

Abstract
Background
There are substantial advances in diagnosis and treatment for idiopathic pulmonary fibrosis (IPF), but without much evidence available on recent mortality and survival trends.
Methods
A narrative synthesis approach was used to investigate the mortality trends, then meta-analyses for survival trends were carried out based on various time periods.
Results
Six studies reported the mortality data for IPF in 22 countries, and 62 studies (covering 63 307 patients from 20 countries) reported survival data for IPF. Age-standardised mortality for IPF varied from ∼0.5 to ∼12 per 100 000 population per year after year 2000. There were increased mortality trends for IPF in Australia, Brazil, Belgium, Canada, Czech Republic, Finland, France, Germany, Hungary, Italy, Lithuania, the Netherlands, Poland, Portugal, Spain, Sweden and UK, while Austria, Croatia, Denmark, Romania and the USA showed decreased mortality trends. The overall 3-year and 5-year cumulative survival rates (CSRs) were 61.8% (95% CI 58.7–64.9; I2=97.1%) and 45.6% (95% CI 41.5–49.7; I2=97.7%), respectively. Prior to 2010, the pooled 3-year CSR was 59.9% (95% CI 55.8–64.1; I2=95.8%), then not significantly (p=0.067) increased to 66.2% (95% CI 62.9–69.5; I2=92.6%) in the 2010s decade. After excluding three studies in which no patients received antifibrotics after year 2010, the pooled 3-year CSRs significantly (p=0.039) increased to 67.4% (95% CI 63.9–70.9; I2=93.1%) in the 2010s decade.
Discussion
IPF is a diagnosis associated with high mortality. There was no observed increasing survival trend for patients with IPF before year 2010, with then a switch to an improvement, which is probably multifactorial.
Short abstract
Mortality of IPF has varied worldwide from ∼0.5 to ∼12 per 100 000 population per year since 2000 and survival of IPF did not change before 2010, after which it improved, which can be attributable to multiple factors https://bit.ly/3qEDSPt
Background
Idiopathic pulmonary fibrosis (IPF), although relatively uncommon, is a progressive interstitial lung disease, with poor prognosis and high mortality risk [1]. Since the affected population is largely over 65 years old with a male predominance, in the more elderly population more specifically the impact of IPF is considerably greater [2]. Estimated incidence rates of IPF showed increased trends ranging from ∼3 to ∼9 per 100 000 population per year between 1998 and 2012 in Europe and North America [3]. Only a limited number of ecological studies [4] (i.e., at population level) of the mortality of IPF have been published worldwide.
A systematic review [3] reported only eight ecological studies and found estimated mortality rates of IPF ranging from around 1 to 14 per 100 000 population per year in various countries between 1979 and 2012. However, the worldwide variation of mortality rates for IPF reported by Hutchinson et al. [3] in 2015 may have been influenced by widespread use of differing International Classification of Diseases (ICD) codes (such as ICD-8 517, ICD-9 515, ICD-9 516.3 and ICD-10 J84.1), death certificates using either IPF as underlying cause of death or as part of multi-cause deaths, and not differentiating between crude and age-standardised disease rates. Most recently, Khor et al. [5] in 2020 conducted a systematic review and meta-analysis of prognosis for patients with IPF in cohort studies or in the control arm of recent drug trials, who were followed for at least 12 months and were not treated with antifibrotic therapies. Although the mean survival time of patients with IPF has been estimated as 4 years from diagnosis [5], survival trends for IPF in various time periods are not well described.
Recently, management guidelines for diagnosis [6, 7] have been updated, and treatment of IPF now focuses on the new antifibrotic medications (pirfenidone and nintedanib) [8, 9] that may slow progression of the disease but without much evidence available on mortality or any overall impact on survival rates. We aimed to update the last systematic review in 2015 [3] and investigate the recent mortality and survival trends for IPF.
Methods
The protocol of this study was registered at PROSPERO (registration number: CRD 42020151288; www.crd.york.ac.uk/Prospero/) on September 18, 2019. During the manuscript review process, we were prompted to make some valid changes to update the literature search, exclude conference abstracts and conduct a meta-analysis of survival using various diagnostic criteria from the protocol. This systematic review was reported in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement [10], and the PRISMA Checklist is presented in supplementary table S1.
Search strategy and databases
The search strategy involved several combinations of “idiopathic pulmonary fibrosis”, “mortality”, “survival” and their synonyms. The detailed search strategy is outlined in supplementary table S2. Databases including PubMed, EMBASE (via Ovid) and Scopus were searched for eligible studies. Non-English language papers were translated using Google translator platform. Further, a key word search of Google Scholar was performed to detect potential additional studies. The searches included all studies published on or before November 1, 2021. The reference lists from the included studies and two previous systematic reviews [3, 5] were reviewed.
Study selection and eligibility
Studies that met the following criteria were included based on “PICOS” algorithms:
1) Patients with the diagnosis of IPF: mortality statistics using ICD-10 J84.1 (other interstitial pulmonary diseases with fibrosis) as the diagnostic criteria and regarding IPF as the underlying cause of death (UCD); survival statistics using ICD codes or clinical guidelines as diagnostic criteria.
2) Interventions: no specific requirement.
3) Comparators: no specific requirement.
4) Outcomes: annual mortality rates for IPF at a population-based level; 3-year or 5-year CSRs for IPF.
5) Study designs: ecological studies for mortality rates; ecological or cohort studies, followed for at least 3 years for CSRs.
6) Without language limitations.
Exclusion criteria were listed as follows:
1) Participants did not represent the general population of patients with IPF (e.g., focused only on patients with IPF with acute exacerbations).
2) Studies without reporting the annual mortality rates or CSRs of IPF, or without required data to calculate these outcomes.
3) Survival time reported from onset of symptoms to death without reporting survival time from diagnosis, as used in many studies.
4) Duration of follow-up <3 years.
5) Death certificates using IPF as part of multi-cause deaths.
6) Randomised controlled trials (RCTs), reviews, letters, commentaries, editorials, case reports and conference abstracts.
7) Non-human studies.
The screening process for eligible studies was performed using Covidence (Veritas Health Innovation, Melbourne, Australia; www.covidence.org). First, all search results from the databases were imported into Covidence to remove the duplicates. Second, using just titles and abstracts of records, potentially eligible studies were assessed by two co-authors (Q.Z. and I.A.C.) independently, based on inclusion criteria. Third, full text studies were further screened by the same two co-authors independently, based on exclusion criteria. All discrepancies were discussed with a third co-author (A.J.P.) to obtain consensus.
Quality assessment
One co-author (Q.Z.) assessed each included study according to the established tool, and the other co-author (I.A.C.) independently validated the results. No validated study appraisal for evaluating quality of epidemiological studies of IPF exists, so we summarised the various criteria used by previous studies [3, 7, 11–15] and established a new tool with a total of 26 items for quality assessment, which includes two parts: criteria for case definition of IPF (13 items) and study methodology for epidemiological studies (13 items). Detailed method of quality assessment was presented in supplementary table S3, and the outcomes of quality score were expressed as a percentage with interquartile range (IQR).
Data extraction
For data extraction, one co-author (Q.Z.) extracted all specific information including: first author, year of publication, median year studied where patients were included across multiple years, country, sample size, age, sex (percentage of males), ethnicity, smoking (percentage of patients with smoking history), pack-years of smoking, family history of interstitial lung diseases, forced vital capacity % predicted, diffusing capacity of the lung for carbon monoxide (DLCO) % predicted, body mass index, 6-min walk distance, adequacy of case definition, percentage of patients without any therapy, percentage of patients with now recognised harmful therapies, percentage of patients with new antifibrotic therapies, source of data (such as from single centre, national registry and national database), duration of follow-up, study design, annual country-specific mortality rates and survival-related outcomes (3-year or 5-year CSRs). Supplementary table S4 shows development of diagnostic criteria for IPF based on ICD codes. Although ICD-10 code J84.1 may include other idiopathic interstitial pneumonias (IIPs), it is the most specific code for IPF to present global mortality statistics in the study timeframe [3]. Therefore, we used the cut-off of year 2000 to show recent mortality trends for IPF. The cut-off of year 2010 was used to describe survival trends for IPF corresponding to substantial advances in diagnosis [7] and treatment [8, 9] for IPF after year 2010. Studies were either distributed to a antifibrotics group if they reported participants explicitly taking antifibrotics, or to a non-antifibrotics group if they reported other therapies. The classification of antifibrotics (effective therapies) and non-antifibrotics (no, ineffective or harmful therapies) were determined according to the Richeldi et al. study [2]. All data from individual studies were entered into a pre-designed Microsoft Excel Worksheet and then were validated by another co-author (J.A.C.). Again, all discrepancies were discussed and resolved with the third co-author (A.J.P.) by consensus.
