blood and lymphatic endothelial SR-BI are a crucial participant in the process (Fig. 1a).
Fig. 1.
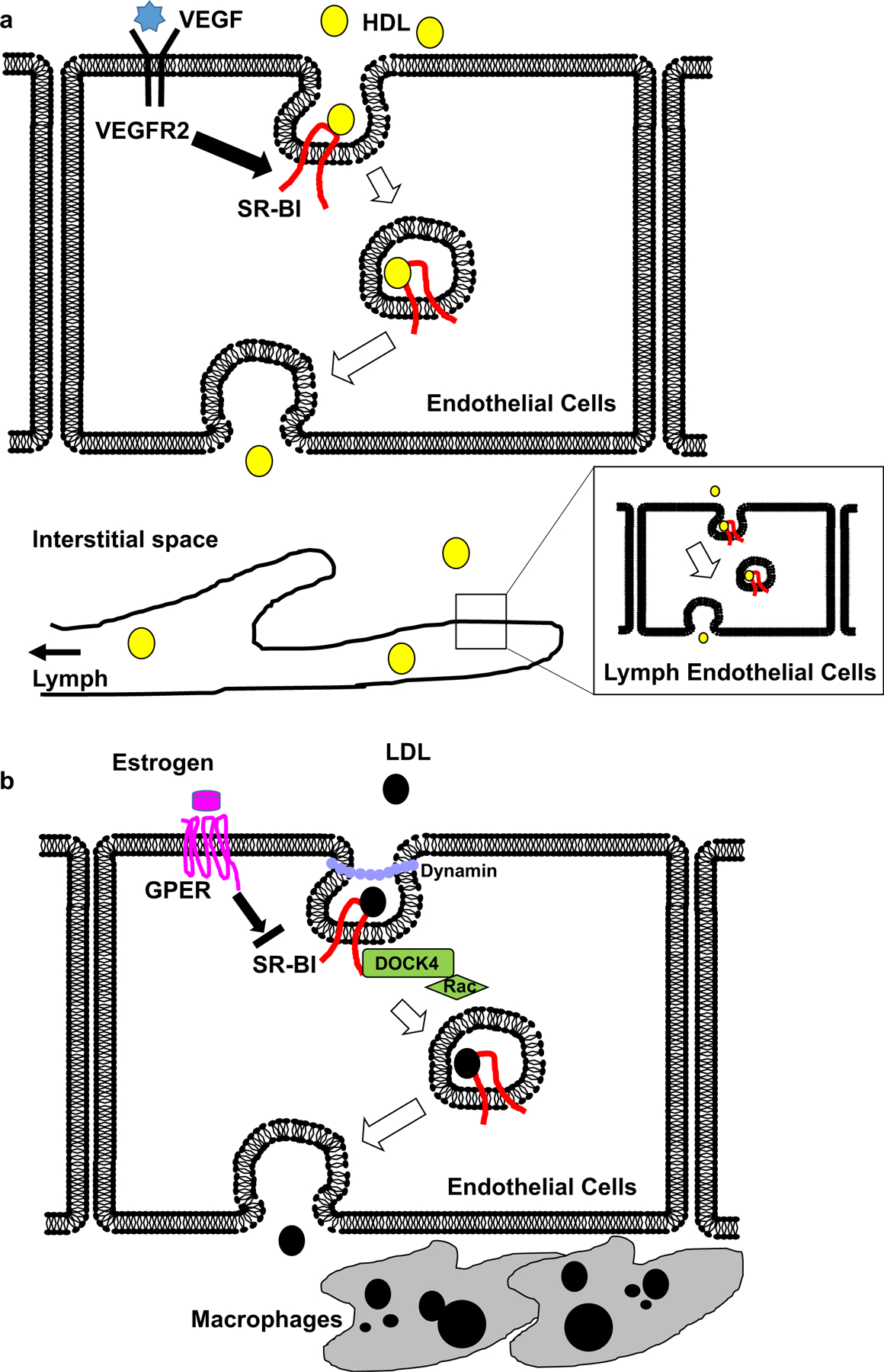
Dichotomous role of endothelial SR-BI as a transporter of both HDL and LDL. a Circulating HDL binds to SR-BI on the luminal surface of endothelial cells, and HDL-SR-BI complex is transported via a vesicular trafficking pathway. VEGF-VEGFR2 signaling promotes HDL transport by upregulating plasma membrane SR-BI. HDL is then released in the interstitial space, where it is further delivered to the lymphatic system through lymphatic endothelial cells in an SR-BI-dependent manner, as a critical step of the reverse cholesterol transport. b SR-BI facilitates internalization of LDL in a dynamin-dependent manner. LDL binding to SR-BI recruits DOCK4 to the C-terminal cytoplasmic domain of the receptor, which in turn promotes LDL internalization by Rac activation. Estrogen binding to GPER negatively regulates LDL transcytosis through downregulation of SR-BI. LDL-SR-BI complex is transported across endothelium to be released in the sub-intimal space, where it is engulfed by macrophages to contribute to atherogenesis
Endothelial SR-BI and LDL Transport
As a number of studies established SR-BI as a mediator of HDL uptake and intracellular transport in endothelial cells, it is relatively recent that studies have started to emerge testing whether SR-BI mediates transcytosis of LDL to deliver the circulating atherogenic lipoproteins into the subendothelial space, where they are engulfed by macrophages that become foam cells to promote the formation of atherosclerotic lesions. Warren L. Lee’s group initially uncovered an unexpected role for endothelial SR-BI in LDL transcytosis [21]. They first perfused mouse aortas ex vivo with LDL and small molecular weight dextran to monitor transendothelial and paracellular transport, respectively. They detected LDL accumulation in the subendothelial space, indicating that LDL transcytosis occurs in intact vessels. No intimal dextran was observed despite its smaller size. They further confirmed that LDL transcytosis occurs in polarized human coronary