

SUMMARY
DOT1L methylates histone H3 lysine 79 during transcriptional elongation and is stimulated by ubiquitylation of histone H2B lysine 120 (H2BK120ub) in a classical trans-histone crosstalk pathway. Aberrant genomic localization of DOT1L is implicated in mixed lineage leukemia (MLL)-rearranged leukemias, an aggressive subset of leukemias that lacks effective targeted treatments. Despite recent atomic structures of DOT1L in complex with H2BK120ub nucleosomes, fundamental questions remain as to how DOT1L-ubiquitin and DOT1L-nucleosome acidic patch interactions observed in these structures contribute to nucleosome binding and methylation by DOT1L. Here, we combine bulk and single-molecule biophysical measurements with cancer cell biology to show that ubiquitin and cofactor binding drive conformational changes to stimulate DOT1L activity. Using structure-guided mutations, we demonstrate that ubiquitin and nucleosome acidic patch binding by DOT1L are required for cell proliferation in the MV4; 11 leukemia model, providing proof of principle for MLL targeted therapeutic strategies.
Graphical abstract
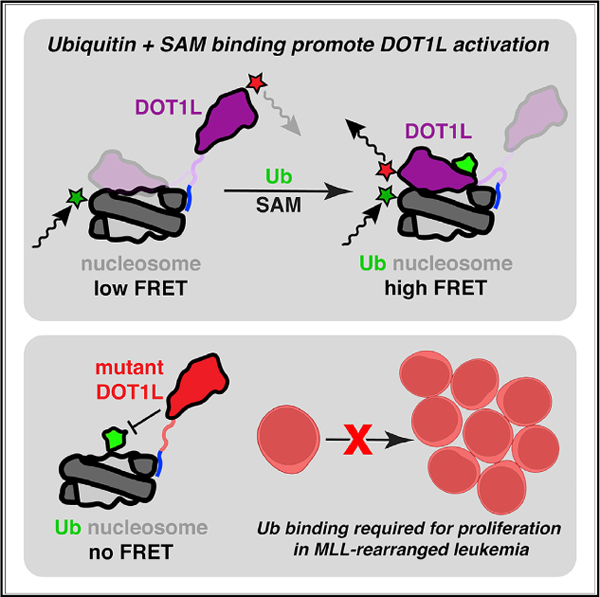
In brief
DOT1L is a histone lysine methyltransferase that is stimulated by H2BK120 ubiquitylation and is central to the pathogenesis of leukemia. Here, Spangler et al. show how ubiquitin and cofactor binding by DOT1L accelerate conformational changes to promote H3K79 methylation. Disruption of the DOT1L-ubiquitin interaction blocks cell proliferation in MLL-rearranged leukemia.
INTRODUCTION
Histone lysine methyltransferase DOT1L (disruptor of telomeric silencing 1-like) is the sole depositor of mono-, di-, and trimethylation on histone H3 lysine 79 (H3K79me1, me2, me3) across the human genome (Feng et al., 2002; Jones et al., 2008). H3K79 methylation is predominantly observed in gene bodies, and levels are highly correlated with transcriptional activity (Wood et al., 2018). DOT1L has emerged as a promising therapeutic target in mixed lineage leukemia (MLL)-rearranged leukemias, an aggressive subset of leukemias with particularly poor prognosis and limited treatment options (Slany, 2016). In MLL-rearranged leukemias, translocation of the MLL1/KMT2A gene with one of many transcriptional elongation factors leads to DOT1L mislocalization to and H3K79 hypermethylation of hematopoietic developmental regulatory genes, including homeobox (HOX) genes (Milne et al., 2005; Okada et al., 2005; Slany, 2016; Zhao et al., 2021). Aberrant activation of these genes is oncogenic and dependent on DOT1L catalytic activity.
The H3K79 methyltransferase activity of DOT1L is directly stimulated by intranucleosomal histone H2B lysine 120 ubiquitylation (H2BK120ub) in a classical histone crosstalk pathway (McGinty et al., 2008). Much like H3K79 methylation, H2BK120ub is present at HOX genes targeted by DOT1L in leukemia, and the H2BK120-targeting ubiquitin E3 ligase is required for MLL-fusion-induced leukemogenesis (Wang et al., 2013; Zhu et al., 2005). Recently, our group and several other groups used cryoelectron microscopy (cryo-EM) to solve the structure of the DOT1L catalytic domain bound to a site-specifically ubiquitylated nucleosome (Anderson et al., 2019; Jang et al., 2019; Valencia-Sánchez et al., 2019; Worden et al., 2019; Yao et al., 2019). Collectively, these structures and accompanying biochemical studies revealed a set of multivalent interactions with the H2BK120ub nucleosome that position DOT1L for H3K79 methylation. DOT1L binds to the H2A-H2B acidic patch on the nucleosome disk surface using a loop containing two arginines, R282 and R278, and simultaneously engages ubiquitin through a hydrophobic interaction requiring DOT1L I290 and F326 (Anderson et al., 2019; Jang et al., 2019; Valencia-Sánchez et al., 2019; Worden et al., 2019; Yao et al., 2019) (Figure 1A). DOT1L also binds to a basic sequence in the H4 tail with an acidic cleft adjacent to its active site (Worden et al., 2019). Finally, though not visualized in cryo-EM reconstructions, a lysine-rich region (LRR) C-terminal to the DOT1L catalytic domain has been shown to be necessary for nucleosome binding and methylation, presumably through non-specific anchoring to nucleosomal DNA (Min et al., 2003) (Figure 1B).
Figure 1. Bulk FRET measurements demonstrate modest contribution by DOT1L-nucleosome acidic patch interaction.
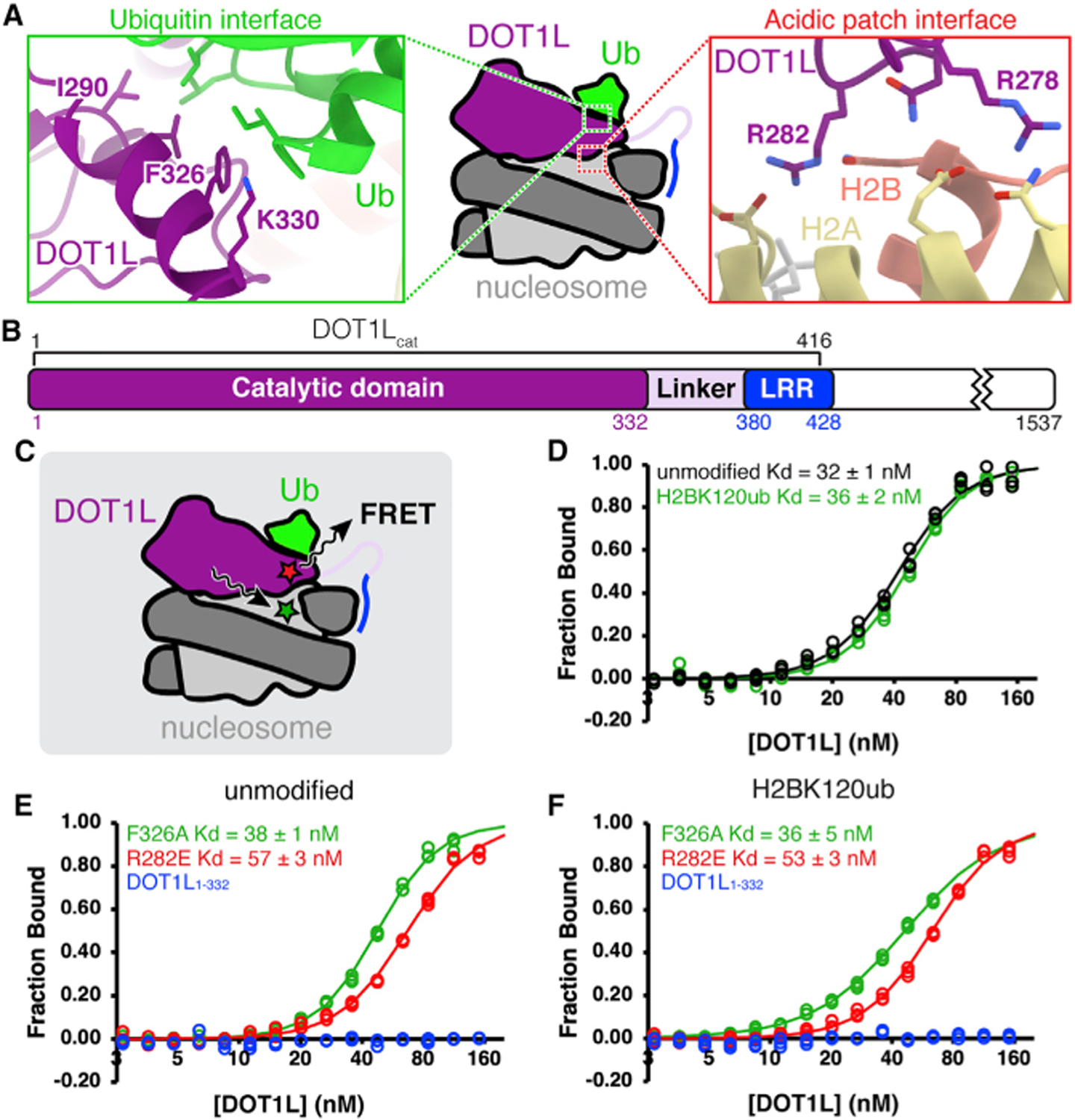
(A) DOT1L-H2BK120ub nucleosome interaction interfaces (PDB: 6NN6). Ub = ubiquitin.
(B) Protein domain map of human DOT1L, with DOT1Lcat region used for binding assays denoted. LRR = lysine-rich region.
(C) Experimental setup for bulk FRET binding assays. FRET between Alexa Fluor 488-labeled nucleosomes (green star) and Atto647N-labeled DOT1L (red star) used to quantitate DOT1L-nucleosome binding affinity.
(D) Normalized binding curves and fits for wild-type DOT1Lcat binding to unmodified or H2BK120ub nucleosomes.
(E and F) Normalized binding curves and fits for mutants of DOT1Lcat or DOT1L1–332 binding to (E) unmodified or (F) H2BK120ub nucleosomes. All nucleosomes contain an H3K79Nle substitution. Individual data points are shown for three independent titrations.
See also Figure S1.
Cryo-EM structures of the DOT1L catalytic domain bound to an H2BK120ub nucleosome containing the native H3K79 target residue revealed conformational heterogeneity of DOT1L relative to the nucleosome, hinging from the points of contact between DOT1L and the acidic patch and ubiquitin (Anderson et al., 2019). Near the hinge, molecular details of the DOT1L-acidic patch and DOT1L-ubiquitin interfaces were clearly visualized. However, the N-terminal region of DOT1L containing the catalytic site located farther from the hinge exhibited more heterogeneous conformations and incomplete engagement of H3K79. We and others interpreted this binding mode as a pre- or post-catalytic state that is ‘‘poised’’ for catalysis (Anderson et al., 2019; Jang et al., 2019; Worden et al., 2019). An ‘‘active’’ conformation was stabilized by mutation of the DOT1L target lysine residue to a norleucine (H3K79Nle) (Worden et al., 2019). In this conformation, the H3K79Nle loop flips out from the nucleosome surface, makes contacts with basic residues in the H4 tail, and positions the norleucine in the catalytic pocket of DOT1L. Several groups also determined structures of DOT1L bound to unmodified nucleosomes in which DOT1L is positioned similarly, but its density is very poorly resolved (Jang et al., 2019; Valencia-Sánchez et al., 2019; Yao et al., 2019). This suggests more conformational heterogeneity in DOT1L when ubiquitin is not present, even in cross-linked complexes.
Despite visualization of DOT1L bound to both unmodified and H2BK120ub nucleosomes, many questions remain with regard to how multivalent interactions between DOT1L and the nucleosome coordinate productive substrate recognition and what triggers the DOT1L poised-to-active transition. Moreover, it is unknown how DOT1L-acidic patch and DOT1L-ubiquitin interfaces contribute to pathogenesis of MLL-rearranged leukemias. As resistance mechanisms of DOT1L catalytic inhibitors currently in clinical trials have been observed (Campbell et al., 2017), dissecting DOT1L-nucleosome multivalency in MLL-rearranged leukemias is critical to identify alternative strategies for DOT1L inhibition. Here, we used structure-guided mutations as precision tools to disrupt individual DOT1L-nucleosome interfaces across biological scales. Using bulk and single-molecule binding experiments, we showed that general DOT1L-nucleosome localization is driven by electrostatic interactions with an LRR of DOT1L. However, both the ubiquitin- and acidic patch-binding surfaces of DOT1L are essential for stable, local acidic patch engagement. We identified a multistate binding pathway for the DOT1L-nucleosome complex in which DOT1L cofactor S-adenosyl methionine (SAM) accelerates a conformational transition to more productive DOT1L binding modes. Finally, we showed in an MLL-rearranged leukemia cell model that disruption of ubiquitin-, acidic patch-, or SAM-binding surfaces of DOT1L leads to severe loss of H3K79 methylation and decreased cell proliferation. Together, these data suggested a mechanistic model for DOT1L-nucleosome binding and defined the dependence of DOT1L activity and leukemia cell proliferation on two key interfaces between DOT1L and H2BK120ub nucleosomes.
