

Abstract
LGL leukemia is a rare chronic lymphoproliferative disorder of cytotoxic lymphocytes which can be immunophenotypically either T cell or NK cell-derived. According to the World Health Organization classification, it can be divided into three subtypes: chronic T-cell leukemia and chronic natural killer cell lymphocytosis, and aggressive natural killer cell LGL leukemia. Clonal proliferation of large granular lymphocytes can be because of stimulation of various molecular pathways namely JAK-STAT3 pathway, FAS/FAS-L pathway, RAS-RAF-1-MEK1-ERK pathway, PI3K/AKT pathway, NF-KB pathway, and Sphingolipid Rheostat pathways. The most common clinical features presenting with this leukemia are neutropenia, anemia, thrombocytopenia. This leukemia is also associated with various autoimmune conditions. It usually has an indolent course except for the aggressive NK cell LGL leukemia. The cause of death in the indolent cases was mostly due to infectious complications related to the neutropenia associated with the disease. The rarity of the disease coupled with the availability of only a handful of clinical trials has been a hindrance to the development of a specific treatment. Most of the cases are managed with immunomodulators. The advances in the knowledge of molecular pathways associated with the disease have brought few targeted therapies into the limelight. We discuss here the evolution, epidemiology, demographic profile, pathophysiology, differential diagnosis, the available treatment options along with the survival and prognostic variables which may help us in better understanding and better management of the disease and hopefully, paving the way for a targeted clinical approach.
Keywords: LGL leukemia, NK cell, T cell
Introduction
Large granular lymphocyte (LGL) is a morphologically distinct lymphoid subtype that is larger than most circulating lymphocytes and has characteristic azurophilic granules containing acid hydrolases. On electron microscopy, these lymphocytes have cytoplasmic inclusion bodies arranged in parallel tubular arrays [1,2]. These parallel tubular arrays correspond to the prominent cytoplasmic azurophilic granules visible on light microscopy.
LGL leukemia was first described in 1975-77 as chronic lymphocytic leukemia of T-cell origin associated with neutropenia and splenomegaly [1,2]. It was finally renamed as large granular lymphocytic leukemia in 1989, because of the clonal proliferation of LGL [3]. This leukemia came to be established as a rare heterogeneous clonal lymphoproliferative disorder of mature post-thymic T- or natural killer (NK)-cells.
It was Loughran TP who described two clinical subtypes of LGL leukemia, one associated with an increased number of LGLs but not clinically aggressive and a chronic clinical course, and the other subtype, with clonal LGL proliferation and an acute clinical presentation with massive organomegaly and systemic involvement [4]. Based on immunophenotyping, the clonal LGL leukemias can either be T-LGL which are CD3 and CD8 positive cytotoxic T-cells and NK-LGL which are negative for CD3. The diagnosis of LGL leukemia is made when these cells are persistently elevated in the peripheral blood for more than 6 months duration and no cause has been identified.
However, the majority of the patients with increased proliferation of LGL are clinically asymptomatic and therefore, tend to have a chronic clinical course. X-linked gene analyses have supported a polyclonal LGL lymphocytosis in such cases [4].
The French-American-British classification described LGL leukemia as a distinct entity in the group of chronic T-lymphoid leukemia [5]. Later, the revised European-American classification of lymphoid neoplasms (REAL) kept them as a distinct entity among the peripheral T-cell and NK-cell leukemia [6].
The World Health Organization (WHO) classification of neoplastic diseases of the hematopoietic and lymphoid tissues 2001 recognized these entities and classified LGL leukemia into two subtypes; type 1: T-cell LGL leukemia and type 2: aggressive NK-cell leukemia [7]. The 2008 version of WHO introduced a new provisional subtype of NK-cell leukemia which clinically was associated with a more chronic NK-cell proliferation and this was called chronic lymphoproliferative disorder of NK cells [8]. The latest version of this classification, revised edition 2016 highlighted the importance of mutation of STAT3 and STAT5 in these subtypes and emphasized how the expression of STAT5 is associated with a more aggressive clinical outcome (Table 1) [9]. It also focused on the three subtypes of leukemia which can come with morphological LGL, viz, T-LGL, Aggressive NK-cell leukemia, and Chronic lymphoproliferative disorder of NK cell type [9]. The recently documented findings of this leukemia have helped in better understanding the ethnicity prevalence, especially concerning aggressive NK-cell leukemia. We shall discuss in brief the epidemiology, relevant etiological factors responsible, the current diagnostic protocol used, its pros and cons, and probable suggestions for such cases.
Table 1.
Changes in the nomenclature of LGL Leukemia with time
| FAB 1989 | REAL 1994 | WHO 2001 | WHO 2008 | WHO 2016 |
|---|---|---|---|---|
| Chronic T lymphoid leukemias | T-cell and putative NK-cell neoplasms | Mature T and NK cell neoplasm | Mature T and NK cell neoplasm | Mature T and NK cell neoplasm |
| T-cell CLL or LGL leukemia | T-cell chronic lymphocytic leukemia/prolymphocytic | T-cell LGL leukemia | T-cell LGL leukemia | T-cell LGL leukemia |
| T prolymphocytic leukemia | LGL leukemia | Aggressive NK-cell leukemia | Chronic lymphoproliferative disorder of NK cells | Chronic lymphoproliferative disorder of NK cells |
| T-cell type | ||||
| NK-cell type | ||||
| Adult T-cell leukemia/lymphoma | Aggressive NK-cell leukemia | Aggressive NK-cell leukemia | ||
| Sezary syndrome | Highlighted the importance of STAT mutation on clinicopathological behaviour |
Epidemiology
LGL leukemia is a rare disorder constituting 2-5% of all chronic lymphoproliferative diseases in the US and Europe and 5-6% of all cases in the Asian population [10]. Recent demographic studies of European and North American cohorts place the average incidence of LGL leukemia at 0.2-0.72 per million persons per year [11].
The incidence is approximately the same in both males and females [12]. The median age at presentation is the middle age group (55-60 years) [13] and it is less common in pediatric age groups [12]. The aggressive variant is more common in the Asian continent [14]. Females are diagnosed at a younger age compared with males [15]. In a US-based population study, 14% of patients were under the age of 50 years at the time of diagnosis [15], in contrast to another French registry database that reported 26% patients <50 years of age [16].
The affiliation of LGL leukemia with autoimmune diseases creates a hindrance in the interpretation of the actual incidence of this leukemia as many patients have already received steroids and/or immunosuppressive therapies for the primary autoimmune diseases diagnosed before, concurrently, or after the diagnosis of the LGL leukemia. Nevertheless, certain ethnicities notably Asians of Japanese, Koreans, and Taiwanese descent have established predilection towards the form of Aggressive NK cell-leukemia [9]. No definite sex predilection is found within these ethnic populations. However, the age group affected is younger, earlier by a decade or two from the other spectrum of LGL leukemia.
Etiopathogenesis
The exact cause of the clonal proliferation of LGLs is not known. However, it is suggested that the initial step in clonal LGL proliferation is an antigen-driven mechanism. This antigen activation first leads to polyclonal proliferation of LGLs, then through mutation in various pathways, there is an oligoclonal proliferation of these LGLs. This switch from polyclonal population to oligoclonal population is well documented in various studies [17-19]. The pathology behind the development of LGL leukemia is dysregulation in the pathways involved in activation-induced cell death (AICD). This abnormal clonal expansion of antigen-primed mature LGLs that successfully escaped AICD, keeps the leukemic LGLs alive thereby functioning as killer cells and memory effector T cells. Certain antigens and cytokines are responsible for clonal expansion of LGLs by utilization of various pathways described in the following sections, and also in Table 2. Zhang et al. also formulated the Boolean dynamic model of the signaling pathway network, which was able to identify the potential causes and key regulators of long-term survival of clonal LGLs [20].
Table 2.
