

Abstract
Microsatellites are repeated DNA sequences of 3–6 nucleotides highly variable in length and sequence and that have important roles in genomes regulation and evolution. However, expansion of a subset of these microsatellites over a threshold size is responsible of more than 50 human genetic diseases. Interestingly, some of these disorders are caused by expansions of similar sequences, sizes and localizations and present striking similarities in clinical manifestations and histopathological features, which suggest a common mechanism of disease. Notably, five identical CGG repeat expansions, but located in different genes, are the causes of fragile X-associated tremor/ataxia syndrome (FXTAS), neuronal intranuclear inclusion disease (NIID), oculopharyngodistal myopathy type 1 to 3 (OPDM1-3) and oculopharyngeal myopathy with leukoencephalopathy (OPML), which are neuromuscular and neurodegenerative syndromes with overlapping symptoms and similar histopathological features, notably the presence of characteristic eosinophilic ubiquitin-positive intranuclear inclusions. In this review we summarize recent finding in neuronal intranuclear inclusion disease and FXTAS, where the causing CGG expansions were found to be embedded within small upstream ORFs (uORFs), resulting in their translation into novel proteins containing a stretch of polyglycine (polyG). Importantly, expression of these polyG proteins is toxic in animal models and is sufficient to reproduce the formation of ubiquitin-positive intranuclear inclusions. These data suggest the existence of a novel class of human genetic pathology, the polyG diseases, and question whether a similar mechanism may exist in other diseases, notably in OPDM and OPML.
Keywords: microsatellite, protein aggregates toxicity, RAN translation, neurodegeneration, trinucleotide repeat disorders
Introduction
DNA short tandem repeats (STR), also known as microsatellites, are short sequences of 3–6 nucleotides repeated multiple time so that they occupy as much as 2–3% of the human genome. These microsatellites are highly variable in size and sequence and have important roles in genomes regulation and evolution. However, expansion of a subset of these microsatellite sequences over a threshold size can also be the leading cause of human genetic diseases, and 2021 marks the 30th anniversary of the discovery of the two first pathogenic trinucleotide expansions, namely CGG and CAG repeats located in the fragile X mental retardation (FMR1) and androgen receptor (AR) genes and that cause fragile X syndrome (FXS) and spinal and bulbar muscular atrophy (SBMA), respectively (Fu et al., 1991; Kremer et al., 1991; La Spada et al., 1991; Oberlé et al., 1991; Verkerk et al., 1991). Since then, expansion of tri-, tetra-, penta- and hexa-nucleotide sequences were identified in more than 50 neurodevelopmental, neuromuscular and neurodegenerative genetic disorders (review in Malik et al., 2021a; Depienne and Mandel, 2021; Table 1). These microsatellite mutations are highly variable with expansion lengths changing among populations, generation and individuals, and even according to tissues, cells and/or age. Thus, in a subgroup of microsatellite diseases where clinical manifestations correlate with expansion sizes, symptoms may greatly diverge between individuals and/or between generations, resulting in atypical heritability such as anticipation (increased disease severity and/or decreased age of onset with increased repeat size among generations) or its opposite mechanism, contraction where the expansion may shrink below its pathogenic threshold size from one generation to another. Moreover, these repeat expansions can present a profound genetic founder effect, restricting observation of some of these mutations to specific human populations. For example, the G4C2 repeat expansion in the C9ORF72 gene, which causes amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), is amply observed in North Europe and North America but rare in other populations; while a CGG repeat expansion in the NOTCH2NLC gene that causes neuronal intranuclear inclusion disease (NIID) is increasingly reported in Asian populations, but rare in Europe. Finally, these microsatellite expansions can be located in either non-transcribed and non-coding sequences, transcribed but untranslated regions, or within translated sequences, and 30 years of researches have now unveiled that these expansions are pathogenic by both loss and/or gain of function mechanisms at the DNA, RNA, and protein levels (review in Malik et al., 2021a; Depienne and Mandel, 2021; Table 1).
TABLE 1.
Repeat expansion diseases, sorted by their proposed pathogenic mechanism.
| Proposed mechanism | Disease | Gene | Localization | Repeat | Normal | Pathogenic | Reference |
|---|---|---|---|---|---|---|---|
| size | size | ||||||
| LOF | BSS | XYLT1 | Promoter | CGG | 9–20 | 120–800 | LaCroix et al. (2019) |
| LOF | FXS | FMR1 | 5′ UTR | CGG | 5–50 | >200 | Verkerk et al. (1991); Oberlé et al. (1991); Fu et al. (1991) |
| LOF | FRAXE | AFF2 | 5′ UTR | CCG | 4–39 | 200–900 | Knight et al. (1993) |
| LOF | EPM1 | CSTB | 5′ UTR | C4GC4GCG | 2–3 | 30–75 | Lalioti et al., 1997 |
| LOF | GDPAG | GLS | 5′ UTR | GCA | 8–16 | 680–1400 | Van Kuilenburg et al. (2019) |
| LOF | FRDA | FXN | Intron | GAA | 5–34 | 65–1300 | Campuzano et al. (1996) |
| LOF | XDP | TAF1 | Intron | C3TCT | absent | 30–55 | Bragg et al. (2017) |
| polyAla | SPD1 | HOXD13 | Exon | GCG | 15 | 24 | Akarsu et al. (1996) |
| polyAla | BCCD | RUNX2 | Exon | GCN | 17 | 27 | Mundlos et al. (1997) |
| polyAla | HFGS | HOXA13 | Exon | GCN | 12–18 | 18–30 | Goodman et al. (2000) |
| polyAla | BPES | FOXL2 | Exon | GCN | 14 | 19–24 | De Baere et al. (2001) |
| polyAla | HPE5 | ZIC2 | Exon | GCN | 15 | 25 | Brown et al. (2001) |
| polyAla | EIEE1 | ARX | Exon | GCN | 12–16 | 20–23 | Stromme et al. (2002) |
| polyAla | MRGH | SOX3 | Exon | GCN | 15 | 26 | Laumonnier et al. (2002) |
| polyAla | CCHS | PHOX2B | Exon | GCN | 20 | 25–29 | Amiel et al. (2003) |
| polyAla | OPMD | PABPN1 | Exon | GCG | 6–10 | 11–18 | Brais et al. (1998) |
| polyQ | SBMA | AR | Exon | CAG | 9–36 | 38–68 | La Spada et al. (1991) |
| polyQ | DRPLA | ATN1 | Exon | CAG | 3–35 | 48–93 | Koide et al. (1994); Nagafuchi et al. (1994) |
| polyQ | HD | HTT | Exon | CAG | 6–35 | 36–200 | Huntington’s Collaborative Group (1993) |
| polyQ | HDL2 | JPH3 AS | Exon | CAG | 6–28 | 41–58 | Margolis et al. (2001) |
| polyQ | SCA1 | ATXN1 | Exon | CAG | 6–38 | 39–88 | Orr et al. (1993) |
| polyQ | SCA2 | ATXN2 | Exon | CAG | 13–31 | 32–500 | Pulst et al. (1996) |
| polyQ | SCA3 | ATXN3 | Exon | CAG | 12–44 | 55–87 | Kawaguchi et al. (1994) |
| polyQ | SCA6 | CACNA1A | Exon | CAG | 4–18 | 20–33 | Zhuchenko et al. (1997) |
| polyQ | SCA7 | ATXN7 | Exon | CAG | 4–33 | 37–460 | Lindblad et al. (1996) |
| polyQ | SCA8 | ATXN8 | Exon | CAG | 15–50 | 74–250 | Koob et al. (1999) |
| polyQ | SCA17 | TBP | Exon | CAG | 25–40 | 43–66 | Koide et al. (1999) |
| ? | SCA12 | PPP2R2B | 5′ UTR | CAG | 4–32 | 43–78 | Holmes et al. (1999) |
| polyGly | FXTAS | FMR1 | 5′ UTR | CGG | 5–50 | 55–200 | Hagerman et al. (2001) |
| polyGly | NIID | NOTCH2NLC | 5′ UTR | CGG | 7–60 | 60–200 | Ishiura et al. (2019); Sone et al. (2019); Tian et al. (2019); Deng et al. (2019) |
| ? | FXPOI | FMR1 | 5′ UTR | CGG | 5–50 | 55–200 | Conway et al. (1998) |
| ? | OPML | LOC642361 | LncRNA | CGG | 3–16 | 50–60 | Ishiura et al. (2019) |
| ? | OPDM1 | LRP12 | 5′ UTR | CGG | 13–45 | 80–130 | Ishiura et al. (2019) |
| ? | OPDM2 | GIPC1 | 5′ UTR | CGG | 12–32 | 70–120 | Deng et al. (2020) |
| ? | OPDM3 | NOTCH2NLC | 5′ UTR | CGG | 7–60 | 60–200 | Yu et al. (2021) |
| RAN | ALS/FTD | C9ORF72 | Intron | G4C2 | 3–25 | >30 | Dejesus-Hernandez et al. (2011); Renton et al. (2011) |
| RAN | SCA36 | NOP56 | Intron | G3C2T | 5–14 | 650–2,500 | Kobayashi et al. (2011) |
| RAN | SCA31 | BEAN1 | Intron | G2A2T | variable | 110–760 | Sato et al. (2009) |
| ? | CANVAS | RFC1 | Intron | G3A2 | variable | 400–2000 | Cortese et al. (2019); Rafehi et al. (2019) |
| RNA | DM1 | DMPK | 3′ UTR | CTG | 5–37 | 50–10,000 | Mahadevan et al. (1992); Brook et al. (1992); Fu et al. (1992) |
| RNA | DM2 | CNBP | Intron | CCTG | 11–30 | 50–11,000 | Liquori et al. (2001) |
| RNA | FECD3 | TCF4 | Intron | CTG | 5–31 | >50 | Mootha et al. (2014) |
| ? | FAME1 | SAMD12 | Intron | TTTCA | absent | 440–3,680 | Ishiura et al. (2018) |
| ? | FAME2 | STARD7 | Intron | TTTCA | absent | >660–730 | Corbett et al. (2019) |
| ? | FAME3 | MARCHF6 | Intron | TTTCA | absent | >660–2,800 | Florian et al. (2019) |
| ? | FAME4 | YEATS2 | Intron | TTTCA | absent | >500 | Yeetong et al. (2019) |
| ? | FAME6 | TNRC6A | Intron | TTTCA | absent | >400 | Ishiura et al. (2018) |
| ? | FAME7 | RAPGEF2 | Intron | TTTCA | absent | >500 | Ishiura et al. (2018) |
| ? | SCA10 | ATXN10 | Intron | TTCTA | 10–32 | 280–4,500 | Matsuura et al. (2000) |
| ? | SCA37 | DAB1 | Intron | TTTCA | absent | 31–75 | Seixas et al. (2017) |
LOF, loss of function mechanism; polyAla, polyalanine; polyGly, polyglycine; polyQ, polyglutamine; RAN, repeat non-ATG, translation; ALS, amyotrophic lateral sclerosis; BCCD, brachydactyly and cleidocranial dysplasia; BPES, blepharophimosis, ptosis and epicanthus inversus; BSS, Baratela-Scott syndrome; CANVAS, cerebellar ataxia, neuropathy and vestibular areflexia syndrome; CCHS, congenital central hypoventilation syndrome; DM1, myotonic dystrophy type 1; DM2, myotonic dystrophy type 2; DRPLA, dentatorubral-pallidoluysian atrophy; EIEE1, early infantile epileptic encephalopathy type 1; EPM1, progressive myoclonus epilepsy type 1; FAME, familial adult myoclonic epilepsy; FECD3, Fuchs endothelial corneal dystrophy type 3; FRAXE, fragile XE, syndrome; FRDA, Friedreich ataxia; FTD, frontotemporal dementia/; FXPOI, Fragile X-associated premature ovarian infertility; FXS, fragile X syndrome; FXTAS, fragile X-associated tremor ataxia syndrome; GDPAG, global developmental delay, progressive ataxia and elevated glutamine; HD, Huntington disease; HDL2, Huntington disease-like 2; HFGS, hand-foot-genital syndrome; HPE5, holoprosencephaly type 5; MRGH, mental retardation with isolated growth hormone deficiency; NIID, neuronal intranuclear inclusion disease; OPDM, oculopharyngodistal myopathy type; OPMD, oculopharyngeal muscular dystrophy; OPML, oculopharyngeal myopathy with leukoencephalopathy; SBMA, spinal and bulbar muscular atrophy; SPD1, synpolydactyly type 1; SCA, spinocerebellar ataxia; XDP, X-linked dystonia parkinsonism.
First, repeat expansions located within or close to promoters can promote DNA epigenetic changes that inhibit transcription, resulting in reduced expression of the encoded protein. As an indication that these expansions are pathogenic through a loss of function mechanism, identical clinical manifestations can be observed in individuals carrying genetic deletions and/or loss of function coding mutations of the gene hosting these microsatellites. Archetype of expansions shutting down gene expression are the GGC, CGG, and GAA expanded repeats located within the promoter, 5′UTR and first intron of the XYLT1, FMR1, and frataxin genes and associated with Baratela-Scott syndrome (BBS), fragile X syndrome (FXS), and Friedreich ataxia (FA), respectively, (Fu et al., 1991; Oberlé et al., 1991; Verkerk et al., 1991; Kremer et al., 1991; Campuzano et al., 1996; laCroix et al., 2019; Table 1).
Second, these expansions can be pathogenic at the RNA level through a toxic RNA gain of function mechanism. In these disorders, large repeat expansions are transcribed into pathogenic RNA that accumulate into nuclear RNA foci, which recruit and consequently alter the localization and function of specific RNA binding proteins, ultimately resulting in multiple RNA processing defects that are responsible of the diverse clinical manifestations observed in these diseases (review in Wheeler and Thornton, 2007; Morriss and Cooper, 2017; Sznajder and Swanson, 2019). This RNA gain of function mechanism is best exemplified by the titration of the Muscleblind-like 1 and 2 (MBNL) RNA binding proteins into nuclear RNA foci constituted of CUG or CCUG RNA repeat expansions in myotonic dystrophy type 1 and 2 (DM1 and 2), as well as in Fuchs endothelial corneal dystrophy (FECD) (Miller et al., 2000; Fardaei et al., 2002; Wieben et al., 2017; Rong et al., 2019). These three disorders are caused by similar mutations, namely large expansions of CTG, CCTG, and CTG repeats embedded within the 3′UTR of DMPK or within introns of the CNBP and TCF4 genes, respectively, (Brook et al., 1992; Fu et al., 1992; Harley et al., 1992; Mahadevan et al., 1992; Liquori et al., 2001; Wieben et al., 2012; Mootha et al., 2014; Table 1).
Third, these repeat expansions can be translated into pathogenic proteins containing a stretch of repeated amino acids, resulting in a toxic gain of function, a loss of function, a dominant negative effect and/or a mix of these mechanisms for the protein hosting the expansion. Importantly, translation of these repeat expansions can occur through two main mechanisms. The first one is based on classical translation initiation at a canonical AUG, or alternatively at a near-cognate (CUG, GUG, UUG, or ACG), start codon, and thus results in expression of a pathogenic protein encoded by one predominant coding frame. The second mechanism, named repeat-associated non-AUG (RAN) translation, uses unconventional translation initiation that starts directly within the repeat expansion, resulting in expression of diverse proteins encoded in the three possible frames (Zu et al., 2011; review in Gao et al., 2017; Cleary et al., 2018; Nguyen et al., 2019). Archetype of this protein gain of function mechanism is the polyglutamine (polyQ) group of diseases, where expansions of CAG repeats, embedded within the main ORF of various genes (Huntingtin, Androgen receptor, Atrophin-1, ATXN1 to 3, 6, 7, and 17) (review in Lieberman et al., 2019), or alternatively within small ATG-driven ORFs located in ill-defined transcribed genetic regions (JPH3AS, ATXN8) (Moseley et al., 2006; Wilburn et al., 2011), are translated in polyglutamine-containing proteins that form inclusions and are toxic for neuronal cells, causing several neurodegenerative diseases with similar histopathological characteristic and related symptoms (Huntington’s disease and Huntington disease-like 2, spinal-bulbar muscular atrophy, dentatorubral-pallidoluysian atrophy, spinocerebellar ataxia 1–3, 6–8, and 17; Table 1). This example illustrates that repeat expansions with similar sequences, sizes, and genetic localizations can be pathogenic by a common molecular mechanism, ultimately resulting in a group of diseases with related clinical symptoms and remarkably similar histopathological features. In that aspect, progress in long-read and whole human genome sequencing has recently unveiled a dozen of novel pathogenic repeat expansions with striking similarities in sequence and localization and that can be divided in two groups (review in Ishiura and Tsuji, 2020). First, six identical expansions of ATTTC repeats that are all localized within introns, but of various gene, are responsible of familial adult myoclonic epilepsy (FAME) 1–4, 6, and 7 (Ishiura et al., 2018; Corbett et al., 2019; Florian et al., 2019; Yeetong et al., 2019). Second and topic of this review, several identical CGG repeat expansions, embedded within the 5′UTR of different genes or in a long non coding RNA (lncRNA), were recently identified as the causes of fragile X-associated tremor/ataxia syndrome (FXTAS), neuronal intranuclear inclusion disease (NIID), oculopharyngodistal myopathy type 1 to 3 (OPDM), and oculopharyngeal myopathy with leukoencephalopathy (OPML) (Hagerman et al., 2001; Ishiura et al., 2019; Sone et al., 2019; Deng et al., 2019; Tian et al., 2019; Deng et al., 2020; Xi et al., 2021; Table 1 and Figure 1). In addition to their similar genetic cause, FXTAS, NIID, OPDM, and OPML present overlapping clinical manifestations and similar histopathological features, including the presence of characteristic eosinophilic ubiquitin-positive nuclear inclusions (NIs), suggesting that these diseases belong to a continuum of neuromuscular and neurodegenerative diseases potentially caused by a common molecular mechanism (Figure 1).