RESULTS
Acidic patch binding contributes only modestly to DOT1L-nucleosome affinity
Previous biochemical studies present conflicting data with respect to the importance of DOT1L-acidic patch and DOT1L-ubiquitin interactions for nucleosome binding and methylation. On one hand, mutation of the critical acidic patch-binding arginine in DOT1L, R282, alone or together with R278, nearly abolishes H3K79 methylation of both H2BK120ub and unmodified nucleosomes (Anderson et al., 2019; Jang et al., 2019; Valencia-Sánchez et al., 2019; Worden et al., 2019; Yao et al., 2019). On the other hand, semi-quantitative electrophoretic mobility shift assays (EMSAs) show equivalent nucleosome-binding affinities for wild-type or R278E/R282E mutant DOT1L (Jang et al., 2019; Valencia-Sánchez et al., 2019). Moreover, ubiquitylation of H2BK120 stimulates DOT1L methylation of nucleosomes at least 6-fold compared with unmodified H2B (Anderson et al., 2019; McGinty et al., 2008). Mutation of DOT1L F326 at the DOT1L-ubiquitin interface decreases DOT1L-mediated methylation of H2BK120ub nucleosomes to levels comparable to those observed on unmodified nucleosomes while having no significant effect on DOT1L activity toward unmodified nucleosomes (Anderson et al., 2019; Jang et al., 2019; Valencia-Sánchez et al., 2019; Worden et al., 2019; Yao et al., 2019). However, in EMSAs, wild-type and F326A mutant DOT1L bind to unmodified and ubiquitylated nucleosomes all with similar affinities (Jang et al., 2019; Valencia-Sánchez et al., 2019).
To better understand the molecular mechanisms governing how DOT1L engages a nucleosome substrate, we established in-solution fluorescence assays for both global and local DOT1L-nucleosome binding. First, we designed a bulk fluorescence resonance energy transfer (FRET) assay to measure the nucleosome affinity of DOT1L and structure-guided DOT1L mutants in solution (Figure 1C). In selecting positions for site-specific labeling of the nucleosome and DOT1L with a FRET donor and acceptor, respectively, we wanted the positions of the fluorophores to be in close proximity to each other to allow for efficient FRET, but far enough from the DOT1L-nucleosome interface to not interfere with binding. Given the minimal conformational heterogeneity that we observed near the DOT1L-acidic patch interface in our cryo-EM data, we selected DOT1L N324 and histone H2A L65 for fluorophore attachment; these positions are near the acidic patch interface, but on the opposite side of DOT1L from the ubiquitin binding surface, are solvent exposed in the complex structure, and are approximately 20 Å apart from each other (Figures 1C and S1A). Since the first 416 residues of DOT1L (DOT1Lcat) are sufficient for nucleosome binding and ubiq-uitin-dependent H3K79 methylation, we used this truncation for our binding studies. We prepared DOT1Lcat fluorescently labeled with Atto647N (acceptor) at the N324 position and a nucleosome fluorescently labeled with Alexa Fluor 488 (donor) at the L65 position of H2A. As we previously showed that DOT1Lcat purifies bound to SAM, we used H3K79Nle nucleosomes that lack the side chain amino group of lysine, thereby preventing enzymatic turnover that could have confounding effects on FRET measurements (Figures S2A and S2B).
Using our FRET assay, we first measured DOT1Lcat affinity for unmodified and H2BK120ub nucleosomes to be equivalent at 32–36 nM, suggesting that H2BK120 ubiquitylation does not contribute to nucleosome binding affinity in solution, similar to previous observations using EMSAs (Figure 1D). We next examined the consequences of DOT1L mutations at the acidic patch and ubiquitin binding surfaces. The DOT1L F326A mutation that prevents ubiquitin interaction had no significant effect on DOT1L binding to either unmodified or H2BK120ub nucleosomes (Figures 1E and 1F). However, we observed a 1.5-fold decrease in nucleosome binding affinity for the R282E charge swap DOT1L mutant at the acidic patch interface regardless of the ubiquitylation status of the nucleosome (Figures 1E and 1F). Similar to previously reported EMSAs, we detected no nucleosome binding for the DOT1L1–332 truncation that includes only the catalytic domain and lacks the LRR (Figures 1E and 1F).
Owing to pseudosymmetry of the nucleosome, there are two DOT1L binding sites per nucleosome. Previously, EMSAs of DOT1L-nucleosome complexes showed three or more shifted nucleosome bands, likely representing the expected 1:1 and 2:1 DOT1L-nucleosome complexes but also 3:1 and higher, super-stoichiometric complexes (Jang et al., 2019; Valencia-Sánchez et al., 2019). We envisioned that using a FRET assay that is inherently distance sensitive would allow us to monitor acidic patch-engaged complexes over non-specifically bound complexes that would place the FRET fluorophores at distances larger than 20 Å . However, we observed similar unnormalized, corrected FRET intensities for wild-type and R282E DOT1L titrations, the latter of which should be unable to bind the acidic patch. This suggests that nucleosome binding is likely dominated by LRR-DNA interactions and that these interactions alone localize DOT1L close enough to produce FRET in our assay. We hypothesized that these non-specific interactions are not productive for methylation but enable more transient productive binding modes at the acidic patch surface that orient DOT1L into a poised conformation. As such, mutation of the acidic patch interface has limited contributions to overall DOT1L-nucleosome affinity.
DOT1L acidic patch binding is dependent on ubiquitin interaction
To deconvolute specific binding of DOT1L to the acidic patch on the nucleosome disk face from non-specific complexes formed by LRR-DNA interactions alone, we needed a complementary position-sensitive binding assay. For this purpose, we adapted a fluorescence (de)quenching assay that detects local environmental changes surrounding a fluorophore owing to complex formation (Winkler et al., 2012). Scouting of positions for fluorophore conjugation on the nucleosome demonstrated that H2A L65C, when conjugated with Alexa Fluor 488 as used for our bulk FRET studies, exhibited maximal dequenching with DOT1L titration into H2BK120ub nucleosomes, allowing apparent binding affinities for acidic patch binding to be directly measured (Figures 2A and S1B).
Figure 2. DOT1L-H2BK120ub nucleosome acidic patch interaction is dependent on ubiquitin binding by DOT1L.
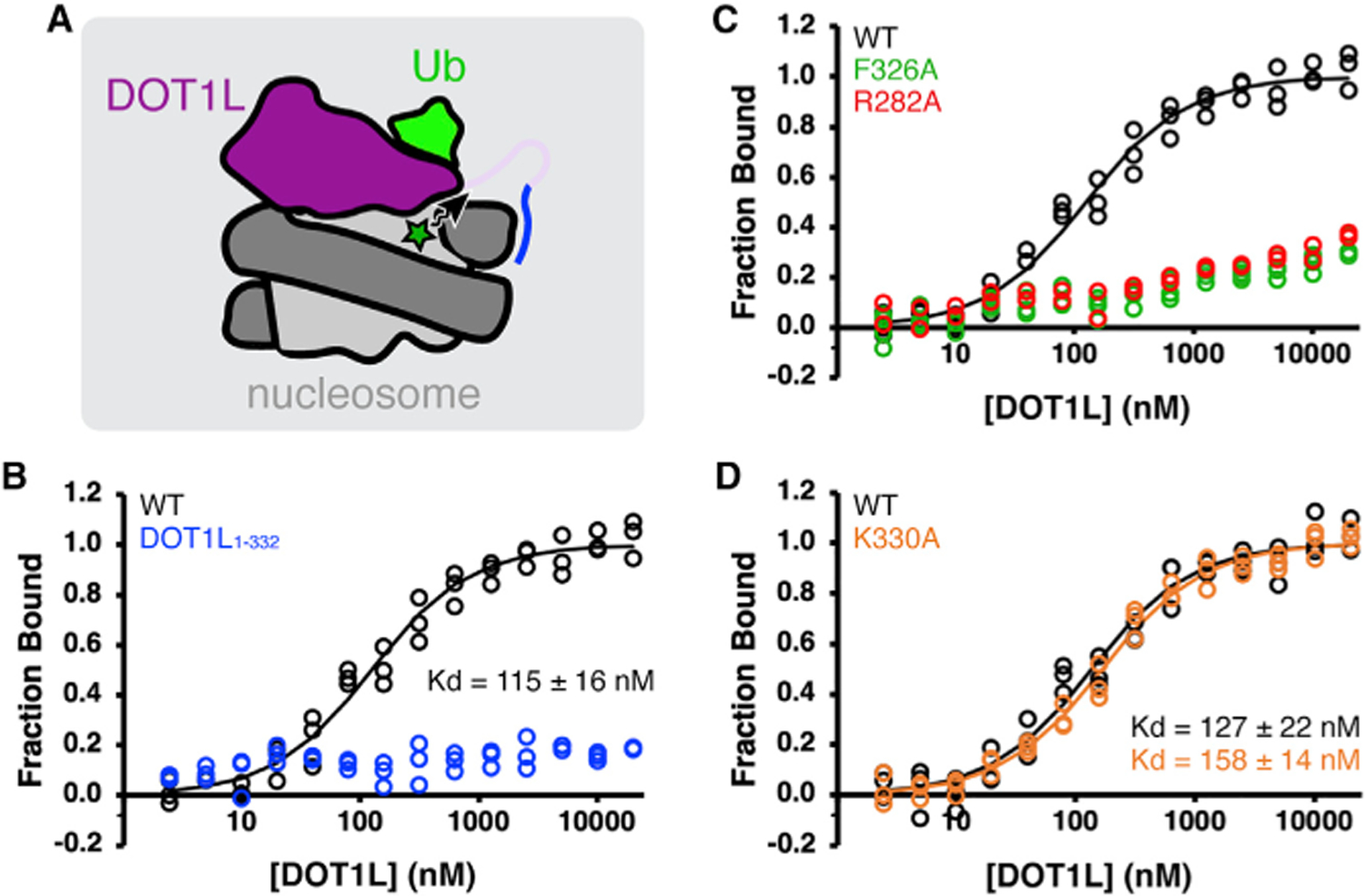
(A) Experimental setup for fluorescence (de) quenching binding assays. DOT1L binding in vicinity of Alexa Fluor 488 (green star) induces a change in fluorescence used to quantitate local DOT1L binding to nucleosome acidic patch. Ub = ubiquitin.
(B–D) Normalized binding curves and fits for interaction between H2BK120ub nucleosomes and (B) wild-type (WT) DOT1Lcat and DOT1L1–332, (C)DOT1L F326A and R282A, or (D) DOT1L K330A.
All H2BK120ub nucleosomes contain an H3K79Nle substitution. Individual data points are shown for three independent titrations.
See also Figures S1 and S2.
DOT1Lcat binds to the acidic patch of H2BK120ub nucleosomes with about a 3-fold lower affinity (~120 nM) in our (de) quenching assay compared with the global nucleosome binding affinity measured by our FRET assay (Figure 2B). Nucleosome acidic patch binding by DOT1L is dependent on the LRR, as DOT1L1–332-acidic patch binding was undetectable up to 10 µM (Figure 2B). Importantly, DOT1L R282A, which lacks the acidic patch binding arginine that is critical for H3K79 methylation, is also unable to bind to the H2BK120ub nucleosome acidic patch (Figure 2C) despite the more severe charge swap mutant protein at the same position binding to nucleosomes at 53 nM affinity in bulk FRET assays (Figures 1E and 1F). We were also unable to detect acidic patch binding by DOT1L with an F326A mutation, which disrupts the DOT1L-ubiquitin interaction (Figure 2C). This result suggests that when H2BK120 is ubiquitylated, ubiquitin engagement by DOT1L is required for acidic patch binding. To rule out the possibility that all mutations in this area of DOT1L preclude detection of acidic patch binding, we prepared and tested a DOT1L K330A mutant in the (de) quenching assay. The DOT1L K330 residue is near the ubiquitin interface, and we previously determined this mutation to have no effect on catalytic activity (Anderson et al., 2019). Indeed, DOT1L K330A binds the acidic patch of H2BK120ub nucleosomes with near wild-type affinity (Figure 2D).
We next attempted to measure acidic patch binding to unmodified nucleosomes. Although fluorescence dequenching was observed for DOT1L binding to H2BK120ub nucleosomes, we observed modest fluorescence quenching on unmodified nucleosomes (Figure S2A). The fluorescence quenching appeared to be non-specific, as neither R282A nor F326A mutations influenced the observed fluorescence change. As structures of the DOT1L-unmodified nucleosome complex have been solved by cryo-EM, we expect that DOT1L can engage the acidic patch in the absence of H2BK120 ubiquitylation, even if less stably. It is possible that H2BK120ub nucleosomes in the unbound state have a different environment surrounding the fluorophore owing to nearby attachment of ubiquitin or that the DOT1L-acidic patch interaction with unmodified nucleosomes is too transient to be detected in this assay. Notably, these fluorescence (de) quenching observations were consistent when measuring DOT1L binding to either native H3K79 or H3K79Nle nucleosomes in both H2BK120ub and unmodified H2B contexts (Figures S2A and S2B).