artery endothelial cells in culture. An assay was then developed to quantify transcytosis of DiI-LDL in real time using total internal reflection fluorescence microscopy. DiI-LDL transcytosis was inhibited by excess unlabeled LDL, whereas degradation of the LDL receptor by PCSK9 had no effect, indicating that the LDL receptor plays a minimum role in LDL transcytosis. Instead, LDL co-localized partially with SR-BI and overexpression of SR-BI increased LDL transcytosis, whereas knockdown by siRNA significantly reduced it. Furthermore, excess HDL, the canonical SR-BI ligand, decreased LDL transcytosis, consistent with a receptor-mediated process rather than non-specific diffusion across paracellular routes. Incubation with Dyngo4A, a specific chemical inhibitor of the GTPase dynamin, abrogated the process. More recently, the same group demonstrated that estrogen suppresses LDL transcytosis in cultured human endothelial cells [59•]. LDL transcytosis was significantly higher in cells derived from men compared with those from premenopausal women. Estrogen treatment attenuated LDL transcytosis in endothelial cells from male but not female donors. They found that estrogen caused downregulation of endothelial SR-BI, and overexpression of SR-BI was sufficient to restore LDL transcytosis. Compared to endothelial cells, treatment with estrogen had no impact on SR-BI expression in hepatocarcinoma-derived HepG2 cells. Inhibition of estrogen receptor α or β had no effect on estrogen-mediated attenuation of LDL transcytosis. Instead, estrogen’s effect on LDL transcytosis was blocked by depletion of the G protein– coupled estrogen receptor 1 (GPER, also known as G protein–coupled receptor 30). GPER was found to be enriched in endothelial cells compared with hepatocytes and it has been known to signal via transactivation of the epidermal growth factor receptors (EGFR) [60]; inhibition of EGFR prevented the effect of estrogen on SR-BI and LDL transcytosis. The study also found that SR-BI protein expression was significantly higher in human coronary artery endothelial cells from male compared with premenopausal female donors.
