

Abstract
Background
The ideal participants for Alzheimer's disease (AD) clinical trials would show cognitive decline in the absence of treatment (i.e., placebo arm) and would also respond to the therapeutic intervention.
Objective
To investigate if predictive models can be an effective tool for identifying and excluding people unlikely to show cognitive decline as an enrichment strategy in AD trials.
Method
We used data from the placebo arms of two phase 3, double‐blind trials, EXPEDITION and EXPEDITION2. Patients had 18 months of follow‐up. Based on the longitudinal data from the placebo arm, we classified participants into two groups: one showed cognitive decline (any negative slope) and the other showed no cognitive decline (slope is zero or positive) on the Alzheimer's Disease Assessment Scale–Cognitive subscale (ADAS‐cog). We used baseline data for EXPEDITION to train regression‐based classifiers and machine learning classifiers to estimate probability of cognitive decline. Models were applied to EXPEDITION2 data to assess predicted performance in an independent sample. Features used in predictive models included baseline demographics, apolipoprotein E ε4 genotype, neuropsychological scores, functional scores, and volumetric magnetic resonance imaging.
Result
In EXPEDITION, 46.3% of placebo‐treated patients showed no cognitive decline and the proportion was similar in EXPEDITION2 (45.6%). Models had high sensitivity and modest specificity in both the training (EXPEDITION) and replication samples (EXPEDITION2) for detecting the stable group. Positive predictive value of models was higher than the base prevalence of cognitive decline, and negative predictive value of models were higher than the base rate of participants who had stable cognition.
Conclusion
Excluding persons with AD unlikely to decline from the active and placebo arms of clinical trials using predictive models may boost the power of AD trials through selective inclusion of participants expected to decline.
Keywords: Alzheimer's disease, anti‐amyloid monoclonal antibody, clinical trials, cognitive decline, machine learning, predictive analytics
1. INTRODUCTION
Alzheimer's disease (AD) is the most common primary neurodegenerative disorder of late life. The costs and burden of caring for patients with AD is expected to expand as the population ages. 1 AD drug development has been extraordinarily difficult, with a failure rate of clinical trials of more than 99% over the last two decades. 2 These failures have led to debate about the potential deficiencies in our understanding of the pathogenesis of AD and potential pitfalls in diagnosis, choice of therapeutic targets, development of drug candidates, and design of clinical trials. 3
Biological and phenotypic heterogeneity poses a major challenge in selection of eligible participants, in the identification of therapeutic targets, and in estimation of trial sample sizes. 4 Individual rates of cognitive decline are highly variable from person to person with AD. 5 For a disease‐modifying therapy (DMT), treatment is expected to slow cognitive decline relative to more rapid decline in the placebo group. But not all placebo‐treated patients decline within the timeframe of AD trials, which reduces the power to detect differences in rate of decline between the active treatment group and the placebo group. 6 We define decline as any worsening of cognitive status or functional status at final follow‐up time. If people not likely to decline are enrolled in studies of potentially DMTs, treatment effects may be underestimated or completely missed due to lack of significant progression in the placebo arm.
Lack of decline in some participants enrolled in trials might be due to absence of AD pathology or contribution of non‐AD pathology to cognitive impairment. Studies of amyloid imaging in patients recruited to clinical trials show that up to 50% of cases with mild cognitive impairment (MCI) and 20% of those with mild dementia do not have a measurable amyloid plaque burden and do not meet biomarker criteria for AD. 7 While using neuroimaging and cerebrospinal fluid (CSF) studies to detect presence of AD pathology reduces biological heterogeneity, it does not account for other sources of heterogeneity in rate of decline of individuals in treatment trials. 8 , 9 AD pathology often co‐exists with other pathologies that contribute to neurodegeneration, cognitive decline, and ultimately dementia. 10 In fact, post mortem studies have consistently shown that in persons with dementia, >50% had mixed pathologies and multiple pathological diagnosis (AD, vascular dementia, Parkinson's disease, and Lewy body disease) at the time of death. 10 , 11 Therefore, even using biomarkers to confirm presence of AD pathology (i.e., amyloid or tau) prior to enrollment of participants in a trial will not entirely eliminate the problem imposed by natural biological heterogeneity.
One strategy to approach this problem and simultaneously boost the power of trials is to enroll only individuals likely to show cognitive progression based on data‐driven predictive models. 9 While models for quantitative risk prediction for AD have been available for many years, these models have not been incorporated into the design of AD clinical trials. Approaches to predicting disease progression vary widely along at least three key dimensions: the operational definition of the outcome being predicted, the candidate indicators used for prediction, and the nature of the statistical model used to make the prediction.
In this study, we aimed to determine the proportion of placebo‐treated patients who do not decline in two AD trials to develop models that predict lack of decline in placebo‐treated patients in one trial based on either logistic regression (LR) or machine learning (ML) models and to validate these models in an independent study. To accomplish these three goals, we used data from the placebo arm of two identical phase 3 trials of solanezumab versus placebo for the treatment of mild‐to‐moderate AD. In EXPEDITION we developed predictive models to estimate rate of cognitive decline in persons with AD. In EXPEDITION2 we validated these models. We then consider the implications of our findings for the design and powering of AD treatment trials.
2. METHODS
2.1. Study design and participants
Data from the placebo arm of two phase 3 clinical trials of solanezumab for mild‐to‐moderate AD were used for this study: EXPEDITION and EXPEDITION2. These trials were conducted by Eli Lilly and Company between May 2009 and June 2012 to evaluate the efficacy of solanezumab, a humanized monoclonal antibody that preferentially binds to soluble forms of amyloid, for treatment of AD. These trials did not demonstrate significant differences between active drug and placebo on cognition or functional ability of study participants. 12 The research protocol was approved by the institutional review board at each institution where the trial was conducted, and all participants provided written informed consent.
Details of these studies, including recruitment and methods, have been reported. 12 In brief, both trials involved otherwise healthy patients, 55 years of age or older, who had mild‐to‐moderate AD without depression. Diagnosis followed the criteria of the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association. 13 Mild‐to‐moderate status was determined on the basis of a score of 16 to 26 on the Mini‐Mental State Examination (MMSE; score range, 0–30, with higher scores indicating better cognitive function). 14 The absence of depression was documented on the basis of a score of 6 or less on the Geriatric Depression Scale (GDS; score range, 0–15, with higher scores indicating more severe depression). 15 Participants were randomly assigned to receive solanezumab or placebo. Only data from the placebo arm of the trial were used. Concurrent treatment with other Food and Drug Administration (FDA)‐approved treatments for AD (cholinesterase inhibitors and/or memantine) was allowed. Additional inclusion criteria for the current analysis required baseline magnetic resonance imaging (MRI) measures and completion of study (18 months of follow‐up). A total of 506 patients in EXPEDITION and 519 patients in EXPEDITION2 were assigned to receive placebo; 365 patients from EXPEDITION and 395 patients from EXPEDITION2 met criteria for our analysis.