Statistical analyses
STATA (STATA 16.1; Stata Corp, College Station, TX, USA) was used for all data analyses and graphing. A narrative synthesis approach was used for the current mortality trends. The random-effects model was selected and applied to summarise the overall effective values of 3-year and 5-year CSRs considering the high between-studies heterogeneity (defined as Higgins's I2>50%) [16]. Three-year or 5-year CSRs were reconstructed from Kaplan–Meier survival curves if studies did not report data directly [17]. If the 95% confidence intervals of CSRs were not provided, the following formula was used for calculating: 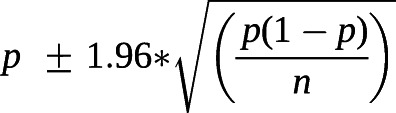 , in which p was defined as CSRs in each included study and n represented the sample size [18]. Non-overlap of the 95% confidence intervals between two subgroups indicates statistical significance, and meta-regression techniques based on random-effects models were used to further test the difference between subgroups if there is a small overlap of the 95% confidence intervals [19].
, in which p was defined as CSRs in each included study and n represented the sample size [18]. Non-overlap of the 95% confidence intervals between two subgroups indicates statistical significance, and meta-regression techniques based on random-effects models were used to further test the difference between subgroups if there is a small overlap of the 95% confidence intervals [19].
Survival trends for IPF were carried out based on various time periods (before 2010, and 2010s). Subgroup analyses for survival outcomes of IPF by various diagnostic criteria (2011 American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Association (ATS/ERS/JRS/ALAT) guideline, 2000 or 2002 ATS/ERS guideline, and other criteria) and treatment (non-antifibrotics, and antifibrotics) were conducted to show diagnostic and therapeutic advances, respectively. Sensitivity analyses for survival outcomes by excluding the studies with extreme data were also performed. In addition, univariate meta-regression was used to investigate the association between age at diagnosis and median year studied. Publication bias and small study effects were explored by using funnel plots and Egger's test [20].
Results
Eligible studies
A total of 14 170 records were retrieved from database searching and hand searching (figure 1). After excluding duplicates, 9588 potentially relevant studies remained for further title and abstract screening. 348 studies were included and assessed for eligibility, and 68 studies [21–88] were finally included in the qualitative analyses. However, only 62 studies [27–88] with sufficient data were eligible for the meta-analyses.
FIGURE 1.

Flow diagram of search progress, informed by PRISMA guidelines.
Study characteristics and quality assessment
Table 1 summarises the characteristics of included studies reporting mortality for IPF. Six studies [21–26] reporting mortality of IPF between 2000 and 2019 were all ecological studies from 22 different countries, with 18 (82%) from Europe (Austria, Belgium, Croatia, Czech Republic, Denmark, Finland, France, Germany, Hungary, Italy, Lithuania, the Netherlands, Poland, Portugal, Romania, Spain, Sweden and UK), two from North America (USA and Canada), one from Oceania (Australia) and one from South America (Brazil). Data on mortality statistics for IPF were from national statistics agencies [21–23, 26], WHO mortality database [25] and regional statistics agencies [24].
TABLE 1.
Summary characteristics of included studies related to mortality for idiopathic pulmonary fibrosis using various ICD codes
| First author (year) [ref.] | Country/region | Years studied | Data sources | Standard population | Incidence (per 100 000) | Mortality trends |
| Algranti (2017) [21] | Brazil | 2000–2014 | National statistics agencies | 2010 Brazilian population | Overall: 0.46–1.10# | Increased |
| Hutchinson (2014) [22] | England and Wales | 2001–2012 | National statistics agencies | 2013 European population | Overall: 4.33–6.90¶; 6.09–8.28# | Increased |
| Australia | 2000–2011 | Overall: 2.56–3.47¶; 4.23–5.08# | Increased | |||
| Canada | 2000–2011 | Overall: 3.06–4.60¶; 5.09–6.38# | Increased | |||
| Spain | 2000–2012 | Overall: 2.78–4.09¶; 3.51–4.64# | Increased | |||
| USA | 2000–2010 | Overall: 3.48–4.12¶; 5.62–6.16# | Increased | |||
| Jeganathan (2021) [23] | USA | 2004–2017 | National health statistics | 2000 US population | Overall: 4.22–3.64# | Decreased |
| Marcon (2021) [24] | Italy | 2008–2019 | Regional statistics agencies | 2013 European population | Males: 2.80#; females: 1.70# | Increased in males aged ≥85 years |
| Marshall (2018) [25] | Austria | 2002–2013 | WHO mortality database | 2013 European population | Males: 2.56–2.34#; females: 0.96–1.29# | Decreased (males only) |
| Belgium | 2001–2013 | Males: 2.63–4.15#; females: 1.43–1.88# | Increased | |||
| Croatia | 2001–2013 | Males: 0.51–0.39#; females: 0.13–0.49# | Decreased (males only) | |||
| Czech Republic | 2001–2013 | Males: 0.77–2.13#; females: 0.46–1.16# | Increased | |||
| Denmark | 2001–2013 | Males: 3.28–1.73#; females: 1.39–0.63# | Decreased | |||
| Finland | 2001–2013 | Males: 4.43–7.36#; females: 2.92–3.62# | Increased | |||
| France | 2001–2013 | Males: 2.63–3.97#; females: 1.27–1.68# | Increased | |||
| Germany | 2001–2013 | Males: 2.80–4.46#; females: 1.43–2.08# | Increased | |||
| Hungary | 2001–2013 | Males: 1.72–2.66#; females: 0.97–1.39# | Increased | |||
| Lithuania | 2001–2013 | Males: 0.24–0.85#; females: 0.10–0.24# | Increased | |||
| The Netherlands | 2001–2013 | Males: 3.56–4.81#; females: 1.61–1.82# | Increased | |||
| Poland | 2001–2013 | Males: 0.75–1.28#; females: 0.44–0.68# | Increased | |||
| Portugal | 2002–2013 | Males: 2.11–4.77#; females: 1.35–2.25# | Increased | |||
| Romania | 2001–2013 | Males: 0.60–0.64#; females: 0.34–0.25# | Decreased (females only) | |||
| Spain | 2001–2013 | Males: 4.81–6.06#; females: 3.02–3.35# | Increased | |||
| Sweden | 2001–2013 | Males: 4.61–6.46#; females: 2.11–2.59# | Increased | |||
| UK | 2001–2013 | Males: 8.16–12.01#; females: 3.61–5.63# | Increased | |||
| Navaratnam (2011) [26] | UK | 2000–2008 | National statistics agencies | 2008 UK population | Overall: 4.40–5.10# | Increased |
Case definition was based on the ICD-10 J84.1 (other interstitial pulmonary diseases with fibrosis) and underlying causes death in all included studies. WHO: World Health Organization; ICD-n: International Classification of Diseases, nth Revision. #: age-standardised rate; ¶: crude mortality rate.
Table 2 shows the characteristics of included studies reporting survival outcomes for IPF. The 62 studies [27–88] reporting survival outcomes of IPF between 1964 and 2017 (these dates indicating the median year of the studies being undertaken) covered 63 307 patients with IPF from 20 different countries, with 90% (n=56) of these studies conducted in Japan (n=9), Korea (n=8), Europe (n=19) and North America (n=20). Most of the survival studies (n=58) were cohort studies. One study [68] including two independent cohorts reported survival outcomes of IPF.
TABLE 2.
