

Abstract
Interferons are an ancient and well-conserved group of inflammatory cytokines most famous for their role in viral immunity. A decade ago, we discovered that interferons also play an important role in the biology of hematopoietic stem cells (HSCs), which are responsible for lifelong blood production. Though we have learned a great deal about the role of interferons on HSC quiescence, differentiation, and self-renewal, there remains some controversy regarding how interferons impact these stem cells, with differing conclusions depending on experimental models and clinical context. Here, we review the contradictory roles of Type 1 and 2 interferons in hematopoiesis. Specifically, we highlight the roles of interferons in embryonic and adult hematopoiesis, along with short-term and long-term adaptive and maladaptive responses to inflammation. We discuss experimental challenges in the study of these powerful yet short-lived cytokines and strategies to address those challenges. We further review the contribution by interferons to disease states including bone marrow failure and aplastic anemia as well as their therapeutic use to treat myeloproliferative neoplasms and viral infections, including SARS-CoV2. Understanding the opposing effects of interferons on hematopoiesis will elucidate immune responses and bone marrow failure syndromes, and future therapeutic approaches for patients undergoing HSC transplantation or fighting infectious diseases and cancer.
Introduction
Interferons (IFNs) are a group of signaling proteins primarily made and released by immune cells upon interaction with pathogens or cancer cells, but can be produced by most other cell types [1, 2]. Interferon production can also be induced endogenously by sensing self-ligands (i.e. dsDNA and dsRNA) or through TLR agonists [3]. These cellularly intrinsic stimuli induce the cGAS/STING (DNA) or RIG-I and MDA5 (RNA) pathways to maintain steady-state interferon production during homeostasis [3]. While IFNs are expressed at low levels in the body at baseline, members of the IFN family are induced to differing degrees depending on the stimulus, therefore their role in the immune response is dynamic and pathogen-specific [1, 2]. Once produced and secreted by immune cells, IFNs modulate activity of the immune system by inducing transcription of interferon signaling genes (ISGs) [1–3]. A complete understanding of the impact of IFNs on the immune system and hematopoiesis — the generation of all circulating, mature blood cells — promises to uncover important therapeutic opportunities for therapeutic use of IFNs in immunology and oncology.
Ten years ago, we discovered both IFNα and IFNγ can influence the biology of hematopoietic stem and progenitor cells (HSPCs) [4, 5]. This discovery created a field that has yielded contradictory evidence about how IFNs influence hematopoietic stem cell (HSC) development and maintenance (Figure 1). In the short term, IFN signaling can be beneficial to induce HSC division and differentiation, increase myelopoiesis, and enhance immune responses to acute infections [4, 6–8]. However, these same molecules are capable of causing deleterious long-term effects like stem cell exhaustion and selection pressure for clonal stem cells [9–11]. In this review, we will dissect the dual roles IFNs play in the hematopoietic system, both under steady state as well as upon inflammatory conditions, including host responses to infection, aging, and cancer. We will focus on how the chronicity of signaling contribute to differential and context-specific effects by IFNα and IFNγ on HSCs (Figure 1). Finally, we will discuss potential new avenues for IFN vaccination and therapeutics to modulate host immune response when faced with novel diseases including pandemic SARS-CoV-2 infection (Figure 3).
Figure 1: The multifaceted impact of Interferons on HSCs.
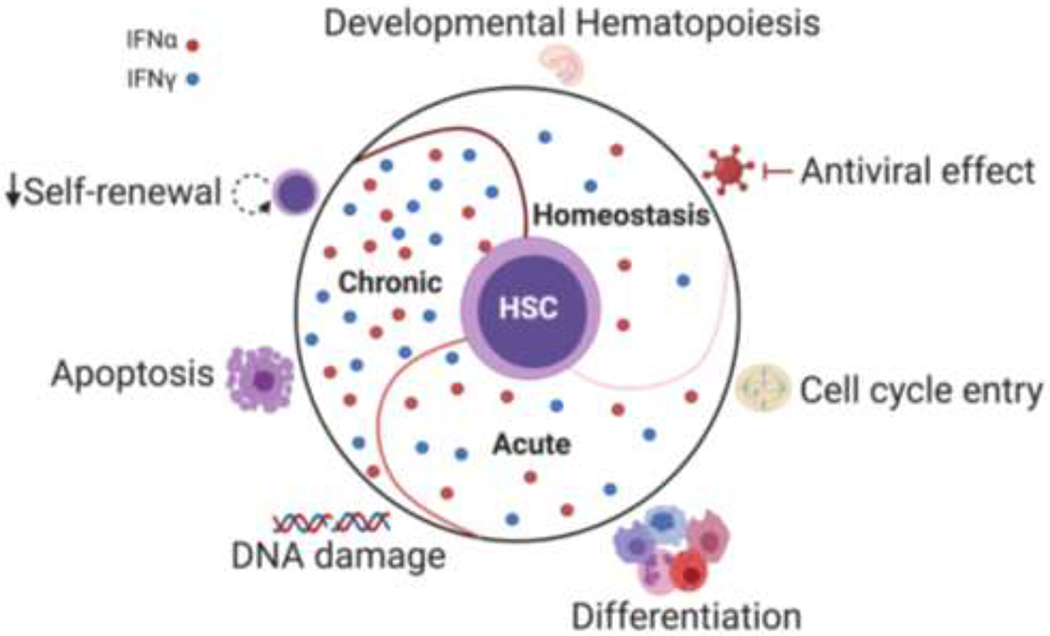
During homeostasis, basal IFN signaling plays a role in regulating HSC emergence during early development. In response to acute IFNs exposure, HSCs exit their quiescent state, start cycling, and show myeloid biased differentiation. Chronic exposure to IFNs can lead to increased DNA damage, activation of apoptotic pathways, as well as compromised self-renewal and repopulation capacity. Thus, the role of IFNs on HSCs biology is complex and highly dependent on the stimulation conditions.
Figure 3: The balance of interferon (IFN) signaling during homeostasis and disease.
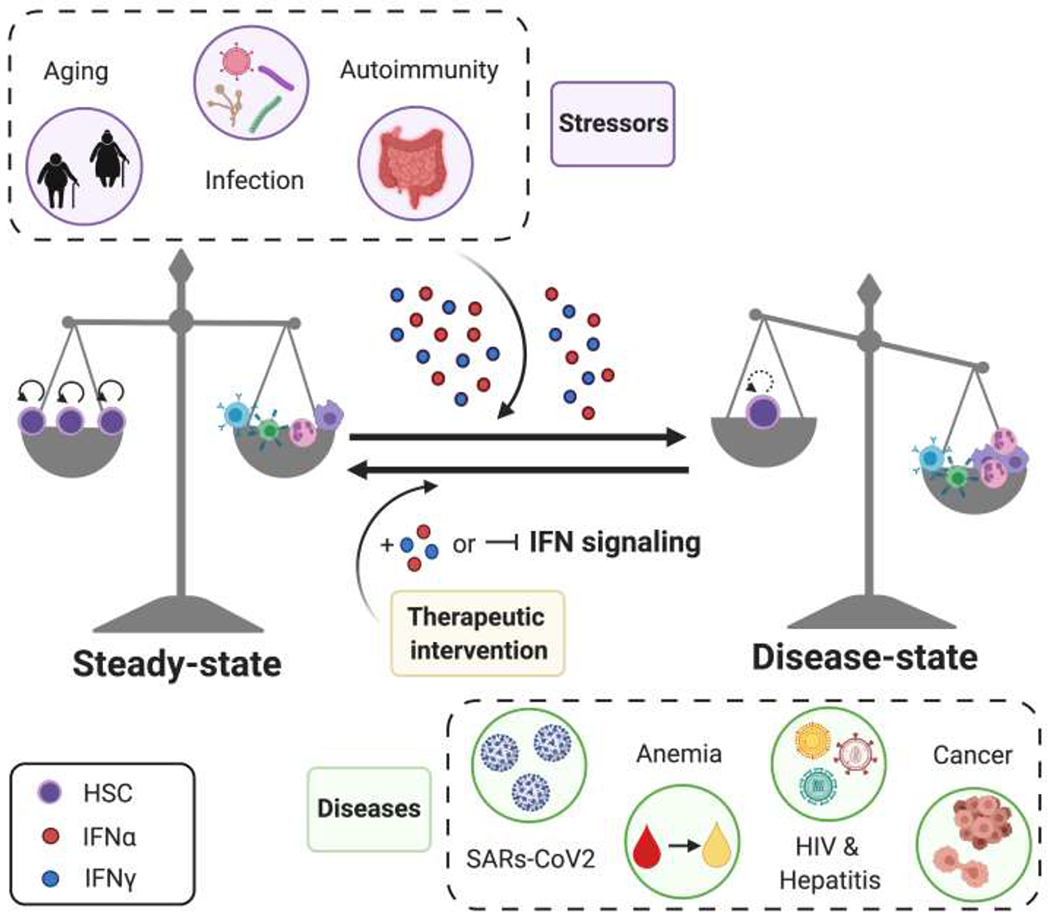
At steady state, HSCs maintain a balance of self-renewal and differentiation into mature blood cells. However, during external and internal stressors that increase IFNα or IFNγ signaling e.g. infection, aging, and autoimmunity, homeostasis is lost. With excessive IFN signaling, HSCs lose their self-renewal capability and differentiate. Modulating IFN signaling by treating patients with recombinant IFNs or blocking IFN signaling pathways have proven to be effective treatment strategies.