More recently, Huang and his colleagues demonstrated that SR-BI in endothelial cells mediates the delivery of LDL into arteries and its accumulation by artery wall macrophages, contributing to promotion atherosclerosis [14••]. Using mice lacking SR-BI selectively in the endothelium (SR-BI△EC), they found that, compared with SR-BIfl/fl controls, both male and female SR-BI△EC mice displayed markedly decreased formation of hyperlipidemia-induced atherosclerosis. The endothelial deletion of SR-BI did not alter circulating total cholesterol, triglyceride, or HDL levels and it also did not change vascular inflammation. Instead, they discovered that DiI-labeled native LDL and oxLDL incorporation into the aorta in vivo was diminished in SR-BI△EC mice, indicating that the previous finding [21] that there is less LDL accumulation in ex vivo perfused arteries from global SR-BI−/− mice is likely to have been related to loss of the receptor in the endothelium. Furthermore, using cultured human aortic endothelial cells, they identified the molecular basis for LDL trafficking by SR-BI. Studies using RNAi-mediated silencing and neutralizing antibodies indicated that SR-BI, LDL receptor, and cluster of differentiation 36 (CD36), the known LDL binding receptors in endothelial cells, all promote LDL uptake by human endothelial cells; however, they found that only SR-BI is required for LDL transcytosis. In contrast to these classical receptors for LDL, downregulation of ALK1, to which LDL binding and transport has been previously described [61], reduced LDL transcytosis to a similar degree by the selective loss of SR-BI, and there was a further decline with their concurrent knockdown. Thus, both SR-BI and ALK1 mediate LDL transcytosis in endothelial cells likely through different mechanisms. Moreover, using a series of deletion and point mutants of SR-BI, they showed that the ability of SR-BI to facilitate LDL transcytosis requires eight amino acids (487–494, IQAYSESL) in the C-terminal cytoplasmic domain of the receptor. Through analysis of the proteins in endothelial cells that were pulled down with wild-type SR-BI using liquid chromatography and tandem mass spectrometry, they identified DOCK4 (guanine nucleotide exchange factor dedicator of cytokinesis 4) as a necessary partner of SR-BI for LDL transcytosis. DOCK4 is a membrane-associated cytoplasmic protein that functions as a guanine nucleotide exchange factor and a participant in actin cytoskeleton regulation, and it has been shown to mediate PDGF receptor internalization in fibroblasts [62–64]. Further studies in cultured endothelial cells showed that LDL binding to SR-BI increases DOCK4 recruitment to the cytoplasmic domain and that DOCK4 promotes internalization of SR-BI through activation of the small GTPase Rac1. Interestingly, in mice compared with the atherosclerosis-resistant greater curvature of the aortic arch, mRNA levels of both SR-BI and DOCK4 were increased in the atherosclerosis-prone lesser curvature before lesion formation. To seek human relevance, they compared gene expression levels of SR-BI and DOCK4 in human atherosclerotic versus normal arteries in three publicly available independent human cohorts (Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/)). The analysis of the datasets revealed that expression of both SR-BI and DOCK4 transcripts was greater in atherosclerotic arteries than normal arteries.
Collectively, these recent studies provide evidence that LDL transcytosis is a key mechanism that drives atherosclerosis, and that this process is actively regulated by SR-BI in the endothelium (Fig. 1b).
Conclusions
The recent findings that are summarized in this review highlight the importance of endothelial SR-BI in regulating lipoprotein transcytosis to influence atherosclerosis (Fig. 1). Although these studies have identified several molecules in addition to SR-BI that participate in the transcytosis process, there are a multitude of questions that remain to be addressed.
The study by Huang et al. has implicated DOCK4 and Rac1 in the initial step of LDL internalization. However, the exact route of LDL-SR-BI in transcytotic pathway is unknown. Several early studies have suggested the involvement of both clathrin-mediated endocytosis and non-coated caveolae (also called plasmalemmal vesicle)-mediated endocytosis in the initial step of transcytosis across the endothelium [30, 65]. It was reported that LDL endocytosis in endothelial cells devoid of caveolin-1 (Cav-1), a structural component of caveolae, was lower than in wild-type cells, supporting a role of caveolae in LDL transcytosis [66, 67]. Furthermore, global Cav-1 knockout mice exhibited reduced LDL infiltration into the artery wall and a drastic reduction of atherosclerotic plaques, whereas endothelial cell-specific re-expressing Cav-1 in these mice reversed both effects [31, 68]. SR-BI has been shown to be localized within caveolae in cultured endothelial cells [69]; however, other studies dispute the involvement of caveolae in LDL transport [52, 70]. Furthermore, in addition to dynamin and DOCK4, what molecules are involved in budding and fusion of transcytotic vesicles, as well as exocytotic events at the basolateral surface of endothelium? Distinctive members of Rab GTPase family members are known to regulate early, late, and recycling vesicular trafficking [71], but Rab proteins specifically involved in transcytosis are not yet identified. The recent finding that SR-BI co-localizes with LDL in trafficking vesicles in mouse aortic endothelial cells [14••] may provide a rationale to perform biochemical and proteomic analysis of purified “transcytotic vesicles” to further characterize them and to identify protein components.
