

Abstract
Colorectal cancer (CRC) remains one of the main causes of cancer death in developed countries. Yet, it is potentially preventable, by removing the precursor lesions - adenomas or serrated lesions. Several studies proved that this intervention reduces CRC mortality and that the first colonoscopy’s results can guide surveillance strategies. More recently, it became clear that several carcinogenesis pathways may lead to sporadic CRC. CRC is a heterogeneous disease, characterized by multiple molecular subtypes. Three main pathways have been implicated in the development of CRC: Chromosomal instability, microsatellite instability, and the “serrated” pathways, with overlapping features between them. This and other molecular and genetic based CRC classifications are known to have clinical implications, spanning from familial risk assessment to therapy choices. The authors review basic science data and provide insight on current implications for the management of patients with CRC.
Keywords: Colorectal cancer, Carcinogenesis pathways, Microsatellite instability, APC, KRAS, BRAF
Core Tip: Colorectal cancer (CRC) is a major cause of cancer death worldwide. It is a heterogeneous entity and its molecular and genetic features have clinical implications. Three main carcinogenesis pathways, with some overlapping features, are now known to lead to CRC: Chromosomal instability, microsatellite instability, and the “serrated” pathways. Their features, namely, microsatellite instability status and BRAF or KRAS mutation status, among others, have to be studied to assess familial cancer risk and to make adequate therapy choices. Ongoing research will potentially even enlarge basic science’s importance for clinical practice.
INTRODUCTION
Colorectal cancer (CRC) is the third most frequently diagnosed cancer in the USA and Asia, and the second in Europe[1,2], being one of the leading causes of cancer death worldwide[3]. Sporadic colorectal cancer represents about 70% of all cases and only 5% are related to known hereditary conditions such as Lynch syndrome (LS) and familiar adenomatous polyposis (FAP). The remaining cases have apparent familial predisposition with no identifiable single germline mutations[4].
Adenomas and serrated lesions are the precursors of the vast majority of CRCs and their number/characteristics at baseline screening colonoscopy allow the definition of adequate surveillance programs after polypectomy, with an impact on survival for over more than 10 years[5]. It is reasonable to think that if we understand the molecular mechanisms underlying the appearance of these lesions, we can be even more effective in identifying grades and temporal windows of risk and in designing individualized strategies for preventing and treating CRC.
Colorectal cancer origin
Colonic stem cells (CSC) are now known to be located at the base of the crypt (cells initially identified in 1974 and called “crypt base columnar cells” - CBCC)[6]. In the normal setting, the division of a stem cell does not generate a new stem cell and another cell committed to differentiation; on the contrary, each division usually generates two cells with the same destination, either stemness or differentiation. This is a random phenomenon of “neutral competition”[7], through which certain lineages are lost (when the two daughter cells progress to differentiation, as progenitors in the transit-amplifying zone) and crypts evolve to clonality, constitutionally.
Since 2007, CSC have also been known to be responsible for the generation of the entire CRC population[8,9]. Certain mutations, namely, in the WNT pathway, may confer selective advantage to the stem cells, granting them a greater potential for subsequent clonal progression in the crypt. The WNT pathway is the main responsible for the proliferation and maintenance of stem cells in the colonic epithelium. However, its level of activity is modulated by several factors, such as NF-kB, KRAS, and the NOTCH signaling pathway[10,11]. In the clonal competition process, either an APC loss (with WNT pathway activation) or a KRAS activation (which apparently leads to increased cell division) may confer a selective advantage. In the specific context of inflammatory bowel diseases, the loss of p53 function can also create a selection advantage for the mutated stem cell[10].
Carcinogenesis pathways in colorectal cancer
In 1990, Fearon and Vogelstein published an important paper about colorectal carcinogenesis[12]. The authors stated the need for the accumulation of several mutations in oncogenes and/or tumour suppressor genes for the development of a CRC. Although certain sequences of events are more frequent, it is the accumulation of mutations, more than its order, that leads to the biologic properties of the CRC[12,13]. The authors also found that although most tumours have mutations in the same key genes, additional mutations in several other genes occur in highly variable frequencies, which may explain some of the heterogeneity in the biologic properties of tumours found in clinical practice[14].
In fact, the CRC molecular characterization done by the Cancer Genome Atlas Network found an altered WNT signaling pathway in 93% of tumours, but it also described two broadly distinct groups of tumours: The “hypermutated” (more than 12 mutations per 106 bases) and the “non-hypermutated” (less than 12 mutations per 106 bases)[15].
Based on gene expression profiles, a classification system comprising four consensus molecular subtypes (CMS 1-4) was created, each having typical histologic and clinical features[16-19]. In Figure 1, we can see a classification system using the consensus molecular subtypes (CMS 1-4), CIMP (CpG island methylator phenotype), and microsatellite instability (MSI) status. The “non-hypermutated” tumours seem to correspond to group 4 and the “hypermutated” tumours to group 1 in the CRC classification proposed by Jass[20].
Figure 1.

Classification system using the consensus molecular subtypes (1-4), CpG island methylator phenotype, and microsatellite instability status. CMS: Consensus molecular subtypes; CIMP: CpG island methylator phenotype; MSI: Microsatellite instability.
