

Abstract
The compatibility of photochemistry with solid-phase peptide synthesis is demonstrated via photochemical hydroalkylation to form C(sp3)-C(sp3) bonds between on-resin Giese acceptors and redox-active esters. Both iridium-based photocatalysts and Hantszch ester led to high yields, with final reaction conditions producing full conversions within 30 minutes under ambient conditions. The chemistry is compatible with a broad range of peptide sidechains, redox-active esters, and resin. These conditions represent the first example of photochemical peptide modifications on resin.
Graphical Abstract
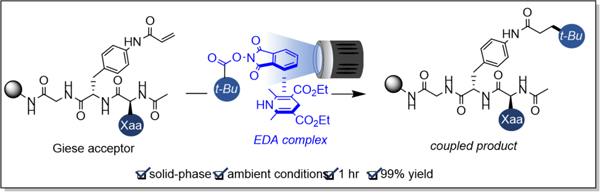
Since the advent of solid-phase peptide synthesis (SPPS) in 1963, over 60 peptide pharmaceuticals have been approved in the US, Europe, and Japan, more than 150 peptides are currently undergoing clinical development, and over 260 have been investigated in human clinical trials.1 SPPS depends greatly on the adaptation of solution-phase reactions to solid-phase conditions. As examples, since the 1990s, C-C bond-forming transformations such as the Stille,2 Heck,3 Suzuki-Miyaura,4 and Sonogashira5 reactions have been adapted to solid-phase by both industry and academia. Additionally, in 1996, the first example of solid-phase ruthenium-catalyzed metathesis was reported, and soon after, the first solid-phase peptide ring-closing metathesis (RCM) reaction was communicated.6,7 Since that time, RCM has been extensively used to form hydrocarbon-stapled peptides in biotherapeutics.8 Although peptide macrocycles generated via RCM can be reduced to C(sp3)-C(sp3) bonds, few direct routes to such structures exist under mild conditions.
Recently, Baran et al. reported two nickel-catalyzed decarboxylative conjugate addition procedures and demonstrated their compatibility with solid-phase conditions.9,10 Both procedures utilize nickel-catalyzed cross-coupling mechanisms to forge C(sp3)-C(sp3) bonds under inert conditions and required 8–16 h reaction times. Given the importance of solid-phase peptide C(sp3)-C(sp3) bond formation, an alternate route through a complementary mechanistic pathway was deemed desirable.
Although photochemistry on peptides and proteins has been a very active field,11,12 to our knowledge, no studies on the compatibility of photochemistry with on-resin peptides have been reported. The application of photochemistry to solid-phase peptide synthesis offers several distinct advantages over extant protocols. First, photo-chemically induced reactions involving open-shell intermediates exhibit extraordinary toleration of diverse functional groups. Additionally, light-mediated transformations typically operate at room temperature and permit the use of buffers or aqueous mixtures that offer biocompatible reaction conditions. The reliance on photon flux also allows the exclusion of light to quench reactions and control reaction progression precisely and conveniently.13 Finally, dual photocatalytic cycles have enabled multi-component reactions, ideal for making diverse peptide macrocycles and bicycles in a single, mild synthetic step.14
With a goal to elaborate peptides by photoinduced Giese-type reactions on solid support, several procedures were assessed. In the seminal solution phase, photosensitized decarboxylative Giese addition reported by Okada,15 redox-active esters (RAEs) were employed as radical precursors and coupled with Giese acceptors in the presence of 1-benzyl-1,4-dihydronicotinamide as a reductant and Ru(bpy)3Cl2 as a photocatalyst in aqueous THF.15 Up to 69% yields were reported. However, the scope of redox-active esters was limited to three hydrocarbons. Given the structural complexity of polypeptides, reaction conditions with high functional group tolerance were imperative. Furthermore, the Okada reaction conditions utilized an aqueous reaction medium, which was expected to be challenging for nonpolar peptide substrates.
In 2015, Overman et al. reported a solution-phase decarboxylative Giese-type addition involving a reductive single electron transfer (SET) of redox-active esters via photoredox catalysis.16 The contribution by Overman and coworkers involved the photochemical coupling of various Giese acceptors and tertiary radicals generated from corresponding redox-active esters at room temperature and in dichloromethane as solvent.16 The reaction employed Hantzsch ester (diethyl 1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate, HE), Ru(bpy)3(BF4)2, and i-Pr2NEt, and featured a broad substrate scope for both coupling partners. A visible light-induced approach to the synthesis and chemical modification of solid-phase peptides was thus initiated based on Overman’s mild photochemical hydroalkylation.