SAM binding promotes conformational rearrangement of DOT1L-nucleosome complex
Our bulk fluorescence measurements highlighted the importance of the DOT1L-ubiquitin interaction for DOT1L binding to the H2BK120ub nucleosome acidic patch. However, these experiments were blind to the higher-resolution conformational changes, such as the transformation from an acidic patch engaged, poised conformation, observed in most cryo-EM structures of the DOT1L-nucleosome complex, to an active conformation required for methylation. Therefore, we established a single-molecule FRET (smFRET) system to observe the conformational states explored by individual DOT1L molecules upon acidic patch binding. Rather than conjugating fluorophores in conformationally homogeneous locations near the acidic patch as we did for bulk FRET measurements, we opted for positions on the opposite side of DOT1L and the nucleosome that would be more sensitive to conformational transitions inferred from cryo-EM data (Figure 3A). We labeled nucleosomal DNA with Cy3 (donor) on the thymidine base at a position +15 bp from the nucleosome dyad. This location is near H3K79 and underneath the N-terminal region of the DOT1L catalytic domain in the complex. We then labeled DOT1Lcat with At-to647N (acceptor) at E69, which is located in an a-helix at the DOT1L N-terminus that is stably positioned within DOT1L. These positions maximize the FRET signal sensitivity to DOT1L conformational changes (Figure S1C). According to the distances between these labeling positions in the poised and active states (47 Å versus 41 Å based on Cα to N3 distance measurement using poised [PDB: 6NOG] and active [PDB: 6NJ9] structures, respectively), the actively bound state would result in a significantly higher FRET efficiency than the poised state. Using the Förster radius between Cy3 and Atto647N, the estimated FRET efficiencies calculated with the standard equation (Wu and Brand, 1994) are 0.5 and 0.7 for the poised and active states, respectively. Therefore, we anticipated that the highest FRET efficiency state would correspond to the active conformation, and mid-range or lower FRET efficiencies would correspond to the poised and other less tightly bound states. With nucleosomes containing native H3K79, DOT1L-H2BK120ub nucleosome binding events could not be detected in our single-molecule FRET experiments, likely occurring within a time window too early for the experimental setup to detect (data not shown). However, when using H2BK120ub nucleosomes containing H3K79Nle, which prevents DOT1L enzymatic turnover, we observed binding events with a broad distribution of FRET efficiency values (Figures 3A and 3B).
Figure 3. DOT1L lysine-rich region, ubiquitin- and acidic patch-binding, and SAM stabilize higher FRET DOT1L-nucleosome conformations in single-molecule studies.
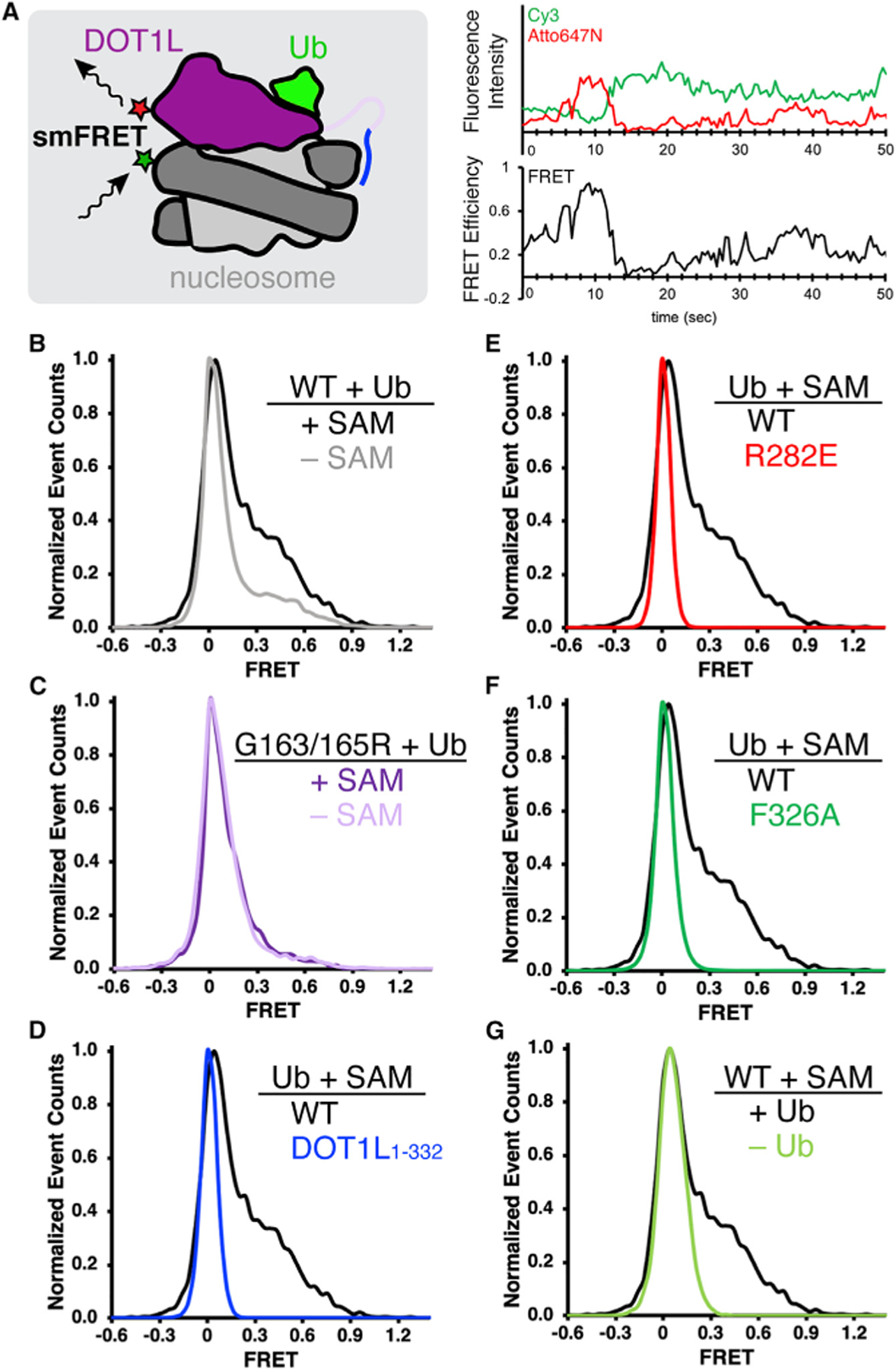
(A) Experimental setup for single-molecule FRET (smFRET) measurements (left), where FRET between Cy3-labeled nucleosomes (green star) and Atto647N-labeled DOT1L (red star) detects DOT1L-nucleosome binding conformation. An example of fluorescence intensity and smFRET time trajectories shows dynamic DOT1L-nucleosome binding. Ub = ubiquitin.
(B–F) (B and C) Normalized histograms comparing DOT1L-H2BK120ub nucleosome interactions with and without SAM, using (B) wild-type (WT) DOT1Lcat (n = 161 and 141 smFRET time trajectories for with and without SAM, respectively) or (C) SAM-binding deficient G163/165R DOT1L (n = 123 and 88 for with and without SAM, respectively), or (D–F) Normalized histograms comparing wild-type DOT1L-H2BK120ub interactions in the presence of SAM with (D) DOT1L1–332 (n = 868), (E) DOT1L R282E acidic patch interface mutant (n = 3,348), or (F) DOT1L ubiquitin interface F326A mutant (n = 2,882).
(G) Normalized histogram comparing wild-type DOT1L binding to H2BK120ub or unmodified nucleosomes (n = 1,251) in the presence of SAM.
See also Figures S1 and S3.
With an smFRET system of the DOT1L-nucleosome complex established, we made a series of targeted perturbations to the system to better understand DOT1L conformational dynamics and to assign the observed FRET efficiencies to functional states. First, we wanted to determine whether SAM, the cofactor that serves as the methyl group donor for DOT1L, impacted DOT1L-nucleosome binding or conformational state. We observed more high FRET binding events of DOT1Lcat to H2BK120ub nucleosomes in the presence of 40 µM SAM, indicating that the SAM cofactor plays a role in enhancing nucleosome binding and/or stabilizing the higher FRET efficiency binding modes of DOT1L (Figure 3B). Importantly, a double G163R and G165R (G163/ 165R) mutant of DOT1L that is unable to bind SAM showed no increase in higher FRET efficiency events upon SAM addition, providing further evidence of SAM-specific effects (Figure 3C). We next tested the DOT1L1–332 truncation that is unable to bind H2BK120ub nucleosomes in bulk assays and observed only a tight distribution of very low FRET events even in the presence of SAM (Figure 3D). Based on our bulk FRET experiments, we conclude that these very low FRET events primarily reflect spatially overlapped, non-binding background signal. Similar tight distributions of very low FRET events were detected for H2BK120ub nucleosome binding by R282A and F326A mutant versions of DOT1L, which prevent acidic patch and ubiquitin interactions, respectively, in the presence of SAM (Figures 3E and 3F). These mutations do not preclude high-affinity nucleosome binding through DOT1L LRR-DNA interactions, but do block acidic patch engagement of H2BK120ub nucleosomes as assessed by fluorescence dequenching. It is therefore likely that these non-specific nucleosomal DNA binding events result in zero or very low FRET signals that cannot be detected in the smFRET measurements.
We next compared the binding of DOT1Lcat to unmodified versus H2BK120ub nucleosomes. Unlike with H2BK120ub nucleosomes, we observed no distribution of higher FRET binding events with wild-type DOT1L and unmodified nucleosomes even in the presence of SAM (Figure 3G). This demonstrates that DOT1L samples higher FRET states only in the presence of H2BK120 ubiquitylation. However, the distribution of lower FRET events was broader for wild-type DOT1L with unmodified nucleosomes than DOT1L R282A or F326A mutants with H2BK120ub nucleosomes (Figures 3E–3G and S3). This likely results from DOT1L binding of the acidic patch of unmodified nucleosomes and suggests that such interactions are disfavored on H2BK120ub nucleosomes in the context of the DOT1L F326A mutation that disrupts the DOT1L-ubiquitin interaction.
Our single-molecule FRET datasets were best fit to a four-state binding model, consisting of zero-, low-, mid-, and high-FRET binding states. The mid- and high-FRET binding states were observed only with wild-type or G163/165R DOT1L binding to ubiquitylated nucleosomes and were detected both in the presence and in the absence of SAM (Figures 4A, 4B, S4A, and S4C). Low-FRET binding states were observed for these as well as for wild-type DOT1L binding to unmodified nucleosomes (Figures 4A–4C and S4A–S4C), and zero-FRET binding states should exist in all experimental setups as explained above (Figures 4A–4D and S4A–S4F). Transitions between FRET states typically proceeded in a stepwise manner, with zero- to low-FRET, low- to mid-FRET, and mid- to high-FRET transitions more common than transitions that skip intermediate states as are evident in the transition heat maps (Figures 4 and S4A). We observed similar trends in transitions from higher to lower FRET states. Consistent with differences in FRET histograms (Figure S3), we observed two separate islands of FRET transitions from zero- to low- FRET and from low- back to zero-FRET states for wild-type DOT1L on unmodified nucleosomes (Figure 4C). In contrast, only one main peak centered on zero-FRET was observed for DOT1L(R282E) and DOT1L(F326A) on H2K120ub nucleosomes (Figure 4D and S4E). Hidden Markov modeling allowed for determination of the FRET efficiencies and the rate constants describing transitions between the FRET states for wild-type and G163/165R SAM-binding deficient mutant DOT1L in complex with H2BK120ub nucleosomes with and without SAM. Importantly, the mid- and high-FRET states identified by modeling wild-type DOT1L-H2BK120 nucleosome complexes in the presence of SAM had FRET efficiencies of 0.45 ± 0.02 and 0.77 ± 0.03, respectively, which closely match the theoretical FRET efficiency values predicted based on the poised and active structures of the DOT1L-H2BK120ub nucleosome complex. Notably, we observed a 2-fold increase in the rate constant for the transition from low- to mid-FRET states for wild-type DOT1L in the presence of SAM, accompanied by a decrease in the rate constant for the mid- to high-FRET state transition (Figure 4E). SAM-dependent changes in rate constants were not observed for the G163/165R mutant, which exhibited kinetics similar to wild-type DOT1L in the absence of SAM (Figure 4F).
Figure 4. Four-state DOT1L-nucleosome interaction observed by single-molecule FRET.