The still most widely used classification for CRC origin distinguishes three pathways that, in fact, have some overlapping features: Chromosomal instability (CIN), MSI, and serrated pathways[13].
CIN: CIN is characterized by chromosome changes that include somatic copy number alterations caused by deletions, loss of aneuploidy, insertions, and amplifications. It was recently subdivided into three CMS: CMS 2, epithelial, with marked WNT and MYC activation; CMS 3, epithelial with important metabolic activation; CMS 4, mesenchymal, with TGF-β activation, stromal invasion, and angiogenesis[21]. This pathway is observed in about 65-70% of colorectal tumours and usually associated with karyotypic abnormalities (loss of heterozygosity at chromosome arm 18q in 70% of CRC), with APC (in 80% of CIN tumours) and TP53 (in 60% of CIN tumours) mutations and with KRAS activating mutations (in 40% of CRC)[13].
MSI: MSI is characterized by a high frequency of genomic copy number variations, and corresponds to CMS 1 and the hypermutated tumours subgroup. It occurs in the presence of abnormal mismatch repair (MMR) proteins (MLH1, MSH2, MSH6, or PMS2), caused by sporadic epigenetic silencing (most commonly through gene promotor hypermethylation) or constitutional mutations (Lynch syndrome)[21]. This pathway is observed in about 15% of sporadic tumours and in most CRC in LS. The most common cause of the MSI phenotype is somatic - the epigenetic silencing of MLH1 due to promoter hypermethylation (usually associated with BRAF mutation and CIMP-high status, a clear example of pathways’ overlap). After the occurrence of MSI, the expression of an inability to correct DNA replication defects, colorectal carcinogenesis progresses more rapidly than through the CIN pathway (1 to 3 years in contrast to 10 years or more)[13].
Serrated pathway: This pathway is characterized by a phenotype of DNA hypermethylation at specific regulatory sites (CpG islands) in the promoter regions of genes - the CIMP[13]. When this hypermethylation affects tumour suppressor genes, it leads to their silencing, promoting carcinogenesis. This pathway is responsible for nearly 15% of CRC and is commonly associated with BRAF mutation (usually the first detected event), after which it can follow different routes. It can converge with the MSI pathway, through inactivation of MMR genes or it can overlap with the CIN pathway, through TP53 mutations and WNT or TGFβ signalling pathway activation (resulting in MSS or MSI-L tumours)[13,22].
Other pathways: With the increasing advances in technology, new subgroups of tumours are being identified - an example is the identification of DNA polymerase protein mutations (POLE and POLD1), that led to the description of a new molecular pathway, characterized by a hypermutated phenotype without MSI[13].
Molecular and genetic features in colorectal cancer screening
Currently, the predominant CRC screening tools are fecal occult blood testing (FOBT), endoscopic evaluation (colonoscopy or sigmoidoscopy), and CT colonography. CRC screening has proven to reduce the risk of CRC associated mortality[23]; however, there are multiple limitations regarding test performances, and lack of access or compliance.
Several biomarkers have been investigated for their use in CRC screening, namely, DNA, proteins, and RNA (messenger or micro-RNA). These new non-invasive markers have the potential to improve screening by improving sensitivity, compliance, and accessibility. The detection of these biomarkers in blood, urine, and stool in people with colon polyps or CRC has been assessed and, to date, the most accurate tests are based on stool samples. This is explained by the abundant exfoliation of neoplastic cells from polyps and CRC into the mucocellular layer of the colonic lumen[24]. From all the options, only DNA-based markers have been used in clinical testing so far.
Multiple stool DNA-based markers have been evaluated but only the Cologuard multitarget stool DNA (MT-sDNA) test has been approved for clinical use - approved for CRC screening in asymptomatic individuals with ages between 50 and 84 years (United States Food and Drug Administration - US FDA). This test detects a combination of gene mutations (KRAS), methylated DNA markers, and fecal immunochemical test (FIT) and has demonstrated the best clinical performance of CRC marker screening to date. In a recent study, MT-sDNA test proved to have an overall CRC detection rate similar to colonoscopy and a superior sensitivity (but lower specificity) when compared to FIT, for the detection of advanced adenomas and CRC. However, 10% of patients with positive MT-sDNA have no polyps or CRC when they undergo colonoscopy[24,25]. Overall, models using 3-year screening intervals predict a very high program sensitivity.
Although CRC screening using stool based molecular markers is more and more a reality, there are also multiple promising assays in development regarding plasma molecular markers. For example, plasma detection of methylated SEPT9 (a gene more frequently methylated in CRC vs normal colon tissue) is currently available in China, USA, and Europe for CRC screening. However, these tests have suboptimal sensitivity for screening for colon polyps and early CRC compared to currently available screening tests[24]. It is hypothesized that plasma and urine marker detection may depend on CRC vascular invasion, which would limit the detection of precursor and early lesions.
Circulating tumor cells, circulating tumor DNA, and serum, fecal, and salivary microRNAs and long non-coding RNAs are all potential biomarkers in this emerging area and their role in CRC screening remains to be established[26].
