

Abstract
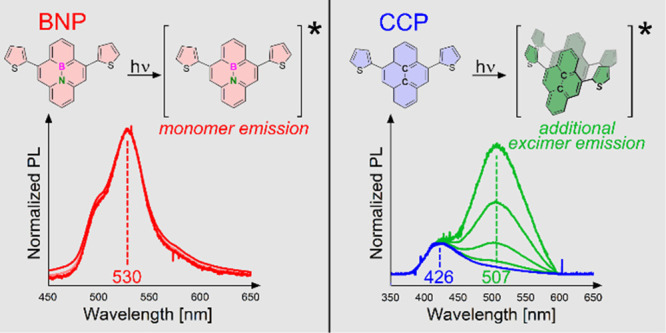
Boron–nitrogen substitutions in polycyclic aromatic hydrocarbons (PAHs) have a strong impact on the optical properties of the molecules due to a significantly more heterogeneous electron distribution. However, besides these single-molecule properties, the observed optical properties of PAHs critically depend on the degree of intermolecular interactions such as π–π-stacking, dipolar interactions, or the formation of dimers in the excited state. Pyrene is the most prominent example showing the latter as it exhibits a broadened and strongly bathochromically shifted emission band at high concentrations in solution compared to the respective monomers. In the solid state, the impact of intermolecular interactions is even higher as it determines the crystal packing crucially. In this work, a thiophene-flanked BN-pyrene (BNP) was synthesized and compared with its all-carbon analogue (CCP) in solution and in the solid state by means of crystallography, NMR spectroscopy, UV–vis spectroscopy, and photoluminescence (PL) spectroscopy. In solution, PL spectroscopy revealed the solvent-dependent presence of excimers of CCP at high concentrations. In contrast, no excimers were found in BNP. Clear differences were also observed in the single-crystal packing motifs. While CCP revealed overlapped pyrene planes with centroid distances in the range of classical π-stacking interactions, the BNP scaffolds were displaced and significantly more spatially separated.
Introduction
Boron–nitrogen-substituted aromatic systems have attracted great interest over the last decade as this formal replacement offers a method to significantly change the physical, chemical, and optoelectronic properties of the molecules. However, the geometrical structures remain similar, at least in the gas phase or in a non-aggregated form in solution.1−4
Consequently, the research on the potential applications of BN-aromatics has intensified, and the main emphasis of the current research has especially been placed on the fields of materials science5,6 and organic electronics.7−10 Other areas have also started to benefit from these developments in the main group chemistry. For instance, BN-substituted pharmaceuticals,11 the BN-analogues of polystyrene and poly(vinylnaphthalene) with small aromatic BN-units as side groups,12−15 and BN-aromatics as ligands for the main group elements and transition metals were investigated.16−18 The increased interest has prompted the development of numerous synthetic approaches giving facilitated access to substituted BN-aromatics.19−24
A BN-substitution in polycyclic aromatic hydrocarbons (PAHs) may not only alter the electronic structure of the single molecules but can also induce the changes in the morphology and aggregation behavior. This in itself results in strongly divergent optical properties25 and provides a powerful tool for synthesizing materials with a precisely attuned photophysical behavior.
Recently, the first organic photovoltaics,10 organic field-effect transistors,7,26 and organic light-emitting diodes (OLEDs)8,9 containing BN-substituted compounds have been reported. Compared to the carbonaceous analogues, the highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO)-gaps of monocyclic azaborinines are usually decreased,27 while fused-ring BN-systems do not allow such a generalized statement. On the one hand, the majority of reports showed that a BN-unit induces a slight decrease of the optical band gaps in comparison with all-carbon analogues.25,28−30 On the other hand, several BN-acenes with particularly stabilized HOMO levels have been presented, which are consequently less prone to oxidation.31,32 Furthermore, a number of studies showed largely increased quantum yields of BN-PAHs due to decreased non-radiative photochemistry pathways33 and excellent thermal stabilities up to 400 °C, which are comparable with their carbonaceous congeners, illustrating the high application potential of these structures.29,32,34−38 The position of the BN-motif inside the molecule proved to be a crucial determinant of the above-mentioned photophysical properties.39−41
One of the best investigated PAHs is pyrene, which undergoes photoluminescence (PL) in the visible spectrum (λem monomer = 384 nm in 0.01 mg mL–1 cyclohexane solution, full band half-width: 1900 cm–1) with moderate quantum yields of its unsubstituted form (φ = 0.32).42,43 With the aim of tuning its optical properties, enormous effort has been made to develop reactions to selectively substitute pyrene asymmetrically and symmetrically.44−47 In fact, substituted pyrenes may display bathochromically shifted emission bands and largely elevated quantum yields.48,49 Therefore, they have become prominent target structures for a variety of applications in materials science.50−54
Pyrene is the textbook example of a PAH that forms excited dimers (excimers) at increased concentrations, resulting from the close interaction between one pyrene molecule in the electronically excited state and the other one in the ground state in coplanar orientation. The unusually high monomer lifetime (up to τf, monomer = 680 ns in degassed solutions),55 caused by a forbidden S1 → S0 transition, favors diffusion-controlled excimer formation.56 The other key factors are the planar and rigid geometry of the molecule57 as well as the strongly stabilizing short- and long-range interactions of the excited and non-excited pyrene in the optimum stacking orientation.58 Consequently, excimer formation occurs before the excited monomer would decay into the electronic ground state. Overall, these determinants induce a low excimer formation energy of pyrene (0.34 eV).55
Pyrene excimers show characteristic red-shifted (Δλem = 105 nm), broadened, and featureless PL emission bands (λem excimer = 489 mm in 1.0 mg mL–1 cyclohexane solution,43 full band half-width: 3800 cm–1) and separate lifetime components (τexcimer = 90 ns).55,58 Numerous works investigated the aggregation-dependent emission and showed that pyrenes may serve as fluorescence probes in biomolecules,59 for the detection of environmental changes such as the pH value and temperature60,61 or the detection of guest molecules such as heavy metal ions.62,63
However, for many optoelectronic applications, excimer formation can be disadvantageous. Above all, it promotes fluorescence quenching64 and limits exciton transfer processes in organic semiconductors.65 Furthermore, the broadened shape of the emission spectrum can be undesirable if an OLED with a distinctly specified emission wavelength is desired45 because this feature prevents a precise tuning of the emission color and may decrease the fluorescence efficiency in the solid or film state.45 Therefore, the number of patents featuring pyrene and its derivatives is lower66 compared to other molecules commonly applied in optoelectronics, despite its structural simplicity. In contrast, in pyrenes and other PAHs with suitably large and/or sterically demanding substituents, excimer formation may be suppressed as a close approximation of the π-planes is impeded for steric reasons and the overlap of the π-planes is incomplete.49,58,67−69
To date, four types of BN-substitution patterns in pyrenes have been reported, with one or two BN-moieties in peripheral or internal positions (Figure 1).70−74 The different isomers impressively illustrated the strong influence of the position of the BN-substitutions on the electronic structure and the optical properties as mirrored by very different absorption maxima. According to the discussed unique aggregation behavior of pyrene, the question arises how a BN-pyrene can be characterized in terms of excimer formation capability in solution and the morphologies in the solid state compared to its all-carbon analogue. One of the studies71 described that sterically demanding mesityl-substituents at the boron atom as in molecule 3 impeded the formation of excimers. Another study reveals that a dimer of 4 did not form excimers in solution, as well.75 However, in both cases, it was not evaluated in what manner the BN-unit contributed to this effect compared to a CC-pyrene with an equal substitution pattern.