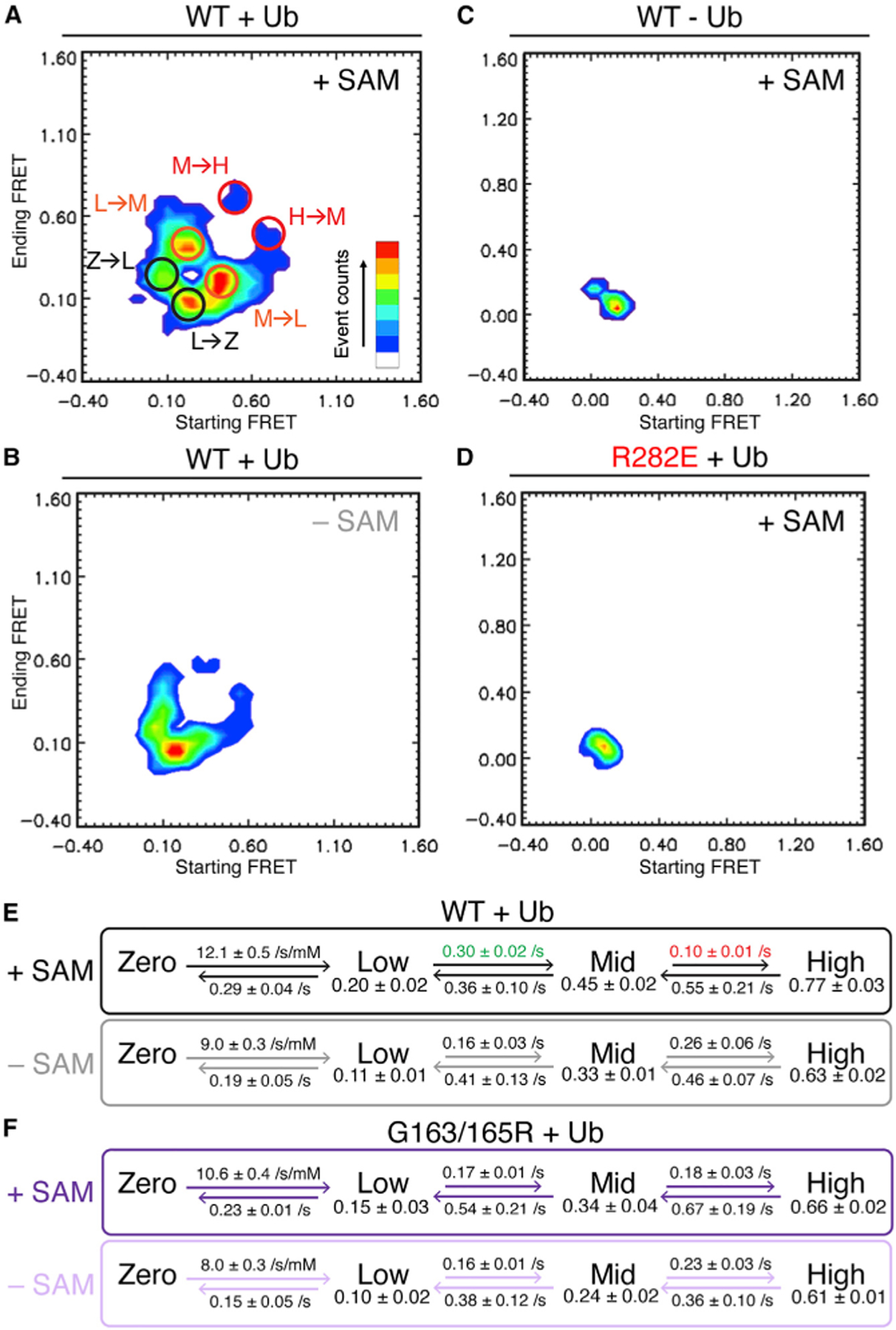
(A) Heatmap displaying starting versus ending FRET of individual events for wild-type (WT) DOT1L with H2BK120ub nucleosomes in the presence of SAM. Circles denote common transitions between zero-FRET (Z), low-FRET (L), mid-FRET (M), and high-FRET (H) states as determined by four-state mechanistic model. Ub = ubiquitin.
(B) Heatmap as in (A) for WT DOT1L-H2BK120ub interaction in the absence of SAM.
(C) Heatmap as in (A) for WT DOT1L-unmodified nucleosome interaction in the presence of SAM.
(D) Heatmap as in (A) for DOT1L R282E-H2BK120ub nucleosome interaction in the presence of SAM.
(E and F) Four-state mechanistic model for FRET values and rate constants for transitions between the zero-, low-, mid-, and high-FRET states for DOT1L binding to H2BK120ub nucleosomes with and without SAM using (E) WT DOT1L or (F) SAM-binding deficient G163/165R DOT1L. Modeled FRET efficiencies listed below FRET states. Indicated errors are standard deviations from multiple independent analyses. The numbers of DOT1L-nucleosome complexes analyzed are {47, 54, 60} and {36, 45, 60}, respectively, for WT DOT1L with and without SAM, and {40, 34, 49} and {39, 49}, respectively, for G163/ 165R DOT1L with and without SAM.
See also Figure S4.
Ubiquitin and acidic patch interfaces in DOT1L are essential for proliferation of MLL-rearranged leukemia cells
With a sophisticated understanding of the consequences of structure-guided mutations of DOT1L in vitro, we were equipped to assess the effects of DOT1L mutations in cells. Based on in vitro bulk and single-molecule binding experiments, we hypothesized that DOT1L mutations that disrupt the ubiquitin or nucleosome acidic patch interactions would interfere with the productive binding of DOT1L to nucleosomes and therefore affect H3K79 methylation in cells. To test this hypothesis, we used MV4; 11 cells, a widely utilized MLL-rearranged (MLL-AF4) acute leukemia model (Daigle et al., 2011). We first established a conditional DOT1L knockout (KO) system with a doxycycline-inducible Cas9 and a stably expressed DOT1L-targeting single guide RNA (sgRNA). In the absence of exogenous DOT1L, induction of DOT1L KO resulted in anticipated decreases of endogenous DOT1L and global H3K79me2 levels in MV4; 11 cells, with the latter barely detected 6 days after KO (Figure 5A left [+ versus – lanes], Figure S5A). Introduction of an exogenous HA-tagged wild-type murine Dot1L (>99% sequence identity between murine Dot1L and human DOT1L in catalytic domain) before KO of endogenous DOT1L efficiently restored bulk H3K79me2 to levels seen in mock-treated cells (Figure 5A, right [+ versus – lanes], Figure S5A), thus validating the rescue with exogenous murine Dot1L. In contrast, expression of exogenous murine Dot1L mutants deficient in acidic patch binding (R282A or R282E), ubiquitin binding (F326A or I290A), or SAM binding (G163/165R) failed to rescue the H3K79me2 loss caused by KO of the endogenous DOT1L, even though the mutants were expressed at similar or higher levels compared with wild-type Dot1L (Figures 5B, S5B, and S5C). Meanwhile, a control mutant Dot1L, which carries a K330A point mutation that is near the ubiquitin binding interface in DOT1L but does not affect nucleosome binding or methylation in vitro, was able to rescue H3K79me2 to wild-type levels (Figure 5B). We next assessed cell proliferation for unrescued and rescued cells over a period of 14 days after induction of DOT1L KO. In the absence of exogenous Dot1L, MV4; 11 cell proliferation dropped sharply over the time period, consistent with the previously reported role for DOT1L in sustaining growth of MLL-rearranged leukemia cells (Chen et al., 2015; Daigle et al., 2011) (Figure 5C). While pre-rescue with wild-type Dot1L or the K330A control mutant largely rescued the proliferation defects caused by DOT1L loss, no rescue was observed by acidic patch-, ubiquitin-, or SAM binding-deficient mutants of Dot1L (Figure 5C), suggesting that DOT1L-acidic patch and DOT1L-ubiquitin binding are absolutely required and equally important for leukemia cell proliferation.
Figure 5. DOT1L-ubiquitin and DOT1L-acidic patch interfaces are both required for DOT1L-mediated H3K79me2 and cell proliferation in MLL-rearranged acute leukemia cells.
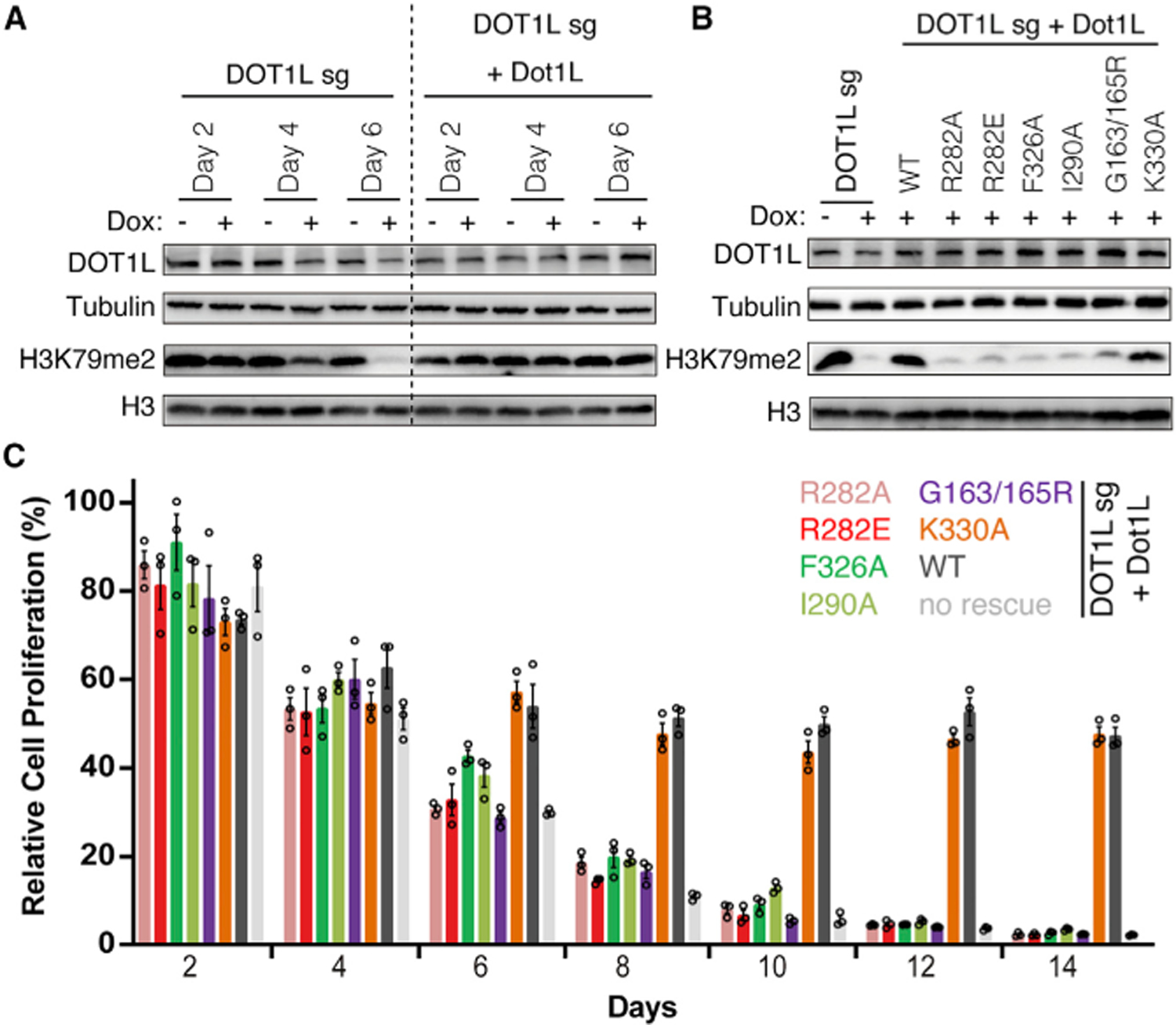
(A) Western blots for the indicated proteins in MV4; 11 cells, for parental lines (left) or those prerescued with wild-type (WT) murine Dot1L (right; this exogenous Dot1L cDNA is resistant to the sgRNA used for targeting human DOT1L [DOT1L sg]), after mock treatment (−) or doxycycline-induced depletion of endogenous DOT1L by CRISPR/Cas9 (+) for 2, 4, or 6 days.
(B) Rescue of the endogenous DOT1L depletion-related H3K79me2 loss by exogenous WT or mutant Dot1L, as indicated. Lanes 1–2 show effect of DOT1L loss in the absence of exogenous Dot1L.
(C) Relative cell proliferation following depletion of endogenous DOT1L in MV4; 11 cells, which were either parental lines (no rescue) or those prerescued with exogenous WT or mutant Dot1L, as indicated. Relative proliferation plotted on y axis after normalizing to the cell number of the culture under the doxycycline-treated condition against cell number without doxycycline treatment and then normalized to day 0 (n = 3 biological replicates; presented as mean ± SE).
See also Figure S5.
We examined expression of three DOT1L-targeted genes (MEIS1, HOXA10, and PBX3) following KO of DOT1L in MV4; 11 cells. The expression levels of MEIS1, HOXA10, and PBX3 all were significantly reduced upon sgRNA-mediated disruption of endogenous DOT1L. This decrease of DOT1L target gene expression was reversed by pre-rescue with wild-type or K330A control mutant Dot1L (Figures 6A–6C). A higher expression of the Dot1L K330A mutant was observed relative to wild-type Dot1L (Figures S5B and S5C), and K330A mutant rescue consistently resulted in higher levels of MEIS1 and HOXA10 expression (Figures 6A–6C). Exogenous Dot1L mutant proteins that are defective in acidic patch, ubiquitin, or SAM binding failed to rescue expression of these DOT1L-targeted genes (Figures 6A–6C). Importantly, wild-type Dot1L and all Dot1L mutants bound tightly to chromatin and could barely be detected in the soluble fraction in cell fractionation experiments (Figure 6D). These data collectively establish the importance of the DOT1L-ubiquitin and DOT1L-acidic patch interactions for H3K79 methylation and expression of known cancer driver genes in leukemia cells, but not chromatin binding.
Figure 6. DOT1L target gene expression is decreased in leukemia cells upon disruption of DOT1L-ubiquitin or DOT1L-acidic patch interfaces despite chromatin association.