Diagnostic, therapeutic, and prognostic implications of CRC molecular and genetic features
MSI: As previously stated, MSI is a major pathogenic pathway implicated in CRC development. The diagnosis of MSI is usually done by polymerase chain reaction (PCR) analysis of five microsatellite markers (the Bethesda panel) in both tumour and normal tissue - tumours with MSI in two or more of the markers are classified as MSI-High (MSI-H). However, immunohistochemistry (IHC) analysis of MMR protein expression has proven to identify around 95% of MSI-H tumours (in the remaining 5%, mutations that affect protein function but not its antigenicity may have happened). This technique is more widely available and does not require normal tissue samples[13](Figure 2).
Figure 2.

Eosin/hematoxylin staining images of colonic neoplasia (Low power objective - 10 ×). A and B: Maintained expression of MSH2 in colonic neoplasia; C and D: Loss of expression of MLH1 and PMS2.
There are multiple implications for the evaluation of MSI status in CRC.
Identifying patients with LS: The MSI status can guide the clinician to better identify patients who will benefit from genetic testing. Studies have shown that almost 17% of all MSI-H tumours happen in LS patients[27], with several of them previously unidentified. Patients with LS may benefit from a more radical surgery due to a higher risk of metachronous cancers and require different surveillance protocols after CRC treatment[13,28,29]. Moreover, the identification of an LS patient may potentially lead to screening and cancer prevention in several other family members. IHC has a significant role in the selection of patients for genetic testing, since the identification of tumours with absent MSH2/MSH6 protein expression should directly lead to referral for genetic testing, while MLH1/PMS2 loss should be followed by testing for BRAF V600E mutation and/or gene promoter hypermethylation[21](Figure 3). Since most MSI-H tumours with absent MLH1/PMS2 protein expression are due to somatic changes, genetic testing can be omitted in most of these patients.
Figure 3.
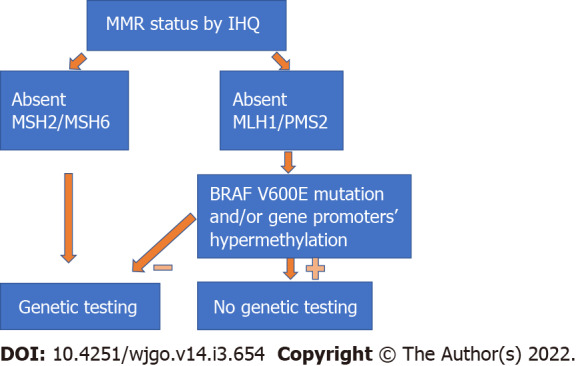
Algorithm for selection of patients for genetic testing (mismatch repair genes). Family history of cancer may indicate genetic testing regardless of immunohistochemistry analysis results. IHQ: Immunohistochemistry; MMR: Mismatch repair.
Different responses to standard chemotherapy: Several studies indicate that MSI tumours may have a reduced response to 5-FU chemotherapy[13,30-32]. Stage II MSI tumours have a better prognosis than MSS tumours and they probably do not benefit from adjuvant chemotherapy, namely, with 5-FU[13,33]. However, data are more conflicting for stage III tumours, where MSI status does not seem to significantly influence response to 5-FU, especially when oxaliplatin is added to the regimen. Regarding irinotecan, data are scarce, but MSI tumours seem to be sensitive[21].
Different responses to immune checkpoint inhibitors: Tumours with MMR deficiency produce several abnormal proteins which seem to elicit antigen-driven immune responses. Perhaps as an adaptative mechanism, these tumours also show increased expression of several immune checkpoints. As a consequence, MSI CRC metastatic tumours have better response and survival patterns when immune checkpoint inhibitors (ICI) are used (as opposed with the disappointing results in non-MSI tumours). Despite a good response to ICI like pembrolizumab and nivolumab, almost 50% of the patients will progress during this therapy and there are no biomarkers available to predict this response. Tumour mutation burden (TMB) is a good predictor of response to ICI and, although they are rare, POLE/D1 mutations can lead to high TMB tumours that are MSS but may still show good response to ICI[22].
Different overall prognosis: MSI tumours are generally considered to have a better prognosis, with less lymph node metastasis and synchronous liver metastasis. However, the prognostic meaning of MSI is modulated by several factors, and their interactions. Tumour stage is an example of this heterogeneity - while stage II MSI cancers have a better prognosis, metastatic MSI CRC globally have a worst prognosis than MSS ones[21,34-37]. However, the grade of the tumour-infiltrating lymphocyte (TIL) response, the BRAF mutation status, or the MSI origin (LS vs sporadic) all interfere with the impact of MSI on prognosis[13,22].
BRAF
BRAF encodes a serine-threonine protein kinase that is a regulator of the MAPK pathway. It is an important oncogene that plays a central role in cancer initiation and progression. BRAF mutations are strongly associated with MSI and hypermutated tumours in the CMS 1[38,39].
BRAF testing is almost mandatory in the metastatic CRC patient population, since it has both prognostic and therapeutic implications.
Multiple studies reported negative prognostic value of the BRAF V600E mutation in patients with stage II, III, or IV CRC[40,41]. Other BRAF mutations, much less frequent, are associated with different clinical and histologic characteristics and have a better prognosis. The prognostic meaning of BRAF mutations may, again, be modulated by other factors, such as tumour stage or MSI status[42]. In fact, while MSS BRAF V600E mutated tumours seem to have the worst prognosis, BRAF V600E mutation in MSI tumours may have different meanings at distinct stages[43].
