

Abstract
OBJECTIVE
Type 2 diabetes (T2D) has heterogeneous patient clinical characteristics and outcomes. In previous work, we investigated the genetic basis of this heterogeneity by clustering 94 T2D genetic loci using their associations with 47 diabetes-related traits and identified five clusters, termed β-cell, proinsulin, obesity, lipodystrophy, and liver/lipid. The relationship between these clusters and individual-level metabolic disease outcomes has not been assessed.
RESEARCH DESIGN AND METHODS
Here we constructed individual-level partitioned polygenic scores (pPS) for these five clusters in 12 studies from the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium and the UK Biobank (n = 454,193) and tested for cross-sectional association with T2D-related outcomes, including blood pressure, renal function, insulin use, age at T2D diagnosis, and coronary artery disease (CAD).
RESULTS
Despite all clusters containing T2D risk-increasing alleles, they had differential associations with metabolic outcomes. Increased obesity and lipodystrophy cluster pPS, which had opposite directions of association with measures of adiposity, were both significantly associated with increased blood pressure and hypertension. The lipodystrophy and liver/lipid cluster pPS were each associated with CAD, with increasing and decreasing effects, respectively. An increased liver/lipid cluster pPS was also significantly associated with reduced renal function. The liver/lipid cluster includes known loci linked to liver lipid metabolism (e.g., GCKR, PNPLA3, and TM6SF2), and these findings suggest that cardiovascular disease risk and renal function may be impacted by these loci through their shared disease pathway.
CONCLUSIONS
Our findings support that genetically driven pathways leading to T2D also predispose differentially to clinical outcomes.
Introduction
Type 2 diabetes (T2D) affects >400 million individuals worldwide (1) and is a major health and economic burden, largely due to the complications of the disease (2). It is well appreciated that there are multiple environmental and genetic risk factors for T2D and also that there is marked heterogeneity observed in patients with T2D (3,4). Greater understanding of the biological pathways precipitating T2D onset and its complications could help improve patient management.
Polygenic scores, which aggregate common genetic variation across the genome, predict risk of T2D with an area under the receiver operator characteristic curve of ∼0.70 and suggest that the 5% of individuals with the highest scores have an approximately threefold increased risk of T2D compared with the remainder of the population (3,5,6). Recently, we and others have investigated methods to partition the T2D polygenic score into “process-specific” subset scores. Two recent publications used cluster analysis of variant trait-associations to identify a shared set of five clusters of T2D genetic loci representing mechanisms of insulin deficiency and insulin resistance (7,8). Briefly, of these five clusters, two were defined by associated traits related to insulin production and secretion (termed “β-cell” and “proinsulin,” respectively), while three were associated with insulin resistance (“obesity,” “lipodystrophy,” “liver/lipid”) (3).
Additionally, Udler et al. (7) showed that the cluster-specific partitioned polygenic risk scores (pPS) generated using the clusters allowed a genetics-driven approach to help reduce the heterogeneity seen in individuals with T2D. For example, the “lipodystrophy” cluster was defined by GWAS trait associations with reduced leptin levels, reduced BMI, increased fasting insulin adjusted for BMI, increased serum triglycerides, and reduced HDL cholesterol mirroring clinical features seen in monogenic forms of lipodystrophy. Individuals with T2D and an increased pPS for the lipodystrophy cluster likewise had significantly higher fasting C-peptide and serum triglyceride levels but lower BMI and HDL cholesterol levels compared with other individuals with T2D (7). There was also a preliminary analysis performed suggesting that the clusters had differing associations with population-level risk of outcomes (which were not included in the cluster derivation), such as coronary artery disease (CAD), systolic blood pressure (SBP), diastolic blood pressure (DBP), and renal function; however, these outcome analyses were performed only using summary statistics from genome-wide association studies (GWAS) and did not involve individual level data (7), therefore limiting ability to adjust association models for custom covariates, such as T2D status, and to analyze extremes of the pPS for risk of metabolic outcomes.
A critical question in assessing clinical relevance of these pPS in personalized T2D patient care is whether they are associated with clinical metabolic disease outcomes using individual-level data. We undertook a large-scale multicohort study using individual-level data to test whether the partitioned T2D genetic scores derived by Udler et al. (7) were associated with clinical outcomes in up to 454,193 participants, including 25,015 individuals with T2D, across the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium cohorts and the UK Biobank (UKBB).
Research Design and Methods
Study Populations
Our analyses included the UKBB and 12 studies from the CHARGE Consortium: Age, Gene/Environment Susceptibility (AGES) Study (9), Atherosclerosis Risk in Communities (ARIC) Study (10), Institute for Personalized Medicine (IPM) BioMe Biobank (11), Doetinchem Cohort Study (12), Framingham Heart Study (FHS) (13–15), Genetic Epidemiology Network of Arteriopathy (GENOA) Study (16,17), Health, Aging and Body Composition (Health ABC) Study (18), Mass General Brigham Biobank (MGB) (19), Multi-Ethnic Study of Atherosclerosis (MESA) (20), Netherlands Epidemiology in Obesity (NEO) Study (21), PROspective Study of Pravastatin in the Elderly at Risk (PROSPER) (22–24), and Rotterdam Study (25). All analyses were restricted to individuals of European ancestry. Descriptive statistics for each study are included in Supplementary Table 1.
All studies obtained relevant participant consent and Institutional Review Board approval. Analysis of UKBB was performed under application 27892. Analysis of the data was approved by the Mass General Brigham (formerly Partners) Institutional Review Board (Boston, MA).
Genotyping and Imputation Methods
Information about genotyping and imputation approaches taken by each cohort is available in Supplementary Table 1.
Generation of pPS
The pPS were derived using results from a published analysis of 94 genetic variants associated with increased T2D risk, which clustered variants based on variant-trait associations and allowed variants to contribute to all clusters (7). The traits included in that analysis were glycemic, anthropometric, and laboratory continuous-trait measures, but notably, no measures of blood pressure, renal function, or disease outcomes. In this present study, we generated polygenic scores for each cluster using the T2D risk alleles and cluster weights, incorporating the top-weighted loci in each cluster (weight ≥0.75) (Supplementary Table 2).
