

Abstract
Herein, we describe the design, synthesis and deciphering of the key characteristics of the structure activity relationship (SAR) of trifluoromethyloxadiazole (TFMO) bearing class-IIa HDAC inhibitors. Our medicinal chemistry campaign of 23 compounds identified compound 1 as a highly potent inhibitor with sub nM affinity to class-IIa HDAC4 isoform. Therefore, We radiolabeled compound 1 (named thereafter as NT160) with [18F]fluoride thus producing the identical [18F]-NT160 as a diagnostic tool for positron emission tomography (PET). [18F]-NT160 was produced in high radiochemical purity (>95%), moderate radiochemical yield (2−5%) and moderate molar activity in the range of 0.30−0.85 GBq/umol (8.0−23.0 mCi/umol). We also established that [18F]-NT160 can cross the blood brain barrier and bind to class-IIa HDACs in vivo. The combination of [18F]-NT160 and 1 represent a novel theranostic pair using the same molecule to enable diagnostic PET imaging with [18F]-NT160 followed by targeted therapy with NT160.
Keywords: High affinity class-IIa HDACs inhibitor, NT160, PET imaging, Radiochemistry, Brain imaging
1. Introduction
The class-IIa histone deacetylases (class-IIa HDACs) are a sub-family of four members (HDACs: 4, 5, 7 and 9) of the HDAC family of 18 enzymes. Class-IIa HDAC dysregulation in the brain is relevant across diverse human disorders of the central nervous system (CNS) such as stroke [1–4], Huntington’s [5–7] and Alzheimer’s diseases [8–10] which underscore their immense potential for molecular imaging and targeted therapy. Currently, non-invasive imaging biomarkers for quantifying the class-IIa HDAC expression in tumors and in the CNS are lacking. Therefore, there is an immense need to identify highly potent and selective class-IIa HDAC inhibitors suitable for theranostic development that can be used simultaneously for positron emission tomography (PET) and targeted therapy of cancer and the disorders of the CNS. Therefore, our current work is aimed at filling this diagnostic and therapeutic gap by developing highly potent and specific class-IIa HDAC inhibitors.
Despite intensive efforts by many research groups, radiolabeled HDAC inhibitors with either [18F]-or [11C]-PET radiotracers such as [18F]-SAHA [11], [18F]-FESAHA [12], and [11C]-MS-275 [13] and other tracers [14] showed poor blood-brain barrier (BBB) permeability, thus limiting their utility for brain imaging. [11C]-martinostat was the first successful class-I HDAC (HDAC1, 2 and 3) inhibitor-based tracer [15–17]. The same group also reported PET imaging in rodents and nonhuman primates with a brain-penetrant and semi-selective HDAC6, a class-IIb HDAC inhibitor [18]. However, class-IIa HDACs’ involvement in cancer or CNS diseases are distinct. Therefore, there is an immense need for a class-IIa HDAC specific PET tracer for the molecular imaging of cancer and the disorders of the CNS.
Recently TMP195 a trifluoromethyl-oxadiazole (TFMO) containing molecule (Fig. 1) was reported to exhibit potent class-IIa HDACs inhibition [19,20]. TMP195 was also reported to exhibit excellent non-cell-based inhibition of HDAC7 and 9 with IC50 in low nM range and moderate inhibition of HDAC4 and 5 (IC50 > 100 nM). TMP195 met our initial criteria for a lead PET tracer development such as high affinity to class-IIa and low affinity to other HDAC isoforms and classes. Therefore, we recently reported a novel late stage method to radiolabel the TFMO moiety with [18F]fluoride and extended its utility to efficiently produce [18F]TMP195 21.
Fig. 1.
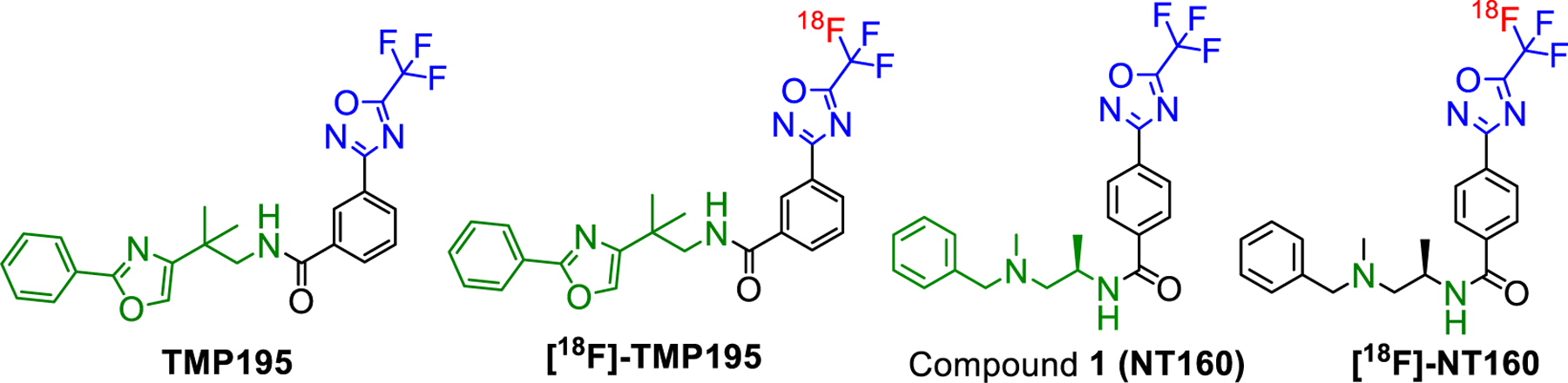
Structures of TMP195/[18F]TMP195 and NT160/[18F]-NT160 a radiolabeled highly potent inhibitor of class-IIa HDACs.
Our SAR campaign successfully identified several candidates for PET tracer development. We utilized our late-stage radiolabeling route to incorporate the F-18 into the TFMO containing molecules thus producing exact radiolabeled molecules. We radiolabeled several potent inhibitors with [18F]fluoride which were produced in high radiochemical purity (>95%), moderate radiochemical yield (2−5%) and moderate molar activity in the range of 0.30−0.85 GBq/ umol (8.0−23.0 mCi/umol). Also, [18F]-NT160 demonstrated the ability to cross the BBB and binds class-IIa HDAC in vivo.
2. Results
We initially examined TMP195 as a lead compound for theranostic development. Therefore, we profiled indigenous class-IIa HDAC inhibition with TMP195 in multiple cancer cell lines (Table 1) and concluded that TMP195 exhibited poor functional affinity to class-IIa HDACs (IC50 > 800 nM), despite having been reported to bind to indigenous class-IIa HDACs with an IC50 of 300 nM in THP-1 cells [ [19]]. We cannot explain the discrepancy between our results and the previous report. However, our data is consistent among several cell types (Table 1).
Table 1.
IC50 (nM) values for HDACs inhibition with TMP195 in a panel of cancer cells using class-distinguishing fluorogenic substrates whole-cell assay.
| |
Cell type |
|||
|---|---|---|---|---|
| THP-1 | Panc-1 | L3.6 | HT-29 | |
| Class IIa HDACs | 887 | 888 ± 230 | 893 ± 225 | 1004 ± 216 |
| Class I/IIb HDACs | >5000 | >5000 | >5000 | >5000 |
Given our interest in developing PET tracers for cancer and the CNS, we recently reported a novel late stage method to radiolabel the TFMO moiety with [18F]fluoride and extended its utility to produce [18F]TMP195 [21]. Next, we synthesized a focused library of new molecules with the aim to improve affinity to class-IIa HDACs. Therefore, we utilized TMP195 as a lead candidate for PET tracer development and performed a focused structure activity relationship and developed a focused library of class-IIa HDAC inhibitors to identify new PET tracer candidates with an improved in vitro profile compared to TMP195.
TMP195 (Fig. 2) contains three distinctive pharmacophore motifs suitable for focused SAR studies: (1) cap moiety (green) that interacts with the surface of the HDAC enzyme; (2) the linker moiety (black) that occupies a hydrophobic channel, and (3) a zinc binding moiety (blue) that interacts with the zinc ion at the bottom of the catalytic pocket [ [19]].
Fig. 2.