The Interferons
Discovered in 1957, IFNs are a class of signaling molecules that trigger host defense mechanisms [1, 2, 12]. Although they were initially named based on their ability to “interfere” with viral infection of cells, it is now known that IFNs can impact a variety of different cellular processes. Each type of IFN is classified according to multiple criteria including sequence identity, genetic loci, cell of origin, receptor distribution, and downstream responses [1]. The three main types of IFNs are Type I IFNs (includes IFNα and IFNβ), Type II IFN (IFNγ), and Type III IFNs (IFN-like cytokines and IFNλ) [1, 2, 12].
Type I IFNs are the largest and best-characterized group of IFNs and are composed of several classes, with IFNα and IFNβ being the most well-defined. IFNα is predominantly produced by plasmacytoid dendritic cells (DCs), whereas IFNβ is produced by most hematopoietic cell types [1–3]. Type I IFNs are one of the first cytokines produced during a virus infection in response to pattern recognition receptor (PRR) stimulation [1, 12]. Best known for their ability to restrict viral replication in infected cells and promote an antiviral state in bystander cells, these IFNs are central regulators of innate immunity [1, 2]. In addition to their role in evading viral infections, Type I IFNs also promote antigen presentation, natural killer (NK) cell function, and activation of adaptive immune cells [1, 2, 12]. Both IFNα and IFNβ bind to a heterodimeric transmembrane receptor termed IFNα receptor (IFNAR), thereby activating Signal Transducer and Activator of Transcription (STATs) proteins, which then translocate as a STAT1/STAT2 heterodimer to the nucleus and bind to specific elements to regulate gene transcription [13].
IFNγ is the sole member of the type II IFN family and acts as a mediator of the host immune system and anti-tumor responses [1, 2]. IFNγ is secreted primarily by T cells and NK cells in response to Th1-inducing pathogens but it has also been shown to be secreted by professional antigen presenting cells (APCs), B cells and NKT cells [1, 2, 12]. The cellular source of IFNγ secretion corresponds to unique functions in response to pathogens, with NK cells secreting IFNγ early during infection and T cells subsequently producing the majority of IFNγ in the adaptive immune response [1, 12]. IFNγ is considered a pro-inflammatory cytokine whose production can be stimulated by cytokines produced by APCs, (i.e. IL-12, IL-15 and IL-18), type I IFNs including IFNγ, and pathogen associated molecular patterns (PAMPs) [1, 2]. Alternatively, negative regulators of IFNγ production include IL-4, IL-10, TNFβ, and glucocorticoids [13]. IFNγ binds to its receptor (IFNGR), which is composed of both an alpha (IFNGR1) and a beta chain (IFNGR2) [1, 12]. IFNGR1 can mediate biological responses to IFNγ alone, independent of the presence of IFNGR2 [1, 2]. Intracellular signaling through IFNGR is mediated via STAT1 homodimers [1, 12]. Thus, the signaling pathways utilized by IFNα and IFNγ are overlapping, and synergistic, as IFNγ signaling can induce production of IFNα.
IFNs as therapeutics and modulators of the immune system
Although IFNs are critical for host immune responses, their therapeutic use has not advanced over the past few decades. IFNα was the first IFN and the first cytokine to be produced by the pharmaceutical industry and has been used to treat chronic myeloid leukemia (CML), melanoma, myeloproliferative neoplasms, and chronic Hepatitis B and C infections [1, 14, 15]. As a therapy, IFNα can induce apoptosis, inhibit proliferation and cell cycle progression, and promote the terminal differentiation of cancer cells by activating IFNAR signaling [1, 2, 12]. However, IFNα therapy has a short half-life in the bloodstream, high myelotoxicity, and paradoxical immunosuppressive effects [1, 12].
Based on work showing potency against intracellular pathogens, activation of NK cells, and recruitment and stimulation of other immune cells like neutrophils to generate a superoxide-laden respiratory burst, IFNγ was thought to have high therapeutic potential as an immune-modulatory drug [1, 12]. Currently, IFNγ is used to treat chronic granulomatous disease and osteopetrosis, a rare osteosclerotic bone disease [2]. However, IFNγ therapies are associated with flu-like side effects, including fever, headaches, nausea, and bone pain [1, 12]. The severity of these symptoms has been correlated with the dose.
Despite these limitations, IFNs remain an important consideration in the treatment of infections and malignancies either alone or in combination with other therapeutic modalities. For example, the combination of pegylated IFNα with antivirals including entecavir or ribavirin has been investigated in multiple clinical trials to treat chronic hepatitis B and C [16–19]. Combination therapy with pegylated IFNα is also being investigated for symptomatic HIV infection for both primary infections and in combination with hepatitis infection [21]. Notably, exploratory combination therapy for solid tumor (e.g. renal cell carcinoma, malignant melanoma) and hematological malignancies (e.g. chronic myeloid leukemia, multiple myeloma) are currently being investigated (www.clinicaltrials.gov). Harnessing the ability of interferons to modulate the host immune system should continue to be pursued, particularly with the introduction of innovative immunotherapy strategies including monoclonal antibodies and CAR-T cells.
IFN signaling during developmental hematopoiesis
Although fetal life is considered to be immunologically protected, several studies have demonstrated that IFNs play a critical role in developmental hematopoiesis. Li et al. published the first evidence of the involvement of IFNs in developmental hematopoiesis, showing that mouse embryos lacking IFNγ or IFNα signaling had significantly fewer HSPCs at the aorto-gonado-mesonephros (AGM) stage of development [21]. Furthermore, the number of lymphoid progenitors (LPs) was significantly reduced in the IFN KO embryos, though erythroid-myeloid progenitors (EMP) were not affected, correlating with a robust innate immune/inflammatory gene expression signature in these cells. It was later shown that IFNα signaling also plays a role in the functional maturation of AGM HSCs [22]. Indeed, treating AGM HSCs with IFNα enhances their long-term engraftment and donor chimerism, indicating improved function, while AGM HSCs lacking IFNAR show reduced donor chimerism. Of interest, limiting dilution experiments showed that treating AGM HSCs with IFNγ but not IFNα increased stem cell frequency, whereas FL fetal liver cells from Ifnar−/− but not Ifngr−/− had lower engraftment potential [22]. A recent study also demonstrates a strong type I interferon response in the late prenatal period that promotes HSC expansion [24]. Results from these studies suggest that IFNα promotes functional maturation and expansion of AGM HSCs while IFNγ plays a role in HSC emergence.