Molecular pathways associated with LGL leukemia
| Pathways | Mechanism of action | Reference |
|---|---|---|
| JAK-STAT3 Signalling Pathway | Somatic activating mutations in the activation domain | [27,28] |
| FAS/FAS-L-mediated Pathway | Cells are resistant to FAS induced AICD | [31] |
| RAS-RAF-1-MEK1-ERK Pathway | Mutation of RAS causes dysregulation of oncogenes | [34] |
| PI3K/AKT Pathway | through phosphorylation of AKT involved in cell survival | [37] |
| NF-KB Pathway | through enhancement of anti-apoptotic function of Bcl2 | [38] |
| Sphingolipid Rheostat Pathway | Anti-apoptotic S1P levels are increased and its pro-apoptotic metabolite ceramide is decreased | [40] |
The various ways of stimulation of LGL are as follows.
Viral antigen-induced LGLs activation
Starkebaum G et al. established the relationship between LGL leukemia and the HTLV1 virus. The proliferation of clonal mature LGLs is the result of chronic antigen stimulation of LGL by the HTLV1 virus. Seropositivity is seen toward its envelope protein BA21 and is seen in 30% to 50% of all patients [21,22]. Studies establishing the relationship between LGL leukemia and seropositivity for Hepatitis C virus, Epstein-Barr virus (EBV), and Cytomegalovirus (CMV) are also reported [23,24].
The Aggressive NK cell-Leukemia has a strong association with EBV infection and their predilection in Asian ethnicities can further be established by the incidence of chronic active EBV infection (CAEBV) in them. This form of leukemia is known to evolve from CAEBV. The WHO revised 2016 edition has categorized CAEBV infection of T and NK-cell type under the headings of systemic CAEBV and indolent, localized forms of hydroa-vacciniforme lymphoproliferative disorder and severe mosquito bite allergy [9]. CAEBV has racial predilection of being found more in the Asian population of Japanese, Korean and Taiwanese descent. It is also seen in Latin Americans but rarely in western and African nations. Children and adolescents are more commonly affected than adults. The genetic polymorphism in genes related to the EBV immune response in these ethnicities has been suggested to play a pivotal role in their susceptibility despite being immunocompetent [9].
Cytokines induced LGLs activation
IL-15 and PDGF have an important role in the activation and uncontrolled proliferation of mature LGLs. IL-15 signals through its receptor IL-15 receptor alpha to activate downstream signals including JAK-STAT and MAPK pathways [25]. These signals lead to activation of the Bcl2 family of anti-apoptotic proteins and suppression of pro-apoptotic factors like Bid and Bim. This further leads to inhibition of apoptosis and prolonged survival.
PDGF signals through a receptor tyrosine kinase (PDGFR) to activate survival pathways including PI3K-AKT and MAPK pathways [26]. This pathway acts through an autocrine loop, thereby leading to longer survival of these cells.
Molecular pathways involved in the pathogenesis of LGL leukemia
JAK-STAT3 signalling pathway
This signaling pathway transmits the extracellular signals into the cell. JAK receptor has a tyrosine kinase activity, which on binding to its ligand (IL-15) undergoes autophosphorylation which further initializes STAT protein binding and phosphorylation by JAK and thereby undergoes dimerization. STAT dimers then enter the nucleus and regulate gene expression, especially genes related to cell survival. Mutation of this pathway has been demonstrated as a fundamental feature in the pathogenesis of LGL leukemia, promoting LGL survival [27]. Activated domain mutations of the STAT3 gene are seen in 30-40% of LGL leukemia patients [27,28].
Teramo A et al., demonstrated that proinflammatory cytokine IL-6 induces, while suppressor of cytokine signaling-3 (SOCS3) inhibits the Jak-Stat3 pathway in LGL leukemia patients [29]. Also, the levels of IL-6 are increased, whereas SOCS3 messenger RNA and protein levels were significantly decreased in LGL leukemia. Mutant STAT3 may be predictive of poor prognosis and contribute to the development of associated disorders like aplastic anemia and myelodysplastic syndrome [28,30].
FAS/FAS-L mediated pathway
Fas and Fas-L signaling pathways normally induce apoptosis and play an important role in the regulation of the immune system. It acts through the activation of caspase-dependent apoptosis. This is the major mechanism through which LGLs induce cell death of foreign cells. Normal LGLs once activated due to an infection, are resistant to apoptosis, and as the infection subsides these activated LGLs are eliminated through FAS-FAS-L pathway activation-induced cell death (AICD). Unlike normal LGLs, leukemic LGLs are resistant to AICD. According to Liu JK et al., a cleaved product of FAS known as soluble FAS can block AICD by interfering with the normal binding of Fas-L to its receptor [31]. Fas-L is responsible for hematopoietic suppression through apoptosis of neutrophil precursors in the bone marrow. This is responsible for the development of neutropenia, the characteristic clinical presentation in patients with LGL leukemia [32].
RAS-RAF-1-MEK1-ERK pathway
The RAS pathway is involved in the survival, proliferation, senescence, and differentiation of normal cells. Activation of RAS causes activation of RAF by phosphorylation, followed by activation of the MEK and ERK sequentially. Mutation of RAS causes abnormal cellular signaling, deregulation of gene expression, and oncogenesis [33]. Overactivation of the Ras pathway plays an important role in the survival signaling of LGLs. Co-activation of Ras and ERK was found in LGL leukemia [34]. Blockade of either ERK or Ras activity may restore Fas sensitivity in leukemic LGLs [35]. This can be used as a probable therapeutic target.
PI3K/AKT pathway
PI3K is a downstream member of the growth factor receptor with tyrosine kinase activity, which acts through phosphorylation of AKT and hence its activation. Activated AKT takes part in cell survival, proliferation, and growth [36]. Overexpression of the PI3K-AKT signaling pathway has been found in T-LGL cells and is associated with apoptosis inhibition [37].
NF-KB pathway
NF-KB is a transcription factor that is engaged in the survival of immune cells. It remains silent by binding to its inhibitors known as inhibitors of NF-KB in resting conditions. Upon activation, NF-KB gets translocated to the nucleus where it regulates the expression of various genes. NF-KB is activated in LGL leukemia and it acts through enhancing the anti-apoptotic function of Bcl-2 proteins. According to studies, it also acts through the PI3K-AKT pathway to prevent apoptosis through Mcl-1, independently of STAT3 [38].
Sphingolipid rheostat pathway
Sphingolipids, Sphingosine-1-Phosphate (S1P) and ceramide are interconvertible metabolites that exist in a balanced state in the blood. Increased S1P level is responsible for increased survival of the cells and when its metabolite ceramide levels are increased, it puts in the signal for apoptosis [39]. In patients with LGL leukemia, there is an increased level of S1P [39]. Inhibition of acid ceramidase (converter of ceramide to sphingosine), into a rat model of LGL leukemia, leads to apoptosis of leukemic LGLs [40].
The molecular pathways associated with LGL leukemia are summarised in Table 2. The antigens, cytokines, and molecular pathways involved in the pathogenesis of LGL leukemia are illustrated in Figure 1.
Figure 1.