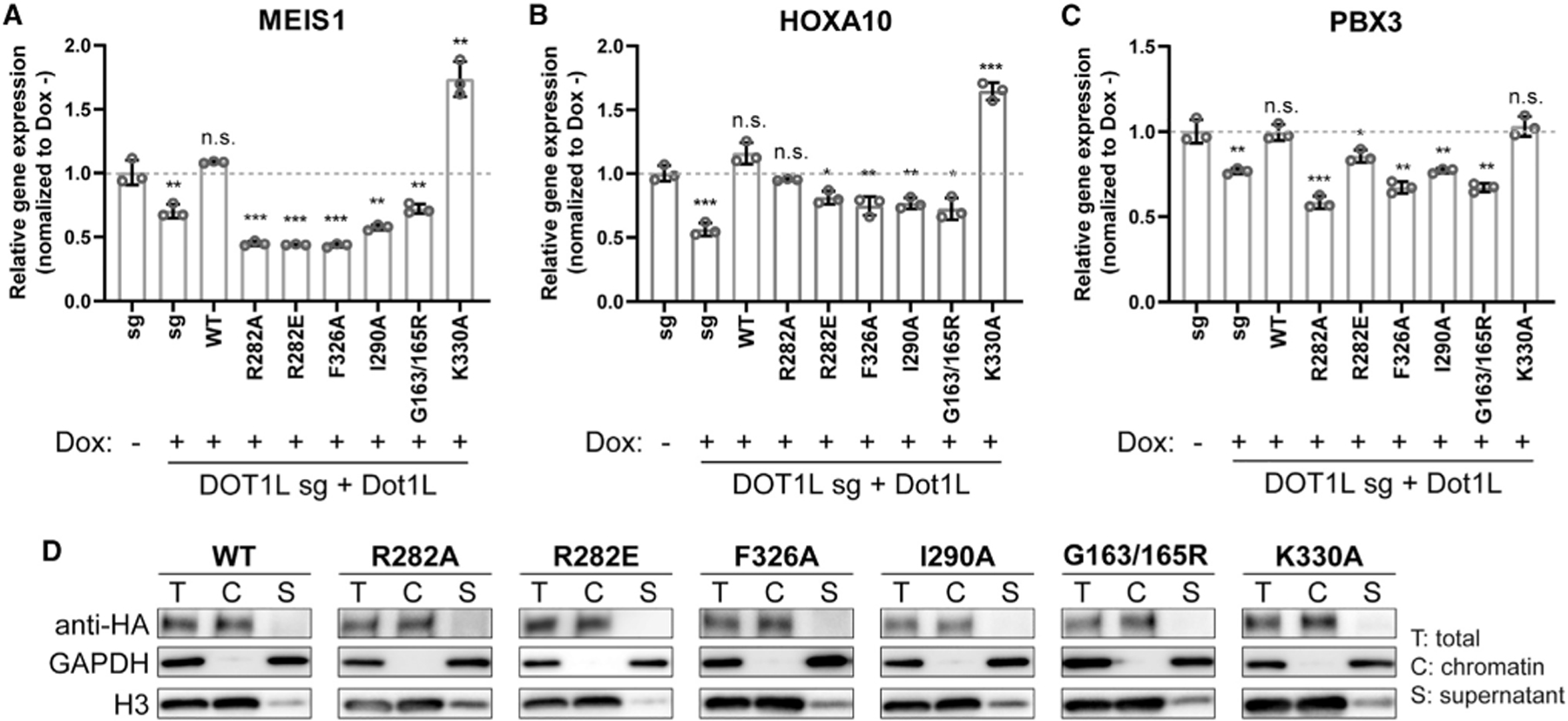
(A–C) Relative MEIS1 (A), HOXA10 (B), and PBX3 (C) expression in MV4; 11 cells, either parental (columns 1–2) or pre-rescued with wild-type (WT) or the indicated mutant Dot1L (columns 3–9), after an 8-day induction of CRISPR/Cas9-mediated depletion of endogenous DOT1L. Relative expression level is plotted on the y axis after normalizing RT-qPCR signals to those of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and then to the mock-treated cells (n = 3 biological replicates; presented as mean ± SD). *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant as determined using a two-tailed t test. Dotted lines denote relative gene expression = 1.0.
(D) Chromatin fractionation of MV4; 11 cells followed by immunoblotting for HA-tagged WT or mutant Dot1L. GAPDH and H3 serve as controls of cell fractionation. T, total cell sample; C, chromatin-bound fraction; S, soluble fraction (supernatant).
DISCUSSION
Recent cryo-EM structures of DOT1L bound to H2BK120ub nucleosomes revealed key interactions between DOT1L and both the nucleosome acidic patch and the ubiquitin that are critical for DOT1L-mediated H3K79 methylation in vitro (Anderson et al., 2019; Jang et al., 2019; Valencia-Sánchez et al., 2019; Worden et al., 2019; Yao et al., 2019). Nevertheless, EMSAs demonstrated that H2BK120 ubiquitylation and the DOT1L-acidic patch and DOT1L-ubiquitin interactions contribute negligibly to overall nucleosome binding (Jang et al., 2019; Valencia-Sánchez et al., 2019). We paired global and local bulk fluorescence binding assays with single-molecule FRET experiments to establish that localized interaction of DOT1L with the nucleosome disk surface is equally dependent on acidic patch engagement and ubiquitin binding.
With bulk FRET binding experiments, we were able to monitor global nucleosome binding by DOT1L at equilibrium. We confirmed that DOT1L-nucleosome complex formation is dependent on an LRR C-terminal to the catalytic methyltransferase domain of DOT1L (Min et al., 2003). Although the DOT1L fragment used for all published DOT1L-nucleosome complex structures contains the LRR, no structures clearly resolved density for DOT1L past residue 332, which marks the end of the catalytic domain. It has been hypothesized that the LRR mediates nucleosome binding by DOT1L through non-specific electrostatic interactions, especially with nucleosomal DNA (Min et al., 2003). The non-specific nature of these interactions makes them inherently heterogeneous and therefore difficult to resolve in cryo-EM reconstructions. However, the LRR-DNA interactions placed the fluorophores in our FRET experiments within a close enough distance for a FRET signal to be detected. We believe that these non-specific DNA interactions enable nucleosome disk binding that is required for H3K79 methylation. Similar to previous reports, the H2B ubiquitylation state and the DOT1L-ubiquitin interaction did not contribute to global DOT1L-nucleosome binding affinity (Jang et al., 2019; Valencia-Sánchez et al., 2019). However, we observed a 1.5-fold reduction in affinity between DOT1L and the nucleosome owing to mutation of R282 that DOT1L uses to engage the nucleosome acidic patch. Of note, previous EMSA-based experiments failed to detect an affinity change resulting from mutating this arginine. We speculate that this discrepancy is due to the more quantitative nature of our FRET assay and/or differences in assay conditions (e.g., in-gel versus in-solution).
The H2A-H2B acidic patch is a major hub for chromatin binding that is used in roughly half of nucleosome interactions (McGinty and Tan, 2021; Skrajna et al., 2020). Structures of chromatin proteins in complex with the nucleosome show that acidic patch binding proteins almost invariably use one or more arginines to dock on the acidic patch surface. By far, the most common type of acidic patch-binding arginine is the ‘‘arginine anchor,’’ which inserts into a cavity surrounded by the H2A E61, D90, and E92 side chains (McGinty and Tan, 2016, 2021). DOT1L is a rare instance of an acidic patch binding protein that does not have an arginine anchor. Rather, DOT1L has two variant arginines, most importantly the R282 type 1 variant arginine that interacts with H2A E56 (McGinty and Tan, 2021). A 1.5-fold reduction in global nucleosome binding affinity is a modest effect of mutating an essential acidic patch-binding arginine when compared with studies of other acidic patch binding proteins that use arginine anchors (Barbera et al., 2006; England et al., 2010; McGinty et al., 2014). Overall, our results raise the possibility that variant type arginines could be important for weaker acidic patch interactions, especially in the absence of an arginine anchor. We observed minimal changes in raw FRET signal upon mutating DOT1L R282. Given the design of the FRET fluorophore pair, we would expect much higher FRET for acidic patch engaged complexes than complexes in which DOT1L is only non-specifically anchored to nucleosomal DNA through its LRR, resulting in an ensemble of structures with larger average interfluorophore distances. Yet, the R282E mutation has little effect on bulk FRET signal. This supports a model of DOT1L-acidic patch binding where the nonspecific DNA binding mode dominates the ensemble of binding states at equilibrium but is accompanied by transient interactions with the acidic patch, which are further stabilized by interaction with H2BK120ub.
By using a position-specific fluorescence (de)quenching binding assay, we specifically probed DOT1L engagement of the acidic patch on the nucleosomal disk face and found that DOT1L residues that directly interact with both ubiquitin and the acidic patch are equally important for stable acidic patch interaction with H2BK120ub nucleosomes. Mutation of DOT1L R282, which directly engages with the acidic patch, or F326, which is essential for ubiquitin-dependent stimulation of DOT1L (Anderson et al., 2019; Jang et al., 2019; Valencia-Sánchez et al., 2019; Worden et al., 2019; Yao et al., 2019), resulted in complete loss of acidic patch binding. The dependence on F326 indicates that without the ability to properly engage ubiquitin, DOT1L can no longer stably bind the acidic patch of H2BK120ub nucleosomes (Figure 7A). This is consistent with our previous demonstration of an acidic patch gatekeeping effect of H2BK120ub, which on its own prevents binding of the LANA peptide to the acidic patch (Anderson et al., 2019), and suggests that the H2BK120ub modification may play a broader regulatory role in safeguarding access to this nucleosome interaction hotspot.
Figure 7. Ubiquitin and SAM stabilize DOT1L-nucleosome binding and increase access to active high-FRET state.
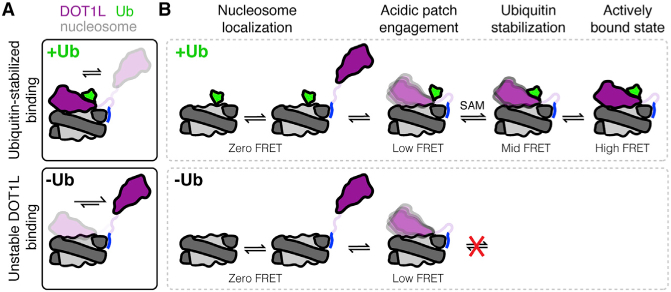
(A) DOT1L binding to unmodified nucleosomes is heterogeneous and unstable, whereas DOT1L binding to H2BK120ub nucleosomes is stabilized by the direct ubiquitin-DOT1L interaction. Ub = ubiquitin.
(B) Model of DOT1L-nucleosome binding states as observed in smFRET experiments on H2BK120ub (top) and unmodified (bottom) nucleosomes.
In contrast to our results with H2BK120ub nucleosomes, we did not observe signal for DOT1L binding to the acidic patch of unmodified nucleosomes. While low level quenching was detected with DOT1L titration, the observation was R282-independent, indicating that any observed signal was due only to non-specific nucleosome interactions. We cannot rule out that DOT1L is binding the acidic patch of unmodified nucleosomes in these assays and is not detectable either because of different fluorescence starting states between the unmodifi and H2BK120ub nucleosomes (e.g., ubiquitin may be positioned near the acidic patch changing the starting fluorescence, and this effect could be reversed as DOT1L binds ubiquitin and lifts it off the nucleosome surface and away from the fluorophore) or because DOT1L engagement of the nucleosome acidic patch without ubiquitin is significantly less stable or more dynamic and therefore fails to cause a measurable change in average fluorescence. While several cryo-EM structures of DOT1L bound to the acidic patch on unmodifi nucleosomes have been determined, these structures all required cross-linking of the complex and still resulted in very low resolution density for DOT1L (Jang et al., 2019; Valencia-Sánchez et al., 2019; Yao et al., 2019). We assume that cross-linking stabilizes acidic patch binding by DOT1L and when combined with computational purification of bound complexes during cryo-EM data analysis, allows visualization of a blurry DOT1L density bound to the nucleosome acidic patch.
On a single-molecule level, we found that the DOT1L-nucleosome interaction follows a four-state mechanism (Figure 7B). According to our results, there must be non-specific zero-FRET (or undetectably low FRET) binding modes in all experimental conditions and even upon removal of the DOT1L LRR that includes background and/or non-specific electrostatic interactions between DOT1L and the nucleosome. The zero-FRET state transitions to a low-FRET state that is dependent on DOT1L variant arginine R282. This state was accessible on unmodified and ubiquitylated nucleosomes, suggesting that even without ubiquitin, acidic patch engagement is possible to some extent. However, the F326A mutation prevented DOT1L from achieving the low-FRET state on H2BK120ub nucleosomes, further establishing that acidic patch binding of H2BK120ub nucleosomes requires DOT1L to interact directly with ubiquitin. Mid-FRET and high-FRET states were accessible only by wild-type DOT1L on H2BK120ub nucleosomes. These states therefore represent binding conformations in which DOT1L is both engaged with the acidic patch and stabilized by ubiquitin. We favor a model in which the mid-FRET state represents DOT1L in the poised conformation observed in cryo-EM structures with H2BK120ub nucleosomes. This state is acidic patch-bound and stabilized by ubiquitin binding but is still dynamic owing to lack of tethering to H3K79. In the high-FRET state, DOT1L is rigidified by binding of H3K79Nle in the active site as observed in the active state structure (Worden et al., 2019). We also find that the cofactor SAM plays a role in helping to promote access to these higher FRET conformations, potentially by driving a conformational change in DOT1L. These FRET assignments are further supported by the high- and mid-FRET values of the wild-type DOT1L-nucleosome complexes in the presence of SAM, 0.77 ± 0.03 and 0.45 ± 0.02, respectively, in good agreement with the estimated FRET values of 0.7 and 0.5 based on the active and poised state structures.