Secondly, the cumulative studies demonstrated dichotomous functions of endothelial SR-BI in atherosclerosis; a mediator of anti-inflammatory, anti-atherogenic actions of HDL and a facilitator of LDL transport during an initial phase of atherogenesis. Because HDL and LDL shares binding sites on the extracellular domain of SR-BI [72] and that excess HDL competes transcytosis of LDL [21], circulating concentrations of the lipoproteins may influence which functions of SR-BI play a dominant role. Deciphering this enigma will be important to further consider therapeutic interventions targeting either endothelial SR-BI or the transcytosis process. Recently a considerable advancement in endothelial targeting of therapeutic cargos has been made, using monoclonal antibody or nanoparticles [73, 74]. It may be feasible to target VEGF inhibitors selectively to endothelium to evaluate its effect on LDL uptake and hyperlipidemia-induced atherosclerosis in rodent models. Also, targeted administration of SR-BI neutralizing antibodies or its inhibitor such as BLT-1 to inhibit selective function of SR-BI in endocytosis without influencing its main functions in the liver may provide an answer to the enigma, as well as possible novel therapeutic opportunities for atherosclerosis prevention.
Lastly, it is critically important to investigate whether endothelial SR-BI and its function as a lipoprotein transporter influence atherosclerosis process in humans. Genome-wide association studies identified human SNPs in the SR-BI (SCARB1) genes that are associated with HDL-cholesterol levels and cardiovascular disease risks [75–79]; however, these human phenotypes are likely due to the receptor’s dominant function in the liver. The work by Huang et al. has found in multiple human cohorts that both SR-BI and DOCK4 gene expression are upregulated in atherosclerotic aortas compared to normal arteries [14••]. More studies are warranted to determine whether increased expressions of SR-BI and DOCK4 contribute to atherogenesis and to identify the transcriptional machinery responsible for the mRNA upregulation in endothelial cells.
Recent single-cell RNA sequencing studies have revealed that CD36, another member of scavenger receptor family which shares high structural homology with SR-BI, serves as a unique marker for a subpopulation of endothelial cells in mouse aortas [80–82]. One of the studies further demonstrated that the subpopulation of endothelial cells represented by the expression of CD36 preferentially located in the greater curvature of the aortic root. This is in contrast to the localization of SR-BI mRNA, which showed higher expression in the lesser curvature [14••]. It will be important to determine in human arteries whether SR-BI and other molecules implicated in transcytosis are component of a subpopulation of endothelial cells, and if so, whether the subset of endothelium is responsible for uptake of LDL or HDL by endothelial cells to cause or prevent formation of atherosclerotic plaques.
Footnotes
Compliance with Ethical Standards
Conflict of Interest All authors declare no conflict of interest.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Acton SL, Scherer PE, Lodish HF, Krieger M. Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J Biol Chem. 1994;269(33):21003–9. [PubMed] [Google Scholar]
- 2.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271(5248):518–20. [DOI] [PubMed] [Google Scholar]
- 3.Krieger M Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J Clin Invest. 2001;108(6):793–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mineo C Lipoprotein receptor signaling in atherosclerosis. Cardiovasc Res 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braun A, Trigatti BL, Post MJ, Sato K, Simons M, Edelberg JM, et al. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ Res. 2002;90(3):270–6. [DOI] [PubMed] [Google Scholar]