BRAF V600E mutation is known to be associated with intrinsic chemoresistance[43]. Regarding targeted therapy, multiple studies also reported that BRAF mutation is associated with cetuximab and panitumumab resistance. Although this association is not as strong as the known influence of the KRAS status, and still controversial, it is thought that BRAF mutated patients probably do not receive much benefit from being treated with these two drugs[44,45]. Therefore, some authors advocate the triplet FOLFOXIRI in combination with bevacizumab as first-line therapy for stage IV BRAF V600E mutated tumours. Due to the low numbers of these tumours in clinical trials, the clinical impact of this strategy remains yet to be demonstrated and the benefit of antiangiogenic drugs like bevacizumab in this subgroup of patients lacks positive results with statistical significance[43].
Finally, several BRAF inhibitors (iBRAF) are now available and have demonstrated important results in several other cancers, starting from melanoma. However, results in CRC were largely disappointing, due to the different carcinogenesis pathways involved. Strategies to overcome these limitations are being developed, mostly by using combinations with standard chemotherapy, targeted agents, and/or immunotherapy.
KRAS status
KRAS protein is a self-inactivating signal. When it binds to a tyrosine kinase receptor such as epidermal growth factor receptor (EGFR), it leads to the activation of the RAS-RAF-MEK kinase pathway. KRAS activating mutations lead to oncogenesis. KRAS mutations are frequently found in MSS tumours in the CMS 3 CIN subgroup[2]. The assessment of the KRAS status is also crucial because it may have prognostic and therapeutic implications.
Some studies associated the KRAS mutations with a worse prognosis in the unresectable metastatic setting. However, conflicting results are yet not sufficient to recommend the evaluation of the KRAS status for prognostication[46].
The predictive value of the KRAS status when choosing therapy in stage IV CRC is, however, undisputed. KRAS exon 2 mutation is associated with intrinsic resistance to anti-EGFR antibodies. In KRAS exon 2 wild type patients, KRAS exons 3 and 4 mutations (as the less common NRAS exons 2, 3, and 4 mutations) have also been shown to be associated with intrinsic resistance to anti-EGFR antibodies (cetuximab and panitumumab), and CRC patients with these mutation have a worse overall survival when they receive anti-EGFR antibodies, either as monotherapy or combined with traditional chemotherapy[47,48]
Anti-EGFR antibodies have been used for the treatment of metastatic CRC since 2004. More recently, both cetuximab and panitumumab have been approved as first line treatments for BRAF/KRAS/NRAS wild type patients, with a demonstrated increase in overall survival, response rate, and progression-free survival. However, there are still patients with the above tumour genotype that cannot obtain these benefits or who experience rapid drug resistance and disease progression[47,48].
In KRAS wild-type patients, KRAS mutant clones frequently emerge and lead to secondary resistance to anti-EGFR therapy[49].
Ongoing studies are investigating the use of targeted agent combinations to overcome both primary and acquired resistance to therapy due to RAS mutations.
Also at research stage, Adagrasib, an oral drug that selectively binds and irreversibly inhibits KRAS with a specific mutation, has shown promising results in CRC patients in a phase I/II trial[50,51].
Finally, there is also recent evidence showing that metformin may be useful as an antitumor agent in KRAS mutated CRC[52].
APC status
The WNT signaling pathway is an important mediator of stem cell activation and is the most commonly dysregulated oncogenic pathway in CRC. APC, a crucial element in this pathway, is the most commonly mutated gene in sporadic CRC - this mutation is an early event in 80-85% of cases[53]. So far, however, attempts to use WNT inhibitors as therapy for CRC have failed, mostly due to adverse effects[53].
Recent data has shown that different APC mutations can lead to different prognoses in CRC patients[53,54]. For instance, C-terminal APC mutation led to a shorter survival, as opposed to N-terminal APC mutations - it was suggested that these could be used as prognosis markers (with no therapeutic implications so far)[54]. Research on molecules that target specific types of APC mutations is also currently ongoing[55].
TILs
Although highly correlated with dMMR status (MSI-H status), there is evidence that TILs are an independent predictor of outcome in CRC patients[56]. Several lines of data support the fact that the host immunologic response (evaluated by histology) against the tumour is a good prognostic indicator. Elevated lymphocytic reaction to CRC is associated with better oncologic outcomes[57-60]. Extensive lymphocytic infiltration is more common in MSI than MSS tumours. The relation between TIL and MSI status can help us even more to discriminate which CRC patients will have a better prognosis. Based on this premise, a TIL/MMR-based classification was created to distinguish the prognosis of CRC subtypes in patients with stage II and III tumours. TIL-low status identifies a clinically aggressive phenotype despite the MSI status[56]. Although these data seem interesting, there is not enough evidence yet to support the utilization of TIL evaluation or TIL/MMR-status in clinical practice for prognostic stratification.
Liquid biopsy in CRC
The term liquid biopsy refers to the isolation and analysis of tumour-derived material from blood or other bodily fluids[61]. In CRC, potential applications range from diagnosis to therapeutic monitoring. The current limitations in its use for screening have already been discussed.
Regarding prognosis, Diehl et al[62] found that cell-free DNA (cfDNA) analysis after surgery for CRC accurately predicted relapse, by identifying patients with otherwise undetectable residual disease. If validated, this information could also be used to select patients for adjuvant chemotherapy.