Quality Control
The following analyses were performed for consistency across cohorts and quality control. Frequencies of the alleles used in generating the pPS were compared with those from the MGB Biobank to prevent accidental allele swapping. The median of the pPS (Supplementary Table 3) was compared with the median in MGB Biobank; all values were in high concordance. Cluster pPS associations were performed with continuous traits that were previously found to be significantly associated with the clusters (7); each cohort calculated associations between the pPS for the five clusters and BMI, HDL cholesterol, and triglycerides in all individuals adjusted for T2D status as well as the subset of individuals designated as T2D case subjects. These models were adjusted for age, sex, and the first five principal components of ancestry. Directionality of the regression estimates was compared with the published results to validate the results provided by each cohort (Supplementary Table 4). Cohort level results’ SE were also plotted against square root of sample size to identify any potential outliers.
Regression Models
Each participating cohort calculated individual-level pPS for the five clusters, as described above. The pPS were tested for association with the following cross-sectional outcomes: CAD, estimated glomerular filtration rate using creatinine (eGFR-creatinine), chronic kidney disease (CKD), SBP, DBP, and hypertension (HTN). Insulin use and age at T2D diagnosis were also tested in the subset of T2D case subjects. Continuous traits with outlier values were winsorized at five SDs from the mean. Outcome definitions had some cohort-specific variability (Supplementary Table 1) but generally aligned with the following criteria: CAD was defined as recognized myocardial infarction or other related mortality; eGFR-creatinine was computed via the Chronic Kidney Disease Epidemiology Collaboration creatinine equation in mL/min; CKD was defined as eGFR values <60 mL/min; HTN was defined as having SBP >140 mmHg or DBP >90 mmHg, with cohort-specific adjustment for blood pressure medications; and T2D was defined as use of diabetes medication, fasting glucose ≥7 mmol/L, 2-h glucose ≥11.1 mmol/L, or glycated hemoglobin (HbA1c) ≥6.5%. See Supplementary Table 1 for complete definitions of all measures.
Within each cohort, we calculated associations between these T2D-related traits and the pPS for each of the five clusters. Regression models, logistic or linear depending on the outcome, were adjusted for age, sex, T2D case subject/control subject status, and the first five principal components of ancestry.
Continuous pPS Model Equation
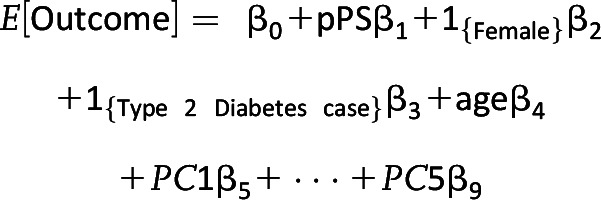 |
For a second analysis, individuals were defined as extreme for a given cluster if their pPS values were larger than the 90th percentile value in a reference population, chosen to be the MBG Biobank cohort. We ran models evaluating the effect of an individual being extreme in a given cluster. Such models included all five indicator variables for being extreme, one for each cluster, in addition to age, sex, T2D case subject/control subject status, and the first five principal components of ancestry in addition to a global test of the effect of falling in the extreme of any cluster. For these extreme analyses on binary outcomes, cohorts were only included if they had at least 500 individuals experiencing the outcome.
Extreme pPS Model Equation
 |
where 1{Condition} is an indicator function equal to 1 if the participant has the condition and 0 otherwise.
Meta-analysis
For each T2D-related trait and outcome, cohorts provided estimates from their study-level regression coefficients. Effect estimates and SEs were used to perform a meta-analysis using an inverse-variance weighted approach (fixed-effect). The R package “meta” version 4.18-2 was used to combine estimates of effect and produce an overall association test. Heterogeneity of effects across cohorts was assessed using the I2 statistic (26). Statistical significance of the association with pPS was evaluated using a 0.0005 level, based on a Bonferroni adjustment for performing 100 tests, including eight outcomes (two of which were specific to T2D case subjects only), five clusters, and three subsamples (all individuals with adjustment for T2D case subject/control subject status, all individuals without adjustment for T2D case subject/control subject status, and then in T2D case subjects only).
We report as main results the cluster-outcome associations that reached the Bonferroni level of significance (P ≤ 0.0005) in at least one analysis (T2D-adjusted, T2D-unadjusted, or T2D case subject only). Given the possibility of collider bias, as described below, we highlighted findings significant in the T2D-adjusted model that also had nominal significance (P < 0.05) in the T2D-unadjusted analysis.
Cohorts also reported estimates and covariances of β-coefficients from regressions including the five indicators for individuals being extremes in a given cluster. To combine these results, a multiple parameter meta-analysis (27) was performed using the “mvmeta” package in R. Moreover, to jointly assess the significance of the extreme variables, a Wald test was performed using the combined estimates and their estimated covariance matrices. Statistical significance of extreme pPS indicators was evaluated using a Bonferroni adjusted level of 0.0025, which controls type I error at 0.05 after performing 20 tests. There were 20 sets of estimates because there were eight outcomes, two of which were specific to T2D case subjects only, and three samples: 1) all individuals with adjustment for T2D case subject/control subject status, 2) all individuals without adjustment for T2D case subject/control subject status, and 3) T2D case subjects only.
Evaluating the Extent of Collider Bias
We acknowledge the possibility that our analyses are susceptible to collider bias (a spurious association that arises from adjustment on a collider). More specifically, we were interested in the association between pPS and T2D-related traits, which, arguably, share a covariate affecting these variables: T2D case subject/control subject status. Therefore, to evaluate the extent of collider bias, we compared each model with and without adjustment for T2D case subject/control subject status and also in case subjects only. If we observed an association between the pPS and an outcome in a model with T2D case subject/control subject status adjustment but did not observe the association without adjustment (P > 0.05), we considered that association to be indicative of collider bias (28).