2.1.1. Clinical outcome measures
Outcome measures for the trials included the 11‐ or 14‐item cognitive subscale of the Alzheimer's Disease Assessment Scale (ADAS‐Cog11 [score range, 0–70] and ADAS‐Cog14 [score range, 0–90], with higher scores indicating greater cognitive impairment) 16 and the Alzheimer's Disease Cooperative Study–Activities of Daily Living (ADCS‐ADL) scale (score range, 0–78, with lower scores indicating worse functioning). 17 Therefore, based on the primary endpoints of trials (ADAS‐Cog11 and ADCS‐ADL), primary outcomes for our analysis are defined as follows:
Change in cognitive status: based on longitudinal ADAS‐Cog11 data at both 15 and 18 months of follow‐up participants were divided into two groups: (1) stable cognition (SC) group, which included individuals with either no change in cognitive function or improvement in cognitive function (i.e., ADAS‐Cog1115 months – ADAS‐Cog11baseline ≤ 0 OR ADAS‐Cog1118 months – ADAS‐Cog11baseline ≤ 0); and (2) declining cognition (DC) group, which included individuals that showed decline in cognitive function at both 15 and 18 months of follow‐up (i.e., ADAS‐Cog15months – ADAS‐Cogbaseline > 0 AND ADAS‐Cog18months – ADAS‐Cogbaseline > 0).
Change in functional status: based on longitudinal ADCS‐ADL data at both 15 and 18 months of follow‐up participants were divided into two groups: (1) stable function (SF) group, which included individuals with either no change in functional status or improvement in functional status (i.e., ADCS‐ADL15 months – ADCS‐ADLbaseline ≥ 0 OR ADCS‐ADL18 months – ADCS‐ADLbaseline ≥ 0); and (2) declining function (DF) group, which included individuals that showed decline in functional function at both 15 and 18 months of follow‐up (i.e., ADCS‐ADL15months – ADCS‐ADL baseline < 0 AND ADCS‐ADL18months – ADCS‐ADLbaseline < 0).
RESEARCH IN CONTEXT
Systematic Review: The ideal participants for Alzheimer's disease (AD) clinical trials would show cognitive decline in the absence of treatment (i.e., placebo arm) and would also respond to the therapeutic intervention. Identifying such participants for AD trials has proven to be challenging.
Interpretation: Our study indicates that by using baseline data and predictive models (i.e., regression‐based or machine learning classifiers), we can effectively predict disease progression in a trial population. These models could be used to improve patient selection and enrich AD trials.
Future Directions: Recent AD trials have moved toward using amyloid and tau biomarkers as part of enrollment criteria. Future research should be conducted using multimodal data from new clinical trials, which have collected comprehensive biomarker data, to explore validity and generalizability of these models.
Most measures of cognition and function, including ADAS‐Cog and ADCS‐ADL, are prone to measurement error and day‐to‐day (or visit‐to‐visit) variability. Therefore, to decrease influence of measurement error and variability on our outcome, we opted to use measures of ADAS‐Cog and ADCS‐ADL at both 15 and 18 months of follow‐up (as summarized above) to differentiate true cognitive or functional decline from false detection of decline.
While using the method described above captures statistically significant changes in ADAS‐Cog scores, this may not equate to clinically relevant changes. 18 There is no clear gold standard to define cognitive status outcomes for “decliners versus stable,“ but clinically and from the patient's viewpoint, any decline in cognition is undesirable. Figure 1 depicts the rate of cognitive decline based on ADAS‐Cog at different follow‐up times. Median change in ADAS‐Cog at 5, 9, 12, 15, and 18 months of follow‐up was 0, 1, 1, 3, and 3, respectively. It has been suggested that 3 or more points decline on the ADAS‐Cog may be an appropriate minimal clinically relevant change (MCRC) that should be used in clinical studies. 18 Therefore, in a supplementary analysis, we used the following definition for cognitive status outcome to develop and validate the predictive models:
MCRC in cognitive status outcome: Based on longitudinal ADAS‐Cog11 data at both 15 and 18 months of follow‐up participants were divided into two groups: (1) SC group, which included individuals with either no change in cognitive function or improvement in cognitive function (i.e., ADAS‐Cog1115 months – ADAS‐cog11baseline < 4 OR ADAS‐Cog1118 months – ADAS‐Cog11baseline < 4) and (2) DC group, which included individuals that showed decline in cognitive function at both 15 and 18 months of follow‐up (i.e., ADAS‐Cog15months – ADAS‐Cogbaseline ≥ 3 AND ADAS‐Cog18months – ADAS‐Cogbaseline ≥ 3).
FIGURE 1.

Change in Alzheimer's Disease Assessment Scale–Cognitive subscale score at different follow‐up timeframes
2.2. Study features and predictors
The following measures were available at screening or baseline (initial visit after enrollment) visits for most patients who met the criteria for inclusion in this study and were used in predictive models:
Demographics: age, sex, years of formal education
Genomics: number of apolipoprotein E (APOE) ε4 alleles (0, 1, 2)
Clinical characteristics: GDS, concurrent treatment with cholinesterase inhibitors and/or memantine (yes or no), Clinical Dementia Rating Sum of Boxes (CDR‐SB) 19
Cognitive measures: ADAS‐Cog11, MMSE total score at screening visit, MMSE total score at baseline visit
Functional measures: ADCS‐ADL, the European Quality of Life—5 Dimensions (EQ‐5D) scale (Proxy version), 20 the Quality of Life in Alzheimer's Disease (QOL‐AD) scale 21
MRI measures: left hippocampal volume, right hippocampal volume, ventricular volume (sum of lateral and third ventricles), and total brain volume.