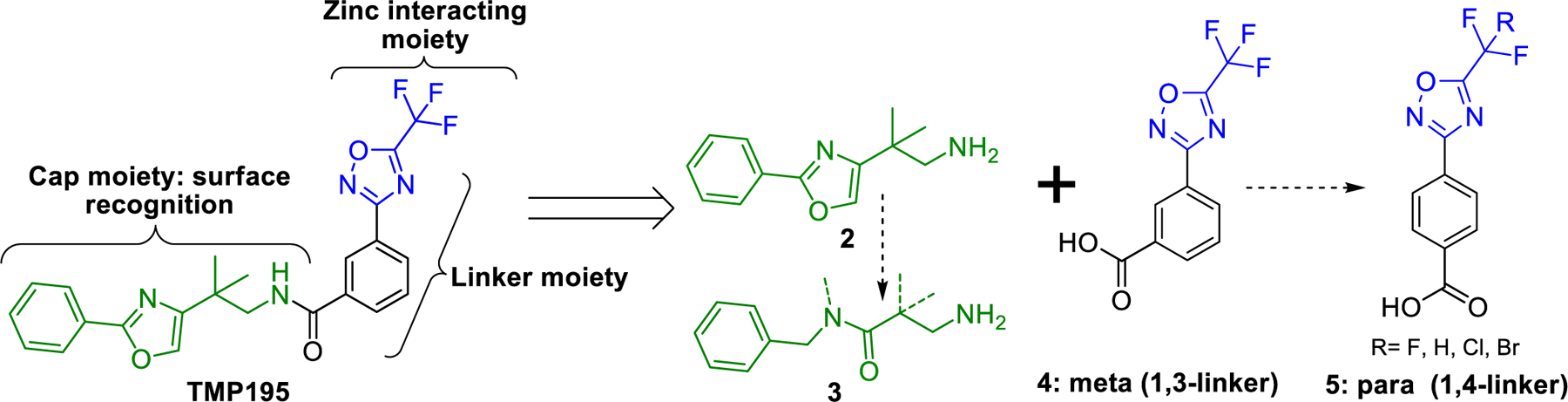
Pharmacophore moieties of TMP195 and retrosynthetic design to focused SAR study.
Based on our extensive experience working with HDAC inhibitors and substrates, we theorized that the cap moiety is the most amenable to structural modification through SAR. Therefore, we started our SAR campaign with mimetics of the cap moiety 2 of TMP195 (Fig. 2) and selected moieties that were less rigid and less lipophilic than 2 to improve aqueous solubility. We also theorized that the1,4-linker which mimics the 6-carbon chain linker (also similar to natural lysine substrates) in class-IIa HDAC substrates that we developed previously [ [22]] will improve inhibition to class-IIa HDACs and tune selectivity toward HDAC4 and 5 which are the isoforms most implicated in brain disorders [ [23]]. We synthesized compounds 6−12 by coupling the appropriate amine with the benzoic acid 4 or 5 using (1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) and N-methyl morpholine (NMM) as coupling agents as shown in Scheme 1.
Scheme 1.
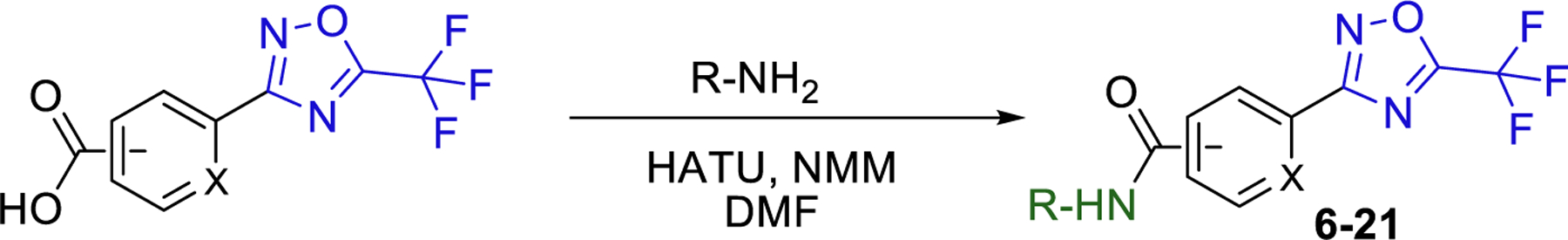
A) General synthetic scheme for coupling reactions and B) Structures of 6−21.
We selected the HT-29 cells to screen our compounds for inhibition of indigenous class-IIa HDACs using class-distinguishing substrates. The utility of whole cell assay allows for rapid and inexpensive screening of a large library of inhibitors. HT-29 colon cancer cells are fast growing and can be readily cultured in high confuencey thus facilitating fast screening of our inhibitor candidates. Compounds of interest identified from the whole cell assay were further analyzed for HDAC isoform specific inhibition against recombinant human HDACs 1e11. TMP195 was used as a control for class-IIa inhibition and SAHA (Vorinostat) was used as a control for class-I/IIb HDACs. SAHA showed modest activity against class-I/IIb HDACs and was not active against class-IIa HDACs.
Compounds 6−8 that contain the 1,3-phenyl linker displayed worse affinity (IC50 > 50 mM) to class-IIa HDACs compared to TMP195 as shown in Table 2. It is likely that the loss of affinity is due to the disruption of the U-shaped configuration provided by the 1,3-phenyl linker combined with the rigid phenyl-oxazol cap moiety 2 (green coded, Fig. 2) that was described previously to induce selectivity to class-IIa HDAC-7 and 9 and further confirmed by crystallography [ [19]]. We also synthesized several compounds (data not shown) that contain the 1,3-phenyl linker with no success at improving class-IIa HDAC affinity over TMP195. It is likely that TMP195 presents a unique U-shaped confirmation that cannot be matched with other 1,3-phenyl linker containing compounds. Therefore, we focused our efforts on a compound that contains the 1,4-phenyl linker which proved to be an effective strategy for improving affinity to class-IIa HDACs and tuning selectivity to HDAC4 and 5.
Table 2.
IC50 values for HDACs inhibition in HT-29 cells using class-distinguishing fluorogenic substrates whole-cell assay..
| |||
|---|---|---|---|
| # | R | Class-IIa (uM) | Class-I/IIb (uM) |
|
SAHA
TMP-195 |
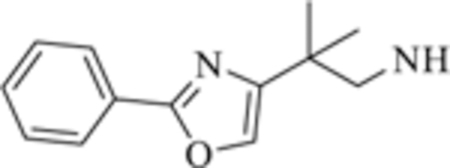
|
>12.0
1.0 ± 0.20 |
0.95 ± 0.11
>50 |
| 6 |
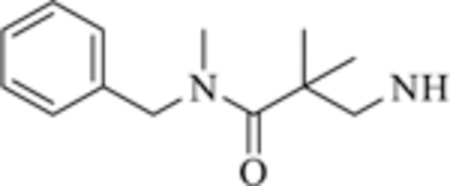
|
>50 | >50 |
| 7 |
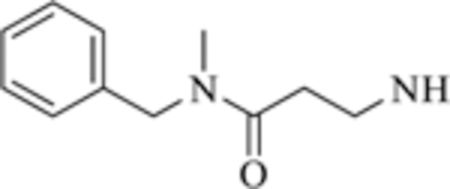
|
>50 | >50 |
| 8 |
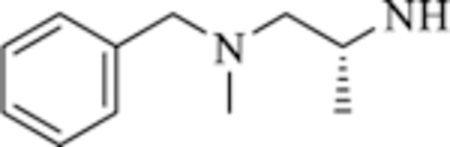
|
>50 | >50 |
Based on these data, we pressed on with further optimization of the cap moiety by synthesizing candidates that contain the 1,4-linker. We also synthesized a few compounds with pyridine linker in an attempt to improve aqueous solubility. We synthesized the new candidate inhibitors (compounds: 9−21) by coupling the appropriate amine with the appropriate 4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)benzoic acid (1,4-phenyl linker) similar to Scheme 1. Indeed, except for compounds 9 and 15, we found that compounds with 1,4-phenyl linker were relatively better inhibitors against class-IIa HDAC4 and 5 than TMP195 (Table 3), thus confirming our hypothesis that the 1,4-phenyl linker might induce a better class-IIa HDAC inhibition and tune selectivity toward HDAC4 and 5. Similarly, the phenyl ring can be easily replaced with a pyridyl ring to produce new inhibitor/radiotracer candidates with improved aqueous solubility and to study the impact of changes at linker moiety on the overall affinity to class-IIa HDACs. The new inhibitor candidates were screened side-by-side with TMP195 and SAHA as controls.
Table 3.
IC50 values for HDACs inhibition in HT-29 cells using class-distinguishing fluorogenic substrates whole-cell assay..