Figure 1.

Four series of boron–nitrogen-substituted pyrenes as presented so far.
In the pioneering synthesis of the internally BN-substituted pyrenes by Piers and co-workers,70 unsubstituted phenyl- and alkyl-substituted BN-pyrenes (1a–e) were presented (Figure 1).
While the crystal structure of the unsubstituted BN-pyrene 4a shows a head-tail packing due to the compensation of internal dipoles, an unsubstituted CC-pyrene exhibits a sandwich herringbone pattern.76 Alkyl-substituted BN-pyrene 4e exhibits parallel sheets with a negligible overlap and therefore a lack of π-stacking. This is considered an unfavorable feature with respect to optoelectronic applications as it lowers the solid-state charge carrier mobilities.77
Based on these works, we hypothesized that the introduction of more suitable substituents should overcome these limitations and implement additional benefits. On the one hand, the potential side groups should not add a large steric hindrance to the system as this might lead to a reduced stacking behavior. On the other hand, the substituents should significantly improve the solubility in organic solvents since PAHs of this size often suffer from low solubility. Furthermore, our goal was to extend the conjugated system and assess how this alteration would affect the absorption and emission maxima. To retain the possibility of post-functionalizations, the substituent should also be easily modifiable. Based on these design principles, we selected thienyl substituents as they fulfill these requirements best and exhibit well-established reactivity.
In this work, we describe the synthesis of internally BN-substituted 5,9-dithienyl-10b-aza-10c-borapyrene BNP and its respective CC-analogue CCP. Furthermore, we present an in-depth investigation of their properties and aggregation dynamics in solution and in the solid state by means of crystallography, NMR and diffusion-ordered NMR spectroscopy (DOSY), stationary and steady-state absorption and PL spectroscopy, and time-correlated single-photon counting (TCSPC). Our mechanistic hypotheses were further supported by density functional theory (DFT) calculations.
Results and Discussion
Syntheses
The synthetic route toward BNP is based on a report by Piers and co-workers (Scheme 1).70 The syntheses were initiated by a Sonogashira cross-coupling reaction of dibromopyridine 5 and ethynylthiophene 6, affording bis(ethynylthienyl)pyridine 7 in a yield of 53%. Boracycle 8, which was required for the final formation of BNP, was synthesized in four steps following the literature procedures.
Scheme 1. Synthesis of BNP Involving the Concluding Reaction of Ethynylpyridine 7 and Boracycle 8.

For that purpose, diethynylpentane 9(78) and di(n-butyl)stannane (10)79 were reacted to form the stannacycle 11.80 A transmetalation of the latter with boron trichloride afforded boracycle 8 (for experimental details, see the Supporting Information, Section 2).78 After the reaction of pyridine 7 and the boracycle 8 including a platinum-catalyzed electrophilic cyclization at 115 °C for 15 h in a microwave reactor, the desired BNP was isolated as a yellow crystalline solid in a yield of 60%.
The synthesis of CCP is based on a procedure presented by Schreiner and co-workers (Scheme 2).81 The first key intermediate to be synthesized was 2,6-dibromo-1,1′-biphenyl (13). A Suzuki–Miyaura cross-coupling of 1,3-dibromo-2-iodobenzene (12)82 and phenylboronic acid afforded biphenyl 13 in a yield of 59%. For the synthesis of the complementary nucleophile 14, a trimethylstannyl group was attached to ethynylthiophene 6 via lithiation, followed by transmetalation with trimethyltin chloride, yielding the stannane 14 in 45% yield.
Scheme 2. Synthesis of CCP Based on a Stille Cross-Coupling Reaction and Subsequent Electrophilic Ring Closure of Biphenyl 15.

The tin functionality was chosen to access the sterically hindered positions at biphenyl 13 and to ensure a complete conversion into the desired product 15. Via a Stille cross-coupling reaction between biphenyl 13 and thiophene 14, bis(thienylethynyl)biphenyl 15 was synthesized in an excellent yield. The concluding electrocyclic ring closure was then performed by heating the reactants with platinum(II) chloride in toluene for 2 d at 120 °C, affording CCP as a colorless solid in 36% yield.
While CCP was stable under ambient conditions for an indefinite period, BNP underwent slow decomposition, which was accelerated in nucleophilic solvents such as water or methanol.
Crystal Structure Analysis
Needle-shaped crystals of BNP and CCP were obtained by slow evaporation of toluene. The crystal structure of BNP (CCDC deposition number: 2040669) was determined via a conventional single-crystal X-ray diffraction experiment at 100 K using a Mo-Kα in-house microfocus source.
To obtain the structure of CCP (CCDC deposition number: 2040670), a synchrotron X-ray diffraction experiment was conducted at 20 K using the beamline BL02B1 of SPring-8, Japan. For further crystallographic and measurement details, see the Supporting Information (Section 4).
The pyrene scaffolds of BNP and CCP are planar, and the orientations of the peripheral thiophene rings with respect to the pyrene scaffolds are similar (φ = 45° for BNP and φ = 48° for CCP, Figure 2a,d). However, the dihedral angles between the two planes of the thienyl units differ significantly (φ = 81.8° for BNP and φ = 28.5° for CCP). This has an enormous impact on the crystal packing motifs. Although both BNP and CCP form herringbone patterns such as the reported alkyl-substituted BN-pyrene 4e and the unsubstituted pyrene, the molecule–molecule stacking behavior differs significantly. Figure 2b,e reveals that the pyrene units are displaced with respect to each other in BNP, while they overlap in CCP. In contrast, the unsubstituted BN-pyrene 4a forms head-to-tail π-stacks with opposed boron and nitrogen atoms. Different from that, the distorted thienyl units in BNP obviously prevent a close approximation of the molecules, at which the attracting electrostatic forces between boron and nitrogen would dominate the crystal packing.
Figure 2.