As normalization of data has proven to improve performance of machine learning models and is essential when predictors have different ranges, 22 all continuous variables were standardized using the following formula: ; where X = score, μ = mean, and σ = standard deviation. The MRI measures had a normal distribution in our sample and therefore converting measures using the standard score was deemed appropriate for our purpose. Total intracranial volume (TICV) was not available for patients; therefore, MRI measures were not corrected for ICV.
2.3. Data analysis
2.3.1. Predictive models and feature selection
The goal of predictive modeling was to partition the patients enrolled in the study into groups, one that did not show cognitive decline or functional decline during the timeframe of trial (18 months) and another group that showed disease progression and declining cognition or function during the same timeframe. Two different methods were used for prediction:
Standard (traditional) multivariate analysis using LR. Definitions of “traditional” multivariate statistical modeling for prediction and its differences with ML have been discussed at length in the literature, yet the distinction is not clear‐cut. 23 Studies comparing performance of ML models versus traditional multivariate models such as LRs have shown mixed results. 24 , 25 Purposeful feature selection is particularly important for LR models as multicollinearity and high dimensionality of features has proven to substantially decrease performance of these models. 26 Therefore, to reduce the number of predictors, we then created a parsimonious model by sequentially removing variables from the full model. A backward selection procedure was applied to select predictors into the final logistic model. A Bayes information criterion (BIC) versus model size plot was created to determine the number of variables in the final parsimonious model. 27 Furthermore, we made sure that additional variables in the model did not significantly improve the C‐statistic, and there was also a minimal loss in the calibration with this approach.
Machine learning predictive model. While many ML models have proven to be effective tools for predictions of outcomes in AD, in a previous study we showed that the ensemble ML models perform better than other ML models in predicting clinical outcomes. 28 Therefore, we used an ensemble linear discriminant (ELD) classifier for prediction of outcomes in this study. ELD is among the family of classification methods known as ensemble learning, in which the output of an ensemble of simple and low‐accuracy classifiers trained on subsets of features are combined (e.g., by weighted average of the individual decisions), so that the resulting ensemble decision rule has a higher accuracy than that obtained by each of the individual classifiers. 29 , 30 Details of this approach are provided elsewhere. 31
Statistical analysis was performed using SPSS (IBM, Inc., version 20) and MATLAB (version 2020a).
2.3.2. Analytical approach
We used baseline data from the EXPEDITION trial to train classifiers to contrast between individuals based on their longitudinal cognitive outcome (i.e., cognitive subgroups of SC and DC). Subsequently, trained models were validated using baseline data from EXPEDITION2 data. Predicted outcome of models were compared with actual longitudinal data at 15 and 18 months of follow‐up. General analytical approach for training and validation of models to contrast subgroups based on cognitive function is summarized in Figure 2. A similar approach was used to train and validate models to classify individuals based on their functional status to stable function and declining functional subgroups. The overall performance of each model was calculated based on the percentage of correct classification (accuracy), sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV).
FIGURE 2.
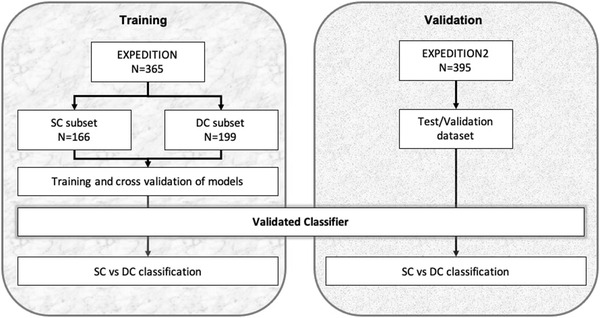
Study design. Data from EXPEDITION trial was used for training and data from EXPEDITION2 trial was used for validation. Participants were classified to two groups based on the longitudinal change in ADAS‐Cog score at 15 months and confirmed at 18 months of follow‐up (see text for details). Models were trained to classify participants of training dataset (left block). Subsequently, the newly developed model was applied to the validation dataset to predict if they will have decline in cognition or will remain cognitively stable in longitudinal follow‐up (right block). ADAS‐Cog, Alzheimer's Disease Assessment Scale–Cognitive subscale; SC, stable cognition; DC, declining cognition
The results from predictive models were compared to actual longitudinal data at 12 and 23 months of follow‐up. The overall performance of each model was calculated based on the percentage of correct classification (accuracy), sensitivity, specificity, PPV, and NPV. A McNemar test was used to compare the pairwise performance of classifications at 95% confidence level (α = 0.05): , where S ab refers to the samples correctly classified in classification a, but incorrectly classified in classification b, and S ba indicates the samples that are misclassified in classification a, but correctly classified in classification b. 32
3. RESULTS
3.1. Baseline characteristics
There were no significant differences between the placebo arm of EXPEDITION 1 and EXPEDTION 2 with respect to age, sex, or educational level (Table 1). Across groups, 55.2% to 60.5% of participants were APOE ε4 positive, with no significant imbalances. Approximately 90% of the patients in EXPEDITION and 93% of the patients in EXPEDITION2 were being treated with cholinesterase inhibitors, memantine, or both at baseline. Both studies included patients with mild‐to‐moderate AD, with a mean (± standard deviation [SD]) MMSE score of 21.0 ± 3.2 in EXPEDITION and 20.8 ± 3.6 in EXPEDITON 2. In the placebo arm of EXPEDITION, 53.7% of participants who completed the trial showed cognitive decline (Figure 3) and 60.5% showed functional decline. In the placebo arm of EXPEDITION2 among those who completed the trial, 54.4% of patients showed cognitive decline and 65.3% showed functional decline.
TABLE 1.