We began our studies using Chem Matrix Rink Amide resin, which features a PEG-based polymer, rather than polystyrene, to avoid potentially reactive aromatics. Aromatic Giese acceptor 1 was employed for ease of analysis by LC-MS and 1H NMR. Acrylamide was chosen as the electron deficient olefin over acrylate for its ease of incorporation into peptides. Installation of the solid-phase Giese acceptor began with synthesis of Fmoc-(4-acrylamide)-Phe-OH, which was then incorporated onto resin. The Fmoc group was deprotected using non-nucleophilic DBU, and subsequently the substrate was acylated on the N-terminus. Alternatively designed as a late-stage peptide modification, Fmoc-(4-Trt-amino)-Phe-OH was incorporated onto the resin. After N-terminal deprotection and acylation, the Trt group was removed, and the aniline was reacted with acrylic acid and (1-cyano-2-ethoxy-2-oxoethylidene-aminooxy)dimethylamino-morpholino-carbenium hexafluorophos-phate (COMU). The purity of crude material in this protocol was 86% as judged by analytical HPLC (SI, S15).
Attempts to optimize the coupling of on-resin olefin (1) with 1-adamantyl N-(acyloxy)phthalimide (2a) rapidly generated fruitful conditions with Ru(bpy)3(PF6)2 as photocatalyst and i-Pr2NEt as reductant in DMF with blue LED irradiation at room temperature. These conditions produced the coupled product 3a with a 30% conversion (Table 1, entry 1). Examination of photocatalysts identified [Ir{dF(CF3)ppy}2(bpy)]PF6 (E1/2 M*/M- = +1.32 V)17 as a suitable catalyst for the solid-phase photocoupling (Table 1, entries 2–5). A similar conversion was observed using the Rink amide polystyrene resin (Table 1, entry 6). Lowering the reaction time below 16 h proved detrimental to the reaction yield (SI, S7). The use of THF as a solvent, which was advantageous for the coupling of tert-alkyl N-phthalimidoyl oxalates,18 decreased the conversion to 3a to 89% (Table 1, entry 7). In the absence of light, no product was observed (Table 1, entry 8). Excluding the photocatalyst or i-Pr2NEt resulted in product formation with substantially diminished yields (Table 1, entries 9–10), demonstrating that the superstoichiometric amine reductant was ineffective at fragmenting redox-active esters. Comparable results in the absence of the photocatalyst were observed by Okada and co-workers in the initial decarboxylative Michael addition report.15 The addition of Hantzsch ester as a reductant generated superior yields (Table 1, entry 11) particularly for primary re-dox-active esters (SI, S8). A similar yield and conversion were produced in the absence of the photocatalyst (SI, S8).
Table 1.
Optimization of Solid-Phase Photocatalytic Hydroalkylation Conditions
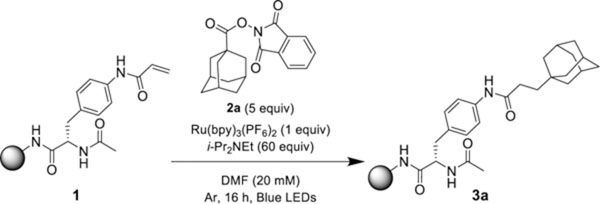
| |||
|---|---|---|---|
| entry | deviation from initial conditions | 3 (P/ISa) | 3 (conv, %b) |
| 1 | none | 0.26 | 30 |
| 2 | [Ir{dF(CF3)ppy}2(bpy)]PF6 | 0.38 | 100 |
| 3 | [Ir{dF(CF3)ppy}2(dtbbpy)]PF6 | 0.31 | 88 |
| 4 | 4CzIPNc | 0.14 | 59 |
| 5 | Cl-4CzIPNd | 0.10 | 31 |
| 6 | entry 2 with polystyrene resin | 0.38 | 100 |
| 7 | THF | 0.24 | 89 |
| 8 | no light | 0.00 | 0 |
| 9 | no i-Pr2NEt | 0.03 | 7 |
| 10 | no photocatalyst | 0.09 | 14 |
| 11 | 5 equiv Hantzsch ester | 0.40 | 100 |
Product-to-internal standard ratio as determined by LC/MS.
Conversion to 3a as determined by LC/MS. PC = photocatalyst.
1,2,3,5-Tetrakis(carbazol-9-yl)-4,6-dicyanobenzene.
