

Abstract
Genetic disorders with predominant central nervous system white matter abnormalities (CNS WMAs), also called leukodystrophies, are heterogeneous entities. We ascertained 117 individuals with CNS WMAs from 104 unrelated families. Targeted genetic testing was carried out in 16 families and 13 of them received a diagnosis. Chromosomal microarray (CMA) was performed for three families and one received a diagnosis. Mendeliome sequencing was used for testing 11 families and all received a diagnosis. Whole exome sequencing (WES) was performed in 80 families and was diagnostic in 52 (65%). Singleton WES was diagnostic for 50/75 (66.67%) families. Overall, genetic diagnoses were obtained in 77 families (74.03%). Twenty-two of 47 distinct disorders observed in this cohort have not been reported in Indian individuals previously. Notably, disorders of nuclear mitochondrial pathology were most frequent (9 disorders in 20 families). Thirty-seven of 75 (49.33%) disease-causing variants are novel. To sum up, the present cohort describes the phenotypic and genotypic spectrum of genetic disorders with CNS WMAs in our population. It demonstrates WES, especially singleton WES, as an efficient tool in the diagnosis of these heterogeneous entities. It also highlights possible founder events and recurrent disease-causing variants in our population and their implications on the testing strategy.
Keywords: next generation sequencing, whole exome sequencing, white matter disease, leukoencephalopathy
Graphical Abstract
Introduction
Disorders with predominant central nervous system white matter abnormalities (CNS WMAs), also called leukodystrophies (LD), are a large group of heterogeneous disorders.1–5 Traditionally, all genetic disorders with CNS WMAs were referred to as leukodystrophies. The Global Leukodystrophy Initiative6 grouped these disorders into leukodystrophies (LDs) and genetic leukoencephalopathies. As per this classification, LDs included disorders with primary glial cell and myelin sheath pathology of genetic etiology. Genetic leukoencephalopathies were defined as genetic disorders with significant CNS WMAs but did not meet the inclusion criteria of leukodystrophy. Another classification centred on pathological changes and pathogenetic mechanisms7 focuses on primary involvement of any white matter component viz. myelin, oligodendrocytes, astrocytes, microglia, axons, and blood vessels. However, in absence of neuropathological and pathomechanism evidence for several disorders, the concept of primary/true white matter involvement remains uncertain. Thus, the experts have now brought all genetic disorders with CNS WMAs under the umbrella term of leukodystrophies, irrespective of structural, cellular, molecular involvement associated with these disorders.4
Owing to extreme heterogeneity, the clinical findings of these disorders is very variable.3 Most disorders have pediatric onset, and few disorders manifest in adulthood. Clinically, they may present with neuroregression after an initial period of normal development or global developmental delay. Seizures, tone abnormalities (hypotonia progressing to hypertonia and spasticity) and cerebellar signs are other commonly noted neurological features. A handful of these disorders may be diagnosed clinically with the aid of extra-neurological clinical features including dental, endocrine, ophthalmologic findings. Radiologically, these disorders can be categorised into hypo/delayed myelination and other white matter abnormalities including de/dysmyelinating changes, cysts and calcifications.
Most of the disorders with CNS WMAs are of monogenic etiology. A small proportion of disorders have underlying chromosomal abnormalities, microdeletions/duplications and pathogenic variants in mitochondrial genome.3 Though some of these disorders can be diagnosed clinically, majority present with heterogeneous and overlapping clinical features. Initially, diagnosis was achieved through detailed clinical evaluation, neuropathological and metabolic investigations.2 Thereafter, white matter abnormalities pattern recognition on magnetic resonance imaging (MRI) brain emerged as a more robust diagnostic aid. Considering the heterogeneous monogenic etiology, currently next generation sequencing (NGS) based genomic testing, especially whole exome sequencing (WES), has become a rapid and efficient tool for diagnosis for disorders with CNS WMAs.5
The wide availability of WES has made molecular diagnosis of single gene disorders much more equitable across most population groups globally especially with the increasing availability of datasets of population specific genomic variants from diverse populations.8 Systematic phenotypic and genomic studies for heterogeneous disorders from underrepresented populations are likely to inform the prevalence, clinical and genomic spectrum, efficiency and yield of tests, thus reducing the global disparities. Previously, cohorts of individuals with disorders with CNS WMAs have been reported from different populations.5, 9–19 We hereby report on the phenotypic and genotypic spectrum of individuals with CNS WMAs from India.
Methods
The cohort includes individuals with CNS WMAs who underwent genetic/genomic testing at the Department of Medical Genetics, Kasturba Medical College and Hospital, Manipal (KMC Manipal) from June 2015 to December 2020. These individuals were evaluated at KMC Manipal and 19 other centres across India (supplementary data). Families were counselled for genetic testing and participation in the research studies. The protocols were approved by the institutional ethics committee at Kasturba Medical College, Manipal, India as per the declaration of Helsinki. An informed consent was obtained from all participants. Genetic testing included targeted genetic testing and broad-spectrum genomic testing. Targeted genetic testing consisted of Sanger sequencing of exonic and flanking intronic regions of disease associated genes, Sanger sequencing of probable founder variants, multiplex ligation-dependent probe amplification and gap-PCR.
Genomic tests employed for testing included WES, Mendeliome sequencing and chromosomal microarray (CMA). CMA was performed using the Affymetrix CytoScan™ 750K array (Thermo Fisher Scientific, Inc.; Waltham, MA, USA). Resulting data was analysed using Chromosome Analysis Suite (ChAS) v.4.2.1. WES and Mendeliome sequencing were performed using multiple sequencing platforms from Illumina Inc. USA to generate 100–150bp paired-end reads (Table S1). Details of downstream WES data processing including capture kits, data processing and annotation using the established in-house pipeline is provided in supplementary information (Supplementary Materials and Methods). Customised bespoke perl scripts were used to generate allele frequencies from in-house WES data. These counts were integrated when the in-house WES cohort consisted of nearly 200 exomes which later increased to 1455 exomes. Allele frequencies, homozygous and heterozygous counts were also taken from gnomAD v.2 data and integrated in the pipeline. Sample VCF files were annotated against these allele frequencies, homozygous and heterozygous counts. Genomic variants were filtered based on in-house and gnomAD allele frequencies and prioritized based on predicted in silico scores and concordance with the phenotype observed in affected individuals.