Follow-up Analyses in UKBB
To further contextualize some of our results, additional analyses were run in the UKBB. The following renal outcomes were assessed in the continuous pPS model with T2D adjustment: serum creatinine, serum cystatin C, eGFR-cystatin C, urine albumin-to-creatinine ratio (UACR), and serum albumin (UKBB fields provided in Supplementary Table 5). A measure of eGFR over time (eGFR-slope) was calculated for both of the above eGFR measures by calculating the difference between eGFR at two different assessment center visits divided by the time between visits (N = 13,000 individuals; median time between visits was 4.4 years). Additionally, for the primary outcomes available in all individuals (CAD, eGFR-creatinine, CKD, SBP, DBP, and HTN), as well as the renal-specific outcomes just described, interactions of T2D and cluster pPS were assessed by adding a multiplicative interaction term to the continuous pPS models. Finally, to evaluate the proportion of variability explained in each outcome, a partial r2 value was calculated for all outcome-cluster regression models including validation traits by calculating the difference in r2 between the full model with the cluster pPS and the covariate-only model. Nagelkerke pseudo-r2 was used for logistic regression of binary outcomes.
Results
Among the 454,193 participants from the 13 cohorts, 46% were men, mean age was 58.3 years, and 25,015 (5.5%) had T2D (Table 1).
Table 1.
Overview of cohort information
| Study | N * | Male, n (%) | Age, years | CAD | eGFR, mL/min | CKD | SBP, mmHg | DBP, mmHg | HTN | T2D |
|---|---|---|---|---|---|---|---|---|---|---|
| AGES | 3,215 | 1,352 (42) | 76.4 (5.5) | 782 | 63.8 (15.4) | 1,274 | 152.1 (22.2) | 80.4 (10.5) | 2,553 | 369 |
| ARIC | 9,344 | 4,406 (47.1) | 54.3 (5.7) | 467 | 99.5 (12.5) | 89 | 122.2 (19.4) | 74.1 (11.5) | 2,498 | 812 |
| BioMe | 8,668 | 4,042 (46.6) | 62.5 (11.8) | 1,690 | 77.8 (23.1) | 541 | 154 (20.8) | 91(11.8) | 2,808 | 922 |
| Doetinchem | 4,080 | 1,909 (46.7) | 60.2 (9.3) | 100 | 95.2 (10.4) | NA† | 136.3 (20.5) | 83.6 (18.5) | 1,526 | 147 |
| FHS | 7,145 | 3,251 (45.5) | 56.5 (13.6) | 273 | 88.8 (15.5) | 275 | 127.7 (21.8) | 76.7 (10.9) | 2,596 | 852 |
| GENOA | 1,391 | 612 (43.9) | 55.5 (10.9) | 108 | 79.6 (16.5) | 165 | 142.5 (19.8) | 84.6 (10.7) | 1,303 | 151 |
| Health ABC | 1,639 | 868 (52.9) | 73.8 (2.8) | 227 | 66.6 (13.3) | 492 | 141.1 (22.4) | 74.9 (12.1) | 1,026 | 231 |
| MGB | 18,127 | 8,659 (47.7) | 62.8 (13.2) | 2,059 | 77.9 (22.7) | 3,370 | 135.4 (19.5) | 80.5 (11.8) | 9,926 | 3,546 |
| MESA | 2,685 | 1,285 (47.8) | 62.7 (10.2) | 254 | 75.8 (17.0) | 344 | 128.2 (23.1) | 75 (12.5) | 1,165 | 173 |
| NEO | 5,705 | 2,735 (47.9) | 56.9 (5.9) | 127 | 86.1 (12.5) | 141 | 137.5 (19.3) | 88.2 (11.6) | 3,193 | 576 |
| PROSPER | 5,244 | 2,524 (48.1) | 75.3 (3.4) | 708 | 72 (21.4) | 1,549 | 165.6 (23.2) | 91 (12.4) | 3,257 | 544 |
| Rotterdam | 8,809 | 3,821 (43.3) | 65.5 (9.9) | 1,003 | 78.8 (16.1) | 1,060 | 144.0 (22.0) | 80.7 (11.9) | 6,482 | 1,050 |
| UKBB | 378,141 | 174,131 (46) | 57.4 (8.0) | 13,286 | 90.5 (13.1) | 7,722 | 141.1 (20.7) | 84.3 (11.3) | 198,189 | 15,642 |
| Combined | 454,193 | 209,595 (46.1) | 58.3 | 21,084 | 89.1 | 17,022 | 140.8 | 83.9 | 236,522 | 25,015 |
Distribution of variables by cohort including sample size (N), age, sex (n male), number of individuals with T2D, and cross-sectional outcomes: CAD, eGFR, CKD, SBP, DBP, and HTN. Continuous variables represented as mean (SD); categorical variables represented as count (%). NA, not available.
Maximum sample size for all outcomes/covariates.
Outcome not available in this cohort.
Using the pPS for each of the five T2D genetic clusters, as previously defined in Udler et al. (7), the associations with expected clinical features were replicated in our study participants: increased obesity cluster pPS was associated with increased BMI; increased lipodystrophy cluster pPS with decreased BMI, decreased HDL cholesterol, and increased triglycerides; and increased liver/lipid cluster pPS with decreased triglycerides (P < 10−10 for all associations) (Supplementary Table 4).
We identified several significant cluster pPS-outcome associations involving all five clusters and SBP, DBP, HTN, CAD, eGFR-creatinine, and CKD outcomes (Fig. 1, Table 2, and Supplementary Tables 6–8).
Figure 1.

Forest plot of cluster pPS associations with SBP, SBP, HTN, CAD, eGFR, and CKD. The error bars represent 95% CIs. **Results met the Bonferroni-corrected significance threshold (0.0005) in both T2D-adjusted and T2D-unadjusted analyses. *Results that met adjusted significance in only one model, as defined in research design and methods. Detailed results in Table 2 and Supplementary Tables 6 and 7.
Table 2.