- 6.Kozarsky KF, Donahee MH, Glick JM, Krieger M, Rader DJ. Gene transfer and hepatic overexpression of the HDL receptor SR-BI reduces atherosclerosis in the cholesterol-fed LDL receptor-deficient mouse. Arterioscler Thromb Vasc Biol. 2000;20(3):721–7. [DOI] [PubMed] [Google Scholar]
- 7.Krieger M, Kozarsky K. Influence of the HDL receptor SR-BI on atherosclerosis. Curr Opin Lipidol. 1999;10(6):491–7. [DOI] [PubMed] [Google Scholar]
- 8.Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci U S A. 1997;94(23):12610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuller M, Dadoo O, Serkis V, Abutouk D, MacDonald M, Dhingani N, et al. The effects of diet on occlusive coronary artery atherosclerosis and myocardial infarction in scavenger receptor class B, type 1/low-density lipoprotein receptor double knockout mice. Arterioscler Thromb Vasc Biol. 2014;34(11):2394–403. [DOI] [PubMed] [Google Scholar]
- 10.Huby T, Doucet C, Dachet C, Ouzilleau B, Ueda Y, Afzal V, et al. Knockdown expression and hepatic deficiency reveal an atheroprotective role for SR-BI in liver and peripheral tissues. J Clin Invest. 2006;116(10):2767–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tao H, Yancey PG, Babaev VR, Blakemore JL, Zhang Y, Ding L, et al. Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. J Lipid Res. 2015;56(8):1449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Covey SD, Krieger M, Wang W, Penman M, Trigatti BL. Scavenger receptor class B type I-mediated protection against atherosclerosis in LDL receptor-negative mice involves its expression in bone marrow-derived cells. Arterioscler Thromb Vasc Biol. 2003;23(9):1589–94. [DOI] [PubMed] [Google Scholar]
- 13.Pei Y, Chen X, Aboutouk D, Fuller MT, Dadoo O, Yu P, et al. SR-BI in bone marrow derived cells protects mice from diet induced coronary artery atherosclerosis and myocardial infarction. PLoS One. 2013;8(8):e72492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang L, Chambliss KL, Gao X, Yuhanna IS, Behling-Kelly E, Bergaya S, et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature. 2019;569(7757):565–9 •• The first in vivo evidence that endothelial SR-BI plays an atherogenic role through its ability to transport LDL across endothelium via DOCK4 and Rac1.
- 15.Zhang W, Yancey PG, Su YR, Babaev VR, Zhang Y, Fazio S, et al. Inactivation of macrophage scavenger receptor class B type I promotes atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2003;108(18):2258–63. [DOI] [PubMed] [Google Scholar]
- 16.Van EM, Bos IS, Hildebrand RB, Van Rij BT, van Berkel TJ. Dual role for scavenger receptor class B, type I on bone marrow-derived cells in atherosclerotic lesion development. Am J Pathol. 2004;165(3):785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mineo C, Shaul PW. Functions of scavenger receptor class B, type I in atherosclerosis. Curr Opin Lipidol. 2012;23(5):487–93. [DOI] [PubMed] [Google Scholar]
- 18.Mineo C, Shaul PW. Novel biological functions of high-density lipoprotein cholesterol. Circ Res. 2012;111(8):1079–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mineo C, Shaul PW. Regulation of signal transduction by HDL. J Lipid Res. 2013;54(9):2315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang X, Sessa WC, Fernandez-Hernando C. Endothelial transcytosis of lipoproteins in atherosclerosis. Front Cardiovasc Med. 2018;5:130. •• A comprehensive review on lipoprotein transport in endothelial cells.
- 21.Armstrong SM, Sugiyama MG, Fung KY, Gao Y, Wang C, Levy AS, et al. A novel assay uncovers an unexpected role for SR-BI in LDL transcytosis. Cardiovasc Res. 2015;108(2):268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zanoni P, Velagapudi S, Yalcinkaya M, Rohrer L, von Eckardstein A. Endocytosis of lipoproteins. Atherosclerosis. 2018;275:273–95. [DOI] [PubMed] [Google Scholar]
- 23.Nordestgaard BG. The vascular endothelial barrier–selective retention of lipoproteins. Curr Opin Lipidol. 1996;7(5):269–73. [DOI] [PubMed] [Google Scholar]