2,4,5,6-Tetrakis-(3,6-dichloro-9H-carbazol-9-yl)isophthalonitrile
After incorporating the Hantzsch ester, a reaction time screen revealed full conversions within 30 minutes (SI, S9). Control studies produced similar NMR yields under air and without the amine base (Table 2, entries 1–3). Conversion of 100% was also observed using Rink amide polystyrene resin (Table 2, entry 4).Comparable catalyst-free conditions were reported in the original Okada report, as well as by Overman and Shang in the solution phase.15,16,19 No product was formed in the absence of light or Hantzsch ester (Table 2, entries 4–5). Increasing the number of equivalents of Hantzsch ester and redox-active ester resulted in no improvements, while reducing the equivalents of these reactants led to inconsistent results (SI, S10).
Table 2.
Development of Solid-Phase, Metal-Free Photochemical Hydroalkylation Conditions
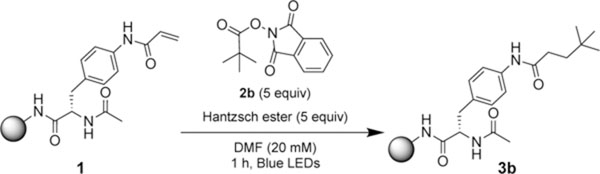
| |||
|---|---|---|---|
| entry | deviation from initial conditions | 3b (conva) | 3b (% yielda) |
| 1 | none | 100 | 90 |
| 2 | under argon | 100 | 88 |
| 3 | under argon with DIPEA | 100 | 90 |
| 4 | Rink amide polystyrene resin | 100 | 90 |
| 5 | no Hantzsch ester | 0 | 0 |
| 6 | no light | 0 | 0 |
| 7 | >5 equiv HE and RAE | no improvement | no improvement |
| 8 | <5 equiv HE and RAE | inconsistent conv | inconsistent yield |
Product yield and conversion determined by crude NMR.
Although Overman proposed a photocatalytic reduction of the redox-active esters (Scheme 1A),16,20 recent investigations have revealed that the reduction takes place largely via a light-induced charge transfer within an electron donor-acceptor (EDA) complex formed between the Hantzsch ester and the redox-active ester (Scheme1B).21 Photoactivation of the EDA complex induces an inner sphere electron transfer from the Hantzsch ester to the redox-active ester, leading to carbon-centered radical III via hemolytic fragmentation and decarboxylation. The radical then adds to the electron-deficient olefin and generates intermediate IV, which abstracts a hydrogen atom from the oxidized Hantzsch ester to generate pyridinium salts to furnish the desired hydroalkylation product. Because of the speed, cost, and operational simplicity of the catalyst-free conditions, they were utilized to explore the scope of the process.
Scheme 1. Mechanistic View of the Light-Mediated Decarboxylative Hydroalkylation.

(A) Envisioned mechanism for the photocatalytic decarboxylative hydroalkylation. (B)Mechanistic view of the catalyst-free photochemical decarboxylative hydroalkylation.
The performance of the hydroalkylation using a series of simple redox-active esters was first investigated (Scheme 2). The on-resin Giese acceptor coupled efficiently with primary, secondary, and tertiary alkyl RAE partners. Tertiary (3b) and secondary (3f) carboncentered radicals performed nearly identically, while their primary analogues produced diminished yields, except for α-benzyloxy radical (3i) and Boc-β-alanine (3p). Cyclic redox-active esters, including adamantyl (3a), 3-cyclopentenyl (3d), and cyclohexyl (3e), all converted to the desired product with excellent yields. NHPI esters featuring nitrogen- (3h, 3p), oxygen- (3n), and sulfur-based (3k, 3o) heterocyclic moieties were also compatible with the hydroalkylation protocol.
Scheme 2. Scope of Solid-Phase Photochemical Hydroalkylationa.