The utility in therapeutic monitoring has been exemplified in a study by Siravegna et al[63], who found that clones with KRAS mutations that lead to secondary resistance to anti-EGFR antibodies may lose dominance after therapy withdrawal and that this can be detected by cfDNA analysis, predicting a benefit of reinstitution of anti-EGFR therapy in these patients.
Although several other promising studies are available, liquid biopsy use in CRC still needs standardization of methods and validation in multicentric prospective trials.
CONCLUSION
CRC is a heterogeneous entity and its molecular and genetic subtypes have significant implications, from familial risk assessment to therapeutic choices.
Regarding the most used classification for CRC origin, there are three important oncogenic pathways: CIN, MSI, and serrated pathways. They have different clinical and molecular/genetic characteristics. The MSI status, BRAF, KRAS, and APC mutation status, and the presence of TILs are the most studied tumour features and those more extensively correlated to clinical data. The combination of MSI status and BRAF mutation status can be used to help identify patients with SL. However, tumour molecular and genetic analyses are now also known to predict response to chemotherapy or to immune checkpoint inhibitors and to affect prognosis. Finally, DNA-based markers have already undergone clinical testing in the field of CRC screening and were shown to be useful.
Clinicians should be aware of the major known carcinogenesis pathways and most commonly mutated genes, since some clinical implications are already proven and several others are currently under investigation.
Footnotes
Conflict-of-interest statement: The authors have no relevant conflicts of interest to declare.
Provenance and peer review: Invited article; Externally peer reviewed.
Peer-review model: Single blind
Corresponding Author's Membership in Professional Societies: American Gastroenterological Association, 1248915.
Peer-review started: September 27, 2021
First decision: December 4, 2021
Article in press: February 20, 2022
Specialty type: Oncology
Country/Territory of origin: Portugal
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): B, B, B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Gao W, Liu Z, Wan X, Xu K, Zhang Y S-Editor: Wang LL L-Editor: Wang TQ P-Editor: Wang LL
Contributor Information
Pedro Currais, Department of Gastroenterology, Instituto Portugues de Oncologia de Lisboa Francisco Gentil, Lisboa 1099-023, Portugal.
Isadora Rosa, Department of Gastroenterology, Instituto Portugues de Oncologia de Lisboa Francisco Gentil, Lisboa 1099-023, Portugal. isarosa@ipolisboa.min-saude.pt.
Isabel Claro, Department of Gastroenterology, Instituto Portugues de Oncologia de Lisboa Francisco Gentil, Lisboa 1099-023, Portugal.
References
- 1.Kasi A, Handa S, Bhatti S, Umar S, Bansal A, Sun W. Molecular Pathogenesis and Classification of Colorectal Carcinoma. Curr Colorectal Cancer Rep. 2020;16:97–106. doi: 10.1007/s11888-020-00458-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruffinelli JC, Santos Vivas C, Sanz-Pamplona R, Moreno V. New advances in the clinical management of RAS and BRAF mutant colorectal cancer patients. Expert Rev Gastroenterol Hepatol. 2021;15:65–79. doi: 10.1080/17474124.2021.1826305. [DOI] [PubMed] [Google Scholar]
- 3.Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D, Bray F. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374–1403. doi: 10.1016/j.ejca.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 4.Kastrinos F, Samadder NJ, Burt RW. Use of Family History and Genetic Testing to Determine Risk of Colorectal Cancer. Gastroenterology. 2020;158:389–403. doi: 10.1053/j.gastro.2019.11.029. [DOI] [PubMed] [Google Scholar]
- 5.Ballegooijen Van, Hankey B, Shi W, Bond J. Colonoscopy polipectomy and long term prevention of colorectal death. N Engl J Med. 2012;366:687–696. doi: 10.1056/NEJMoa1100370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng H. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. IV. Paneth cells. Am J Anat. 1974;141:521–535. doi: 10.1002/aja.1001410406. [DOI] [PubMed] [Google Scholar]
- 7.Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, Clevers H. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 8.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 9.O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 10.Vermeulen L, Morrissey E, van der Heijden M, Nicholson AM, Sottoriva A, Buczacki S, Kemp R, Tavaré S, Winton DJ. Defining stem cell dynamics in models of intestinal tumor initiation. Science. 2013;342:995–998. doi: 10.1126/science.1243148. [DOI] [PubMed] [Google Scholar]