Results of pPS on outcomes that reached study-wide significance
| Without T2D adjustment | With T2D adjustment | ||||
|---|---|---|---|---|---|
| Cluster | Outcome | Effect estimate per average pPS allele | P value | Effect estimate per average pPS allele | P value |
| Obesity | SBP | 0.68 mmHg | 8.1 × 10−17 | 0.56 | 7.9 × 10−11 |
| Obesity | DBP | 0.44 mmHg | 1.9 × 10−19 | 0.38 | 1.5 × 10−13 |
| Obesity | Hypertension | OR 1.08 | 1.1 × 10−18 | 1.07 | 1.3 × 10−11 |
| Lipodystrophy | SBP | 2.43 mmHg | 8.9 × 10−39 | 1.8 | 4.7 × 10−20 |
| Lipodystrophy | DBP | 0.95 mmHg | 1.8 × 10−17 | 0.73 | 6.9 × 10−10 |
| Lipodystrophy | Hypertension | OR 1.30 | 1.3 × 10−34 | 1.20 | 6.8 × 10−15 |
| Lipodystrophy | CAD | OR 1.32 | 1.5 × 10−08 | 1.11 | 5.8 × 10−02 |
| Liver/lipid | CAD | OR 0.94 | 2.7 × 10−03 | 0.92 | 1.2 × 10−04 |
| Liver/lipid | eGFR | −0.87 mL/min | 5.0 × 10−78 | −0.81 | 9.4 × 10−61 |
| Liver/lipid | CKD | OR 1.08 | 1.5 × 10−04 | 1.06 | 2.1 × 10−02 |
| β-Cell | SBP | 1.43 mmHg | 3.6 × 10−11 | 0.35 | 1.3 × 10−01 |
| β-Cell | DBP | −0.13 mmHg | 3.1 × 10−01 | −0.61 | 9.3 × 10−06 |
| β-Cell | HTN | OR 1.15 | 2.6 × 10−08 | 1.00 | 8.6 × 10−01 |
| β-Cell | CAD | OR 1.41 | 1.7 × 10−09 | 1.12 | 7.7 × 10−02 |
| Proinsulin | DBP | −0.18 mmHg | 4.4 × 10−03 | −0.25 | 1.8 × 10−04 |
Cluster-outcome models with a significantly associated pPS. Effect estimates are in terms of outcome units per average weighted alleles in the pPS. Bold indicates a significant result at the 0.0005 Bonferroni-corrected threshold.
Increased obesity cluster pPS and lipodystrophy cluster pPS were significantly associated with higher measures of blood pressure: for obesity pPS: SBP (β = 0.68 mmHg, P = 8.1 × 10−17), DBP (β = 0.44 mmHg, P = 1.9 × 10−19), and risk of HTN (odds ratio [OR] = 1.08, P = 1.1 × 10−18); for lipodystrophy pPS: SBP (β = 2.43 mmHg, P = 8.9 × 10−39), DBP (β = 0.95 mmHg, P = 1.8 × 10−17), and HTN (OR = 1.30, P = 1.3 × 10−34), results reported as change per average weighted allele pPS in all participants without T2D adjustment. Results remained significant with T2D adjustment. Only the lipodystrophy-HTN analysis showed evidence of heterogeneity across cohorts with I2 > 0.5 (Supplementary Table 7). Thus, the obesity and lipodystrophy cluster pPS were both associated with increased blood pressure, despite these cluster pPS having opposite directions of effect from each other for metrics of body fat composition such as BMI (Supplementary Table 4) (7). In contrast, the proinsulin cluster was associated with reduced DBP in the T2D-adjusted model (β = −0.25, P = 1.8 × 10−4), with some attenuation, but still residual signal in the unadjusted model (β = −0.18, P = 4.4 × 10−3) (Fig. 1 and Supplementary Tables 6–8).
We observed significant associations with CAD risk for the liver/lipid and lipodystrophy pPS but in opposite directions (Fig. 1 and Supplementary Tables 6–8), highlighting the value of partitioning T2D loci. The liver/lipid pPS was associated with reduced risk of CAD (OR = 0.92, P = 1.2 × 10−4 in T2D-adjusted analysis). The association remained nominally significant in the T2D-unadjusted and case subject-only analyses (P < 0.05 for both). In contrast, the lipodystrophy pPS was significantly associated with increased CAD risk in the model unadjusted for T2D (OR = 1.32, P = 1.5 × 10−8), although with some evidence of heterogeneity with I2 > 0.5 (Supplementary Table 7), but lost significance in the T2D-adjusted and case subject-only analyses.
We also found that an increased liver/lipid cluster pPS was significantly associated with decreased eGFR-creatinine, indicative of reduced renal function (β = −0.87 mL/min, P = 5.0 × 10−78 for change per weighted allele pPS in T2D-unadjusted model). This result remained significant with T2D adjustment (Table 2 and Supplementary Table 7). This association was attenuated, but remained significant in the UKBB even after removal of the top-weighted locus in the cluster, GCKR rs780094, which is known to be especially pleiotropic (29) (UKBB change per weighted allele = −0.91 mL/min, P = 1.4 × 10−7 after removing GCKR locus vs. before β = −0.80, P = 8.1 × 10−54). This cluster pPS was associated with CKD, defined by most cohorts as eGFR ≤60 mL/min, with a more significant association observed for the T2D-unadjusted model (OR per weighted allele = 1.09, P = 1.5 × 10−4) than the T2D-adjusted model (OR = 1.06, P = 0.02), suggesting that T2D partially mediated the association of this cluster with CKD.
To further contextualize the association of the liver/lipid pPS with eGFR-creatinine, we assessed the association of this cluster pPS with other biomarkers related to renal function in all UKBB participants: serum creatinine, cystatin-C, eGFR-cystatin, and UACR (Supplementary Table 5). Compared with serum creatinine, which is used in the equation to derive eGFR-creatinine, cystatin-C is less influenced by muscle mass, sex, and age (30,31). We found that increased pPS for the liver/lipid cluster was associated with both increased serum creatinine and cystatin-C in the UKBB (change per weighted allele pPS β = 0.92, P = 4.2 × 10−68 and β = 0.006, P = 1.1 × 10−20, respectively, in T2D-unadjusted analyses) as well as reduced eGFR-cystatin (P = 9.1 × 10−25), further supporting the notion that increased genetic risk for this cluster was associated with reduced renal function. Interestingly, the liver/lipid pPS was associated with reduced UACR (β = −0.02, P = 3.6 × 10−8 in T2D-unadjusted analysis) in contrast to the increased ratios typically seen with T2D-induced renal injury. This prompted us to look at the liver/lipid cluster’s association with serum albumin, which was also significantly reduced (β = −0.02, P = 8.9 × 10−89 in T2D-unadjusted analysis), perhaps indicating the reduced UACR relates to reduced liver production of albumin. All results remained significant in the T2D-adjusted analyses. Finally, we assessed whether the liver/lipid pPS was associated with reduced renal function over time, as measured by slopes of eGFR-creatinine and eGFR-cystatin between study visits (median 4.4 years), and found no association for either (P > 0.05).