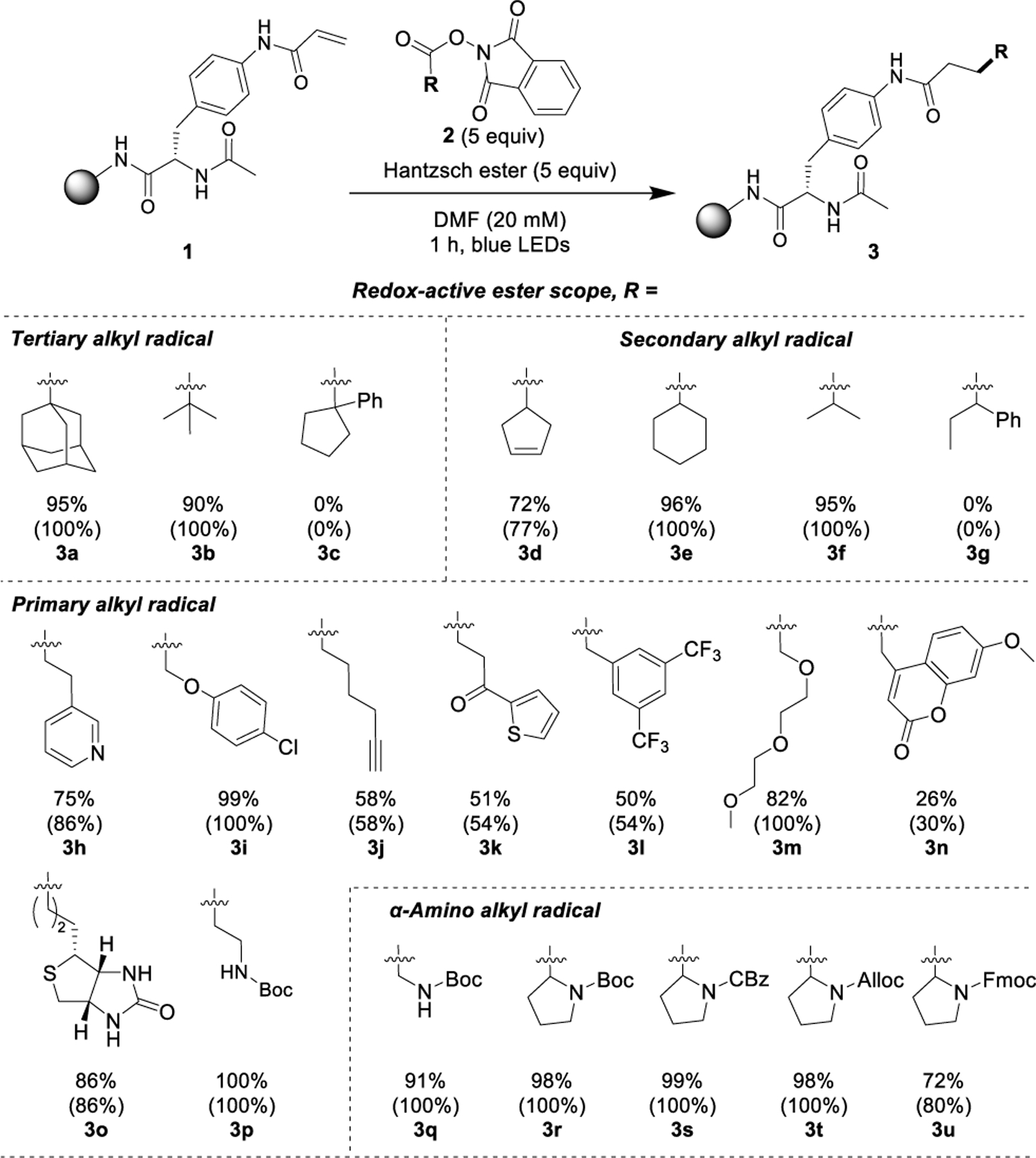
aYields determined by quantitative 1H NMR; conversions determined by quantitative 1H NMR and reported in parentheses.
Cycloadditions between terminal alkynes and azides (“click chemistry”) are widely utilized for bioorthogonal transformations in which considerations of reaction efficiency, chemical inertness, cost, and compatibility with aqueous conditions are paramount. An alkynyl-substituted RAE was synthesized and found to deliver a 58% yield of 3j. The moderate yield was expected because of the difficulty of generating primary radicals as well as their reduced nucleophilicities. Furthermore, reports of terminal alkynes undergoing decarbox-ylative radical addition to form alkenes suggested a loss of reactant may be partially responsible as well.22,23 The interaction between biotin and streptavidin is one of the strongest in nature and widely utilized in biology and biochemistry (Kd = 10−15 M).24 A biotinyl-substituted RAE was synthesized and found to react with the solid-phase substrate in 86% yield; high given the yields of other primary RAEs (3o).