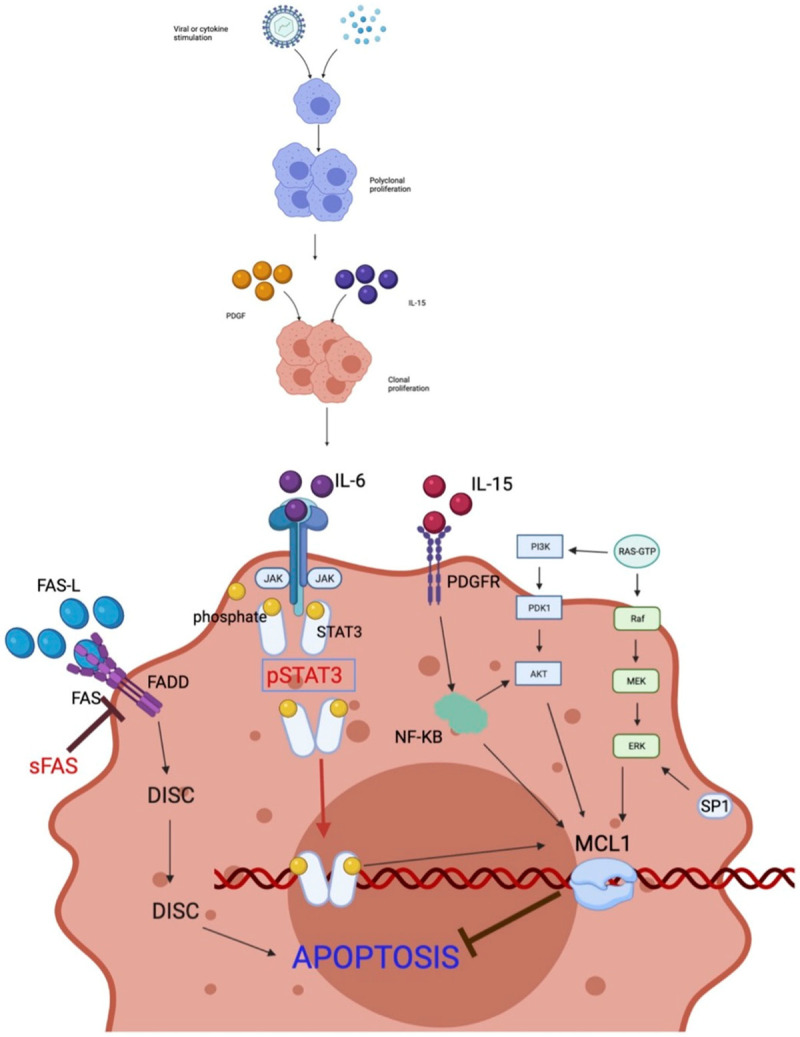
Antigen, Cytokines and Molecular pathways involved in the pathogenesis of LGLs. Mainly HTLV1 virus and cytokines like IL15 and PDGF acts as an antigen stimulus for polyclonal proliferation of LGLs this is followed by acquisition of mutations at various molecular pathways leading to clonal proliferation and increased survival of these cells. The major pathways involved are described here. JAK-STAT3 Signalling Pathway: STAT3 is continuously activated in LGL leukemia, it inhibits apoptosis by increasing the transcription of Mcl-1 which is an anti-apoptotic protein; FAS/FAS-L-mediated Pathway: LGL leukemia cells are resistant to FAS mediated apoptosis, soluble cleaved product of FAS (sFAS) is interferes with the anti-apoptotic action of FAS; RAS-RAF-1-MEK1-ERK Pathway: This pathway acts by modulating the transcription of Mcl-1, this pathway is upregulated in LGL leukemia and is associated with inhibition of apoptosis; PI3K/AKT Pathway: Increased activity of this is seen in LGL leukemia, it also controls apoptosis through Mcl-1 and is responsible for inhibition of apoptosis; NF-KB Pathway: activity is increased in LGL leukemia and it acts through downstream of the PI3K-AKT pathway to prevent apoptosis; Sphingolipid Rheostat Pathway: SP1 level is increased in LGL leukemia and is responsible for increased survival of the cells and when its metabolite ceramide levels are increased, it puts in the signal for apoptosis. It acts through downstream of ERK pathways and prevents apoptosis.
Clinical features
It is commonly seen around the 7th decade of life (median age 60 years) except the aggressive NK-cell leukemia which is found more in the younger age group (median age 40 years) of Asian descents. One-third of patients are usually asymptomatic at the time of presentation. Fatigue is the most common clinical finding and is seen in more than half of the patients.
Pure red cell aplasia (PRCA) (seen in 68% of cases [41]) accompanies LGL leukemia more commonly in Asians while in western countries, neutropenia is more common [42]. The clinical features viz, anemia due to PRCA and recurrent infections due to neutropenia are the most common clinical presentations [12]. Infections mostly involve skin, oropharynx, sinuses, and perirectal areas, but may also become more severe and result in pneumonia or sepsis.
Thrombocytopenia is less common than both neutropenia and anemia in both Asian and western countries [41,42]. Neutropenia, anemia, or pancytopenia may present at different points of time throughout the disease or may occur simultaneously [43]. However, B symptoms are rarely seen [16].
In aggressive NK-cell leukemia, the presence of hepatomegaly, lymphadenopathy, uncommon cutaneous lesions, and common effusions may be found. The disease may further be complicated by coagulopathy, hemophagocytic syndrome or, multiorgan failure [9].
LGL leukemia is frequently associated with autoimmune disorders. Rheumatoid arthritis is the most common association, but other associations include ulcerative colitis, Sjogren’s syndrome, systemic lupus erythematosus, etc. [13]. Autoimmune disorders associated with LGL leukemia are enumerated in Table 3.
Table 3.
Autoimmune disorder associated with LGL leukemia
| Autoimmune disorder associated | Percentages of cases (%) | Reference |
|---|---|---|
| Rheumatoid arthritis (RA) and Felty’s syndrome | 25-33% | [44] |
| Pulmonary artery hypertension (PAH) | rare | [45] |
| Sjogren syndrome | 50% | [46] |
| Autoimmune thyroiditis (Hashimoto’s disease) | 13% | [46] |
| Systemic lupus erythematosus (SLE) | 6% | [46] |
| Vasculitis | 7% | [47] |
| Autoimmune cytopenia | [48] | |
| Anemia | 31% | |
| Neutropenia | 31% | |
| Thrombocytopenia | 12% |
Splenomegaly is reported with a frequency varying from 20% to 50%, however, occurrences of hepatomegaly and lymphadenopathy are relatively low [49,50]. It is associated with some hematological malignancies like chronic lymphocytic leukemia, hairy cell leukemia, follicular lymphoma, mantle cell lymphoma, Hodgkin lymphoma, monoclonal gammopathy of uncertain significance, myeloma, B cell acute lymphoblastic leukemia, chronic myeloid leukemia, myelodysplastic syndrome, and also non-malignant conditions like aplastic anemia and paroxysmal nocturnal hemoglobinuria [51-55]. This has been depicted in Table 4.
Table 4.
Haematological conditions associated with LGL leukemia
| Haematological malignancy | Percentages of cases (%) | Reference |
|---|---|---|
| Monoclonal gammopathy of unknown significance (MGUS) | 19% | [51,52] |
| Chronic Lymphocytic Leukaemia | 8% | [51,52] |
| Hairy Cell Leukaemia | 2% | [53] |
| Follicular Lymphoma | 2% | [51,52] |
| Mantle cell lymphoma | 2% | [51,52] |
| Myelodysplastic Syndrome | 7% | [54] |
| Aplastic Anemia | 4% | [55] |
| Paroxysmal Nocturnal Haemoglobinuria | 2% | [51] |
Hematological features
LGL leukemia was previously diagnosed when the LGL count exceeded 0.5 × 109/L for a period of more than 6 months [4]. Diagnosis can also be considered when circulating large granular lymphocytes count is less than 0.5 × 109/L, if these cells are clonal and the patient exhibits typical morphology, immunophenotype, clinical or hematologic presentations (concomitant autoimmune diseases such as rheumatoid arthritis, splenomegaly), or cytopenias associated with a clonal LGL marrow infiltration [12,16]. The characteristic finding is the presence of lymphocytosis which is seen in ~50% of the cases [16]. Most patients are neutropenic, which is responsible for its characteristic clinical presentation with frequent recurrent infections. There is no direct correlation between the extent of LGL marrow infiltration and the degree of neutropenia. It was explained by Liu JH et al. that the reason behind this neutropenia was Fas/Fas-ligand-dependent mechanism [32]. Anemia is very common [53]. This anemia can be both Coombs test positive or negative. Coombs-positive anemia is because of autoimmune hemolytic anemia seen in ~30% of cases [41]. Coombs negative anemia is mostly because of PRCA, which is seen in 19% of cases [41]. Anemia in PRCA is associated with low reticulocyte count and about 20% of these cases are transfusion-dependent [12]. Thrombocytopenia is usually moderate, observed in 15%-35% of patients, and does not require any platelet transfusions [53]. Table 5 shows the hematological parameters frequently associated with LGL leukemia.
Table 5.