Demographic and baseline clinical characteristics of the patients
| EXPEDITION | EXPEDITION2 | |||||
|---|---|---|---|---|---|---|
| Characteristic 1 | All | Decliner a | Stable a | All | Decliner a | Stable a |
| Sample size, N (%) |
365 (100) |
199 (53.7) |
166 (46.3) |
395 (100) |
215 (54.4) |
180 (45.6) |
| Age (year) | 74.5 ± 7.8 | 74.5 ± 7.5 | 74.5 ± 8.2 | 72.1 ± 7.7 | 71.4 ± 8.1 | 72.9 ± 7.2 |
| Education (year) | 12.7 ± 3.9 | 12.9 ± 4.0 | 12.5 ± 3.8 | 11.6 ± 4.1 | 12.0 ± 4.0 | 11.2 ± 4.1 |
| Sex, men, N (%) |
156 (42.7) |
92 (46.9) |
64 (37.9) |
177 (44.8) |
103 (47.9) |
74 (41.1) |
| APOE ε4 carrier, N % |
221 (60.5) |
113 (57.5) |
108 (63.5) |
218 (55.2) |
125 (58.1) |
93 (51.7) |
| Antidementia therapy, N (%) |
329 (90.1) |
185 (64.4) |
144 (85.2) |
366 (92.7) |
200 (93.0) |
166 (92.2) |
| GDS | 1.7 ± 1.4 | 1.6 ± 1.4 | 1.8 ± 1.5 | 2.2 ± 1.6 | 2.2 ± 1.6 | 2.1 ± 1.5 |
| MMSE score | 21.0 ± 3.2 | 20.4 ± 3.1 | 21.8 ± 3.1 | 20.8 ± 3.6 | 20.6 ± 3.5 | 21.2 ± 3.5 |
| ADAS‐Cog11 score | 21.2 ± 8.1 | 22.0 ± 8.7 | 20.3 ± 7.3 | 22.4 ± 3.6 | 20.7 ± 9.2 | 22.1 ± 8.5 |
| ADCS‐ADL score | 62.5 ± 10.7 | 60.9 ± 11.0 | 64.3 ± 10.2 | 60.7 ± 12.3 | 59.0 ± 12.8 | 62.8 ± 11.4 |
| Total brain volume (cm3 ) | 1005.3 ± 105.9 | 997.8 ± 110.7 | 1013.0 ± 106.7 | 1009 ± 106.0 | 1006.5 ± 104.2 | 1012.3 ± 108.2 |
Note . Plus–minus values are means ± SD.
Based on change in ADAS‐Cog11 score at 15 and 18 months.
Abbreviations: ADAS‐cog, Alzheimer's Disease Assessment Scale Cognitive subscale; ADCS‐ADL, Alzheimer's Disease Cooperative Study–Activities of Daily Living; APOE, apolipoprotein E; GDS, Geriatric Depression Scale; MMSE, Mini‐Mental State Examination; SD, standard deviation.
FIGURE 3.
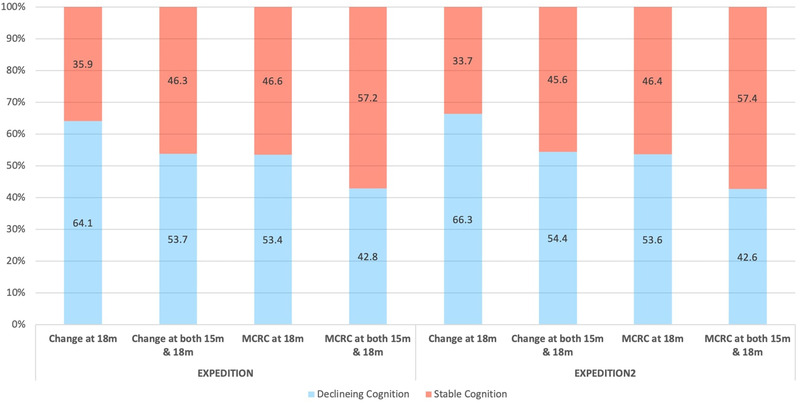
Percentage of patients with Declining or Stable Cognition based on any change on ADAS‐Cog score or minimal clinically relevant change (MCRC) on Alzheimer's Disease Assessment Scale–Cognitive subscale score
3.2. Models for predicting cognitive outcomes
The results for performance of all models developed for prediction of cognitive outcomes are summarized in Table 2. Based on the backward selection criteria, a total of five variables were selected for the final LR model including age, MMSE (at screening visit), CDR, ADAS‐Cog11, and ventricular volume. The relative importance of each predictor in classification by LR model in the training sample was 34.3% (age), 8.6% (MMSE), 17.4% (CDR‐SB), 12.4% (ADAS‐Cog11), and 27.3% (ventricular volume). In EXPEDITION (training sample), LR models achieved a sensitivity of 67.8% and specificity of 59.1% in classifying patients to cognitively stable and decliner groups. In EXPEDITON2 (validation sample), the model showed sensitivity and specificity of 64.2% and 53.3%, respectively. ELD models showed comparable performance in classification. In EXPEDITON, ELD models showed sensitivity of 71.4% and specificity of 55.0%, and in EXPEDITON2, models showed sensitivity of 68.8% and specificity of 51.7%. There was no significant difference between classification performance of LR and ELD models in validation sample (McNemar Chi‐square with Yates's correction = 0.16, P = .68).
TABLE 2.
Performance of predictive models in classifying patients with stable cognition from patients with declining cognition
| Model | Sample | Sensitivity, % (95% CI) | Specificity, % (95% CI) | PPV, % (95% CI) | NPV, % (95% CI) | AUC | Base rate, % a |
|---|---|---|---|---|---|---|---|
| LR |
EXPEDITION (training) |
67.8 (60.8–74.3) |
59.1 (62.4–66.6) |
65.8 (61.4–70.3) |
61.3 (55.6–66.8) |
0.68 | 53.7 |
|
EXPEDITION2 (validation) |
64.2 (57.4–70.6) |
53.3 (45.8–60.8) |
62.2 (57.7–66.4) |
55.5 (49.9+1.0) |
0.62 | 54.4 | |
| ELD |
EXPEDITION (training) |
71.4 (64.5–77.6) |
55.0 (47.2–65.6) |
64.8 (60.4–69.0) |
62.4 (56.2–68.3) |
0.66 | 53.7 |
|
EXPEDITION2 (validation) |
68.4 (61.7–74.5) |
51.7 (44.1–59.2) |
62.8 (58.6–66.8) |
57.7 (51.8–63.5) |
0.62 | 54.4 |
Base rate of cognitive decline (change of > 0 from baseline ADAS‐cog score) in the sample using longitudinal data at both 15 and 18 months.
Abbreviations: AUC, area under the curve; CI, confidence interval; ELD, ensemble linear discriminant model; LR, logistic regression; NPV, negative predictive value; PPV, positive predictive value.
In a supplementary analysis, we used MCRC of three or more on the ADAS‐Cog as the outcome of models. Based on this outcome, 42.8% of patients in the EXPEDITION trial and 42.6% of patients in the EXPEDITION2 trial showed cognitive decline (Figure 3). Using this new outcome led to decrease in sensitivity of models and increase in specificity. In the training sample (EXPEDITION), LR models had sensitivity of 47.4% and specificity of 76.2% in classifying patients to cognitively stable and decliner groups and in the validation sample (EXPEDITON2), the model showed sensitivity and specificity of 54.2% and 71.8%, respectively (Table 3). ELD models showed sensitivity of 45.4% and specificity of 80.6% in the training sample (EXPEDITON), and sensitivity of 49.4% and specificity of 75.9%. There was no significant difference between classification performance of LR and ELD models in validation sample (McNemar Chi‐square with Yates's correction = 0.01, P = .84).