The role of IFN signaling in embryonic HSPC development is conserved in zebrafish. As in mice, loss of IFN signaling did not affect EMP numbers in zebrafish embryos [21], highlighting the specificity of IFNs on HSC and LP development. In addition, zebrafish morphants lacking IFNγ or its receptor had reduced expression of runx1, an important marker of HSPCs in the AGM region [27], thus supporting the significance of IFNγ in HSC emergence across species. Furthermore, Sawamiphak et al. showed that IFNγ overexpression was sufficient to rescue loss of HSCs in two models (Notch signaling knockdown and blood loss) [27]. Similarly, zebrafish embryos lacking the IFNα ortholog (IFNφ) had fewer HSPCs and thymocytes, whereas knockdown of Irf2a, a negative regulator of IFN signaling, had more of these cells. In zebrafish, IFNγ acts through STAT3 signaling, whereas the effect of IFNα in AGM HSPCs was mediated solely through STAT1 [27]. These studies suggest that IFNs signal through distinct downstream molecules during developmental hematopoiesis, likely contributing to their separate roles.
The role of IFNs during developmental hematopoiesis is conserved not only in zebrafish and mice but also in humans, as IFN signaling is active in human fetal HSPCs [21]. The study by Li et al. indicated that fetal macrophages act as local source of IFNs to promote the production of HSCs and LPs [21]. However, which cells produce IFNs during early stages of development in the AGM remains an important open question.
Impact of IFNs on adult hematopoiesis
IFNs have long been known for their anti-proliferative, apoptotic and cytotoxic properties in multiple cell types, consistent with their role as anti-viral agents [28]. In 1962, Paucker first described the effect of IFNs on hematopoietic cells, showing G0-G1 cell cycle arrest and a decline in the growth of both malignant and non-malignant cell lines following IFN exposure [29]. A dual nature of IFNs has been suggested more recently, where, in addition to their inhibitory properties, they also stimulate immune cells [2]. In contrast to the well-described roles of IFNs in immune cell biology, studies addressing how they affect HSCs have only recently begun and results appear complex and contradictory. IFNs have been shown to both induce cell division of HSCs and impair their self-renewal capacity and long-term persistence [4, 5, 9, 26] (Figure 1).
Initial reports by Gidali et al showed that in vitro treatment of murine HSCs with Type 1 IFNs inhibited proliferation by reducing the number of colony forming units (CFUs) in the S phase [31]. Similar results were observed with parallel in vivo experiments carried out with polyI:C, a synthetic double stranded RNA that mimics viral infections by inducing IFNα production. Although polyI:C induces a response similar to IFN it is important to note their mechanism of signaling is different. Because polyI:C signals through intracellular sensors, it is possible polyI:C treatment can lead to additional effects that differ from a solely IFN response. Moreover, mice overexpressing IFNγ showed reduced numbers and frequencies of hematopoietic progenitors [28], whereas mice lacking Irf2 showed increased numbers of proliferating HSCs [33]. Several studies then focused on the role of IFNs as regulators of HSPCs and showed that both IFNα and IFNγ exert anti-proliferative properties on many different bone marrow-derived HSPCs in vitro [3, 30, 31]. These suppressive effects were mediated by activation of protein kinase Cn (PKCn) and p38 mitogen activated protein kinase [32] [33]. Thus, IFNs were considered suppressors of hematopoiesis, which was further supported by a study showing that treatment of human HSCs in culture with IFNγ compromised their self-renewal properties [34]. Note that the readout in all of these studies was a CFU assay, which measures the potential of a cell to divide, but not necessarily to self-renew. Moreover, the frequent use of the term “proliferation” as a synonym for cell division in many of these studies is confusing, as it can be mistaken to indicate an increase in stem cell number. However, stem cells can undergo division without proliferating, particularly when those divisions result in differentiation as opposed to self-renewal. It is therefore important to specify the actual assays performed to analyze the state of HSCs in order to better understand the impact of IFNs on HSC biology.
In more recent years, we and others have shown that treatment of mice with IFNα, polyI:C, or IFNγ induces cell cycle entry of quiescent HSCs [4, 5, 35]. Furthermore, infections with Mycobacterium avium, a bacterium, or murine cytomegalovirus (MCMV) strongly induce IFNγ or IFNα, respectively, in the bone marrow, leading to increased cell division of the HSC pool. Nevertheless, the HSC pool does not increase in number despite their exit from quiescence and increased cell division following IFN exposure in vivo [8, 35] as a result of impaired self-renewal [30]. It is thus more accurate to describe the IFN-induced HSC exit out of quiescence as an increase in cell division rather than an increase in cellular proliferation. Moreover, although IFNs are able to induce cell cycle activation of HSCs in vivo, this effect was not recapitulated in vitro regardless of the concentration of IFNs used and even when co-cultured with bone marrow or stromal cells [39]. Results of in vivo versus in vitro studies can be highly disparate because removal of HSCs from their niche stresses the cells and alters their response to IFN treatment. Despite advances in stromal co-culturing and other in vitro technologies, in vitro assays remain inadequate to replicate the contribution of the bone marrow niche in supporting HSC biology.
Many cell types in the bone marrow can respond to IFNs and thus might play a role in mediating IFN-induced activation of HSCs in vivo. The cellular composition of the niche — made up of osteoblasts, neurons, endothelial cells, mesenchymal stromal cells (MSCs), and hematopoietic cells such as megakaryocytes and monocytes, among others — plays an important role in HSC maintenance and homeostasis [36, 37]. The concept that inflammatory stress-induced alterations in stromal cells can affect the hematopoietic process was proposed several years ago [7]. For example, IL-6 secretion of MSCs has been suggested to indirectly mediate IFNγ-induced myelopoiesis on HSPCs [38]. Furthermore, IFNγ can alter MSC biology by inhibiting proliferation and differentiation and enhancing immunosuppressive properties [43]. However, how IFN-responding MSCs impact HSC biology in their niche remains unclear.
Macrophages in the bone marrow also respond to IFNγ, resulting in reduced HSC numbers, both at steady state as well as under infection [40]. Furthermore, IFNγ can induce alterations in HSC-niche adhesive interactions. For example, αvβ3 integrin enhances the suppressive effects of IFNγ on HSC function through enhanced STAT1 phosphorylation [45]. We have recently shown that IFNα can affect bone marrow vessel permeability; as endothelial cells can be activated in response to acute IFNα exposure in vivo, and this is in part mediated by signaling from IFNα-stimulated HSCs [42]. In addition, IFNα can impact megakaryocyte distribution in the bone marrow niche [47]. Furthermore, IFNα leads to increased expression of ECM components in HSCs, suggesting alterations in the connection of HSCs with the ECM in the niche [44]. These data provide evidence for crosstalk between HSCs and niche components under inflammatory stress conditions, and might thus explain, at least in part, the discrepant impact of in vitro and in vivo IFN treatments on HSC biology.
Differing responses by hematopoietic cells to IFNs may also reflect differences in cell types analyzed. Studies showing anti-proliferative effects of IFNs were mostly done on total bone marrow or a stem and progenitor cell enriched population, defined as a “LSK” (lineage-negative, cKit+, Sca1+). Moreover, inflammatory stress in the bone marrow is accompanied by changes in stem cell marker expression, complicating the molecular and functional identification of HSCs under stress. IFN-induced signaling increases expression of the stem cell marker Sca1, leading not only to strong up-regulation of Sca-1 on HSCs [5, 35], but also to its expression on common myeloid progenitors (CMPs) and granulocyte-monocyte progenitors (GMPs) that normally do not express Sca1, shifting these cells into the LSK gate (Figure 2). IFN treatment also slightly alters expression of other stem cell markers including CD48 and CD150 (Figure 2). [39]. Therefore, stringent markers are critical to exclude other cell types and enhance interpretation of findings related to HSC function, particularly when HSCs are exposed to inflammatory stressors.