We also observed significant associations for the β-cell pPS with a number of outcomes that were only significant for a single model and lost significance with either adjustment for T2D (SBP, HTN, CAD) or removing T2D adjustment (DBP) (Table 2 and Supplementary Tables 6 and 7). These results therefore likely indicate either complete mediation by T2D (for SBP, HTN, CAD) or collider bias (DBP).
In the T2D-case subject only analyses, most of the above findings were directionally consistent, but no results reached study-wide significance in this subset (Supplementary Table 8).
Extreme Polygenic Risk
In addition to considering each pPS as a continuous trait, we also analyzed the extremes of each pPS as a way of studying individuals with the highest scores in one or more clusters, which has the potential to categorize individuals and advance individual-level clinical translation for clinical decision making. We defined participants as being extreme for a given cluster if their pPS was at or about the 90th percentile in the MGB Biobank, a chosen reference population.
In the global test including the individuals with extreme pPS for any cluster, we observed significant associations with four outcomes (Supplementary Table 9). The results of the extreme pPS analysis were overall consistent with the continuous pPS results, showing that individuals with extreme pPS values for any the five clusters had significantly different estimates of eGFR-creatinine, DBP, SBP, and risk of HTN compared with the rest of the population. These results appear to be driven by the same significant cluster outcome relationships observed in the continuous pPS results (Supplementary Tables 9–11).
Conclusions
We have analyzed partitioned T2D polygenic scores derived from genetic clustering of T2D loci (7) and assessed associations with clinical outcomes in up to 454,193 participants across the CHARGE Consortium studies and the UKBB. We find significant cross-sectional associations for cluster outcome associations, including all clusters and SBP, DBP, HTN, eGFR-creatinine, CKD, and CAD. These differential associations observed with partitioning the full T2D polygenic score into cluster-specific polygenic scores speak to the benefit of identifying subsets of loci through cluster analysis for improved understanding of disease biology. Our results also have important potential implications if pathways are targeted for drug development. At the same time, the proportions of variance explained by each cluster pPS for any given outcome were small, generally <0.1% (Supplementary Tables 6–8), indicating that the polygenic scores in and of themselves have limited predictive ability, such that clinical utility is not yet established.
The finding that both increased obesity pPS and lipodystrophy pPS are associated with increased measures of blood pressure is particularly interesting since the two polygenic scores are associated with the opposite direction of effect from each other for metrics of body fat composition such as BMI (Supplementary Table 4) (7). We found that adjusting for BMI completely attenuated all blood pressure associations for the obesity pPS as well as for the proinsulin pPS, but not for the lipodystrophy pPS (Supplementary Table 12). As both obesity and lipodystrophy clusters are associated with metrics of insulin resistance (7), the results support that genetic factors increasing both T2D risk and blood pressure may relate to different mechanisms of insulin resistance, with the mechanism for the obesity cluster, but not lipodystrophy cluster, explained by increased whole-body adiposity.
The lipodystrophy cluster has been hypothesized to relate instead to abnormal compartmentalization of fat. Within the lipodystrophy cluster, one of the top-weighted loci includes the putative causal gene PPARG, known to cause familial partial lipodystrophy, a Mendelian disease characterized by abnormal fat distribution with reduced subcutaneous and greater ectopic adiposity as well as increased insulin resistance and HTN (32). The lipodystrophy pPS in the present analysis is similar to another pPS developed by Yaghootkar et al. (33,34) that involved genetic loci associated with body fat distribution. This “body fat distribution cluster” was also associated with increased measures of SBP and DBP.
Within the obesity cluster, MC4R, is likewise a top-weighted locus and putative causal gene that is known to cause monogenic obesity and increased insulin resistance; however, interestingly, individuals with loss-of-function mutations in MC4R have been found to have lower blood pressure compared with age- and BMI-matched control subjects (35,36). For the common variant tagging the MC4R locus included in the obesity cluster pPS (rs12970134), the T2D risk-increasing allele was not associated with HTN in the UKBB (P > 0.05 in all models). The obesity cluster’s association with HTN was therefore driven by other loci in the cluster, particularly FTO and NRXN3 (HTN in both T2D-adjusted and -unadjusted UKBB models P < 10−5). Therefore, multiple pathways increasing risk of obesity and T2D may exist with differing impact on HTN that could be further delineated in the future with additional loci and relevant physiological trait inputs into the genetic clustering.
Despite all cluster alleles increasing T2D risk, we observed two clusters with opposite directions of effect on CAD risk: the lipodystrophy pPS increasing CAD risk and the liver/lipid pPS reducing risk. The lipodystrophy cluster’s significant association with CAD in the T2D-unadjusted model was attenuated with T2D adjustment, potentially indicating that this cluster’s impact on CAD risk is mediated by T2D. As mentioned above, the lipodystrophy cluster is similar to the Yaghootkar et al. “body fat distribution cluster,” which was also associated with increased CAD risk in a T2D-unadjusted model (33,34). Notably, a defining trait feature of both the lipodystrophy and liver/lipid clusters is serum triglyceride levels, with the lipodystrophy cluster significantly associated with increased triglyceride levels and the liver/lipid cluster with decreased levels (Supplementary Table 4) (7). Adjusting for serum triglycerides in the regression model only partially attenuated the associations of both clusters with CAD (Supplementary Table 12), suggesting that this biomarker alone does not capture the effect of the clusters on CAD risk.
The association of increased liver/lipid pPS with multiple measures of reduced renal function and reduced UACR was unexpected and intriguing. The loci in the liver/lipid cluster include GCKR, PNPLA3, and TM6SF2, which all share a common mechanism of involvement in liver lipid metabolism (37–39), and one of the driving traits defining this cluster was again serum triglycerides (Supplementary Table 4) (7). We therefore investigated whether adjustment for serum triglycerides would attenuate the cluster’s associations with eGFR and CKD, but it did not (Supplementary Table 12). Additionally, T2D status did not interact with liver/lipid pPS on measures of renal function, with the exception of UACR (Supplementary Table 13). Of note, a recent analysis clustering individuals with diabetes based on clinical measures identified a severe insulin resistance diabetes subgroup, which portended an increased risk of CKD and fatty liver disease (4); when the genetics of ∼1,000 individuals in the severe insulin resistance diabetes subgroup were studied, there was enrichment for obesity and lipodystrophy genetic cluster alleles, but no enrichment for liver/lipid alleles was detected (40). Thus, it remains unclear how the liver/lipid genetic cluster, presumably related to liver lipid metabolism and increased liver-mediated insulin resistance, also predisposes to reduced renal function, and merits further investigation.