Region of homozygosity (ROH) analysis was performed in individuals with homozygous disease-causing variants to determine whether these variants were present within an ROH. Also, the WES/Mendeliome data of individuals with recurrent variants was manually examined to identify identical haplotype for a possibility of a founder event. Copy number variant analysis was performed for all individuals with no SNV (Single Nucleotide Variant) of clinical significance upon WES data analysis. Detailed methodology of CNV (Copy Number Variant) calling, ROH mapping and analysis is described in supplementary section.
Results
We ascertained 135 individuals with predominant CNS WMAs from 120 families between June 2015 and December 2020. Of these, families with findings of novel disease-gene associations20–23, extremely rare disorders with CNS WMAs24–29, probable founder variants20, 22, 23, 30 and locus and allelic heterogeneity associated with mitochondrial disorders31 in 18 individuals from 16 unrelated families have been published earlier and are provided in Table S5.
The present cohort consists of 117 individuals from 104 unrelated families with CNS WMAs. Seventy-two individuals from 62 unrelated families were recruited from our centre and forty-five individuals from 42 families were clinically evaluated at other centres across India (Supplementary section). All families are of Indian origin except one family (F82) from Bangladesh. Sixty-eight individuals (58.11%) were males and 49 (41.88%) were females. Consanguinity was noted in 54.80% (57/104) of the families. The age ranged from new-born to 59 years, however a majority (n=111, 94.87%) of individuals were of paediatric age.
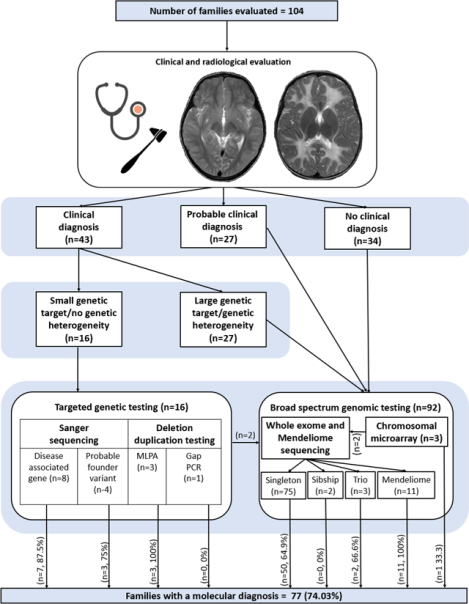
On neuroimaging, 24 families (23.07%) presented with deficient myelination and 80 families (76.92%) had other white matter abnormalities. The specific type of white matter abnormalities are described in the case summaries and Table S2 and S3 in supplementary section. Based on clinical and neuroimaging findings, a clinical diagnosis could be made in 43 families (41.34%). A probable diagnosis was available in 27 families (25.96%) and no clinical diagnosis could be ascertained in 34 families (32.69%).
Targeted genetic testing was performed in a total of 16 families, thirteen of these received a genetic diagnosis. Sanger sequencing of exonic and flanking intronic regions of small sized genes was performed for eight families viz. GALC (F16), GCDH (F20), BTD (F23), ASPA (F52) and ABCD1 (F40, F85, F94), seven of these families received diagnosis. Sanger sequencing of probable founder variants was performed in four families for the variants ISCA1; NM_030940.3; c.259G>A (F28 and F93) and RNASEH2C; NM_032193.3; c.205C>T (F98 and F56), three of these families received diagnosis. Deletion/duplication analysis for PLP1 (Pelizaeus-Merzbacher disease, MIM# 312080) carried out in three families (F37, F95 and F96) by multiplex ligation dependent probe amplification was diagnostic. Gap-PCR for common 30kb deletion in GALC (Krabbe disease, MIM# 245200) carried out in one family (F72) was non-diagnostic. The three families undiagnosed by targeted genetic testing (F20, F56 and F72) underwent singleton WES. CMA and WES were performed in similarly affected siblings (sibship) for one family (F28) and rendered diagnoses of 18q deletion syndrome and partial 4q trisomy. CMA was non-diagnostic for two families (F29 and F82) and was followed up with singleton WES. WES was performed in a total of 80 families with singleton in 75, parent-child trio in three (F25, F60, F69) and sibships in two families (F27, F67). WES was diagnostic in a total of 52/80 (65%) families of which 50/75 (66.67%) were diagnosed by singleton (Table S2 and S3). Mendeliome sequencing was done for eleven families (F64, F75, F84, F86, F97, F99, F100, F101, F102, F103 and F104), all of them received diagnosis. In total, 77 families (74.03%) received genetic diagnosis (Figure 1A, Table S2, S3, S4). Segregation analysis was carried out in all families (supplementary section). No CNVs of clinical significance were detected from WES data analysis. Detailed results are provided is supplementary section.
Figure 1.