Haematological parameters associated with LGL leukemia
Serological features
There are various serological abnormalities associated with LGL leukemia; the most common being an increase in Fas-L which is seen in approximately 90% of cases and is responsible for abnormal proliferation and clinical features associated with LGL leukemia [12]. These patients are often found to have neutropenia with the resolution of the neutropenia accompanying normalization of serum Fas-L levels [32]. The second most common abnormality associated with this disorder is raised Rheumatoid factor, which is seen in 40-60% of the cases according to various studies [4,12,54]. Anti-neutrophil antibodies were seen in 40% of the cases but their significance in the pathogenesis of LGL leukemia is still inconclusive [12,54]. Beta2-microglobulin is raised in about 70% of cases [56,57]. Table 6 enlists the serological and biochemical parameters associated with LGL leukemia.
Table 6.
Serological and biochemical parameters associated with LGL leukemia
Diagnostic criteria used for treatment of LGL
Clinical suspicion should be made in settings of unexplained neutropenia, recurrent infections, anemia, lymphocytosis, or autoimmune conditions. In these cases, morphological examination of peripheral blood should be done to determine the presence of large granular lymphocytes on the peripheral blood smear. Along with peripheral blood examination, flow-cytometric analysis using CD3, CD5, CD4, CD8, CD16, CD56, CD57, TCRγδ rearrangements, STAT3 protein, STAT5, Vb repertoire, and killer cell immunoglobulin-like receptor (KIR) phenotyping should be done. Molecular genetics for STAT3/STAT5 rearrangement can help in making the diagnosis. Cytotoxic markers like TIA1, granzyme B, and M are positive. KIR expression has an important role in ruling out CLPD of NK cell type. KIR expression is abnormal, with restricted expression or lack of detection in them.
Based on these findings, LGLs can be polyclonal which indicates reactive LGL proliferation secondary to viral infection (such as CMV, HIV, EBV). Oligoclonal or clonal LGL proliferation may occur as a post-transplant lymphoproliferative disorder as well as in the company of other hematological malignancies.
Whenever clonal expansion of LGLs occurs, diagnosis of T or NK LGL leukemia with clonal TCRyδ should be considered. However, additional tests for STAT3-5b/NFKB mutation, Vb repertoire, and KIR phenotyping should be performed for prognostication and targeted therapy.
Some cases with the presence of pancytopenia and lymphopenia having low LGL count around 0.5-1 × 10^9/L, no blood clonal evidence, and unmutated STAT3 are labeled as “Grey zone cases”. In these cases, bone marrow biopsy with immunohistochemical analysis should be done to classify them into either Interstitial and intravascular; Infiltration of clusters of CD3+/CD8+ Granzyme B/TiA1+ LGL or Lymphoid nodules of B and T CD4 cells. For such cases also, STAT3-5b/NFKB mutation, Vb repertoire, and KIR phenotyping should be evaluated, if not done earlier, for their prognostication.
The revised 2016 edition of WHO provides the cut-off of LGL in peripheral blood with a lymphocytosis of 2-20 × 109/L for a duration of 6 months without any known cause and on immunophenotyping has the particular phenotype of T or NK-cell in its criteria of diagnosis. It also mentions that in cases of lymphocytosis <2 × 109/L, keeping in consideration the associated clinical features, the diagnosis can be made. CLPD of NK-cell type has been labeled as a provisional entity due to its chronic clinical course [9]. The aggressive NK-cell leukemia has a strong association with EBV infection (85-100% cases are reported positive for EBV). Genetic profile studies can be recommended for such patients. Del (6) (q21q25) and 11q deletion have been reported in them.
Cytological features
Large granular lymphocytes (LGL) are cytotoxic cells of large size (2-3 times the size of mature lymphocytes), with abundant cytoplasm containing typical azurophilic granules, and a reniform or round nucleus with mature/condensed chromatin. These cells contain cytotoxic granulations of perforin and granzyme and express apoptosis signals through death receptors (Fas and TRAIL). They normally represent 10-15% of peripheral blood (PB) mononuclear cells, where they are involved in cell-mediated cytotoxicity. Figure 2 represents the micrograph of an LGL as seen in the peripheral blood smear.
Figure 2.
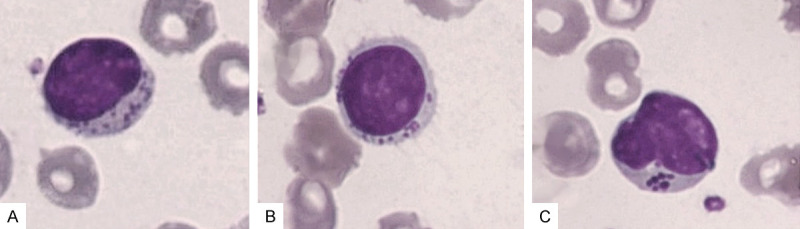
A-C. Shows LGL in peripheral blood smear, stained with May Grunwald Giemsa stain and viewed under 100 × magnification.
Histological features
Bone marrow
Bone marrow (BM) shows mainly interstitial and intra-sinusoidal infiltrate of CD8+ T cells [58], in association with a reactive non-malignant lymphoid population containing polyclonal B and T cells. Myeloid series of cells may show maturation arrest [54]. Erythroid series of cells are usually increased. Immunohistochemistry is usually needed to identify the CD8+ T cells or, the CD3 epsilon + NK cells.
Spleen
Characteristic findings in the spleen include lymphocytic red pulp cord infiltration with preservation of the white pulp. The white pulp may show expansion of its germinal center.
Liver
The liver characteristically shows prominent sinusoid and portal infiltration.
Skin
Skin shows perivascular lymphoid infiltrate in the upper dermis. No epidermal changes were seen [3].
Immunophenotyping
Immunophenotypically LGLs can be divided into T-LGLs and NK-LGLs.
T-LGLs
According to Semezato et al., 80% of cases of T-LGLs are positive for CD3 and CD8. The cells usually show restriction for TCRα/β. They are also positive for CD16, CD57 and are negative for CD4, CD56, and CD28 [59]. Uncommonly, they can be CD8 negative and CD4 positive or can be double negative for CD4 and CD8 with restriction for TCRγδ.
Only a few cases of TCRγδ positive LGL leukemia have been described but they usually have the same clinical phenotype as TCRα/β as they are associated with neutropenia, rheumatoid arthritis, PRCA, etc. [60,61]. They are positive for cytotoxic proteins, TIA 1, granzyme B and granzyme M.
Few patients show expression of CD56. These patients present at a younger age group and were associated with a more aggressive clinical course and poorer prognosis [62]. About 50% of cases are positive for CD94/NKG2 and KIR [63]. Only a single isoform of KIR is present in an individual. This can also be used as a marker of clonality. The flow cytometric analysis of a typical case of T CD3+ LGL leukemia is shown in Figure 3.
Figure 3.
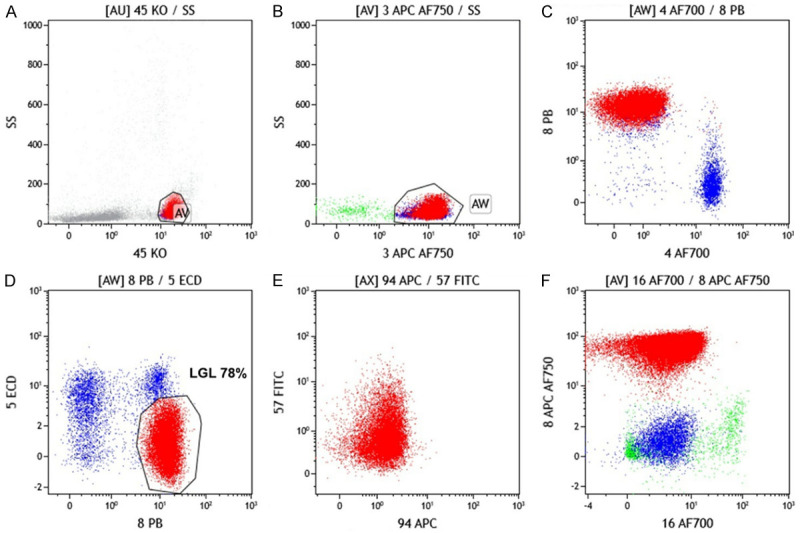
Flow cytometric analysis of a typical case of T CD3+ LGL leukemia. A. Lymphocytes are gated on CD45 versus side scatter. B. CD3+ T cells are gated. C. Majority (~84%) CD3+ cells are CD8+. D. CD3+CD8+ are showing dim to negative expression of CD5. E. CD3+CD8+ are showing negative expression of CD94 and CD57. F. CD3+CD8+ are showing negative expression of CD16.