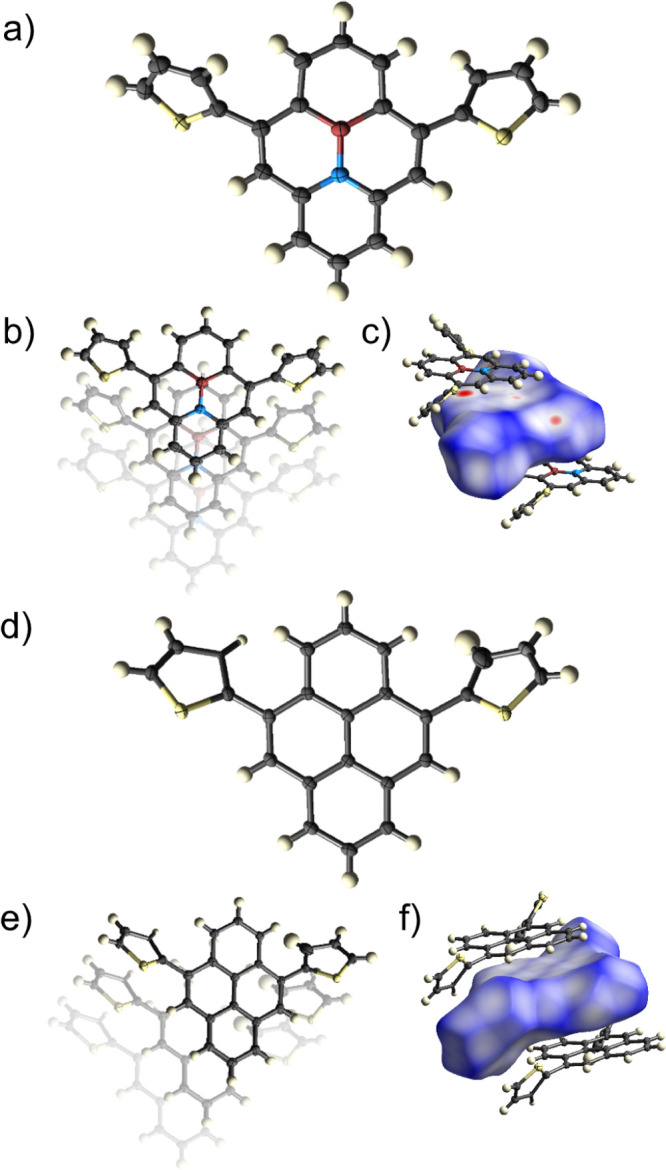
Solid-state molecular structures of BNP (a) and CCP (d) as well as the crystal packing of BNP (b) and CCP (e). Also presented are the Hirshfeld surfaces with the dnorm color coded onto them on a scale from −0.14 (red), 0 (white), to 1.26 (blue) [BNP: (c), CCP: (f)]. Only the major disorder component is shown for both BNP and CCP. The atomic displacement parameters are at the 50% probability level.
To further analyze this, a Hirshfeld surface analysis83 was performed. The displaced crystal packing in BNP leads to some distinct close atom–atom contacts (red spots on the Hirshfeld surface) in BNP, in particular a B···C contact of 3.4 Å distance (Figure 2c).
In CCP, there is no such specific close atom–atom contact, visualized by the absence of any red marker on the Hirshfeld surface. The pyrene centroid–centroid distance in CCP is 4.0 Å, whereas it is 4.8 Å in BNP. The plane-to-plane distances are equal (3.5 Å for both BNP and CCP), a value which was reported for unsubstituted pyrene and BN-pyrene as well.70,84 Consequently, in BNP and CCP, a charge carrier transport between the pyrene planes should be possible.68,70,85,86 While the overlap area of two stacked molecules of BNP is 35%, it is largely elevated in CCP (58%), which is comparable with unsubstituted pyrene (60%) and a number of substituted pyrenes that throughout exhibit excimer formation in the solid state.68 We will discuss the consequences of this overlap angle on the optical properties later in the article.
The fingerprint plots shown in the Supporting Information (Section 4, Figures S9 and S10) confirm the different nature of interactions in both crystal structures with close C···C contacts (indicative of π–π interactions) accounting for 19% of all contacts in CCP, while they are only half as frequent in BNP (9.5%, including contacts involving B and N atoms).
The nature of the forces behind the crystal packing contacts can be elucidated with a model energy analysis, here based on calculations at the B3LYP87,88 /6-31G(d,p)89 level of theory. The analysis shows the total interaction energy of all molecular pairs in the first coordination sphere in relation to the asymmetric unit and the corresponding electrostatic, dispersion, polarization, and Pauli repulsion terms (see the Supporting Information, Section 4, Tables S2 and S3 for a summary of these energy terms).90BNP is more stabilized than CCP by 14 kJ mol–1 in its crystal packing among the first coordination sphere. This stabilization derives from the significantly higher electrostatic component, which is a direct consequence of the BN-substitution. In turn, the dispersion term is slightly higher in CCP than in BNP. The interaction (plus its symmetry-related inverse) depicted in Figure 2c,f, where the Hirshfeld surface encloses the asymmetric unit, is the most energetically dominant one in both compounds. In both cases, dispersion forces are the main stabilizing factor, but they are 25 kJ mol–1 higher in CCP than in BNP, while in BNP, the electrostatic term is 9 kJ mol–1 higher than in CCP. This indicates that the mechanism of charge transfer in the solid state is different for the two compounds, with considerable consequences for the spectroscopic properties as discussed below.
Summarizing the discussed features, BNP and CCP are structurally very similar as single molecules, and both form herringbone patterns with a plane distance of 3.5 Å. Although a dominance of electrostatic interactions of boron and nitrogen atoms in the neighboring molecules of BNP as a structure-determining motif is prevented, significant differences in the centroid–centroid distances, overlap areas, and dihedral angles involving the thienyl units are found, which must be a consequence of the increased electrostatic term in BNP.
NMR Spectroscopy
All signals in the 1H and 13C{1H} NMR spectra of CCP and BNP were located in the aromatic region. As a main trend in the 1H NMR spectrum of BNP (solvent: C6D6), the chemical shifts of the outer ring protons differed significantly both in comparison with each other and with their counterparts in CCP. While the proton resonances in the pyridine ring (Figure 3, H-a and H-b) are shifted upfield, these are shifted downfield in the borabenzene ring (Figure 3, H-d and H-e). It is moreover significant that the H-h of BNP is considerably shifted downfield due to the sterical interaction with the borabenzene ring.
Figure 3.

Comparison of the proton resonances in the respective 1H NMR spectra of BNP (top) and CCP (bottom). For better perceptibility, the signals of the solvent (C6D6) were omitted.
This indicates the high electronic influence of electropositive boron and electronegative nitrogen in close proximity as it determines the degree of aromaticity (see the section on calculations).
Interestingly, the shifts of the outer ring protons of BNP were far smaller compared to those of CCP, when chloroform was chosen as the solvent (see the Supporting Information, Section 3.14 and Figure S1). This could arise from increased charge compensation, as chloroform is a moderate H-bond donor, and is an indication of a different solvent-dependent behavior of both molecules.