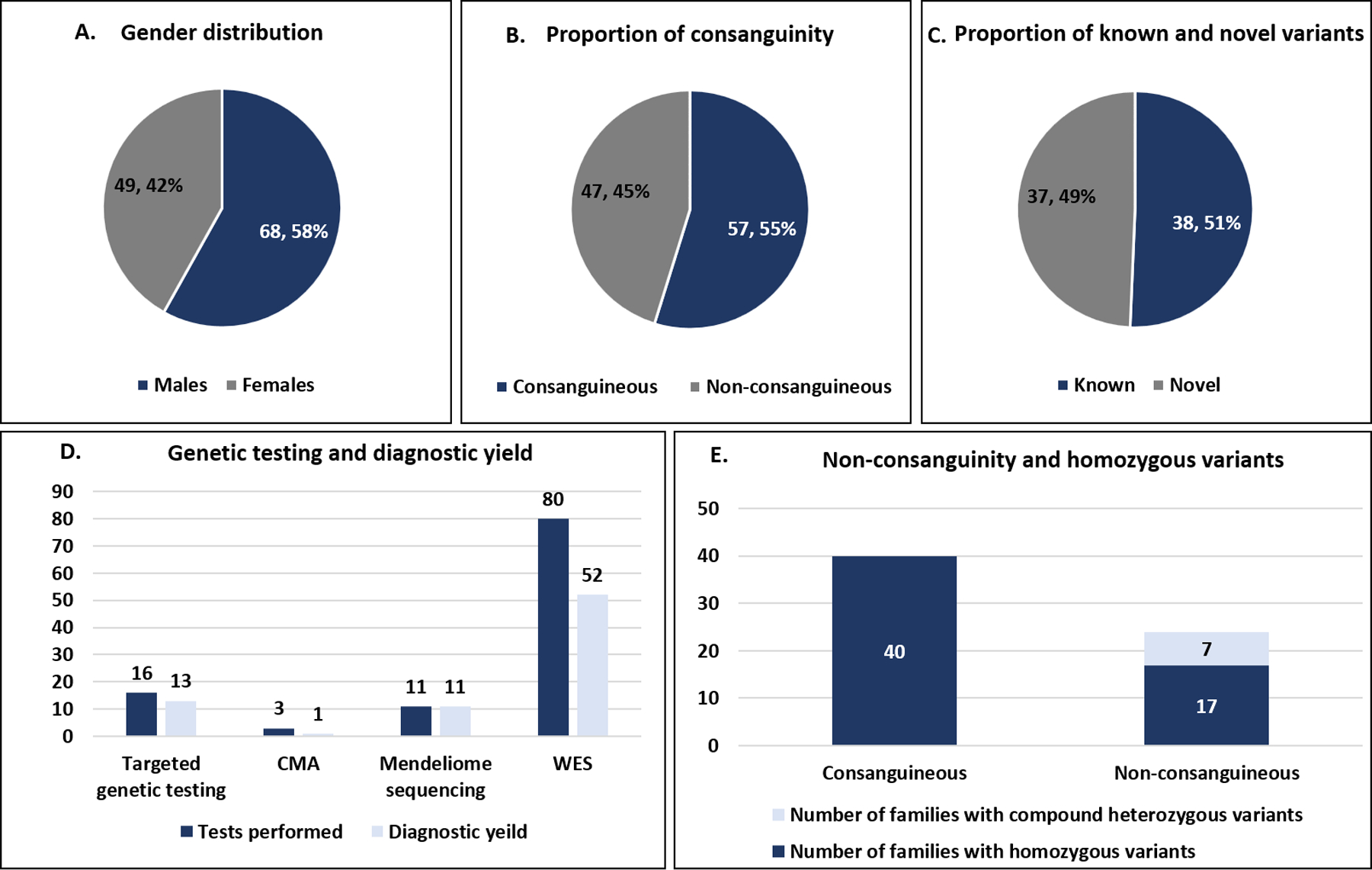
Gender distribution (A) proportion of consanguinity (B) proportion of novel and known variants (C) genetic testing performed and diagnostic yield (D) homozygous variants in consanguineous and non-consanguineous families observed in the present cohort. CMA- chromosomal microarray, WES- Whole exome sequencing.
A total of 47 disorders with CNS WMAs were observed in 77 families (Table 1). Of these, 76 families had monogenic disorders and one had microdeletion syndrome (18q deletion syndrome, F27). Thirty-seven of these disorders were autosomal recessive, 4 were autosomal dominant and 6 were X-linked disorders. Of the monogenic disorders, the spectrum of disorders consisted of myelin protein defects (n=1), cellular organelle defects including mitochondrial (n=9), lysosomal (n=5) and peroxisomal (n=1) dysfunctions, metabolic defects including amino acid and organic acid (n=5), carbohydrate (n=1) metabolism, membrane transport and homeostasis defects including disorders of intracellular vesicular transport (n=1) and ion and water homeostasis (n=4), defect in DNA replication and transcription regulation (n=2), DNA repair (n=2), mRNA translation (n=2), RNA modification and editing (n=1), cell-cell adhesion defects (n=1) and cell cycle differentiation and apoptosis (n=4). Eight disorders of diverse etiologies are clubbed under the miscellaneous category.
Table 1.
Spectrum of monogenic disorders with CNS WMAs observed in the cohort (n=46)
| Pathogenesis | Disorder (MIM#; Gene) | |
|---|---|---|
| Myelin protein defect (n=1) | Pelizaeus -Merzbacher disease (312080; PLP1)* | |
| Subcellular organelle dysfunction (14) | Mitochondrial dysfunction (n=9) | Perrault syndrome 3 (614129; CLPP), Leukodystrophy, hypomyelinating, 18, (618404; DEGS1), Multiple mitochondrial Dysfunctional syndrome 5 (617613; ISCA1)*, Mitochondrial complex I deficiency (252010; NDUFV1, NDUFV2)*, Pyruvate-dehydrogenase E1-alpha deficiency, (312170; PDHA1)*, Thiamine metabolism dysfunction syndrome 2 (biotin- or thiamine-responsive encephalopathy type 2) (607483; SLC19A3)*, Mitochondrial DNA depletion syndrome 5 (encephalomyopathic with or without methylmalonic aciduria) (612073; SUCLA2). Leigh syndrome, due to COX IV deficiency (256000; SURF1)*, Mitochondrial DNA depletion syndrome 1 (MNGIE type) (603041; TYMP)* |
| Lysosomal dysfunction (n=5) | Metachromatic leukodystrophy (250100; ARSA)*, Krabbe disease (245200; GALC), GM2-gangliosidosis, several forms; Tay-Sachs disease (272800; HEXA)*, Niemann-Pick disease, type C1 (257220; NPC1), Neuronal ceroid lipofiscinosis type 1 (256730; PPT1)* | |
| Peroxisomal dysfunction (n=1) | X- adrenoleukodystrophy (300100; ABCD1)* | |
| Metabolic disorders (n=6) | Amino acid & organic acid metabolism (n=5) | Maple Syrup Urine disease type 1a (248600; BCKDHA)*, Canavan disease (608034; ASPA)*, 3-methylglutaconic aciduria, type I (250950; AUH), Glutaric aciduria type 1 (231670; GCDH)*, Homocystinuria due to MTHFR deficiency (236250; MTHFR)* |
| Carbohydrate metabolism (n=1) | Galactosemia (230400; GALT)* | |
| Membrane Transport and homeostasis (n=5) | Intracellular vesicular transport (n=1) | Spastic paraplegia 11 (604360; SPG11)* |
| Ion and water homeostasis (n=4) | Menkes disease (309400; ATP7A), FKTN related dystroglycanopathy (253800; FKTN)*, Merosin deficient congenital muscular dystrophy (607855; LAMA2)*, Megalencephalic leukoencephalopathy with subcortical cysts (604004; MLC1)* | |
| DNA replication and transcription regulation (n=2) | Short stature, brachydactyly, intellectual developmental disability, and seizures (617157; PRMT7), Aicardi-Goutières syndrome type 3 (610329, RNASEH2C)* | |
| DNA repair mechanism defect (n=2) | Intellectual disorder with speech delay, autism and dysmorphic facies (618672; CNOT3), Aicardi-Goutières syndrome type 1 (225750; TREX1) | |
| mRNA translation (n=2) | Leukoencephalopathy with vanishing white matter (603896; EIF2B1, EIF2B4, EIF2B5)*, Leukodystrophy, hypomyelinating, 7, with or without oligodontia and/or hypogonadotropic hypogonadism (607694; POLR3A)* | |
| RNA modification and editing (n=1) | Muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies, type A, 11) (615181; B3GALNT2) | |