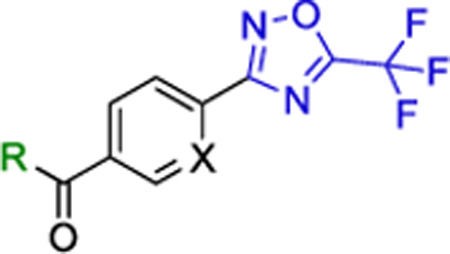
| ||||
|---|---|---|---|---|
| # | R | X | Class-Ila (uM) | Class-I/IIb (uM) |
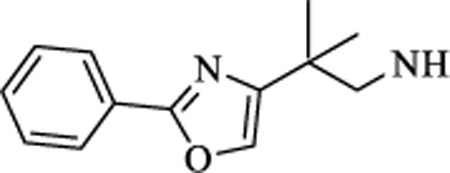
| 9 |
|
CH | >5 | >15 |
| 10 |
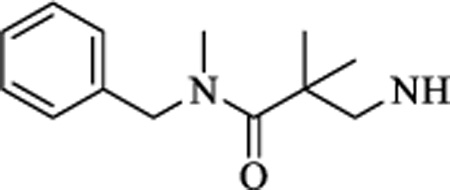
|
CH | 0.933 ± 0.26 | >15 |
| 11 |
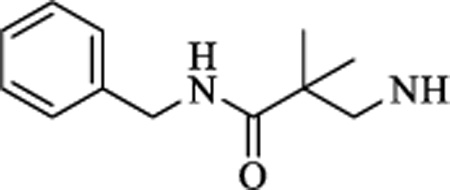
|
CH | 0.768 ± 0.19 | >15 |
| 12 |
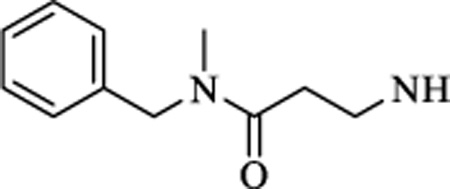
|
CH | 0.905 ± 0.31 | >15 |
| 13 |
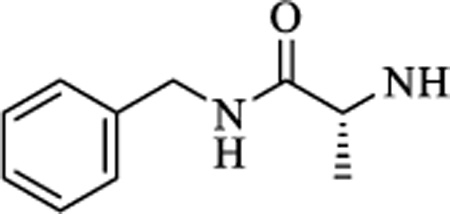
|
CH | 0.953 ± 0.31 | >15 |
| 14 |
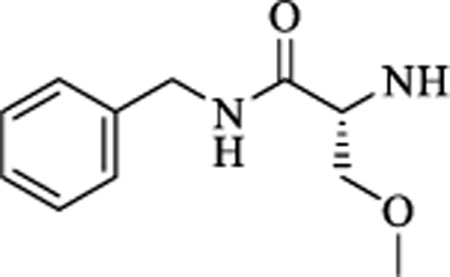
|
CH | 0.537 ± 0.15 | >15 |
| 15 |
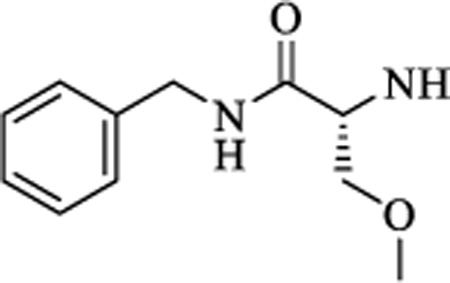
|
N | 1.710 ± 0.33 | >15 |
| 16 |
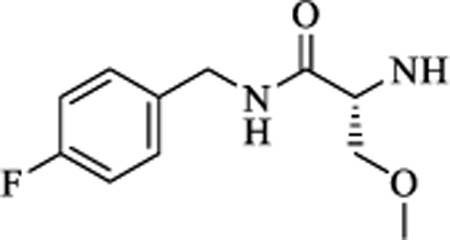
|
CH | 0.233±0.11 | >15 |
| 17 |
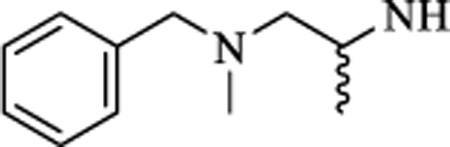
|
CH | 0.395 ± 0.14 | >50 |
| 18 |
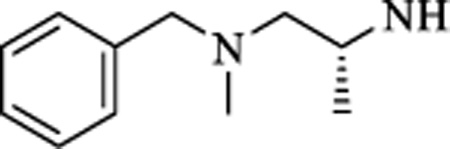
|
N | 0.147 ± 0.07 | >50 |
| 19 |
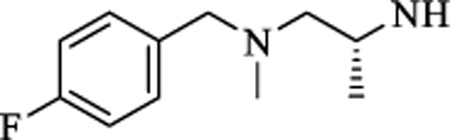
|
CH | 0.059 ± 0.016 | >15 |
| 1 (NT160) |
|
CH | 0.046 ±0.015 | >15 |
These above findings paved the way for pursuing candidates with the less rigid cap moiety. It was apparent that benzylic amino acid cap moieties showed promise as a novel approach to improve affinity to class-IIa HDACs. This is not surprising considering that the lysine moiety on histones is the native substrate of HDACs. Therefore, we initially focused our approach on alanine and serine derivatives. Compound 12 which contains a glycine-based cap moiety displayed affinity comparable to that of TMP195. However, 13 which contains a racemic alanine cap moiety was less active yet the D-alanine containing cap (compound 14) exhibited significantly higher class-IIa HDAC inhibition. Building on these findings, we theorized that higher HDAC4 and 5 inhibitions can be achieved by incorporating the D-amino acids alanine and serine in the cap moieties as shown for compound 14−21. Overall, we found that the amino acid alanine containing compounds were better inhibitors than the amino acid serine containing cap moieties. Introducing a pyridine linker reduced the affinity as shown for compounds 15 and 18. Utilizing the commercially available cap moiety of 17 which is a D,L-alanine derivative led to significant improvement in affinity to class-IIa HDACs. Again, the R-isomer (also commercially available) as the cap moiety of 1 was found to be superior to the racemic 17. In fact, NT160 was superior to TMP195 in HT-29 cell (Fig. 3) and exhibited a remarkably high inhibition (low to sub nM) against class-IIa HDACs including HDAC7 and 9 as shown in Table 4.
Fig. 3.
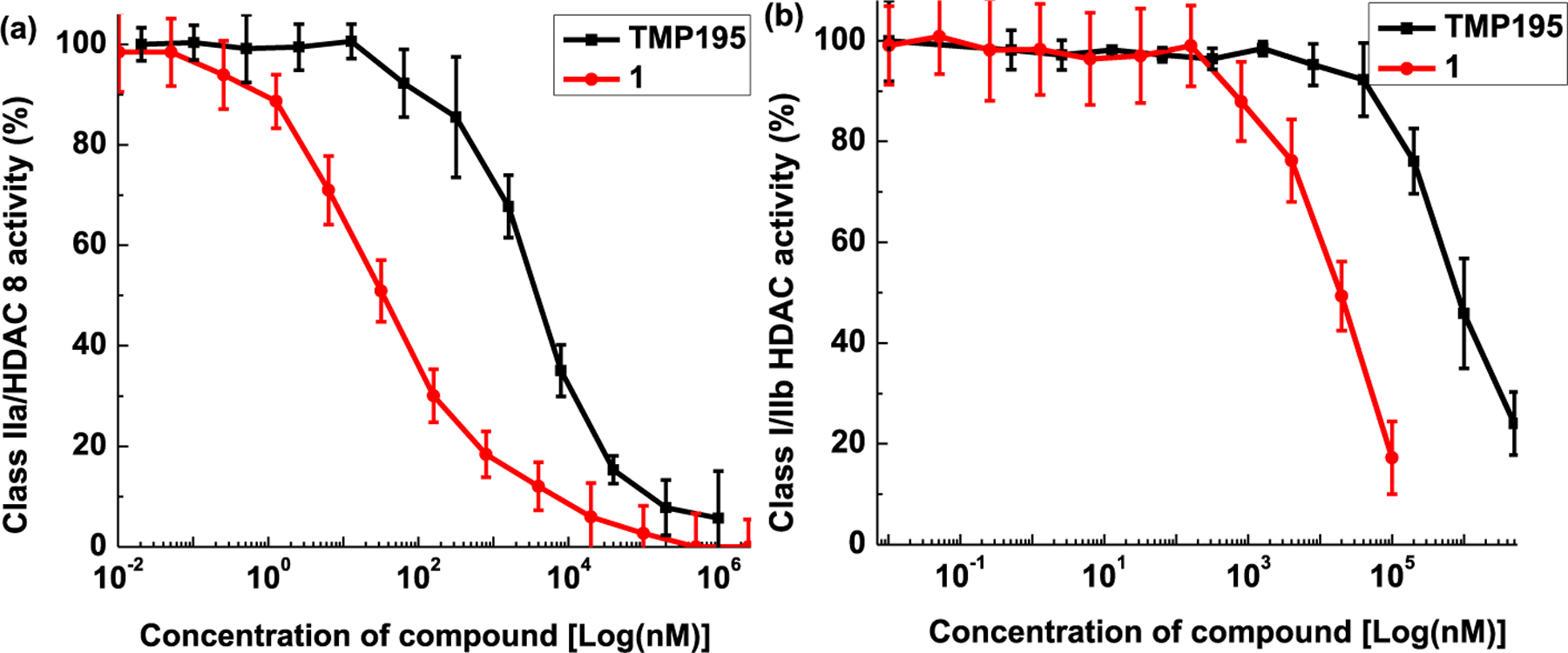
Side-by-side comparison of selective HDAC class inhibition by TMP195 and NT160 in HT-29 cell line.