FIGURE 1.
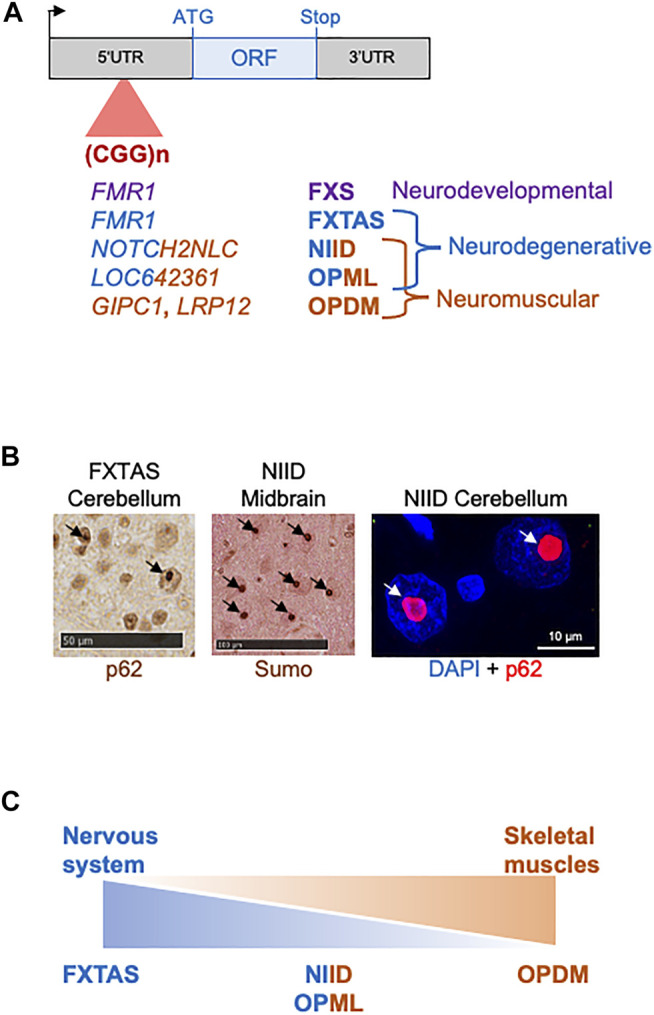
CGG repeat expansions cause a spectrum of disease. (A) Identical CGG repeat expansions embedded within the 5′UTR of different genes cause various neurodevelopmental, neuromuscular and neurodegenerative disorders. (B) Brain sections of individuals with FXTAS or NIID show identical p62-or sumo-positive intranuclear inclusions. (C) FXTAS, NIID, OPML, and OPDM may belong to a continuum of neuromuscular and neurodegenerative disorders.
Fragile X-Associated Tremor/Ataxia Syndrome
Fragile X-associated tremor/ataxia syndrome (FXTAS, OMIM: 300623) is a late-onset X-linked neurodegenerative disorder originally described in grandfathers of infants with fragile X (Hagerman et al., 2001). Due to its X-linkage, FXTAS mostly affect males, while females are protected from overt neurodegeneration by random X-chromosome inactivation of the mutant allele, but are nonetheless at risk of developing another disease, fragile X-associated primary ovarian insufficiency (FXPOI; Conway et al., 1998). FXTAS is characterized by variable progressive intention tremor, gait ataxia and dementia, frequently accompanied by progressive cognitive decline, parkinsonism, peripheral neuropathy, and autonomic dysfunctions (Jacquemont et al., 2003). Neuroradiological features of FXTAS include mild brain atrophy and white matter lesions with T2 hyperintensities in the middle cerebellar peduncle, as well as in the splenium of the corpus callosum, in the pons, insula and periventricular white matter. Furthermore, FXTAS is characterized by the presence of large eosinophilic ubiquitin-, p62-, and sumo-positive intranuclear inclusions in both neurons and astrocytes across the nervous system, as well as in non-nervous tissues (Greco et al., 2006; Hunsaker et al., 2011; Ariza et al., 2016). However, these inclusions proved to be negative for polyQ or any other common protein inclusion markers of neurodegenerative disorders (Synuclein, Tau, ß-amyloid, TDP43, and FUS, etc.) and they origin remained mysterious until recently (Todd et al., 2013).
FXTAS is caused by a limited expansion (called premutation) of 55–200 CGG repeats localized in the 5′UTR of the FMR1 gene (Hagerman et al., 2001). Interestingly, a longer expansion (>200 CGG repeats, named full mutation) causes the neurodevelopmental fragile X mental retardation syndrome, while typical individuals carry less than 45 CGG repeats, with 30 repeats being the most common allele. Importantly, the instable nature of this genetic mutation, notably repeat expansion during meiosis, explains the anticipation (also known as the Sherman paradox) observed in fragile X families where the clinical manifestations occur at progressively earlier age and/or with increasing severity in successive generations (Sherman et al., 1984; Sherman et al., 1985; Fu et al., 1991). At the molecular level, in fragile X syndrome, the full mutation promotes DNA epigenetic changes leading to transcriptional silencing of the FMR1 promoter and thus, loss of expression of the FMR1-encoded protein, FMRP, ultimately resulting in autism and intellectual disability in male. In female, FMRP haploinsufficiency is less marked due to expression by the second unmethylated FMR1 allele. In contrast, premutation expansions (55–200 CGG) enhance transcription, resulting in 2 to 8-fold increased levels of FMR1 mRNA in individuals with FXTAS (Tassone et al., 2000; Kenneson et al., 2001; Tassone et al., 2007). In spite of this, FMRP expression are often decreased or at near-normal level due to the reduced translation efficiency of transcripts carrying elongated CGG repeats (Feng et al., 1995). Importantly, the sole expression of expanded CGG repeats, embedded in the 5′UTR of FMR1, in cell or animal models is sufficient to cause neuronal cell dysfunctions and to induce formation of the typical FXTAS intranuclear inclusions (Jin et al., 2003; Willemsen et al., 2003; Arocena et al., 2005; Entezam et al., 2007; Hashem et al., 2009; Todd et al., 2013; Hukema et al., 2015). These pioneering studies indicate that FXTAS, in contrary to the fragile X syndrome, is not due to a reduce expression of FMRP and to a loss of function mechanism, but is more likely caused by an RNA and/or protein gain of function mechanism.
About a toxic RNA gain of function mechanism in FXTAS, in vitro assays revealed that multiple RNA binding proteins bind to CGG RNA repeats, including Pura, hnRNP A2, SAM68, CUGBP1, TDP43, and Drosha/DGCR8, among others (Iwahashi et al., 2006; Jin et al., 2007; Sellier et al., 2010; Sofola et al., 2007; Sellier et al., 2013; Cid-Samper et al., 2018). However, whether CGG expansions are toxic at the RNA level in FXTAS is unclear. It notably remains to clarify whether these CGG RNA repeats deplete a sufficient amount of these RNA binding proteins, so that they loss their mobility and functions below a pathological threshold. As a further note of caution, many studies, including ours, were performed with CGG repeats deleted of any natural FMR1 sequence and that consequently accumulate artificially in nuclei in some cell models, potentially titrating RNA binding proteins in nuclear RNA foci (Sellier et al., 2010). However, expanded CGG repeats embedded in their natural FMR1 5′UTR are exported into the cytoplasm and thus may not accumulate sufficiently into the nucleus to drive a nuclear RNA gain of function toxicity mechanism (Sellier et al., 2017). In that aspect, it remains to test whether exported CGG RNA repeats may accumulate into cytoplasmic RNA granules and titrate specific RNA binding proteins. Interestingly and notwithstanding their level of titration, these RNA binding proteins may also chaperone CGG RNA repeats to modulate their stability, localization and translation, resulting in diminished toxicity, thus illuminating a potential therapeutic strategy for FXTAS (Jin et al., 2007; Qurashi et al., 2011; He et al., 2014; Malik et al., 2021b).
Concerning a pathogenic protein gain of function in FXTAS, studies of cell and animal models coupled to mass spectrometry analyses revealed that the FMR1 CGG expansion is translated into a novel protein, where each GGC triplet encodes for a glycine, resulting in a small polyglycine-containing protein (polyG), which was named FMRpolyG (Todd et al., 2013). Of interest, translation of the FMR1 CGG repeats occurs in one principal frame (glycine) among the three possible (alanine, glycine and arginine), but in absence of any ATG start codon. Instead, translation initiation takes place at an ACG near-cognate codon located 36 nucleotides upstream of the CGG repeats and in the glycine frame (Kearse et al., 2016; Sellier et al., 2017, Figure 2). Near-cognate initiation codons are codons differing from the cognate AUG start codon by one nucleotide, but that can still initiate translation through mispairing with the initiator methionine-tRNA. In vitro experiments and large-scale ribosome profiling revealed that predominantly four near-cognate start codons (CUG, GUG, UUG, and ACG) are tolerated and can initiate translation, despite fidelity control, notably by the eIF1 protein (Clements et al., 1988; Kozak, 1989; Peabody, 1989, Ingolia et al., 2011; Lee et al., 2012; review in Kearse and wilusz, 2017). However, translation initiation at near-cognate codons is 5 to 10-fold less efficient compared to initiation at an AUG start codon, and is decisively dependent of surrounding secondary RNA structures and sequences, notably the Kozak sequence (G/ACCAUGG) that borders the start codon (Kozak, 1981; Kozak, 1990). Further experiments revealed that FMRpolyG translation follows a classical m7G cap-dependent ribosome scanning mechanism, and that the CGG repeat expansion is located within a small upstream ORF overlapping and negatively-regulating translation of the main FMR1 ORF, and FMRP (Kearse et al., 2016; Sellier et al., 2017; Rodriguez et al., 2020; Figure 2). Upstream ORFs (uORFs) are short open reading frames located upstream of the main protein-encoding ORF, and are thus embedded in the 5′UTR sequence of gene. Whole genome analyses revealed that most mammalian genes contain uORFs, with their majority (>75%) initiating at near-cognate codons (Ingolia et al., 2011; Lee et al., 2012; Fields et al., 2015; Johnstone et al., 2016). As a consequence of the 5′ to 3′ ribosome scanning mechanism, a ribosome translating an uORF and dissociating at its stop codon may not re-initiate to a nearby downstream ORF, even initiated by a canonical AUG start codon. Thus, presence of upstream ORFs may negatively regulate, yet not systematically, translation of downstream ORFs. However, due to the low efficiency of translation initiation at near-cognate start codons, ribosome may bypass an uORF and instead initiates at the main downstream ORF start codon, a process known as leaky scanning translation (review in Hinnebusch et al., 2016; Kearse and Wilusz, 2017; Gao et al., 2017). As uORFs act predominantly as translation negative cis-regulatory RNA elements, they are rarely expressing functional proteins and the vast majority of uORFs encode for small and instable peptides that are hardly detectable. Consistent with this, in control condition with no repeat expansions (∼30 CGG) the FMR1 uORF encodes for a small protein of 83 amino acids (∼7 kDa), hardly detectable without inhibition of the proteasomal degradation pathway (Sellier et al., 2017). In contrast, expansions over 60–70 CGG repeats in FXTAS lengthen this uORF, resulting in expression of a protein with a polyglycine stretch, FMRpolyG, which is stable and readily detectable (Todd et al., 2013; Kearse et al., 2016; Sellier et al., 2017, Figure 2). Of interest, the FMRpolyG protein with an expansion over ∼30 glycine repeats is prone to aggregation and expression of FMRpolyG in various cell and animal models leads to the formation of cytoplasmic and intranuclear aggregates, positives for ubiquitin and p62 (Todd et al., 2013; Hukema et al., 2015; Sellier et al., 2017; Derbis et al., 2018; Hoem et al., 2019; Wenzel et al., 2019). Importantly, various mouse or rabbit antibodies independently developed and directed against FMRpolyG revealed presence of this protein within the typical eosinophilic ubiquitin-positive intranuclear inclusions in cell and tissue sections of individuals with FXTAS (Todd et al., 2013; Buijsen et al., 2014; Sellier et al., 2017; Bonapace et al., 2019; Krans et al., 2019; Dijkstra et al., 2021). Of technical interest, these antibodies are directed against the N- or C-terminal parts of FMRpolyG, but none are directly targeting the polyglycine stretch. However, recent mass-spectrometry analyses of FXTAS brain samples revealed only trace amount of FMRpolyG, casting important questions on the expression level of this protein in individuals with FXTAS (Ma et al., 2019; Holm et al., 2021). Such low levels of FMRpolyG found in proteomic analyses may originate from the high propensity of this protein to aggregates (Sellier et al., 2017), so that little FMRpolyG is present in brain extract or in fluids of individuals with FXTAS. Overall, these proteomic studies have important clinical consequences as they temper relevance of FMRpolyG as an easily quantifiable biomarker for FXTAS (Ma et al., 2019; Holm et al., 2021). Finally, expression of FMRpolyG in cell and animal models is toxic, and Drosophila or mice expressing FMRpolyG show locomotor alterations, neuronal cell loss and ultimately, premature death of these animals (Todd et al., 2013; Sellier et al., 2017). However, the mechanism of FMRpolyG toxicity is currently unclear (review in Boivin et al., 2018; Glineburg et al., 2018), with proposed alteration of the proteasome and degradation pathway (Oh et al., 2015), of the nuclear architecture and nucleocytoplasmic transport (Sellier et al., 2017), of mitochondrial functions (Gohel et al., 2019) and/or through binding to FMR1 CGG RNA repeats (Asamitsu et al., 2021). Furthermore, it is unclear whether FMRpolyG toxicity is related to its propensity to form large cellular aggregates and/or to its localization and notably whether its import into nuclei is required for its pathogenicity. Similarly, whether FMRpolyG toxicity is mainly triggered by its polyglycine stretch, or whether bordering N- or C-terminal amino acid sequences contribute to FMRpolyG pathogenicity is currently ill-defined (Sellier et al., 2017).
FIGURE 2.

CGG repeat expansions are translated into polyG proteins in FXTAS and NIID. (A) Scheme of FMR1 indicating FMRpolyG upstream ORF and FMRP main ORF localization. (B) Scheme of NOTCH2NLC indicating uN2CpolyG upstream ORF and NOTCH2NLC (abbreviated N2C) main ORF localization. Initiation codons and stop codons are indicated in red for uORFs and in blue for the main ORFs. Near-cognate initiations codons are indicated in bold yellow. (C) Protein sequences of FMRpolyG and uN2CpolyG show no similitude beyond their polyglycine stretch. (D) Sequences of the putative uORFs embedded within NOTCH2, NOTCHNLA, B and C 5′UTRs. Variant amino acids are indicated in bold. The sequence required for uN2C to interact with KU70/KU80 is indicated in blue.
In parallel to the translation initiation of a CGG repeats-containing uORF at a near-cognate start codon resulting in expression of the toxic FMRpolyG protein, RAN translation where initiation occurs directly within the CGG expansion and in all three frames was also proposed as a leading pathogenic mechanism in FXTAS (Todd et al., 2013). While modest to no translation of CGG repeats was found in the arginine frame, which is the frame of the downstream FMRP ORF, assays using highly-sensitive nanoluciferase detection methods revealed some translation of the expanded CGG repeats into the alanine frame, resulting in expression of a novel polyalanine containing protein that was named FMRpolyA (Todd et al., 2013; Oh et al., 2015; Kearse et al., 2016). However, independently developed antibodies directed against FMRpolyA failed to detect significant accumulation of this protein in cells and tissues of individuals with FXTAS, suggesting that FMRpolyA is not a major component of the intranuclear inclusions typical of this disease (Sellier et al., 2017; Krans et al., 2019). Furthermore, a mouse model expressing an expansion of CGG repeats but deleted of the FMR1 5′UTR sequence that contains the near-cognate start codon initiating FMRpolyG translation shows no inclusions and no locomotor or neurodegenerative phenotype. In contrast, mice expressing the same CGG expansion, but with its upstream natural FMR1 near-cognate codon, show numerous FMRpolyG inclusions and neurodegeneration (Sellier et al., 2017). These animal models suggest that RAN translation initiating inside the CGG repeat track is not sufficiently efficient to drive inclusion formation and neuronal cell death, at least in the limited time frame of mouse longevity. Similarly, these models suggest that the sole expression of CGG repeats at the RNA level, and thus an RNA gain of function mechanism, is not sufficient to drive neuropathogenicity in these animals.