| Cell-cell adhesion (n=1) | MASA syndrome and CRASH syndrome (303350; L1CAM) | |
| Cell cycle and differentiation and apoptosis (n=4) | Neuronal ceroid lipofuscinosis (600143; CLN8), CARASIL syndrome (600142; HTRA1)*, Coffin Lowry syndrome (303600; RPS6KA3), familial hemophagocytic lymphohistiocytosis 2, (603553, PRF1) | |
| Miscellaneous (n=7) | Biotinidase deficiency (253260; BTD)*, Leukodystrophy, hypomyelinating, 5 (610532; FAM126A), Alexander disease (203450; GFAP)*, Epileptic encephalopathy, early infantile, 27 (616139; GRIN2B)*, Methylmalonic aciduria, vitamin B12-responsive, due to defect in synthesis of adenosylcobalamin, cblB complementation type (251110; MMAB)*, Warburg micro syndrome (614222; RAB18), Epilepsy, hearing loss and mental retardation syndrome (616577; SPATA5) | |
These disorders have been previously reported in Indian population.
We observed 75 causative variants in the present cohort, of which 73 were single nucleotide variants and two were copy number variants. Of the 73 single nucleotide variants observed in 51 genes in families with monogenic disorders, 44 were missense, 10 were indels, 9 were splicing, 8 were stopgain, one stoploss and one synonymous variant. According to the standards and guidelines for the interpretation of sequence variants by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology, 36 of these variants were classified as pathogenic, 28 as likely pathogenic and 9 as variants of uncertain clinical significance. Thirty-seven of 75 (49.33%) among the observed variants in this cohort are novel. Of the families with single nucleotide variants, fifty-seven (89.06%) had homozygous variants, 7 families had compound heterozygous variants, 5 families had hemizygous variants, 4 families had de novo heterozygous variants. Of the 46 individuals with homozygous disease-causing variants identified by WES, 39 (84.78%) individuals had respective variants within an ROH. The observed sizes of the ROH harbouring disease-causing variants varied from 1 Mb to 51.7 Mb with mean size of 16 Mb (Table S6). A probable founder variant was noted in two families with maple syrup urine disease type 1a (F1 and F2) among others published previously.
Discussion
The present cohort, though far from being representative of the complete spectrum of disorders with CNS WMAs in the Indian population, illustrates the well-known clinical and genomic heterogeneity of these disorders. Several disorders with CNS WMAs have been reported from India earlier including a recently reported a cohort of 50 individuals with relatively common and recognisable disorders of CNS WMAs.12 Multiple case series describing the genotypic spectrum of relatively common and clinically recognisable disorders like metachromatic leukodystrophy (67 families),32, 33 megalencephalic leukoencephalopathy with subcortical cysts (39 families)34 and Tay-Sachs disease (28 families)35 have been published. Multiple case reports describe individuals with hypomyelination with hypodontia and hypogonadotropic hypogonadism,36 Krabbe disease,37 Alexander disease,38 vanishing white matter disease,39 Canavan disease,40 leukoencephalopathy with brainstem and spinal cord involvement,41 glutaric aciduria,42 GM1 gangliosidosis,43 Coats plus syndrome,44 ribose 5-phosphate isomerase deficiency,45 TUB4A1 related hypomyelinating leukodystrophy,46 homocystinuria due to MTHFR deficiency47 and adult onset disorders like cerebral autosomal-dominant arteriopathy with subcortical infarcts (CADASIL),48 CARASIL,49 diffuse hereditary leukoencephalopathy with spheroids50 and X-linked adrenoleukodystrophy.51–53 Rare disorders with CNS WMAs like LYRM7 related mitochondrial complex III deficiency with cavitation leukoencephalopathy53 and SLC33A1 related Huppke-Brendel syndrome with hypomyelination and cerebellar hypoplasia54 have also been reported. Also, individuals with atypical presentations of previously well-known disorders have also been reported like Canavan disease with microcephaly.40 We add an additional twenty-two distinct disorders with CNS WMAs through this cohort which have not been reported in Indian individuals previously (Table 1).