Figure 2: Inflammatory stress in the bone marrow leads to increased Sca1 expression on HSCs and progenitors.
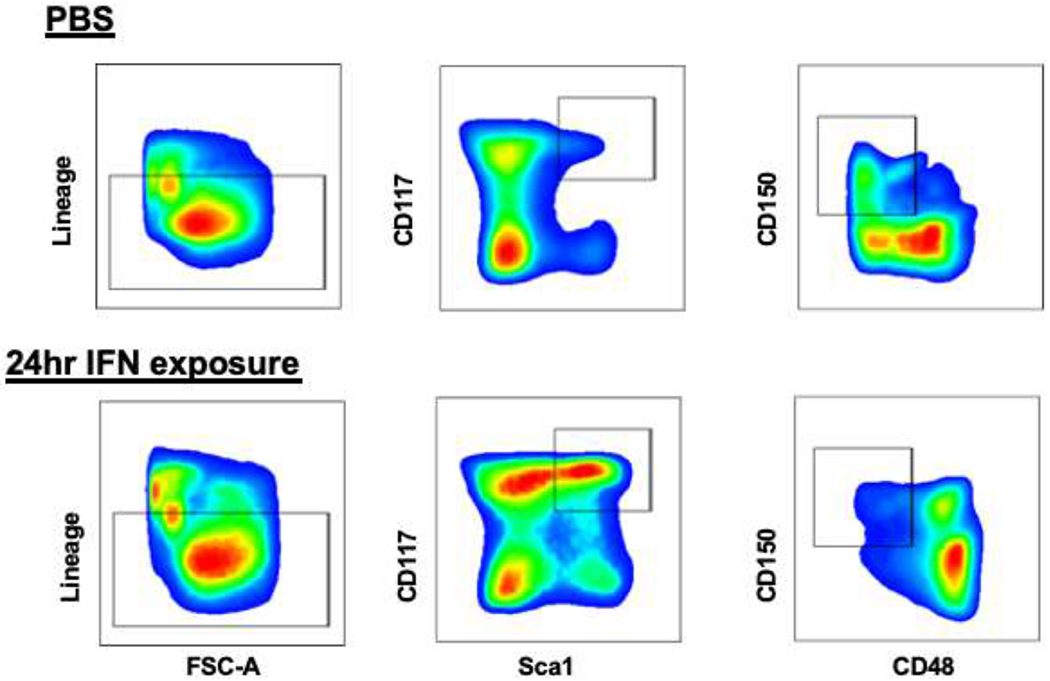
Compared to PBS treated controls, bone marrow cells treated with recombinant IFN show increased levels of Sca1 expression, a stem cell marker commonly used to define hematopoietic stem and progenitor enriched cell populations. Under inflammatory stress, Sca1 is not only further upregulated on HSCs but also expressed on CMPs and GMPs, which typically do not express Sca1 in the steady state. The upregulation of Sca1 on these cells causes a shift of these cells into the LSK gate. Furthermore, IFN treatment modulates the expression of other stem cell markers like CD150 and CD48. Thus, to accurately determine the effects of inflammatory stressors on HSCs and progenitors, it is critical to use further markers to define these populations.
Several recent studies have suggested a combination of EPCR and CD34 together with the SLAM markers, cKit and lineage markers, provide highly stringent enrichment for functional HSC following inflammatory stress [45]. In support of this finding, previous work suggested IFN-exposed SLAM HSCs are functionally impaired [5, 35]. However, Fgd5-mCherry+ SLAM HSCs, highly enriched for EPCR+/CD34−, from polyI:C-treated mice showed comparable reconstitution to untreated cells [46]. Thus, a careful analysis of the phenotypic markers in combination with functional studies is essential to interpret how IFN treatment affects HSC biology.
In addition to their impact on HSC cell cycling, IFNs also have distinct effects on HSC differentiation. During infection-associated emergency myelopoiesis, IFNγ was identified as a potential mediator of stress-induced myeloid specification. IFNγ can act specifically on myeloid-biased HSCs and induce myeloid differentiation via activation of the transcription factors Batf2 and C/EBPβ [8, 9]. Moreover, IFNγ treatment leads to elevated expression of the monocyte promoting transcription factor PU.1 and inhibits granulocyte colony stimulating factor (G-CSF) induced activation of STAT3, an essential transcription factor for emergency granulopoiesis [47, 48]. In support of this, IFNγ-deficient mice infected with viruses that typically induce monopoiesis in wildtype mice, such as lymphocytic choriomeningitis virus [49], displayed extreme granulocytosis and a strong increase in neutrophil development [51] [54]. Even though no parallel analysis was performed with IFNα, there is no clear evidence so far that IFNα promotes HSC myeloid differentiation. Nevertheless, we have shown that IFNα rapidly activates a posttranscriptional megakaryopoietic program in stem-like megakaryocyte-committed progenitor (SL-MkPs), leading to efficient replenishment of platelets that are lost during inflammatory insults [51].
Tonic IFN stimulation has a time and context-dependent impact on HSC responses. In contrast to acute IFNα-stimulation, chronic IFNα treatment compromised HSC repopulation capacity and a return to quiescence, protecting HSCs from IFNα-induced apoptosis [39]. On the other hand, HSC cell cycle activity remains high with continuous IFNγ treatment [9]. Thus, chronic exposure to IFNα but not IFNγ appears to induce survival mechanisms in HSCs. However, there is a window of time during which in vivo treatment of IFNs leads to HSC exit from quiescence and promote cell division. In addition, IFNγ plays a role in the demand-adapted hematopoiesis to replenish myeloid cells lost during acute inflammatory conditions [9]. Interestingly, recent evidence has shown that IFNγ is involved in the process of trained immunity by educating HSCs to generate trained monocytes, thereby conferring long-term functional reprogramming of innate immune cells [52]. Even though demand-adapted myelopoiesis is not observed following IFNα treatment, high levels of IFN signaling in HSCs do alter their chromatin structure and protect them from viral infections [57].
In summary, many studies have described the impact of IFN treatment on HSC biology, however differences in read out, in vitro and in vivo settings, and cell populations analyzed lead to seemingly contradictory data. In the sections below, we discuss the impact of timing, dose, and environment on IFN-mediated effects on HSCs and hematopoiesis.
Short-term vs. long-term effects of interferon signaling on hematopoiesis
Here, we will describe the differing effects of short- and long-term IFN in variety disease contexts (Figure 3).
Infection
Persistent pro-inflammatory cytokine signaling during infections such as can result in loss of HSCs and bone marrow failure [54, 55]. Therefore, it is critical to study how infection-based bone marrow suppression occurs, particularly the influence of IFNs on cell homeostasis, to develop novel therapeutics to maintain the HSC pool and prevent stem cell exhaustion.
Multiple infection models show that IFN signaling disrupts HSC quiescence. Mycobacterium avium infection induces persistent IFNγ signaling and increases the proportion of dividing LT-HSCs [4, 8]. Similar results have been described in response to another intracellular bacteria, Ehrlichia muris, or upon viral infection with LCMV or MCMV [7, 19, 50]. However, despite the increased cell division rate, HSC engraftment was decreased after M. avium or MCMV infection, reflecting impaired self-renewal [8, 9, 56]. We showed that IFNγ signaling induced by M. avium infection caused myeloid-biased differentiation and infection-mediated depletion of HSCs [10]. Thus, on one hand inflammation-mediated HSC differentiation may enable prompt responses to infection, but on the other it could negatively impact the generation of lasting immunity and maintenance of the stem cell pool.