Summary characteristics of included studies related to survival outcomes in idiopathic pulmonary fibrosis
| First author (year) [ref.] | Country/region | n | Year studied | Time periods | Diagnostic criteria | Treatment | Age years | 3-year CSRs (%) | 5-year CSRs (%) |
| Adegunsoye (2020) [27] | USA | 240 | 2010–2019 | 2010s | 2011 guideline | Antifibrotics | NA | 62.5 | NA |
| Aggarwal (2017) [28] | USA | 81 | 1985–2014 | 2000s | 2011 guideline | Non-antifibrotics | 63±8.4 | 81.6 | 59.0 |
| Akyil (2016) [29] | Turkey | 92 | 2005–2013 | 2000s | 2011 guideline | Non-antifibrotics | 63.5±10.0 | 45.5 | 30.7 |
| Alakhras (2007) [30] | USA | 197 | 1994–1996 | 1990s | Other criteria | Non-antifibrotics | 71.4±8.9 | 60.8 | NA |
| Alhamad (2008) [31] | Saudi Arabia | 61 | 1996–2005 | 2000s | 2002 guideline | Non-antifibrotics | 54.7±15.2 | 92.8 | 73.7 |
| Antoniou (2020) [32] | Greece | 244 | 2013–2018 | 2010s | 2011 guideline | Antifibrotics | 71.8±7.5 | 59.4 | 58.0 |
| Araki (2003) [33] | Japan | 86 | 1978–1997 | Before 1990 | Other criteria | Non-antifibrotics | 80.5±6.6 | 57.3 | 35.2 |
| Bando (2014) [34] | Japan | 321 | 2006–2010 | 2000s | 2011 guideline | Non-antifibrotics | NA | 73.1 | 59.3 |
| Barlo (2009)# [35] | Netherlands | 113 | 1998–2007 | 2000s | 2002 guideline | Non-antifibrotics | 69±12.7 | 74.8 | 27.1 |
| Bjoraker (1998) [36] | USA | 104 | 1967–1985 | Before 1990 | Other criteria | Non-antifibrotics | 61.7±10.6 | 60.7 | 42.0 |
| Cai (2014) [37] | China | 210 | 1999–2007 | 2000s | 2002 guideline | Non-antifibrotics | 64±10.0 | 46.9 | 39.0 |
| Collard (2004) [38] | US | 82 | 1984–2002 | 1990s | 2000 guideline | Non-antifibrotics | 66.5±7.4 | 62.4 | 42.8 |
| Costabel (2017) [39] | USA | 1058 | 2008–2015 | 2010s | 2011 guideline | Antifibrotics | 68.5±7.5 | 79.3 | 60.5 |
| Doubkova (2017) [40] | Czech Republic | 118 | 2012–2016 | 2010s | 2011 guideline | Antifibrotics | NA | 77.9 | 62.6 |
| Douglas (2000) [41] | USA | 487 | 1994–1996 | 1990s | Other criteria | Non-antifibrotics | NA | 52.1 | NA |
| Fernández Pérez (2010) [42] | USA | 47 | 1997–2005 | 2000s | 2002 guideline | Non-antifibrotics | 73.5±7.9 | 61.9 | 32.5 |
| Gao (2021) [43] | Sweden | 540 | 2014–2020 | 2010s | 2011 guideline | Antifibrotics | 72.7±7.5 | 70.0 | 52.0 |
| Guiot (2018) [44] | Belgium | 82 | 2009–2017 | 2010s | 2011 guideline | Non-antifibrotics | 71.1±9.4 | 57.0 | 38.6 |
| Hamada (2007) [45] | Japan | 61 | 1991–2004 | 1990s | 2000 guideline | Non-antifibrotics | 62.0±8.0 | 64.5 | 47.1 |
| Hopkins (2016) [46] | Canada | 1151 | 2007–2011 | 2000s | Other criteria | Non-antifibrotics | 68.1±11.1 | 63.2 | NA |
| Jacob (2017) [47] | UK | 272 | 2007–2011 | 2000s | 2011 guideline | Non-antifibrotics | NA | 41.8 | 22.5 |
| Jeon (2006) [48] | Korea | 88 | 1996–2002 | 1990s | 2000 guideline | Non-antifibrotics | 60.3±7.5 | 57.0 | 41.0 |
| Jo (2017) [49] | Australia | 647 | 2012–2016 | 2010s | 2011 guideline | Antifibrotics | 70.9±8.5 | 63.0 | NA |
| Kang (2020) [50] | Korea | 948 | 2004–2017 | 2010s | 2011 guideline | Antifibrotics | 65.8±8.3 | 57.8 | 39.0 |
| Kärkkäinen (2017) [51] | Finland | 132 | 2002–2012 | 2000s | Other criteria | Non-antifibrotics | 70.5±9.8 | 56.4 | 36.7 |
| Kaunisto (2019) [52] | Finland | 453 | 2011–2015 | 2010s | 2011 guideline | Antifibrotics | 73.0±9.0 | 70.0 | 45.0 |
| Kim (2012) [54] | Korea | 67 | 1996–2007 | 2000s | 2011 guideline | Non-antifibrotics | 69.9±9.9 | 86.5 | 78.3 |
| Kim (2015) [53] | Korea | 268 | 2005–2009 | 2000s | 2011 guideline | Non-antifibrotics | 65.9±9.6 | 69.0 | 53.9 |
| Ko (2021) [55] | Korea | 42 777 | 2006–2016 | 2010s | Other criteria | Antifibrotics | 64.6±13.8 | 71.9 | 62.9 |
| Kondoh (2005) [56] | Japan | 27 | 1991–1998 | 1990s | 2000 guideline | Non-antifibrotics | 56±10.9 | 62.4 | 40.8 |
| Koo (2016) [57] | Korea | 1663 | 2003–2007 | 2000s | 2002 guideline | Non-antifibrotics | NA | 62.6 | 49.2 |
| Kreuter (2016) [58] | Germany | 272 | 2004–2012 | 2000s | 2011 guideline | Non-antifibrotics | 68.5±9.0 | 54.8 | 40.8 |
| Kurashima (2010) [59] | Japan | 362 | 1997–2006 | 2000s | Other criteria | Non-antifibrotics | 72.9±8.1 | 79.6 | 69.4 |
| Lai (2019) [60] | Taiwan | 114 | 2006–2016 | 2010s | 2011 guideline | Non-antifibrotics | 77.8±9.4 | 53.0 | 37.5 |
| Lassenius (2019) [61] | Finland | 266 | 2005–2017 | 2010s | Other criteria | Non-antifibrotics | 74.3±8.5 | 66.2 | 47.0 |
| Le Rouzic (2015) [62] | France | 66 | 2000–2010 | 2000s | 2000 guideline | Non-antifibrotics | NA | 53.5 | 34.9 |
| Lindell (2015) [63] | USA | 404 | 2000–2012 | 2000s | Other criteria | Non-antifibrotics | 71.5±9.2 | 41.8 | 31.0 |
| Mancuzo (2018) [64] | Brazil | 70 | 1993–2017 | 2000s | 2011 guideline | Non-antifibrotics | 71.9±6.4 | 67.2 | 41.4 |
| Mapel (1998) [65] | USA | 209 | 1988–1992 | 1990s | Other criteria | Non-antifibrotics | 71.7±12.3 | 73.0 | 64.0 |
| Margaritopoulos (2018) [66] | Greece | 82 | 2011–2016 | 2010s | 2011 guideline | Antifibrotics | 74.9±11.0 | 73.0 | 54.7 |
| Mejia (2009) [67] | Mexico | 110 | 1996–2006 | 2000s | 2000 guideline | Non-antifibrotics | 63.0±10.0 | 42.0 | NA |
| Moon (2021)¶ [68] | Korea | 689 | 2000–2008 | 2000s | 2000 guideline | Non-antifibrotics | 68.0±9.0 | 50.2 | NA |
| Moon (2021)¶ [68] | Korea | 656 | 2010–2018 | 2010s | 2011 guideline | Antifibrotics | 68.0±8.0 | 70.5 | NA |
| Mura (2012) [69] | Italy | 70 | 2005–2007 | 2000s | 2000 guideline | Non-antifibrotics | 67.0±8.0 | 54.0 | NA |
| Nadrous (2004) [70] | US | 476 | 1994–1996 | 1990s | Other criteria | Non-antifibrotics | 70.6±9.0 | 47.7 | NA |
| Nathan (2020) [71] | USA | 436 | 2007–2016 | 2010s | 2011 guideline | Antifibrotics | 67.0±8.9 | 58.0 | 34.4 |
| Natsuizaka (2014) [72] | Japan | 553 | 2003–2007 | 2000s | 2000 guideline | Non-antifibrotics | 70.0±9.0 | 49.2 | 33.4 |
| Nicholson (2000) [73] | USA | 78 | 1978–1989 | Before 1990 | Other criteria | Non-antifibrotics | 57.2±7.1 | 62.1 | 41.3 |
| Ogawa (2018) [74] | Japan | 46 | 2009–2014 | 2010s | 2011 guideline | Antifibrotics | NA | 53.2 | NA |
| Reid (2015) [75] | Germany | 27 | 2005–2009 | 2000s | 2000 guideline | Non-antifibrotics | NA | 63.1 | 33.5 |
| Ryerson (2013) [76] | USA | 192 | 2000–2012 | 2000s | 2011 guideline | Non-antifibrotics | 69.9±8.7 | 47.5 | 24.1 |
| Shin (2008) [77] | USA | 108 | 1996–2004 | 2000s | Other criteria | Non-antifibrotics | 63.0±7.4 | NA | 54.1 |
| Strand (2014) [78] | USA | 321 | 1985–2011 | 1990s | 2000 guideline | Non-antifibrotics | 66.1±9.1 | 64.9 | 44.9 |
| Strongman (2018) [79] | UK | 555 | 2000–2012 | 2000s | Other criteria | Non-antifibrotics | NA | NA | 32.0 |
| Su (2011) [80] | USA | 148 | 2002–2009 | 2000s | 2002 guideline | Non-antifibrotics | 68.6±12.1 | 61.0 | 53.0 |