Owing to extreme heterogeneity of disorders with CNS WMAs, clinical diagnosis necessitates clues from age at onset, family pedigree, associated systemic findings, neuroimaging pattern recognition for white matter abnormalities along with other CNS malformations and metabolic findings.3 Also, clinical diagnosis of these disorders depends largely on the experience and expertise of the individuals/team.2, 4 Permanent and significant hypomyelination is a good clue for clinical diagnosis.55 Marked deficiency of myelin as seen in Pelizaeus-Merzbacher disease (F37, F95, F96) and Pelizaeus-Merzbacher-like disorders provides a good clinical handle. Characteristic MRI patterns provided clinical diagnosis in several families including those with megalencephalic leukoencephalopathy with subcortical cysts (F6), Leigh disease (F44, F55, F77, F84), Krabbe disease (F16, F31, F63, F72), glutaricaciduria (F17, F20), multiple mitochondrial dysfunctions syndrome 5 (F28, F93), Canavan disease (F52), vanishing white matter disease (F76, F93), Alexander disease (F99) and aided selection of appropriate genetic test. In a few families, a combination of MRI imaging combined with other clinical findings provided good handle for clinical diagnosis. Adult age at onset along with characteristic neuroimaging findings aided in a clinical diagnosis of adrenoleukodystrophy (F40) and CARASIL/CADASIL (F57). A combination of systemic clinical features with neuroimaging findings aided in a clinical diagnosis of hypomyelinating leukodystrophy with or without oligodontia and/or hypogonadotropic hypogonadism (4H) (F19) and hypomyelination with congenital cataract (F18), Menkes disease (F15) and biotinidase deficiency (F23). Biochemical tests along with white matter abnormalities pattern recognition were crucial for diagnosis in several families such as in elevated creatine phosphokinase in merosin deficient congenital muscular dystrophy (F8, F14, F74, F86) and elevated lactate levels in mitochondrial disorders (F44, F55, F77, F84). Often non-specific CNS WMAs may be seen as an associated finding in recognisable syndromes as was observed in Coffin-Lowry syndrome (F92) in this cohort.56 We also observed hypomyelination in an individual with Warburg Micro syndrome (F70), not reported in the literature till date (Figure S1).
Genetic testing has emerged as the first-tier testing for disorders with CNS WMAs. Targeted genetic tests can be employed in individuals with clinical diagnoses and small target region to test either common/recurrent or founder variants or genes of small size. We used these tests for disorders like X-linked adrenoleukodystrophy and biotinidase deficiency caused by defects in small sized genes, Pelizaeus–Merzbacher disease with recurrent pathogenic variations and in ISCA1 and RNASEH2C for probable founder variants. Cohorts of individuals with targeted testing including restriction fragment length polymorphisms, amplification-refractory mutation system based polymerase chain reaction, and Sanger sequencing for common and recognisable phenotypes, especially for recurrent genetic variants have been reported from India previously.32, 33 The diagnostic yield of targeted testing is likely to be variable and dependent on the cohort selection as well as the clinical expertise available with the team. Richards et al reported the diagnostic yield of targeted genetic testing in a cohort of individuals with leukodystrophies to be 34% with average time of diagnosis being one-and-half years.9 Another study reported the yield of targeted testing as high as 64% (16/25).10
Appropriate administration of broad-spectrum next generation sequencing based tests, particularly WES, has not only proved to be rapid but also cost effective for diagnosis of these disorders.9 The in-house collation WES data of 1455 unrelated individuals with monogenic disorders at our centre aided in generation of allele frequencies, homozygote counts, and heterozygote counts for population specific genomic variants in addition to those from gnomAD v.2 dataset. These counts proved highly efficient in filtering and prioritization of causative variants. We employed WES as an early line of testing in most families. Several families underwent WES and not targeted testing despite a clinical diagnosis due to presence of genetic heterogeneity and/or a large target region for sequencing. The diagnostic yield in families with a clinical diagnosis was substantially high (96.29%; 26/27) and lower in families with a probable (70.37%, 19/27) or no diagnosis (47.05%, 16/34). Overall, diagnostic yield of WES and Mendeliome sequencing was noted to be 65% and 100% respectively in the present cohort. These findings underscore the efficacy of broad-spectrum genetic tests and also highlight the variability in diagnostic yield in the presence/absence of a clinical diagnosis. In other cohorts worldwide the diagnostic yield of WES as a first tier test is noted to be as high as 73–90%.10, 18 19 In a cohort undiagnosed by targeted testing, Vanderver et al reported diagnostic yield of WES to be 42% (30/71).5
NGS based gene panel for LDs have reported variable diagnostic yield (44–60%).10–12 There is a rapid surge in number of LDs being identified. In 2015, 237 genes were known to be implicated in pathogenesis of disorders with CNS WMAs2, 6 which increased to 317 by 2019.4 By 2020, 29 additional genes were added.3, 4 Thus, determining an exhaustive and up-to-date list of genes associated with pathogenesis of disorders with CNS WMAs poses a challenge in developing a comprehensive panel. Recently, a cohort employing genome sequencing (GS) has shown to increase rate of diagnosis by 14% in individuals undiagnosed with WES.14 A proof of concept demonstrated early application of GS, in conjunction with radiologic, enzymatic, biochemical analyte and chromosomal testing as a rapid test with diagnostic rate of (76.5%) in individuals with LDs which is comparable to that of WES as a first line test.17
Consanguinity was noted to be 54.80%, 58.50% and 31%, in the present cohort, previously reported Indian, and Saudi Arabian cohorts respectively. The data on consanguinity is not available for cohorts reported from other populations. Mode of inheritance was noted to be largely autosomal recessive in Indian12 and Saudi Arabian cohorts10, 19 viz., 87–92% as compared to 60–72%, in other populations.5 In the current cohort, 64/76 (84.21%) families had autosomal recessive disorders. Similarly, 80% of Indian families in previously reported cohort12 and 92% of Saudi Arabian families19 with autosomal recessive disorders had disease-causing variants in a homozygous state. In contrast, 21% to 32% of the families with autosomal recessive disorders had disease-causing variants in a homozygous state in previously reported cohorts of CNS WMAs from mixed and northern European descent, African American, African, Asian, and Latin American origin.5, 14, 17 Also, 17/57 (29.82%) families in our cohort which had homozygous variants were clinically documented to be non-consanguineous (Figure 1B). Hence, the high rate of homozygous variants in Indian and Saudi Arabian population underscores the possible effects of inbreeding and founder events other than consanguinity in the Indian population.57
Occurrence of at least four founder variants and several recurrent mutations have been observed in causative genes for disorders with CNS WMAs reported from India previously.58, 59 A well-established founder variant c.135dupC in MLC1 (Megalencephalic leukoencephalopathy with subcortical cysts) is noted in Indian Aggarwal community.59 We observed recurrent variants in six genes in twenty families. Of these, probable founder events were noted in nine families and four genes, including cases published previously.20–23, 30 The present cohort adds 37 novel disease-causing variants to the genotypic spectrum of disorders with CNS WMAs.