- 24.Glass CK, Witztum JL. Atherosclerosis. The road ahead. Cell. 2001;104(4):503–16. [DOI] [PubMed] [Google Scholar]
- 25.Linton MF, Yancey PG, Davies SS, Jerome WG, Linton EF, Song WL, et al. The role of lipids and lipoproteins in atherosclerosis. Endotext 2019. [Google Scholar]
- 26.Fung KYY, Fairn GD, Lee WL. Transcellular vesicular transport in epithelial and endothelial cells: challenges and opportunities. Traffic. 2018;19(1):5–18. [DOI] [PubMed] [Google Scholar]
- 27.Rahimi N Defenders and challengers of endothelial barrier function. Front Immunol. 2017;8:1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279–367. [DOI] [PubMed] [Google Scholar]
- 29.Tabas I, Garcia-Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209(1):13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vasile E, Simionescu M, Simionescu N. Visualization of the binding, endocytosis, and transcytosis of low-density lipoprotein in the arterial endothelium in situ. J Cell Biol. 1983;96(6):1677–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez-Hernando C, Yu J, Suarez Y, Rahner C, Davalos A, Lasuncion MA, et al. Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell Metab. 2009;10(1):48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandez-Hernando C, Yu J, Davalos A, Prendergast J, Sessa WC. Endothelial-specific overexpression of caveolin-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2010;177(2):998–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rader DJ, Hovingh GK. HDL and cardiovascular disease. Lancet. 2014;384(9943):618–25. [DOI] [PubMed] [Google Scholar]
- 34.Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371(25):2383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krieger M Charting the fate of the “good cholesterol”: identification and characterization of the high-density lipoprotein receptor SR-BI. Annu Rev Biochem. 1999;68:523–58. [DOI] [PubMed] [Google Scholar]
- 36.Linton MRF, Yancey PG, Davies SS, Jerome WG, Linton EF, Song WL, et al. The role of lipids and lipoproteins in atherosclerosis. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, et al. , editors. Endotext. South Dartmouth (MA) 2000. [Google Scholar]
- 37.Rohrer L, Ohnsorg PM, Lehner M, Landolt F, Rinninger F, von Eckardstein A. High-density lipoprotein transport through aortic endothelial cells involves scavenger receptor BI and ATP-binding cassette transporter Gl. Circ Res. 2009;104(10):1142–50. [DOI] [PubMed] [Google Scholar]
- 38. Velagapudi S, Yalcinkaya M, Piemontese A, Meier R, Norrelykke SF, Perisa D, et al. VEGF-A regulates cellular localization of SR-BI as well as transendothelial transport of HDL but not LDL. Arterioscler Thromb Vase Biol. 2017;37(5):794–803 • The work discovered inhibitors of VEGF-R2 signaling suppress HDL transcytosis by modulating SR-BI cellular localization.
- 39.Glass C, Pittman RC, Weinstein DB, Steinberg D. Dissociation of tissue uptake of cholesterol ester from that of apoprotein A-I of rat plasma high density lipoprotein: selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc Natl Acad Sci USA. 1983;80(17):5435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knecht TP, Pittman RC. A plasma membrane pool of cholesteryl esters that may mediate the selective uptake of cholesteryl esters from high-density lipoproteins. Biochim Biophys Acta. 1989;1002(3):365–75. [DOI] [PubMed] [Google Scholar]
- 41.Pittman RC, Knecht TP, Rosenbaum MS, Taylor CA Jr. A nonendocytotic mechanism for the selective uptake of high density lipoprotein-associated cholesterol esters. J Biol Chem. 1987;262(6):2443–50. [PubMed] [Google Scholar]
- 42.Silver DL, Wang N, Xiao X, Tall AR. High density lipoprotein (HDL) particle uptake mediated by scavenger receptor class B type 1 results in selective sorting of HDL cholesterol from protein and polarized cholesterol secretion. J Biol Chem. 2001;276(27):25287–93. [DOI] [PubMed] [Google Scholar]