Overall, finding that CGG repeats are translated into a novel and toxic polyglycine-containing protein clarifies the origin of the intranuclear inclusions and the pathogenic mechanism at play in FXTAS. Furthermore, these results support a novel model of pathogenicity in human diseases, where repeat expansions located in ill-defined upstream ORFs can be decoded into novel and toxic proteins through translation initiation at near-cognate start codons. Interestingly, a similar mechanism was recently identified in amyotrophic lateral sclerosis and frontotemporal dementia, where an expansion of G4C2 repeats located in the first intron of the C9ORF72 gene is translated into a polyglycine-alanine dipeptide repeat (polyGA DPR) protein through translation initiation at a CUG near-cognate start codon located 24 nucleotides upstream of the repeats (Green et al., 2017; Tabet et al., 2018; Sonobe et al., 2018; Almeida et al., 2019; Boivin et al., 2020; Sonobe et al., 2021; Figure 3). It remains to determine whether these examples of translation initiation at near-cognate start codons are two isolated cases or reflect a more global mechanism of disease.
FIGURE 3.

Repeat expansions located in “non-coding” regions are nonetheless translated. (A) CGG repeat expansions embedded in FMR1 and NOTCH2NLC 5′UTR are translated into polyglycine-containing proteins in NIID and FXTAS, respectively. (B) G3C2T and G4C2 repeats embedded in the first unspliced intron of the NOP56 and C9ORF72 genes are translated into poly(glycine-proline) and poly(glycine-alanine)-containing proteins in SCA36 and ALS/FTD, respectively. (C) CAG repeats and G2C4 repeats embedded in ATXN8, JPH3AS and C9ORF72AS long “non-coding” RNAs are translated into polyglutamine- or poly(glycine-proline)-containing proteins in SCA8, HDL2, and ALS/FTD, respectively. Initiation codons and stop codons of the ORFs containing the pathogenic expansion are indicated in red, while they are indicated in blue for the main ORFs. Near-cognate initiations codons are indicated in bold yellow.
Neuronal Intranuclear Inclusion Disease
Neuronal intranuclear inclusion disease (NIID, OMIM: 603472), also known as intranuclear inclusion body disease (INIBD) or neuronal intranuclear hyaline inclusion disease (NIHID), is a rare autosomal dominant genetic disorder with variable age of onset, from infant to late adult (Lindenberg et al., 1968; Michaud and Gilbert, 1981; Munoz-Garcia and Ludwin, 1986; Takahashi-Fujigasaki, 2003; Sone et al., 2016). The clinical manifestations of NIID are highly heterogenous and generally comprise variable skeletal muscle weakness associated with variable dysfunctions of the central and peripheral nervous systems, including cerebellar ataxia, parkinsonism, cognitive decline, peripheral neuropathy, and autonomic dysfunctions (Takahashi-Fujigasaki, 2003; Sone et al., 2016; Takahashi-Fujigasaki et al., 2016). However, atypical NIID clinical manifestations are increasingly described, including leukoencephalopathy, essential tremor, multiple system atrophy, retinal changes and retinopathy, amyotrophic lateral sclerosis, Alzheimer, or Parkinson disease (Okubo et al., 2019; Fang et al., 2020; Ma et al., 2020; Nakamura et al., 2020; Ng et al., 2020a; Yuan et al., 2020; Shi et al., 2021; Yang et al., 2022). Furthermore, there is increasing report of various acute symptoms of the central nervous system associated with NIID, including stroke-like episodes, migraine-like attack, epileptic seizures and/or encephalitic episodes; as well as evidences of clinical manifestations from other systems (Chen H. et al., 2020). Overall, these recent clinical observations suggest that NIID covers a much larger-than-previously-thought spectrum of clinical manifestations potentially extending to almost all systems (review in Cao Y. et al., 2021; Cao L. et al., 2021; Fan et al., 2022).
As its name imply, NIID is characterized by the presence of large eosinophilic ubiquitin-, p62-, and sumo-positive intranuclear inclusions. These aggregates are present in both neurons and glial cells in the central and peripheral nervous systems, as well as in various other tissues, including the skin, which observation can be determinant to confirm diagnosis of this multifaced syndrome (Patel et al., 1985; Kimber et al., 1998; Pountney et al., 2003; Liu et al., 2008; Sone et al., 2011; Mori et al., 2012; Chen H. et al., 2020; Zhang et al., 2021). Importantly, these intranuclear inclusions are virtually identical and indistinguishable to those observed in FXTAS (Gelpi et al., 2017; Lim et al., 2020; Toko et al., 2021). Similarly, brain MRI of individuals with NIID reveal abnormalities reminiscent of FXTAS with mild brain atrophy and white matter lesions, including T2-weighted hyperintensities in the middle cerebellar peduncle and high-intensity signals in the corticomedullary junction (Gelpi et al., 2017; Sugiyama et al., 2017; Ng et al., 2020b).
The genetic cause of NIID was recently identified as a limited expansion of ∼60 to 200–300 CGG repeats in the 5′UTR of the NOTCH2NLC gene (Sone et al., 2019; Ishiura et al., 2019; Deng et al., 2019; Tian et al., 2019). Of interest, an identical expansion of CGG repeats in NOTCH2NLC was also recently identified as the genetic cause of oculopharyngodistal myopathy type 3 (OPDM3) with variable neurological manifestations (Ogasawara et al., 2020; Yu et al., 2021). These studies highlight the variability of clinical manifestations and the ensuing complexity to diagnose these syndromes, as well as the potential overlap between OPDM and NIID. Furthermore, NOTCH2NLC CGG expansion is mostly reported in individuals of Asian origin, but rarely observed in European NIID cases, despite clinical similitudes and the presence of identical intranuclear inclusions (Chen Z. et al., 2020), suggesting a founder effect for this mutation, but also that NIID is genetically heterogenous with other mutations awaiting to be identified. The Notch 2 N-terminal like (NOTCH2NL) A, B and C genes are hominid-specific genes located on chromosome 1q21.1, which result from the duplication of the promoter region and N-terminal part (exons 1–5) of Notch2. As a consequence of this partial duplication, the NOTCH2NL proteins contain six epidermal growth factor (EGF)-like domains, but are deleted of the middle transmembrane and C-terminal cytoplasmic domains of Notch2. Moreover, these proteins also lack the N-terminal Notch2 peptide signal due to a modified ATG start site localization in NOTCH2NLA and C, and an intronic nucleotide variation changing NOTCH2NLB intron 1 splice acceptor by 8 nucleotides and thus modifying NOTCH2NLB ATG start site accordingly. Important to hominid brain size evolution, NOTCH2NL proteins regulate Notch signaling and expand human cortical progenitors. Furthermore, 1q21.1 genomic deletions or duplications potentially encompassing the NOTCH2NLA and B genes are associated with neurodevelopmental syndromes with microcephaly or macrocephaly, respectively, (Fiddes et al., 2018; Suzuki et al., 2018). These seminal studies highlight the importance of the NOTCH2NL proteins for neuronal progenitor proliferation and brain size expansion in human evolution, but question how a CGG repeat expansion embedded within the 5′UTR of NOTCHNLC can be pathogenic. First, a loss of function of the NOTCH2NLC (abbreviated N2C) protein is unlikely as the CGG repeats are located 140 nucleotides ahead of the ATG start site of this protein, and as NOTCH2NLC mRNA levels are found unaltered, or even increased, in blood, brain, fibroblasts and muscle samples of individuals with NIID or OPDM3 (Sone et al., 2019; Ishiura et al., 2019; Tian et al., 2019; Yu et al., 2021). Moreover, the NOTCHNLC protein is most likely expressed at very low level due to the presence of six ATG-driven upstream ORFs inhibiting its translation initiation, questioning the physiological importance of this protein (Zhong et al., 2021). Second, an RNA gain of function mechanism has been considered for NIID, with some evidences of CGG RNA accumulation in RNA foci, but with unclear pathological consequences (Yu et al., 2021; Deng et al., 2021). Third, translation of these CGG repeats into a potentially toxic protein has been recently observed in two independent reports (Boivin et al., 2021; Zhong et al., 2021; Figure 2). Importantly, these studies revealed that the NOTCH2NLC CGG repeats are embedded into a small upstream ORF, which is translated from a canonical ATG start site located 15 nucleotides upstream of the CGG repeats. This uORF was hence named uN2C for upstream of the NOTCH2NLC ORF. As a results of this ATG translation initiation, each GGC triplet encodes for a glycine, resulting in expression of a polyglycine-containing protein, which was named either uN2CpolyG (Boivin et al., 2021) or N2NLCpolyG (Zhong et al., 2021). Despite different names and independent identification, these two proteins are strictly identical (Figure 2). As with the majority of upstream ORFs, in control condition the N2C uORF with no expansion (∼12 CGG) encodes for a small protein (56 amino acids, ∼6 kDa), which is unstable and hardly detectable without inhibiting degradation pathways (Boivin et al., 2021). In contrast, CGG repeat expansion increases the length of this uORF, resulting in expression of a polyglycine-containing protein that is now stable and detectable. Importantly, mouse monoclonal antibodies directed against two different amino acid sequences, both located in the C-terminal part, of uN2CpolyG revealed presence of this protein in the typical ubiquitin- and p62-positive intranuclear inclusions in skin and brain sections of individuals with NIID (Boivin et al., 2021). These results were recently confirmed using an independently-developed rabbit polyclonal antibody directed against a stretch of 12 glycine followed by 11 amino acids from the uN2C sequence (Zhong et al., 2021). Furthermore, expression of this polyG protein in cell and animal models drives the formation of cytoplasmic and intranuclear inclusions, positive for ubiquitin, sumo and p62. Finally, expression of uN2CpolyG in mice is toxic, resulting in locomotor alterations, neuronal cell loss and reduced lifespan (Boivin et al., 2021). However, by which mechanism this polyG protein is toxic is unclear. Immunoprecipitation followed by mass spectrometry analyze indicate that the normal uN2C protein with a normal stretch of polyglycine (∼12 repeats) interacts with the Ku70 and Ku80 proteins, which form a scaffold for the non-homologous end joining (NHEJ) repair of DNA double-strand breaks. Interestingly, expression of uN2C (with a control number of CGG repeats) boosts the repair of DNA double strand breaks (Boivin et al., 2021). These results indicate that the small uN2C protein is potentially a novel regulator of DNA damage response, suggesting that the NOTCH2NL genes may encode different proteins with complementary functions for human brain development. In this model, the NOTCH2NLB gene, encoding the NOTCH2NLB protein, would regulate Notch signaling to promote extensive neural progenitor proliferation (Fiddes et al., 2018; Suzuki et al., 2018), but at the cost of potentially generating multiple DNA damages. In contrast, the NOTCH2NLC gene contains six ATG-driven upstream ORFs that reduce expression of the NOTCH2NLC protein (Zhong et al., 2021), but the first of these uORFs encodes for a small uN2C protein that may protect neuronal cells from DNA damages by stimulating the NHEJ repair mechanism. Of interest, NOTCH2, NOTCH2NLA and B also contains 1, 5 and 2 potential ATG-driven uORFs, respectively, but with protein sequences different from uN2C and are thus unable to interacts with the Ku70 and 80 proteins (Boivin et al., 2021; Figure 2). Note that alike NOTCH2NLC, NOTCH2NLA contains multiple ATG-driven uORFs, which may silence expression of the downstream NOTCH2NLA ORF, questioning the expression and relevance of the NOTCH2NLA and NOTCH2NLC proteins in physiological conditions.
In NIID, the polyglycine expansion alters the interaction of uN2C with the Ku proteins and reduces its DNA repair activity. However, this reduced function is likely compensated by the second NOTCH2NLC allele and is not sufficient to drive overt DNA repair alterations, as no clear evidence of DNA damage accumulation was observed in mice expressing the uN2CpolyG protein or in brain sections of NIID individuals (Boivin et al., 2021). Interestingly, expression of this polyG protein promotes alterations of the nucleocytoplasmic transport (Zhong et al., 2021), which is reminiscent of the pathogenicity described for FMRpolyG in FXTAS (Sellier et al., 2017), as well as for DPR proteins expressed from the G4C2 repeats in the C9ORF72 gene in ALS/FTD (Jovičić et al., 2015; Hayes et al., 2020). However, it remains to investigate by what molecular mechanisms the uN2CpolyG and FMRpolyG proteins may dysregulate nucleocytoplasmic traffic. It also remains to determine whether RAN translation of the NOTCH2NLC repeats, with initiation within the CGG expansion and protein expression in all three frames, may occur and contribute to the pathogenicity in NIID.
Overall, these studies suggest the existence of a novel class of human genetic disorder, the polyG diseases, where expanded CGG repeats are embedded in small upstream ORFs and consequently, are translated into novel polyglycine-containing proteins that are toxic and forms ubiquitin-positive inclusions (Todd et al., 2013; Sellier et al., 2017; Boivin et al., 2021; Zhong et al., 2021). It remains to determine whether this group of disease is limited to NIID and FXTAS, or whether it may include other pathologies with related symptoms and/or similar histopathological features. Candidate pathologies may include mutations, yet to be identified, in NIID cases negative for the NOTCH2NLC mutation, but that nevertheless present the typical clinical and histopathological features of that syndrome (Chen Z. et al., 2020). Similarly, it remains to explore whether translation of expanded CGG repeats, yet to be identified, may underlie the presence of intranuclear inclusions of unknown origins (review in JM Woulfe, 2007), notably Marinesco bodies, which are eosinophilic ubiquitin-positive nuclear inclusions found in pigmented neurons of the substantia nigra and locus ceruleus in aged human brain (Yuen and Baxter 1963; Odagiri et al., 2012). Finally, the recent identification of CGG repeat expansions in the OPDM and OPML neuromuscular disorders, which are characterized by the presence of eosinophilic ubiquitin-positive intranuclear inclusions, may potentially represent novel examples of polyG diseases.
Oculopharyngodistal Myopathies
Oculopharyngodistal myopathy (OPDM, OMIM: 164310) is a rare adult-onset and slowly progressive autosomal dominant disease characterized by ptosis, external ophthalmoplegia, dysphagia and dysarthria, associated to facial and distal limb muscle weakness (Satoyoshi and Kinoshita 1977; Uyama et al., 1998; Minami et al., 2001; van der Sluijs et al., 2004; Lu et al., 2008; Durmus et al., 2011; Zhao et al., 2015). Serum creatine kinase levels are usually normal or mildly increased in individuals with OPDM, and skeletal muscles shows moderate fibrosis and small angular fibers. Besides these non-specific myopathic changes, OPDM histopathology is characterized by the presence of cytoplasmic rimmed vacuoles (RVs) and typical eosinophilic nuclear inclusions (NIs), which are both p62-and ubiquitin-positive (Zhao et al., 2015; Deng et al., 2020; Saito et al., 2020; Kumutpongpanich et al., 2021; Matsubara et al., 2021; Xi et al., 2021). Of interest, these intranuclear inclusions are also observed in skin sections of individuals with OPDM cases (Ogasawara et al., 2021), and are reminiscent of the typical inclusions observed in FXTAS and NIID. Importantly, the mutations causing OPDM were recently identified as similar expansions of ∼70 to 200–300 CGG repeats located within the 5′UTR of two different genes, LRP12 and GIPC1 (Ishiura et al., 2019; Deng et al., 2020; Kumutpongpanich et al., 2021; Xi et al., 2021). Furthermore, an expansion of CGG repeats in the 5′UTR of the NOTCH2NLC gene was identified in OPDM cases with variable neurological manifestations (Ogasawara et al., 2020; Yu et al., 2021), and an expansion of CGG repeats within the 5′UTR of the RILPL1 gene was recently reported in medRxiv in affected individuals of a large Chinese family with OPDM (Yang et al., 2021). Consequently, OPDM is now distinguished by its genetic cause with oculopharyngodistal myopathy type 1 (OPDM1) caused by a CGG repeat expansion in LRP12, OPDM type 2 is caused by a CGG expansion in GIPC1, OPDM3/NIID1 is caused by a CGG expansion in NOTCH2NLC and OPDM4 is potentially associated with a CGG expansion in RILPL1. There is probably a founder effect for these mutations as the CGG expansion in LRP12 is more prevalent in Japan compared to China, while an inverse distribution is observed for the CGG expansion in GIPC1.