In view of lack of a uniform classification and sub-classification of these disorders, comparison of cohorts across diverse populations is challenging. Till date, ten cohorts of individuals with CNS WMAs have been reported in both paediatric and adult age group across different populations (mixed and northern European descent, African American, Arab, African, Asian, Latin American, Argentinian, Chinese, Malay, Filipino, Japanese, South Arabian and Indian). The cohorts are significantly heterogeneous with respect to the disorders included5, 10, 12, 14, 17 Two of these cohorts13, 19 only reported the disorders listed as LDs by the Global leukodystrophy Initiative (GLIA).6 The common disorders noted are adrenoleukodystrophy, CADASIL and vanishing white matter disease in Argentinian cohort, Krabbe disease in Chinese cohort and metachromatic leukodystrophy in Saudi Arabian populations respectively. Aicardi-Goutières syndrome was noted to be most common disorders based on variant allele frequencies in local Saudi Arabian Genetic database.19, 60 In the adult cohorts, most common disorders noted were NOTCH3 related cerebral arteriopathy with subcortical infarcts and leukoencephalopathy (MIM# 125310) and CSF1R related diffuse hereditary leukoencephalopathy with spheroids (MIM# 221820). We categorized monogenic disorders observed in this cohort based on known cellular or molecular pathomechanisms. Of these, the disorders leading to cellular organelle dysfunction, particularly mitochondrial disorders were the most common entities observed in the cohort (Table 1).
There are a few limitations of the present study. Individuals with CNS WMAs without genetic testing could not be included in the cohort, thus limiting our understanding of epidemiology of these disorders. Chromosomal microarray could be done only in a few individuals. Further, a parent-child trio WES, sibship WES, genome sequencing including mitochondrial genome sequencing in those undiagnosed by singleton WES, if performed, would have added to the diagnostic yield of the cohort.
In conclusion, the spectrum of disorders with CNS WMAs is ever widening and the need for diagnosis is unmet worldwide. Considering the high proportion of monogenic recessive disorders contributing to the genetic etiology of these disorders, we propose singleton WES/Mendeliome sequencing as an early line of testing. Availability of more comprehensive cohorts especially from a country with a large population as India would inform the incidence, prevalence, recurrent variants, and founder variants, resulting in devising better testing strategies for these disorders. The increasing pool of disease-causing variants would aid in delineating pathogenicity of several novel genomic variants and providing genetic diagnosis in our population as well as globally.
Supplementary Material
Acknowledgements
We thank all the patients and families for participating in this study. This work was supported by Department of Health Research funded the project titled “Clinical and molecular characterization of leukodystrophies in Indian children” (V.25011/379/2015-GIA/HR) and National Institutes of Health funded projects, ‘Genetic Diagnosis of Heritable Neurodevelopmental Disorders in India: Investigating the Use of Whole Exome Sequencing and Genetic Counselling to Address the High Burden of Neurodevelopmental Disorders’(1R21NS094047-01) and ‘Genetic Diagnosis of Neurodevelopmental Disorders in India’ (Grant ID - 1R01HD093570-01A1).
Footnotes
Ethics declaration
Ethics approval was obtained from the institutional ethics committee at Kasturba Medical College; Kasturba Hospital Institutional Ethics Committee, Manipal. All participants were de-identified and informed consents were obtained as approved by the ethics committee.
Conflict of Interest
The authors declare no conflict of interest.
Data Availability
Data will be made available upon reasonable request to the corresponding author.
References
- 1.Kevelam SH, Steenweg ME, Srivastava S, et al. Update on Leukodystrophies: A Historical Perspective and Adapted Definition. Neuropediatrics. 2016;47(6):349–354. [DOI] [PubMed] [Google Scholar]
- 2.Parikh S, Bernard G, Leventer RJ, et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol Genet Metab. 2015;114(4):501–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shukla A, Kaur P, Narayanan DL, do Rosario MC, Kadavigere R, Girisha KM. Genetic disorders with central nervous system white matter abnormalities: An update. Clin Genet. 2021;99(1):119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Knaap MS, Schiffmann R, Mochel F, Wolf NI. Diagnosis, prognosis, and treatment of leukodystrophies. Lancet Neurol. 2019;18(10):962–972. [DOI] [PubMed] [Google Scholar]
- 5.Vanderver A, Simons C, Helman G, et al. Whole exome sequencing in patients with white matter abnormalities. Ann Neurol. 2016;79(6):1031–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanderver A, Prust M, Tonduti D, et al. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol Genet Metab. 2015;114(4):494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Knaap MS, Bugiani M. Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol. 2017;134(3):351–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sirugo G, Williams SM, Tishkoff SA. The Missing Diversity in Human Genetic Studies. Cell. 21 2019;177(1):26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richards J, Korgenski EK, Taft RJ, Vanderver A, Bonkowsky JL. Targeted leukodystrophy diagnosis based on charges and yields for testing. Am J Med Genet A. 2015;167a(11):2541–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen L, Manín A, Medina N, et al. Argentinian clinical genomics in a leukodystrophies and genetic leukoencephalopathies cohort: Diagnostic yield in our first 9 years. Ann Hum Genet. 2020;84(1):11–28. [DOI] [PubMed] [Google Scholar]