TABLE 3.
Performance of predictive models in classifying patients with stable functional status from patients with declining functional status
| Model | Sample | Sensitivity, % (95% CI) | Specificity, % (95% CI) | PPV, % (95% CI) | NPV, % (95% CI) | AUC | Base rate, % a |
|---|---|---|---|---|---|---|---|
| LR |
EXPEDITION (training) |
81.4 (75.6–86.3) |
41.0 (32.9–49.4) |
60.5 (55.3–65.6) |
59.0 (50.6–66.9) |
0.67 | 60.5 |
|
EXPEDITION2 (validation) |
74.4 (98.6–79.6) |
46.7 (38.2–55.4) |
72.4 (68.9–75.6) |
49.2 (42.4–56.0) |
0.63 | 65.3 | |
| ELD |
EXPEDITION (training) |
83.7 (78.2–88.3) |
30.5 (23.2–38.7) |
64.9 (62.1–67.6) |
55.0 (45.4–64.2) |
0.65 | 60.5 |
|
EXPEDITION2 (validation) |
84.9 (79.9–89.0) |
37.2 (29.1–45.9) |
71.8 (68.9–74.5) |
56.7 (47.6–65.2) |
0.68 | 65.3 |
Base rate of functional decline (change of < 0 from baseline ADCS‐ADL score) in the sample at both 15 and 18 months
Abbreviations: ADCS‐ADL, Alzheimer's Disease Cooperative Study–Activities of Daily Living; AUC, area under the curve; CI, confidence interval; ELD, ensemble linear discriminant model; LR, logistic regression; NPV, negative predictive value; PPV, positive predictive value.
4. MODELS FOR PREDICTING FUNCTIONAL OUTCOMES
Results of models developed for prediction of functional outcomes are summarized in Table 4. Four features were selected for LR models for prediction of functional outcome, which included CDR‐SB, ADAS‐Cog11, ADCS‐ADL, and ventricular volume. The relative importance of each predictor in classification by the LR model in the training sample was 16.0% (CDR‐SB), 25.7% (ADAS‐Cog11), 21.9% (ADCS‐ADL), and 36.4% (ventricular volume). Sensitivity and specificity of LR models in the training sample (EXPEDITION) were 81.4% and 41.0%, respectively. Sensitivity and specificity of LR models in the validation sample (EXPEDITION2) were 74.4% and 46.7%, respectively. ELD model in the training sample showed classification sensitivity of 83.7% and specificity of 30.5%. In the validation sample, ELD models had sensitivity of 84.9 and specifity of 37.2%. There was no significant difference between classification performance of LR and ELD models in the validation sample (McNemar chi‐square with Yates's correction = 0.19, P = .16).
TABLE 4.
Performance of predictive models in classifying patients with clinically meaningful cognitive decline from patients with no meaningful decline cognition
| Model | Sample | Sensitivity, % (95% CI) | Specificity, % (95% CI) | PPV, % (95% CI) | NPV, % (95% CI) | AUC | Base rate, % a |
|---|---|---|---|---|---|---|---|
| LR |
EXPEDITION (training) |
47.4 (39.3–55.6) |
76.2 (69.8–81.8) |
59.8 (52.6–66.7) |
66.0 (62.1–69.6) |
0.67 | 42.8% |
|
EXPEDITION2 (validation) |
54.2 (46.3–62.0) |
71.8 (65.5–77.6) |
58.8 (52.6–64.8) |
67.9 (63.8–71.8) |
0.63 | 42.6% | |
| ELD |
EXPEDITION (training) |
45.4 (37.4–53.7) |
80.6 (74.5–85.7) |
63.6 (55.8–70.8) |
66.4 (62.8–69.8) |
0.67 | 42.8% |
|
EXPEDITION2 (validation) |
49.4 (41.6–57.3) |
75.9 (69.8–81.3) |
60.3 (37.6–47.6) |
66.9 (63.1–70.5) |
0.63 |
42.6% |
Base rate of clinically meaningful cognitive decline (change of ≥3 from baseline ADAS‐Cog score) in the sample using longitudinal data at both 15 and 18 months
Abbreviations: ADAS‐Cog, Alzheimer's Disease Assessment Scale—Cognitive subscale; AUC, area under the curve; CI, confidence interval; ELD, ensemble linear discriminant model; LR, logistic regression; NPV, negative predictive value; PPV, positive predictive value.
5. DISCUSSION
In this study, we showed that models developed using baseline data from one trial can effectively predict probability of longitudinal cognitive or functional decline in that trial and in an independent trial. Predictive performance of LR classifiers and ELD models were similar. When cognitive outcome was defined as any cognitive decline observed on ADAS‐Cog score, models for prediction of cognitive decline had PPVs ranging from 62.8% to 65.8%, which was approximately 10% higher than base rate of cognitive decline observed in the longitudinal follow‐up. When cognitive outcome was defined as minimal clinically significant decline based on ADAS‐Cog score, models for prediction of cognitive decline had PPVs ranging from 58.8% to 63.6%, which was at least 15% higher than base rate of clinically significant cognitive decline observed in the longitudinal follow‐up.
The idea of targeting a subgroup of AD patients in trials to assess treatment effects is not new. 33 In fact, in recent years many drug trials for prodromal AD or mild‐to‐moderate AD have been recruiting patients with an inclusion criterion based on amyloid positron emission tomography (PET) positivity 34 or CSF amyloid and tau. 35 This approach increases efficacy of trials by decreasing heterogeneity in the study population and ensuring that the target for the treatment drug (e.g., amyloid) is present in the enrolled patients. Although enrollment criteria have greatly evolved over the last decades, becoming stricter with the addition of in vivo biomarkers as part of inclusion criteria, up to 40% of enrolled participants do not show disease progression during the timeframe of study. 9 Inclusion of such participants decreases the power of trials in finding effective treatments. Predictive models such as those developed in current study can increase this power by enriching in those most likely to progress during the course of the trial.