| Sugino (2014) [81] | Japan | 108 | 2003–2010 | 2000s | 2000 guideline | Non-antifibrotics | 71.4±6.7 | 53.8 | 31.6 |
| Tarride (2018) [82] | Canada | 1,673 | 2006–2011 | 2000s | Other criteria | Non-antifibrotics | 76.8±12.0 | 37.4 | NA |
| Tran (2020) [83] | Europe | 1620 | 1996–2008 | 2000s | 2011 guideline | Non-antifibrotics | 67.6±8.9 | 65.5 | 46.4 |
| Turner-Warwick (1980) [84] | UK | 181 | 1955–1973 | Before 1990 | Other criteria | Non-antifibrotics | 57.6±11.3 | 57.7 | 43.8 |
| Vietri (2020) [85] | Italy | 91 | 2011–2013 | 2010s | 2011 guideline | Antifibrotics | 68.5±7.7 | 67.5 | NA |
| Watanabe (2019) [86] | Japan | 32 | 2008–2018 | 2010s | 2011 guideline | Antifibrotics | NA | 74.6 | 49.8 |
| Zhang (2016) [87] | China | 192 | 2001–2013 | 2000s | 2011 guideline | Non-antifibrotics | 66.0±8.5 | NA | 55.5 |
| Zurkova (2019) [88] | Czech Republic | 383 | 2012–2017 | 2010s | 2011 guideline | Antifibrotics | NA | NA | 47.1 |
Age values are presented as mean±sd. Data in bold are extracted from Kaplan–Meier curves. IPF: idiopathic pulmonary fibrosis; 2000 guideline: 2000 ATS/ERS guideline; 2002 guideline: 2002 ATS/ERS guideline; 2011 guideline: 2011 ATS/ERS/JRS/LATA guideline; Other criteria: all other diagnostic criteria combined (such as clinical, radiographic and biopsy criteria); ATS: American Thoracic Society; ERS: European Respiratory Society; JRS: Japanese Respiratory Society; LATA: Latin American Thoracic Association; n: number of participants; NA: not applicable; CSRs: cumulative survival rates. #: non-English (Netherlandish) study; ¶: one study including two independent cohorts.
In terms of quality assessment, a detailed scoring for each study has been provided in supplementary table S5. The median index of quality score for cohort studies (69.2%) was higher than ecological studies (50.0%) due to cohort studies having robust case definition criteria (clinical guidelines) compared to ecological studies (ICD codes). Median index of the quality score was 69.2% (IQR 65.4–73.1) for all included studies (supplementary figure S1). Only one study [46] was low quality, while 68% (n=46) and 31% (n=21) of all included studies were ranked as moderate and high level of quality, respectively.
Mortality trends for IPF in various countries
The six ecological studies reporting mortality rates of IPF since the year 2000 used a relatively narrower case definition of IPF (ICD-10 J84.1) and regarded IPF as the UCD. These data suggested that crude mortality rates have increased from 2 to 7 per 100 000 population per year in five regions (England and Wales, Australia, Canada, Spain and the USA) between 2000 and 2012 (table 1). Age-standardised mortality for IPF varied from ∼0.5 to ∼12 per 100 000 population per year in 22 different countries, being lowest in Brazil, Croatia, Czech Republic, Lithuania, Poland and Romania, and being highest in the UK. There were increased mortality trends for IPF in Australia, Brazil, Belgium, Canada, Czech Republic, Finland, France, Germany, Hungary, Italy (males aged ≥85 years only), Lithuania, the Netherlands, Poland, Portugal, Spain, Sweden and the UK, while Austria (males only), Croatia (males only), Denmark, Romania (females only) and the USA (between 2004 and 2017) showed decreased mortality trends.
Survival trends for IPF in various time periods
The overall 3-year CSRs (based on 59 studies with 62 069 patients) and 5-year CSRs (based on 50 studies with 56 774 patients) were 61.8% (95% CI 58.7–64.9; I2=97.1%) and 45.6% (95% CI 41.5–49.7; I2=97.7%), respectively (table 3). Prior to 2010, the pooled 3-year and 5-year CSRs were 59.9% (95% CI 55.8–64.1; I2=95.8%) and 44.1% (95% CI 39.9–48.3; I2=93.7%), then increased to 66.2% (95% CI 62.9–69.5; I2=92.6%) and 49.3% (95% CI 42.7–55.9; I2=97.7%) in the 2010s, respectively. However, a test for difference between two subgroups (before 2010 versus 2010s) was not statistically significant (p=0.067 for 3-year CSRs and p=0.203 for 5-year CSRs). After excluding three studies [44, 60, 61] in which no patients received antifibrotics after year 2010, the overall 3-year and 5-year CSRs remained consistent, while the pooled 3-year CSRs significantly increased to 67.4% (95% CI 63.9–70.9; I2=93.1%) in the 2010s after test for difference between two subgroups (p=0.039). Figure 2 shows that the pooled 3-year and 5-year CSRs remained consistently low before 2010, following by an improving picture in the 2010s decade.
TABLE 3.
Subgroup analyses for pooled analyses of survival by various time periods
| Baseline analyses | Sensitivity analyses# | |||||
| n | CSRs (95% CI) | I2 (%) | n | CSRs (95% CI) | I2 (%) | |
| 3-year CSRs | ||||||
| Overall | 59 | 61.8 (58.7–64.9) | 97.1 | 56 | 61.9 (58.7–65.1) | 97.2 |
| Before 2010 | 41 | 59.9 (55.8–64.1) | 95.8 | 41 | 59.9 (55.8–64.1) | 95.8 |
| 2010s | 18 | 66.2 (58.7–64.9) | 92.6 | 15 | 67.4 (63.9–70.9) | 93.1 |
| Test for difference¶ | p=0.067 | p=0.039 | ||||
| 5-year CSRs | ||||||
| Overall | 50 | 45.6 (41.5–49.7) | 97.7 | 47 | 45.9 (41.6–50.1) | 97.8 |
| Before 2010 | 36 | 44.1 (39.9–48.3) | 93.7 | 36 | 44.1 (39.9–48.3) | 93.7 |
| 2010s | 14 | 49.3 (42.7–49.7) | 97.7 | 11 | 51.4 (44.1–58.7) | 93.9 |
| Test for difference¶ | p=0.203 | p=0.106 | ||||
I2 >50% represents high between-studies heterogeneity. CSRs: cumulative survival rates; n: number of included studies. #: exclusion of three studies in which no patients received antifibrotics after year 2010; ¶: test for difference between subgroups (before 2010 versus 2010s).
FIGURE 2.

a) and c) Subgroup analyses for survival rates by various time periods; b) and d) after exclusion of three studies in which no patients received antifibrotics after year 2010. I2>50% represents high between-studies heterogeneity. IPF: idiopathic pulmonary fibrosis; n: number of included studies.
Subgroup analysis by various treatment and diagnostic criteria
Figure 3 presents the outcomes of the pooled 3-year and 5-year CSRs by the various pharmaceutical regimens. Patients taking antifibrotics (67.4%; 95% CI 63.9–70.9; I2=93.1%) had significantly (p=0.032) higher pooled 3-year CSRs than those taking non-antifibrotics (59.8%; 95% CI 59.8–63.8; I2 =95.5%). Similar trend was found for the 5-year CSRs (patients taking antifibrotics: 51.4% (95% CI 44.1–58.7; I2=97.9%) versus those taking non-antifibrotics: 43.9% (95% CI 39.9–47.8; I2 =93.4%)) (p=0.084). In addition, there were no significant associations between various diagnostic criteria and CSRs, and those associations remained consistent after exclusion of 16 studies [27, 32, 39, 40, 43, 49, 50, 52, 55, 66, 68, 71, 74, 85, 86, 88] in which patients received antifibrotics (table 4).