- 11.Snippert HJ, Schepers AG, van Es JH, Simons BD, Clevers H. Biased competition between Lgr5 intestinal stem cells driven by oncogenic mutation induces clonal expansion. EMBO Rep. 2014;15:62–69. doi: 10.1002/embr.201337799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knudson AG Jr. Hereditary cancer, oncogenes, and antioncogenes. Cancer Res. 1985;45:1437–1443. [PubMed] [Google Scholar]
- 13.Nguyen LH, Goel A, Chung DC. Pathways of Colorectal Carcinogenesis. Gastroenterology. 2020;158:291–302. doi: 10.1053/j.gastro.2019.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dwyer ML, Colombo A, Bozzi G. Retrieval technique of a PTCA guidewire. G Ital Cardiol. 1989;19:170–172. [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Sousa E Melo F, Wang X, Jansen M, Fessler E, Trinh A, de Rooij LP, de Jong JH, de Boer OJ, van Leersum R, Bijlsma MF, Rodermond H, van der Heijden M, van Noesel CJ, Tuynman JB, Dekker E, Markowetz F, Medema JP, Vermeulen L. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med. 2013;19:614–618. doi: 10.1038/nm.3174. [DOI] [PubMed] [Google Scholar]
- 17.Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, Ostos LC, Lannon WA, Grotzinger C, Del Rio M, Lhermitte B, Olshen AB, Wiedenmann B, Cantley LC, Gray JW, Hanahan D. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, Bot BM, Morris JS, Simon IM, Gerster S, Fessler E, De Sousa E Melo F, Missiaglia E, Ramay H, Barras D, Homicsko K, Maru D, Manyam GC, Broom B, Boige V, Perez-Villamil B, Laderas T, Salazar R, Gray JW, Hanahan D, Tabernero J, Bernards R, Friend SH, Laurent-Puig P, Medema JP, Sadanandam A, Wessels L, Delorenzi M, Kopetz S, Vermeulen L, Tejpar S. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marisa L, de Reyniès A, Duval A, Selves J, Gaub MP, Vescovo L, Etienne-Grimaldi MC, Schiappa R, Guenot D, Ayadi M, Kirzin S, Chazal M, Fléjou JF, Benchimol D, Berger A, Lagarde A, Pencreach E, Piard F, Elias D, Parc Y, Olschwang S, Milano G, Laurent-Puig P, Boige V. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453. doi: 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113–130. doi: 10.1111/j.1365-2559.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- 21.Ryan E, Sheahan K, Creavin B, Mohan HM, Winter DC. The current value of determining the mismatch repair status of colorectal cancer: A rationale for routine testing. Crit Rev Oncol Hematol. 2017;116:38–57. doi: 10.1016/j.critrevonc.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 22.Cohen R, Rousseau B, Vidal J, Colle R, Diaz LA Jr, André T. Immune Checkpoint Inhibition in Colorectal Cancer: Microsatellite Instability and Beyond. Target Oncol. 2020;15:11–24. doi: 10.1007/s11523-019-00690-0. [DOI] [PubMed] [Google Scholar]
- 23.Ahlquist DA. Molecular detection of colorectal neoplasia. Gastroenterology. 2010;138:2127–2139. doi: 10.1053/j.gastro.2010.01.055. [DOI] [PubMed] [Google Scholar]
- 24.Dickinson BT, Kisiel J, Ahlquist DA, Grady WM. Molecular markers for colorectal cancer screening. Gut. 2015;64:1485–1494. doi: 10.1136/gutjnl-2014-308075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imperiale TF, Ransohoff DF, Itzkowitz SH. Multitarget stool DNA testing for colorectal-cancer screening. N Engl J Med. 2014;371:187–188. doi: 10.1056/NEJMc1405215. [DOI] [PubMed] [Google Scholar]
- 26.Vacante M, Ciuni R, Basile F, Biondi A. The Liquid Biopsy in the Management of Colorectal Cancer: An Overview. Biomedicines. 2020;8 doi: 10.3390/biomedicines8090308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Latham A, Srinivasan P, Kemel Y, Shia J, Bandlamudi C, Mandelker D, Middha S, Hechtman J, Zehir A, Dubard-Gault M, Tran C, Stewart C, Sheehan M, Penson A, DeLair D, Yaeger R, Vijai J, Mukherjee S, Galle J, Dickson MA, Janjigian Y, O'Reilly EM, Segal N, Saltz LB, Reidy-Lagunes D, Varghese AM, Bajorin D, Carlo MI, Cadoo K, Walsh MF, Weiser M, Aguilar JG, Klimstra DS, Diaz LA Jr, Baselga J, Zhang L, Ladanyi M, Hyman DM, Solit DB, Robson ME, Taylor BS, Offit K, Berger MF, Stadler ZK. Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J Clin Oncol. 2019;37:286–295. doi: 10.1200/JCO.18.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heneghan HM, Martin ST, Winter DC. Segmental vs extended colectomy in the management of hereditary nonpolyposis colorectal cancer: a systematic review and meta-analysis. Colorectal Dis. 2015;17:382–389. doi: 10.1111/codi.12868. [DOI] [PubMed] [Google Scholar]
- 29.Järvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomäki P, De La Chapelle A, Mecklin JP. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–834. doi: 10.1016/s0016-5085(00)70168-5. [DOI] [PubMed] [Google Scholar]