We then performed diffusion-ordered NMR spectroscopy (DOSY) experiments of BNP in CDCl3 at concentrations of 9.33 and 0.15 mg mL–1, which was close to the lower detection limit of the NMR spectrometer. The solvodynamic radius was determined to be 9.69 ± 6.7 × 10–3 Å and 10.05 ± 6.0 × 10–2 Å (see the Supporting Information, Section 3.15, Figures S2 and S3, and Table S2). Therefore, it can be assumed that there is no concentration effect on aggregation in the studied range, and there is no evidence of the presence of different ground-state aggregates. As the nature of aggregations was unequivocally proven for CCP (see next chapters), these measurements were not performed for this analogue.
Optical Spectroscopy in Solution
The most significant data from the optical measurements are denoted in Table 1.91
Table 1. Photophysical Properties of BNP and CCP in DCM Solution and in the Solid State.
| Compound | λabs (DCM) [nm]a,b | ε (CHCl3) [mol–1 L–1 cm–1] | λem (DCM)[nm]a,b | Stokes shift (DCM) [cm–1] | Φem (DCM) | FLT (DCM) [ns] | λem (solid) [nm]a | Φem (solid) | FLT (solid) [ns] |
|---|---|---|---|---|---|---|---|---|---|
| BNP | 270, 335, 452 | 15 486 | 531 | 3300 | 0.03 | 11.8d | 541 | <0.01 | 2.4g |
| CCP | 283, 351 | 23 052 | 403, 494c | 3700 | 0.05 | 3.7,e 7.3f | 432, ∼500 | 0.03 | 0.6,h 6.4i |
Values in bold represent the intensity maxima.
c ∼ 1 × 10–3 mg mL–1.
The local maximum of CCP at λem = 494 nm was only found at c = 10 mg mL–1.
λem = 530 nm.
λem = 420 nm.
λem = 570 nm.
λem = 540 nm.
λem = 432 nm.
λem = 486 nm.
The stationary absorption and PL spectra were recorded from DCM solutions at c ∼ 1 × 10–3 mg mL–1. Regarding the absorption of BNP and CCP, the intensity maximum of BNP was red-shifted by Δλabs = 52 nm to λabs = 335 nm compared to the highest intensity band of CCP at λabs = 283 nm (Figure 4).
Figure 4.
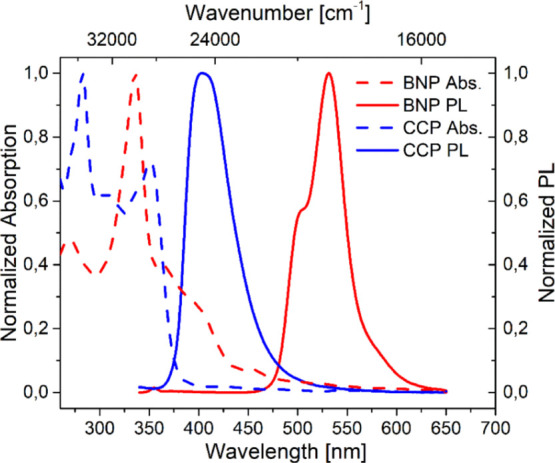
Normalized absorption (dashed lines) and emission (continuous lines) spectra of BNP (red) and CCP (blue) measured in DCM at c ∼ 1 × 10–3 mg mL–1. The excitation wavelengths were λex = 300 nm (CCP) and λex = 330 nm (BNP).
The bathochromic shift in both the absorption and emission has been reported for BN-substituted pyrenes70 and is a result of the different electronic structures of CCP and BNP (for a detailed discussion of this effect, see the section on calculations). The lowest energy local maximum of CCP was found at λabs = 351 nm.
The observed band shapes differed significantly. While BNP displayed one major absorption band with a high intensity (ε = 15 486 mol–1 L cm–1 at λabs = 336 nm, see the Supporting Information, Section 5.1 and Figure S14, the differences in absorption maximum are due to the solvent used, which was chloroform), the absorption curve of CCP was broader, and a protruding maximum intensity band was not present (ε = 23 052 mol–1 L cm–1 at λabs = 283 nm, see the Supporting Information, Section 5.1 and Figure S15).
The PL spectrum of CCP showed a broad, featureless emission band with an intensity maximum at λem = 403 nm, in contrast to the vibronic fine structuring of unsubstituted pyrenes with five exposed bands56 and the four exposed bands of BN-pyrene.70 For several mono-, di-, and tetrathienyl substitutes of pyrene, this merging effect of the emission bands of the pyrene unit and the thienyl substituents has been reported92,93 as a result of the decreasing energies of non-radiative deactivations. This observation is caused by a uniform delocalization of the frontier molecular orbitals among the pyrenyl and thienyl units.94,95
For BNP, the intensity maximum was found at λem = 531 nm, and in contrast to CCP, two shoulder signals were observable (λem ∼ 500 and 575 nm). This slight vibronic resolution is a first indication that the emissive species is monomeric. With regard to the presented diphenyl-substituted BN-pyrene (λem = 489 and 522 nm),70 only a slight red shift of Δλem = 9 nm was determined. In addition, the emission band of BNP was narrowed compared to CCP, as evidenced by a full width at half-maximum of 1950 cm–1 for BNP compared to 3080 cm–1 for CCP.
In comparison with BN-pyrene (λabs = 321 nm and λem = 514 nm in DCM)70 and CC-pyrene (λabs = 335 nm and λem = 384 nm in cyclohexane),43 the peripheral thienyl units induce a bathochromic shift of both absorption and emission of about Δλ = 15–20 nm in all cases, reflecting an effective mixing of the respective frontier orbitals of pyrene and thiophene that are involved in the transitions in solution.
The quantum yields of both BNP (Φ = 0.03) and CCP (Φ = 0.05) were decreased compared to their unsubstituted congeners (Φ = 0.15 for BN-pyrene and Φ = 0.32 for CC-pyrene), which is a consequence of both the increased atomic mass and the formation of additional weak hydrogen bonds of the sulfur atom of the thienyl unit (the S–H interactions account for 12.6% in BNP and 14.7% in CCP, see the Supporting Information, Section 4 and Figures S10 and S11).96,97
In order to analyze the different species in the ground and excited state, steady-state absorption and PL measurements were performed as a function of concentration (Figure 5, a,b: BNP; c,d: CCP).
Figure 5.

Normalized concentration-dependent steady-state absorption (a,c) and emission (b,d) spectra for BNP (a,b) and CCP (c,d) solutions in DCM. The excitation wavelength was λex = 330 nm.
Both BNP and CCP solutions in DCM showed no significant alteration in the absorption curves upon concentration changes. Hence, the aggregation behavior of both compounds in the ground state does not depend on the concentrations in the studied region.
An examination of the PL of CCP revealed an emerging broadened band at λem = 494 nm at increased concentrations. Such behavior is characteristic for excimer formation in all carbon pyrenes.55 The PL spectrum of BNP remained nearly unchanged at different concentrations, hinting at the absence of excimers at increased concentrations.