The importance of the ubiquitin and acidic patch recognition surfaces of DOT1L for DOT1L nucleosome binding and methylation in vitro present two therapeutic strategies for inhibiting DOT1L activity in MLL, where DOT1L activity has been shown to be essential for leukemogenesis (Okada et al., 2005). Using precision structure-guided DOT1L mutations, we show that acidic patch- and ubiquitin-binding surfaces of DOT1L are essential for bulk levels of H3K79me2 and proliferation in an MV4; 11 cell model of MLL-rearranged acute leukemia even though they do not prevent chromatin engagement. These results indicate that the ubiquitin-dependent activity of DOT1L is truly the disease-relevant activity of DOT1L and suggests that targeting these DOT1L interaction surfaces may be an effective strategy for treatment of MLL-rearranged leukemias.
Limitations of the study
Owing to an inability to detect acidic patch binding by DOT1L to unmodified nucleosomes using our fluorescence (de) quenching assay, we could not measure local acidic patch binding affinities in the absence of H2BK120ub. There were also two limitations of our rescue experiments in MV4; 11 cells. First, our attempts to express sgRNA-resistant exogenous human DOT1L were not successful; therefore, we used mouse Dot1L instead. Mouse and human orthologs are nearly identical, with only two amino acid differences in the 332-amino acid catalytic domain. Second, we were unable to detect exogenous Dot1L at targeted genomic loci by chromatin immuno-precipitation (ChIP)-PCR with an anti-HA antibody, indicating a technical difficulty. While we know that wild-type Dot1L and all Dot1L mutants are tightly bound to chromatin based on chromatin fractionation, we cannot rule out mutation-induced genomic mislocalization.
STAR☆METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Robert McGinty (rmcginty@email.unc.edu).
Materials availability
Unique reagents generated in this study will be available without restrictions upon request.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and tissue culture
The MV4;11 cells (American Tissue Culture Collection [ATCC], CRL-9591, male) were cultured in the RPMI 1640 base medium supplemented with 10% of fetal bovine serum (FBS) and 1% of penicillin and streptomycin. Authentication of cell line identities, including those of parental and derived lines, was ensured by the Tissue Culture Facility affiliated to UNC Lineberger Comprehensive Cancer Center (LCCC) with genetic signature profiling and fingerprinting analyses. Every 1–2 months, a routine examination of cell lines for possible mycoplasma contamination was performed with a commercially available detection kit (Lonza).
METHOD DETAILS
Protein expression and purification
DOT1Lcat (human DOT1L residues 1–416) was expressed and purified as previously described (Anderson et al., 2019). Briefly, protein was expressed in E. coli BL21(DE3)pLysS cells at 18°C as a Step-tag-6xHis fusion, purified using TALON Metal Affinity resin, the affinity tag was cleaved with TEV protease, and the protein was further purified using cation exchange chromatography with Source S resin. All DOT1L point mutants were cloned by site-directed mutagenesis and the DOT1L1–332 truncation was cloned by traditional ligation cloning. DOT1L mutants and truncations were analyzed by 18% SDS denaturing protein gel to confirm sample purity and quantitation (Figures S6A and S6B). Mutant histone H2A(L65C) was cloned by site-directed mutagenesis. All recombinant human histones were expressed and purified as previously described (Anderson et al., 2019; Luger et al., 1997, 1999), except for mutant histone H3 (K79Nle), which was expressed separately as previously described (Worden et al., 2019). Briefly, individual histones were expressed in inclusion bodies using E. coli BL21(DE3)pLysS cells at 37°C. Histones were extracted from inclusions bodies and refolded into H2A-H2B dimers or H3-H4 tetramers by combining equimolar amounts of histones and refolding by dialysis from unfolding buffer (20 mM Tris-Cl, pH 8.0, 10 mM DTT, 7 M guanidine-HCl) into refolding buffer (10 mM HEPES, pH 7.5, 100 mM NaCl, 10 mM 2-mercaptoethanol). Histone complexes were purified by cation exchange chromatography with Source S resin. H3 (K79Nle) was expressed using the H3.2 (K79M) mutant in methionine auxotrophic E. coli B834(DE3)pLysS cells at 37°C. Cells were grown in minimal media supplemented with all amino acids. Prior to induction, cells were washed with media without amino acids and then resuspended in media supplemented with all amino acids, replacing methionine with norleucine. H3 (K79Nle) was incorporated into H3-H4 tetramers as described above. Site-specifically ubiquitylated human H2BK120ub(G76A) was prepared by protein semi-synthesis exactly as previously described (Anderson et al., 2019) and is referred to simply as H2BK120ub. Briefly, a recombinant ubiquitin(1–75)-MES (MES = mercaptoethanesulfonate) thioester was ligated to an H2B C-terminal synthetic peptide (Thz-VTK(C)YTSSK, Thz = thiazolidine). The Thz was converted to a cysteine in situ using methoxylamine. The product was purified by C18 reverse phase chromatography (Vydac). The resultant ubiquitylated H2B C-terminal peptide was ligated to a recombinant H2B(1–116)-MES thioester, the product was desulfurized in situ using radical-mediated desulfurization, and the desulfurized product, H2BK120ub(G76A), was purified by C18 reverse phase chromatography. Recombinant protein thioesters were prepared using split Npu inteins and cleavage with MESNa.
Preparation of fluorescent proteins
Atto647N-fluorescently labeled DOT1Lcat was prepared by individually mutating all three native cysteines present in DOT1Lcat to alanine (C45A, C75A, and C179A), before incorporating a cysteine mutation at the desired labeling position (N324C or E69C for bulk FRET assay or single-molecule FRET, respectively). The respective DOT1L mutant was then incubated at 20 µM with a three-fold molar excess of Atto647N maleimide (Sigma) in labeling buffer (20 mM Tris-Cl, 300 mM NaCl, 0.2 mM TCEP, pH 7.5) for 20–60 minutes before ion exchange purification using a Source S resin (GE Healthcare). Labeled DOT1L and mutants were confirmed on an 18% SDS denaturing protein gel (Figure S6A). AlexaFluor488-labeled histone H2A was prepared by incubating histone dimers containing mutant H2A(L65C) and either unmodified H2B or H2BK120ub at 2 mg/mL with one molar equivalent of Alex-aFluor488 maleimide (Thermo Fisher) in histone labeling buffer (20 mM Tris-Cl, 25 mM NaCl, 7 M guanidine-Cl, 0.2 mM TCEP, pH 7.5) for 2 hours at 4°C. After 2 hours, one more molar equivalent of AlexaFluor488 maleimide was added and the labeling reaction was incubated for an additional 2 hours at 4°C. The reaction was quenched by addition of DTT to 20 mM before overnight dialysis into reconstitution buffer (10 mM HEPES, 50 mM NaCl, 10 mM BME, pH 7.5), followed by purification of the dimer over Source S resin as previously reported (Anderson et al., 2019).
Nucleosome reconstitution
Nucleosomes used in FRET and (de)quenching binding assays were reconstituted as previously described (Anderson et al., 2019; Luger et al., 1997, 1999) using AlexaFluor488-labeled histone dimer containing either unmodified H2B or H2BK120ub, wild type H3.2-H4 tetramer, and a 147 bp DNA fragment of the Widom 601 nucleosome positioning sequence (Lowary and Widom, 1998; Makde et al., 2010). Briefly, H2A-H2B dimers, H3-H4 tetramers, and DNA were combined in 2.8:1:1.1 molar ratios and dialyzed from high salt buffer (10 mM Tris-Cl, pH 7.5, 2 M KCl, 1 mM EDTA, 1 mM DTT) into low salt buffer (10 mM Tris-Cl, pH 7.5, 250 mM KCl, 1 mM EDTA, 1 mM DTT) using a gradient dialysis pump. Nucleosomes were purified by anion exchange chromatography using Source Q resin. Labeling and quantitation of fluorescently labeled nucleosomes used in FRET and (de)quenching binding assays were confirmed by 10% native acrylamide and 18% SDS denaturing protein gels (Figure S6C). Nucleosomes used for single-molecule FRET experiments were reconstituted by combining histone octamers reconstituted with unmodified H2B or H2BK120ub with nucleosomal DNA made up of 147 bp of the Widom 601 nucleosome positioning sequence flanked by a 20-nucleotide single-stranded DNA (ssDNA) linker with a biotin at the 5’end. Histone octamers were prepared using wild type H2A, H3.2 (K79Nle), wild type H4 and either unmodified H2B or H2BK120ub as previously described (Luger et al., 1999). Briefly, histones were combined in unfolding buffer in equimolar amounts and dialyzed into octamer refolding buffer (10 mM Tris-Cl, pH 7.5, 2 M NaCl, 1 mM EDTA, 5 mM 2-mercaptoethanol) prior to purification by gel filtration in octamer refolding buffer using a Superdex 200 increase 10/300 column (GE Healthcare). The nucleosomal DNA was generated by ligating five ssDNA fragments with lengths of 40–75 nucleotides (Table S1). A succinimidyl ester functionalized Cy3 fluorophore was conjugated to the amino-modified thymine residue (iAmMC6T) at the +15 position from the nucleosome dyad. Nucleosomes were reconstituted by mixing nucleosomal DNA and histone octamer in a 1:1.5 ratio in nucleosome reconstitution start buffer (10 mM Tris-HCl, 2 M NaCl, 1 mM EDTA, pH 8.0) on ice for 30 min, followed by five successive 1-hour dialysis steps in the same buffer but containing 850 mM, 650 mM, 500 mM, 300 mM, and 1 mM NaCl. The resulting sample was annealed for 1 hour at 54°C. Assembled nucleosomes were confirmed on a 5% native PAGE gel (0.2X TBE polyacrylamide gel) (Figure S6D).
DOT1L-nucleosome bulk FRET binding assay
The bulk FRET binding assay was performed by preparing serial dilutions of Atto647N-labeled DOT1Lcat, DOT1L1–332, DOT1Lcat(R282E), or DOT1Lcat(F326A) and combining 5 µL of each DOT1L dilution with either 5 µL of assay buffer (20 mM Tris-Cl pH 7.5, 50 mM NaCl, 5 mM DTT, 5% glycerol, 0.01% NP40, 0.01% CHAPS, 100 µg/mL BSA) or 20 nM AlexaFluor488-labeled nucleosome in assay buffer. Samples were combined in a 384-well, small volume, non-binding, white microplate (Greiner #784904) and mixed for 60 seconds in a plate shaker. The plate was centrifuged at 1,000 rpm for 60 seconds and incubated covered from light at room temperature for 60 minutes to equilibrate. Plates were analyzed with an EnVision 2103 plate reader, scanning individually for FRET (excitation at 485 nm, emission at 665 nm), acceptor only (excitation at 615 nm, emission at 665 nm), and donor only (excitation at 485 nm, emission at 535 nm) signal. Donor bleed-through and acceptor direct excitation corrections were performed as previously described (Winkler et al., 2012). Briefly, donor bleed-through was corrected by subtracting the product of the donor signal for the donor+acceptor samples and the ratio of FRET signal to donor signal for the donor only sample. Direct acceptor excitation was corrected by subtracting the product of the acceptor signal for the donor+acceptor samples and the ratio of the FRET signal to acceptor signal for acceptor only samples. Corrected FRET values were fit using the fitnlm (Fit nonlinear regression model) function in MATLAB R2021a using the following quadratic binding equation that accounts for ligand depletion and fits the data with a Hill binding coefficient: fun = @(b,x)((b(1)-b(2))*(((x-(((Rtotal+x+b(3))-sqrt(((Rtotal+x+b(3)).^2)-(4*Rtotal.*x)))./2)).^b(4))./((b(3)^ b(4))+((x-(((Rtotal+x+b(3))-sqrt(((Rtotal+x+b(3)).^2)-(4*Rtotal.*x)))./2)).^b(4))))+b(2))where x is the corrected FRET value, Rtotal is the total effective receptor concentration (as each nucleosome provides two binding sites for DOT1L, this is twice the nucleosome concentration, or 20 nM), b(1) and b(2) are the maximum and minimum FRET values, respectively, b(3) is the Kd, and b(4) is the Hill coefficient. Positive cooperativity was indicated by these data, as all curves were fit with a Hill coefficient around 2 (Table S2). FRET binding assays were performed on nucleosomes containing the histone H3 K79Nle substitution. Aggregation of sample mixtures was observed at high DOT1L concentrations, especially for the F326 mutant, which limited the collection of further saturated binding data points along the titration curve. All titration points were performed in triplicate, and each assay was repeated independently at least two times. All samples are discrete samples.
DOT1L-nucleosome (de)quenching binding assay
The (de)quenching binding assay was performed essentially as previously described (Winkler et al., 2012). Briefly, DOT1L serial dilutions were prepared and combined 1:1 with 20 nM AlexaFluor488-labeled nucleosome to a final volume of 20 µL in a 384-well OptiPlate, White Opaque Microplate (PerkinElmer #6007290) in assay buffer (20 mM Tris-Cl pH 7.5, 50 mM NaCl, 5 mM DTT, 5% glycerol, 0.01% NP40, 0.01% CHAPS, 100 µg/mL BSA). After mixing, plates were agitated for 60 seconds on a plate shaker prior to centrifugation at 1,000 rpm for 60 seconds and incubated covered from light at room temperature for 60 minutes. Plates were read with an EnVision 2103 plate reader (excitation at 485 nm, emission at 535 nm). Fluorescence values were fit using the fitnlm (Fit nonlinear regression model) function in MATLAB R2021a using the following quadratic binding equation that accounts for ligand depletion: fun = @(b,x)((b(1)-b(2))*((x-(((Rtotal+x+b(3))-sqrt(((Rtotal+x+b(3)).^2)-(4*Rtotal.*x)))./2))./(b(3)+(x-(((Rtotal+x+b(3))-sqrt(((Rtotal+x+b(3)).^2)-(4*Rtotal.*x)))./2))))+b(2))where x is the fluorescence value, Rtotal is the total effective receptor concentration (as each nucleosome provides two binding sites for DOT1L, this is twice the nucleosome concentration, or 20 nM), b(1) and b(2) are the maximum and minimum fluorescence values, respectively, and b(3) is the Kd. All curves would fit with a Hill coefficient around 1, therefore cooperativity was not indicated by the data and final fits were performed with a fixed Hill coefficient of 1. (De)quenching assays were all performed on both wild type and H3 K79Nle nucleosomes, which showed equivalent results for each DOT1L mutant and truncation (Figures S2A and S2B). All titration points were performed in triplicate, and each assay was repeated independently at least two times (Figures S2C–S2F). DOT1L binding was salt dependent but produced a more robust (de)quenching signal at the lower salt concentration of 50 mM NaCl, allowing more confident quantitation of binding affinity (Figure S2G). Under these binding conditions, equilibrium was reached within 30 minutes at room temperature (Figure S2H). All samples are discrete samples except for time course which was measured repeatedly.