Table 4.
IC50 (nM) values for TMP195 and NT160 obtained using fluorogenic assay against recombinant HDAC proteins.
| # | HDAC4 | HDAC5 | HDAC7 | HDAC9 | Other HDACS |
|---|---|---|---|---|---|
| TMP195 | 128.1 ± 13 | 147.9 ± 22 | 7.7 ± 1.2 | 2.4 ± 0.51 | >5000 |
| 1 (NT160) | 0.08 ± 0.02 | 1.2 ± 0.17 | 1.0 ± 0.2 | 0.9 ± 0.2 | >600 ± 231 |
We also examined the amenability of the TFMO moiety to modifications. We synthesized and evaluated compounds 20−21, which are analogs of NT160. Data in Table 5 indicates that replacing a fluorine with a bromine or chlorine significantly reduced affinity to class-IIa HDACs (see Table 5).
Table 5.
IC50 values for HDACs inhibition in HT-29 cells (compounds 23–25)..
| |||
|---|---|---|---|
| # | R | Class-IIa (nM) | Class-I/IIb (nM) |
| 20 | CF2Cl | >2000 | >2000 |
| 21 | CF2Br | >5000 | >5000 |
2.1. Radiochemistry
The synthesis of the bromo-precursor and subsequent radio-synthesis of [18F]-NT160 is shown in Scheme 2. Radiochemical synthesis of [18F]TMP195 and compound 23 were performed as described in our previous report [21]. Coupling the commercially available (Sigma catalog: ENA448969219) amine 22 with 23 led to bromo-precursor 21 which set the stage for radiolabeling experiments. 18F-NT160 was radio-synthesized by treating 21 with Cs18F/ kryptand at 160 °C for 25 min. The radiochemical yield was 2e5%, radiochemical purity >98% and molar activity range of 0.30−0.85 GBq/umol (8.0−23.0 mCi/umol). Discussion on molar activity was provided in our previous report.
Scheme 2.
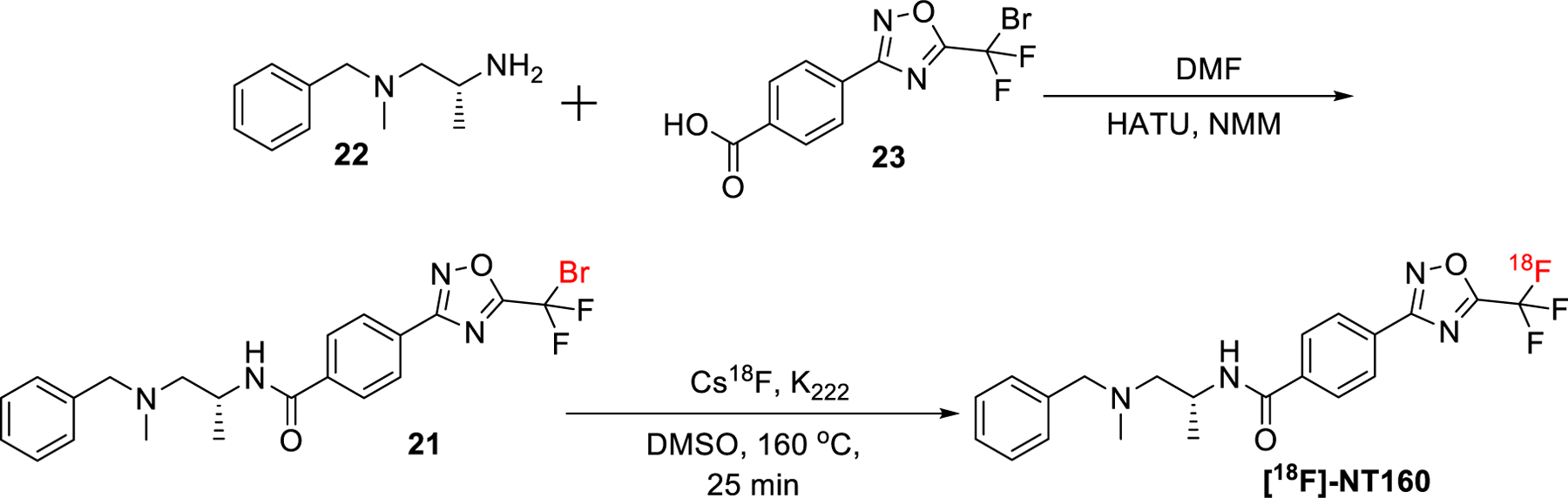
Synthesis of bromo-precursors and subsequent radiosynthesis of [18F]-NT160.
Radiosynthesis, HPLC purification and formulation of [18F]-NT160 was accomplished in less than 90 min [18F]-NT160 was purified using semi-preparative HPLC (C18 column) and its identity was confirmed by co-injection with compound NT160 and both compounds eluted together using analytical HPLC. The molar (specific) activity of [18F]-NT160 was determined from the area under the curve of the tracer attributed to the ultraviolet peak in the HPLC chromatogram against a calibration curve pre-prepared with the unlabeled reference standards.
2.2. PET imaging studies
We initially performed microPET imaging in mice to demonstrate the ability of [18F]TMP195 to penetrate the BBB and accumulate in the brain of a healthy mouse (n = 3), as shown in Fig. 4. Dynamic PET imaging was performed for 60 min. Imaging with [18F]TMP195 in mice demonstrated BBB penetration albeit with relatively low accumulation in the brain maximized at 1.1 %ID/cc in the first 5 min post injection. We also performed PET imaging with [18F]-NT160 in mice for side-by-side comparison with [18F] TMP195. As expected, the PET imaging data clearly demonstrated that [18F]-NT160 was superior and displayed relatively high accumulation in the mice brains maximized at ~5.0 %ID/cc in the first 5 min post injection as shown in Fig. 4 and Table 6.
Fig. 4.
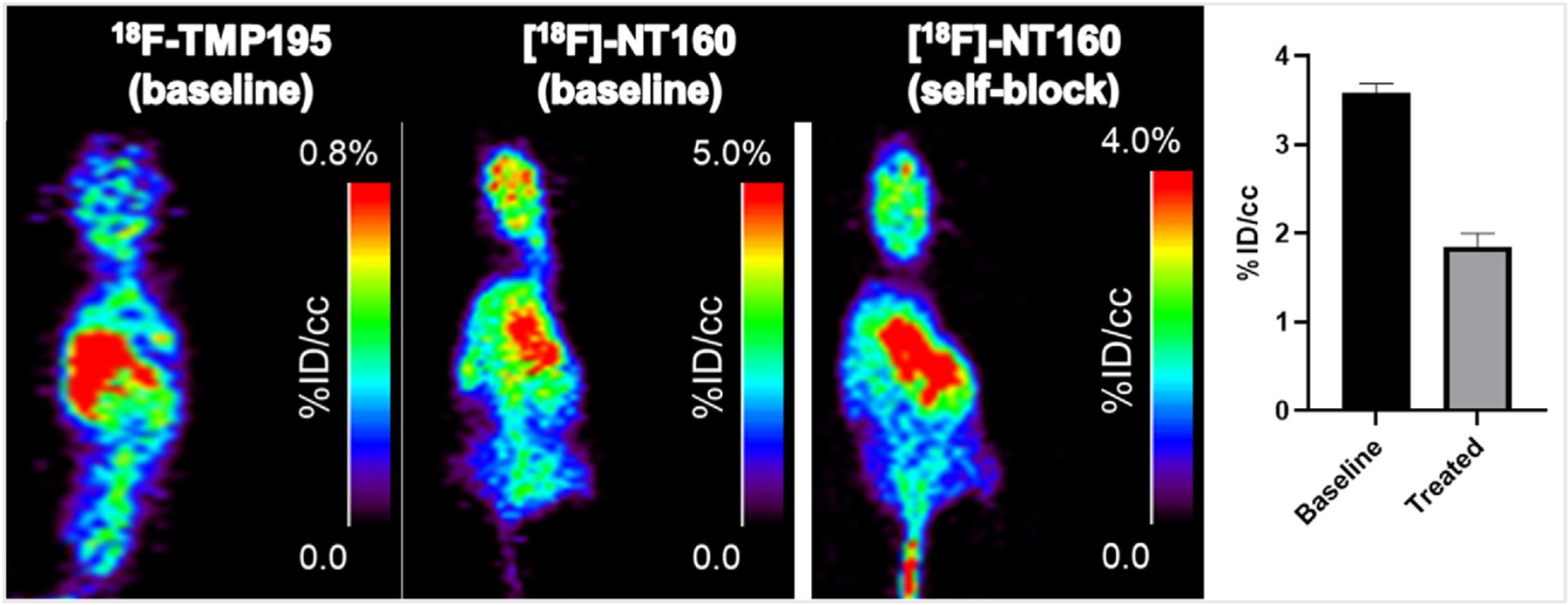
A) Representative PET images with A) [18F]TMP195 and B) [18F]-NT160 in mice and C and D) self-blocking (signal reduced by 47%) of [18F]-NT160 in mice brain with the non-radioactive 1.