Several infection models in mice have now described myeloid differentiation bias and stem cell exhaustion upon increased IFNγ signaling. In our chronic M. avium infection model, depletion of the stem cell pool through terminal differentiation ultimately led to pancytopenia [10]. The stem cell pool also was depleted in the LCMV mouse model of chronic viral infection [20]. In studies looking at systemic E. coli bacterial infection, sonic hedgehog signaling (SHH) was found to be critically important in regulating HSPC activation and reprogramming during the granulopoietic response to serious bacterial infection [61]. Mice with deletions in Gli genes, critical mediators of SHH signaling, showed attenuated activation of marrow HSPC division and production of granulocytes following bacteremia, suggesting that the SHH pathway might be involved in HSC differentiation upon infection [61]. Ehrlichia muris infection increased myelopoiesis, circulating myeloid cells, increased Sca-1 expression and decreased myeloid progenitor cells in mice — all responses dependent on IFNγ but not IFNα signaling [54]. Strikingly, IFNγ promoted the emergence of a specific myelo-lymphoid progenitor that produced myeloid cells to constrain microbe spread in a Plasmodium chabaudi-based murine model of malaria [62]. These findings demonstrate IFNγ signaling, particularly in the context of infection, is a potent mediator of HSC myeloid differentiation and eventual exhaustion in the setting of infection.
In the context of an acute viral infection, IFNα, along with TNFα and lymphotoxin (LT), can transiently alter hematopoiesis, similar to IFNγ. In vesicular stomatitis virus (VSV) and murine cytomegalovirus (MCMV) infection, persistent IFNα signaling induced by non-acute MCMV infection results in a sustained inflammatory state within the bone marrow that is associated with long-lasting impairment of HSC function [56]. Acute viral infection impairs reconstitution potential of LT-HSCs even after resolution of the acute infection, indicating there are lasting reprogramming effects following pathogen clearance. Bone marrow aplasia after LCMV infection was shown to be IFNα-dependent [20]. In contrast, IFNα plays an important role in protecting hematopoiesis during systemic stress responses to the opportunistic fungal pathogen Pneumocystis [63]. The differences in HSC response to IFNα between sterile stimulus models that induce proliferation and viral infection models that lead to stem cell exhaustion imply that the duration and the source of IFNα may play a role in altering the balance of HSC self-renewal and homeostasis.
Interferonopathies
Control of the host immune response is dependent on its ability to be able to discriminate self vs. foreign antigens, including determining whether free genetic material is pathogen or host-derived. In the past few years, studies have shown there are monogenic diseases present within the human population that lead to persistent activation or loss of negative regulation of_the type I (IFNα) interferon response, known as interferonopathies [64]. Type I interferonopathies are comprised of phenotypically heterogenous groups of autoimmune and auto-inflammatory disorders including Aicardi-Goutiéres syndrome (AGS), systemic lupus erythematosus (SLE), and proteosome associated auto-inflammatory disorders (PRASS) [64]. Although most interferonopathies are caused by loss of function mutations, STING-associated vasculopathy onset in infants (SAVI) is caused by gain of function mutations in STING1 [65]. The presentation and severity of auto-inflammation in each patient is typically determined by the specific mutated gene and the protein machinery’s function (e.g. deoxyribonuclease, ribonuclease, dsDNA sensor, proteasome) [64–66]. For example, mutations in STING 1 and TMEM173 are associated with the development of severe lung disease but this is not associated with other type I interferonopathies [64, 65]. In a recent publication Uggenti et al. showed that mutations in genes necessary for the replication-dependent histone pre-mRNA-processing complex in patients with Aicardi-Goutiéres syndrome, indicating nuclear histones are also critical in suppressing the immunogenicity of DNA [66].
As previously mentioned in the introduction of this review, interferon production can be induced via cell intrinsic mechanisms. Interestingly, this persistent IFNα signaling seen in type I interferonopathies do not typically lead to aplastic anemia in humans except in cases of ACP5, implying that this autoinflammation does not lead to decreased stem cell renewal [64]. This is consistent with in vivo studies of transgenic mice that were engineered to overexpress picornavirus RNA-dependent RNA polymerase even in the absence of viral infection [67]. Although these mice have lifelong upregulation of ISGs, these mice are overall healthy and with normal lifespans [67]. Prior work has shown that the bone marrow is able to adapt to persistent IFNα to prevent stem cell exhaustion [39]. However, this is not true for persistent type II interferon signaling. This may explain why most patients with interferonopathies do not develop cytopenias while patients with type II mediated aplastic anemia do. The differential responses of persistent IFNα versus IFNγ is another example of how the in vivo response to interferon signaling is complex and dependent on the modality, context, and duration of interferon exposure.
Aplastic anemia and aging
Depending on the context and duration of exposure, IFNα signaling can have contradictory effects on the hematological system. Although it may be helpful to induce HSC division in certain cases as a response to an infection or other assault on the body, the persistent presence of IFN signaling can lead to HSC exhaustion and overall loss of the stem cell pool, the cause of aplastic anemia [68]. Similarly, while tonic IFNγ signaling is required to maintain baseline homeostasis, dysregulation of IFNγ signaling can lead to disease pathology in disease contexts [2, 4, 9, 61]. As previously mentioned, IFNγ can push HSCs to divide and differentiate but persistent signaling leads to HSC loss and exhaustion. HSC loss can result in bone marrow failure and pancytopenia. These effects have been shown to play a role in the development of aging-related phenotypes in HSCs. Aging is regarded as a natural form of stress to the hematopoietic system. Work done by Michael Milsom’s group showed that extensive treatment with IFNs induces high levels of DNA damage in HSCs as a result of increasing proliferation of these typically quiescent cells [70]. Using a mouse model of Fanconi anemia, they were able to show that extensive exposure to IFNα was sufficient to deplete the HSC pool and promote severe aplastic anemia (SAA) in FA mice [70]. The Fanconi pathway is critical in the response to interstrand cross-links (ICLs); failure to remove these ICLs results in a failure cell division or uncontrolled cell growth due to an inability to make new DNA molecules or lack of DNA repair processes, respectively [71]. Because hematopoietic cells are fairly fast dividing, the blood system of FA patients is particularly affected and increased damage leads to pancytopenia in almost all patients and a higher risk for developing acute myeloid leukemia (AML) and myelodysplasia [72]. However, without IFN-induced stress, FA mice did not develop SAA, even in extreme old age, indicating that HSC quiescence is a protective mechanism that preserves the functionality of the stem cell pool by preventing DNA damage.
As we age, inflammation increases, escalating the risk for aplastic anemia (AA). Although the age of AA patients may vary, it is more common to develop AA beyond the age of 60 [73]. Classically, excessive IFNα and IFNγ signaling have been reported in patients with AA [58]. Patients who have AA have increased IFNγ serum levels due to autoreactive T cells [54, 60]. This dysregulation leads to HSC loss, which is likely linked to a deficiency of HSC self-renewal properties [68]. Classically-defined differentiation patterns of multipotent progenitor cells (MPP) to megakaryocyte erythroid (MEP) and granulocyte macrophage (GMP) progenitors are disrupted in AA due to intrinsic IFNγ signaling, leading to overall decreased circulating blood counts [68].