A number of outcomes had study-wide significant associations with the β-cell cluster pPS, but none replicated at nominal significance in a second analysis model and thus were thought to represent either collider bias (for DBP only seen in the T2D-adjusted model) or complete mediation by T2D (for SBP, HTN, CAD only seen in the T2D-unadjusted model). As the β-cell cluster is the cluster most strongly associated with fasting glucose and HbA1c (7), it is possible that it was particularly impacted by changes in model adjustment for T2D.
While the T2D-case subject only results were overall directionally consistent with the full population analyses (including noncase subjects), none reached study-wide significance. The lack of associations seen in T2D case subjects could potentially reflect the smaller sample size (5.5% of the full data set) and/or noise introduced by looking at a disease population that is likely to have additional other contributions to these outcomes, such as medication use and environmental risk factors. We also did not observe any significant interactions with T2D status and pPS for any outcomes when assessed in the UKBB (Supplementary Table 13), potentially also indicating that each pPS did not impact outcome risk differentially in those with or without T2D. The lack of association seen for the two outcomes restricted to case subjects—age at T2D diagnosis and insulin use—could be due to a true lack of association or, alternatively, may reflect limited power, especially as these particular outcomes can be challenging to accurately capture.
One important limitation of this work is the restriction to individuals of European ancestry. The genetic clusters were originally constructed using studies of individuals of predominantly European ancestry due to availability of trait GWAS summary statistics in this population and to minimize heterogeneity across studies included in the clustering (7). We therefore focused on populations of European ancestries in this present work as an initial discovery analysis; in ongoing work, we are performing cluster analysis using T2D and trait GWAS involving non-European ancestral groups.
Another limitation is the cross-sectional nature of the analysis, which may create a selection/survivor bias. We attempted to look longitudinally at the impact of the clusters on renal function over time (median 4.4 years) in 13,000 UKBB participants with data available, but were unable to detect an association, which could potentially require a longer follow-up interval. Future investigation of the identified outcome associations in prospective studies with incident outcomes will be informative.
In summary, by partitioning T2D genetic loci by proposed mechanistic pathways, we have identified differential associations of loci with T2D-related outcomes. These associations are best seen when analyzing the full pPS as continuous variables, but the same patterns were also noted in analyses of the extremes of the distributions. Thus, there may be subgroups of individuals who can be identified based on genetic pPS as having a higher risk of T2D-related outcomes, although risk of these T2D outcomes conferred to such individuals is predicted to be small in magnitude. Expansion of genetic clusters to include more related variants together conferring large effect sizes would likely be necessary before individual-level clinical translation is possible. Nevertheless, our findings suggest that genetically driven pathways leading to T2D may also predispose to other distinct clinical outcomes, highlighting the benefit of separating T2D risk loci into process-specific genetic clusters and offering insight into mechanisms of disease.
Article Information
Acknowledgments and Funding.
AGES. The authors thank all AGES-Reykjavik study participants and the staff of the Icelandic Heart Association for their contribution to the AGES-Reykjavik study. This AGES-Reykjavik study has been funded by National Institutes of Health (NIH) contract N01-AG-1-2100 and HHSN271201200022C, the National Institute on Aging (NIA) Intramural Research Program, Hjartavernd (the Icelandic Heart Association), and the Althingi (the Icelandic Parliament). VGudm. is supported by the Icelandic Centre for Research (grant no. 184845-051).
ARIC. The authors thank the staff and participants of the ARIC study for their important contributions. The Atherosclerosis Risk in Communities study has been funded in whole or in part with funds from the National Heart, Lung, and Blood Institute (NHLB), NIH, Department of Health and Human Services (contract numbers HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700004I, and HHSN268201700005I), R01HL087641, R01HL059367, and R01HL086694, National Human Genome Research Institute (NHGRI) contract U01HG004402, and NIH contract HHSN268200625226C. Infrastructure was partly supported by the National Institutes of Health Roadmap for Medical Research, a component of the NIH, grant number UL1RR025005.
BioMe. The authors thank all participants in the Mount Sinai Biobank and all of the recruiters who have assisted and continue to assist in data collection and management. The authors are grateful for the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai. The Mount Sinai BioMe Biobank has been supported by The Andrea and Charles Bronfman Philanthropies and in part by Federal funds from the NHLBI (X01HL134588) and NHGRI (U01HG007417). G.N.N. is funded by the NIH NIDDK (R01DK127139, R56DK126930, K23DK107908). R.J.F.L. is funded by NIH NIDDK (R01DK110113, R01DK107786), NHLBI (R01HL142302), and NHGRI (R56HG010297).
Doetinchem. The authors would like to thank the field workers of the Municipal Health Services in Doetinchem (C. te Boekhorst, I. Thus, M. Zwiers, and B. Heusinkveld) for their contribution to the data collection for the current study. Principal investigator is Prof. Dr. Ir. W.M.M. Verschuren, project leader Dr. H.S.J. Picavet, data manager A. Blokstra, and logistic manager P. Vissink. The Doetinchem Cohort Study was financially supported by the Ministry of Health, Welfare and Sport of the Netherlands and the National Institute for Public Health and the Environment.
FHS. Supported by from NHLBI contracts HHSN268201500001I and N01-HC-25195, and its contract with Affymetrix, Inc. for genotyping services (contract number N02-HL-6-4278). The analyses reflect intellectual input and resource development from the Framingham Heart Study investigators participating in the SNP Health Association Resource (SHARe) project. Also supported by NIDDK (R01 DK078616, U01 DK078616, and UM1 DK078616), NHLBI (R01 HL151855), and by the National Institute of General Medical Sciences (NIGMS) Interdisciplinary Training Grant for Biostatisticians (T32 GM74905). C.S., C.T.L., D.D., J.B.M., and J.D. received funding from NIDDK (UM1 DK078616) and NHLBI (R01 HL151855). J.L. received funding from NIGMS (T32GM074905).