We subsequently incorporated an example of a PEGylation (3m) featuring an α-alkoxy group that conveniently aided radical generation. Mini-PEGs are frequently used to tune the lipophilicity of bio-molecules. A coumarin moiety was also reasonably well-tolerated by the reaction protocol (3n). Coumarins are found in a broad range of medicinally relevant natural products, synthetic pharmaceuticals, and fluorescent labels.25,26 The low yield of coumarin 3n is likely a result of the low nucleophilicity of the highly stabilized radical generated (vide infra). Recognition of protein conformers by fluorine NMR necessitates the integration of fluorinated moieties whose chemical shifts are responsive to slight variations in the local dielectric and magnetic shielding environment. To that end, an NHPI ester incorporating a fluorine (19F) NMR tag27,28 was evaluated, with the 3,5-bis(trifluoromethyl)phenyl moiety 3l selected as a model fluorine probe that was successfully coupled with a reasonable yield. A single 19F-NMR peak confirmed the presence of the fluorine (19F) NMR tag (SI, S70).
The amenability of resonance stabilized radicals, which exhibit lower nucleophilicities than localized carbon-centered radicals, was also examined. Unfortunately, the success of substrates generating such stabilized radicals was limited. Products 3l and 3n derived from primary resonance stabilized radicals were obtained in 50% and 26% yields, respectively. Neither secondary nor tertiary benzylic RAE substrates produced product (3c, 3g), perhaps because of the combination of steric and stabilizing resonance effects.