- 30.Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, French AJ, Kabat B, Foster NR, Torri V, Ribic C, Grothey A, Moore M, Zaniboni A, Seitz JF, Sinicrope F, Gallinger S. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28:3219–3226. doi: 10.1200/JCO.2009.27.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hutchins G, Southward K, Handley K, Magill L, Beaumont C, Stahlschmidt J, Richman S, Chambers P, Seymour M, Kerr D, Gray R, Quirke P. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol. 2011;29:1261–1270. doi: 10.1200/JCO.2010.30.1366. [DOI] [PubMed] [Google Scholar]
- 32.Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, Tu D, Redston M, Gallinger S. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ota DM. Colon cancer. Cancer Treat Res. 1997;90:347–356. doi: 10.1007/978-1-4615-6165-1_18. [DOI] [PubMed] [Google Scholar]
- 34.Sinicrope FA, Shi Q, Smyrk TC, Thibodeau SN, Dienstmann R, Guinney J, Bot BM, Tejpar S, Delorenzi M, Goldberg RM, Mahoney M, Sargent DJ, Alberts SR. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. Gastroenterology. 2015;148:88–99. doi: 10.1053/j.gastro.2014.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vasen HF, Blanco I, Aktan-Collan K, Gopie JP, Alonso A, Aretz S, Bernstein I, Bertario L, Burn J, Capella G, Colas C, Engel C, Frayling IM, Genuardi M, Heinimann K, Hes FJ, Hodgson SV, Karagiannis JA, Lalloo F, Lindblom A, Mecklin JP, Møller P, Myrhoj T, Nagengast FM, Parc Y, Ponz de Leon M, Renkonen-Sinisalo L, Sampson JR, Stormorken A, Sijmons RH, Tejpar S, Thomas HJ, Rahner N, Wijnen JT, Järvinen HJ, Möslein G Mallorca group. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013;62:812–823. doi: 10.1136/gutjnl-2012-304356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miles A, McClements PL, Steele RJ, Redeker C, Sevdalis N, Wardle J. The Psychological Impact of a Colorectal Cancer Diagnosis Following a Negative Fecal Occult Blood Test Result. Cancer Epidemiol Biomarkers Prev. 2015;24:1032–1038. doi: 10.1158/1055-9965.EPI-15-0004. [DOI] [PubMed] [Google Scholar]
- 37.Jennings BA, Loke YK, Skinner J, Keane M, Chu GS, Turner R, Epurescu D, Barrett A, Willis G. Evaluating predictive pharmacogenetic signatures of adverse events in colorectal cancer patients treated with fluoropyrimidines. PLoS One. 2013;8:e78053. doi: 10.1371/journal.pone.0078053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonsalves WI, Mahoney MR, Sargent DJ, Nelson GD, Alberts SR, Sinicrope FA, Goldberg RM, Limburg PJ, Thibodeau SN, Grothey A, Hubbard JM, Chan E, Nair S, Berenberg JL, McWilliams RR Alliance for Clinical Trials in Oncology. Patient and tumor characteristics and BRAF and KRAS mutations in colon cancer, NCCTG/Alliance N0147. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lipsyc M, Yaeger R. Impact of somatic mutations on patterns of metastasis in colorectal cancer. J Gastrointest Oncol. 2015;6:645–649. doi: 10.3978/j.issn.2078-6891.2015.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fariña-Sarasqueta A, van Lijnschoten G, Moerland E, Creemers GJ, Lemmens VEPP, Rutten HJT, van den Brule AJC. The BRAF V600E mutation is an independent prognostic factor for survival in stage II and stage III colon cancer patients. Ann Oncol. 2010;21:2396–2402. doi: 10.1093/annonc/mdq258. [DOI] [PubMed] [Google Scholar]
- 41.Morris V, Overman MJ, Jiang ZQ, Garrett C, Agarwal S, Eng C, Kee B, Fogelman D, Dasari A, Wolff R, Maru D, Kopetz S. Progression-free survival remains poor over sequential lines of systemic therapy in patients with BRAF-mutated colorectal cancer. Clin Colorectal Cancer. 2014;13:164–171. doi: 10.1016/j.clcc.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bläker H, Alwers E, Arnold A, Herpel E, Tagscherer KE, Roth W, Jansen L, Walter V, Kloor M, Chang-Claude J, Brenner H, Hoffmeister M. The Association Between Mutations in BRAF and Colorectal Cancer-Specific Survival Depends on Microsatellite Status and Tumor Stage. Clin Gastroenterol Hepatol. 2019;17:455–462.e6. doi: 10.1016/j.cgh.2018.04.015. [DOI] [PubMed] [Google Scholar]
- 43.Caputo F, Santini C, Bardasi C, Cerma K, Casadei-Gardini A, Spallanzani A, Andrikou K, Cascinu S, Gelsomino F. BRAF-Mutated Colorectal Cancer: Clinical and Molecular Insights. Int J Mol Sci. 2019;20 doi: 10.3390/ijms20215369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rowland A, Dias MM, Wiese MD, Kichenadasse G, McKinnon RA, Karapetis CS, Sorich MJ. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br J Cancer. 2015;112:1888–1894. doi: 10.1038/bjc.2015.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pietrantonio F, Petrelli F, Coinu A, Di Bartolomeo M, Borgonovo K, Maggi C, Cabiddu M, Iacovelli R, Bossi I, Lonati V, Ghilardi M, de Braud F, Barni S. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer. 2015;51:587–594. doi: 10.1016/j.ejca.2015.01.054. [DOI] [PubMed] [Google Scholar]
- 46.Labianca R, Nordlinger B, Beretta GD, Mosconi S, Mandalà M, Cervantes A, Arnold D ESMO Guidelines Working Group. Early colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24 Suppl 6:vi64–vi72. doi: 10.1093/annonc/mdt354. [DOI] [PubMed] [Google Scholar]