- 11.Chen Z, Tan YJ, Lian MM, et al. High Diagnostic Utility Incorporating a Targeted Neurodegeneration Gene Panel With MRI Brain Diagnostic Algorithms in Patients With Young-Onset Cognitive Impairment With Leukodystrophy. Front Neurol. 2021;12:631407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parayil Sankaran B, Nagappa M, Chiplunkar S, et al. Leukodystrophies and Genetic Leukoencephalopathies in Children Specified by Exome Sequencing in an Expanded Gene Panel. J Child Neurol. 2020;35(7):433–441. [DOI] [PubMed] [Google Scholar]
- 13.Xie JJ, Ni W, Wei Q, et al. New clinical characteristics and novel pathogenic variants of patients with hereditary leukodystrophies. CNS Neurosci Ther. 2020;26(5):567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helman G, Lajoie BR, Crawford J, et al. Genome sequencing in persistently unsolved white matter disorders. Ann Clin Transl Neurol. 2020;7(1):144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lynch DS, Rodrigues Brandao de Paiva A, Zhang WJ, et al. Clinical and genetic characterization of leukoencephalopathies in adults. Brain. 2017;140(5):1204–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kunii M, Doi H, Ishii Y, et al. Genetic analysis of adult leukoencephalopathy patients using a custom-designed gene panel. Clin Genet. 2018;94(2):232–238. [DOI] [PubMed] [Google Scholar]
- 17.Vanderver A, Bernard G, Helman G, et al. Randomized Clinical Trial of First-Line Genome Sequencing in Pediatric White Matter Disorders. Ann Neurol. 2020;88(2):264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahdieh N, Soveizi M, Tavasoli AR, et al. Genetic testing of leukodystrophies unraveling extensive heterogeneity in a large cohort and report of five common diseases and 38 novel variants. Sci Rep. 2021;11(1):3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alfadhel M, Almuqbil M, Al Mutairi F, et al. The Leukodystrophy Spectrum in Saudi Arabia: Epidemiological, Clinical, Radiological, and Genetic Data. Front Pediatr. 2021;9:633385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shukla A, Kaur P, Girisha KM. Report of the Third Family with Multiple Mitochondrial Dysfunctions Syndrome 5 Caused by the Founder Variant p.(Glu87Lys) in ISCA1. J Pediatr Genet. 2018;7(3):130–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shukla A, Narayanan DL, Kaur P, Girisha KM. ISCA1-Related Multiple Mitochondrial Dysfunctions Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews(®). University of Washington, Seattle: [PubMed] [Google Scholar]
- 22.Shukla A, Hebbar M, Srivastava A, et al. Homozygous p.(Glu87Lys) variant in ISCA1 is associated with a multiple mitochondrial dysfunctions syndrome. J Hum Genet. 2017;62(7):723–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shukla A, Das Bhowmik A, Hebbar M, et al. Homozygosity for a nonsense variant in AIMP2 is associated with a progressive neurodevelopmental disorder with microcephaly, seizures, and spastic quadriparesis. J Hum Genet. 2018;63(1):19–25. [DOI] [PubMed] [Google Scholar]
- 24.Galada C, Hebbar M, Lewis L, et al. Report of four novel variants in ASNS causing asparagine synthetase deficiency and review of literature. Congenit Anom (Kyoto). 2018;58(5):181–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samaddar S, Kaur P, Rajagopal KV, Girisha KM, Shukla A, Sharma S. Spastic Paraplegia Type 56 in a Young Child. Indian J Pediatr. 2020;87(8):650–651. [DOI] [PubMed] [Google Scholar]
- 26.Kaur P, Sharma S, Kadavigere R, Girisha KM, Shukla A. Novel variant p.(Ala102Thr) in SDHB causes mitochondrial complex II deficiency: Case report and review of the literature. Ann Hum Genet. 2020;84(4):345–351. [DOI] [PubMed] [Google Scholar]
- 27.Kaur P, Neethukrishna K, Kumble A, Girisha KM, Shukla A. Identification of a novel homozygous variant confirms ITPA as a developmental and epileptic encephalopathy gene. Am J Med Genet A. 2019;179(5):857–861. [DOI] [PubMed] [Google Scholar]
- 28.Kaur P, Bhavani GS, Raj A, Girisha KM, Shukla A. Homozygous variant, p.(Arg643Trp) in VAC14 causes striatonigral degeneration: report of a novel variant and review of VAC14-related disorders. J Hum Genet. 2019;64(12):1237–1242. [DOI] [PubMed] [Google Scholar]
- 29.Kaur P, Wamelink MMC, van der Knaap MS, Girisha KM, Shukla A. Confirmation of a Rare Genetic Leukoencephalopathy due to a Novel Bi-allelic Variant in RPIA. Eur J Med Genet. 2019;62(8):103708. [DOI] [PubMed] [Google Scholar]
- 30.Hebbar M, Kanthi A, Shrikiran A, et al. p.Arg69Trp in RNASEH2C is a founder variant in three Indian families with Aicardi-Goutières syndrome. Am J Med Genet A. 2018;176(1):156–160. [DOI] [PubMed] [Google Scholar]
- 31.Srivastava A, Srivastava KR, Hebbar M, et al. Genetic diversity of NDUFV1-dependent mitochondrial complex I deficiency. Eur J Hum Genet. 2018;26(11):1582–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Narayanan DL, Matta D, Gupta N, et al. Spectrum of ARSA variations in Asian Indian patients with Arylsulfatase A deficient metachromatic leukodystrophy. J Hum Genet. 2019;64(4):323–331. [DOI] [PubMed] [Google Scholar]
- 33.Shukla P, Vasisht S, Srivastava R, et al. Molecular and structural analysis of metachromatic leukodystrophy patients in Indian population. J Neurol Sci. 2011;301(1–2):38–45. [DOI] [PubMed] [Google Scholar]
- 34.Shukla P, Gupta N, Ghosh M, et al. Molecular genetic studies in Indian patients with megalencephalic leukoencephalopathy. Pediatr Neurol. 2011;44(6):450–8. [DOI] [PubMed] [Google Scholar]