Another aging-related phenomenon is the development of clonal hematopoiesis (CH), wherein a genetically distinct population of HSCs becomes disproportionally represented in the peripheral blood [21, 66, 67]. CH is associated with a set of somatic mutations that give some HSCs a competitive selection advantage over other clones [21, 66–69]. Meisel et al. showed that the expansion of HSCs with somatic Tet2 mutations, associated with pre-leukemic myelo-proliferation (PMP), was dependent on microbe-mediated inflammation [11]. Recently, it has been shown that persistent inflammatory states induced by autoimmune disorders like ulcerative colitis can promote of clonal hematopoiesis. Ulcerative colitis patients had significantly increased IFNγ serum levels and sequencing of the peripheral blood showed a high frequency of mutations in PPM1D and DNMT3A, two of the most common genes affiliated with clonal hematopoiesis in the population [11]. This observation implies that persistent inflammation can contribute to the expansion of stem cell clones, which also can increase overall hematological cancer risk. In a recent study using powerful genomic tools to investigate the transcriptome and epigenome, it was found that aged Lineage−CD34+CD38− (HSC enriched; HSCe) cells undergo age-associated epigenetic reprogramming through redistribution of DNA methylation and reductions in activating histone marks [78]. These changes, particularly seen in developmental and cancer-related pathways, were comparable to those observed in other AML patients regardless of age, indicating that age-associated reprogramming, likely due to increased inflammation, may form a pre-existing condition that fosters the development of age-related AML.
IFNs and hematological cancers
Whereas prolonged exposure to IFNγ and IFNα can lead to HSC exhaustion and AA, recent studies suggest that leukemic stem cells (LSCs) might respond differently. In most leukemias, LSCs are thought to derive from human CD34+/CD38− HSPCs and typically occur at a small frequency [71]. LSCs are also self-renewing, multipotent, quiescent cells with the ability to engraft and initiate human disease in immune-compromised mice [71]. Due to their heightened drug resistance, LSCs are thought to facilitate relapse and thus are an important therapeutic target [71]. IFNγ signaling might promote LSC expansion, since growth factor related expansion of CD34+ cells can be increased by IFNγ in leukemic individuals [72]. In a murine CML model, IFNγ secreted by cytotoxic T cells increased proliferation and induced differentiation of LSCs [81], and proliferation and colony formation of CD34+ cells from CML patients increased upon IFNγ exposure [81]. Thus, these data suggest that, rather than leading to exhaustion, IFNγ may promote LSC expansion. Understanding how IFNγ produced by the host immune system contributes to leukemia progression or resistance will be crucial to develop novel treatment strategies to eliminate LSCs.
IFNα can exert direct anti-tumor effects by limiting the production of growth-promoting cytokines, stimulating apoptosis, inhibiting cellular proliferation, and increasing immunogenicity. IFNα therapy was first introduced to treat hairy cell leukemia, a rare chronic B-cell lymphoid malignancy that causes leukemic cells to have a “hairy” phenotype [82, 83]. The response rate was 80-90%, however the duration of cancer remission was short with a high chance of relapse [82, 83]. IFNα was also used as a first-line treatment for CML patients prior to the introduction of tyrosine kinase inhibitors (TKIs) [1]. A recent study suggests that IFNα can exhaust CML LSCs by CEBPβ-mediated differentiation [76]. IFNα has also been a mainstay of therapy for myeloproliferative neoplasms [77], with some studies suggesting that IFNα may have a differential effect on WT versus JAK2-mutant HSCs, thereby reducing JAK2-mutant hematopoiesis while restoring normal clones [86]. However, similar to IFNγ, IFNα causes adverse side effects that can lead to flu-like symptoms, central nervous system defects, depression, and a 20% chance of developing a secondary cancer [1, 12]. IFNα has also been used to increase blood cell counts in patients who suffer from anemia due to infection or as a result from chemotherapy treatments [1, 12, 75]. Though monotherapy with IFNα was not successful, combination therapy with TKIs or other chemotherapeutics may be a viable treatment option to target LSCs through activation and improved chemosensitivity upon IFNα treatment or by altering the host immune response to LSCs. However, more studies investigating the impact of IFN treatment on LSC function and resistance will be essential to gain knowledge for optimization of treatment protocols.
The future of IFN therapy: Reprogramming HSCs to enhance the host immune response
Throughout this review we have discussed how IFNα and IFNγ exhibit duality in their ability to modulate HSC self-renewal and differentiation, particularly regarding the duration and source of this inflammatory signaling (Figure 1). An exciting avenue currently being pursued is how IFN signaling may function to reprogram HSPCs. Within the past decade, it was discovered that innate immune cells, traditionally thought to have no immunological memory, can mount heightened responses to infectious rechallenge, a phenomenon called trained immunity [79–81]. The existence of trained immunity in humans has been exhibited in epidemiological studies of children vaccinated with live attenuated vaccines including Bacillus Calmette-Guerin (BCG), measles, and oral polio vaccine, who were significantly protected against other upper respiratory infections, leading to decreased mortality [90]. A recent study indicates that HSCs serve as the reservoir for BCG-induced trained immunity, and that this response is IFNγ-signaling dependent [52, 83]. Specifically, HSCs from a vaccinated animal are sufficient to confer improved immunity in naïve transplant recipients, but trained immunity was not transferred if HSCs lacked IFNγ receptor [52]. Further work uncovering the mechanism of this induced protection will not only help to improve methods of vaccination, but also increase our knowledge on how priming influences the BM niche, induces IFN signaling, and reprograms HSCs to respond more efficiently to infectious challenge. The ability to influence HSC differentiation with a myeloid bias could be a powerful tool to improve our host immune response to a wide variety of pathogens. Further work needs to be completed to determine the longevity of this trained response along with important transcriptional and epigenetic regulators required to induce HSC reprogramming.
Strikingly, pegylated IFNα is being used in both prophylactic and therapeutic contexts for the SARS-CoV-2 pandemic with some success. Since the beginning of the COVID-19 pandemic, the levels of IFNα signaling have been correlated with severity of infection. Lower levels of circulating IFNα correlate with increased disease severity likely due to inadequate chemokine signaling to recruit critical immune effector cells to the site of infection and hence a less effective immune response [92]. One study conducted in Wuhan, China found that patients that were treated with IFNα with or without Umifenovir, an antiviral drug, had a significant reduction in detectible virus in the upper respiratory tract and reduced systemic levels of IL-6 and C-reactive protein (CRP), two common inflammatory molecules [85]. While IFNα is currently only recommended as part of an approved clinical trials, it will be interesting to follow current work on the use of IFNα treatment for COVID-19 as well as its potential involvement in COVID19-associated lymphopenia[86, 87].
Conclusions
In this review we have highlighted the effects of IFNs on HSPCs. We have discussed the somewhat paradoxical finding that IFNs can promote HSC division while at the same time impairing function and depleting numbers through increased differentiation and reduced self-renewal. The duration of IFN signaling is thus pivotal to the overall outcome, with short-term effects possibly contributing to improved immunity whereas long-term exposure leading to stem cell exhaustion and aplastic anemia (Figure 1). We highlighted the experimental hurdles that make studying the effects of IFNs on hematopoiesis challenging, particularly with respect to changes in marker expression (Figure 2) and the role of the bone marrow niche in governing HSC responses. Although IFNs are an old drug, continuously expanding knowledge about their role in HSC division, differentiation, and self-renewal may reveal novel opportunities to modulate IFN signaling to treat infectious, inflammatory, and oncologic disorders (Figure 3).
Highlights.