The mechanism of toxicity in OPDM is yet to be identified but a loss of function is unlikely as the CGG repeat expansions are located ahead of the ATG start sites of the GIPC1, LRP12, NOTCH2NLC, and RILPL1 proteins. Furthermore, expression of the GIPC1 protein is unaltered in tissue samples from individuals with OPDM2 (Deng et al., 2020). A mechanism of CGG RNA gain of function and/or of translation into pathogenic proteins remains to be explored, but it is noteworthy that oculopharyngeal muscular dystrophy, a neuromuscular disease with clinical manifestations related to OPDM, is also caused by a GC-rich repeat expansion. Oculopharyngeal muscular dystrophy (OPMD, OMIM: 164300) is a late-onset and slowly progressive autosomal dominant neuromuscular disease characterized by proximal muscle weakness, ptosis, and swallowing difficulty. OPMD is caused by an extension of 1–8 repeats of a GCG trinucleotide stretch located in PABPN1 gene, encoding the polyadenylate-binding protein 2 (PABP2). The N-terminal part of this protein contains a short polyalanine tract (Met-(Ala)10x-Gly-(Ala)2x … ), and in OPMD a small GCG expansion adding 1 to 8 alanine, or a missense mutation of the +12 glycine into an alanine resulting in a contiguous stretch of 13 alanine, leads to a modified PABP2 protein that interfere with the cellular traffic of polyadenylated RNA (Brais et al., 1998; Robinson et al., 2006). This is reminiscent of congenital polyalanine diseases, which are caused by small GCN repeat expansions encoding polyalanine tracts located within eight different transcription factors (Table 1). These elongated polyalanine stretches of relatively small size (<35 repeats) are proposed to cause loss of function and/or dominant negative effect on the transcriptional activities of these factors, ultimately leading to developmental disorders (review in Albrecht and Mundlos, 2005; Shoubridge and Gecz, 2012). Of interest, similar congenital aetiologies are observed in individuals with deletion and/or loss of function mutations in the genes coding for these transcription factors, strengthening the hypothesis that these short polyalanine tracts are pathogenic through a loss of function mechanism. In contrast, the CGG repeat expansions in OPDM1 to 4 are longer and localized in the 5′UTR, and not in the coding sequences of the GIPC1, LRP12, NOTCH2NLC, and RILPL1 proteins. Thus, expansions of ∼70 to 200–300 CGG repeats in OPDM are likely to be pathogenic by a different mechanism than the small (11–18) polyalanine tract that alters PABP2 protein functions in OPMD.
Oculopharyngodistal Myopathy With Leukoencephalopathy: The Missing Piece of a Disease Continuum?
Overall, it is striking to note that the OPDM, FXTAS, and NIID diseases share overlapping clinical manifestations and identical histopathological features, with the presence of characteristic eosinophilic ubiquitin-positive nuclear inclusions (NIs). Furthermore, these diseases are caused by similar genetic causes, namely microsatellite expansions of identical sequence and size (60–70 to 200–300 CGG repeats). These data suggest that these disorders may belong to a continuum of neuromuscular and neurodegenerative diseases (Figure 1). Importantly, this hypothesis is reinforced by the recent discovery that both NIID and OPDM3 can be caused by an identical CGG repeat expansion in NOTCH2NLC (Sone et al., 2019; Ishiura et al., 2019; Deng et al., 2019; Ogasawara et al., 2020; Yu et al., 2021), as well as by the identification of a novel and rare clinical entity with clinical features of both NIID and OPDM, and that was consequently named oculopharyngodistal myopathy with leukoencephalopathy (OPML, OMIM: 618637) (Ishiura et al., 2019). OPML is characterized by muscle dystrophy and weakness, especially of the ocular, pharyngeal, facial, and distal limb muscles, associated with leukoencephalopathy, brain atrophy, and T2 hyperintensity signals in the white matter and sign of tremor and ataxia in some individuals. Interestingly, OPML is caused by an expansion of CGG repeats located within the ill-defined LOC642361/NUTM2B-AS1 genetic region, which is transcribed but predicted as non-coding (Ishiura et al., 2019). Thus OPML, alike FXTAS, OPDM, and NIID, is caused by a CGG repeat expansion embedded in a genetic region predicted as non-coding, questioning the pathogenic mechanisms underlying these diseases. In that aspect, it was recently unveiled that CGG repeat expansions located in the 5′-untranslated regions of the FMR1 or NOTCH2NLC genes are nonetheless translated into toxic polyglycine-containing proteins through canonical translation initiation at canonical AUG, or near-cognate, start codons located upstream of the repeats (Todd et al., 2013; Sellier et al., 2017; Boivin et al., 2021; Zhong et al., 2021; Figure 3). Similarly, G4C2 and G3C2T repeat expansions are translated into toxic poly(glycine-alanine)- and poly(glycine-proline)-containing proteins in ALS/FTD and SCA36, despite these repeats being localized in the first intron of the C9ORF72 and NOP56 genes, respectively. These intronic regions are unspliced in patients and, alike in FXTAS and NIID, repeat expansions are translated through initiation at canonical AUG, or near-cognate, start codons located upstream of the repeats (Green et al., 2017; Tabet et al., 2018; Sonobe et al., 2018; Almeida et al., 2019; Boivin et al., 2020; McEachin et al., 2020; Sonobe et al., 2021; Figure 3). These results are reminiscent of pioneering studies showing that CAG expanded repeats located within the ATXN8 and JPH3AS RNAs, which were initially considered as long non-coding RNAs but were later on found to be carrying small ORFs, are translated through canonical ATG-initiated translation into toxic polyglutamine proteins in SCA8 and HDL2 (Moseley et al., 2006; Wilburn et al., 2011; Figure 3). Finally, it is consistently reported that various repeat expansions can be RAN translated into toxic proteins through initiation directly within the repeats and in all three frames (Zu et al., 2011; review in; Cleary et al., 2018; Nguyen et al., 2019). Thus, whether the CGG repeat expansions causing OPML and OPDM pathologies, which are located in genetic sequences predicted as non-coding, are nonetheless translated into potentially toxic proteins accumulating into ubiquitin-positive inclusions, is an exciting question that remains to be addressed.
Expansion Without Anticipation in NIID and OPDM?
In contrast to familial anticipation and correlation between expansion lengths and disease severity and/or age of onset observed in various microsatellite diseases (FXS, polyQ disorders, and myotonic dystrophy type 1, etc.), there are only few examples of anticipation reported in NIID and OPDM families, and a limited correlation between the size of CGG expansions and the age of onset in OPDM1 and OPDM2, with a r 2 of 0.188 and 0.158, respectively, (Kumutpongpanich et al., 2021; Xi et al., 2021). On the contrary, there are increasing reports of individuals carrying large CGG expansions (>200–300 repeats) in the NOTCH2NLC, GIPC1, and LRP12 genes and who are free of overt clinical manifestations (Deng et al., 2020; Ogasawara et al., 2020; Deng et al., 2021; Fukuda et al., 2021; Kumutpongpanich et al., 2021; Yu et al., 2021). Molecular analyses indicate that these individuals present DNA hypermethylation associated to promoter silencing and loss of expression of the allele carrying these large CGG expansions. These results have important consequences. First, they indicate that a partial reduction in expression and/or haploinsufficiency of the NOTCH2NLC, LRP12, and GIPC1 genes are not associated with overt clinical manifestations, dismissing the hypothesis of a loss of function mechanism in OPDM and NIID. Second, these data also provide some molecular bases to the complex pedigree observed in these diseases, with increasing reports of asymptomatic ascendants who nevertheless carry the causative mutation, but of longer size (over 200–300 CGG repeats) compared to their affected descendants who typically carry an allele with 60 to 200–300 CGG repeats. Interestingly, of the ∼10 currently reported examples of asymptomatic individuals with large expansions and who have transmitted shorter repeats to their NIID or OPDM-affected descendants, all were father to infant transmission cases, suggesting a bias of expansions contraction in male germinal cells (Deng et al., 2020; Deng et al., 2021; Fukuda et al., 2021; Kumutpongpanich et al., 2021; Yu et al., 2021). Of interest, a similar bias of transmission was observed in carriers of the FMR1 CGG repeat premutation where contraction occurred more often in paternal than in maternal transmission (Nolin et al., 2019). Moreover, it is noteworthy that a difference in parental gender transmission is observed in other microsatellite diseases, such as myotonic dystrophy type 1 where large expansions of CTG repeats occurs mainly through maternal transmission (review in Lanni and Pearson, 2019), while in Huntington disease, maternal transmission mainly results in CAG repeat contraction, whereas paternal transmission is associated with CAG repeat expansion (Aziz et al., 2011).
Overall, these studies suggest that in NIID and OPDM, the causing CGG expansions are pathogenic in a narrow range of size, with a lower limit of 60–70 repeats, below which they are likely not long enough to be expressed in stable and toxic proteins, and a “protective” threshold around 200 to 300 CGG repeats, over which carriers of large expansions benefit from the transcriptional silencing of the toxic allele (Figure 4). However, these intriguing observations are currently limited to a small number of clinical cases, and larger studies will be required to refine the correlation between CGG expansion sizes and age of onset and/or clinical severity in these disorders.
FIGURE 4.

Repeat expansion range of toxicity in different microsatellite diseases. CGG repeat expansions are pathogenic between ∼60–70 and 200–300 repeats when they are expressed into toxic polyglycine-containing proteins, but over 200 repeats when they promote DNA epigenetic changes, promoter silencing and a loss of function mechanism, such as in FXS. This range of protein toxicity is to be compared to other translated microsatellite diseases such as polyalanine-containing proteins that are generally toxic with 10–35 GCN repeats, while polyQ proteins are generally pathogenic with longer CAG expansions (∼40–80–200 repeats), with the exception of short polyglutamine stretch altering functions of the calcium voltage-gated channel subunit alpha1A (CACNA1A) in SCA6. In contrast, an RNA gain of function mechanism such as titration of the MBNL RNA binding proteins in DM1, require much longer repeat expansions.
Putative Mechanisms of polyG Proteins Toxicity
FXTAS and NIID are characterized by the translation of CGG repeats into pathogenic FMRpolyG and uN2CpolyG proteins, respectively. These two proteins form similar ubiquitin-positive inclusions, are toxic in cell and animal models and possess an identical polyglycine stretch, suggesting a common mechanism of toxicity. However, this raises numerous questions, notably it is unclear whether these polyG proteins are toxic under their aggregated or soluble forms. Similarly, it remains to explore whether their cellular localization, notably their import into the nucleus, is required for their pathogenicity. In that aspect, overexpression of FMRpolyG or uN2CpolyG in transformed cell lines mainly results in their accumulation in cytoplasmic aggregates, while these proteins are mainly found in intranuclear inclusions in patient tissues. Thus, it remains to clarify the mechanism responsible of polyglycine-containing proteins nuclear import in these diseases. Furthermore, FXTAS and NIID are neurodegenerative diseases, but FMR1 and NOTCHNLC expression are not restricted to neurons, and intranuclear inclusions are widely observed outside of the CNS in individuals with FXTAS and NIID (Greco et al., 2006; Hunsaker et al., 2011; Yamaguchi et al., 2018; Chen H. et al., 2020). However, presences of these inclusions are associated with only limited tissue dysfunctions and/or sub-clinical observations. In consequence, the mechanisms underlying the cellular specificity of FMRpolyG or uN2polyG toxicity remain to be determined. Finally, whether the pathogenicity of these proteins originates uniquely from their polyglycine stretches, or whether there is a toxic contribution of their bordering N- or C-terminal amino acid sequences is ill-defined. In that aspect, expression of a polyglycine protein deleted of any FMR1 or NOTCH2NLC bordering sequence is toxic in cell models, however not as much as the full-length FMRpolyG or uN2polyG proteins (Sellier et al., 2017; Boivin et al., 2021). Furthermore, C-terminal tagging of the FMRpolyG protein by the cherry fluorescent protein prevents its aggregation, while fusion of FMRpolyG with the GFP promotes its stability and increases its expression (Sellier et al., 2017; Derbis et al., 2018). These data suggest that the sequences bordering the polyglycine stretch of FMRpolyG or uN2polyG may contribute to their pathogenicity. However, this remains to be confirmed, notably in animal models.
Conclusion
Translation of CGG repeat expansions into simar polyglycine-containing (polyG) proteins in two neurodegenerative diseases with overlapping clinical manifestations and nearly identical histopathological features, FXTAS and NIID, suggest the existence of a novel class of human genetic disorders, the polyG diseases. This model is inspired by the translation into toxic polyalanine- or polyglutamine-containing proteins of GCN or CAG repeat expansions embedded in the ORFs of diverse genes, resulting in the polyAla or polyQ diseases, respectively. Furthermore, these data confirm that expanded repeats localized in human genomic regions predicted as “non-coding” can nevertheless be translated into novel pathogenic proteins, notably through classical initiation at canonical AUG, or near-cognate, start codons. In that aspect, the similarities of clinical manifestations and histopathological characteristic between FXTAS, NIID, OPDM, and OPML question whether CGG repeats also located in sequences predicted as non-coding, such as the LOC642361/NUTM2B-AS1 locus or the 5′UTR of the LRP12 and GIPC1 genes, are similarly translated into novel and potentially pathogenic polyG proteins. Importantly, a similar mechanism of toxicity for NIID, FXTAS and potentially other diseases, suggests that therapeutic strategies targeting the common CGG repeats/polyglycine-stretch could be of interest for the whole group of polyG diseases (review in Xu et al., 2021). In that aspect, constant efforts during the last decade to develop small pharmacological molecules specifically binding CGG RNA repeats in vitro, and that now efficiently decrease expression of the toxic FMRpolyG in cell and animal models provide exciting therapeutic hope for these disorders (Disney et al., 2012; Qurashi et al., 2012; Tran et al., 2014; Yang et al., 2016; Green et al., 2019; Verma et al., 2020; Asamitsu et al., 2021; Haify et al., 2021; Malik et al., 2021b; Konieczny et al., 2021). In parallel, major advances in antisense oligonucleotides (ASO) chemistry to reduce their toxicity and improve their biodistribution and cellular uptake open new hope to identify ASOs sequences targeting these CGG repeats to promote their RNA degradation and/or block their translation into toxic polyglycine-containing proteins, as recently reported in animal models of FXTAS (Rodriguez et al., 2020; Derbis et al., 2021).
Author Contributions
The manuscript was written and edited by MB and NC-B.