- 35.Sheth J, Mistri M, Sheth F, et al. Burden of lysosomal storage disorders in India: experience of 387 affected children from a single diagnostic facility. JIMD Rep. 2014;12:51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tewari VV, Mehta R, Sreedhar CM, et al. A novel homozygous mutation in POLR3A gene causing 4H syndrome: a case report. BMC Pediatr. 2018;18(1):126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michael SN, Madaan P, Jauhari P, Chakrabarty B, Kumar A, Gulati S. Selective Pyramidal Tract Involvement in Late-Onset Krabbe Disease. Indian J Pediatr. 2019;86(10):970–971. [DOI] [PubMed] [Google Scholar]
- 38.Ramesh K, Sharma S, Kumar A, Salomons GS, van der Knaap MS, Gulati S. Infantile-onset Alexander disease: a genetically proven case with mild clinical course in a 6-year-old Indian boy. J Child Neurol. 2013;28(3):396–8. [DOI] [PubMed] [Google Scholar]
- 39.Sharma S, Ajij M, Singh V, Aneja S. Vanishing white matter disease with mutations in EIF2B5 gene. Indian J Pediatr. 2015;82(1):93–5. [DOI] [PubMed] [Google Scholar]
- 40.Gowda VK, Bhat MD, Srinivasan VM, Prasad C, Benakappa A, Faruq M. A case of Canavan disease with microcephaly. Brain Dev. 2016;38(8):759–62. [DOI] [PubMed] [Google Scholar]
- 41.Sharma S, Sankhyan N, Kumar A, Scheper GC, van der Knaap MS, Gulati S. Leukoencephalopathy with brain stem and spinal cord involvement and high lactate: a genetically proven case without elevated white matter lactate. J Child Neurol. 2011;26(6):773–6. [DOI] [PubMed] [Google Scholar]
- 42.Shah H, Chandarana M, Sheth J, Shah S. A Case Report of Chronic Progressive Pancerebellar Syndrome with Leukoencephalopathy:L-2 Hydroxyglutaric Aciduria. Mov Disord Clin Pract. 2020;7(5):560–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tuteja M, Bidchol AM, Girisha KM, Phadke S. White matter changes in GM1 gangliosidosis. Indian Pediatr. 2015;52(2):155–6. [DOI] [PubMed] [Google Scholar]
- 44.Netravathi M, Kumari R, Kapoor S, et al. Whole exome sequencing in an Indian family links Coats plus syndrome and dextrocardia with a homozygous novel CTC1 and a rare HES7 variation. BMC Med Genet. 2015;16:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naik N, Shah A, Wamelink MMC, van der Knaap MS, Hingwala D. Rare case of ribose 5 phosphate isomerase deficiency with slowly progressive leukoencephalopathy. Neurology. 2017;89(11):1195–1196. [DOI] [PubMed] [Google Scholar]
- 46.Vinayagamani S, Nair SS, Sundaram S. Teaching NeuroImages: Hypomyelinating leukodystrophy with generalized dystonia. Neurology. 2020;94(3):e335–e336. [DOI] [PubMed] [Google Scholar]
- 47.Senthilvelan S, Kandasamy S, Menon RN, et al. Methylenetetrahydrofolate Reductase Deficiency as a Cause of Treatable Adult-onset Leukoencephalopathy and Myelopathy. Clin Neuroradiol. 2020. [DOI] [PubMed] [Google Scholar]
- 48.Yadav S, Bentley P, Srivastava P, Prasad K, Sharma P. The first Indian-origin family with genetically proven cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). J Stroke Cerebrovasc Dis. 2013;22(1):28–31. [DOI] [PubMed] [Google Scholar]
- 49.Preethish-Kumar V, Nozaki H, Tiwari S, et al. CARASIL families from India with 3 novel null mutations in the HTRA1 gene. Neurology. 2017;89(23):2392–2394. [DOI] [PubMed] [Google Scholar]
- 50.Tamhankar PM, Zhu B, Tamhankar VP, et al. A Novel Hypomorphic CSF1R Gene Mutation in the Biallelic State Leading to Fatal Childhood Neurodegeneration. Neuropediatrics. 2020;51(4):302–306. [DOI] [PubMed] [Google Scholar]
- 51.Kumar N, Taneja KK, Kalra V, Behari M, Aneja S, Bansal SK. Genomic profiling identifies novel mutations and SNPs in ABCD1 gene: a molecular, biochemical and clinical analysis of X-ALD cases in India. PLoS One. 2011;6(9):e25094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muranjan M, Karande S, Sankhe S, Eichler S. Childhood cerebral X-linked adrenoleukodystrophy with atypical neuroimaging abnormalities and a novel mutation. J Postgrad Med. 2018;64(1):59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dallabona C, Abbink TE, Carrozzo R, et al. LYRM7 mutations cause a multifocal cavitating leukoencephalopathy with distinct MRI appearance. Brain. 2016;139(Pt 3):782–94. [DOI] [PubMed] [Google Scholar]
- 54.Chiplunkar S, Bindu PS, Nagappa M, et al. Huppke-Brendel syndrome in a seven months old boy with a novel 2-bp deletion in SLC33A1. Metab Brain Dis. 2016;31(5):1195–8. [DOI] [PubMed] [Google Scholar]
- 55.Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72(8):750–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Martinez JE, Wilson GL, et al. A novel RSK2 (RPS6KA3) gene mutation associated with abnormal brain MRI findings in a family with Coffin-Lowry syndrome. Am J Med Genet A. 2006;140(12):1274–9. [DOI] [PubMed] [Google Scholar]
- 57.Nakatsuka N, Moorjani P, Rai N, et al. The promise of discovering population-specific disease-associated genes in South Asia. Nat Genet. 2017;49(9):1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ankala A, Tamhankar PM, Valencia CA, Rayam KK, Kumar MM, Hegde MR. Clinical applications and implications of common and founder mutations in Indian subpopulations. Hum Mutat. 2015;36(1):1–10. [DOI] [PubMed] [Google Scholar]
- 59.Singhal BS, Gorospe JR, Naidu S. Megalencephalic leukoencephalopathy with subcortical cysts. J Child Neurol. 2003;18(9):646–52. [DOI] [PubMed] [Google Scholar]
- 60.Monies D, Abouelhoda M, AlSayed M, et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum Genet. 2017;136(8):921–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available upon reasonable request to the corresponding author.