GENOA. Genotyping was performed at the Mayo Clinic by Stephan T. Turner, MD, Mariza de Andrade, PhD, and Julie Cunningham, PhD. The authors thank Eric Boerwinkle, PhD, and Megan L. Grove, from the Human Genetics Center and Institute of Molecular Medicine and Division of Epidemiology, University of Texas Health Science Center, Houston, Texas, for their help with genotyping. The authors would also like to thank the families that participated in the GENOA study. GENOA was supported by the NIH NBLBI grant numbers HL054457, HL054464, HL054481, HL087660, and HL119443.
Health ABC. This research was supported by NIA contracts N01-AG-6-2101, N01-AG-6-2103, N01-AG-6-2106, NIA grant R01-AG028050, and National Institute of Nursing Research grant R01-NR012459. This research was funded in part by the Intramural Research Program of the NIH, NIA.
MGB. The authors thank Partners HealthCare Biobank for providing samples, genomic data, and health information data. M.S.U. was supported by NIDDK K23DK114551. A.K.M. and Y.C. were supported by NIH NIDDK R03DK118305. J.M.M. is supported by American Diabetes Association Innovative and Clinical Translational Award 1-19-ICTS-068, and by NHGRI, grant FAIN no. U01HG011723. J.B.C. is supported by an NIDDK Pathway to Independence Award K99DK127196. A.L. is supported by grant 2020096 from the Doris Duke Charitable Foundation.
MESA. Genotyping was performed at Affymetrix (Santa Clara, CA) and the Broad Institute of Harvard and Massachusetts Institutes of Technology (Boston, MA) using the Affymetrix Genome-Wide Human SNP Array 6.0. MESA and the MESA SHARe project are conducted and supported by the NHLBI in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, and N01-HC-95169, and National Center for Advancing Translational Sciences grants UL1-TR-000040, UL1-TR-001079, and UL1-TR-001420. Funding for SHARe genotyping was provided by NHLBI contract N02-HL-64278. Also supported in part by the National Center for Advancing Translational Sciences, Clinical and Translational Science Institute CTSI grant UL1TR001881, and the NIDDK Diabetes Research Center grant DK063491 to the Southern California Diabetes Endocrinology Research Center.
NEO. The authors of the NEO study thank all individuals who participated in the NEO study, all participating general practitioners for inviting eligible participants, and all research nurses for collection of the data. The authors thank the NEO study group, Pat van Beelen, Petra Noordijk, and Ingeborg de Jonge, for the coordination, laboratory, and data management of the NEO study. The genotyping in the NEO study was supported by the Centre National de Génotypage (Paris, France), headed by Jean-Francois Deleuze. The NEO study is supported by the participating Departments, the Division, and the Board of Directors of the Leiden University Medical Center, and by the Leiden University, Research Profile Area Vascular and Regenerative Medicine. D.M.-K. is supported by Dutch Science Organization (ZonMW-VENI Grant 916.14.023).
PROSPER. Prof. Dr. J.W. Jukema is an Established Clinical Investigator of the Netherlands Heart Foundation (grant 2001 D 032). Support for genotyping was provided by the Seventh Framework Program of the European commission (grant 223004) and by the Netherlands Genomics Initiative (Netherlands Consortium for Healthy Aging grant 050-060-810).
Rotterdam. The Rotterdam Study is supported by Erasmus MC and Erasmus University Rotterdam, Netherlands Organisation for Scientific Research (NWO), Netherlands Organisation for Health Research and Development (ZonMW), Research Institute for Diseases in the Elderly (RIDE), Netherlands Genomics Initiative, Ministry of Education, Culture and Science, Ministry of Health, Welfare and Sports, European Commission (DG XII), and Municipality of Rotterdam.
Duality of Interest. J.B.M. is an academic associate for Quest Diagnostics. The PROSPER study was supported by an investigator-initiated grant obtained from Bristol-Myers Squibb. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. D.D., J.L., J.B.C., C.S., and M.S.U. performed the analysis. D.D., J.I.R., J.B.M., J.D., and M.S.U. conceived the study. D.D., J.L., and M.S.U. wrote the first draft of the manuscript. F.A., L.F.B., A.B., E.P.B., L.C., Y.-D.I.C., Y.C., P.S.d.V., T.F., M.G., V.Gudm., X.G., N.R.H., D.I., M.A.I., M.K., H.L.L., A.L., J.M.M., A.C.M., G.N.N., M.A.N., R.N., M.P., J.A.S., S.T., P.V., J.Y., W.Z., E.B., M.O.G., V.Gudn., J.W.J., S.L.R.K., R.J.F.L., C.-T.L., A.K.M., D.M.-K., J.S.P., H.S.J.P., N.S., E.M.S., W.M.M.V., K.W.D., and J.C.F. provided data, contributed to data interpretation, and aided in manuscript revision. D.D. and M.S.U. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
D.D., J.L., and J.B.C. contributed equally.
This article contains supplementary material online at https://doi.org/10.2337/figshare.17209511.