Clinical trials in AD generally collect a large set of measures from participants spanning multiple cognitive and functional tests and a variety of different types of biomarker data. These measures have grown in number and quality over the years. Typically, inclusion or exclusion is based on a threshold‐based dichotomous classification of the measures collected in the screening visit and prior to randomization. For example, in some studies, patients are included only if the amyloid level is more than a cutoff value on amyloid PET imaging. This threshold‐based dichotomous classification approach has its own drawbacks, including susceptibility to diagnostic misclassification (i.e., high false positive rate), and increasing need to conduct prescreening on a much larger population. 36 Multivariate predictive models do not solely rely on the absolute value of the features and account for patterns in the relationship between the features, which may improve participant selection and decrease the associated costs and burden of trial by boosting power. Our results indicate that even by using a relatively small feature set consisting of demographics, APOE ε4 status, cognitive and functional measures, and MRI volumetrics, classifiers are effective tools for prediction of clinical disease progression in the setting of trials.
In this study, performance of ELD classifiers was similar to LR models. Previous studies comparing performance of ML models versus conventional multivariate models such as LRs have shown mixed results. 24 , 25 The feature selection process may contribute to the differences in findings among studies. High‐dimensional data can introduce noise to multivariate models, and together with small number of subjects (high feature/subject ratio) can result in over‐fitting. 37 In the current study, for LR‐based models, we purposefully selected the most informative features to avoid multicollinearity and overfitting. We ran LR‐based models without any feature selection and found that performance of LR‐based models significantly decreases (results not shown). One potential advantage of ensemble ML models—such as ELD—is that part of the feature‐selection process is embedded in the models, which decreases the need for additional feature engineering steps.
Previous studies have shown that different measures, such as neuropsychological tests, genomic risk scores, MRI or PET measures, or other CSF and blood‐based biomarkers can predict cognitive trajectories in older adults in different stages of AD. 24 , 38 , 39 , 40 , 41 , 42 Most of these studies use longitudinal data from prospective cohorts of aging and dementia, which have information collected over extended follow‐up periods. However, due to the costs, burden, and regulations, treatment efficacy in clinical trials is usually studied over relatively short timeframes of 18 months to 5 years. In a previous study, 9 we showed that predictive models trained using data from longitudinal cohorts such as Alzheimer's Disease Neuroimaging Initiative (ADNI), could be effectively used in prediction of cognitive trajectories in clinical trials. The current study confirms that classifiers trained using data from one trial can effectively predict cognitive or functional decline in another trial. This approach could be used to boost the power of future trials by inclusion of individuals who are more likely to show disease progression.
We hypothesize that predictive models similar to those presented in this study could be used as part of design of clinical trials to increase the power and decrease sample size requirements. However, application of these models is not limited to prospective studies. Predictive models potentially could be used for post hoc analysis of concluded trials to identify patients who were expected to show decline. We hypothesize that this approach might identify subgroups who showed significant trends toward effectiveness of drugs and in extreme cases it might even revive some of the failed trials, especially if “poor subject selection” was the main reason for their failure. We did not have access to the treatment arm of the trials studied here, but this could be the subject of future studies once full clinical trials data become available. Finally, these predictive models could be used to identify individuals who would benefit the most from primary or secondary prevention using effective treatments that might become available. Recently, the FDA approved using aducanumab, a human monoclonal antibody that selectively targets aggregated amyloid beta for AD. 43 While efficacy of this treatment for prevention of cognitive or functional decline has been questioned by the scientific community, 44 , 45 it is possible that more therapeutic agents that are under development even with low levels of efficacy will get approval from FDA. Treating all AD patients who have the underlying pathology (e.g., amyloid in the case of aducanumab) is logistically impossible and is not cost effective. Once sufficient data from these patients become available, predictive models could be used to identify individuals who would benefit the most from these treatments.
Our study had limitations. First the number of features available for these studies to include in the predictive models were relatively small, which limited our ability to use more complex feature engineering methods. Furthermore, our training sample was relatively small. Another limitation of this study is using the change in a single cognitive test (ADAS‐Cog) and a single functional test (ADCS‐ADL) to classify participants into cognitively stable or declining groups. However, it is well known that, when used alone, these tests are prone to measurement error. 46 These studies were designed and conducted more than 10 years ago, and are somewhat different from the design of current AD trials. Some AD‐related clinical or cognitive measures (i.e., cognitive domains other than memory) and biomarkers (amyloid and tau imaging, or CSF biomarkers) were not collected at all or only collected for a very small proportion of the sample, making it implausible to assess the effect of these important measures in our models. Last, in this study we trained our models to classify those who had SC/SF versus those who had any cognitive/functional decline, which is a conservative approach. It is expected that models achieve even higher performance if the goal was to identify individuals with more rapid cognitive decline.
Despite all limitations, these results are encouraging in showing potential for using predictive models in the design of future clinical studies. These results also raised greater questions for the neuroscience community: Should we revise the clinical diagnostic criteria to decrease clinical heterogeneity in populations being studied? How can we improve our understanding of disease pathophysiology and predict which patients will decline, requiring more urgent intervention, and which patients are expected to remain stable for a long time in whom intervention is less urgent? These questions could be the topic of future studies once more data from recent clinical trials becomes available. More studies should be conducted to validate and expand our findings, especially in the trials with larger feature sets, larger sample size, and longer follow‐up.