NK-LGLs
They are characteristically positive for CD3 epsilon, CD2, CD17, and CD56; but negative for surface CD3, CD4, CD8, and CD57 [44]. They are positive for granzymes M, granzyme B, and TIA1. KIR expression is skewed, CD161 expression is reduced while CD94/NKG expression is bright [64,65].
Differential diagnosis
Reactive LGL cell expansion
Many benign, as well as malignant conditions, are associated with the proliferation of reactive LGLs. These conditions include viral infections, bone marrow transplants, many solid malignancies, and Non-Hodgkin’s Lymphomas. The reactive LGLs are polyclonal and subside as the primary condition subsides. These conditions have no clinical features associated with LGL leukemia [12].
Hepatosplenic T cell lymphoma
It is a T-NHL which can clinically mimic LGL leukemia but morphologically, the cells are immature with opened up chromatin and a thin rim of cytoplasm. Immunophenotypically, it shows restriction for TCRγδ and is negative for CD8, CD57 and granzyme B. Bone marrow involvement are intra-sinusoidal with expansion rather than interstitial [66]. Males of median age of 30 years are more affected with B symptoms.
Peripheral T cell lymphoma-not otherwise specified (PTCL-NOS)
It is a mature nodal and extra-nodal T-NHL, which can mimic LGL leukemia morphologically. Though they are associated with EBV however the association with EBV has been established only in cases of aggressive NK-LGLs [67]. Immunophenotypically they are positive for CD4, and usually negative for CD8. They are also positive for CD15 and may co-express with CD30 [68].
Therapy
Cases with T-LGL leukemia and indolent NK-LGL leukemia are treated in the same manner, however aggressive NK-LGL leukemia are treated more aggressively. For asymptomatic cases, only observation is necessary. However, most of the patients become symptomatic over time and require therapy.
Indication for treatment: Severe neutropenia (<500/µL) with severe infection.
Transfusion dependent anemia.
The algorithm used to treat patients with LGL leukemia is given in Figure 4 and has been described in the sections below.
Figure 4.
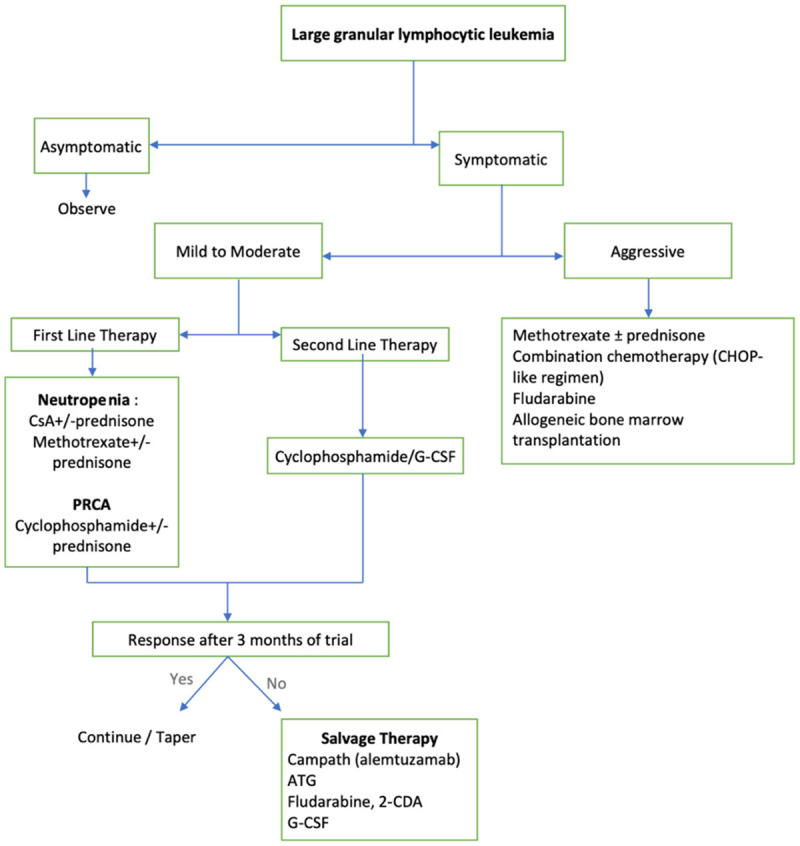
Therapeutic algorithm of LGL leukemia.
For neutropenia, G-CSF has shown significant response in the initial stages of the disease but some patients show no response to the treatment or have a delayed response [69].
Steroids may relieve some symptoms and improve neutropenia, but remissions are usually not durable and eventually, patients also suffer the side effects of steroid therapy [59].
Oral methotrexate has been used successfully for the treatment of LGL leukemia. It is one of the best therapies for neutropenia when used at a dose of 10 mg/m2 orally per week [70]. The median duration of therapy is 1-9 years [30,70]. It has the side effect of interfering with liver and renal function; therefore, regular hepatic and renal check-up is needed.
Cyclosporin is an immunomodulator that can be used as a first-line as well as a second-line drug, alone or in combination with steroid or methotrexate [54,71]. It is used at a dose of 1-1.5 mg/kg orally two times a day. Its response is not permanent and relapses do occur after discontinuing, therefore requiring indefinite administration.
Cyclophosphamide is an alkylating agent it can be successfully used in refractory cases at a dose of 50-100 mg/day per orally. It has a better effect in neutropenia and PRCA as compared to methotrexate, although the response is not permanent and the condition may recur if therapy is stopped. It is associated with side effects like myelosuppression requiring supplementation with G-CSF. It should be used not more than 4 months in cases of non-responders, and 6-12 months in cases of responders [72]. Renal function should be monitored regularly.
Aggressive LGL leukemia has been usually treated with polychemotherapy that can be either CHOP-like (cyclophosphamide, doxorubicin, vincristine, and prednisone-like) or CVP (cyclophosphamide, vincristine, and prednisone) based therapies [12]. They are associated with poor prognosis [62]. HSCT can be used as a potential therapeutic option in rare aggressive and relapsing forms of T-LGL leukemia [73].
Response assessment
Hematologic complete response (CR)
It is defined by normalization of blood counts for more than 4 months-Haemoglobin ≥12 g/dL; Platelets ≥150 × 109/L; ANC ≥1.5 × 109/L and Lymphocytosis ≤4 × 109/L.
Hematologic partial response (PR)
It is defined by - There is improvement in hematological parameters but not meeting the criteria of CR.
Treatment failure
It is defined by - Patient not meeting either criteria of CR or PR for more than 4 months.
Progressive disease (PD)
It is defined by worsening of cytopenia or organomegaly.
Prognosis
Patients with T-LGL leukemia and indolent NK-LGL leukemia are associated with an indolent course. For the initial phase of therapy, most of these patients need no treatment but need observation and follow-ups at regular intervals. Most of these patients have a progressive course when they become symptomatic and need to be treated. These patients usually have an indolent course with the cause of death is mainly secondary to other associated conditions like severe infection secondary to neutropenia. However, only very few patients die due to disease progression.
According to Bareau B et al., around 4% of cases died because of severe infection due to neutropenia and around 2% cases died because of disease progression [16]. The overall 5-year survival is around 75% [74]. However, the prognosis of aggressive NK-LGL leukemia is very poor, which is refractory to most of the therapy options available and the median survival of these cases is only a few weeks [75].
Despite clinical response to all the therapeutic options available presently, relapses are infrequent. This is due to the persistence of the leukemic LGL clone. A better understanding of its etiological evolution and an understanding of targeted therapeutic agents combined with novel and FDA-approved agents in clinical trials in the future may help in giving more consistent responses in these patients.
Conclusion
LGL leukemia has a wide spectrum of clinical presentations varying from an indolent chronic lymphoproliferative disorder to an aggressive NK cell leukemia. The disposition from a reactive polyclonal LGL proliferation through clonal indolent proliferation to an aggressive lymphoma requires more studies for better clarity of its evolution. Even the therapeutic options available require more clinical trials, especially for the aggressive forms.