Funding
Our work is supported by the following grants: ERC-2012-StG 310659, ANR-18-CE16-0019, and FRM EQU202103012936 (NCB); ANR-10-LABX-0030-INRT and ANR-10-IDEX-0002-02 (IGBMC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- Akarsu A., Stoilov I., Yilmaz E., Sayli B. S., Sarfarazi M. (1996). Genomic Structure of HOXD13 Gene: a Nine Polyalanine Duplication Causes Synpolydactyly in Two Unrelated Families. Hum. Mol. Genet. 5 (7), 945–952. 10.1093/hmg/5.7.945 [DOI] [PubMed] [Google Scholar]
- Albrecht A., Mundlos S. (2005). The Other Trinucleotide Repeat: Polyalanine Expansion Disorders. Curr. Opin. Genet. Develop. 15 (3), 285–293. 10.1016/j.gde.2005.04.003 [DOI] [PubMed] [Google Scholar]
- Almeida S., Krishnan G., Rushe M., Gu Y., Kankel M. W., Gao F.-B. (2019). Production of Poly(GA) in C9ORF72 Patient Motor Neurons Derived from Induced Pluripotent Stem Cells. Acta Neuropathol. 138 (6), 1099–1101. 10.1007/s00401-019-02083-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiel J., Laudier B., Attié-Bitach T., Trang H., de Pontual L., Gener B., et al. (2003). Polyalanine Expansion and Frameshift Mutations of the Paired-like Homeobox Gene PHOX2B in Congenital central Hypoventilation Syndrome. Nat. Genet. 33 (4), 459–461. 10.1038/ng1130 [DOI] [PubMed] [Google Scholar]
- Ariza J., Rogers H., Monterrubio A., Reyes-Miranda A., Hagerman P. J., Martínez-Cerdeño V. (2016). A Majority of FXTAS Cases Present with Intranuclear Inclusions within Purkinje Cells. Cerebellum 15 (5), 546–551. 10.1007/s12311-016-0776-y [DOI] [PubMed] [Google Scholar]
- Arocena D. G., Iwahashi C. K., Won N., Beilina A., Ludwig A. L., Tassone F., et al. (2005). Induction of Inclusion Formation and Disruption of Lamin A/C Structure by Premutation CGG-Repeat RNA in Human Cultured Neural Cells. Hum. Mol. Genet. 14, 3661–3671. 10.1093/hmg/ddi394 [DOI] [PubMed] [Google Scholar]
- Asamitsu S., Yabuki Y., Ikenoshita S., Kawakubo K., Kawasaki M., Usuki S., et al. (2021). CGG Repeat RNA G-Quadruplexes Interact with FMRpolyG to Cause Neuronal Dysfunction in Fragile X-Related Tremor/ataxia Syndrome. Sci. Adv. 7 (3), eabd9440. 10.1126/sciadv.abd9440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz N. A., van Belzen M. J., Coops I. D., Belfroid R. D. M., Roos R. A. C. (2011). Parent-of-origin Differences of Mutant HTT CAG Repeat Instability in Huntington's Disease. Eur. J. Med. Genet. 54 (4), e413–e418. 10.1016/j.ejmg.2011.04.002 [DOI] [PubMed] [Google Scholar]
- Boivin M., Pfister V., Gaucherot A., Ruffenach F., Negroni L., Sellier C., et al. (2020). Reduced Autophagy upon C9ORF72 Loss Synergizes with Dipeptide Repeat Protein Toxicity in G4C2 Repeat Expansion Disorders. EMBO J. 39 (4), e100574. 10.15252/embj.2018100574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin M., Deng J., Pfister V., Grandgirard E., Oulad-Abdelghani M., Morlet B., et al. (2021). Translation of GGC Repeat Expansions into a Toxic Polyglycine Protein in NIID Defines a Novel Class of Human Genetic Disorders: The polyG Diseases. Neuron 109 (11), 1825–1835. e5. 10.1016/j.neuron.2021.03.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin M., Willemsen R., Hukema R. K., Sellier C. (2018). Potential Pathogenic Mechanisms Underlying Fragile X Tremor Ataxia Syndrome: RAN Translation And/or RNA Gain-Of-Function? Eur. J. Med. Genet. 61 (11), 674–679. 10.1016/j.ejmg.2017.11.001 [DOI] [PubMed] [Google Scholar]
- Bonapace G., Gullace R., Concolino D., Iannello G., Procopio R., Gagliardi M., et al. (2019). Intracellular FMRpolyG-Hsp70 Complex in Fibroblast Cells from a Patient Affected by Fragile X Tremor Ataxia Syndrome. Heliyon 5 (6), e01954. 10.1016/j.heliyon.2019.e01954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragg D. C., Mangkalaphiban K., Vaine C. A., Kulkarni N. J., Shin D., Yadav R., et al. (2017). Disease Onset in X-Linked Dystonia-Parkinsonism Correlates with Expansion of a Hexameric Repeat within an SVA Retrotransposon in TAF1. Proc. Natl. Acad. Sci. USA 114 (51), E11020–E11028. 10.1073/pnas.1712526114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brais B., Bouchard J. P., Xie Y. G., Rochefort D. L., Chrétien N., Tomé F. M., et al. (1998). Short GCG Expansions in the PABP2 Gene Cause Oculopharyngeal Muscular Dystrophy. Nat. Genet. 18 (2), 164–167. 10.1038/ng0298-164 [DOI] [PubMed] [Google Scholar]
- Brook J. D., McCurrach M. E., Harley H. G., Buckler A. J., Church D., Aburatani H., et al. (1992). Molecular Basis of Myotonic Dystrophy: Expansion of a Trinucleotide (CTG) Repeat at the 3' End of a Transcript Encoding a Protein Kinase Family Member. Cell. 68 (4), 799–808. 10.1016/0092-8674(92)90154-5 [DOI] [PubMed] [Google Scholar]
- Brown L. Y., Odent S., David V., Blayau M., Dubourg C., Apacik C., et al. (2001). Holoprosencephaly Due to Mutations in ZIC2: Alanine Tract Expansion Mutations May Be Caused by Parental Somatic Recombination. Hum. Mol. Genet. 10 (8), 791–796. 10.1093/hmg/10.8.791 [DOI] [PubMed] [Google Scholar]
- Buijsen R. A., Sellier C., Severijnen L. A., Oulad-Abdelghani M., Verhagen R. F., Berman R. F., et al. (2014). FMRpolyG-Positive Inclusions in CNS and Non-CNS Organs of a Fragile X Premutation Carrier with Fragile X-Associated Tremor/ataxia Syndrome. Acta Neuropathol. Commun. 2, 162. 10.1186/s40478-014-0162-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campuzano V., Montermini L., Moltò M. D., Pianese L., Cossée M., Cavalcanti F., et al. (1996). Friedreich's Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion. Science 271 (5254), 1423–1427. 10.1126/science.271.5254.1423 [DOI] [PubMed] [Google Scholar]
- Cao L., Yan Y., Zhao G. (2021). NOTCH2NLC-related Repeat Expansion Disorders: an Expanding Group of Neurodegenerative Disorders. Neurol. Sci. 42 (10), 4055–4062. 10.1007/s10072-021-05498-3 [DOI] [PubMed] [Google Scholar]
- Cao Y., Wu J., Yue Y., Zhang C., Liu S., Zhong P., et al. (2021). Expanding the Clinical Spectrum of Adult-Onset Neuronal Intranuclear Inclusion Disease. Acta Neurol. Belg. [Epub ahead of print]. 10.1007/s13760-021-01622-4 [DOI] [PubMed] [Google Scholar]
- Chen H., Lu L., Wang B., Cui G., Wang X., Wang Y., et al. (2020a). Re-defining the Clinicopathological Spectrum of Neuronal Intranuclear Inclusion Disease. Ann. Clin. Transl Neurol. 7 (10), 1930–1941. 10.1002/acn3.51189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Yan Yau W., Jaunmuktane Z., Tucci A., Sivakumar P., Gagliano Taliun S. A., et al. (2020b). Neuronal Intranuclear Inclusion Disease Is Genetically Heterogeneous. Ann. Clin. Transl Neurol. 7 (9), 1716–1725. 10.1002/acn3.51151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid-Samper F., Gelabert-Baldrich M., Lang B., Lorenzo-Gotor N., Ponti R. D., Severijnen L. W. F. M., et al. (2018). An Integrative Study of Protein-RNA Condensates Identifies Scaffolding RNAs and Reveals Players in Fragile X-Associated Tremor/Ataxia Syndrome. Cell Rep 25 (12), 3422–3434. 10.1016/j.celrep.2018.11.076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary J. D., Pattamatta A., Ranum L. P. W. (2018). Repeat-associated Non-ATG (RAN) Translation. J. Biol. Chem. 293 (42), 16127–16141. 10.1074/jbc.r118.003237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements J. M., Laz T. M., Sherman F. (1988). Efficiency of Translation Initiation by Non-AUG Codons in Saccharomyces cerevisiae . Mol. Cel Biol. 8 (10), 4533–4536. 10.1128/mcb.8.10.4533-4536.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway G. S., Payne N. N., Webb J., Murray A., Jacobs P. A. (1998). Fragile X Premutation Screening in Women with Premature Ovarian Failure. Hum. Reprod. 13, 1184–1187. 10.1093/humrep/13.5.1184 [DOI] [PubMed] [Google Scholar]
- Corbett M. A., Kroes T., Veneziano L., Bennett M. F., Florian R., Schneider A. L., et al. (2019). Intronic ATTTC Repeat Expansions in STARD7 in Familial Adult Myoclonic Epilepsy Linked to Chromosome 2. Nat. Commun. 10 (1), 4920. 10.1038/s41467-019-12671-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese A., Simone R., Sullivan R., Vandrovcova J., Tariq H., Yau W. Y., et al. (2019). Biallelic Expansion of an Intronic Repeat in RFC1 Is a Common Cause of Late-Onset Ataxia. Nat. Genet. 51 (4), 649–658. 10.1038/s41588-019-0372-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Baere E., Dixon M. J., Small K. W., Jabs E. W., Leroy B. P., Devriendt K., et al. (2001). Spectrum of FOXL2 Gene Mutations in Blepharophimosis-Ptosis-Epicanthus Inversus (BPES) Families Demonstrates a Genotype–Phenotype Correlation. Hum. Mol. Genet. 10 (15), 1591–1600. 10.1093/hmg/10.15.1591 [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., et al. (2011). Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 72 (2), 245–256. 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J., Gu M., Miao Y., Yao S., Zhu M., Fang P., et al. (2019). Long-read Sequencing Identified Repeat Expansions in the 5'UTR of the NOTCH2NLC Gene from Chinese Patients with Neuronal Intranuclear Inclusion Disease. J. Med. Genet. 56 (11), 758–764. 10.1136/jmedgenet-2019-106268 [DOI] [PubMed] [Google Scholar]
- Deng J., Yu J., Li P., Luan X., Cao L., Zhao J., et al. (2020). Expansion of GGC Repeat in GIPC1 Is Associated with Oculopharyngodistal Myopathy. Am. J. Hum. Genet. 106 (6), 793–804. 10.1016/j.ajhg.2020.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J., Zhou B., Yu J., Han X., Fu J., Li X., et al. (2021). Genetic Origin of Sporadic Cases and RNA Toxicity in Neuronal Intranuclear Inclusion Disease. J. Med. Genet. 2020, 107649. 10.1136/jmedgenet-2020-107649 [DOI] [PubMed] [Google Scholar]
- Depienne C., Mandel J. L. (2021). 30 Years of Repeat Expansion Disorders: What Have We Learned and what Are the Remaining Challenges? Am. J. Hum. Genet. 108 (5), 764–785. 10.1016/j.ajhg.2021.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbis M., Konieczny P., Walczak A., Sekrecki M., Sobczak K. (2018). Quantitative Evaluation of Toxic Polyglycine Biosynthesis and Aggregation in Cell Models Expressing Expanded CGG Repeats. Front. Genet. 9, 216. 10.3389/fgene.2018.00216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbis M., Kul E., Niewiadomska D., Sekrecki M., Piasecka A., Taylor K., et al. (2021). Short Antisense Oligonucleotides Alleviate the Pleiotropic Toxicity of RNA Harboring Expanded CGG Repeats. Nat. Commun. 12 (1), 1265. 10.1038/s41467-021-21021-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra A. A., Haify S. N., Verwey N. A., Prins N. D., van der Toorn E. C., Rozemuller A. J. M., et al. (2021). Neuropathology of FMR1-Premutation Carriers Presenting with Dementia and Neuropsychiatric Symptoms. Brain Commun. 3 (1), fcab007. 10.1093/braincomms/fcab007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disney M. D., Liu B., Yang W. Y., Sellier C., Tran T., Charlet-Berguerand N., et al. (2012). A Small Molecule that Targets R(CGG)(exp) and Improves Defects in Fragile X-Associated Tremor Ataxia Syndrome. ACS Chem. Biol. 7 (10), 1711–1718. 10.1021/cb300135h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durmus H., Laval S. H., Deymeer F., Parman Y., Kiyan E., Gokyigiti M., et al. (2011). Oculopharyngodistal Myopathy Is a Distinct Entity: Clinical and Genetic Features of 47 Patients. Neurology 76 (3), 227–235. 10.1212/wnl.0b013e318207b043 [DOI] [PubMed] [Google Scholar]
- Entezam A., Biacsi R., Orrison B., Saha T., Hoffman G. E., Grabczyk E., et al. (2007). Regional FMRP Deficits and Large Repeat Expansions into the Full Mutation Range in a New Fragile X Premutation Mouse Model. Gene 395, 125–134. 10.1016/j.gene.2007.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y., Xu Y., Shi C. (2022). NOTCH2NLC-related Disorders: the Widening Spectrum and Genotype-Phenotype Correlation. J. Med. Genet. 59 (1), 1–9. 10.1136/jmedgenet-2021-107883 [DOI] [PubMed] [Google Scholar]
- Fang P., Yu Y., Yao S., Chen S., Zhu M., Chen Y., et al. (2020). Repeat Expansion Scanning of the NOTCH2NLC Gene in Patients with Multiple System Atrophy. Ann. Clin. Transl Neurol. 7 (4), 517–526. 10.1002/acn3.51021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fardaei M., Rogers M. T., Thorpe H. M., Larkin K., Hamshere M. G., Harper P. S., et al. (2002). Three Proteins, MBNL, MBLL and MBXL, Co-localize In Vivo with Nuclear Foci of Expanded-Repeat Transcripts in DM1 and DM2 Cells. Hum. Mol. Genet. 11 (7), 805–814. 10.1093/hmg/11.7.805 [DOI] [PubMed] [Google Scholar]
- Feng Y., Zhang F., Lokey L. K., Chastain J. L., Lakkis L., Eberhart D., et al. (1995). Translational Suppression by Trinucleotide Repeat Expansion at FMR1. Science 268, 731–734. 10.1126/science.7732383 [DOI] [PubMed] [Google Scholar]
- Fiddes I. T., Lodewijk G. A., Mooring M., Bosworth C. M., Ewing A. D., Mantalas G. L., et al. (2018). Human-Specific NOTCH2NL Genes Affect Notch Signaling and Cortical Neurogenesis. Cell. 173 (6), 1356–1369. e22. 10.1016/j.cell.2018.03.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields A. P., Rodriguez E. H., Jovanovic M., Stern-Ginossar N., Haas B. J., Mertins P., et al. (2015). A Regression-Based Analysis of Ribosome-Profiling Data Reveals a Conserved Complexity to Mammalian Translation. Mol. Cel. 60 (5), 816–827. 10.1016/j.molcel.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florian R. T., Kraft F., Leitão E., Kaya S., Klebe S., Magnin E., et al. (2019). Unstable TTTTA/TTTCA Expansions in MARCH6 Are Associated with Familial Adult Myoclonic Epilepsy Type 3. Nat. Commun. 10 (1), 4919. 10.1038/s41467-019-12763-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y. H., Kuhl D. P., Pizzuti A., Pieretti M., Sutcliffe J. S., Richards S., et al. (1991). Variation of the CGG Repeat at the Fragile X Site Results in Genetic Instability: Resolution of the Sherman Paradox. Cell. 67 (6), 1047–1058. 10.1016/0092-8674(91)90283-5 [DOI] [PubMed] [Google Scholar]
- Fu Y. H., Pizzuti A., Fenwick R. G., Jr, King J., Rajnarayan S., Dunne P. W., et al. (1992). An Unstable Triplet Repeat in a Gene Related to Myotonic Muscular Dystrophy. Science 255 (5049), 1256–1258. 10.1126/science.1546326 [DOI] [PubMed] [Google Scholar]
- Fukuda H., Yamaguchi D., Nyquist K., Yabuki Y., Miyatake S., Uchiyama Y., et al. (2021). Father-to-offspring Transmission of Extremely Long NOTCH2NLC Repeat Expansions with Contractions: Genetic and Epigenetic Profiling with Long-Read Sequencing. Clin. Epigenetics 13 (1), 204. 10.1186/s13148-021-01192-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F. B., Richter J. D., Cleveland D. W. (2017). Rethinking Unconventional Translation in Neurodegeneration. Cell. 171 (5), 994–1000. 10.1016/j.cell.2017.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelpi E., Botta-Orfila T., Bodi L., Marti S., Kovacs G., Grau-Rivera O., et al. (2017). Neuronal Intranuclear (Hyaline) Inclusion Disease and Fragile X-Associated Tremor/ataxia Syndrome: a Morphological and Molecular Dilemma. Brain 140 (8), e51. 10.1093/brain/awx156 [DOI] [PubMed] [Google Scholar]