Type I and II Interferons, specifically IFNα and IFNα, play an important role in the self-renewal, quiescence, and differentiation of hematopoietic stem cells (HSCs)
Basal interferon signaling is critical for exit from HSC quiescence during development but the context and duration of interferon signaling in acute and chronic contexts determine IFN’s effect on adult HSC biology
Internal and external stressors including infection, aging, and autoimmunity can lead to persistent IFN signaling and tip the balance of HSC homeostasis
Modulating IFN signaling is a proven treatment modality in patients and has further therapeutic potential for HSC transplantation and infectious diseases
Acknowledgements
This work was supported by the SFB873 and FOR2033 funded by the Deutsche Forschungsgemeinschaft (DFG) (M.A.G.E.) and the Dietmar Hopp Foundation (M.A.G.E.), and by the National Institutes of Health grants R01HL136333, R01AI141716 and R01HL134880 (K.Y.K) and T32 AI053831 and DK092883 (B.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Borden EC, et al. , Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov, 2007. 6(12): p. 975–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee AJ and Ashkar AA, The Dual Nature of Type I and Type II Interferons. Front Immunol, 2018. 9: p. 2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raefsky EL, et al. , Studies of interferon as a regulator of hematopoietic cell proliferation. J Immunol, 1985. 135(4): p. 2507–12. [PubMed] [Google Scholar]
- 4.Baldridge MT, et al. , Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature, 2010. 465(7299): p. 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Essers MA, et al. , IFNalpha activates dormant haematopoietic stem cells in vivo. Nature, 2009. 458(7240): p. 904–8. [DOI] [PubMed] [Google Scholar]
- 6.King KY and Goodell MA, Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nature Publishing Group, 2011: p. 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacNamara KC, et al. , Transient Activation of Fiematopoietic Stem and Progenitor Cells by IFNα during Acute Bacterial Infection. PLoS ONE, 2011. 6(12): p. e28669–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matatall KA, et al. , Type II Interferon Promotes Differentiation of Myeloid-Biased Fiematopoietic Stem Cells. Stem Cells, 2014. 32(11): p. 3023–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matatall KA, et al. , Chronic Infection Depletes Fiematopoietic Stem Cells through Stress-Induced Terminal Differentiation. Cell Reports, 2016. 17(10): p. 2584–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang CRC, et al. , Inflammatory cytokines promote clonal hematopoiesis with specific mutations in ulcerative colitis patients. Exp Hematol, 2019. 80: p. 36–41 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meisel M, et al. , Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature, 2018. 557(7706): p. 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor MW, Interferons, in Viruses and Man: A History of Interactions. 2014. p. 101–119.
- 13.Ivashkiv LB and Donlin LT, Regulation of type I interferon responses. Nat Rev Immunol, 2014. 14(1): p. 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Y, et al. , Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell, 2012. 21(6): p. 723–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang T-H, Chintalacharuvu KR, and Morrison SL, Targeting IFN-α to B Cell Lymphoma by a Tumor-Specific Antibody Elicits Potent Antitumor Activities. The Journal of Immunology, 2007. 179(10): p. 6881–6888. [DOI] [PubMed] [Google Scholar]
- 16.Su TH and Liu CJ, Combination Therapy for Chronic Hepatitis B: Current Updates and Perspectives. Gut Liver, 2017. 11(5): p. 590–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie QL, et al. , The Efficacy and Safety of Entecavir and Interferon Combination Therapy for Chronic Hepatitis B Virus Infection: A Meta-Analysis. PLoS One, 2015. 10(7): p. e0132219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shehab TM, et al. , Effectiveness of interferon alpha-2b and ribavirin combination therapy in the treatment of naive chronic hepatitis C patients in clinical practice. Clin Gastroenterol Hepatol, 2004. 2(5): p. 425–31. [DOI] [PubMed] [Google Scholar]
- 19.Binder D, et al. , Virus-induced transient bone marrow aplasia: major role of interferon-alpha/beta during acute infection with the noncytopathic lymphocytic choriomeningitis virus. J Exp Med, 1997. 185(3): p. 517–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Utay NS and Douek DC, Interferons and HIV Infection: The Good, the Bad, and the Ugly. Pathog Immun, 2016. 1(1): p. 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaiswal S, et al. , Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. New England Journal of Medicine, 2014. 371(26): p. 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim PG, et al. , Interferon-α signaling promotes embryonic HSC maturation. Blood, 2016. 128(2): p. 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sawamiphak S, Kontarakis Z, and Stainier DY, Interferon gamma signaling positively regulates hematopoietic stem cell emergence. Dev Cell, 2014. 31(5): p. 640–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bekisz J, et al. , Antiproliferative Properties of Type I and Type II Interferon. Pharmaceuticals (Basel), 2010. 3(4): p. 994–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paucker K, Cantell K, and Henle W, Quantitative studies on viral interference in suspended L cells. III. Effect of interfering viruses and interferon on the growth rate of cells. Virology, 1962. 17: p. 324–34. [DOI] [PubMed] [Google Scholar]
- 26.de Bruin AM, et al. , Interferon- impairs proliferation of hematopoietic stem cells in mice. Blood, 2013. 121(18): p. 3578–3585. [DOI] [PubMed] [Google Scholar]
- 27.Gidali J, Feher I, and Talas M, Proliferation inhibition of murine pluripotent haemopoietic stem cells by interferon or poly I:C. Cell Tissue Kinet, 1981. 14(1): p. 1–7. [DOI] [PubMed] [Google Scholar]
- 28.Young HA, et al. , Bone marrow and thymus expression of interferon-gamma results in severe B-cell lineage reduction, T-cell lineage alterations, and hematopoietic progenitor deficiencies. Blood, 1997. 89(2): p. 583–95. [PubMed] [Google Scholar]
- 29.Sato T, et al. , Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon–dependent exhaustion. Nature Medicine, 2009. 15(6): p. 696–700. [DOI] [PubMed] [Google Scholar]
- 30.Klimpel GR, Fleischmann WR Jr., and Klimpel KD, Gamma interferon (IFN gamma) and IFN alpha/beta suppress murine myeloid colony formation (CFU-C)N: magnitude of suppression is dependent upon level of colony-stimulating factor (CSF). J Immunol, 1982. 129(1): p. 76–80. [PubMed] [Google Scholar]
- 31.Snoeck HW, et al. , Interferon gamma selectively inhibits very primitive CD342+CD38− and not more mature CD34+CD38+ human hematopoietic progenitor cells. The Journal of Experimental Medicine, 1994. 180(3): p. 1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verma A, et al. , Activation of the p38 mitogen-activated protein kinase mediates the suppressive effects of type I interferons and transforming growth factor-beta on normal hematopoiesis. J Biol Chem, 2002. 277(10): p. 7726–35. [DOI] [PubMed] [Google Scholar]
- 33.Redig AJ, et al. , Activation of protein kinase C{eta} by type I interferons. J Biol Chem, 2009. 284(16): p. 10301–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang L, et al. , IFN-gamma negatively modulates self-renewal of repopulating human hemopoietic stem cells. J Immunol, 2005. 174(2): p. 752–7. [DOI] [PubMed] [Google Scholar]
- 35.Pietras EM, et al. , Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. The Journal of Experimental Medicine, 2014. 211(2): p. 245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morrison SJ and Spradling AC, Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell, 2008. 132(4): p. 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ehninger A and Trumpp A, The bone marrow stem cell niche grows up: mesenchymal stem cells and macrophages move in. The Journal of Experimental Medicine, 2011. 208(3): p. 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schurch CM, Riether C, and Ochsenbein AF, Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell, 2014. 14(4): p. 460–72. [DOI] [PubMed] [Google Scholar]
- 39.Liang C, et al. , Interferon-gamma mediates the immunosuppression of bone marrow mesenchymal stem cells on T-lymphocytes in vitro. Hematology, 2018. 23(1): p. 44–49. [DOI] [PubMed] [Google Scholar]
- 40.McCabe A, et al. , Macrophage-Lineage Cells Negatively Regulate the Hematopoietic Stem Cell Pool in Response to Interferon Gamma at Steady State and During Infection. Stem Cells, 2015. 33(7): p. 2294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Umemoto T, et al. , Integrin αvβ3 enhances the suppressive effect of interferon-γ on hematopoietic stem cells. The EMBO Journal, 2017. 36(16): p. 2390–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prendergast AM, et al. , IFNalpha-mediated remodeling of endothelial cells in the bone marrow niche. Haematologica, 2017. 102(3): p. 445–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Negrotto S, et al. , Expression and functionality of type I interferon receptor in the megakaryocytic lineage. J Thromb Haemost, 2011. 9(12): p. 2477–85. [DOI] [PubMed] [Google Scholar]