FIGURE 3.

Subgroup analyses for cumulative survival rates (CSRs) by various pharmaceutical regimens. a) 3-year CSRs; b) 5-year CSRs. a) Test for difference between subgroups = 0.032; b) test for difference between subgroups = 0.084.
TABLE 4.
Subgroup analyses for pooled analyses of survival by various diagnostic criteria
| Baseline analyses | Sensitivity analyses# | |||||
| n | CSRs (95% CI) | I2 (%) | n | CSRs (95% CI) | I2 (%) | |
| 3-year CSRs | ||||||
| Overall | 59 | 61.8 (58.7–64.9) | 97.1 | 44 | 59.8 (55.9–63.8) | 95.5 |
| 2011 guideline | 26 | 64.7 (60.8–68.6) | 93.2 | 12 | 61.9 (55.0–63.8) | 93.5 |
| 2000 or 2002 guideline | 17 | 60.4 (54.6–66.3) | 91.0 | 17 | 60.4 (54.6–66.3) | 91.0 |
| Other criteria | 16 | 58.6 (50.7–66.5) | 98.9 | 15 | 57.6 (50.4–64.9) | 97.1 |
| Test for difference¶ | p=0.105 | p=0.360 | ||||
| 5-year CSRs | ||||||
| Overall | 50 | 45.6 (41.5–49.7) | 97.7 | 39 | 43.9 (39.9–47.8) | 93.4 |
| 2011 guideline | 23 | 47.3 (42.3–52.2) | 94.7 | 13 | 45.1 (37.5–52.7) | 94.9 |
| 2000 or 2002 guideline | 15 | 41.8 (36.4–47.2) | 87.5 | 15 | 41.8 (36.4–47.2) | 87.5 |
| Other criteria | 12 | 46.7 (37.2–56.2) | 98.1 | 11 | 45.2 (36.0–54.4) | 95.6 |
| Test for difference¶ | p=0.421 | p=0.991 | ||||
I2 >50% represents high between-studies heterogeneity. 2000 guideline: 2000 ATS/ERS guideline; 2002 guideline: 2002 ATS/ERS guideline; 2011 guideline: 2011 ATS/ERS/JRS/LATA guideline; Other criteria: all other diagnostic criteria combined (such as clinical, radiographic and biopsy criteria); ATS: American Thoracic Society; ERS: European Respiratory Society; JRS: Japanese Respiratory Society; LATA: Latin American Thoracic Association; n: number of included studies; CSRs: cumulative survival rates. #: exclusion of 16 studies in which patients received antifibrotics; ¶: test for difference between subgroups.
Association between mean age at diagnosis and median year studied
There were 51 studies reporting mean±sd age at diagnosis, which significantly (p=0.002) increased by 0.26 year (95% CI 0.10–0.41) for each 1-year increase in the median year studied between 1980 and 2020. This association did not change dramatically after removing four outlier studies [31, 36, 84, 88] (the orange circles in figure 4) in a sensitivity analysis.
FIGURE 4.

Association between mean age at diagnosis and median year studied between 1960 and 2020 by using univariate meta-regression. Each size of the bubble depends on the weights in the random-effects models. Orange circles show studies removed for sensitivity analysis with extreme data points or before year 1980. Coef.: coefficient.
Publication bias
Funnel plots (supplementary figure S3) for assessing the influence of each included study on the overall meta-analysis estimates identified several outliers, but Egger's test found no evidence for publication bias for the 3-year CSRs (bias=0.25, p=0.854) or 5-year CSRs (bias= −0.67, p=0.591).
Discussion
We found that the age-standardised mortality rates for IPF ranged from 0.5 to 12 per 100 000 population per year after year 2000, carrying a burden as severe as several cancers including those of oesophagus, pancreas and prostate, but without the same prominence in screening, management, surveillance, research and disease control [89]. Our data suggest no increased survival trend for patients with IPF up to year 2010, while there might be an increase thereafter. Patients with IPF taking antifibrotics had significantly higher long-term survival compared to those not on antifibrotics, which reinforces the beneficial messages from drug-development studies, but this should be interpreted in the context of high heterogeneity.
The lack of age adjustment for much of mortality data has proved to be a significant limitation. Therefore, we described the age-standardised mortality rates for IPF based on a narrower definition (ICD-10 J84.1) and UCD across various countries different from the previous systematic review [3]. We found that age-standardised mortality for IPF has varied worldwide since 2000. There were increased mortality trends for IPF in Brazil [21], Australia [22], Canada [22] and many European countries (Belgium, Canada, Czech Republic, Finland, France, Germany, Hungary, Italy, Lithuania, the Netherlands, Poland, Portugal, Spain, Sweden and the UK) with the exception of Austria, Denmark, Croatia and Romania [24–26]. Hutchinson et al. [22] reported that there was an increased mortality trend (age adjusted for 2013 European population) in the USA ranging from 5.62 to 6.16 per 100 000 population per year between 2000 and 2010, while a more recent study [23] found a decreased mortality trend (age adjusted for 2000 US population) ranging from 4.22 to 3.64 per 100 000 population per year between 2004 and 2017, which may be attributed to a decline in smoking or changes in other environmental and genetic factors.
Recently, Khor et al. [5] conducted a systematic review reporting a mortality of 69% beyond 5 years for patients with IPF without taking antifibrotics based on 170 included studies, and 34 of them were also included in the current study. We had different study aims to those of Khor et al. because: 1) we summarised annual mortality rates for IPF based on population-based studies and presented the changing trends in various countries; and 2) we investigated survival trends over various time periods including both patients with and without antifibrotics.
The lack of evidence for the improvement in the survival trends of IPF up to year 2010 might be explained by two main causes. First, the advanced populations and higher age at diagnosis were used in that earlier era. Nearly 90% of included studies reporting survival outcomes were from countries with ageing populations, with the mean age at diagnosis of IPF having significantly increased over the past six decades. Second, routinely used immunosuppressive combination drugs were used for IPF in that earlier era. Cortisone was first used to treat IPF in 1948 [90] and several subsequent studies [91–93] purported to demonstrate that corticosteroids might improve lung function and prolong survival, so that it became the first-line therapy for IPF essentially from the 1950s. In 2012, multicentre RCTs suggested the significant harmful effects and decreased survival for patients with IPF using the combination of prednisone, azathioprine and N-acetylcysteine compared to those using placebo [94]. Since then, the usage of steroid/immunosuppressive drug combinations has rapidly reduced.
Shortly after the “downfall” of the established steroid/immunosuppressant era, in 2014, a substantial breakthrough was made for two antifibrotic drugs that had been confirmed to be effective in treating IPF through several multicentre RCTs [8, 9]. In 2017, Costabel et al. [39] provided the long-term safety evidence for pirfenidone after following an open-label extension study of RCTs. We found that there may be beneficial effects of antifibrotic therapy on the long-term survival of patients with IPF, which was in accordance with the finding of a systematic review [95], including 8 RCTs and 18 cohort studies, that reported antifibrotic treatment might reduce the risk of all-cause mortality in IPF. However, we did not detect such an association between diagnostic criteria and long-term survival outcomes of patients with IPF.
We can draw several clinical observations from our review. First, IPF carries mortality burdens as bad as several cancers, but with less attention being given to it in general, perhaps because it largely affects a more elderly population and is more insidious and less dramatic at onset. Second, our summaries for the mortality and survival of IPF internationally might help stimulate future studies to consider the issues about surveillance, disease control and development of new therapies. Third, the likely impact at a population level of harmful but widely used treatments in the past for IPF emphasises the vital importance of adequately powered RCTs in guiding IPF therapy. Further, there might be some signals emerging for an improvement in long-term survival related to the relatively newly available antifibrotic drugs for IPF.
Our study, however, is not without limitations. First, although ICD-10 code J84.1 is the most specific code for IPF to present mortality statistics in the study timeframe [3], it may be inherently inaccurate due to the inclusion of other IIPs. Future studies reporting mortality statistics for IPF should use stricter and narrower ICD codes (e.g., ICD-11 CB03.4). Second, patients who were misdiagnosed with IPF may have superior survival due to diagnostic inaccuracies (e.g., ICD codes), which may influence the survival trend for IPF in various time periods. Further, a review with such inherent heterogeneity due to drawing together various types of work worldwide (with different data sources, study designs and study methodologies) makes our conclusions rather provisional. Lastly, studies showing favourable effects of antifibrotic drugs are more likely to have been published in recent years, and there might be reporting biases that better holistic management of patients with IPF might contribute to improved survival.