The abundance of structurally diverse, commercially available amino acids makes them a particularly appealing radical feedstock for peptide modification. They are also attractive building blocks because the amine functional group promotes radical addition and provides a site for further functionalization and peptide elongation. The scope of the coupling using α-amino alkyl radical precursors was therefore evaluated. Couplings employing Boc-, CBz-, Alloc-, and Fmoc-protected prolines all produced excellent yields (3q-3u). Fmoc protection is seldom used in photochemistry, but it is used abundantly with SPPS. Fmoc-proline was well-tolerated, albeit with a slightly diminished yield of 72%.
Toleration of the transformation to structural and functional complexity was further evaluated using a series of tripeptides (Scheme 3). The Giese acceptor was incorporated as a late-stage modification to prevent attack of the N-terminus on the Giese acceptor during peptide elongation. His, Trp, Cys, Met, and Tyr moieties have all participated in photochemistry under various solution-phase conditions,12 but their behavior under similar conditions protected on solid phase has not been tested. In the event, tripeptides incorporating these amino acid derivatives exhibited high conversions and yields.
Scheme 3. Tolerance of Various Amino Acid Side Chainsa.
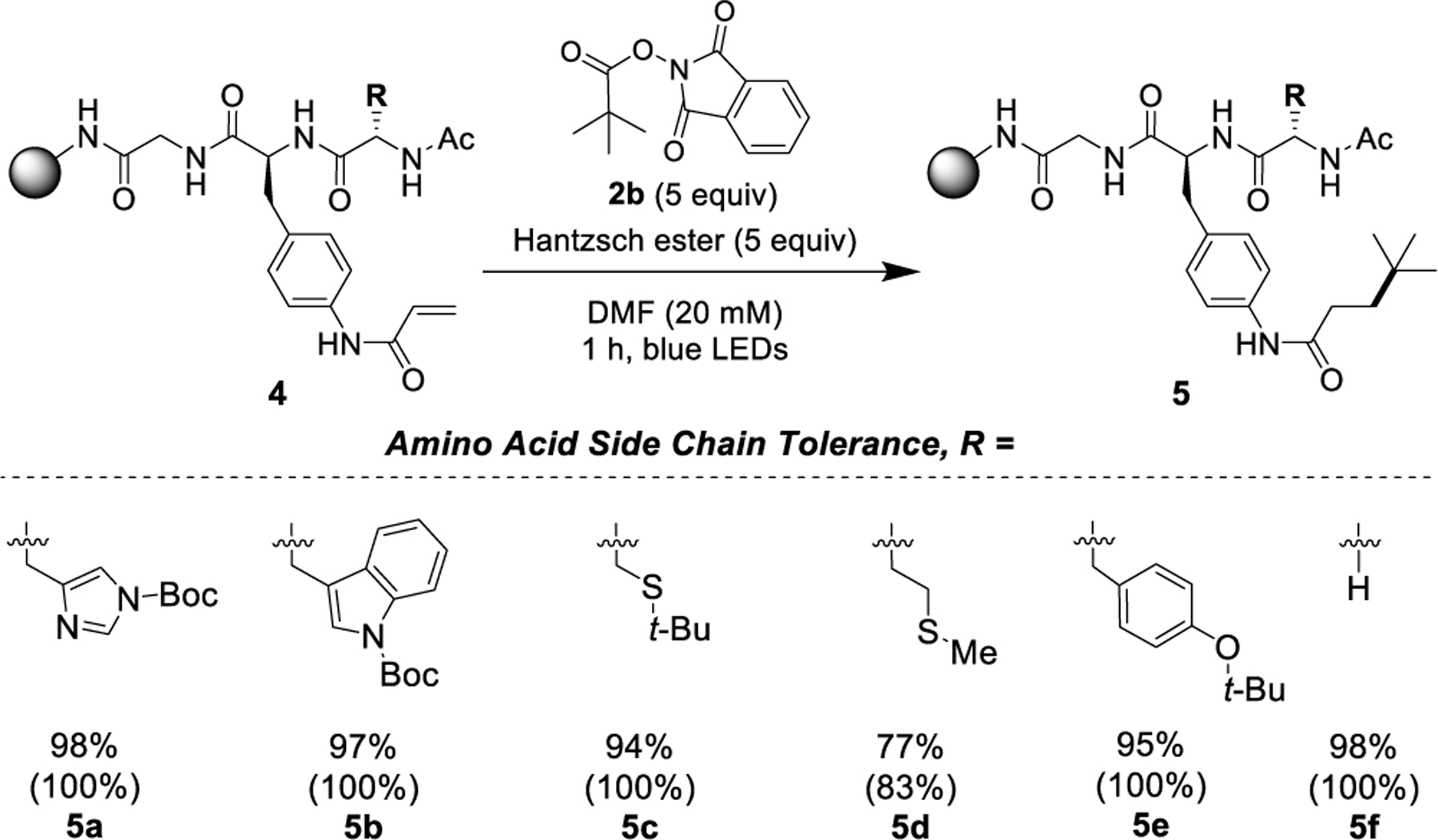
aYields determined by quantitative 1H NMR; conversions determined by quantitative 1H NMR and reported in parenthesis.
The amenability of aliphatic Giese acceptors was then demon-strated by on-resin acrylation of lysine 6 and subsequent reaction with CBz-Pro-NHPI (Scheme 4). A 98% yield was observed. Next, 15-mer collagen-model peptide 7 was synthesized and acylated on the N-terminus. A reaction with CBz-Pro-NHPI led to full conversion by HPLC (SI, S112).
Scheme 4. Tolerance of Aliphatic Giese Acceptors.
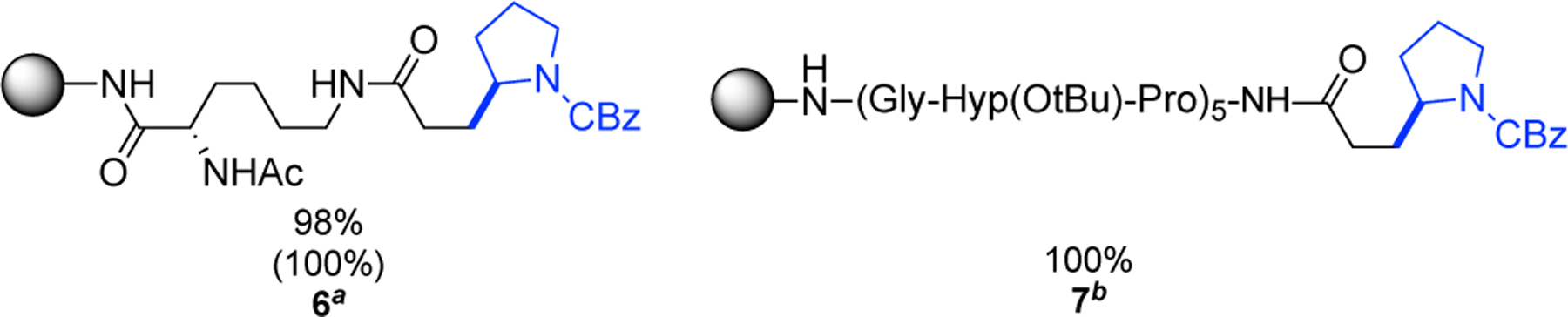
aYield determined by quantitative 1H NMR; conversions determined by quantitative 1H NMR and reported in parenthesis. bConversion determined by HPLC.