The rarity of LGL leukemia with the availability of few clinical trials is an obstacle to the development of a specific treatment. Albeit the extensive use of immunomodulators, the relapse of these cases is at a higher number. Advances in the understanding of the molecular pathways involved in the development of this entity have brought about few targeted therapies in the market. However, there is a substantial need for reducing the relapsed cases and diagnosing them earlier than is ordained. Moreover, the ideal LGL number in the peripheral blood for an invasive procedure like bone marrow investigation to be undertaken without the absence of any clinical features is a dilemma. The recognition of CLPD of NK cell type as a distinct entity and not as provisional, as described by the revised edition of WHO may help in earlier monitoring and diagnosis of these cases. Monitoring such cases will help in better understanding of the pathways involved, especially in specifying the exact pathway which shifts, as they evolved from chronic to a more acute presentation. The alliance of LGL leukemia with autoimmune disorders has been documented in numerous works of literature. The autoimmune condition is of priority, however, the presence of LGL should not be disregarded. The number of LGL in their peripheral blood should be closely monitored as chances of these patients developing LGL leukemia is higher compared to apparently healthy individuals. Future research on this group of patients may bring about a cut-off of LGL for warning and work-up. These are the steps that can be implemented in the future and which will help in better diagnosis of this sporadic leukemia.
We conclude that for the improvement in the survival of these cases, clinical trials with thorough detail on the evolution and the clinical course of the disease are needed.
Disclosure of conflict of interest
None.
References
- 1.Brouet JC, Sasportes M, Flandrin G, Preud’Homme JL, Seligmann M. Chronic lymphocytic leukaemia of T-cell origin immunological and clinical evaluation in eleven patients. Lancet. 1975;306:890–893. doi: 10.1016/s0140-6736(75)92127-3. [DOI] [PubMed] [Google Scholar]
- 2.McKenna RW, Parkin J, Kersey JH, Gajl-Peczalska KJ, Peterson L, Brunning RD. Chronic lymphoproliterative disorder with unusual clinical, morphologic, ultrastructural and membrane surface marker characteristics. Am J Med. 1977;62:588–596. doi: 10.1016/0002-9343(77)90422-3. [DOI] [PubMed] [Google Scholar]
- 3.Agnarsson BA, Loughran TP Jr, Starkebaum G, Kadin ME. The pathology of large granular lymphocyte leukemia. Hum Pathol. 1989;20:643–651. doi: 10.1016/0046-8177(89)90151-2. [DOI] [PubMed] [Google Scholar]
- 4.Lougharn TP. Clonal diseases of large granular lymphocytes. Blood. 1993;82:12–14. [PubMed] [Google Scholar]
- 5.Bennett J, Catovsky D, Daniel M, Flandrin G, Galton D, Gralnick H, Sultan C. Proposals for the classification of chronic (mature) B and T lymphoid leukaemias. French-American-British (FAB) Cooperative Group. J Clin Pathol. 1989;42:567–584. doi: 10.1136/jcp.42.6.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML, Delsol G, De Wolf-Peeters C, Falini B, Gatter KC, et al. Lymphoma classification proposal: clarification. Blood. 1995;85:857–60. [PubMed] [Google Scholar]
- 7.Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, Lister TA, Bloomfield CD. The World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the clinical advisory committee meeting, Airlie House, Virginia, November, 1997. Ann Oncol. 1999;10:1419–1432. doi: 10.1023/a:1008375931236. [DOI] [PubMed] [Google Scholar]
- 8.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. WHO classification of tumours of haematopoietic and lymphoid tissues. France: International Agency for Research on Cancer Lyon; 2008. [Google Scholar]
- 9.Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–2390. doi: 10.1182/blood-2016-01-643569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moignet A, Lamy T. Latest advances in the diagnosis and treatment of large granular lymphocytic leukemia. Am Soc Clin Oncol Educ Book. 2018;38:616–625. doi: 10.1200/EDBK_200689. [DOI] [PubMed] [Google Scholar]
- 11.Cheon H, Dziewulska KH, Moosic KB, Olson KC, Gru AA, Feith DJ, Loughran TP Jr. Advances in the diagnosis and treatment of large granular lymphocytic leukemia. Curr Hematol Malig Rep. 2020;15:103–112. doi: 10.1007/s11899-020-00565-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamy T, Loughran T. Clinical features of large granular lymphocytic leukemia. Semin Hematol. 2003;40:185–195. doi: 10.1016/s0037-1963(03)00133-1. [DOI] [PubMed] [Google Scholar]
- 13.Lamy T, Loughran T. Current concepts: large granular lymphocyte leukemia. Blood Rev. 1999;13:230–240. doi: 10.1054/blre.1999.0118. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki R, Suzumiya J, Nakamura S, Aoki S, Notoya A, Ozaki S, Gondo H, Hino N, Mori H, Sugimori H. Aggressive natural killer-cell leukemia revisited: large granular lymphocyte leukemia of cytotoxic NK cells. Leukemia. 2004;18:763–770. doi: 10.1038/sj.leu.2403262. [DOI] [PubMed] [Google Scholar]
- 15.Shah MV, Hook CC, Call TG, Go RS. A population-based study of large granular lymphocytic leukemia. Blood Cancer J. 2016;6:e455. doi: 10.1038/bcj.2016.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bareau B, Rey J, Hamidou M, Donadieu J, Morcet J, Reman O, Schleinitz N, Tournilhac O, Roussel M, Fest T. Analysis of a French cohort of patients with large granular lymphocyte leukemia: a report on 229 cases. Haematologica. 2010;95:1534. doi: 10.3324/haematol.2009.018481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clemente MJ, Wlodarski MW, Makishima H, Viny AD, Bretschneider I, Shaik M, Bejanyan N, Lichtin AE, Hsi ED, Paquette RL. Clonal drift demonstrates unexpected dynamics of the T-cell repertoire in T-large granular lymphocyte leukemia. Blood. 2011;118:4384–4393. doi: 10.1182/blood-2011-02-338517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinway SN, LeBlanc F, Loughran TP Jr. The pathogenesis and treatment of large granular lymphocyte leukemia. Blood Rev. 2014;28:87–94. doi: 10.1016/j.blre.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamy T, Moignet A, Loughran TP Jr. LGL leukemia: from pathogenesis to treatment. Blood. 2017;129:1082–1094. doi: 10.1182/blood-2016-08-692590. [DOI] [PubMed] [Google Scholar]
- 20.Zhang R, Shah MV, Yang J, Nyland SB, Liu X, Yun JK, Albert R, Loughran TP Jr. Network model of survival signaling in large granular lymphocyte leukemia. Proc Natl Acad Sci U S A. 2008;105:16308–16313. doi: 10.1073/pnas.0806447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Starkebaum G, Loughran TP Jr, Kalyanaraman VS, Kadin ME, Kidd PG, Singer JW, Ruscetti FW. Serum reactivity to human T-cell leukaemia/lymphoma virus type I proteins in patients with large granular lymphocytic leukaemia. Lancet. 1987;329:596–599. doi: 10.1016/s0140-6736(87)90236-4. [DOI] [PubMed] [Google Scholar]