Table 6.
Brain uptake of [18F]TMP195 and [18F]-NT160 (%ID/cc).
| Time after injection | ||||
|---|---|---|---|---|
| Tracer | 2 min | 10 min | 30 min | 60 min |
| [ 18 F]TMP195 | 1.10 ± 0.05 | 1.03 ± 0.05 | 0.89 ± 0.035 | 0.80 ± 0.04 |
| [18F]-NT160 (Baseline) | 4.77 ± 0.24 | 3.74 ± 0.21 | 3.06 ± 0.19 | 2.86 ± 0.19 |
| [18F]-NT160 (blocked) | 3.66 ± 0.21 | 2.31 ± 0.16 | 1.64 ± 0.4 | 1.46 ± 0.4 |
Also, self-blocking by administering a cold dose (1.0 mg/kg) of the non-radioactive NT160 (competitive binding) significantly reduced the uptake of [18F]-NT160 in the mice brains in vivo by ~47% compared to baseline (Fig. 4C and D), thus demonstrating specific binding to class-IIa HDACs in vivo. Unfortunately, the spatial resolution of the uPET does not permit quantitative imaging of the relatively small mice brain; therefore, a follow up imaging study with [18F]-NT160 in the rat’s brain is needed to determine accurate regional biodistribution of the [18F]-NT160 in the brain. These studies are ongoing and will be reported in due course. Moreover, higher molar activity values may be needed to enhance the specific binding in vivo (i.e. improved in vivo blocking).
3. Discussion
Given our interest in developing theranostics for molecular imaging and targeted therapy of class-IIa HDACS, we profiled TMP195 in vitro (this work) and radiosynthesized [18F]TMP195 [21]. While TMP195 met our initial criteria for a PET tracer development such as high affinity to class-IIa and low affinity to other HDAC isoforms and classes, TMP195 exhibited poor in vitro functional affinity (Table 1) and [18F]TMP195 exhibited poor physiochemical properties and poor in vivo profile (unpublished data). Therefore, we performed a structure activity relationship (SAR) study and developed a library of class-IIa HDAC inhibitors to identify new theranostic candidates with improved in vitro profiles compared to TMP195. Our work led us to compounds NT160 which exhibited superior affinity to class-IIa HDACs compared to TMP195. During the course of work the compounds NT160 and 18 reported here have been disclosed in a patent application [ [24]]. However, our SAR profile is novel and our characterization of NT160 in vitro is extensive. Moreover, our work led to several important findings concerning the key characteristic of SAR, and we are the first to radiolabel NT160 with [18F]fluoride thus paving the way for the development of the next generation of class-IIa HDAC theranostics for PET imaging and targeted therapy.
We found that compounds with the 1,4-phenyl linker were better inhibitors against class-IIa HDAC4 and 5 when compared to the 1,3-phenyl linker. In fact, we synthesized and evaluated a significant number of compounds with the 1,3-phenyl linker; however, none were superior to TMP195 (data not shown). By far, the most promising approach to class-IIa HDAC inhibition is the systematic change to the cap moiety. Unsurprisingly, the amino acid-based cap moieties showed promise as a novel approach to improve affinity to class-IIa HDACs, considering that histones represent the cap moiety in the native substrate of HDACs. Also, we found that the R-isomers are generally better than the S-isomers or racemic mixtures.
The most surprising finding is the significant loss of affinity of 20 and 21. Our data is in line with a recent report which also demonstrated that replacing the trifluoro-with difluoro-moiety to significantly reduce the affinity to class-IIa HDACs [25]. The high selectivity of TFMO to class-IIa HDACs combined with reducing its affinity to class-I/IIb HDACs can be explained by the relatively larger size (volume) of catalytic cavity of class-IIa HDACs due the substitution of His976 (class IIa) for Tyr306 (class-I/IIb) in the catalytic site [26]. This change not only resulted in negligible intrinsic deacetylase activity of class-IIa HDACs but changed in size of the active cavity. Class-I/IIb HDACs have a smaller catalytic cavity than class-IIa enzymes; therefore, they cannot accommodate the relatively larger TFMO moiety which led to diminished affinity. In contrast the larger size of the catalytic cavity of class-IIa HDAC can readily accommodate the larger TFMO moiety in their binding site [26,27]. Notably, the low affinity of 21 is highly desirable, since 21 is the radiolabeling precursor for producing [18F]-NT160; therefore, the chemical excipient from 21 that might be present in the final radioactive dose will not compete with the radioactive tracer at the class-IIa HDAC active site in vivo.
4. Conclusions
In summary, we synthesized and evaluated a focused library of class-IIa HDAC inhibitors. We delineated the key characteristics of the structure activity relationship (SAR) which identified compound 1 (NT160) as a highly potent and highly selective inhibitor of class-IIa HDACs. Preliminary non-invasive PET imaging studies with [18F]-NT160 in mice demonstrated, for the first-time evidence of brain entry and specific binding to class-IIa HDAC in vivo. Altogether, our current studies will pave the way for the synthesis of the next generation of class-IIa HDAC inhibitor-based theranostics for diagnosis and targeted treatment with class-IIa HDAC inhibitors. These studies are ongoing and will be reported in due course.
5. Materials and methods
Biochemical assay
We screened the tracer candidates against recombinant human HDACs 1e11 using 9 dilutions using the commercially available HDACs enzyme assay kits (BPS Bioscience, Inc.) as described in the manufacturer protocol. The IC50 values are shown in the corresponding tables. The class IIa HDAC fluorogenic assay functions in a two-step process: first, the class IIa HDAC removes the trifluoroacetylate (TFA) from TFA-lysine substrate to generate free lysine which is recognized by trypsin and releases a fluorescent fluorophore (intact substrate is not recognized by trypsin) that can be detected using microplate reader [19]. We determined the IC50 using nonlinear fit curves (GraphPad Prism) (% HDAC activity inhibition vs. compound concentration).
Cell-based deacetylase assay.
The cell-based assay was conducted similar to the previous work [19,28]. Briefly, compounds were diluted in 5 µL DMSO (11-point concentration curve, 5% final DMSO) and were added to 96 wells with positive (5 µM Trichostatin A (TSA)) and negative control (5 µL DMSO) wells. HT-29 cells (obtained from ATCC) with the addition of either 100 µM Boc-Lys-TFA (class-IIa selective substrate) or 200 µM Boc-Lys-Ac (class-I/IIb selective substrate) were plated into 96 well plates at 200,000 cells/well in 45 µL cellular assay buffer (RPMI without phenol red, 0.1% Fetal Bovine Serum) and were incubated for 3 h at 37 °C. The deacetylation achieved by the addition of 50 µL https://bpsbioscience.com/hdac-assay-developer-50030. Click or tap if you trust this link.”>HDAC developer solution (2.5 mg/mL trypsin in DMEM without Fetal Bovine Serum and 10% tween 80) for a further 1 h to sensitize the substrate and to lyse the cells. Fluorescent counts were read with microplate reader at an excitation wavelength of 360 nm and detection of emitted light of 460 nm.
6. Chemical synthesis
General information.
Solvents and starting material were obtained from commercial sources and were used as received. Thinlayer chromatography (TLC) was performed on pre-coated Kieselgel 60 F254 glass plates (Merck, Darmstadt, Germany) and aluminum-backed, pre-coated silica gel plates (Sorbent Technologies, Inc., Norcross, GA). 1H, 13C, and 19F NMR spectroscopy were performed using 400 MHz Bruker instrument. High resolution mass spectroscopy (HRMS) was performed using Agilent 1260HPLC/G6224A TOF MS. High-Performance Liquid Chromatography (HPLC) was performed with a 1260 series pump (Agilent Technologies, Stuttgart, Germany) with a built-in UV detector operated at 250 nm and a radioactivity detector with a single-channel analyzer (labLogic). Chemical and radiochemical purity were determined using analytical C18 column (4.6 × 150 mm, ASCENTIS RP-AMIDE, Sigma). HPLC was performed with 70% acetonitrile and 30% ammonium acetate buffer (20 mM) for solvents at a flow of 1 mL/min for all compounds except for compound 20 in which 80%acetonitrile and 20% ammonium acetate buffer (20 mM) was used. Enantiomeric excess (ee) was determined with Astec® CHIROBIOTIC® T Chiral HPLC Column, 5 µm particle size, L × I.D. 25 cm × 10 mm using 70% acetonitrile and 30% ammonium acetate buffer (20 mM) at a flow of 1 mL/min.