- 47.Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 48.De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, Biesmans B, Van Laethem JL, Peeters M, Humblet Y, Van Cutsem E, Tejpar S. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19:508–515. doi: 10.1093/annonc/mdm496. [DOI] [PubMed] [Google Scholar]
- 49.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, Veronese S, Zanon C, Sartore-Bianchi A, Gambacorta M, Gallicchio M, Vakiani E, Boscaro V, Medico E, Weiser M, Siena S, Di Nicolantonio F, Solit D, Bardelli A. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jänne P, Rybkin I, Spira A, Riely G, Papadopoulos K, Sabari J. KRYSTAL-1: Activity and Safety of Adagrasib (MRTX849) in Advanced/ Metastatic Non-Small-Cell Lung Cancer (NSCLC) Harboring KRAS G12C Mutation. Eur J Cancer. 2020;138:S1–2. [Google Scholar]
- 51.Riely G, Ou S, Rybkin I, Spira A, Papadopoulos K, Sabari J. KRYSTAL-1: Activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (Pts) with advanced non-small cell lung cancer (NSCLC) harboring KRASG12C mutation. J Thorac Oncol. 2021;16:S751–S752. [Google Scholar]
- 52.Xie J, Xia L, Xiang W, He W, Yin H, Wang F, Gao T, Qi W, Yang Z, Yang X, Zhou T, Gao G. Metformin selectively inhibits metastatic colorectal cancer with the KRAS mutation by intracellular accumulation through silencing MATE1. Proc Natl Acad Sci U S A. 2020;117:13012–13022. doi: 10.1073/pnas.1918845117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang L, Shay JW. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. J Natl Cancer Inst. 2017;109 doi: 10.1093/jnci/djw332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mondaca S, Walch H, Nandakumar S, Chatila WK, Schultz N, Yaeger R. Specific Mutations in APC, but Not Alterations in DNA Damage Response, Associate With Outcomes of Patients With Metastatic Colorectal Cancer. Gastroenterology. 2020;159:1975–1978.e4. doi: 10.1053/j.gastro.2020.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang W, Zhang L, Morlock L, Williams NS, Shay JW, De Brabander JK. Design and Synthesis of TASIN Analogues Specifically Targeting Colorectal Cancer Cell Lines with Mutant Adenomatous Polyposis Coli (APC) J Med Chem. 2019;62:5217–5241. doi: 10.1021/acs.jmedchem.9b00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Williams DS, Mouradov D, Jorissen RN, Newman MR, Amini E, Nickless DK, Teague JA, Fang CG, Palmieri M, Parsons MJ, Sakthianandeswaren A, Li S, Ward RL, Hawkins NJ, Faragher I, Jones IT, Gibbs P, Sieber OM. Lymphocytic response to tumour and deficient DNA mismatch repair identify subtypes of stage II/III colorectal cancer associated with patient outcomes. Gut. 2019;68:465–474. doi: 10.1136/gutjnl-2017-315664. [DOI] [PubMed] [Google Scholar]
- 57.Klintrup K, Mäkinen JM, Kauppila S, Väre PO, Melkko J, Tuominen H, Tuppurainen K, Mäkelä J, Karttunen TJ, Mäkinen MJ. Inflammation and prognosis in colorectal cancer. Eur J Cancer. 2005;41:2645–2654. doi: 10.1016/j.ejca.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 58.Ogino S, Nosho K, Irahara N, Meyerhardt JA, Baba Y, Shima K, Glickman JN, Ferrone CR, Mino-Kenudson M, Tanaka N, Dranoff G, Giovannucci EL, Fuchs CS. Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clin Cancer Res. 2009;15:6412–6420. doi: 10.1158/1078-0432.CCR-09-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mei Z, Liu Y, Liu C, Cui A, Liang Z, Wang G, Peng H, Cui L, Li C. Tumour-infiltrating inflammation and prognosis in colorectal cancer: systematic review and meta-analysis. Br J Cancer. 2014;110:1595–1605. doi: 10.1038/bjc.2014.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ropponen KM, Eskelinen MJ, Lipponen PK, Alhava E, Kosma VM. Prognostic value of tumour-infiltrating lymphocytes (TILs) in colorectal cancer. J Pathol. 1997;182:318–324. doi: 10.1002/(SICI)1096-9896(199707)182:3<318::AID-PATH862>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 61.Corcoran RB, Chabner BA. Application of Cell-free DNA Analysis to Cancer Treatment. N Engl J Med. 2018;379:1754–1765. doi: 10.1056/NEJMra1706174. [DOI] [PubMed] [Google Scholar]
- 62.Lindforss U, Zetterquist H, Papadogiannakis N, Olivecrona H. Persistence of K-ras mutations in plasma after colorectal tumor resection. Anticancer Res. 2005;25:657–661. [PubMed] [Google Scholar]
- 63.Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, Ponzetti A, Cremolini C, Amatu A, Lauricella C, Lamba S, Hobor S, Avallone A, Valtorta E, Rospo G, Medico E, Motta V, Antoniotti C, Tatangelo F, Bellosillo B, Veronese S, Budillon A, Montagut C, Racca P, Marsoni S, Falcone A, Corcoran RB, Di Nicolantonio F, Loupakis F, Siena S, Sartore-Bianchi A, Bardelli A. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:795–801. doi: 10.1038/nm.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]