In summary, the development of the photochemical hydroalkylation of electron-deficient olefins has been reported in the solid phase. These conditions offer mild, expeditious, and operationally simple routes for introducing C(sp3)-C(sp3) bonds to peptides on resin. They also represent the first example of a light-mediated peptide modification in the solid-phase and demonstrate the potential for further elaboration of polypeptides on solid support.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful for the financial support provided by NIGMS (R35 GM 131680) and NSF (CHE-1952583) to G.M., NIH supplement awards 3R01GM118510–03S1 and 3R01GM087605–06S1, and Vagelos Institute for Energy Science and Technology for supporting the purchase of the NMRs used in this study. M.A.E. is grateful for the NIH research supplement award. We thank Dr. Charles W. Ross, III (University of Pennsylvania) for obtaining HRMS data.
Footnotes
The authors declare no competing financial interest.
Contributor Information
Mahmoud Elkhalifa, Roy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6323, United States.
Michael B. Elbaum, Roy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6323, United States.
David M. Chenoweth, Roy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6323, United States
Gary A. Molander, Roy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6323, United States
REFERENCES
- (1).Lau JL; Dunn MK Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorganic Med. Chem 2018, 26, 2700–2707. 10.1016/j.bmc.2017.06.052. [DOI] [PubMed] [Google Scholar]
- (2).Deshpande MS Formation of Carbon-Carbon Bond on Solid Support: Application of the Stille Reaction. Tetrahedron Lett 1994, 35, 5613–5614. 10.1016/S0040-4039(00)77260-1. [DOI] [Google Scholar]
- (3).Yu KL; Deshpande MS; Vyas DM Heck Reactions in Solid Phase Synthesis. Tetrahedron Lett 1994, 35, 8919–8922. 10.1016/0040-4039(94)88389-0. [DOI] [Google Scholar]
- (4).Frenette R; Friesen RW Biaryl Synthesis via Suzuki Coupling on a Solid Support. Tetrahedron Lett 1994, 35, 9177–9180. 10.1016/0040-4039(94)88458-7. [DOI] [Google Scholar]
- (5).Young JK; Nelson JC; Moore JS Synthesis of Sequence Specific Phenylacetylene Oligomers on an Insoluble Solid Support. J. Am. Chem. Soc 1994, 116, 10841–10842. 10.1021/ja00102a082. [DOI] [Google Scholar]
- (6).Schuster M; Pernerstorfer J; Blechert S Ruthenium-Catalyzed Metathesis of Polymer-Bound Olefins. Angew. Chemie Int. Ed. English 1996, 35, 1979–1980. 10.1002/anie.199619791. [DOI] [Google Scholar]
- (7).Miller SJ; Blackwell HE; Grubbs RH Application of Ring-Closing Metathesis to the Synthesis of Rigidified Amino Acids and Peptides. J. Am. Chem. Soc 1996, 118, 9606–9614. 10.1021/ja961626l. [DOI] [Google Scholar]
- (8).Walensky LD; Bird GH Hydrocarbon-Stapled Peptides: Principles, Practice, and Progress. J. Med. Chem 2014, 57, 6275–6288. 10.1021/jm4011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Qin T; Cornella J; Li C; Malins LR; Edwards JT; Kawamura S; Maxwell BD; Eastgate MD; Baran PS A General Alkyl-Alkyl Cross-Coupling Enabled by Redox-Active Esters and Alkylzinc Reagents. Science 2016, 352 (6287), 801–805. 10.1126/science.aaf6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Qin T; Malins LR; Edwards JT; Merchant RR; Novak AJE; Zhong JZ; Mills RB; Yan M; Yuan C; Eastgate MD; Baran PS Nickel-Catalyzed Barton Decarboxylation and Giese Reactions: A Practical Take on Classic Transforms. Angew. Chemie 2017, 129, 266–271. 10.1002/ange.201609662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Liu JQ; Shatskiy A; Matsuura BS; Kärkäs MD Recent Advances in Photoredox Catalysis Enabled Functionalization of α-Amino Acids and Peptides: Concepts, Strategies and Mechanisms. Synth 2019, 51, 2759–2791. 10.1055/s-0037-1611852. [DOI] [Google Scholar]