- 22.Yang J, Epling-Burnette PK, Painter JS, Zou J, Bai F, Wei S, Loughran TP Jr. Antigen activation and impaired Fas-induced death-inducing signaling complex formation in T-large-granular lymphocyte leukemia. Blood. 2008;111:1610–1616. doi: 10.1182/blood-2007-06-093823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poullot E, Bouscary D, Guyader D, Ghandour C, Roussel M, Fest T, Houot R, Lamy T. Large granular lymphocyte leukemia associated with hepatitis C virus infection and B cell lymphoma: improvement after antiviral therapy. Leuk Lymphoma. 2013;54:1797–1799. doi: 10.3109/10428194.2012.752486. [DOI] [PubMed] [Google Scholar]
- 24.Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist. 2004;9:247–258. doi: 10.1634/theoncologist.9-3-247. [DOI] [PubMed] [Google Scholar]
- 25.Lodolce JP, Burkett PR, Koka RM, Boone DL, Ma A. Regulation of lymphoid homeostasis by interleukin-15. Cytokine Growth Factor Rev. 2002;13:429–439. doi: 10.1016/s1359-6101(02)00029-1. [DOI] [PubMed] [Google Scholar]
- 26.Yang J, Liu X, Nyland SB, Zhang R, Ryland LK, Broeg K, Baab KT, Jarbadan NR, Irby R, Loughran TP Jr. Platelet-derived growth factor mediates survival of leukemic large granular lymphocytes via an autocrine regulatory pathway. Blood. 2010;115:51–60. doi: 10.1182/blood-2009-06-223719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmäki H, Andersson EI, Lagström S, Clemente MJ, Olson T, Jalkanen SE. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366:1905–1913. doi: 10.1056/NEJMoa1114885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jerez A, Clemente MJ, Makishima H, Koskela H, LeBlanc F, Peng Ng K, Olson T, Przychodzen B, Afable M, Gomez-Segui I. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 2012;120:3048–3057. doi: 10.1182/blood-2012-06-435297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teramo A, Gattazzo C, Passeri F, Lico A, Tasca G, Cabrelle A, Martini V, Frezzato F, Trimarco V, Ave E. Intrinsic and extrinsic mechanisms contribute to maintain the JAK/STAT pathway aberrantly activated in T-type large granular lymphocyte leukemia. Blood. 2013;121:3843–3854. doi: 10.1182/blood-2012-07-441378. [DOI] [PubMed] [Google Scholar]
- 30.Jerez A, Clemente MJ, Makishima H, Rajala H, Gómez-Seguí I, Olson T, McGraw K, Przychodzen B, Kulasekararaj A, Afable M. STAT3 mutations indicate the presence of subclinical T-cell clones in a subset of aplastic anemia and myelodysplastic syndrome patients. Blood. 2013;122:2453–2459. doi: 10.1182/blood-2013-04-494930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu JH, Wei S, Lamy T, Li Y, Epling-Burnette P, Djeu JY, Loughran TP Jr. Blockade of Fas-dependent apoptosis by soluble Fas in LGL leukemia. Blood. 2002;100:1449–1453. [PubMed] [Google Scholar]
- 32.Liu JH, Wei S, Lamy T, Epling-Burnette P, Starkebaum G, Djeu JY, Loughran TP Jr. Chronic neutropenia mediated by fas ligand. Blood. 2000;95:3219–3222. [PubMed] [Google Scholar]
- 33.Ülkü AS, Der CJ. Ras signaling, deregulation of gene expression and oncogenesis. Semin Cancer Biol. 2004;15:189–208. [PubMed] [Google Scholar]
- 34.Mizutani N, Ito H, Hagiwara K, Kobayashi M, Hoshikawa A, Nishida Y, Takagi A, Kojima T, Suzuki M, Osawa Y. Involvement of KRAS G12A mutation in the IL-2-independent growth of a human T-LGL leukemia cell line, PLT-2. Nagoya J Med Sci. 2012;74:261–71. [PMC free article] [PubMed] [Google Scholar]
- 35.Epling-Burnette PK, Bai F, Wei S, Chaurasia P, Painter JS, Olashaw N, Hamilton A, Sebti S, Djeu JY, Loughran TP. ERK couples chronic survival of NK cells to constitutively activated Ras in lymphoproliferative disease of granular lymphocytes (LDGL) Oncogene. 2004;23:9220–9229. doi: 10.1038/sj.onc.1208122. [DOI] [PubMed] [Google Scholar]
- 36.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 37.Schade AE, Powers JJ, Wlodarski MW, Maciejewski JP. Phosphatidylinositol-3-phosphate kinase pathway activation protects leukemic large granular lymphocytes from undergoing homeostatic apoptosis. Blood. 2006;107:4834–4840. doi: 10.1182/blood-2005-08-3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang R, Shah MV, Yang J, Nyland SB, Liu X, Yun JK, Albert R, Loughran TP Jr. Network model of survival signalling in large granular lymphocyte leukemia. Proc Natl Acad Sci U S A. 2008;105:16308–16313. doi: 10.1073/pnas.0806447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shah MV, Zhang R, Irby R, Kothapalli R, Liu X, Arrington T, Frank B, Lee NH, Loughran TP Jr. Molecular profiling of LGL leukemia reveals role of sphingolipid signaling in survival of cytotoxic lymphocytes. Blood. 2008;112:770–781. doi: 10.1182/blood-2007-11-121871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X, Ryland L, Yang J, Liao A, Aliaga C, Watts R, Tan SF, Kaiser J, Shanmugavelandy SS, Rogers A. Targeting of survivin by nanoliposomal ceramide induces complete remission in a rat model of NK-LGL leukemia. Blood. 2010;116:4192–4201. doi: 10.1182/blood-2010-02-271080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kwong Y, Wong K. Association of pure red cell aplasia with T large granular lymphocyte leukaemia. J Clin Patholy. 1998;51:672–675. doi: 10.1136/jcp.51.9.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aribi A, Huh Y, Keating M, O’Brien S, Ferrajoli A, Faderl S, Wierda W, Kantarjian H, Ravandi F. T-cell large granular lymphocytic (T-LGL) leukemia: experience in a single institution over 8 years. Leuk Res. 2007;31:939–945. doi: 10.1016/j.leukres.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 43.Akashi K, Shibuya T, Taniguchi S, Hayashi S, Iwasaki H, Teshima T, Takamatsu Y, Gondo H, Okamura T, Harada M. Multiple autoimmune haemopoietic disorders and insidious clonal proliferation of large granular lymphocytes. Br J Haematol. 1999;107:670–673. doi: 10.1046/j.1365-2141.1999.01734.x. [DOI] [PubMed] [Google Scholar]
- 44.Sokol L, Loughran TP Jr. Large granular lymphocyte leukemia. Oncologist. 2006;11:263–273. doi: 10.1634/theoncologist.11-3-263. [DOI] [PubMed] [Google Scholar]
- 45.Epling-Burnette P, Sokol L, Chen X, Bai F, Zhou J, Blaskovich MA, Zou J, Painter JS, Edwards TD, Moscinski L. Clinical improvement by farnesyltransferase inhibition in NK large granular lymphocyte leukemia associated with imbalanced NK receptor signaling. Blood. 2008;112:4694–4698. doi: 10.1182/blood-2008-02-136382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shvidel L, Duksin C, Tzimanis A, Shtalrid M, Klepfish A, Sigler E, Haran M, Eilat E, Berrebi A. Cytokine release by activated T-cells in large granular lymphocytic leukemia associated with autoimmune disorders. Hematol J. 2002;3:32–37. doi: 10.1038/sj.thj.6200149. [DOI] [PubMed] [Google Scholar]
- 47.Vanness ER, Davis MD, Tefferi A. Cutaneous findings associated with chronic natural killer cell lymphocytosis. Int J Dermatol. 2002;41:852–857. doi: 10.1046/j.1365-4362.2002.01671.x. [DOI] [PubMed] [Google Scholar]
- 48.Rabbani GR, Phyliky RL, Tefferi A. A long-term study of patients with chronic natural killer cell lymphocytosis. Br J Haematol. 1999;106:960–966. doi: 10.1046/j.1365-2141.1999.01624.x. [DOI] [PubMed] [Google Scholar]
- 49.Agnarsson BA, Loughran TP Jr, Starkebaum G, Kadin ME. The pathology of large granular lymphocyte leukemia. Hum Pathol. 1989;20:643–651. doi: 10.1016/0046-8177(89)90151-2. [DOI] [PubMed] [Google Scholar]