TMP195 and [18F]TMP195 were prepared similar to the previously described procedures [19,21].
General procedure for amine/carboxylic acid coupling reactions.
The acid (1.0 eq) and HATU (1.2 eq.) in DMF (1.0−3.0 mL) were stirred for 15 min followed by simultaneous addition of the amine (1.2 eq) and NMM (excess: ~1.0 mL). The reaction mixture was stirred for 3 h. The DMF was removed under vacuum (~70 °C water bath) and the residue was purified by column chromatography followed by trituration in cold pentane to afford the final products in 50−70% yield.
(R)-N-(1-(benzyl(methyl)amino)propan-2-yl)-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide (1: NT160). 1H NMR (CDCl3, 400 MHz) δ 8.21 (d, J = 8.22 Hz, 2H), 7.90 (d, J = 8.26 Hz, 2H), 7.32 (m, 5H), 6.59 (s, 1H), 4.18 (m, 1H), 3.67 (d, J = 13.04 Hz, 1H), 3.47 (d, J = 13.04 Hz, 1H), 2.55 (m, 1H), 2.42 (m, 1H), 2.32 (s, 3H), 1.31 (d, J = 6.05 Hz, 3H). 19F NMR (CDCl3, 376.5 MHz), δ −65.31. HRMS: Calculated for C21H22F3N4O2 [M H]+ 419.1689, Found: 419.1686. Chemical purity >98% (HPLC).
N-(3-(benzyl(methyl)amino)-2,2-dimethyl-3-oxopropyl)-3-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide (6) 1H NMR (CDCl3, 400 MHz) δ 8.57 (s, 1H), 8.26 (d, J = 8.15 Hz, 1H), 7.99 (d, J = 8.15 Hz, 1H), 7.63 (t, J = 8.15 Hz, 1H), 7.50 (m, 1H), 7.32 (m, 5H), 7.21 (d, J = 8.15 Hz, 2H), 4.67 (s, 2H), 3.71 (d, J = 8.15 Hz, 2H), 3.05 (s, 3H), 1.44 (s, 6H), 19F NMR (CDCl3, 376.5 MHz) δ −65.01. HRMS: HRMS: Calculated for C23H24F3N4O3 [M H]+ 461.1795, Found: 461.1794. Chemical purity: 96.9% (HPLC).
N-(3-(benzyl(methyl)amino)-3-oxopropyl)-3-(5-(tri-fluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide (7). 1H NMR (CDCl3, 400 MHz) δ 8.50 (m, 1H), 8.27 (d, J = 8.22 Hz, 1H), 8.01 (d, J = 8.26 Hz, 1H), 7.63 (t, J = 8.22 Hz, 1H), 7.32 (m, 4H), 7.23 (d, J = 7.35 Hz, 1H), 7.16 (d, J = 7.35 Hz, 1H), 4.59 (d, J = 31.00 Hz, 2H), 3.85 (m, 2H), 2.98 (d, J = 24.53 Hz, 2H), 2.75 (t, J = 6.25 Hz, 2H). 19F NMR (CDCl3, 376.5 MHz) δ −65.31. HRMS: Calculated for C21H20F3N4O3 [M+H]+ 433.1482, Found: 433.1483. Chemical purity: 97.6% (HPLC).
(R)-N-(1-(benzyl(methyl)amino)propan-2-yl)-3-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide (8). 1H NMR (CDCl3, 400 MHz) δ 8.50 (s, 1H), 8.29 (d, J = 8.22 Hz, 1H), 8.05 (d, J = 8.26 Hz, 1H), 7.63 (t, J = 8.22 Hz, 1H), 7.32 (m, 5H), 6.65 (1, 1H), 4.20 (m, 1H), 3.67 (d, J = 13.04 Hz, 1H), 3.48 (d, J = 13.04 Hz, 1H), 2.55 (m, 1H), 2.42 (m, 1H), 2.32 (s, 3H), 1.31 (d, J = 6.05 Hz, 3H). 19F NMR (CDCl3, 376.5 MHz), δ −65.31. HRMS: Calculated for C21H22F3N4O2 [M+H]+ 419.1689, Found: 419.1690. Chemical purity: 98% (HPLC).
N-(2-methyl-2-(2-phenyloxazol-4-yl)propyl)-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide (9). 1H NMR (CDCl3, 400 MHz) δ 8.24 (d, J = 8.88 Hz, 2H), 8.10 (d, J = 8.88 Hz, 2H), 8.07 (s, 1H), 7.50 (m, 5H), 3.66 (d, J = 4.00 Hz, 2H), 1.44 (s, 6H). 19F NMR (CDCl3, 376.5 MHz), δ −65.30. HRMS: Calculated for C23H20F3N4O3 [M+H]+ 457.1482, Found: 457.1480. Chemical purity: 97% (HPLC).
N-(3-(benzylamino)-2,2-dimethyl-3-oxopropyl)-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide (10). 1H NMR (CDCl3, 400 MHz) δ 8.21 (d, J = 8.22 Hz, 2H), 7.96 (d, J = 8.26 Hz, 2H), 7.56 (t, J = 8.22 Hz, 1H), 7.32 (m, 3H), 7.23 (d, J = 7.35 Hz, 2H), 4.68 (s, 2H), 3.69 (d, J = 8.22 Hz, 2H), 3.05 (s, 3H), 1.44 (s, 6H), 19F NMR (CDCl3, 376.5 MHz) δ −65.01. HRMS: HRMS: Calculated for C23H24F3N4O3 [M+H]+ 462.1795, Found: 461.1791. Chemical purity: 97% (HPLC).
N-(3-(benzylamino)-2,2-dimethyl-3-oxopropyl)-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide (11). 1H NMR (CDCl3, 400 MHz) δ 8.20 (d, J = 8.57 Hz, 2H), 7.94 (d, J = 8.57 Hz, 2H), 7.55 (m, 1H), 7.30 (m, 5H), 6.17 (m, 1H), 4.49 (d, J = 6.17 Hz, 2H), 3.62 (d, J = 6.17 Hz, 2H), 1.33 (s, 6H). 19F NMR (CDCl3, 376.5 MHz), δ −65.31. HRMS: Calculated for C22H22F3N4O3 [M+H]+ 447.1639, Found: 447.1637. Chemical purity >99% (HPLC).
N-(3-(benzyl(methyl)amino)-3-oxopropyl)-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide (12). 1H NMR (CDCl3, 400 MHz) δ 8.21 (d, J = 8.25 Hz, 2H), 7.96 (dd, J1 = 8.25 Hz, J2 = 2.15 Hz, 2H), 7.48 (m, 1H), 7.30 (m, 3H), 7.24 (d, J = 7.35 Hz, 1H), 7.21 (d, J = 7.35 Hz, 1H), 4.56 (d, J = 35.05 Hz, 2H), 3.87 (m, 2H), 2.99 (d, J = 24.53 Hz, 2H), 2.74 (t, J = 6.25 Hz, 2H). 19F NMR (CDCl3, 376.5 MHz) δ 65.15. HRMS: Calculated for C21H20F3N4O3 [M+H]+ 433.1482, Found: 433.1485. Chemical purity >99.6% (HPLC).
(R)-N-(1-(benzylamino)-1-oxopropan-2-yl)-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide(13). 1H NMR (CDCl3, 400 MHz) δ 8.21 (d, J = 8.03 Hz, 2H), 7.94 (d, J = 8.03 Hz, 2H), 7.32 (m, 5H), 7.09 (d, J = 8.03 Hz, 1H), 6.58 (m, 1H), 4.76 (m, 1H), 4.50 (m, 2H), 1.58 (d, J = 7.03 Hz, 3H). 19F NMR (CDCl3, 376.5 MHz), δ −65.32. HRMS: Calculated for C20H19F3N4O3 [M+H]+ 419.1326, Found: 419.1321. Chemical purity: 98.9% (HPLC). Enantiomeric excess (ee): 81.0% (Chiral HPLC).
(R)-N-(1-(benzylamino)-3-methoxy-1-oxopropan-2-yl)-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide (14). 1H NMR (CDCl3, 400 MHz) δ 8.24 (d, J = 9.04 Hz, 2H), 8.00 (d, J = 9.05 Hz, 2H), 7.32 (m, 5H), 6.88 (m, 1H), 4.76 (m, 1H), 4.56 (m, 2H), 3.96 (dd, J = 9.18 Hz, 1H), 3,58 (t, J = 9.18 Hz, 1H), 3.47 (s, 3H). 19F NMR (CDCl3, 376.5 MHz), δ 64.71. HRMS: Calculated for C21H20F3N4O4 [M+H]+ 449.1431, Found: 449.1429. Chemical purity: 99.2% (HPLC). Enantiomeric excess (ee): 99.3% (Chiral HPLC).