To analyze a potential solvatochromism and possible aggregation behavior, concentration-dependent PL measurements were performed in solvents of different polarities and dielectric constants (cyclohexane, DCM, ethyl acetate, and methanol, see the Supporting Information, Section 5.2 and Figure S16).
For BNP, a variation of the solvent did not have a notable impact on the emission maxima and only a minor impact on the intensity of the shoulder signal at λem ∼500 nm (Figure 6, DCM and methanol as examples).
Figure 6.
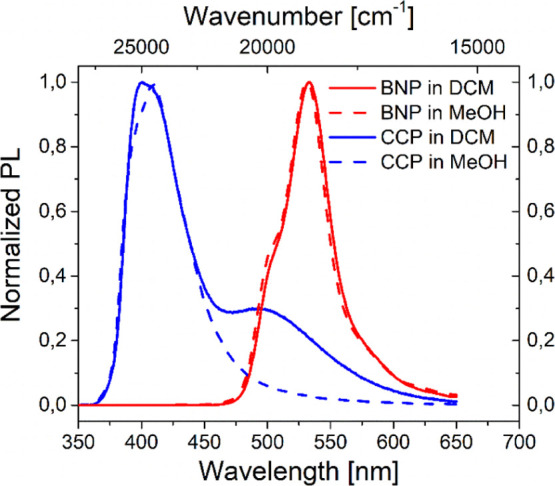
Normalized PL spectra of BNP and CCP in DCM and methanol at c = 10 mg mL–1.
As excitation spectra were unchanged up to concentrations as low as c = 4.3 × 10–6 mg mL–1 besides that (see the Supporting Information, Section 5.5 and Figure S24), the presence of a hypsochromically shifted ground-state aggregate in solution is unlikely. Contrary to Piers’ work,70 no bathochromically shifted band was identified; therefore, excimer formation is not plausible.
In contrast, the intensity of the characteristic excimer emission band of CCP was significantly solvent dependent, as it was absent in methanol and most intense in DCM. This could be a solubility issue, as all tested solvents except DCM did not allow an entire dissolution at c ≥ 1 mg mL–1. Furthermore, methanol may cause more intense interactions of the thienyl units, also leading to decreased pyrene π–π-interactions.98 In all solvents at different concentrations, the monomer emission band of CCP remained nearly unchanged.
The PL lifetimes of CCP and BNP were determined at different concentrations by TCSPC experiments using non-degassed DCM as the solvent (Figure 7). In TCSPC measurements, the emitted single photons are counted as a function of time, resulting in a photon distribution over time.
Figure 7.
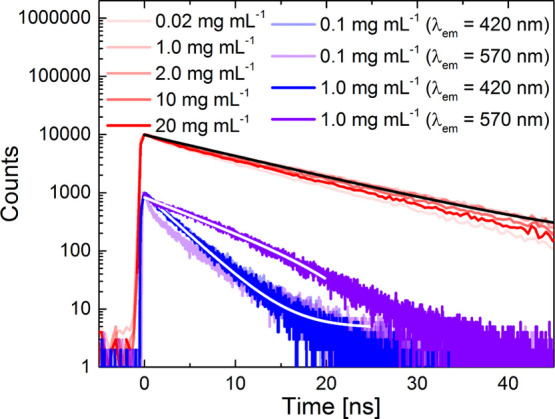
TCSPC measurements of BNP (red, λex = 355 nm and λem = 530 nm) and CCP (blue and violet tones, λex = 371 nm and λem = 420 and 570 nm) in DCM solutions and regarding lifetime fittings (black/white lines). The dynamic range of the measurements was 103 (CCP) and 104 (BNP), which explains the different onset values of the respective curves.
The lifetimes (FLT) of BNP (τ = 11.8 ns) and CCP (τ = 3.7 ns), determined by fitting the decay curves at c = 0.1 mg mL–1, differed remarkably (Figure 7). However, both of them followed single exponential decays in broad sections of the curves.
The relative brevity of the lifetimes is an immediate consequence of the thienyl units in 4,10-positions, which contribute to most of the natural transition orbitals (NTOs) that are involved in the fluorescence process (see the section on calculations and the Supporting Information, Section 6.2 and Figure S29) and enhance the symmetry-forbidden S1 → S0 transitions. In contrast, unsubstituted CC-pyrene (τ up to 680 ns)58 and unsubstituted BN-pyrene (τ = 70 ns)70 exhibit largely increased lifetimes, which can doubtlessly be attributed to forbidden transitions at least for CC-pyrene.69
As oxygen is a well-known quencher of fluorescence,55,99 we also measured the PL lifetimes in degassed DCM (see the Supporting Information, Section 5.3 and Figures S17 and S18). As expected, the lifetimes were increased, however only in the range of 4–14%, indicating that oxygen did not lead to major quenching.
Recording the lifetime curves of CCP at different concentrations (c = 0.1 and 1.0 mg mL–1) allowed us to analyze the decays of both lifetime components (λem = 420 nm for the excited monomer and λem = 570 nm for the excimer), deriving from the existence of excimers only at higher concentrations. While only monomer decay was observed at c = 0.1 mg mL–1, the excimer decay became apparent at c = 1.0 mg mL–1 (τ = 7.3 ns) when setting the emission wavelength to λem = 570 nm. In contrast, only one lifetime of BNP was observed when screening low to medium concentrations (Figure 7). This suggests the presence of only one emissive species and contradicts the presence of excimers. When the concentration became very high, a notable decrease of the lifetimes was observed due to enhanced collision-induced quenching. All lifetimes were independent of the fluence, which was tested by setting it to values between 25 and 500 μW (see the Supporting Information, Section 5.3 and Figure S19).
To obtain an explicit representation of the differences in excimer formation, we measured the sample emission curves at different time delays using an intensified charge-coupled device (iCCD) setup. The excitation light pulse at λex = 355 nm results from the double frequency of λex = 710 nm, generated by a non-collinear optical parametric amplifier. We then plotted separate emission curves for a range of lifetime periods as well as separate lifetime curves for different wavelength sections. The excimer emission could be visualized by iCCD images.
In highly diluted solutions of CCP (c = 0.05 mg mL–1), no excimer formation could be detected as the decays were comparable for four investigated emission wavelengths, and the emission curve remained almost unchanged for all lifetime sections (Figure 8).
Figure 8.