CONFLICT OF INTEREST
Ali Ezzati has served on the advisory board of Eisai. Christos Davatzikos receives research support from the following sources unrelated to this manuscript: NIH: R01NS042645, R01MH112070, R01NS042645, U24CA189523; and medical legal consulting work unrelated to this paper. David Wolk received grants from NIH, Merck, Biogen, and Eli Lilly. All payments have been to the University of Pennsylvania. He has received consulting fees from GE Healthcare and Neuronix and honoraria from MH Life Sciences. He also received Alzheimer's Association support to attend to AAIC in 2018 and 2019. He has served on advisory board of Functional Neuromodulation. He has received PET tracer from Eli Lilly for research (no payments were made). Charlie Hal receives research support from NIH and National Institute of Occupational Safety and Health for work unrelated to this manuscript. He receives payments from Washington University, St. Louis, for serving on External Advisory Committee for National Institute of Aging‐sponsored research, University of Iowa, for serving on Data Safety Monitoring Committee for National Institute of Aging‐sponsored research, from National Institutes of Health for serving on Study Section and Special Emphasis Panels for reviewing grant applications. He is also the unpaid director and secretary, Torah and Nature Foundation. His institution received loan of Thorasys tremoFlo C‐100 airwave oscillometer, Thorasys Thoracic Medical Systems Inc., www.thorasys.com, for a pilot study. The device was returned. Chris Habeck receives research support from NIA/NIH unrelated to this publication. He is an advisor for the Clinical trial “Motor Imagery Intervention” run by Helena Blumen at Albert Einstein College of Medicine, no payments are made. He received honorarium for Alzheimer's Research & Prevention Foundation Invited Presentation. Richard B. Lipton receives research support from the following sources unrelated to this manuscript: NIH: 2PO1 AG003949 (mPI), 5U10 NS077308 (PI), R21 AG056920 (Investigator), 1RF1 AG057531 (Site PI), RF1 AG054548 (Investigator), 1RO1 AG048642 (Investigator), R56 AG057548 (Investigator), U01062370 (Investigator), RO1 AG060933 (Investigator), RO1 AG062622 (Investigator), 1UG3FD006795 (mPI), 1U24NS113847 (Investigator), K23 NS09610 (Mentor), K23AG049466 (Mentor), K23 NS107643 (Mentor). He also receives support from the Migraine Research Foundation and the National Headache Foundation. He serves on the editorial board of Neurology, is a senior advisor to Headache, and associate editor for Cephalalgia. He has reviewed for the NIA and NINDS; holds stock options in eNeura Therapeutics and Biohaven Holdings; serves as consultant, advisory board member, or has received honoraria from: Abbvie (Allergan), American Academy of Neurology, American Headache Society, Amgen, Avanir, Biohaven, Biovision, Boston Scientific, Reddy's (Promius), Electrocore, Eli Lilly, eNeura Therapeutics, Equinox, GlaxoSmithKline, Grifols, Lundbeck (Alder), Merck, Pernix, Pfizer, Supernus, Teva, Trigemina, Vector, Vedanta. He receives royalties from Wolff's Headache seventh and eighth edition, Oxford Press University, 2009, Wiley and Informa. He receives consulting fees from Impel NeuroPharma and Novartis and has stock or options in Control M.
ACKNOWLEDGMENTS
Lilly makes patient‐level data available from Lilly‐sponsored studies on marketed drugs for approved uses following approval by regulators in the US and EU and after the primary manuscript describing the results has been accepted for publication, whichever is later. Lilly is one of several companies that provide this access through the website clinicalstudydatarequest.com. Qualified researchers can submit research proposals and request anonymized data to test new hypotheses. Lilly's data‐sharing policies are provided on the clinicalstudydatarequest.com site under the Study Sponsors page. Authors of this study were supported in part by grants from the National Institute of Health (NIA K23 AG063993, Ali Ezzati; 2PO1 AG003949, Richard B. Lipton); the Alzheimer's Association (2019‐AACSF‐641329; Ali Ezzati); Cure Alzheimer Fund (Ali Ezzati & Richard B. Lipton), and the Leonard and Sylvia Marx Foundation (Richard B. Lipton).
Ezzati A, Davatzikos C, Wolk DA, et al. Application of predictive models in boosting power of Alzheimer's disease clinical trials: A post hoc analysis of phase 3 solanezumab trials. Alzheimer's Dement. 2022;8:e12223. 10.1002/trc2.12223
Data used in these analyses are from the following Eli‐Lilly trials: EXPEDITION (ClinicalTrials.gov Identifier: NCT00905372), and EXPEDITION2 (ClinicalTrials.gov Identifier: NCT00904683)
REFERENCES
- 1. Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 2013;9:63‐75.e62. [DOI] [PubMed] [Google Scholar]
- 2. Cummings JL, Morstorf T, Zhong K. Alzheimer's disease drug‐development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cummings J. Lessons learned from Alzheimer disease: clinical trials with negative outcomes. Clin Transl Sci. 2018;11:147‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ferreira D, Nordberg A, Westman E. Biological subtypes of Alzheimer disease: a systematic review and meta‐analysis. Neurology. 2020;94:436‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tschanz JT, Corcoran CD, Schwartz S, et al. Progression of cognitive, functional, and neuropsychiatric symptom domains in a population cohort with Alzheimer dementia: the Cache County Dementia Progression study. Am J Geriatr Psychiatry. 2011;19:532‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frölich L, Ashwood T, Nilsson J, Eckerwall G, Investigators S. Effects of AZD3480 on cognition in patients with mild‐to‐moderate Alzheimer's disease: a phase IIb dose‐finding study. J Alzheimers Dis. 2011;24:363‐374. [DOI] [PubMed] [Google Scholar]
- 7. Sevigny J, Suhy J, Chiao P, et al. Amyloid PET screening for enrichment of early‐stage Alzheimer disease clinical trials. Alzheimer Dis Assoc Disord. 2016;30:1‐7. [DOI] [PubMed] [Google Scholar]
- 8. Salloway S, Sperling R & Fox NC. Two phase 3 trials of bapineuzumab in mild‐to‐moderate Alzheimer's disease. New England J Med. 2014;370(4):322‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ezzati A, Lipton RB. Machine learning predictive models can improve efficacy of clinical trials for Alzheimer's disease 1, 2. J Alzheimers Dis. 2020:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McAleese KE, Colloby SJ, Thomas AJ, et al. Concomitant neurodegenerative pathologies contribute to the transition from mild cognitive impairment to dementia. Alzheimers Dement. 2021. [DOI] [PubMed] [Google Scholar]
- 11. Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community‐dwelling older persons. Neurology. 2007;69:2197‐2204. [DOI] [PubMed] [Google Scholar]