In conclusion, IPF is a diagnosis associated with high mortality, similar to that seen in several cancers, though there is much less recognition of IPF in the population, press or research funding agendas. Lack of improvement in survival trends for IPF worldwide before 2010 may be related to changing age profiles at diagnosis or the prevailing therapeutic regimens, which have since been proven to have negative effects. Substantial therapeutic advances after 2010 might have contributed to the increased survival trends. Further, there might be some signals emerging for an improvement in long-term survival related specifically to the newly available antifibrotic drugs.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00591-2021.SUPPLEMENT (679.8KB, pdf)
Acknowledgement
We thank all authors of reviewed studies for the effective and timely communication.
Provenance: Submitted article, peer reviewed.
Author contribution: I.A. Cox, Q. Zheng, B. de Graaff and A.J. Palmer worked on protocol development. Q. Zheng, I.A, Cox and J.A. Campbell collected data. Q. Zheng, E.H. Walters, Q. Xia, P. Otahal, I.A. Cox and A.J. Palmer analysed and interpreted data. Q. Zheng wrote the draft of manuscript. All authors contributed to review and agreed the manuscript.
Conflict of interest: T.J. Corte reports grants, personal fees and nonfinancial support from Boehringer Ingelheim and Hoffman La Rochel grants and personal fees from Bristol Myers Squibb; grants from Avalyn Pharma and Biogen; and personal fees from Promedior, outside the submitted work. A.K.Y. Teoh reports conference fees from Boehringer Ingelheim and speaker fees from Roche outside the submitted work. The other co-authors declare no competing interests.
Support statement: This study is funded through the NHMRC Centre of Research Excellence in Pulmonary Fibrosis (GNT1116371); and by foundation partner Boehringer Ingelheim, and programme partners Roche and Galapagos. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med 2018; 378: 1811–1823. doi: 10.1056/NEJMra1705751 [DOI] [PubMed] [Google Scholar]
- 2.Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. The Lancet 2017; 389: 1941–1952. doi: 10.1016/S0140-6736(17)30866-8 [DOI] [PubMed] [Google Scholar]
- 3.Hutchinson J, Fogarty A, Hubbard R, et al. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46: 795–806. doi: 10.1183/09031936.00185114 [DOI] [PubMed] [Google Scholar]
- 4.Levin KA. Study design VI – ecological studies. Evid Based Dent 2006; 7: 108. doi: 10.1038/sj.ebd.6400454 [DOI] [PubMed] [Google Scholar]
- 5.Khor YH, Ng Y, Barnes H, et al. Prognosis of idiopathic pulmonary fibrosis without anti-fibrotic therapy: a systematic review. Eur Respir Rev 2020; 29: 190158. doi: 10.1183/16000617.0158-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med 2018; 6: 138–153. doi: 10.1016/S2213-2600(17)30433-2 [DOI] [PubMed] [Google Scholar]
- 7.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183: 788–824. doi: 10.1164/rccm.2009-040GL [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.King TE, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2083–2092. doi: 10.1056/NEJMoa1402582 [DOI] [PubMed] [Google Scholar]
- 9.Richeldi L, Du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2071–2082. doi: 10.1056/NEJMoa1402584 [DOI] [PubMed] [Google Scholar]
- 10.Moher D, Liberati A, Tetzlaff J, et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 2009; 6: e1000097. doi: 10.1371/journal.pmed.1000097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2018; 198: e44–e68. doi: 10.1164/rccm.201807-1255ST [DOI] [PubMed] [Google Scholar]
- 12.Nakamura Y, Suda T. Idiopathic pulmonary fibrosis: diagnosis and clinical manifestations. Clin Med Insights Circ Respir Pulm Med 2015; 9: Suppl. 1, 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lomas A, Leonardi-Bee J, Bath-Hextall F. A systematic review of worldwide incidence of nonmelanoma skin cancer. Br J Dermatol 2012; 166: 1069–1080. doi: 10.1111/j.1365-2133.2012.10830.x [DOI] [PubMed] [Google Scholar]
- 14.Prins J, Blanker MH, Bohnen AM, et al. Prevalence of erectile dysfunction: a systematic review of population-based studies. Int J Impot Res 2002; 14: 422–432. doi: 10.1038/sj.ijir.3900905 [DOI] [PubMed] [Google Scholar]
- 15.Munn Z, Moola S, Riitano D, et al. The development of a critical appraisal tool for use in systematic reviews addressing questions of prevalence. Int J Health Policy Manag 2014; 3: 123–128. doi: 10.15171/ijhpm.2014.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sedgwick P. Meta-analyses: heterogeneity and subgroup analysis. BMJ 2013; 346: f4040. [DOI] [PubMed] [Google Scholar]
- 17.Guyot P, Ades A, Ouwens MJ, et al. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol 2012; 12: 9. doi: 10.1186/1471-2288-12-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borkowf CB. Constructing binomial confidence intervals with near nominal coverage by adding a single imaginary failure or success. Stat Med 2006; 25: 3679–3695. doi: 10.1002/sim.2469 [DOI] [PubMed] [Google Scholar]
- 19.Higgins JPT GS. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0. The Cochrane Collaboration, 2011. https://handbook-5-1.cochrane.org/ Date last accessed: 1 March 2020. Date last updated: March 2011.
- 20.Egger M, Smith GD, Schneider M, et al. Bias in meta-analysis detected by a simple, graphical test. BMJ 1997; 315: 629–634. doi: 10.1136/bmj.315.7109.629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Algranti E, Saito CA, Silva D, et al. Mortality from idiopathic pulmonary fibrosis: a temporal trend analysis in Brazil, 1979–2014. J Bras Pneumol 2017; 43: 445–450. doi: 10.1590/s1806-37562017000000035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hutchinson JP, McKeever TM, Fogarty AW, et al. Increasing global mortality from idiopathic pulmonary fibrosis in the twenty-first century. Ann Am Thorac Soc 2014; 11: 1176–1185. doi: 10.1513/AnnalsATS.201404-145OC [DOI] [PubMed] [Google Scholar]
- 23.Jeganathan N, Smith RA, Sathananthan M. Mortality trends of idiopathic pulmonary fibrosis in the United States from 2004 through 2017. Chest 2021; 159: 228–238. doi: 10.1016/j.chest.2020.08.016 [DOI] [PubMed] [Google Scholar]
- 24.Marcon A, Schievano E, Fedeli U. Mortality associated with idiopathic pulmonary fibrosis in Northeastern Italy, 2008–2020: a multiple cause of death analysis. Int J Environ Res Public Health 2021; 18: 7249. doi: 10.3390/ijerph18147249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marshall DC, Salciccioli JD, Shea BS, et al. Trends in mortality from idiopathic pulmonary fibrosis in the European Union: an observational study of the WHO mortality database from 2001–2013. Eur Respir J 2018; 51: 1701603. doi: 10.1183/13993003.01603-2017 [DOI] [PubMed] [Google Scholar]
- 26.Navaratnam V, Fleming KM, West J, et al. The rising incidence of idiopathic pulmonary fibrosis in the UK. Thorax 2011; 66: 462–467. doi: 10.1136/thx.2010.148031 [DOI] [PubMed] [Google Scholar]
- 27.Adegunsoye A, Alqalyoobi S, Linderholm A, et al. Circulating plasma biomarkers of survival in antifibrotic-treated patients with idiopathic pulmonary fibrosis. Chest 2020; 158: 1526–1534. doi: 10.1016/j.chest.2020.04.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aggarwal R, McBurney C, Schneider F, et al. Myositis-associated usual interstitial pneumonia has a better survival than idiopathic pulmonary fibrosis. Rheumatology 2017; 56: 384–389. [DOI] [PubMed] [Google Scholar]
- 29.Akyil FT, Sevim T, Akman C, et al. The predictors of mortality in IPF: does emphysema change the prognosis? Sarcoidosis Vasc Diffuse Lung Dis 2016; 33: 267–274. [PubMed] [Google Scholar]
- 30.Alakhras M, Decker PA, Nadrous HF, et al. Body mass index and mortality in patients with idiopathic pulmonary fibrosis. Chest 2007; 131: 1448–1453. doi: 10.1378/chest.06-2784 [DOI] [PubMed] [Google Scholar]