(R)-N-(1-(benzylamino)-3-methoxy-1-oxopropan-2-yl)-6-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)nicotinamide (15). 1H NMR (CDCl3, 400 MHz) δ 9.25 (s, 1H), 8.32 (m, 2H), 7.47 (m, 1H), 7.24 (m, 5H), 6.91 (m, 1H), 4.78 (m, 1H), 4.56 (m, 2H), 3.94 (dd, J = 9.18 Hz, 1H), 3,61 (t, J = 9.18 Hz, 1H), 3.47 (s, 3H). 19F NMR (CDCl3, 376.5 MHz), δ 64.71. HRMS: Calculated for C20H19F3N5O4 [M+H]+ 450.1384, Found: 450.1384. Chemical purity: 99.2% (HPLC). Enantiomeric excess (ee): 95.3% (Chiral HPLC).
(R)-N-(1-(4-fluorobenzylamino)-3-methoxy-1-oxopropan-2-yl)-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide (16). 1H NMR (CDCl3, 400 MHz) δ 8.24 (d, J = 9.04 Hz, 2H), 8.00 (d, J = 9.05 Hz, 2H), 7.32 (m, 5H), 6.88 (m, 1H), 4.76 (m, 1H), 4.56 (m, 2H), 3.96 (dd, J = 9.18 Hz, 1H), 3,58 (t, J = 9.18 Hz, 1H), 3.47 (s, 3H). 19F NMR (CDCl3, 376.5 MHz), δ −65.32, −114.70. HRMS: Calculated for C21H19F4N4O4 [M+H]+ 467.1337, Found: 467.1338. Chemical purity: 98.6% (HPLC). Enantiomeric excess (ee): 98.0% (Chiral HPLC).
N-(1-(benzyl(methyl)amino)propan-2-yl)-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide (17). 1H NMR (CDCl3, 400 MHz) δ 8.21 (d, J = 8.16 Hz, 2H), 7.94 (d, J = 7.76 Hz, 2H), 7.33 (m, 5H), 6.80 (m, 1H), 4.22 (m, 1H), 3.70 (d, J = 12.66 Hz, 1H), 3.54 (d, J = 12.74 Hz, 1H), 2.66 (m, 1H), 2.48 (m, 1H), 2.36 (s, 3H), 1.32 (d, J = 7.66 Hz, 3H). 19F NMR (CDCl3, 376.5 MHz), δ −65.32. HRMS: Calculated for C21H22F3N4O2 [M+H]+ 419.1689, Found: 419.1688. Chemical purity: 98.5% (HPLC). Enantiomeric excess (ee): 95.0% (Chiral HPLC).
(R)-N-(1-(benzyl(methyl)amino)propan-2-yl)-6-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)nicotinamide (18). 1H NMR (CDCl3, 400 MHz) δ 9.16 (s, 1H), 8.31 (m, 2H), 7.24 (m, 5H), 6.88 (m, 1H), 4.25 (m, 1H), 3.69 (d, J = 12.66 Hz, 1H), 3.51 (d, J = 12.74 Hz, 1H), 2.63 (m, 1H), 2.45 (m, 1H), 2.36 (s, 3H), 1.33 (d, J = 7.66 Hz, 3H). 19F NMR (CDCl3, 376.5 MHz), δ −65.16. HRMS: Calculated for C21H22F3N5O2 [M+H]+ 419.1689 Found: 419.1688. Chemical purity: 95% (HPLC). Enantiomeric excess (ee): 96.0% (Chiral HPLC).
(R)-N-(1-((4-fluorobenzyl)(methyl)amino)propan-2-yl)-4-(5- (trifluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide (19). 1H NMR (CDCl3, 400 MHz) δ 8.21 (d, J = 8.16 Hz, 2H), 7.94 (d, J = 7.76 Hz, 2H), 7.29 (m, 2H), 7.01 (t, J = 8.75 Hz, 2H) 6.80 (m, 1H), 4.22 (m, 1H), 3.66 (d, J = 12.66 Hz, 1H), 3.53 (d, J = 12.74 Hz, 1H), 2.66 (m, 1H), 2.48 (m, 1H), 2.34 (s, 3H), 1.32 (d, J = 7.66 Hz, 3H). 19F NMR (CDCl3, 376.5 MHz), δ −65.32, −114.70. Calculated for C21H21F4N4O2 [M+H]+ 447.1595, Found: 447.1594. Chemical purity: 96.5% (HPLC). Enantiomeric excess (ee): 94.1% (Chiral HPLC).
(R)-N-(1-(benzyl(methyl)amino)propan-2-yl)-4-(5-(chlorodifluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide (20). 1H NMR (CDCl3, 400 MHz) δ 8.21 (d, J = 8.27 Hz, 2H), 7.92 (d, J = 8.26 Hz, 2H), 7.32 (m, 5H), 6.79 (1, 1H), 4.21 (m, 1H), 3.69 (d, J = 13.04 Hz, 1H), 3.50 (d, J = 13.04 Hz, 1H), 2.63 (m, 1H), 2.44 (m, 1H), 2.34 (s, 3H), 1.32 (d, J = 6.05 Hz, 3H). 19F NMR (CDCl3, 376.5 MHz), δ −51.94. HRMS: Calculated for C21H22BrF2N4O2 [M+H]+ 435.1394, Found: 435.1395. Chemical purity: 98.4% (HPLC).
(R)-N-(1-(benzyl(methyl)amino)propan-2-yl)-4-(5-(bromodifluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide (21). δ1H NMR (CDCl3, 400 MHz) δ 8.21 (d, J = 8.27 Hz, 2H), 7.92 (d, J = 8.26 Hz, 2H), 7.32 (m, 5H), 6.79 (1, 1H), 4.21 (m, 1H), 3.69 (d, J = 13.04 Hz, 1H), 3.50 (d, J = 13.04 Hz, 1H), 2.63 (m, 1H), 2.44 (m, 1H), 2.34 (s, 3H), 1.32 (d, J = 6.05 Hz, 3H). 19F NMR (CDCl3, 376.5 MHz), δ _51.94. HRMS: Calculated for C21H22BrF2N4O2 [M+H]+ 479.0889, Found: 479.0886. Chemical purity: 96.9% (HPLC).
6.1. Radiochemistry
The solution of [18F] was purchased from NCM USA (Bronx, NY). The [18F] is trapped on a QMA cartridge and then eluted with 1.0−1.2 mL of a solution that contains kryptofix/Cs2CO3 [Cs2CO3 (40 mg) and kryptofix (100 mg)] to a V-vial (Wheaton) with 92−96% recovery. The solvent was removed under a stream of Argon at 110 °C. Water residue was removed aziotropically with the addition of acetonitrile (3 × 1.0 mL) and repeated drying under a stream of Argon at 110 °C.
A solution of the bromo-precursor (6e8 mg) in the appropriate solvent (i.e. DMSO) (0.4 mL) was added to the dried K[18F]/kryptofix or Cs[18F]/kryptofix and the mixture was heated at 160 °C for 25 min. The reaction mixture was cooled and passed through a silica gel cartridge (waters, 900 mg) and eluted with 30% methanol in dichloromethane (2.5 mL). After evaporating of the solvent under a stream of argon at 60−80 °C, the residue was redissolved in the appropriate HPLC solvent and purified by semipreparative HPLC.
(R)-N-(1-(benzyl(methyl)amino)propan-2-yl)-4-(5-([18F]-tri-fluoromethyl)-1,2,4-oxadiazol-3-yl)benzamide ([18F]-NT160) was isolated with 67% acetonitrile/ammonium acetate buffer (20 mM) solution in 17−19 min. The solvent was evaporated under reduced pressure and was redissolved in 20%ethanol/saline for animal injection.
6.2. Authentication of the radioactive tracers
The radioactive product was co-injected with an authentic non-radiolabeled 1 (NT160) into an analytical HPLC to confirm its purity and identity. The radiochemical purity was >95%. The radioactive peak was detected with a radioactivity detector co-injected with the relevant authentic cold compound which was detected with ultraviolet detector (250 nM) using analytical HPLC.
6.3. Molar activity
The specific activity was determined from the area under the curve of the tracer that is attributed to the ultraviolet peak in the HPLC chromatogram against a calibration curve pre-prepared with the unlabeled reference standard. Molar activity of our tracers ranged from 0.30 to 0.85 GBq/umol (8.0e23.0 mCi/umol) which is remarkably high for [18F]trifluoromethyl moiety. It is important to note that we are currently purchasing F-18 from a commercial source with significant decay prior to the start of our radiochemical experiment (~3 half-lives). We expect to obtain significantly higher molar activities for our tracers once our cyclotron becomes operational. Furthermore, automated synthesis is expected to further improve the radiochemical yield and molar activity due to more efficient synthesis and shorter overall production time. Moreover, starting with a higher amount of radioactivity may also further improve the yield and molar activities.