DOT1L measurements using single-molecule FRET
The single-molecule FRET (smFRET) measurements were performed on the surface of a quartz microscope slide that was cleaned thoroughly and coated with biotinylated polyethylene glycol (PEG) and a lipid bilayer. Microscope slides were prepared following previously published protocols (J. Lee et al., 2019; Yue et al., 2016). Briefly, thoroughly cleaned slides were immersed in the mixture of 8 mg Biotin-PEG-silane (3,400 MW) and 10 mg PEG-silane (2,000 MW) dissolved in 50 mL acetonitrile, sonicated for 15 minutes, and subsequently washed with water and sonicated twice for 5 minutes each in water. The sparsely PEGylated slides were coated with a 1,2-dioleoyl-snglycero-3-phosphocholine (DOPC, Avanti Polar Lipids, Alabaster, AL) lipid bilayer. To accomplish this, a chloroform solution of DOPC was dried with mild N2 flow followed by vacuum-drying for 2 hours. Dried DOPC was suspended in 10 mM Tris pH 7.8 and 100 mM NaCl at a lipid concentration of 2 mg/mL. The suspension was sonicated with an Ultrasonic cell disruptor (Sonifier 550, Branson Ultrasonics) using a microtip (Branson Ultrasonics) until the lipid suspension became a clear solution. The solution was extruded through a synthetic fiber membrane with an 80 nm pore size (Avanti Polar Lipids, Alabaster, AL). The liposome solution was monodisperse with a particle diameter of 50 nm as determined with a particle size analyzer (90 Plus, Brookhaven Instruments Corp., Holtsville, NY) compared against deionized water. The liposome solution was aliquoted and flash-frozen with liquid N2 for long-term storage at –80°C. To deposit a lipid bilayer on the slide surface, the slide was incubated with a 1 mg/mL DOPC liposome solution for 45 min.
Nucleosomes were immobilized on slides via biotin-streptavidin conjugation. Prior to immobilization of nucleosomes, the flow cell chamber was incubated with 0.1 mg/ml streptavidin for 15 minutes at room temperature followed by a washing step with wash buffer (10 mM Tris, 100 mM NaCl, pH 7.8). 10 nM Atto647N-labelled DOT1L and DOT1L mutants with or without 40 µM SAM were prepared in imaging buffer (100 mM Tris-HCl, 100 mM KCl, 25 mM MgCl2, 250 mM NaCl, 5 mM DTT, 25% Glycerol, 0.25 % Tween 20, 0.4 unit/mL protocatechuate 3,4-dioxygenase, 4 mM protocatechuic acid, 1 mM Trolox, pH 8.0) and introduced onto the slide with 50 pM immobilized nucleosomes.
The smFRET measurements were performed on a prism-coupled total internal reflection fluorescence microscope (TIRFM) (J. Lee et al., 2019). After adding imaging buffer containing Atto647N-labelled DOT1L, Cy3 was excited with a 532 nm laser (150 mW, CrystalLaser, Reno, NV), and fluorescence signals from both Cy3 and Atto647N were recorded with an electron multiplying CCD (EMCCD) camera (iXonEM+897 Andor Technology, Belfast, Ireland) at a 10 Hz frame rate with 100 ms integration over a 30-minute period. FRET signals were recorded as a series of fluorescence images forming a movie. The time series of fluorescence intensities of Cy3 and Atto647N were obtained from the series of fluorescence images and were plotted against the elapsed time. FRET efficiency at each time point was estimated with a formula, IA /(ID + IA) where I is fluorescence intensity for the FRET donor (ID) and acceptor (IA), respectively. We corrected the residual background fluorescence by measuring the donor and acceptor intensities after they photo-bleached. The constructed time series of FRET efficiencies per donor/acceptor pair, or their FRET efficiency time trajectory, reveals how the distance between the FRET pair is changing over time. From the FRET time trajectories, we extracted kinetics information of the nucleosome-DOT1L interaction. To determine the kinetic rate constants of all possible transitions among FRET states we analyzed the smFRET time trajectories with hidden Markov models (HMM), details of which are previously reported (T.-H. Lee, 2009). Briefly, we optimized the average FRET efficiencies of the FRET states and the transition rates among them with 3-, 4-, 5-, and 6-state models. We used one Gaussian distribution per state for optimization. We tested two and three Gaussian distributions per state and found no difference. After reaching 4 states, no more valid states were identified. The extra states in the 5- and 6-state models showed zero FRET efficiencies, confirming 4-state dynamics. The two-dimensional FRET transition histograms further confirm up to 4 states in the DOT1L-nucleosome complexes.
Antibodies and immunoblotting
Total cell lysate was prepared and used for western blotting as described previously (Fan et al., 2020). Briefly, cells were lysed in lysis buffer (50 mM Tris-Cl, pH 8.0, 150 mM NaCl, 1% IGEPAL CA-630), followed by sonication and high-speed centrifugation, and supernatants were collected. Identical amounts of the extracted protein samples were loaded onto SDS–PAGE gels for immunoblotting analysis. Antibodies used in this study include DOT1L (Abclonal, A12329), tubulin (Cell Signaling Technology, 2146), HA-Tag (Cell Signaling Technology, 3724), H3 (Cell Signaling Technology, 9715), and H3K79me2 (Millipore, 04–835). All western blots were performed at least two or three times.
Plasmid construction
The cDNA for mouse Dot1L was obtained from the DNASU Plasmid Repository (Reference sequence: NM_199322.1) and subsequently cloned into the MSCV-neo retroviral expression vector (Clontech) with an N-terminal HA tag by Gibson assembly. Point mutations were introduced by site-directed mutagenesis. All plasmids used were confirmed by full sequencing before use. The sgRNA targeting the human DOT1L gene (5’- GCTGAGACTGAAGTCGCCCG-3’) was designed based on the online CRISPR sgRNA design tool (GenScript; https://www.genscript.com/gRNA-database.html) and cloned into the LRG2.1 lentiviral vector co-expressing GFP (Tarumoto et al., 2018) (Addgene #108098) as previously described (Sanjana et al., 2014; Shalem et al., 2014). Briefly, the synthesized sgRNA oligos were annealed and ligated into a BsmBI-digested LRG2.1 vector and verified by sequencing.
CRISPR/Cas9-mediated DOT1L knockout and rescue
MV4;11 cells bearing the stable expression of a doxycycline (Dox)-inducible Cas9 (MV4;11-iCas9) were a gift from Drs. Xiaobing Shi and Hong Wen (Van Andel Research Institute) (Wan et al., 2017). Preparation of the lentivirus and MSCV-based retrovirus was performed as previously described (Ahn et al., 2021). In brief, the packaging plasmids and sgRNA-containing LRG2.1 plasmid were co-transfected into 293FT cells using Lipofectamine 3000 (Thermo Fisher Scientific) for virus packaging, with the supernatant collected two days post-transfection. MV4;11-iCas9 cells were then infected with the sgRNA lentivirus and GFP-positive cells were sorted five days post-infection. Such MV4;11-iCas9 cells with stable transduction of the DOT1L sgRNA were subjected to treatment with 1 µg/mL Dox to induce expression of Cas9, and DOT1L KO efficiency was evaluated by immunoblotting with DOT1L and H3K79me2 antibodies. For generating human cancer cell lines with the pre-rescue of DOT1L, we stably transduced the MSCV-neo retrovirus containing murine Dot1L, with its cDNA resistant to the sgRNA used for targeting human DOT1L (5’-GCTGAGACTGAAGTCGCCCG-3’) due to sequence difference of human and murine Dot1L cDNAs, into the above MV4;11-iCas9 cells carrying the DOT1L sgRNA, followed by drug selection for two weeks and western blotting to verify stable expression of exogenous Dot1L.
Cell proliferation assays
Leukemia cells grown in suspension were seeded at a density of 3 × 105 /mL in triplicate in 24-well plates and quantification was conducted as previously described (Xu et al., 2015; Yu et al., 2021). Media was changed every two days. Cells were passaged after thorough mixing of the cultures by pipetting up and down multiple times and diluting to maintain cell density under 1 × 106 /mL at all times. Cells were counted using a TC10 automated cell counter (Bio-Rad) every two days. All counted samples are discrete samples from three biological replicates.
Quantitative RT-qPCR
Total RNA extraction was prepared with the RNeasy Mini Kit (Qiagen #74104) according to manufacturer’s recommendations. 1 µg of total RNA was used for reverse transcription with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Then, real-time PCR was performed using a QuantStudio 6 Flex Real-Time PCR platform (Thermo Fisher Scientific) and Power SYBR Green Master Mix (Bio-Rad). The comparative CT method was used to calculate relative gene expression by comparing the CT value of each target gene to that of an internal control GAPDH (2∆∆CT) as follows: ∆∆CT = CT(target gene)–CT(GAPDH) (Xu et al., 2015; Yu et al., 2021). Primers used for RT-qPCR are listed in Table S3. Three discrete biological replicates were measured for each sample. Two-tailed t-tests were used to determine p-values.
Cell fractionation
Fractionation of cells for the assessment of chromatin association by DOT1L was conducted as previously described (Li et al., 2021). In brief, cells were collected and washed twice with cold PBS, followed by centrifugation at 300 g for 5 min at 4°C. After removal of supernatant, cell pellet was resuspended in CSK buffer (10 mM PIPES pH 7.0, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 0.5% Triton X-100, supplemented with freshly added cocktail of protease and phosphatase inhibitors) and incubated on ice for 15 min, followed by centrifugation at 1,500 g for 10 min. The supernatant and pellet represented the soluble and chromatin-bound fractions, respectively. The pellet was then washed once with CSK buffer and resuspended in gel loading buffer. Samples were boiled for 10 minutes, equivalent amounts of sample were loaded on SDS-PAGE gels, and subjected to western blot analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
All bulk fluorescence binding experiments were performed with triplicate titrations. Triplicate samples from discrete biological replicates were analyzed for cell proliferation and RT-qPCR experiments and are shown as means and standard errors or standard deviations as indicated in figure legends. Statistical significance was determined as described in figure legends using two-tailed t-tests and p-value thresholds are listed in figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Rabbit anti-DOT1L/KMT4 | Abclonal | Cat#A12329; RRID:AB_2861655 |
| Rabbit anti-β-tubulin | Cell Signaling Technology | Cat#2146; RRID:AB_2210545 |
| Rabbit anti-HA tag | Cell Signaling Technology | Cat#3724; RRID:AB_1549585 |
| Rabbit anti-Histone H3 | Cell Signaling Technology | Cat#9715; RRID:AB_331563 |
| Rabbit anti-H3K79me2 | Millipore | Cat#04–835; RRID:AB_1587126 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| E. coli BL21(DE3)pLysS | Novagen | Cat#69388–3 |
| E. coli B834(DE3)pLysS | Novagen | Cat#69042–3 |
| E. coli HB101 | ATCC | Cat#67593 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Atto647N maleimide | Sigma | Cat#05316 |
| AlexaFluor488 maleimide | Fisher | Cat#A10254 |
| TALON Metal Affinity Resin | Clontech | Cat.#635669 |
| Source S resin | GE Healthcare | Cat.#17094405 |
| Source Q resin | GE Healthcare | Cat.#17094705 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| MV4;11 | ATCC; gifts from gift from Drs. Xiaobing Shi and Hong Wen; Wan et al. (2017) | Cat#CRL-9591 |
|
| ||
| Oligonucleotides | ||
|
| ||
| smFRET nucleosome oligonucleotides (for oligonucleotides used to prepare nucleosomes for smFRET, see Table S1) | This study | N/A |
| RT-qPCR primers (for RT-qPCR primers, see Table S3) | This study | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| Plasmid expressing DOT1Lcat | Anderson et al. (2019) | pST50Tr-STRaHISNhDot1t1x1 |
| Plasmid expressing DOT1L(1–332) | This study | pST50Tr-STRaHISNhDot1t3 |
| Plasmid expressing DOT1Lcat(R282A) | Anderson et al. (2019) | pST50Tr-STRaHISNhDot1t1x4 |
| Plasmid expressing DOT1Lcat(F326A) | Anderson et al. (2019) | pST50Tr-STRaHISNhDot1t1x6 |
| Plasmid expressing DOT1Lcat(K330A) | Anderson et al. (2019) | pST50Tr-STRaHISNhDot1t1x5 |
| Plasmid expressing DOT1Lcat(C45A, C75A, C179A, N324C) | This study | pST50Tr-STRaHISNhDot1t1x30 |
| Plasmid expressing DOT1Lcat(R282E, C45A, C75A, C179A, N324C) | This study | pST50Tr-STRaHISNhDot1t1x33 |
| Plasmid expressing DOT1Lcat(F326A, C45A, C75A, C179A, N324C) | This study | pST50Tr-STRaHISNhDot1t1x31 |
| Plasmid expressing DOT1L(1–332, C45A, C75A, C179A, N324C) | This study | pST50Tr-STRaHISNhDot1t3x30 |
| Plasmid expressing DOT1Lcat(C45A, C75A, C179A, E69C) | This study | pST50Tr-STRaHISNhDot1t1x26 |
| Plasmid expressing DOT1Lcat(R282E, C45A, C75A, C179A, E69C) | This study | pST50Tr-STRaHISNhDot1t1x37 |
| Plasmid expressing DOT1Lcat(F326A, C45A, C75A, C179A, E69C) | This study | pST50Tr-STRaHISNhDot1t1x39 |
| Plasmid expressing DOT1Lcat(G163R, G165R, C45A, C75A, C179A, E69C) | This study | pST50Tr-STRaHISNhDot1t1x38 |
| Plasmid expressing DOT1L(1–332, C45A, C75A, C179A, E69C) | This study | pST50Tr-STRaHISNhDot1t3x26 |
| Plasmid expressing H2A(L65C) | This study | pST50Trc4-hH2A.Dx11 |
| Plasmid expressing H3(K79M) | This study | pST50Tr-hH3.2x26 |
| Lentiviral plasmid expressing DOT1L-targeted sgRNA | This study | LRG2.1-sgDOT1L |
| Retroviral plasmid expression murine Dot1L | This study | pMSCVneo-KHAmDot1L |
| Retroviral plasmid expression murine Dot1L(R282A) | This study | pMSCVneo-KHAmDot1Lx1 |
| Retroviral plasmid expression murine Dot1L(R282E) | This study | pMSCVneo-KHAmDot1Lx2 |
| Retroviral plasmid expression murine Dot1L(F326A) | This study | pMSCVneo-KHAmDot1Lx4 |
| Retroviral plasmid expression murine Dot1L(I290A) | This study | pMSCVneo-KHAmDot1Lx5 |
| Retroviral plasmid expression murine Dot1L(K330A) | This study | pMSCVneo-KHAmDot1Lx9 |
| Retroviral plasmid expression murine Dot1L(G163R/G165R) | This study | pMSCVneo-KKHAmDot1Lx7 |
|
| ||
| Software and algorithms | ||
|
| ||
| Microsoft Excel | Microsoft | https://products.office.com/en-us/excel |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Matlab R2021a | Mathworks | https://www.mathworks.com |
Highlights.