- 31.Alhamad EH, Masood M, Shaik SA, et al. Clinical and functional outcomes in Middle Eastern patients with idiopathic pulmonary fibrosis. Clin Respir J 2008; 2: 220–226. doi: 10.1111/j.1752-699X.2008.00070.x [DOI] [PubMed] [Google Scholar]
- 32.Antoniou K, Markopoulou K, Tzouvelekis A, et al. Efficacy and safety of nintedanib in a Greek multicentre idiopathic pulmonary fibrosis registry: a retrospective, observational, cohort study. ERJ Open Res 2020; 6: 00172-02019. doi: 10.1183/23120541.00172-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Araki T, Katsura H, Sawabe M, et al. A clinical study of idiopathic pulmonary fibrosis based on autopsy studies in elderly patients. Intern Med 2003; 42: 483–489. doi: 10.2169/internalmedicine.42.483 [DOI] [PubMed] [Google Scholar]
- 34.Bando M, Sugiyama Y, Azuma A, et al. A prospective survey of idiopathic interstitial pneumonias in a web registry in Japan. Respir Investig 2015; 53: 51–59. doi: 10.1016/j.resinv.2014.11.001 [DOI] [PubMed] [Google Scholar]
- 35.Barlo NP, Van Moorsel CHM, Van Den Bosch JMM, et al. Idiopathic pulmonary fibrosis: description of a Dutch cohort. Ned Tijdschr Geneeskd 2009; 153: 1260–1265. [PubMed] [Google Scholar]
- 36.Bjoraker JA, Ryu JH, Edwin MK, et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998; 157: 199–203. doi: 10.1164/ajrccm.157.1.9704130 [DOI] [PubMed] [Google Scholar]
- 37.Cai MT, Zhu M, Ban CJ, et al. Clinical features and outcomes of 210 patients with idiopathic pulmonary fibrosis. Chin Med J (Engl) 2014; 127: 1868–1873. [PubMed] [Google Scholar]
- 38.Collard HR, Ryu JH, Douglas WW, et al. Combined corticosteroid and cyclophosphamide therapy does not alter survival in idiopathic pulmonary fibrosis. Chest 2004; 125: 2169–2174. doi: 10.1378/chest.125.6.2169 [DOI] [PubMed] [Google Scholar]
- 39.Costabel U, Albera C, Lancaster LH, et al. An open-label study of the long-term safety of Pirfenidone in patients with idiopathic pulmonary fibrosis (RECAP). Respiration 2017; 94: 408–415. doi: 10.1159/000479976 [DOI] [PubMed] [Google Scholar]
- 40.Doubková M, Švancara J, Svoboda M, et al. EMPIRE Registry, Czech Part: Impact of demographics, pulmonary function and HRCT on survival and clinical course in idiopathic pulmonary fibrosis. Clin Respir J 2018; 12: 1526–1535. doi: 10.1111/crj.12700 [DOI] [PubMed] [Google Scholar]
- 41.Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med 2000; 161: 1172–1178. doi: 10.1164/ajrccm.161.4.9907002 [DOI] [PubMed] [Google Scholar]
- 42.Fernández Pérez ER, Daniels CE, St. Sauver J, et al. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis. Chest 2010; 137: 129–137. doi: 10.1378/chest.09-1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao J, Kalafatis D, Carlson L, et al. Baseline characteristics and survival of patients of idiopathic pulmonary fibrosis: a longitudinal analysis of the Swedish IPF Registry. Respir Res 2021; 22: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guiot J, Duysinx B, Seidel L, et al. Clinical experience in idiopathic pulmonary fibrosis: a retrospective study. Acta Clin Belg 2018; 73: 139–143. doi: 10.1080/17843286.2017.1399228 [DOI] [PubMed] [Google Scholar]
- 45.Hamada K, Nagai S, Tanaka S, et al. Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest 2007; 131: 650–656. doi: 10.1378/chest.06-1466 [DOI] [PubMed] [Google Scholar]
- 46.Hopkins RB, Burke N, Fell C, et al. Epidemiology and survival of idiopathic pulmonary fibrosis from national data in Canada. Eur Respir J 2016; 48: 187–195. doi: 10.1183/13993003.01504-2015 [DOI] [PubMed] [Google Scholar]
- 47.Jacob J, Bartholmai BJ, Rajagopalan S, et al. Functional and prognostic effects when emphysema complicates idiopathic pulmonary fibrosis. Eur Respir J 2017; 50: 1700379. doi: 10.1183/13993003.00379-2017 [DOI] [PubMed] [Google Scholar]
- 48.Jeon K, Chung MP, Lee KS, et al. Prognostic factors and causes of death in Korean patients with idiopathic pulmonary fibrosis. Respir Med 2006; 100: 451–457. doi: 10.1016/j.rmed.2005.06.013 [DOI] [PubMed] [Google Scholar]
- 49.Jo HE, Glaspole I, Grainge C, et al. Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian Idiopathic Pulmonary Fibrosis Registry. Eur Respir J 2017; 49: 1601592. doi: 10.1183/13993003.01592-2016 [DOI] [PubMed] [Google Scholar]
- 50.Kang J, Han M, Song JW. Antifibrotic treatment improves clinical outcomes in patients with idiopathic pulmonary fibrosis: a propensity score matching analysis. Sci Rep 2020; 10: 15620. doi: 10.1038/s41598-020-72607-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kärkkäinen M, Kettunen H-P, Nurmi H, et al. Effect of smoking and comorbidities on survival in idiopathic pulmonary fibrosis. Respir Res 2017; 18: 160. doi: 10.1186/s12931-017-0642-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaunisto J, Salomaa E-R, Hodgson U, et al. Demographics and survival of patients with idiopathic pulmonary fibrosis in the Finnish IPF registry. ERJ Open Res 2019; 5: 00170-02018. doi: 10.1183/23120541.00170-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim ES, Choi SM, Lee J, et al. Validation of the GAP score in Korean patients with idiopathic pulmonary fibrosis. Chest 2015; 147: 430–437. doi: 10.1378/chest.14-0453 [DOI] [PubMed] [Google Scholar]
- 54.Kim JH, Lee JH, Ryu YJ, et al. Clinical predictors of survival in idiopathic pulmonary fibrosis. Tuberc Respir Dis (Seoul) 2012; 73: 162–168. doi: 10.4046/trd.2012.73.3.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ko SJ, Choi SM, Han K-D, et al. All-cause mortality of patients with idiopathic pulmonary fibrosis: a nationwide population-based cohort study in Korea. Sci Rep 2021; 11: 15145. doi: 10.1038/s41598-021-94655-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kondoh Y, Taniguchi H, Yokoi T, et al. Cyclophosphamide and low-dose prednisolone in idiopathic pulmonary fibrosis and fibrosing nonspecific interstitial pneumonia. Eur Respir J 2005; 25: 528–533. doi: 10.1183/09031936.05.00071004 [DOI] [PubMed] [Google Scholar]
- 57.Koo S-M, Uh S-T, Kim DS, et al. Relationship between survival and age in patients with idiopathic pulmonary fibrosis. J Thorac Dis 2016; 8: 3255–3264. doi: 10.21037/jtd.2016.11.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kreuter M, Ehlers-Tenenbaum S, Palmowski K, et al. Impact of comorbidities on mortality in patients with idiopathic pulmonary fibrosis. PLoS ONE 2016; 11: e0151425. doi: 10.1371/journal.pone.0151425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurashima K, Takayanagi N, Tsuchiya N, et al. The effect of emphysema on lung function and survival in patients with idiopathic pulmonary fibrosis. Respirology 2010; 15: 843–848. doi: 10.1111/j.1440-1843.2010.01778.x [DOI] [PubMed] [Google Scholar]
- 60.Lai RS, Chen CF, Chu KA, et al. The effect of emphysema on survival in patients with idiopathic pulmonary fibrosis: a retrospective study in Taiwan. J Chin Med Assoc 2019; 82: 922–928. doi: 10.1097/JCMA.0000000000000201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lassenius MI, Toppila I, Pöntynen N, et al. Forced Vital Capacity (FVC) decline, mortality and healthcare resource utilization in idiopathic pulmonary fibrosis. Eur Clin Respir J 2020; 7: 1702618. doi: 10.1080/20018525.2019.1702618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Le Rouzic O, Bendaoud S, Chenivesse C, et al. Prognostic value of the initial chest high-resolution CT pattern in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2015; 32: 353–359. [PubMed] [Google Scholar]