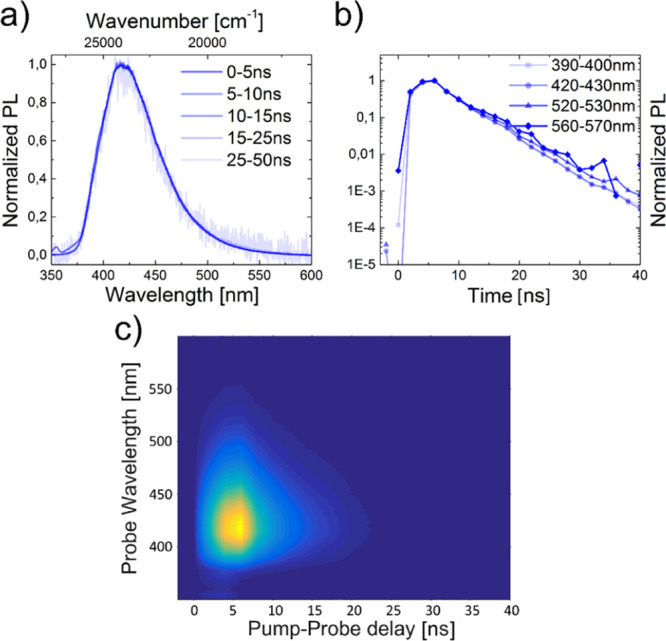
Segmented emission spectra (a) and lifetime curves (b) as well as iCCD images (c) of CCP at c = 0.05 mg mL–1 and an excitation wavelength of λex = 355 nm. The solvent was non-degassed DCM.
In contrast, when the concentration was increased to c = 5 mg mL–1, the emission decays at different wavelengths diverged (Figure 9).
Figure 9.
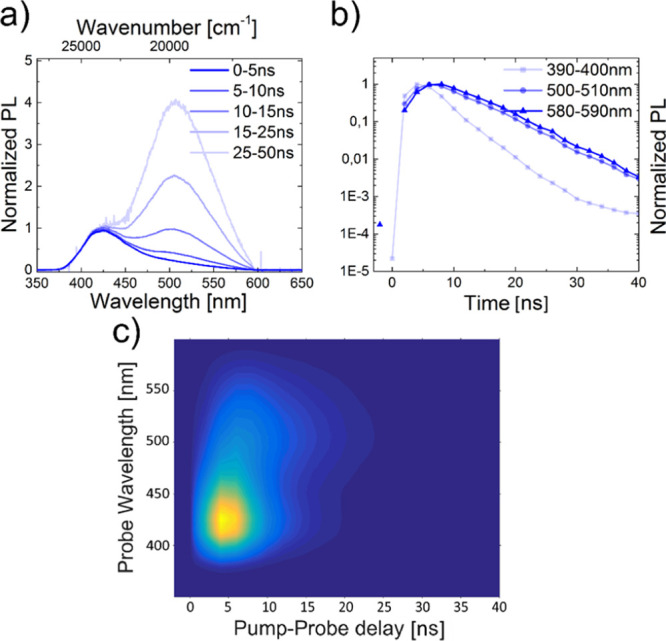
Segmented emission spectra (a) and lifetime curves (b) as well as iCCD images (c) of CCP at c = 5.0 mg mL–1 and an excitation wavelength of λex = 355 nm. The solvent was non-degassed DCM.
While the component of the emission spectrum at λem = 390–400 nm decayed with a lifetime of τ ∼ 3–4 ns following a single exponential decay, the components at λem = 500–510 nm and λem = 580–590 nm decayed much more slowly (Figure 9b). Furthermore, the lower energy band at λem = 507 nm became the most intense band when displaying the emission components that are associated with long lifetimes (Figure 9a). The results strongly support excimer formation of CCP at high concentrations, coexisting with monomers.
In contrast, no secondary emission band was present for BNP for both investigated concentrations (c = 0.2 and 5.0 mg mL–1). Moreover, no significant differences were found in the lifetimes of the different sections of emission wavelengths (Figures 10 and 11).
Figure 10.
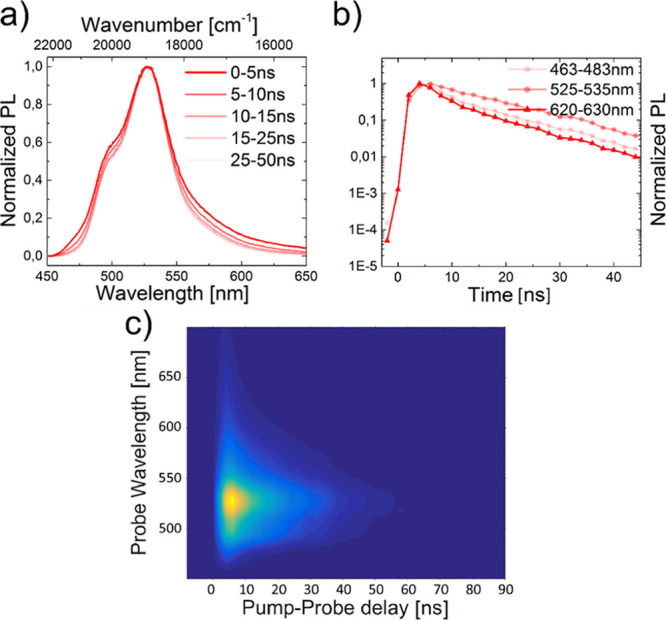
Segmented emission spectra (a) and lifetime curves (b) as well as iCCD images (c) of BNP at c = 0.2 mg mL–1 and an excitation wavelength of λex = 355 nm. The solvent was non-degassed DCM.
Figure 11.
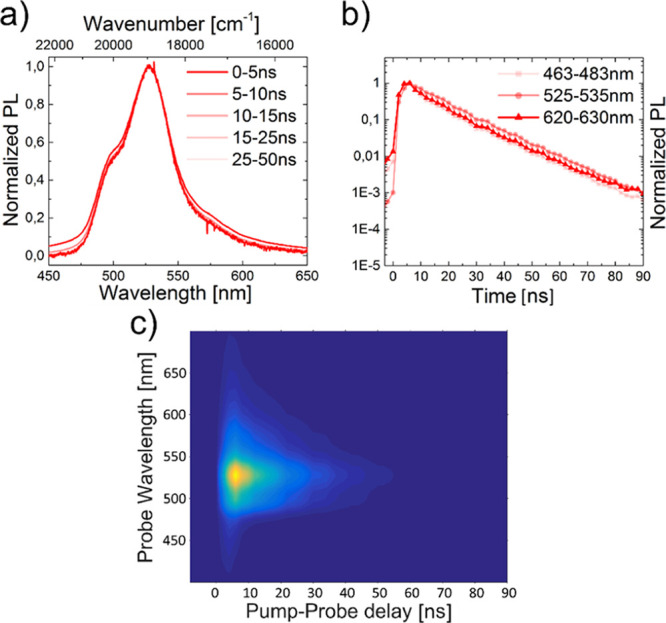
Segmented emission spectra (a) and lifetime curves (b) as well as iCCD images (c) of BNP at c = 5.0 mg mL–1 and an excitation wavelength of λex = 355 nm. The solvent was non-degassed DCM.
Optical Spectroscopy in the Solid State
In general, the absorption bands in solution were well reflected by the results from solid-state measurements (see the Supporting Information, Section 5.4 and Figure S22), with negligible deviations of the intensity maxima. However, in contrast to CCP, the bands of BNP were significantly broadened.