References
- 1. Saeedi P, Petersohn I, Salpea P, et al.; IDF Diabetes Atlas Committee . Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract 2019;157:107843. [DOI] [PubMed] [Google Scholar]
- 2. American Diabetes Association . Economic costs of diabetes in the U.S. in 2017. Diabetes Care 2018;41:917–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Udler MS, McCarthy MI, Florez JC, Mahajan A. Genetic risk scores for diabetes diagnosis and precision medicine. Endocr Rev 2019;40:1500–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ahlqvist E, Storm P, Käräjämäki A, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol 2018;6:361–369 [DOI] [PubMed] [Google Scholar]
- 5. Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018;50:1219–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mahajan A, Taliun D, Thurner M, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 2018;50:1505–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Udler MS, Kim J, von Grotthuss M, et al.; Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: a soft clustering analysis. PLoS Med 2018;15:e1002654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mahajan A, Wessel J, Willems SM, et al.; ExomeBP Consortium; MAGIC Consortium; GIANT Consortium . Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat Genet 2018;50:559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harris TB, Launer LJ, Eiriksdottir G, et al. Age, gene/environment susceptibility-Reykjavik Study: multidisciplinary applied phenomics. Am J Epidemiol 2007;165:1076–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. The ARIC Investigators . The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am J Epidemiol 1989;129:687–702 [PubMed] [Google Scholar]
- 11. Tayo BO, Teil M, Tong L, et al. Genetic background of patients from a university medical center in Manhattan: implications for personalized medicine. PLoS One 2011;6:e19166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Verschuren WM, Blokstra A, Picavet HS, Smit HA. Cohort profile: the Doetinchem Cohort Study. Int J Epidemiol 2008;37:1236–1241 [DOI] [PubMed] [Google Scholar]
- 13. Splansky GL, Corey D, Yang Q, et al. The third generation cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol 2007;165:1328–1335 [DOI] [PubMed] [Google Scholar]
- 14. Dawber TR, Meadors GF, Moore FE Jr. Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health Nations Health 1951;41:279–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol 1979;110:281–290 [DOI] [PubMed] [Google Scholar]
- 16. ; FBPP Investigators . Multi-center genetic study of hypertension: the Family Blood Pressure Program (FBPP). Hypertension 2002;39:3–9 [DOI] [PubMed] [Google Scholar]
- 17. Daniels PR, Kardia SL, Hanis CL, et al.; Genetic Epidemiology Network of Arteriopathy study . Familial aggregation of hypertension treatment and control in the Genetic Epidemiology Network of Arteriopathy (GENOA) study. Am J Med 2004;116:676–681 [DOI] [PubMed] [Google Scholar]
- 18. Franse LV, Di Bari M, Shorr RI, et al.; Health, Aging, and Body Composition Study Group . Type 2 diabetes in older well-functioning people: who is undiagnosed? Data from the Health, Aging, and Body Composition study. Diabetes Care 2001;24:2065–2070 [DOI] [PubMed] [Google Scholar]
- 19. Boutin NT, Mathieu K, Hoffnagle AG, et al. Implementation of electronic consent at a biobank: an opportunity for precision medicine research. J Pers Med 2016;6:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bild DE, Bluemke DA, Burke GL, et al. Multi-Ethnic Study of Atherosclerosis: objectives and design. Am J Epidemiol 2002;156:871–881 [DOI] [PubMed] [Google Scholar]
- 21. de Mutsert R, den Heijer M, Rabelink TJ, et al. The Netherlands Epidemiology of Obesity (NEO) study: study design and data collection. Eur J Epidemiol 2013;28:513–523 [DOI] [PubMed] [Google Scholar]
- 22. Shepherd J, Blauw GJ, Murphy MB, et al. The design of a prospective study of Pravastatin in the Elderly at Risk (PROSPER). PROSPER Study Group. PROspective Study of Pravastatin in the Elderly at Risk. Am J Cardiol 1999;84:1192–1197 [DOI] [PubMed] [Google Scholar]
- 23. Shepherd J, Blauw GJ, Murphy MB, et al.; PROSPER study group. PROspective Study of Pravastatin in the Elderly at Risk . Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002;360:1623–1630 [DOI] [PubMed] [Google Scholar]
- 24. Trompet S, de Craen AJ, Postmus I, et al.; PROSPER Study Group . Replication of LDL GWAs hits in PROSPER/PHASE as validation for future (pharmaco)genetic analyses. BMC Med Genet 2011;12:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ikram MA, Brusselle GGO, Murad SD, et al. The Rotterdam Study: 2018 update on objectives, design and main results. Eur J Epidemiol 2017;32:807–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med 2002;21:1539–1558 [DOI] [PubMed] [Google Scholar]
- 27. Becker BJ, Wu MJ. The synthesis of regression slopes in meta-analysis. Stat Sci 2007;22:414–429 [Google Scholar]
- 28. Cole SR, Platt RW, Schisterman EF, et al. Illustrating bias due to conditioning on a collider. Int J Epidemiol 2010;39:417–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brouwers MCGJ, Jacobs C, Bast A, Stehouwer CDA, Schaper NC. Modulation of glucokinase regulatory protein: a double-edged sword? Trends Mol Med 2015;21:583–594 [DOI] [PubMed] [Google Scholar]
- 30. Randers E, Erlandsen EJ. Serum cystatin C as an endogenous marker of the renal function--a review. Clin Chem Lab Med 1999;37:389–395 [DOI] [PubMed] [Google Scholar]
- 31. Coll E, Botey A, Alvarez L, et al. Serum cystatin C as a new marker for noninvasive estimation of glomerular filtration rate and as a marker for early renal impairment. Am J Kidney Dis 2000;36:29–34 [DOI] [PubMed] [Google Scholar]
- 32. Nolis T. Exploring the pathophysiology behind the more common genetic and acquired lipodystrophies. J Hum Genet 2014;59:16–23 [DOI] [PubMed] [Google Scholar]
- 33. Yaghootkar H, Lotta LA, Tyrrell J, et al. Genetic evidence for a link between favorable adiposity and lower risk of type 2 diabetes, hypertension, and heart disease. Diabetes 2016;65:2448–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yaghootkar H, Scott RA, White CC, et al. Genetic evidence for a normal-weight “metabolically obese” phenotype linking insulin resistance, hypertension, coronary artery disease, and type 2 diabetes. Diabetes 2014;63:4369–4377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Greenfield JR, Miller JW, Keogh JM, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med 2009;360:44–52 [DOI] [PubMed] [Google Scholar]
- 36. Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med 2003;348:1085–1095 [DOI] [PubMed] [Google Scholar]
- 37. Carr RM, Ahima RS. Pathophysiology of lipid droplet proteins in liver diseases. Exp Cell Res 2016;340:187–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kozlitina J, Smagris E, Stender S, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Speliotes EK, Yerges-Armstrong LM, Wu J, et al.; NASH CRN; GIANT Consortium; MAGIC Investigators; GOLD Consortium . Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 2011;7:e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mansour Aly D, Dwivedi OP, Prasad RB, et al.; Regeneron Genetics Center . Genome-wide association analyses highlight etiological differences underlying newly defined subtypes of diabetes. Nat Genet 2021;53:1534–1542 [DOI] [PubMed] [Google Scholar]