- Glineburg M. R., Todd P. K., Charlet-Berguerand N., Sellier C. (2018). Repeat-associated Non-AUG (RAN) Translation and Other Molecular Mechanisms in Fragile X Tremor Ataxia Syndrome. Brain Res. 1693 (Pt A), 43–54. 10.1016/j.brainres.2018.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohel D., Sripada L., Prajapati P., Singh K., Roy M., Kotadia D., et al. (2019). FMRpolyG Alters Mitochondrial Transcripts Level and Respiratory Chain Complex Assembly in Fragile X Associated Tremor/ataxia Syndrome [FXTAS]. Biochim. Biophys. Acta Mol. Basis Dis. 1865 (6), 1379–1388. 10.1016/j.bbadis.2019.02.010 [DOI] [PubMed] [Google Scholar]
- Goodman F. R., Bacchelli C., Brady A. F., Brueton L. A., Fryns J. P., Mortlock D. P., et al. (2000). Novel HOXA13 Mutations and the Phenotypic Spectrum of Hand-Foot-Genital Syndrome. Am. J. Hum. Genet. 67 (1), 197–202. 10.1086/302961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco C. M., Berman R. F., Martin R. M., Tassone F., Schwartz P. H., Chang A., et al. (2006). Neuropathology of Fragile X-Associated Tremor/ataxia Syndrome (FXTAS). Brain 1, 243–255. 10.1093/brain/awh683 [DOI] [PubMed] [Google Scholar]
- Green K. M., Glineburg M. R., Kearse M. G., Flores B. N., Linsalata A. E., Fedak S. J., et al. (2017). RAN Translation at C9orf72-Associated Repeat Expansions Is Selectively Enhanced by the Integrated Stress Response. Nat. Commun. 8 (1), 2005. 10.1038/s41467-017-02200-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green K. M., Sheth U. J., Flores B. N., Wright S. E., Sutter A. B., Kearse M. G., et al. (2019). High-throughput Screening Yields Several Small-Molecule Inhibitors of Repeat-Associated Non-AUG Translation. J. Biol. Chem. 294 (49), 18624–18638. 10.1074/jbc.ra119.009951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman R. J., Leehey M., Heinrichs W., Tassone F., Wilson R., Hills J., et al. (2001). Intention Tremor, Parkinsonism, and Generalized Brain Atrophy in Male Carriers of Fragile X. Neurology 1, 127–130. 10.1212/wnl.57.1.127 [DOI] [PubMed] [Google Scholar]
- Haify S. N., Buijsen R. A. M., Verwegen L., Severijnen L. W. F. M., de Boer H., Boumeester V., et al. (2021). Small Molecule 1a Reduces FMRpolyG-Mediated Toxicity in In Vitro and In Vivo Models for FMR1 Premutation. Hum. Mol. Genet. 30 (17), 1632–1648. 10.1093/hmg/ddab143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley H. G., Brook J. D., Rundle S. A., Crow S., Reardon W., Buckler A. J., et al. (1992). Expansion of an Unstable DNA Region and Phenotypic Variation in Myotonic Dystrophy. Nature 355 (6360), 545–546. 10.1038/355545a0 [DOI] [PubMed] [Google Scholar]
- Hashem V., Galloway J. N., Mori M., Willemsen R., Oostra B. A., Paylor R., et al. (2009). Ectopic Expression of CGG Containing mRNA Is Neurotoxic in Mammals. Hum. Mol. Genet. 18, 2443–2451. 10.1093/hmg/ddp182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes L. R., Duan L., Bowen K., Kalab P., Rothstein J. D. (2020). C9orf72 Arginine-Rich Dipeptide Repeat Proteins Disrupt Karyopherin-Mediated Nuclear Import. Elife 9, e51685. 10.7554/eLife.51685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F., Krans A., Freibaum B. D., Taylor J. P., Todd P. K. (2014). TDP-43 Suppresses CGG Repeat-Induced Neurotoxicity through Interactions with HnRNP A2/B1. Hum. Mol. Genet. 23 (19), 5036–5051. 10.1093/hmg/ddu216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch A. G., Ivanov I. P., Sonenberg N. (2016). Translational Control by 5'-untranslated Regions of Eukaryotic mRNAs. Science 352 (6292), 1413–1416. 10.1126/science.aad9868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoem G., Bowitz Larsen K., Øvervatn A., Brech A., Lamark T., Sjøttem E., et al. (2019). The FMRpolyGlycine Protein Mediates Aggregate Formation and Toxicity Independent of the CGG mRNA Hairpin in a Cellular Model for FXTAS. Front. Genet. 10, 249. 10.3389/fgene.2019.00249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm K. N., Herren A. W., Taylor S. L., Randol J. L., Kim K., Espinal G., et al. (2021). Human Cerebral Cortex Proteome of Fragile X-Associated Tremor/Ataxia Syndrome. Front. Mol. Biosci. 7, 600840. 10.3389/fmolb.2020.600840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes S. E., O'Hearn E. E., McInnis M. G., Gorelick-Feldman D. A., Kleiderlein J. J., Callahan C., et al. (1999). Expansion of a Novel CAG Trinucleotide Repeat in the 5' Region of PPP2R2B Is Associated with SCA12. Nat. Genet. 23 (4), 391–392. 10.1038/70493 [DOI] [PubMed] [Google Scholar]
- Hukema R. K., Buijsen R. A., Schonewille M., Raske C., Severijnen L. A., Nieuwenhuizen-Bakker I., et al. (2015). Reversibility of Neuropathology and Motor Deficits in an Inducible Mouse Model for FXTAS. Hum. Mol. Genet. 24 (17), 4948–4957. 10.1093/hmg/ddv216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsaker M. R., Greco C. M., Spath M. A., Smits A. P., Navarro C. S., Tassone F., et al. (2011). Widespread Non-central Nervous System Organ Pathology in Fragile X Premutation Carriers with Fragile X-Associated Tremor/ataxia Syndrome and CGG Knock-In Mice. Acta Neuropathol. 122 (4), 467–479. 10.1007/s00401-011-0860-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N. T., Lareau L. F., Weissman J. S. (2011). Ribosome Profiling of Mouse Embryonic Stem Cells Reveals the Complexity and Dynamics of Mammalian Proteomes. Cell. 147 (4), 789–802. 10.1016/j.cell.2011.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiura H., Doi K., Mitsui J., Yoshimura J., Matsukawa M. K., Fujiyama A., et al. (2018). Expansions of Intronic TTTCA and TTTTA Repeats in Benign Adult Familial Myoclonic Epilepsy. Nat. Genet. 50 (4), 581–590. 10.1038/s41588-018-0067-2 [DOI] [PubMed] [Google Scholar]
- Ishiura H., Shibata S., Yoshimura J., Suzuki Y., Qu W., Doi K., et al. (2019). Noncoding CGG Repeat Expansions in Neuronal Intranuclear Inclusion Disease, Oculopharyngodistal Myopathy and an Overlapping Disease. Nat. Genet. 51 (8), 1222–1232. 10.1038/s41588-019-0458-z [DOI] [PubMed] [Google Scholar]
- Ishiura H., Tsuji S. (2020). Advances in Repeat Expansion Diseases and a New Concept of Repeat Motif-Phenotype Correlation. Curr. Opin. Genet. Dev. 65, 176–185. 10.1016/j.gde.2020.05.029 [DOI] [PubMed] [Google Scholar]
- Iwahashi C. K., Yasui D. H., An H. J., Greco C. M., Tassone F., Nannen K., et al. (2006). Protein Composition of the Intranuclear Inclusions of FXTAS. Brain 129, 256–271. 10.1093/brain/awh650 [DOI] [PubMed] [Google Scholar]
- Jacquemont S., Hagerman R. J., Leehey M., Grigsby J., Zhang L., Brunberg J. A., et al. (2003). Fragile X Premutation Tremor/ataxia Syndrome: Molecular, Clinical, and Neuroimaging Correlates. Am. J. Hum. Genet. 4, 869–878. 10.1086/374321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P., Duan R., Qurashi A., Qin Y., Tian D., Rosser T. C., et al. (2007). Pur Alpha Binds to rCGG Repeats and Modulates Repeat-Mediated Neurodegeneration in a Drosophila Model of Fragile X Tremor/ataxia Syndrome. Neuron 55, 556–564. 10.1016/j.neuron.2007.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P., Zarnescu D. C., Zhang F., Pearson C. E., Lucchesi J. C., Moses K., et al. (2003). RNA-mediated Neurodegeneration Caused by the Fragile X Premutation rCGG Repeats in Drosophila. Neuron 39, 739–747. 10.1016/s0896-6273(03)00533-6 [DOI] [PubMed] [Google Scholar]
- Johnstone T. G., Bazzini A. A., Giraldez A. J. (2016). Upstream ORFs Are Prevalent Translational Repressors in Vertebrates. EMBO J. 35 (7), 706–723. 10.15252/embj.201592759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovičić A., Mertens J., Boeynaems S., Bogaert E., Chai N., Yamada S. B., et al. (2015). Van Den Bosch L, Robberecht W, Gitler AD. Modifiers of C9orf72 Dipeptide Repeat Toxicity Connect Nucleocytoplasmic Transport Defects to FTD/ALS. Nat. Neurosci. 18 (9), 1226–1229. 10.1038/nn.4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y., Okamoto T., Taniwaki M., Aizawa M., Inoue M., Katayama S., et al. (1994). CAG Expansions in a Novel Gene for Machado-Joseph Disease at Chromosome 14q32.1. Nat. Genet. 8 (3), 221–228. 10.1038/ng1194-221 [DOI] [PubMed] [Google Scholar]
- Kearse M. G., Green K. M., Krans A., Rodriguez C. M., Linsalata A. E., Goldstrohm A. C., et al. (2016). CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-dependent Scanning Mechanism of Initiation to Produce Toxic Proteins. Mol. Cel. 62 (2), 314–322. 10.1016/j.molcel.2016.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M. G., Wilusz J. E. (2017). Non-AUG Translation: a New Start for Protein Synthesis in Eukaryotes. Genes Dev. 31 (17), 1717–1731. 10.1101/gad.305250.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenneson A., Zhang F., Hagedorn C. H., Warren S. T. (2001). Reduced FMRP and Increased FMR1 Transcription Is Proportionally Associated with CGG Repeat Number in Intermediate-Length and Premutation Carriers. Hum. Mol. Genet. 10 (14), 1449–1454. 10.1093/hmg/10.14.1449 [DOI] [PubMed] [Google Scholar]
- Kimber T. E., Blumbergs P. C., Rice J. P., Hallpike J. F., Edis R., Thompson P. D., et al. (1998). Familial Neuronal Intranuclear Inclusion Disease with Ubiquitin Positive Inclusions. J. Neurol. Sci. 160 (1), 33–40. 10.1016/s0022-510x(98)00169-5 [DOI] [PubMed] [Google Scholar]
- Knight S. J., Flannery A. V., Hirst M. C., Campbell L., Christodoulou Z., Phelps S. R., et al. (1993). Trinucleotide Repeat Amplification and Hypermethylation of a CpG Island in FRAXE Mental Retardation. Cell. 74 (1), 127–134. 10.1016/0092-8674(93)90300-f [DOI] [PubMed] [Google Scholar]
- Kobayashi H., Abe K., Matsuura T., Ikeda Y., Hitomi T., Akechi Y., et al. (2011). Expansion of Intronic GGCCTG Hexanucleotide Repeat in NOP56 Causes SCA36, a Type of Spinocerebellar Ataxia Accompanied by Motor Neuron Involvement. Am. J. Hum. Genet. 89 (1), 121–130. 10.1016/j.ajhg.2011.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide R., Ikeuchi T., Onodera O., Tanaka H., Igarashi S., Endo K., et al. (1994). Unstable Expansion of CAG Repeat in Hereditary Dentatorubral-Pallidoluysian Atrophy (DRPLA). Nat. Genet. 6 (1), 9–13. 10.1038/ng0194-9 [DOI] [PubMed] [Google Scholar]
- Koide R., Kobayashi S., Shimohata T., Ikeuchi T., Maruyama M., Saito M., et al. (1999). A Neurological Disease Caused by an Expanded CAG Trinucleotide Repeat in the TATA-Binding Protein Gene: a New Polyglutamine Disease? Hum. Mol. Genet. 8 (11), 2047–2053. 10.1093/hmg/8.11.2047 [DOI] [PubMed] [Google Scholar]
- Konieczny P., Mukherjee S., Stepniak-Konieczna E., Taylor K., Niewiadomska D., Piasecka A., et al. (2021). Cyclic Mismatch Binding Ligands Interact with Disease-Associated CGG Trinucleotide Repeats in RNA and Suppress Their Translation. Nucleic Acids Res. 49 (16), 9479–9495. 10.1093/nar/gkab669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob M. D., Moseley M. L., Schut L. J., Benzow K. A., Bird T. D., Day J. W., et al. (1999). An Untranslated CTG Expansion Causes a Novel Form of Spinocerebellar Ataxia (SCA8). Nat. Genet. 21 (4), 379–384. 10.1038/7710 [DOI] [PubMed] [Google Scholar]
- Kozak M. (1989). Context Effects and Inefficient Initiation at Non-AUG Codons in Eucaryotic Cell-free Translation Systems. Mol. Cel Biol. 9 (11), 5073–5080. 10.1128/mcb.9.11.5073-5080.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. (1990). Downstream Secondary Structure Facilitates Recognition of Initiator Codons by Eukaryotic Ribosomes. Proc. Natl. Acad. Sci. U S A. 87 (21), 8301–8305. 10.1073/pnas.87.21.8301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. (1981). Possible Role of Flanking Nucleotides in Recognition of the AUG Initiator Codon by Eukaryotic Ribosomes. Nucleic Acids Res. 9 (20), 5233–5252. 10.1093/nar/9.20.5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krans A., Skariah G., Zhang Y., Bayly B., Todd P. K. (2019). Neuropathology of RAN Translation Proteins in Fragile X-Associated Tremor/ataxia Syndrome. Acta Neuropathol. Commun. 7 (1), 152. 10.1186/s40478-019-0782-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer E. J., Pritchard M., Lynch M., Yu S., Holman K., Baker E., et al. (1991). Mapping of DNA Instability at the Fragile X to a Trinucleotide Repeat Sequence p(CCG)n. Science 252 (5013), 1711–1714. 10.1126/science.1675488 [DOI] [PubMed] [Google Scholar]
- Kumutpongpanich T., Ogasawara M., Ozaki A., Ishiura H., Tsuji S., Minami N., et al. (2021). Clinicopathologic Features of Oculopharyngodistal Myopathy with LRP12 CGG Repeat Expansions Compared with Other Oculopharyngodistal Myopathy Subtypes. JAMA Neurol. 78 (7), 853–863. 10.1001/jamaneurol.2021.1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Spada A. R., Wilson E. M., Lubahn D. B., Harding A. E., Fischbeck K. H. (1991). Androgen Receptor Gene Mutations in X-Linked Spinal and Bulbar Muscular Atrophy. Nature 352 (6330), 77–79. 10.1038/352077a0 [DOI] [PubMed] [Google Scholar]
- LaCroix A. J., Stabley D., Sahraoui R., Adam M. P., Mehaffey M., Kernan K., et al. (2019). GGC Repeat Expansion and Exon 1 Methylation of XYLT1 Is a Common Pathogenic Variant in Baratela-Scott Syndrome. Am. J. Hum. Genet. 104 (1), 35–44. University of Washington Center for Mendelian Genomics, Mefford HC, Sol-Church K. 10.1016/j.ajhg.2018.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalioti M. D., Scott H. S., Buresi C., Rossier C., Bottani A., Morris M. A., et al. (1997). Dodecamer Repeat Expansion in Cystatin B Gene in Progressive Myoclonus Epilepsy. Nature 386 (6627), 847–851. 10.1038/386847a0 [DOI] [PubMed] [Google Scholar]
- Lanni S., Pearson C. E. (2019). Molecular Genetics of Congenital Myotonic Dystrophy. Neurobiol. Dis. 132, 104533. 10.1016/j.nbd.2019.104533 [DOI] [PubMed] [Google Scholar]
- Laumonnier F., Ronce N., Hamel B. C., Thomas P., Lespinasse J., Raynaud M., et al. (2002). Transcription Factor SOX3 Is Involved in X-Linked Mental Retardation with Growth Hormone Deficiency. Am. J. Hum. Genet. 71 (6), 1450–1455. 10.1086/344661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Liu B., Lee S., Huang S. X., Shen B., Qian S. B. (2012). Global Mapping of Translation Initiation Sites in Mammalian Cells at Single-Nucleotide Resolution. Proc. Natl. Acad. Sci. U S A. 109 (37), E2424–E2432. 10.1073/pnas.1207846109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman A. P., Shakkottai V. G., Albin R. L. (2019). Polyglutamine Repeats in Neurodegenerative Diseases. Annu. Rev. Pathol. 14, 1–27. 10.1146/annurev-pathmechdis-012418-012857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S. Y., Ishiura H., Ramli N., Shibata S., Almansour M. A., Tan A. H., et al. (2020). Adult-onset Neuronal Intranuclear Inclusion Disease Mimicking Fragile X-Associated Tremor-Ataxia Syndrome in Ethnic Chinese Patients. Parkinsonism Relat. Disord. 74, 25–27. 10.1016/j.parkreldis.2020.03.025 [DOI] [PubMed] [Google Scholar]