- 63.Lindell KO, Liang Z, Hoffman LA, et al. Palliative care and location of death in decedents with idiopathic pulmonary fibrosis. Chest 2015; 147: 423–429. doi: 10.1378/chest.14-1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mancuzo EV, Soares MR, Pereira CAC. Six-minute walk distance and survival time in patients with idiopathic pulmonary fibrosis in Brazil. J Bras Pneumol 2018; 44: 267–272. doi: 10.1590/s1806-37562018000000049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mapel DW, Hunt WC, Utton R, et al. Idiopathic pulmonary fibrosis: survival in population based and hospital based cohorts. Thorax 1998; 53: 469–476. doi: 10.1136/thx.53.6.469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Margaritopoulos GA, Trachalaki A, Wells AU, et al. Pirfenidone improves survival in IPF: results from a real-life study. BMC Pulm Med 2018; 18: 177. doi: 10.1186/s12890-018-0736-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mejia M, Carrillo G, Rojas-Serrano J, et al. Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest 2009; 136: 10–15. doi: 10.1378/chest.08-2306 [DOI] [PubMed] [Google Scholar]
- 68.Moon SW, Kim SY, Chung MP, et al. Longitudinal changes in clinical features, management, and outcomes of idiopathic pulmonary fibrosis. A Nationwide Cohort Study. Ann Am Thorac Soc 2021; 18: 780–787. doi: 10.1513/AnnalsATS.202005-451OC [DOI] [PubMed] [Google Scholar]
- 69.Mura M, Porretta MA, Bargagli E, et al. Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: a 3-year prospective study. Eur Respir J 2012; 40: 101–109. doi: 10.1183/09031936.00106011 [DOI] [PubMed] [Google Scholar]
- 70.Nadrous HF, Ryu JH, Douglas WW, et al. Impact of angiotensin-converting enzyme inhibitors and statins on survival in idiopathic pulmonary fibrosis. Chest 2004; 126: 438–446. doi: 10.1016/S0012-3692(15)31155-7 [DOI] [PubMed] [Google Scholar]
- 71.Nathan SD, Brown AW, Mogulkoc N, et al. The association between white blood cell count and outcomes in patients with idiopathic pulmonary fibrosis. Respir Med 2020; 170: 106068. doi: 10.1016/j.rmed.2020.106068 [DOI] [PubMed] [Google Scholar]
- 72.Natsuizaka M, Chiba H, Kuronuma K, et al. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am J Respir Crit Care Med 2014; 190: 773–779. doi: 10.1164/rccm.201403-0566OC [DOI] [PubMed] [Google Scholar]
- 73.Nicholson AG, Colby TV, Dubois RM, et al. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162: 2213–2217. doi: 10.1164/ajrccm.162.6.2003049 [DOI] [PubMed] [Google Scholar]
- 74.Ogawa K, Miyamoto A, Hanada S, et al. The efficacy and safety of long-term Pirfenidone therapy in patients with idiopathic pulmonary fibrosis. Intern Med 2018; 57: 2813–2818. doi: 10.2169/internalmedicine.0559-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reid T, Vennelle M, McKinley M, et al. Sleep-disordered breathing and idiopathic pulmonary fibrosis: is there an association? Sleep Breath 2015; 19: 719–721. doi: 10.1007/s11325-014-1117-3 [DOI] [PubMed] [Google Scholar]
- 76.Ryerson CJ, Hartman T, Elicker BM, et al. Clinical features and outcomes in combined pulmonary fibrosis and emphysema in idiopathic pulmonary fibrosis. Chest 2013; 144: 234–240. doi: 10.1378/chest.12-2403 [DOI] [PubMed] [Google Scholar]
- 77.Shin KM, Lee KS, Chung MP, et al. Prognostic determinants among clinical, thin-section CT, and histopathologic findings for fibrotic idiopathic interstitial pneumonias: tertiary hospital study. Radiology 2008; 249: 328–337. doi: 10.1148/radiol.2483071378 [DOI] [PubMed] [Google Scholar]
- 78.Strand MJ, Sprunger D, Cosgrove GP, et al. Pulmonary function and survival in idiopathic vs secondary usual interstitial pneumonia. Chest 2014; 146: 775–785. doi: 10.1378/chest.13-2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Strongman H, Kausar I, Maher TM. Incidence, prevalence, and survival of patients with idiopathic pulmonary fibrosis in the UK. Adv Ther 2018; 35: 724–736. doi: 10.1007/s12325-018-0693-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Su R, Bennett M, Jacobs S, et al. An analysis of connective tissue disease-associated interstitial lung disease at a US Tertiary Care Center: better survival in patients with systemic sclerosis. J Rheumatol 2011; 38: 693–701. doi: 10.3899/jrheum.100675 [DOI] [PubMed] [Google Scholar]
- 81.Sugino K, Ishida F, Kikuchi N, et al. Comparison of clinical characteristics and prognostic factors of combined pulmonary fibrosis and emphysema versus idiopathic pulmonary fibrosis alone. Respirology 2014; 19: 239–245. doi: 10.1111/resp.12207 [DOI] [PubMed] [Google Scholar]
- 82.Tarride JE, Hopkins RB, Burke N, et al. Clinical and economic burden of idiopathic pulmonary fibrosis in Quebec, Canada. Clinicoecon Outcomes Res 2018; 10: 127–137. doi: 10.2147/CEOR.S154323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tran T, Šterclová M, Mogulkoc N, et al. The European MultiPartner IPF registry (EMPIRE): validating long-term prognostic factors in idiopathic pulmonary fibrosis. Respir Res 2020; 21: 11. doi: 10.1186/s12931-019-1271-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Turner-Warwick M, Burrows B, Johnson A. Cryptogenic fibrosing alveolitis: clinical features and their influence on survival. Thorax 1980; 35: 171–180. doi: 10.1136/thx.35.3.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vietri L, Cameli P, Perruzza M, et al. Pirfenidone in idiopathic pulmonary fibrosis: real-life experience in the referral centre of Siena. Ther Adv Respir Dis 2020; 14: 1753466620906326. doi: 10.1177/1753466620906326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Watanabe T, Minezawa T, Hasegawa M, et al. Prognosis of pulmonary fibrosis presenting with a usual interstitial pneumonia pattern on computed tomography in patients with myeloperoxidase anti-neutrophil cytoplasmic antibody-related nephritis: a retrospective single-center study. BMC Pulm Med 2019; 19: 194. doi: 10.1186/s12890-019-0969-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang L, Zhang C, Dong F, et al. Combined pulmonary fibrosis and emphysema: a retrospective analysis of clinical characteristics, treatment and prognosis. BMC Pulm Med 2016; 16: 137. doi: 10.1186/s12890-016-0300-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zurkova M, Kriegova E, Kolek V, et al. Effect of pirfenidone on lung function decline and survival: 5-yr experience from a real-life IPF cohort from the Czech EMPIRE registry. Respir Res 2019; 20: 16. doi: 10.1186/s12931-019-0977-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015; 136: E359–E386. doi: 10.1002/ijc.29210 [DOI] [PubMed] [Google Scholar]
- 90.Mapel DW, Samet JM, Coultas DB. Corticosteroids and the treatment of idiopathic pulmonary fibrosis. Chest 1996; 110: 1058–1067. doi: 10.1378/chest.110.4.1058 [DOI] [PubMed] [Google Scholar]
- 91.Stack BHR, Choo-Kang YFJ, Heard BE. The prognosis of cryptogenic fibrosing alveolitis. Thorax 1972; 27: 535–542. doi: 10.1136/thx.27.5.535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Scadding JG. Chronic diffuse interstitial fibrosis of the lungs. Br Med J 1960; 1: 443–450. doi: 10.1136/bmj.1.5171.443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Turner-Warwick M, Burrows B, Johnson A. Cryptogenic fibrosing alveolitis: response to corticosteroid treatment and its effect on survival. Thorax 1980; 35: 593–599. doi: 10.1136/thx.35.8.593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Idiopathic Pulmonary Fibrosis Clinical Research Network . Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366: 1968–1977. doi: 10.1056/NEJMoa1113354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Petnak T, Lertjitbanjong P, Thongprayoon C, et al. Impact of antifibrotic therapy on mortality and acute exacerbation in idiopathic pulmonary fibrosis: a systematic review and meta-analysis. Chest 2021; 160: 1751–1763. doi: 10.1016/j.chest.2021.06.049 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00591-2021.SUPPLEMENT (679.8KB, pdf)