Comprehensive mechanistic analysis of DOT1L activation by H2BK120 ubiquitylation
DOT1L must bind ubiquitin to stably engage nucleosome acidic patch
SAM cofactor accelerates DOT1L-nucleosome conformational change for methylation
Ubiquitin binding by DOT1L is required for MLL-rearranged leukemia proliferation
ACKNOWLEDGMENTS
MV4; 11 cells bearing inducible Cas9 (MV4; 11-iCas9) were a kind gift of Drs. X. Shi and H. Wen. We thank UNC’s facilities, including the Flow Cytometry Core and the Tissue Culture Facility, for their professional assistance with this work. We also thank the McGinty, Lee, and Wang laboratories for helpful discussion and critical comments on the manuscript. This work was funded by NIH grants (R35GM133498 to R.K.M.; F99CA253730 to C.J.S.; R01GM123164 and R01GM130793 to T.-H.L.; R01CA211336 and R01CA215284 to G.G.W.). The cores affiliated to UNC Cancer Center are supported in part by the UNC Line-berger Comprehensive Cancer Center Core Support Grant P30CA016086. G.G.W. is an American Cancer Society (ACS) Research Scholar and a Leukemia and Lymphoma Society (LLS) Scholar. R.K.M. is a Searle Scholar and a Pew-Stewart Scholar for Cancer Research. Graphical abstract was created in part with BioRender.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110369.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahn JH, Davis ES, Daugird TA, Zhao S, Quiroga IY, Uryu H, Li J, Storey AJ, Tsai Y-H, Keeley DP, et al. (2021). Phase separation drives aberrant chromatin looping and cancer development. Nature 595, 591–595. 10.1038/s41586-021-03662-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CJ, Baird MR, Hsu A, Barbour EH, Koyama Y, Borgnia MJ, and McGinty RK (2019). Structural basis for recognition of ubiquitylated nucleosome by Dot1L methyltransferase. Cell Rep 26, 1681–1690.e5. 10.1016/j.celrep.2019.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, and Kaye KM (2006). The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 311, 856–861. 10.1126/science.1120541. [DOI] [PubMed] [Google Scholar]
- Campbell CT, Haladyna JN, Drubin DA, Thomson TM, Maria MJ, Yamauchi T, Waters NJ, Olhava EJ, Pollock RM, Smith JJ, et al. (2017). Mechanisms of pinometostat (EPZ-5676) treatment-emergent resistance in MLL-rearranged leukemia. Mol. Cancer Ther 16, 1669–1679. 10.1158/1535-7163.MCT-16-0693. [DOI] [PubMed] [Google Scholar]
- Chen C-W, Koche RP, Sinha AU, Deshpande AJ, Zhu N, Eng R, Doench JG, Xu H, Chu SH, Qi J, et al. (2015). DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat. Med 21, 335–343. 10.1038/nm.3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, et al. (2011). Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20, 53–65. 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England JR, Huang J, Jennings MJ, Makde RD, and Tan S (2010). RCC1 Uses a conformationally diverse loop region to interact with the nucleosome: a model for the RCC1–nucleosome complex. J. Mol. Biol 398, 518–529. 10.1016/j.jmb.2010.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Lu J, Guo Y, Li D, Zhang Z-M, Tsai Y-H, Pi W-C, Ahn JH, Gong W, Xiang Y, et al. (2020). BAHCC1 binds H3K27me3 via a conserved BAH module to mediate gene silencing and oncogenesis. Nat. Genet 52, 1384–1396. 10.1038/s41588-020-00729-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, and Zhang Y (2002). Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol 12, 1052–1058. [DOI] [PubMed] [Google Scholar]
- Jang S, Kang C, Yang H-S, Jung T, Hebert H, Chung KY, Kim SJ, Hohng S, and Song J-J (2019). Structural basis of recognition and destabilization of the histone H2B ubiquitinated nucleosome by the DOT1L histone H3 Lys79 methyltransferase. Genes Dev 33, 620–625. 10.1101/gad.323790.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B, Su H, Bhat A, Lei H, Bajko J, Hevi S, Baltus GA, Kadam S, Zhai H, Valdez R, et al. (2008). The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet 4, e1000190. 10.1371/journal.pgen.1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T-H (2009). Extracting kinetics information from single-molecule fluorescence resonance energy transfer data using hidden Markov models. J. Phys. Chem. B 113, 11535–11542. 10.1021/jp903831z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Crickard JB, Reese JC, and Lee T-H (2019). Single-molecule FRET method to investigate the dynamics of transcription elongation through the nucleosome by RNA polymerase II. Methods 159–160, 51–58. 10.1016/j.ymeth.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Galbo PM, Gong W, Storey AJ, Tsai Y-H, Yu X, Ahn JH, Guo Y, Mackintosh SG, Edmondson RD, et al. (2021). ZMYND11-MBTD1 induces leukemogenesis through hijacking NuA4/TIP60 acetyltransferase complex and a PWWP-mediated chromatin association mechanism. Nat. Commun 12, 1045–1118. 10.1038/s41467-021-21357-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, and Widom J (1998). New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol 276, 19–42. 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, Flaus AJ, Waye MM, and Richmond TJ (1997). Characterization of nucleosome core particles containing histone proteins made in bacteria. J. Mol. Biol 272, 301–311. 10.1006/jmbi.1997.1235. [DOI] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, and Richmond TJ (1999). Preparation of nucleosome core particle from recombinant histones. Methods Enzymol 304, 3–19. [DOI] [PubMed] [Google Scholar]
- Makde RD, England JR, Yennawar HP, and Tan S (2010). Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature 467, 562–566. 10.1038/nature09321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty RK, and Tan S (2016). Recognition of the nucleosome by chromatin factors and enzymes. Curr. Opin. Struct. Biol 37, 54–61. 10.1016/j.sbi.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty RK, and Tan S (2021). Principles of nucleosome recognition by chromatin factors and enzymes. Curr. Opin. Struct. Biol 71, 16–26. 10.1016/j.sbi.2021.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty RK, Kim J, Chatterjee C, Roeder RG, and Muir TW (2008). Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature 453, 812–816. 10.1038/nature06906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty RK, Henrici RC, and Tan S (2014). Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 514, 591–596. 10.1038/nature13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Martin ME, Brock HW, Slany RK, and Hess JL (2005). Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer Res 65, 11367–11374. 10.1158/0008-5472.CAN-05-1041. [DOI] [PubMed] [Google Scholar]
- Min J, Feng Q, Li Z, Zhang Y, and Xu R-M (2003). Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 112, 711–723. [DOI] [PubMed] [Google Scholar]
- Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, and Zhang Y (2005). hDOT1L links histone methylation to leukemogenesis. Cell 121, 167–178. 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrajna A, Goldfarb D, Kedziora KM, Cousins EM, Grant GD, Spangler CJ, Barbour EH, Yan X, Hathaway NA, Brown NG, et al. (2020). Comprehensive nucleosome interactome screen establishes fundamental principles of nucleosome binding. Nucleic Acids Res 389, 251. 10.1093/nar/gkaa544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slany RK (2016). The molecular mechanics of mixed lineage leukemia. Oncogene 35, 5215–5223. 10.1038/onc.2016.30. [DOI] [PubMed] [Google Scholar]
- Tarumoto Y, Lu B, Somerville TDD, Huang Y-H, Milazzo JP, Wu XS, Klingbeil O, El Demerdash O, Shi J, and Vakoc CR (2018). LKB1, salt-inducible kinases, and MEF2C are linked dependencies in acute myeloid leukemia. Mol. Cell 69, 1017–1027.e6. 10.1016/j.molcel.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia-Sánchez MI, De Ioannes P, Wang M, Vasilyev N, Chen R, Nudler E, Armache J-P, and Armache K-J (2019). Structural basis of Dot1L stimulation by histone H2B lysine 120 ubiquitination. Mol. Cell 74, 1010–1019.e6. 10.1016/j.molcel.2019.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Wen H, Li Y, Lyu J, Xi Y, Hoshii T, Joseph JK, Wang X, Loh Y-HE, Erb MA, et al. (2017). ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 543, 265–269. 10.1038/nature21687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E, Kawaoka S, Yu M, Shi J, Ni T, Yang W, Zhu J, Roeder RG, and Vakoc CR (2013). Histone H2B ubiquitin ligase RNF20 is required for MLL-rearranged leukemia. Proc. Natl. Acad. Sci. U S A 110, 3901–3906. 10.1073/pnas.1301045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DD, Luger K, and Hieb AR (2012). Quantifying chromatin-associated interactions: the HI-FI system. Methods Enzymol 512, 243–274. 10.1016/B978-0-12-391940-3.00011-1. [DOI] [PubMed] [Google Scholar]
- Wood K, Tellier M, and Murphy S (2018). DOT1L and H3K79 methylation in transcription and genomic stability. Biomolecules 8, 11. 10.3390/biom8010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worden EJ, Hoffmann NA, Hicks CW, and Wolberger C (2019). Mechanism of cross-talk between H2B ubiquitination and H3 methylation by Dot1L. Cell 176, 1490–1501.e12. 10.1016/j.cell.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, and Brand L (1994). Resonance energy transfer: methods and applications. Anal. Biochem 218, 1–13. 10.1006/abio.1994.1134. [DOI] [PubMed] [Google Scholar]
- Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, Allison DF, Cai L, Rockowitz S, Liu S, et al. (2015). Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood 125, 346–357. 10.1182/blood-2014-06-581082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T, Jing W, Hu Z, Tan M, Cao M, Wang Q, Li Y, Yuan G, Lei M, and Huang J (2019). Structural basis of the crosstalk between histone H2B monoubiquitination and H3 lysine 79 methylation on nucleosome. Cell Res 29, 330–333. 10.1038/s41422-019-0146-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Li D, Kottur J, Shen Y, Kim HS, Park K-S, Tsai Y-H, Gong W, Wang J, Suzuki K, et al. (2021). A selective WDR5 degrader inhibits acute myeloid leukemia in patient-derived mouse models. Sci. Transl. Med 13, eabj1578. 10.1126/scitranslmed.abj1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue H, Fang H, Wei S, Hayes JJ, and Lee T-H (2016). Single-molecule studies of the linker histone H1 binding to DNA and the nucleosome. Biochemistry 55, 2069–2077. 10.1021/acs.biochem.5b01247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Allis CD, and Wang GG (2021). The language of chromatin modification in human cancers. Nat. Rev. Cancer 21, 413–430. 10.1038/s41568-021-00357-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Zheng Y, Pham A-D, Mandal SS, Erdjument-Bromage H, Tempst P, and Reinberg D (2005). Monoubiquitination of human histone H2B: the factors involved and their roles in HOX gene regulation. Mol. Cell 20, 601–611. 10.1016/j.molcel.2005.09.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.