- Lindblad K., Savontaus M. L., Stevanin G., Holmberg M., Digre K., Zander C., et al. (1996). An Expanded CAG Repeat Sequence in Spinocerebellar Ataxia Type 7. Genome Res. 6 (10), 965–971. 10.1101/gr.6.10.965 [DOI] [PubMed] [Google Scholar]
- Lindenberg R., Rubinstein L. J., Herman M. M., Haydon G. B. (1968). A Light and Electron Microscopy Study of an Unusual Widespread Nuclear Inclusion Body Disease. A Possible Residuum of an Old Herpesvirus Infection. Acta Neuropathol. 10, 54–73. 10.1007/bf00690510 [DOI] [PubMed] [Google Scholar]
- Liquori C. L., Ricker K., Moseley M. L., Jacobsen J. F., Kress W., Naylor S. L., et al. (2001). Myotonic Dystrophy Type 2 Caused by a CCTG Expansion in Intron 1 of ZNF9. Science 293 (5531), 864–867. 10.1126/science.1062125 [DOI] [PubMed] [Google Scholar]
- Liu Y., Mimuro M., Yoshida M., Hashizume Y., Niwa H., Miyao S., et al. (2008). Inclusion-positive Cell Types in Adult-Onset Intranuclear Inclusion Body Disease: Implications for Clinical Diagnosis. Acta Neuropathol. 116 (6), 615–623. 10.1007/s00401-008-0442-7 [DOI] [PubMed] [Google Scholar]
- Lu H., Luan X., Yuan Y., Dong M., Sun W., Yan C. (2008). The Clinical and Myopathological Features of Oculopharyngodistal Myopathy in a Chinese Family. Neuropathology 28 (6), 599–603. 10.1111/j.1440-1789.2008.00924.x [DOI] [PubMed] [Google Scholar]
- Ma D., Tan Y. J., Ng A. S. L., Ong H. L., Sim W., Lim W. K., et al. (2020). Association of NOTCH2NLC Repeat Expansions with Parkinson Disease. JAMA Neurol. 77 (12), 1559–1563. 10.1001/jamaneurol.2020.3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L., Herren A. W., Espinal G., Randol J., McLaughlin B., Martinez-Cerdeño V., et al. (2019). Composition of the Intranuclear Inclusions of Fragile X-Associated Tremor/Ataxia Syndrome. Acta Neuropathol. Commun. 7 (1), 143. 10.1186/s40478-019-0796-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan M., Tsilfidis C., Sabourin L., Shutler G., Amemiya C., Jansen G., et al. (1992). Myotonic Dystrophy Mutation: an Unstable CTG Repeat in the 3' Untranslated Region of the Gene. Science 255 (5049), 1253–1255. 10.1126/science.1546325 [DOI] [PubMed] [Google Scholar]
- Malik I., Kelley C. P., Wang E. T., Todd P. K. (2021a). Molecular Mechanisms Underlying Nucleotide Repeat Expansion Disorders. Nat. Rev. Mol. Cell Biol. 22 (9), 589–607. 10.1038/s41580-021-00382-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik I., Tseng Y. J., Wright S. E., Zheng K., Ramaiyer P., Green K. M., et al. (2021b). SRSF Protein Kinase 1 Modulates RAN Translation and Suppresses CGG Repeat Toxicity. EMBO Mol. Med. 13 (11), e14163. 10.15252/emmm.202114163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis R. L., O'Hearn E., Rosenblatt A., Willour V., Holmes S. E., Franz M. L., et al. (2001). A Disorder Similar to Huntington's Disease Is Associated with a Novel CAG Repeat Expansion. Ann. Neurol. 50 (6), 373–380. 10.1002/ana.1312 [DOI] [PubMed] [Google Scholar]
- Matsubara T., Saito Y., Kurashige T., Higashihara M., Hasegawa F., Ogasawara M., et al. (2021). Neuropathy/intranuclear Inclusion Bodies in Oculopharyngodistal Myopathy: A Case Report. eNeurologicalSci. 24, 100348. 10.1016/j.ensci.2021.100348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura T., Yamagata T., Burgess D. L., Rasmussen A., Grewal R. P., Watase K., et al. (2000). Large Expansion of the ATTCT Pentanucleotide Repeat in Spinocerebellar Ataxia Type 10. Nat. Genet. 26 (2), 191–194. 10.1038/79911 [DOI] [PubMed] [Google Scholar]
- McEachin Z. T., Gendron T. F., Raj N., García-Murias M., Banerjee A., Purcell R. H., et al. (2020). Chimeric Peptide Species Contribute to Divergent Dipeptide Repeat Pathology in c9ALS/FTD and SCA36. Neuron 107 (2), 292–305. 10.1016/j.neuron.2020.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud J., Gilbert J. J. (1981). Multiple System Atrophy with Neuronal Intranuclear Hyaline Inclusions. Report of a New Case with Light and Electron Microscopic Studies. Acta Neuropathol. 54 (2), 113–119. 10.1007/bf00689403 [DOI] [PubMed] [Google Scholar]
- Miller J. W., Urbinati C. R., Teng-Umnuay P., Stenberg M. G., Byrne B. J., Thornton C. A., et al. (2000). Recruitment of Human Muscleblind Proteins to (CUG)(n) Expansions Associated with Myotonic Dystrophy. EMBO J. 19 (17), 4439–4448. 10.1093/emboj/19.17.4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami N., Ikezoe K., Kuroda H., Nakabayashi H., Satoyoshi E., Nonaka I. (2001). Oculopharyngodistal Myopathy Is Genetically Heterogeneous and Most Cases Are Distinct from Oculopharyngeal Muscular Dystrophy. Neuromuscul. Disord. 11 (8), 699–702. 10.1016/s0960-8966(01)00227-9 [DOI] [PubMed] [Google Scholar]
- Mootha V. V., Gong X., Ku H. C., Xing C. (2014). Association and Familial Segregation of CTG18.1 Trinucleotide Repeat Expansion of TCF4 Gene in Fuchs' Endothelial Corneal Dystrophy. Invest. Ophthalmol. Vis. Sci. 55 (1), 33–42. 10.1167/iovs.13-12611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori F., Tanji K., Odagiri S., Hattori M., Hoshikawa Y., Kono C., et al. (2012). Ubiquitin-related Proteins in Neuronal and Glial Intranuclear Inclusions in Intranuclear Inclusion Body Disease. Pathol. Int. 62 (6), 407–411. 10.1111/j.1440-1827.2012.02812.x [DOI] [PubMed] [Google Scholar]
- Morriss G. R., Cooper T. A. (2017). Protein Sequestration as a normal Function of Long Noncoding RNAs and a Pathogenic Mechanism of RNAs Containing Nucleotide Repeat Expansions. Hum. Genet. 136 (9), 1247–1263. 10.1007/s00439-017-1807-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley M. L., Zu T., Ikeda Y., Gao W., Mosemiller A. K., Daughters R. S., et al. (2006). Bidirectional Expression of CUG and CAG Expansion Transcripts and Intranuclear Polyglutamine Inclusions in Spinocerebellar Ataxia Type 8. Nat. Genet. 38 (7), 758–769. 10.1038/ng1827 [DOI] [PubMed] [Google Scholar]
- Mundlos S., Otto F., Mundlos C., Mulliken J. B., Aylsworth A. S., Albright S., et al. (1997). Mutations Involving the Transcription Factor CBFA1 Cause Cleidocranial Dysplasia. Cell. 89 (5), 773–779. 10.1016/s0092-8674(00)80260-3 [DOI] [PubMed] [Google Scholar]
- Munoz-Garcia D., Ludwin S. K. (1986). Adult-onset Neuronal Intranuclear Hyaline Inclusion Disease. Neurology 36, 785–790. 10.1212/wnl.36.6.785 [DOI] [PubMed] [Google Scholar]
- Nagafuchi S., Yanagisawa H., Sato K., Shirayama T., Ohsaki E., Bundo M., et al. (1994). Dentatorubral and Pallidoluysian Atrophy Expansion of an Unstable CAG Trinucleotide on Chromosome 12p. Nat. Genet. 6 (1), 14–18. 10.1038/ng0194-14 [DOI] [PubMed] [Google Scholar]
- Nakamura N., Tsunoda K., Mitsutake A., Shibata S., Mano T., Nagashima Y., et al. (2020). Clinical Characteristics of Neuronal Intranuclear Inclusion Disease-Related Retinopathy with CGG Repeat Expansions in the NOTCH2NLC Gene. Invest. Ophthalmol. Vis. Sci. 61 (11), 27. 10.1167/iovs.61.11.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng A. S. L., Lim W. K., Xu Z., Ong H. L., Tan Y. J., Sim W. Y., et al. (2020a). NOTCH2NLC GGC Repeat Expansions Are Associated with Sporadic Essential Tremor: Variable Disease Expressivity on Long-Term Follow-Up. Ann. Neurol. 88 (3), 614–618. 10.1002/ana.25803 [DOI] [PubMed] [Google Scholar]
- Ng A. S. L., Xu Z., Chen Z., Tan Y. J., Lim W. K., Ting S. K. S., et al. (2020b). NOTCH2NLC-linked Neuronal Intranuclear Inclusion Body Disease and Fragile X-Associated Tremor/ataxia Syndrome. Brain 143 (8), e69. 10.1093/brain/awaa210 [DOI] [PubMed] [Google Scholar]
- Nguyen L., Cleary J. D., Ranum L. P. W. (2019). Repeat-Associated Non-ATG Translation: Molecular Mechanisms and Contribution to Neurological Disease. Annu. Rev. Neurosci. 42, 227–247. 10.1146/annurev-neuro-070918-050405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolin S. L., Glicksman A., Tortora N., Allen E., Macpherson J., Mila M., et al. (2019). Expansions and Contractions of the FMR1 CGG Repeat in 5,508 Transmissions of normal, Intermediate, and Premutation Alleles. Am. J. Med. Genet. A. 179 (7), 1148–1156. 10.1002/ajmg.a.61165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlé I., Rousseau F., Heitz D., Kretz C., Devys D., Hanauer A., et al. (1991). Instability of a 550-base Pair DNA Segment and Abnormal Methylation in Fragile X Syndrome. Science 252 (5009), 1097–1102. 10.1126/science.252.5009.1097 [DOI] [PubMed] [Google Scholar]
- Odagiri S., Tanji K., Mori F., Kakita A., Takahashi H., Kamitani T., et al. (2012). Immunohistochemical Analysis of Marinesco Bodies, Using Antibodies against Proteins Implicated in the Ubiquitin-Proteasome System, Autophagy and Aggresome Formation. Neuropathology 32 (3), 261–266. 10.1111/j.1440-1789.2011.01267.x [DOI] [PubMed] [Google Scholar]
- Ogasawara M., Eura N., Nagaoka U., Sato T., Arahata H., Hayashi T., et al. (2021). Intranuclear Inclusions in Skin Biopsies Are Not Limited to Neuronal Intranuclear Inclusion Disease but Can Also Be Seen in Oculopharyngodistal Myopathy. Neuropathol. Appl. Neurobiol. [Epub ahead of print]. 10.1111/nan.12787 [DOI] [PubMed] [Google Scholar]
- Ogasawara M., Iida A., Kumutpongpanich T., Ozaki A., Oya Y., Konishi H., et al. (2020). CGG Expansion in NOTCH2NLC Is Associated with Oculopharyngodistal Myopathy with Neurological Manifestations. Acta Neuropathol. Commun. 8 (1), 204. 10.1186/s40478-020-01084-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S. Y., He F., Krans A., Frazer M., Taylor J. P., Paulson H. L., et al. (2015). RAN Translation at CGG Repeats Induces Ubiquitin Proteasome System Impairment in Models of Fragile X-Associated Tremor Ataxia Syndrome. Hum. Mol. Genet. 24 (15), 4317–4326. 10.1093/hmg/ddv165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo M., Doi H., Fukai R., Fujita A., Mitsuhashi S., Hashiguchi S., et al. (2019). GGC Repeat Expansion of NOTCH2NLC in Adult Patients with Leukoencephalopathy. Ann. Neurol. 86 (6), 962–968. 10.1002/ana.25586 [DOI] [PubMed] [Google Scholar]
- Orr H. T., Chung M. Y., Banfi S., Kwiatkowski T. J., Jr, Servadio A., Beaudet A. L., et al. (1993). Expansion of an Unstable Trinucleotide CAG Repeat in Spinocerebellar Ataxia Type 1. Nat. Genet. 4 (3), 221–226. 10.1038/ng0793-221 [DOI] [PubMed] [Google Scholar]
- Patel H., Norman M. G., Perry T. L., Berry K. E. (1985). Multiple System Atrophy with Neuronal Intranuclear Hyaline Inclusions. Report of a Case and Review of the Literature. J. Neurol. Sci. 67 (1), 57–65. 10.1016/0022-510x(85)90022-x [DOI] [PubMed] [Google Scholar]
- Peabody D. S. (1989). Translation Initiation at Non-AUG Triplets in Mammalian Cells. J. Biol. Chem. 264 (9), 5031–5035. 10.1016/s0021-9258(18)83694-8 [DOI] [PubMed] [Google Scholar]
- Pountney D. L., Huang Y., Burns R. J., Haan E., Thompson P. D., Blumbergs P. C., et al. (2003). SUMO-1 marks the Nuclear Inclusions in Familial Neuronal Intranuclear Inclusion Disease. Exp. Neurol. 184 (1), 436–446. 10.1016/j.expneurol.2003.07.004 [DOI] [PubMed] [Google Scholar]
- Pulst S. M., Nechiporuk A., Nechiporuk T., Gispert S., Chen X. N., Lopes-Cendes I., et al. (1996). Moderate Expansion of a Normally Biallelic Trinucleotide Repeat in Spinocerebellar Ataxia Type 2. Nat. Genet. 14 (3), 269–276. 10.1038/ng1196-269 [DOI] [PubMed] [Google Scholar]
- Qurashi A., Li W., Zhou J. Y., Peng J., Jin P. (2011). Nuclear Accumulation of Stress Response mRNAs Contributes to the Neurodegeneration Caused by Fragile X Premutation rCGG Repeats. Plos Genet. 7 (6), e1002102. 10.1371/journal.pgen.1002102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qurashi A., Liu H., Ray L., Nelson D. L., Duan R., Jin P. (2012). Chemical Screen Reveals Small Molecules Suppressing Fragile X Premutation rCGG Repeat-Mediated Neurodegeneration in Drosophila. Hum. Mol. Genet. 21 (9), 2068–2075. 10.1093/hmg/dds024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafehi H., Szmulewicz D. J., Bennett M. F., Sobreira N. L. M., Pope K., Smith K. R., et al. (2019). Bioinformatics-Based Identification of Expanded Repeats: A Non-reference Intronic Pentamer Expansion in RFC1 Causes CANVAS. Am. J. Hum. Genet. 105 (1), 151–165. 10.1016/j.ajhg.2019.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton A. E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J. R., et al. (2011). A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 72 (2), 257–268. 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D. O., Wills A. J., Hammans S. R., Read S. P., Sillibourne J. (2006). Oculopharyngeal Muscular Dystrophy: a point Mutation Which Mimics the Effect of the PABPN1 Gene Triplet Repeat Expansion Mutation. J. Med. Genet. 43 (5), e23. 10.1136/jmg.2005.037598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez C. M., Wright S. E., Kearse M. G., Haenfler J. M., Flores B. N., Liu Y., et al. (2020). A Native Function for RAN Translation and CGG Repeats in Regulating Fragile X Protein Synthesis. Nat. Neurosci. 23 (3), 386–397. 10.1038/s41593-020-0590-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Z., Hu J., Corey D. R., Mootha V. V. (2019). Quantitative Studies of Muscleblind Proteins and Their Interaction with TCF4 RNA Foci Support Involvement in the Mechanism of Fuchs' Dystrophy. Invest. Ophthalmol. Vis. Sci. 60 (12), 3980–3991. 10.1167/iovs.19-27641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito R., Shimizu H., Miura T., Hara N., Mezaki N., Higuchi Y., et al. (2020). Oculopharyngodistal Myopathy with Coexisting Histology of Systemic Neuronal Intranuclear Inclusion Disease: Clinicopathologic Features of an Autopsied Patient Harboring CGG Repeat Expansions in LRP12. Acta Neuropathol. Commun. 8 (1), 75. 10.1186/s40478-020-00945-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N., Amino T., Kobayashi K., Asakawa S., Ishiguro T., Tsunemi T., et al. (2009). Spinocerebellar Ataxia Type 31 Is Associated with "inserted" Penta-Nucleotide Repeats Containing (TGGAA)n. Am. J. Hum. Genet. 85 (5), 544–557. 10.1016/j.ajhg.2009.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoyoshi E., Kinoshita M. (1977). Oculopharyngodistal Myopathy. Arch. Neurol. 34 (2), 89–92. 10.1001/archneur.1977.00500140043007 [DOI] [PubMed] [Google Scholar]