- 43.Rohrl C, Stangl H. HDL endocytosis and resecretion. Biochim Biophys Acta. 2013;1831(11):1626–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pagler TA, Rhode S, Neuhofer A, Laggner H, Strobl W, Hinterndorfer C, et al. SR-BI-mediated high density lipoprotein (HDL) endocytosis leads to HDL resecretion facilitating cholesterol efflux. J Biol Chem. 2006;281(16):11193–204. [DOI] [PubMed] [Google Scholar]
- 45.Rhode S, Breuer A, Hesse J, Sonnleitner M, Pagler TA, Doringer M, et al. Visualization of the uptake of individual HDL particles in living cells via the scavenger receptor class B type I. Cell Biochem Biophys. 2004;41(3):343–56. [DOI] [PubMed] [Google Scholar]
- 46.Sun B, Eckhardt ER, Shetty S, van der Westhuyzen DR, Webb NR. Quantitative analysis of SR-BI-dependent HDL retroendocytosis in hepatocytes and fibroblasts. J Lipid Res. 2006;47(8):1700–13. [DOI] [PubMed] [Google Scholar]
- 47.Wustner D Mathematical analysis of hepatic high density lipoprotein transport based on quantitative imaging data. J Biol Chem. 2005;280(8):6766–79. [DOI] [PubMed] [Google Scholar]
- 48.Wustner D, Mondal M, Huang A, Maxfield FR. Different transport routes for high density lipoprotein and its associated free sterol in polarized hepatic cells. J Lipid Res. 2004;45(3):427–37. [DOI] [PubMed] [Google Scholar]
- 49.Nordestgaard BG, Hjelms E, Stender S, Kjeldsen K. Different efflux pathways for high and low density lipoproteins from porcine aortic intima. Arteriosclerosis. 1990;10(3):477–85. [DOI] [PubMed] [Google Scholar]
- 50.Lim HY, Thiam CH, Yeo KP, Bisoendial R, Hii CS, McGrath KC, et al. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab. 2013;17(5):671–84. [DOI] [PubMed] [Google Scholar]
- 51.Martel C, Li W, Fulp B, Platt AM, Gautier EL, Westerterp M, et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J Clin Invest. 2013;123(4):1571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fung KY, Wang C, Nyegaard S, Heit B, Fairn GD, Lee WL. SR-BI mediated transcytosis of HDL in brain microvascular endothelial cells is independent of caveolin, clathrin, and PDZK1. Front Physiol 2017;8:841. • Following up the group’s earlier works, the study showed in brain microvascular endothelial cells that HDL transcytosis requires dynamin and SR-BI, but not clathrin or caveolin-1.
- 53.Miao L, Okoro EU, Cao Z, Yang H, Motley-Johnson E, Guo Z. High-density lipoprotein-mediated transcellular cholesterol transport in mouse aortic endothelial cells. Biochem Biophys Res Commun. 2015;465(2):256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cooke CJ, Nanjee MN, Stepanova IP, Olszewski WL, Miller NE. Variations in lipid and apolipoprotein concentrations in human leg lymph: effects of posture and physical exercise. Atherosclerosis. 2004;173(1):39–45. [DOI] [PubMed] [Google Scholar]
- 55.Martel C, Randolph GJ. Atherosclerosis and transit of HDL through the lymphatic vasculature. Curr Atheroscler Rep. 2013;15(9):354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fernandez-Hernando C Lymphatic vessels clean up your arteries. J Clin Invest. 2013;123(4):1417–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vaisman BL, Vishnyakova TG, Freeman LA, Amar MJ, Demosky SJ, Liu C, et al. Endothelial expression of scavenger receptor class B, type i protects against development of atherosclerosis in mice. Biomed Res Int. 2015;2015:607120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eklund L, Kangas J, Saharinen P. Angiopoietin-tie signalling in the cardiovascular and lymphatic systems. Clin Sci (Lond) 2017;131(1):87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ghaffari S, Naderi Nabi F, Sugiyama MG, Lee WL. Estrogen inhibits LDL (low-density lipoprotein) transcytosis by human coronary artery endothelial cells via GPER (G-protein-coupled estrogen receptor) and SR-BI (scavenger receptor class B type 1). Arterioscler Thromb Vasc Biol. 2018;38(10):2283–94 • The work demonstrated for the first time that modification of SR-BI expression by a hormone, estrogen, influences LDL transcytosis in cultured endothelial cells.