- 44.Uckelmann H, et al. , Extracellular matrix protein Matrilin-4 regulates stress-induced HSC proliferation via CXCR4. J Exp Med, 2016. 213(10): p. 1961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rabe JL, et al. , CD34 and EPCR coordinately enrich functional murine hematopoietic stem cells under normal and inflammatory conditions. Exp Hematol, 2020. 81: p. 1–15 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bujanover N, et al. , Identification of immune-activated hematopoietic stem cells. Leukemia, 2018. 32(9): p. 2016–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Bruin AM, et al. , IFN induces monopoiesis and inhibits neutrophil development during inflammation. Blood, 2012. 119(6): p. 1543–1554. [DOI] [PubMed] [Google Scholar]
- 48.Zhang H, et al. , STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood, 2010. 116(14): p. 2462–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas SE, et al. , p53 and translation attenuation regulate distinct cell cycle checkpoints during endoplasmic reticulum (ER) stress. J Biol Chem, 2013. 288(11): p. 7606–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MacNamara KC, et al. , Infection-Induced Myelopoiesis during Intracellular Bacterial Infection Is Critically Dependent upon IFN- Signaling. The Journal of Immunology, 2011. 186(2): p. 1032–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haas S, et al. , Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell, 2015. 17(4): p. 422–34. [DOI] [PubMed] [Google Scholar]
- 52.Kaufmann E, et al. , BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell, 2018. 172(1-2): p. 176–190.e19. [DOI] [PubMed] [Google Scholar]
- 53.Wu X, et al. , Intrinsic Immunity Shapes Viral Resistance of Stem Cells. Cell, 2018. 172(3): p. 423–438.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Young NS, Scheinberg P, and Calado RT, Aplastic anemia. Current Opinion in Hematology, 2008. 15(3): p. 162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Young NS and Maciejewski J, The pathophysiology of acquired aplastic anemia. New England Journal of Medicine, 1997. 336(19): p. 1365–1372. [DOI] [PubMed] [Google Scholar]
- 56.Hirche C, et al. , Systemic Virus Infections Differentially Modulate Cell Cycle State and Functionality of Long-Term Hematopoietic Stem Cells In Vivo. Cell Rep, 2017. 19(11): p. 2345–2356. [DOI] [PubMed] [Google Scholar]
- 57.Shi X, et al. , Sonic Hedgehog Signaling Regulates Hematopoietic Stem/Progenitor Cell Activation during the Granulopoietic Response to Systemic Bacterial Infection. Frontiers in Immunology, 2018. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Belyaev NN, et al. , Extramedullary myelopoiesis in malaria depends on mobilization of myeloid-restricted progenitors by IFN-gamma induced chemokines. PLoS Pathog, 2013. 9(6): p. e1003406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prigge JR, et al. , Type I IFNs Act upon Hematopoietic Progenitors To Protect and Maintain Hematopoiesis during Pneumocystis Lung Infection in Mice. The Journal of Immunology, 2015. 195(11): p. 5347–5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith JN, Kanwar VS, and MacNamara KC, Hematopoietic Stem Cell Regulation by Type I and II Interferons in the Pathogenesis of Acquired Aplastic Anemia. Front Immunol, 2016. 7: p. 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.King KY, et al. , Irgm1 protects hematopoietic stem cells by negative regulation of IFN signaling. Blood, 2011. 118(6): p. 1525–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaschutnig P, et al. , The Fanconi anemia pathway is required for efficient repair of stress-induced DNA damage in haematopoietic stem cells. Cell Cycle, 2015. 14(17): p. 2734–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Müller LUW and Williams DA, Finding the needle in the hay stack: Hematopoietic stem cells in Fanconi anemia. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 2009. 668(1-2): p. 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Du W, et al. , Inflammation-Mediated Notch Signaling Skews Fanconi Anemia Hematopoietic Stem Cell Differentiation. The Journal of Immunology, 2013. 191(5): p. 2806–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Young NS and Kaufman DW, The epidemiology of acquired aplastic anemia. Haematologica, 2008. 93(4): p. 489–492. [DOI] [PubMed] [Google Scholar]
- 66.Genovese G, et al. , Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. New England Journal of Medicine, 2014. 371(26): p. 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zink F, et al. , Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood, 2017. 130(6): p. 742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Coombs CC, et al. , Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell, 2017. 21(3): p. 374–382.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wong TN, et al. , Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nature Communications, 2018. 9(1): p. 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.MacLean AL, et al. , Single Cell Phenotyping Reveals Heterogeneity Among Hematopoietic Stem Cells Following Infection. Stem Cells, 2017. 35(11): p. 2292–2304. [DOI] [PubMed] [Google Scholar]
- 71.Vetrie D, Helgason GV, and Copland M, The leukaemia stem cell: similarities, differences and clinical prospects in CML and AML. Nat Rev Cancer, 2020. 20(3): p. 158–173. [DOI] [PubMed] [Google Scholar]
- 72.Madapura HS, et al. , Interferon gamma is a STAT1-dependent direct inducer of BCL6 expression in imatinib-treated chronic myeloid leukemia cells. Oncogene, 2017. 36(32): p. 4619–4628. [DOI] [PubMed] [Google Scholar]
- 73.Schurch C, et al. , Cytotoxic T cells induce proliferation of chronic myeloid leukemia stem cells by secreting interferon-gamma. J Exp Med, 2013. 210(3): p. 605–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bohn J-P, Gastl G, and Steurer M, Long-term treatment of hairy cell leukemia with interferon-α: still a viable therapeutic option, memo - Magazine of European Medical Oncology, 2016. 9(2): p. 63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Silva W.F.d., et al. , Current role of interferon in hairy cell leukemia therapy: a timely decision. Hematology, Transfusion and Cell Therapy, 2019. 41(1): p. 88–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yokota A, et al. , C/EBPbeta is a critical mediator of IFN-alpha-induced exhaustion of chronic myeloid leukemia stem cells. Blood Adv, 2019. 3(3): p. 476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yacoub A, et al. , Pegylated interferon alfa-2a for polycythemia vera or essential thrombocythemia resistant or intolerant to hydroxyurea. Blood, 2019. 134(18): p. 1498–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tashi T, et al. , Pegylated interferon Alfa-2a and hydroxyurea in polycythemia vera and essential thrombocythemia: differential cellular and molecular responses. Nature Publishing Group, 2018. 112: p. 3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fanucchi S and Mhlanga MM, Lnc-ing Trained Immunity to Chromatin Architecture. Frontiers in cell and developmental biology, 2019. 7: p. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Groh L, et al. , Getting to the marrow of trained immunity. Epigenomics, 2018. 10(9): p. 1151–1154. [DOI] [PubMed] [Google Scholar]
- 81.Netea MG and van der Meer JWM, Trained Immunity: An Ancient Way of Remembering. Cell Host and Microbe, 2017. 21(3): p. 297–300. [DOI] [PubMed] [Google Scholar]
- 82.Butkeviciute E, Jones CE, and Smith SG, Heterologous effects of infant BCG vaccination: Potential mechanisms of immunity. Future Microbiology, 2018. 13(10): p. 1193–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Arts RJW, et al. , BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host and Microbe, 2018. 23(1): p. 89–100.e5. [DOI] [PubMed] [Google Scholar]
- 84.Blanco-Melo D, et al. , Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell, 2020: p. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xu P, et al. , Arbidol/IFN-alpha2b therapy for patients with corona virus disease 2019: a retrospective multicenter cohort study. Microbes Infect, 2020. 22(4-5): p. 200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sallenave J-M and Guillot L, Innate Immune Signaling and Proteolytic Pathways in the Resolution or Exacerbation of SARS-CoV-2 in Covid-19: Key Therapeutic Targets? Frontiers in Immunology, 2020. 11: p. 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fathi N and Rezaei N, Lymphopenia in COVID-19: Therapeutic opportunities. Cell Biol Int, 2020. 44(9): p. 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]