Similar PL bands as in solution were also found in the solid state. However, a significantly slower decrease of the PL intensity at the red edge of the spectrum was noted for both derivatives. While a solution of BNP in DCM revealed its emission maximum at λem = 531 nm, the latter was only slightly shifted to λem = 541 nm in the solid state (Figure 12). Moreover, the lifetime was significantly reduced in the solid state (τ = 2.41 ns, see the Supporting Information, Section 5.3 and Figure S20).
Figure 12.
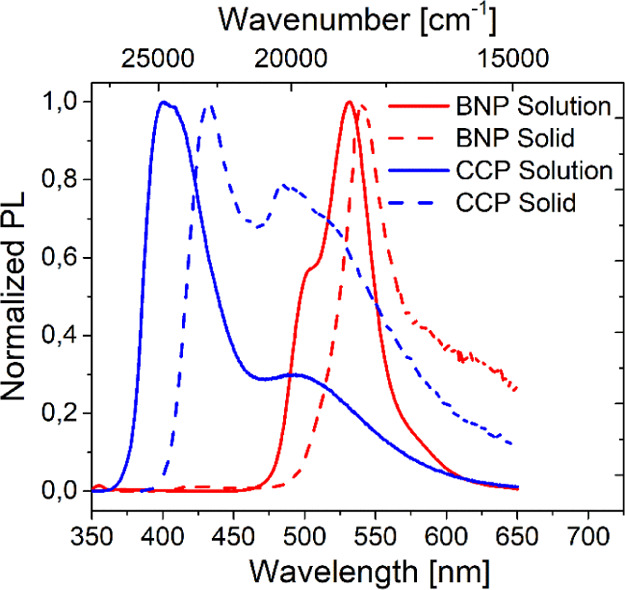
Normalized PL spectra of BNP (red) and CCP (blue) in DCM solution (continuous, c = 10 mg mL–1) and the solid state (dashed).
As no bathochromically shifted bands were present, no evidence is given for the presence of solid-state excimers of BNP. This can be explained by the small interplanar overlap area of 35% as can be seen from the crystal structure.
The solid-state emission spectrum of CCP revealed the presence of both the monomeric emissive species (λem = 432 nm) and the excimer (λem ∼ 500 nm), similar to the results from PL spectroscopy in solution at the highest investigated concentration (c = 10 mg mL–1). Apparently, the large molecule-to-molecule overlap area of 58% allows effective excimer formation in this case. The high intensity of the excimer band in the solid state can be explained by the already closely adjoined molecules (plane-to-plane distance: 3.5 Å in a single crystal), which readily allow excimer formation in contrast to a diffusion-controlled process in solution. Two lifetime components were noticed (τ = 0.64 ns/6.40 ns, see the Supporting Information, Section 5.3 and Figure S21), which support the presence of excimers.
The excitation spectra of BNP and CCP are shown in the Supporting Information (Section 5.4 and Figure S23). The quantum yields in the solid state were low (Φ < 0.01 for BNP and Φ = 0.03 for CCP), which indicates a predominating aggregation-caused quenching (ACQ).100,101
Calculations
We calculated the optimized geometries in S0 and S1, NTOs,102 as well as the absorption and emission spectra of CCP and BNP with ORCA 4.2.0103 using time-dependent DFT (TD-DFT)104−106 at the B3LYP107−109 /cc-pVDZ110 level of theory (for details, see the Supporting Information, Section 6). The absorption spectra were well reproduced as all local maxima were mapped by calculations with deviations of only Δλabs ±15 nm (Figure 13).
Figure 13.

Measured normalized absorption bands and calculated absorption sticks of BNP (a) and CCP (b).
The emission maxima of the calculated bands match with the experimental results in good approximation and even the vibronic bands that suffer from broadening in the experimental spectra were reflected correctly (Figure 14).
Figure 14.
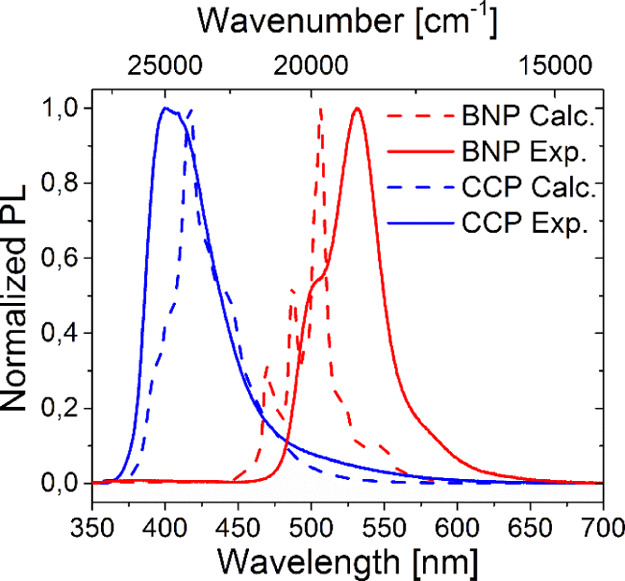
Measured (continuous, solvent: DCM) and calculated (dashed) normalized PL spectra of BNP (red) and CCP (blue).
Furthermore, a decreased full width at half-maximum of BNP compared to CCP was obvious. Both molecules clearly fluoresce out of a π–π* state as evidenced by NTO calculations (see the Supporting Information, Section 6.2 and Figure S29). The results indicate that the decisive factor for the bathochromic shifts in the emission of BNP compared to its carbonaceous counterpart is indeed the different electronic structure, while aggregation effects may play an additional but subordinate role in the solution PL spectra at low concentrations.
NICS(0)111 (nucleus-independent chemical shift) values were calculated at the MP2112 /cc-pVDZ level of theory. The results confirmed the trend that the rings at the long end of the pyrene core were more aromatic (NICS(0) ∼ −12.5 ppm for CCP and −11.9 and −13.7 ppm for BNP) than the rings at the short end (NICS(0) = −4.0 ppm for CCP and −6.8 ppm for BNP); moreover, the latter results pointed out an overall increased aromaticity in BNP.