- Seixas A. I., Loureiro J. R., Costa C., Ordóñez-Ugalde A., Marcelino H., Oliveira C. L., et al. (2017). Pentanucleotide ATTTC Repeat Insertion in the Non-coding Region of DAB1, Mapping to SCA37, Causes Spinocerebellar Ataxia. Am. J. Hum. Genet. 101 (1), 87–103. 10.1016/j.ajhg.2017.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C., Buijsen R. A. M., He F., Natla S., Jung L., Tropel P., et al. (2017). Translation of Expanded CGG Repeats into FMRpolyG Is Pathogenic and May Contribute to Fragile X Tremor Ataxia Syndrome. Neuron 93 (2), 331–347. 10.1016/j.neuron.2016.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C., Rau F., Liu Y., Tassone F., Hukema R. K., Gattoni R., et al. (2010). Sam68 Sequestration and Partial Loss of Function Are Associated with Splicing Alterations in FXTAS Patients. EMBO J. 29, 1248–1261. 10.1038/emboj.2010.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C., Freyermuth F., Tabet R., Tran T., He F., Ruffenach F., et al. (2013). Sequestration of DROSHA and DGCR8 by Expanded CGG RNA Repeats Alters microRNA Processing in Fragile X-Associated Tremor/ataxia Syndrome. Cel Rep 3 (3), 869–880. 10.1016/j.celrep.2013.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman S. L., Jacobs P. A., Morton N. E., Froster-Iskenius U., Howard-Peebles P. N., Nielsen K. B., et al. (1985). Further Segregation Analysis of the Fragile X Syndrome with Special Reference to Transmitting Males. Hum. Genet. 69 (4), 289–299. 10.1007/bf00291644 [DOI] [PubMed] [Google Scholar]
- Sherman S. L., Morton N. E., Jacobs P. A., Turner G. (1984). The Marker (X) Syndrome: a Cytogenetic and Genetic Analysis. Ann. Hum. Genet. 48 (1), 21–37. 10.1111/j.1469-1809.1984.tb00830.x [DOI] [PubMed] [Google Scholar]
- Shi C. H., Fan Y., Yang J., Yuan Y. P., Shen S., Liu F., et al. (2021). NOTCH2NLC Intermediate-Length Repeat Expansions Are Associated with Parkinson Disease. Ann. Neurol. 89 (1), 182–187. 10.1002/ana.25925 [DOI] [PubMed] [Google Scholar]
- Shoubridge C., Gecz J. (2012). Polyalanine Tract Disorders and Neurocognitive Phenotypes. Adv. Exp. Med. Biol. 769, 185–203. 10.1007/978-1-4614-5434-2_12 [DOI] [PubMed] [Google Scholar]
- Sofola O. A., Jin P., Qin Y., Duan R., Liu H., de Haro M., et al. (2007). RNA-binding Proteins hnRNP A2/B1 and CUGBP1 Suppress Fragile X CGG Premutation Repeat-Induced Neurodegeneration in a Drosophila Model of FXTAS. Neuron 55, 565–571. 10.1016/j.neuron.2007.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sone J., Mitsuhashi S., Fujita A., Mizuguchi T., Hamanaka K., Mori K., et al. (2019). Long-read Sequencing Identifies GGC Repeat Expansions in NOTCH2NLC Associated with Neuronal Intranuclear Inclusion Disease. Nat. Genet. 51 (8), 1215–1221. 10.1038/s41588-019-0459-y [DOI] [PubMed] [Google Scholar]
- Sone J., Mori K., Inagaki T., Katsumata R., Takagi S., Yokoi S., et al. (2016). Clinicopathological Features of Adult-Onset Neuronal Intranuclear Inclusion Disease. Brain 139 (Pt 12), 3170–3186. 10.1093/brain/aww249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sone J., Tanaka F., Koike H., Inukai A., Katsuno M., Yoshida M., et al. (2011). Skin Biopsy Is Useful for the Antemortem Diagnosis of Neuronal Intranuclear Inclusion Disease. Neurology 76 (16), 1372–1376. 10.1212/wnl.0b013e3182166e13 [DOI] [PubMed] [Google Scholar]
- Sonobe Y., Aburas J., Krishnan G., Fleming A. C., Ghadge G., Islam P., et al. (2021). Elegans Model of C9orf72-Associated ALS/FTD Uncovers a Conserved Role for eIF2D in RAN Translation. Nat. Commun. 12 (1), 6025. 10.1038/s41467-021-26303-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonobe Y., Ghadge G., Masaki K., Sendoel A., Fuchs E., Roos R. P. (2018). Translation of Dipeptide Repeat Proteins from the C9ORF72 Expanded Repeat Is Associated with Cellular Stress. Neurobiol. Dis. 116, 155–165. 10.1016/j.nbd.2018.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strømme P., Mangelsdorf M. E., Shaw M. A., Lower K. M., Lewis S. M., Bruyere H., et al. (2002). Mutations in the Human Ortholog of Aristaless Cause X-Linked Mental Retardation and Epilepsy. Nat. Genet. 30 (4), 441–445. 10.1038/ng862 [DOI] [PubMed] [Google Scholar]
- Sugiyama A., Sato N., Kimura Y., Maekawa T., Enokizono M., Saito Y., et al. (2017). MR Imaging Features of the Cerebellum in Adult-Onset Neuronal Intranuclear Inclusion Disease: 8 Cases. AJNR Am. J. Neuroradiol 38 (11), 2100–2104. 10.3174/ajnr.a5336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki I. K., Gacquer D., Van Heurck R., Kumar D., Wojno M., Bilheu A., et al. (2018). Human-Specific NOTCH2NL Genes Expand Cortical Neurogenesis through Delta/Notch Regulation. Cell. 173 (6), 1370–1384. e16. 10.1016/j.cell.2018.03.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sznajder Ł. J., Swanson M. S. (2019). Short Tandem Repeat Expansions and RNA-Mediated Pathogenesis in Myotonic Dystrophy. Int. J. Mol. Sci. 20 (13), 3365. 10.3390/ijms20133365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabet R., Schaeffer L., Freyermuth F., Jambeau M., Workman M., Lee C. Z., et al. (2018). CUG Initiation and Frameshifting Enable Production of Dipeptide Repeat Proteins from ALS/FTD C9ORF72 Transcripts. Nat. Commun. 9 (1), 152. 10.1038/s41467-017-02643-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi-Fujigasaki J., Nakano Y., Uchino A., Murayama S. (2016). Adult-onset Neuronal Intranuclear Hyaline Inclusion Disease Is Not Rare in Older Adults. Geriatr. Gerontol. Int. 16 (Suppl. 1), 51–56. 10.1111/ggi.12725 [DOI] [PubMed] [Google Scholar]
- Takahashi-Fujigasaki J. (2003). Neuronal Intranuclear Hyaline Inclusion Disease. Neuropathology 23 (4), 351–359. 10.1046/j.1440-1789.2003.00524.x [DOI] [PubMed] [Google Scholar]
- Tassone F., Beilina A., Carosi C., Albertosi S., Bagni C., Li L., et al. (2007). Elevated FMR1 mRNA in Premutation Carriers Is Due to Increased Transcription. RNA 13 (4), 555–562. 10.1261/rna.280807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F., Hagerman R. J., Loesch D. Z., Lachiewicz A., Taylor A. K., Hagerman P. J. (2000). Fragile X Males with Unmethylated, Full Mutation Trinucleotide Repeat Expansions Have Elevated Levels of FMR1 Messenger RNA. Am. J. Med. Genet. 94, 232–236. [DOI] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group (1993). A Novel Gene Containing a Trinucleotide Repeat that Is Expanded and Unstable on Huntington's Disease Chromosomes. Cell. 72 (6), 971–983. 10.1016/0092-8674(93)90585-e [DOI] [PubMed] [Google Scholar]
- Tian Y., Wang J. L., Huang W., Zeng S., Jiao B., Liu Z., et al. (2019). Expansion of Human-specific GGC Repeat in Neuronal Intranuclear Inclusion Disease-Related Disorders. Am. J. Hum. Genet. 105 (1), 166–176. 10.1016/j.ajhg.2019.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd P. K., Oh S. Y., Krans A., He F., Sellier C., Frazer M., et al. (2013). CGG Repeat-Associated Translation Mediates Neurodegeneration in Fragile X Tremor Ataxia Syndrome. Neuron 78 (3), 440–455. 10.1016/j.neuron.2013.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toko M., Ohshita T., Kurashige T., Morino H., Kume K., Yamashita H., et al. (2021). FXTAS Is Difficult to Differentiate from Neuronal Intranuclear Inclusion Disease through Skin Biopsy: a Case Report. BMC Neurol. 21 (1), 396. 10.1186/s12883-021-02425-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T., Childs-Disney J. L., Liu B., Guan L., Rzuczek S., Disney M. D. (2014). Targeting the R(CGG) Repeats that Cause FXTAS with Modularly Assembled Small Molecules and Oligonucleotides. ACS Chem. Biol. 9 (4), 904–912. 10.1021/cb400875u [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyama E., Uchino M., Chateau D., Tomé F. M. (1998). Autosomal Recessive Oculopharyngodistal Myopathy in Light of Distal Myopathy with Rimmed Vacuoles and Oculopharyngeal Muscular Dystrophy. Neuromuscul. Disord. 8 (2), 119–125. 10.1016/s0960-8966(98)00002-9 [DOI] [PubMed] [Google Scholar]
- van der Sluijs B. M., ter Laak H. J., Scheffer H., van der Maarel S. M., van Engelen B. G. (2004). Autosomal Recessive Oculopharyngodistal Myopathy: a Distinct Phenotypical, Histological, and Genetic Entity. J. Neurol. Neurosurg. Psychiatry 75 (10), 1499–1501. 10.1136/jnnp.2003.025072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kuilenburg A. B. P., Tarailo-Graovac M., Richmond P. A., Drögemöller B. I., Pouladi M. A., Leen R., et al. (2019). Glutaminase Deficiency Caused by Short Tandem Repeat Expansion in GLS. N. Engl. J. Med. 380 (15), 1433–1441. 10.1056/nejmoa1806627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk A. J., Pieretti M., Sutcliffe J. S., Fu Y. H., Kuhl D. P., Pizzuti A., et al. (1991). Identification of a Gene (FMR-1) Containing a CGG Repeat Coincident with a Breakpoint Cluster Region Exhibiting Length Variation in Fragile X Syndrome. Cell. 65 (5), 905–914. 10.1016/0092-8674(91)90397-h [DOI] [PubMed] [Google Scholar]
- Verma A. K., Khan E., Mishra S. K., Mishra A., Charlet-Berguerand N., Kumar A. (2020). Curcumin Regulates the R(CGG)(exp) RNA Hairpin Structure and Ameliorate Defects in Fragile X-Associated Tremor Ataxia Syndrome. Front. Neurosci. 14, 295. 10.3389/fnins.2020.00295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel H. J., Murray K. D., Haify S. N., Hunsaker M. R., Schwartzer J. J., Kim K., et al. (2019). Astroglial-targeted Expression of the Fragile X CGG Repeat Premutation in Mice Yields RAN Translation, Motor Deficits and Possible Evidence for Cell-To-Cell Propagation of FXTAS Pathology. Acta Neuropathol. Commun. 7 (1), 27. 10.1186/s40478-019-0677-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler T. M., Thornton C. A. (2007). Myotonic Dystrophy: RNA-Mediated Muscle Disease. Curr. Opin. Neurol. 20 (5), 572–576. 10.1097/wco.0b013e3282ef6064 [DOI] [PubMed] [Google Scholar]
- Wieben E. D., Aleff R. A., Tang X., Butz M. L., Kalari K. R., Highsmith E. W., et al. (2017). Trinucleotide Repeat Expansion in the Transcription Factor 4 (TCF4) Gene Leads to Widespread mRNA Splicing Changes in Fuchs' Endothelial Corneal Dystrophy. Invest. Ophthalmol. Vis. Sci. 58 (1), 343–352. 10.1167/iovs.16-20900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieben E. D., Aleff R. A., Tosakulwong N., Butz M. L., Highsmith W. E., Edwards A. O., et al. (2012). A Common Trinucleotide Repeat Expansion within the Transcription Factor 4 (TCF4, E2-2) Gene Predicts Fuchs Corneal Dystrophy. PLoS One 7 (11), e49083. 10.1371/journal.pone.0049083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilburn B., Rudnicki D. D., Zhao J., Weitz T. M., Cheng Y., Gu X., et al. (2011). An Antisense CAG Repeat Transcript at JPH3 Locus Mediates Expanded Polyglutamine Protein Toxicity in Huntington's Disease-like 2 Mice. Neuron 70 (3), 427–440. 10.1016/j.neuron.2011.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen R., Hoogeveen-Westerveld M., Reis S., Holstege J., Severijnen L. A., Nieuwenhuizen I. M., et al. (2003). The FMR1 CGG Repeat Mouse Displays Ubiquitin-Positive Intranuclear Neuronal Inclusions; Implications for the Cerebellar Tremor/ataxia Syndrome. Hum. Mol. Genet. 12, 949–959. 10.1093/hmg/ddg114 [DOI] [PubMed] [Google Scholar]
- Woulfe J. M. (2007). Abnormalities of the Nucleus and Nuclear Inclusions in Neurodegenerative Disease: a Work in Progress. Neuropathol. Appl. Neurobiol. 33 (1), 2–42. 10.1111/j.1365-2990.2006.00819.x [DOI] [PubMed] [Google Scholar]
- Xi J., Wang X., Yue D., Dou T., Wu Q., Lu J., et al. (2021). 5' UTR CGG Repeat Expansion in GIPC1 Is Associated with Oculopharyngodistal Myopathy. Brain 144 (2), 601–614. 10.1093/brain/awaa426 [DOI] [PubMed] [Google Scholar]
- Xu K., Li Y., Allen E. G., Jin P. (2021). Therapeutic Development for CGG Repeat Expansion-Associated Neurodegeneration. Front Cel Neurosci 15, 655568. 10.3389/fncel.2021.655568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi N., Mano T., Ohtomo R., Ishiura H., Almansour M. A., Mori H., et al. (2018). An Autopsy Case of Familial Neuronal Intranuclear Inclusion Disease with Dementia and Neuropathy. Intern. Med. 57 (23), 3459–3462. 10.2169/internalmedicine.1141-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Cen Z., Wang L., Chen X., Liu P., Wang H., et al. (2022). Neuronal Intranuclear Inclusion Disease Tremor-Dominant Subtype: A Mimicker of Essential Tremor. Eur. J. Neurol. 29 (2), 450–458. 10.1111/ene.15169 [DOI] [PubMed] [Google Scholar]
- Yang W. Y., He F., Strack R. L., Oh S. Y., Frazer M., Jaffrey S. R., et al. (2016). Disney MD.Small Molecule Recognition and Tools to Study Modulation of R(CGG)(exp) in Fragile X-Associated Tremor Ataxia Syndrome. ACS Chem. Biol. 11 (9), 2456–2465. 10.1021/acschembio.6b00147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Zhang D., Li P., Niu J., Xu D., Guo X., et al. (2021). Expansion of 5’ UTR CGG Repeat in RILPL1 Is Associated with Oculopharyngodistal Myopathy. medRxiv. 10.1101/2021.09.18.21263669 [DOI] [Google Scholar]
- Yeetong P., Pongpanich M., Srichomthong C., Assawapitaksakul A., Shotelersuk V., Tantirukdham N., et al. (2019). TTTCA Repeat Insertions in an Intron of YEATS2 in Benign Adult Familial Myoclonic Epilepsy Type 4. Brain 142 (11), 3360–3366. 10.1093/brain/awz267 [DOI] [PubMed] [Google Scholar]
- Yu J., Deng J., Guo X., Shan J., Luan X., Cao L., et al. (2021). The GGC Repeat Expansion in NOTCH2NLC Is Associated with Oculopharyngodistal Myopathy Type 3. Brain 144 (6), 1819–1832. 10.1093/brain/awab077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y., Liu Z., Hou X., Li W., Ni J., Huang L., et al. (2020). Identification of GGC Repeat Expansion in the NOTCH2NLC Gene in Amyotrophic Lateral Sclerosis. Neurology 95 (24), e3394–e3405. 10.1212/wnl.0000000000010945 [DOI] [PubMed] [Google Scholar]
- Yuen P., Baxter D. W. (1963). The Morphology of Marinesco Bodies (Paranucleolar Corpuscles) in the Melanin-Pigmented Nuclei of the Brain-Stem. J. Neurol. Neurosurg. Psychiatry 26 (2), 178–183. 10.1136/jnnp.26.2.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G. J., Wu D., Zhu Y. X., Ni H. F., Zhang Z. J. (2021). Clinicopathological Features of Neuronal Intranuclear Inclusion Disease Diagnosed by Skin Biopsy. Neurol. Sci. [Epub ahead of print]. 10.1007/s10072-021-05526-2 [DOI] [PubMed] [Google Scholar]
- Zhao J., Liu J., Xiao J., Du J., Que C., Shi X., et al. (2015). Clinical and Muscle Imaging Findings in 14 mainland Chinese Patients with Oculopharyngodistal Myopathy. PLoS One 10 (6), e0128629. 10.1371/journal.pone.0128629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S., Lian Y., Luo W., Luo R., Wu X., Ji J., et al. (2021). Upstream Open reading Frame with NOTCH2NLC GGC Expansion Generates Polyglycine Aggregates and Disrupts Nucleocytoplasmic Transport: Implications for Polyglycine Diseases. Sacta Neuropathol. 142 (6), 1003–1023. 10.1007/s00401-021-02375-3 [DOI] [PubMed] [Google Scholar]
- Zhuchenko O., Bailey J., Bonnen P., Ashizawa T., Stockton D. W., Amos C., et al. (1997). Autosomal Dominant Cerebellar Ataxia (SCA6) Associated with Small Polyglutamine Expansions in the Alpha 1A-voltage-dependent Calcium Channel. Nat. Genet. 15 (1), 62–69. 10.1038/ng0197-62 [DOI] [PubMed] [Google Scholar]
- Zu T., Gibbens B., Doty N. S., Gomes-Pereira M., Huguet A., Stone M. D., et al. (2011). Non-ATG-initiated Translation Directed by Microsatellite Expansions. Proc. Natl. Acad. Sci. U S A. 108 (1), 260–265. 10.1073/pnas.1013343108 [DOI] [PMC free article] [PubMed] [Google Scholar]