- 12. Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild‐to‐moderate Alzheimer's disease. N Engl J Med. 2014;370:311‐321. [DOI] [PubMed] [Google Scholar]
- 13. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group* under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939‐939. [DOI] [PubMed] [Google Scholar]
- 14. Folstein MF, Folstein SE, McHugh PR. Mini‐mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189‐198. [DOI] [PubMed] [Google Scholar]
- 15. Yesavage JA, Sheikh JI. 9/Geriatric depression scale (GDS) recent evidence and development of a shorter version. Clin Gerontol. 1986;5:165‐173. [Google Scholar]
- 16. Mohs RC, Knopman D, Petersen RC, et al. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer's Disease Assessment Scale that broaden its scope. Alzheimer Dis Assoc Disord. 1997;11:S13‐21. [PubMed] [Google Scholar]
- 17. Galasko D, Bennett D, Sano M, et al. An inventory to assess activities of daily living for clinical trials in Alzheimer's disease. Alzheimer Dis Assoc Disord. 1997;11:S33‐39. [PubMed] [Google Scholar]
- 18. Schrag A, Schott JM, Initiative AsDN . What is the clinically relevant change on the ADAS‐Cog?. J Neurol Neurosurg Psychiatry. 2012;83:171‐173. [DOI] [PubMed] [Google Scholar]
- 19. O'Bryant SE, Waring SC, Cullum CM, et al. Staging dementia using Clinical Dementia Rating Scale sum of boxes scores: a Texas Alzheimer's research consortium study. Arch Neurol. 2008;65:1091‐1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herdman M, Gudex C, Lloyd A, et al. Development and preliminary testing of the new five‐level version of EQ‐5D (EQ‐5D‐5L). Quality of Life Research. 2011;20(10):1727‐1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thorgrimsen L, Selwood A, Spector A, et al. Whose quality of life is it anyway? The validity and reliability of the quality of life‐Alzheimer's disease (QoL‐AD) scale. Alzheimer Dis Assoc Disord. 2003;17:201‐208. [DOI] [PubMed] [Google Scholar]
- 22. Liu H, Motoda H. Computational Methods of Feature Selection. CRC Press; 2007. [Google Scholar]
- 23. Breiman L. Statistical modeling: the two cultures (with comments and a rejoinder by the author). Stat Sci. 2001;16:199‐231. [Google Scholar]
- 24. Falahati F, Westman E, Simmons A. Multivariate data analysis and machine learning in Alzheimer's disease with a focus on structural magnetic resonance imaging. J Alzheimers Dis. 2014;41:685‐708. [DOI] [PubMed] [Google Scholar]
- 25. Jie M, Collins GS, Steyerberg EW, Verbakel JY, van Calster B. A systematic review shows no performance benefit of machine learning over logistic regression for clinical prediction models. J Clin Epidemiol. 2019;110:12‐22. [DOI] [PubMed] [Google Scholar]
- 26. Bursac Z, Gauss CH, Williams DK, Hosmer DW. Purposeful selection of variables in logistic regression. Source Code Biol Med. 2008;3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cook NR. Use and misuse of the receiver operating characteristic curve in risk prediction. Circulation. 2007;115:928‐935. [DOI] [PubMed] [Google Scholar]
- 28. Ezzati A, Zammit AR, Harvey DJ, Habeck C, Hall CB, Lipton RB, Initiative AsDN . Optimizing machine learning methods to improve predictive models of Alzheimer's disease. J Alzheimers Dis. 2019;71:1027‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang C, Ma Y. Ensemble Machine Learning: Methods and Applications. Springer; 2012. [Google Scholar]
- 30. Kuncheva LI. Combining Pattern Classifiers: Methods and Algorithms. John Wiley & Sons; 2004. [Google Scholar]
- 31. Ezzati A, Harvey DJ, Habeck C, et al. Predicting Amyloid‐β levels in amnestic mild cognitive impairment using machine learning techniques. J Alzheimers Dis. 2019;73:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dietterich TG. Approximate statistical tests for comparing supervised classification learning algorithms. Neural Comput. 1998;10:1895‐1923. [DOI] [PubMed] [Google Scholar]
- 33. Frisoni GB, Fox NC, Jack CR Jr, Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol. 2010;6:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Honig LS, Vellas B, Woodward M, et al. Trial of solanezumab for mild dementia due to Alzheimer's disease. N Engl J Med. 2018;378:321‐330. [DOI] [PubMed] [Google Scholar]
- 35. Coric V, Salloway S, van Dyck CH, et al. Targeting prodromal Alzheimer disease with avagacestat: a randomized clinical trial. JAMA Neurol. 2015;72:1324‐1333. [DOI] [PubMed] [Google Scholar]
- 36. Shah ND, Steyerberg EW, Kent DM. Big data and predictive analytics: recalibrating expectations. JAMA. 2018;320:27‐28. [DOI] [PubMed] [Google Scholar]
- 37. McEvoy LK, Holland D, Hagler DJ Jr, Fennema‐Notestine C, Brewer JB, Dale AM. Mild cognitive impairment: baseline and longitudinal structural MR imaging measures improve predictive prognosis. Radiology. 2011;259:834‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ezzati A, Zammit AR, Harvey DJ, Habeck C, Hall CB, Lipton RB, Initiative AsDN . Optimizing machine learning methods to improve predictive models of Alzheimer's disease. J Alzheimers Dis. 2019:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ezzati A, Zammit AR, Habeck C, Hall CB, Lipton RB, Initiative AsDN . Detecting biological heterogeneity patterns in ADNI amnestic mild cognitive impairment based on volumetric MRI. Brain Imaging Behav. 2020;14(5), 1792‐1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shaffer JL, Petrella JR, Sheldon FC, et al, Initiative AsDN . Predicting cognitive decline in subjects at risk for Alzheimer disease by using combined cerebrospinal fluid, MR imaging, and PET biomarkers. Radiology. 2013;266:583‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zammit AR, Hall CB, Katz MJ, et al. Class‐specific incidence of all‐cause dementia and Alzheimer's disease: a latent class approach. J Alzheimers Dis. 2018;66:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zammit AR, Muniz‐Terrera G, Katz MJ, et al. Subtypes based on neuropsychological performance predict incident dementia: findings from the Rush Memory and Aging Project. J Alzheimers Dis. 2019;67:125‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature. 2016;537:50‐56. [DOI] [PubMed] [Google Scholar]
- 44. Knopman DS, Jones DT, Greicius MD. Failure to demonstrate efficacy of aducanumab: an analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2021;17:696‐701. [DOI] [PubMed] [Google Scholar]
- 45. Alexander GC, Emerson S, Kesselheim AS. Evaluation of aducanumab for Alzheimer disease: scientific evidence and regulatory review involving efficacy, safety, and futility. JAMA. 2021;325:1717‐1718. [DOI] [PubMed] [Google Scholar]
- 46. Benge JF, Balsis S, Geraci L, Massman PJ, Doody RS. How well do the ADAS‐cog and its subscales measure cognitive dysfunction in Alzheimer's disease?. Dement Geriatr Cogn Disord. 2009;28:63‐69. [DOI] [PubMed] [Google Scholar]