- 50.Mohan SR, Maciejewski JP. Diagnosis and therapy of neutropenia in large granular lymphocyte leukemia. Curr Opin Hematol. 2009;16:27–34. doi: 10.1097/MOH.0b013e32831c8407. [DOI] [PubMed] [Google Scholar]
- 51.Zhang R, Shah MV, Loughran TP Jr. The root of many evils: indolent large granular lymphocyte leukaemia and associated disorders. Hematol Oncol. 2010;28:105–117. doi: 10.1002/hon.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Viny AD, Lichtin A, Pohlman B, Loughran T, Maciejewski J. Chronic B-cell dyscrasias are an important clinical feature of T-LGL leukemia. Leuk Lymphoma. 2008;49:932–938. doi: 10.1080/10428190801932635. [DOI] [PubMed] [Google Scholar]
- 53.Bouroncle BA, Wiseman BK, Doan CA. Leukemic reticuloendotheliosis. Blood. 1958;13:609–630. doi: 10.1182/blood-2016-01-696179. [DOI] [PubMed] [Google Scholar]
- 54.Dhodapkar MV, Li CY, Lust JA, Tefferi A, Phyliky RL. Clinical spectrum of clonal proliferations of T-large granular lymphocytes: a T-cell clonopathy of undetermined significance? Blood. 1994;84:1620–7. [PubMed] [Google Scholar]
- 55.Go RS, Tefferi A, Li CY, Lust JA, Phyliky RL. Lymphoproliferative disease of granular T lymphocytes presenting as aplastic anemia. Blood. 2000;96:3644–3646. [PubMed] [Google Scholar]
- 56.Gentile TC, Wener MH, Starkebaum G, Loughran TP Jr. Humoral immune abnormalities in T-cell large granular lymphocyte leukemia. Leuk Lymphoma. 1996;23:365–370. doi: 10.3109/10428199609054840. [DOI] [PubMed] [Google Scholar]
- 57.Bassan R, Pronesti M, Buzzetti M, Allavena P, Rambaldi A, Mantovani A, Barbui T. Autoimmunity and B-cell dysfunction in chronic proliferative disorders of large granular lymphocytes/natural killer cells. Cancer. 1989;63:90–95. doi: 10.1002/1097-0142(19890101)63:1<90::aid-cncr2820630115>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 58.Morice WG, Kurtin PJ, Tefferi A, Hanson CA. Distinct bone marrow findings in T-cell granular lymphocytic leukemia revealed by paraffin section immunoperoxidase stains for CD8, TIA-1, and granzyme B. Blood. 2002;99:268–274. doi: 10.1182/blood.v99.1.268. [DOI] [PubMed] [Google Scholar]
- 59.Semenzato G, Zambello R, Starkebaum G, Oshimi K, Loughran TP Jr. The lymphoproliferative disease of granular lymphocytes: updated criteria for diagnosis. Blood. 1997;89:256–260. [PubMed] [Google Scholar]
- 60.Sun T, Cohen NS, Marino J, Koduru P, Cuomo J, Henshall J. CD3+, CD4-, CD8- large granular T-cell lymphoproliferative disorder. Am J Hematol. 1991;37:173–178. doi: 10.1002/ajh.2830370308. [DOI] [PubMed] [Google Scholar]
- 61.Morikawa K, Oseko F, Hara J, Kobayashi S, Nakano A, Morikawa S. Functional analysis of clonally expanded CD8, TCRγδ T cells in a patient with chronic T-gamma lymphoproliferative disease. Leuk Res. 1990;14:581–592. doi: 10.1016/0145-2126(90)90011-w. [DOI] [PubMed] [Google Scholar]
- 62.Alekshun TJ, Tao J, Sokol L. Aggressive T-cell large granular lymphocyte leukemia: a case report and review of the literature. Am J Hematol. 2007;82:481–485. doi: 10.1002/ajh.20853. [DOI] [PubMed] [Google Scholar]
- 63.Fischer L, Hummel M, Burmeister T, Schwartz S, Thiel E. Skewed expression of natural-killer (NK)-associated antigens on lymphoproliferations of large granular lymphocytes (LGL) Hematol Oncol. 2006;24:78–85. doi: 10.1002/hon.777. [DOI] [PubMed] [Google Scholar]
- 64.Zambello R, Trentin L, Facco M, Cerutti A, Sancetta R, Milani A, Raimondi R, Tassinari C, Agostini C, Semenzato G. Analysis of the T cell receptor in the lymphoproliferative disease of granular lymphocytes: superantigen activation of clonal CD3+ granular lymphocytes. Cancer Res. 1995;55:6140–6145. [PubMed] [Google Scholar]
- 65.Lima M, Almeida J, Montero AG, Teixeira Mdos A, Queirós ML, Santos AH, Balanzategui A, Estevinho A, Algueró Mdel C, Barcena P, Fonseca S, Amorim ML, Cabeda JM, Pinho L, Gonzalez M, San Miguel J, Justiça B, Orfão A. Clinicobiological, immunophenotypic, and molecular characteristics of monoclonal CD56-/+ dim chronic natural killer cell large granular lymphocytosis. Am J Pathol. 2004;165:1117–1127. doi: 10.1016/s0002-9440(10)63373-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Attygalle AD, Cabeçadas J, Gaulard P, Jaffe ES, de Jong D, Ko YH, Said J, Klapper W. Peripheral T-cell and NK-cell lymphomas and their mimics; taking a step forward-report on the lymphoma workshop of the XVI th meeting of the European Association for Haematopathology and the Society for Hematopathology. Histopathology. 2014;64:171–199. doi: 10.1111/his.12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ishihara S, Okada S, Wakiguchi H, Kurashige T, Hirai K, Kawa-Ha K. Clonal lymphoproliferation following chronic active Epstein-Barr virus infection and hypersensitivity to mosquito bites. Am J Hematol. 1997;54:276–281. doi: 10.1002/(sici)1096-8652(199704)54:4<276::aid-ajh3>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 68.Barry TS, Jaffe ES, Sorbara L, Raffeld M, Pittaluga S. Peripheral T-cell lymphomas expressing CD30 and CD15. Am J Surg Pathol. 2003;27:1513–1522. doi: 10.1097/00000478-200312000-00003. [DOI] [PubMed] [Google Scholar]
- 69.Genvresse I, Späth-Schwalbe E, Lukowsky A, Possinger K. Delayed response to granulocyte colony-stimulating factor (G-CSF) in a case of severe neutropenia associated with large granular lymphocyte (LGL) leukemia. Eur J Haematol. 1998;60:133–134. doi: 10.1111/j.1600-0609.1998.tb01010.x. [DOI] [PubMed] [Google Scholar]
- 70.Loughran TP Jr, Kidd PG, Starkebaum G. Treatment of large granular lymphocyte leukemia with oral low-dose methotrexate. Blood. 1994;84:2164–2170. [PubMed] [Google Scholar]
- 71.Osuji N, Matutes E, Tjonnfjord G, Grech H, Del Giudice I, Wotherspoon A, Swansbury JG, Catovsky D. T-cell large granular lymphocyte leukemia: a report on the treatment of 29 patients and a review of the literature. Cancer. 2006;107:570–578. doi: 10.1002/cncr.22032. [DOI] [PubMed] [Google Scholar]
- 72.Lamy T, Loughran TP Jr. How I treat LGL leukemia. Blood. 2011;117:2764–2774. doi: 10.1182/blood-2010-07-296962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marchand T, Lamy T, Finel H, Arcese W, Choquet S, Finke J, Huynh A, Irrera G, Karakasis D, Konopacki J. Hematopoietic stem cell transplantation for T-cell large granular lymphocyte leukemia: a retrospective study of the European Society for blood and marrow transplantation. Leukemia. 2016;30:1201–1204. doi: 10.1038/leu.2015.256. [DOI] [PubMed] [Google Scholar]
- 74.Dinmohamed A, Brink M, Visser O, Jongen-Lavrencic M. Population-based analyses among 184 patients diagnosed with large granular lymphocyte leukemia in the Netherlands between 2001 and 2013. Leukemia. 2016;30:1449–1451. doi: 10.1038/leu.2016.68. [DOI] [PubMed] [Google Scholar]
- 75.Li C, Tian Y, Wang J, Zhu L, Huang L, Wang N, Xu D, Cao Y, Li J, Zhou J. Abnormal immunophenotype provides a key diagnostic marker: a report of 29 cases of de novo aggressive natural killer cell leukemia. Transl Res. 2014;163:565–577. doi: 10.1016/j.trsl.2014.01.010. [DOI] [PubMed] [Google Scholar]