- (12).Bottecchia C; Noël T Photocatalytic Modification of Amino Acids, Peptides, and Proteins. Chem. - A Eur. J 2019, 25, 26–42. 10.1002/chem.201803074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Su Y; Kuijpers KPL; König N; Shang M; Hessel V; Noël T A Mechanistic Investigation of the Visible-Light Photocatalytic Trifluoromethylation of Heterocycles Using CF3I in Flow. Chem. - A Eur. J 2016, 22, 12295–12300. 10.1002/chem.201602596. [DOI] [PubMed] [Google Scholar]
- (14).Badir SO; Molander GA Developments in Photoredox/Nickel Dual-Catalyzed 1,2-Difunctionalizations. Chem 2020, 6, 1327–1339. 10.1016/j.chempr.2020.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Okada K; Okamoto K; Morita N; Okubo K; Oda M Photosensitized Decarboxylative Michael Addition through N-(Acyloxy)Phthalimides via an Electron-Transfer Mechanism. J. Am. Chem. Soc 1991, 113, 9401–9402. 10.1021/ja00024a074. [DOI] [Google Scholar]
- (16).Pratsch G; Lackner GL; Overman LE Constructing Quaternary Carbons from N-(Acyloxy)Phthalimide Precursors of Tertiary Radicals Using Visible-Light Photocatalysis. J. Org. Chem 2015, 80, 6025–6036. 10.1021/acs.joc.5b00795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kelly CB; Patel NR; Primer DN; Jouffroy M; Tellis JC; Molander GA Preparation of Visible-Light-Activated Metal Complexes and Their Use in Photoredox/Nickel Dual Catalysis. Nat. Protoc 2017, 12, 472–492. 10.1038/nprot.2016.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lackner GL; Quasdorf KW; Overman LE Direct Construction of Quaternary Carbons from Tertiary Alcohols via Photoredox-Catalyzed Fragmentation of Tert-Alkyl N-Phthalimidoyl Oxalates. J. Am. Chem. Soc 2013, 135, 15342–15345. 10.1021/ja408971t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zheng C; Wang GZ; Shang R Catalyst-Free Decarboxylation and Decarboxylative Giese Additions of Alkyl Carboxylates through Photoactivation of Electron Donor-Acceptor Complex. Adv. Synth. Catal 2019, 361, 4500–4505. 10.1002/adsc.201900803. [DOI] [Google Scholar]
- (20).Niu P; Li J; Zhang Y; Huo C One-Electron Reduction of Redox-Active Esters to Generate Carbon-Centered Radicals. European J. Org. Chem 2020, 2020, 5801–5814. 10.1002/ejoc.202000525. [DOI] [Google Scholar]
- (21).Crisenza GEM; Mazzarella D; Melchiorre P Synthetic Methods Driven by the Photoactivity of Electron Donor-Acceptor Complexes. J. Am. Chem. Soc 2020, 142, 5461–5476. 10.1021/jacs.0c01416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Dai GL; Lai SZ; Luo Z; Tang ZY Selective Syntheses of Z- Alkenes via Photocatalyzed Decarboxylative Coupling of N-Hydroxyphthalimide Esters with Terminal Arylalkynes. Org. Lett 2019, 21, 2269–2272. 10.1021/acs.orglett.9b00558. [DOI] [PubMed] [Google Scholar]
- (23).Till NA; Smith RT; MacMillan DWC Decarboxylative Hydroalkylation of Alkynes. J. Am. Chem. Soc 2018, 140, 5701–5705. 10.1021/jacs.8b02834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Delgadillo RF; Mueser TC; Zaleta-Rivera K; Carnes KA; González-Valdez J; Parkhurst LJ Detailed Characterization of the Solution Kinetics and Thermodynamics of Biotin, Biocytin and HABA Binding to Avidin and Streptavidin. PLoS One 2019, 14, e0204194. 10.1371/journal.pone.0204194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Awale S; Okada T; Dibwe DF; Maruyama T; Takahara S; Okada T; Endo S; Toyooka N Design and Synthesis of Functionalized Coumarins as Potential Anti-Austerity Agents That Eliminates Cancer Cells’ Tolerance to Nutrition Starvation. Bioorganic Med. Chem. Lett 2019, 29, 1779–1784. 10.1016/j.bmcl.2019.05.010. [DOI] [PubMed] [Google Scholar]
- (26).Sarker SD; Nahar L Progress in the Chemistry of Naturally Occurring Coumarins; 2017; Vol. 106. 10.1007/978-3-319-59542-9_3. [DOI] [PubMed] [Google Scholar]
- (27).Gimenez D; Phelan A; Murphy CD; Cobb SL 19F NMR as a Tool in Chemical Biology. Beilstein J. Org. Chem 2021, 17, 293–318. 10.3762/BJOC.17.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chrominski M; Baranowski MR; Chmielinski S; Kowalska J; Jemielity J Synthesis of Trifluoromethylated Purine Ribonucleotides and Their Evaluation as 19F NMR Probes. J. Org. Chem 2020, 85, 3440–3453. 10.1021/acs.joc.9b03198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.