Supplementary Material
Acknowledgements
The authors would like to thank the Stony Brook Cancer Center for the start-up support provided to the Turkman Lab. In addition, the research reported in this publication was supported in part by the following grants to N. Turkman: FUSION Award from The Stony Brook Renaissance School of Medicine and the Office of the Vice President for Research (Targeted Research Opportunity Program) and by R01AG067417 from the National Institutes of Health/National Institute on Aging.
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2021.114011.
References
- [1].Kassis H, Shehadah A, Li C, Zhang Y, Cui Y, Roberts C, Sadry N, Liu X, Chopp M, Zhang ZG, Class IIa histone deacetylases affect neuronal remodeling and functional outcome after stroke, Neurochem. Int 96 (2016) 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Faraco G, Pancani T, Formentini L, Mascagni P, Fossati G, Leoni F, Moroni F, Chiarugi, Pharmacological inhibition of histone deacetylases by sub-eroylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain, Mol. Pharmacol 70 (6) (2006) 1876–1884. [DOI] [PubMed] [Google Scholar]
- [3].Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM, Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action, J. Pharmacol. Exp. Therapeut 321 (3) (2007) 892–901. [DOI] [PubMed] [Google Scholar]
- [4].Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM, Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction, J. Neurochem 89 (6) (2004) 1358–1367. [DOI] [PubMed] [Google Scholar]
- [5].Steffan Js BL, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM, Nature 413 (2001) 739–743. [DOI] [PubMed] [Google Scholar]
- [6].Bürli Rw LC, Aziz O, Matthews KL, Yates D, Lyons KA, Beconi M, McAllister G, Breccia P, Stott AJ, Penrose SD, Wall M, Lamers M, Leonard P, Müller I, Richardson CM, Jarvis R, Stones L, Hughes S, Wishart G, Haughan AF, O’Connell C, Mead T, McNeil H, Vann J, Mangette J, Maillard M, Beaumont V, Munoz-Sanjuan I, Dominguez C, J. Med. Chem 56 (2013) 9934–9954. [DOI] [PubMed] [Google Scholar]
- [7].Mielcarek M, C. B, Franklin SA, Smith DL, Woodman B, Marks PA, Bates GP, PLoS One 6 (2011), e27746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shen X, J. C, Li J, Kofler J, Herrup K, eNeuro 3 (2016) pii: ENEURO.0124–15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Neuner SM, L. W, Hoffmann BR, Mozhui K, Kaczorowski CC, Behav. Brain Res 322 (2017) 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sen A, T. N, Alkon DL, J. Neurosci 35 (2015) 7538–7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hendricks JA, Keliher EJ, Marinelli B, Reiner T, Weissleder R, Mazitschek R, In vivo PET imaging of histone deacetylases by 18F-suberoylanilide hydroxamic acid (18F-SAHA), J. Med. Chem 54 (15) (2011) 5576–5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zeglis BM, Pillarsetty N, Divilov V, Blasberg RA, Lewis JS, The synthesis and evaluation of N1-(4-(2-[18F]-fluoroethyl)phenyl)-N8-hydroxyoctanediamide ([18F]-FESAHA), a PET radiotracer designed for the delineation of histone deacetylase expression in cancer, Nucl. Med. Biol 38 (5) (2011) 683–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hooker JM, Kim SW, Alexoff D, Xu Y, Shea C, Reid A, Volkow N, Fowler JS, Histone deacetylase inhibitor, MS-275, exhibits poor brain penetration: PK studies of [C]MS-275 using Positron Emission Tomography, ACS Chem. Neurosci 1 (1) (2010) 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Seo YJ, Muench L, Reid A, Chen J, Kang Y, Hooker JM, Volkow ND, Fowler JS, Kim SW, Radionuclide labeling and evaluation of candidate radioligands for PET imaging of histone deacetylase in the brain, Bioorg. Med. Chem. Lett 23 (24) (2013) 6700–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang C, Schroeder FA, Wey HY, Borra R, Wagner FF, Reis S, Kim SW, Holson EB, Haggarty SJ, Hooker JM, In vivo imaging of histone deacetylases (HDACs) in the central nervous system and major peripheral organs, J. Med. Chem 57 (19) (2014) 7999–8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wey HY, Wang C, Schroeder FA, Logan J, Price JC, Hooker JM, Kinetic analysis and quantification of [11C]martinostat for in vivo HDAC imaging of the brain, ACS Chem. Neurosci 6 (5) (2015) 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Reid AE, Hooker J, Shumay E, Logan J, Shea C, Kim SW, Collins S, Xu Y, Volkow N, Fowler JS, Evaluation of 6-([(18)F]fluoroacetamido)-1-hexanoicanilide for PET imaging of histone deacetylase in the baboon brain, Nucl. Med. Biol 36 (3) (2009) 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Strebl Mg CA, Zhao WN, et al. , ACS Cent. Sci 3 (9) (2017. Sep 27) 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lobera M, Madauss KP, Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, Baloglu E, Trump RP, Head MS, Hofmann GA, Murray-Thompson M, Schwartz B, Chakravorty S, Wu Z, Mander PK, Kruidenier L, Reid RA, Burkhart W, Turunen BJ, Rong JX, Wagner C, Moyer MB, Wells C, Hong X, Moore JT, Williams JD, Soler D, Ghosh S, Nolan MA, Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group, Nat. Chem. Biol 9 (5) (2013) 319–325. [DOI] [PubMed] [Google Scholar]
- [20].Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, Johnson SF, Carrasco RD, Lazo S, Bronson RT, Davis SP, Lobera M, Nolan MA, Letai A, Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages, Nature 543 (7645) (2017) 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Turkman N, Liu D, Pirola I, Novel late-stage radiosynthesis of 5-[18F]-trifluoromethyl-1,2,4-oxadiazole (TFMO) containing molecules for PET imaging, Sci. Rep 11 (1) (2021) 10668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bonomi R, U. M, Shavrin A, Yeh HH, Majhi A, Dewage SW, Najjar A, Lu X, Cisneros GA, Tong WP, Alauddin MM, Liu RS, Mangner TJ, Turkman N, Gelovani JG, PLoS One 10 (2015), e0133512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow CJ, Distribution of histone deacetylases 1–11 in the rat brain, J. Mol. Neurosci. : MN 31 (1) (2007) 47–58. [DOI] [PubMed] [Google Scholar]
- [24].Hebach CJ, E, Kallen J, Ternois JG, Tintelnot-Blomley M, Novel Trifluoromethyl-Oxadiazole Derivatives and Their Use in the Treatment of Disease, WO2013080120, 2013.
- [25].Stott AJ, Maillard MC, Beaumont V, Allcock D, Aziz O, Borchers AH, Blackaby W, Breccia P, Creighton-Gutteridge G, Haughan AF, Jarvis RE, Luckhurst CA, Matthews KL, McAllister G, Pollack S, Saville-Stones E, Van de Poël AJ, Vater HD, Vann J, Williams R, Yates D, Muñoz-Sanjuán I, Dominguez C, Evaluation of 5-(Trifluoromethyl)-1,2,4-oxadiazole-Based class IIa HDAC inhibitors for huntington’s disease, ACS Med. Chem. Lett 12 (3) (2021) 380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bonomi R, Mukhopadhyay U, Shavrin A, Yeh HH, Majhi A, Dewage SW, Najjar A, Lu X, Cisneros GA, Tong WP, Alauddin MM, Liu RS, Mangner TJ, Turkman N, Gelovani JG, Novel histone deacetylase class IIa selective sub-strate radiotracers for PET imaging of epigenetic regulation in the brain, PLoS One 10 (8) (2015), e0133512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jones P, Altamura S, De Francesco R, Gallinari P, Lahm A, Neddermann P, Rowley M, Serafini S, Steinkühler C, Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases, Bioorg. Med. Chem. Lett 18 (6) (2008) 1814–1819. [DOI] [PubMed] [Google Scholar]
- [28].Luckhurst CA, Aziz O, Beaumont V, Bürli RW, Breccia P, Maillard MC, Haughan AF, Lamers M, Leonard P, Matthews KL, Raphy G, Stott AJ, Munoz-Sanjuan I, Thomas B, Wall M, Wishart G, Yates D, Dominguez C, Development and characterization of a CNS-penetrant benzhydryl hydroxamic acid class IIa histone deacetylase inhibitor, Bioorg. Med. Chem. Lett 29 (1) (2019) 83–88. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.