- 60.Barton M, Prossnitz ER. Emerging roles of GPER in diabetes and atherosclerosis. Trends Endocrinol Metab. 2015;26(4):185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kraehling JR, Chidlow JH, Rajagopal C, Sugiyama MG, Fowler JW, Lee MY, et al. Genome-wide RNAi screen reveals ALK1 mediates LDL uptake and transcytosis in endothelial cells. Nat Commun 2016;7:13516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gadea G, Blangy A. Dock-family exchange factors in cell migration and disease. Eur J Cell Biol. 2014;93(10–12):466–77. [DOI] [PubMed] [Google Scholar]
- 63.Yajnik V, Paulding C, Sordella R, McClatchey AI, Saito M, Wahrer DC, et al. DOCK4, a GTPase activator, is disrupted during tumorigenesis. Cell. 2003;112(5):673–84. [DOI] [PubMed] [Google Scholar]
- 64.Kawada K, Upadhyay G, Ferandon S, Janarthanan S, Hall M, Vilardaga JP, et al. Cell migration is regulated by platelet-derived growth factor receptor endocytosis. Mol Cell Biol. 2009;29(16): 4508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ghitescu L, Fixman A, Simionescu M, Simionescu N. Specific binding sites for albumin restricted to plasmalemmal vesicles of continuous capillary endothelium: receptor-mediated transcytosis. J Cell Biol. 1986;102(4):1304–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pavlides S, Gutierrez-Pajares JL, Danilo C, Lisanti MP, Frank PG. Atherosclerosis, caveolae and caveolin-1. Adv Exp Med Biol. 2012;729:127–44. [DOI] [PubMed] [Google Scholar]
- 67.Frank PG, Pavlides S, Cheung MW, Daumer K, Lisanti MP. Role of caveolin-1 in the regulation of lipoprotein metabolism. Am J Physiol Cell Physiol. 2008;295(1):C242–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frank PG, Lee H, Park DS, Tandon NN, Scherer PE, Lisanti MP. Genetic ablation of caveolin-1 confers protection against atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24(1):98–105. [DOI] [PubMed] [Google Scholar]
- 69.Mineo C, Shaul PW. Circulating cardiovascular disease risk factors and signaling in endothelial cell caveolae. Cardiovasc Res. 2006;70(1):31–41. [DOI] [PubMed] [Google Scholar]
- 70.Sun B, Boyanovsky BB, Connelly MA, Shridas P, van der Westhuyzen DR, Webb NR. Distinct mechanisms for OxLDL uptake and cellular trafficking by class B scavenger receptors CD36 and SR-BI. J Lipid Res. 2007;48(12):2560–70. [DOI] [PubMed] [Google Scholar]
- 71.Pfeffer SR. Rab GTPases: master regulators that establish the secretory and endocytic pathways. Mol Biol Cell. 2017;28(6):712–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neculai D, Schwake M, Ravichandran M, Zunke F, Collins RF, Peters J, et al. Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature. 2013;504(7478):172–6. [DOI] [PubMed] [Google Scholar]
- 73.Kiseleva RY, Glassman PM, Greineder CF, Hood ED, Shuvaev VV, Muzykantov VR. Targeting therapeutics to endothelium: are we there yet? Drug Deliv Transl Res 2018;8(4):883–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Glassman PM, Myerson JW, Ferguson LT, Kiseleva RY, Shuvaev VV, Brenner JS, et al. Targeting drug delivery in the vascular system: focus on endothelium. Adv Drug Deliv Rev. 2020;157:96–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466(7307):707–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Webb TR, Erdmann J, Stirrups KE, Stitziel NO, Masca NG, Jansen H, et al. Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J Am Coll Cardiol. 2017;69(7):823–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Howson JMM, Zhao W, Barnes DR, Ho WK, Young R, Paul DS, et al. Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat Genet. 2017;49(7):1113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vergeer M, Korporaal SJ, Franssen R, Meurs I, Out R, Hovingh GK, et al. Genetic variant of the scavenger receptor BI in humans. N Engl J Med. 2011;364(2):136–45. [DOI] [PubMed] [Google Scholar]
- 79.Zanoni P, Khetarpal SA, Larach DB, Hancock-Cerutti WF, Millar JS, Cuchel M, et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science. 2016;351(6278):1166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lukowski SW, Patel J, Andersen SB, Sim SL, Wong HY, Tay J, et al. Single-cell transcriptional profiling of aortic endothelium identifies a hierarchy from endovascular progenitors to differentiated cells. Cell Rep. 2019;27(9):2748–58 e3. [DOI] [PubMed] [Google Scholar]
- 81.Kalluri AS, Vellarikkal SK, Edelman ER, Nguyen L, Subramanian A, Ellinor PT, et al. Single-cell analysis of the normal mouse aorta reveals functionally distinct endothelial cell populations. Circulation. 2019;140(2):147–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Feng W, Chen L, Nguyen PK, Wu SM, Li G. Single cell analysis of endothelial cells identified organ-specific molecular signatures and heart-specific cell populations and molecular features. Front Cardiovasc Med. 2019;6:165. [DOI] [PMC free article] [PubMed] [Google Scholar]