Conclusions
A BN-dithienylpyrene BNP and its internally CC-substituted counterpart CCP were synthesized successfully by employing cross-coupling reactions, transmetalations, and concluding electrophilic cyclizations. While BNP was obtained in a total yield of 23%, the overall yield of CCP was 21%. The crystal packing of CCP is comparable with the unsubstituted and aryl-substituted pyrenes, showing classical π–π interactions and a pyrene–pyrene overlap area of 58%. In contrast, the BN-unit of BNP induces a higher electrostatic term to the attraction forces. As the molecular structures are very similar besides that, the dipolar interactions cause a more displaced crystal packing with a significantly reduced overlap area (35%). However, the role of the distorted thienyl units should not be underestimated, as they may prevent a head-to-tail aggregation of the opposed boron and nitrogen atoms. The differences in the spectro-optical behavior of both compounds are immense. Due to the divergent electronic structures, a bathochromic shift of Δλem = 128 nm for the emission maximum of BNP was observed compared to CCP, corresponding to the results of TD-DFT calculations. Compared to the unsubstituted derivatives, both emission bands are featureless due to the electronic impact of the thienyl substituents, which moreover cause a bathochromic shift of λabs and λem ∼ 15–20 nm. As can be seen from the steady-state experiments, the monomer emission bands of BNP and CCP are barely solvent and concentration dependent. Compared to Piers’ BN-pyrene, no evidence was given that BNP forms excimers in solution. In contrast, CCP showed excimer formation at high concentrations (λem monomer = 403 nm and λem excimer = 494 nm at c = 10.0 mg mL–1), which is a classical feature of pyrenes. The presence of excimers was also proven by TCSPC lifetime measurements, showing a secondary emission decay. Unlike BNP, CCP shows excimer formation in the solid state as well. The differences in excimer formation can clearly be attributed to the significant distinction in the π–π-overlap areas. Therefore, despite having the same plane-to-plane distance and a larger energy gain upon stacking in BNP, it is a different centroid–centroid length, sideway shift of each molecule, and thus a different transition dipole moment that is likely to be the reason for an entirely different excited-state photochemistry. We conclude that a BN-substitution in combination with the implementation of flanking thienyl groups can be a useful tool to suppress excimer formation in pyrene solutions and in the solid state. As BNP can be post-functionalized selectively at the thiophene sites and should be electrochemically stable such as the parent BN-pyrene,70 the generated knowledge could be fundamental for the design of novel materials for the application in organic electronic devices. While the issue of ACQ limits the potential as an electroluminescent compound,113 the investigation of the charge carrier generation and transporting capability of BN-PAHs is of high interest. Especially, an analysis of the competing redox processes at the pyrene scaffold and the thienyl units should be performed in the future. Consequently, the implementation of BNP and related BN-pyrenes into field-effect transistors as well as electropolymerization should be envisaged.
Acknowledgments
Y.A., P.J. Gliese, S.A.W.S., A.S., R.P., L.A.M., and S.G. thank the German Research Foundation (DFG) for funding as part of the Emmy-Noether Program (STA 1195/2-1 and GR 4451/1-1). This research has been supported by the Institutional Strategy of the University of Bremen, funded by the German Excellence Initiative. N.C.B. and P.R. thank the DFG for partial funding under Germany’s Excellence Strategy within the Cluster of Excellence PhoenixD (EXC 2122, Project ID 390833453) and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 714429). A.A.B. is a Royal Society University Research Fellow. J.Z. thanks the China Scholarship Council for the financial support for his Ph.D. studies and the Science and Technology Innovation Program of Hunan Province (2021RC3083). The synchrotron measurements were supported by SPring-8 under proposal no. 2016B1204.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcc.1c08812.
Experimental procedures, crystal data, NMR spectra, additional optical measurements, and calculation details (PDF)
Author Contributions
∥∥ Y.A. and P.J. Gliese contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Campbell P. G.; Marwitz A. J. V.; Liu S.-Y. Recent Advances in Azaborine Chemistry. Angew. Chem., Int. Ed. 2012, 51, 6074–6092. 10.1002/anie.201200063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosdet M. J. D.; Piers W. E. B-N as a C-C Substitute in Aromatic Systems. Can. J. Chem. 2009, 87, 8–29. 10.1139/v08-110. [DOI] [Google Scholar]
- Morgan M. M.; Piers W. E. Efficient Synthetic Methods for the Installation of Boron-Nitrogen Bonds in Conjugated Organic Molecules. Dalton Trans. 2016, 45, 5920–5924. 10.1039/c5dt03991f. [DOI] [PubMed] [Google Scholar]
- Wang J.-Y.; Pei J. BN-Embedded Aromatics for Optoelectronic Applications. Chin. Chem. Lett. 2016, 27, 1139–1146. 10.1016/j.cclet.2016.06.014. [DOI] [Google Scholar]
- Chen G.; Zakharov L. N.; Bowden M. E.; Karkamkar A. J.; Whittemore S. M.; Garner E. B.; Mikulas T. C.; Dixon D. A.; Autrey T.; Liu S.-Y. Bis-BN Cyclohexane: A Remarkably Kinetically Stable Chemical Hydrogen Storage Material. J. Am. Chem. Soc. 2015, 137, 134–137. 10.1021/ja511766p. [DOI] [PubMed] [Google Scholar]
- Campbell P. G.; Zakharov L. N.; Grant D. J.; Dixon D. A.; Liu S.-Y. Hydrogen Storage by Boron–Nitrogen Heterocycles: A Simple Route for Spent Fuel Regeneration. J. Am. Chem. Soc. 2010, 132, 3289–3291. 10.1021/ja9106622. [DOI] [PubMed] [Google Scholar]
- Wang X.-Y.; Lin H.-R.; Lei T.; Yang D.-C.; Zhuang F.-D.; Wang J.-Y.; Yuan S.-C.; Pei J. Azaborine Compounds for Organic Field-Effect Transistors: Efficient Synthesis, Remarkable Stability, and BN Dipole Interactions. Angew. Chem., Int. Ed. 2013, 52, 3117–3120. 10.1002/anie.201209706. [DOI] [PubMed] [Google Scholar]
- Wang X.; Zhang F.; Liu J.; Tang R.; Fu Y.; Wu D.; Xu Q.; Zhuang X.; He G.; Feng X. Ladder-Type BN-Embedded Heteroacenes with Blue Emission. Org. Lett. 2013, 15, 5714–5717. 10.1021/ol402745r. [DOI] [PubMed] [Google Scholar]
- Li G.; Zhao Y.; Li J.; Cao J.; Zhu J.; Sun X. W.; Zhang Q. Synthesis, Characterization, Physical Properties, and OLED Application of Single BN-Fused Perylene Diimide. J. Org. Chem. 2015, 80, 196–203. 10.1021/jo502296z. [DOI] [PubMed] [Google Scholar]
- Zhao R.; Dou C.; Xie Z.; Liu J.; Wang L. Polymer Acceptor Based on B←N Units with Enhanced Electron Mobility for Efficient All-Polymer Solar Cells. Angew. Chem., Int. Ed. 2016, 55, 5313–5317. 10.1002/anie.201601305. [DOI] [PubMed] [Google Scholar]
- Zhao P.; Nettleton D. O.; Karki R. G.; Zécri F. J.; Liu S.-Y. Medicinal Chemistry Profiling of Monocyclic 1,2-Azaborines. ChemMedChem 2017, 12, 358–361. 10.1002/cmdc.201700047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiedemann B.; Gliese P. J.; Hoffmann J.; Lawrence P. G.; Sönnichsen F. D.; Staubitz A. High Molecular Weight Poly(N-methyl-B-vinylazaborine) - A Semi-Inorganic B-N Polystyrene Analogue. Chem. Commun. 2017, 53, 7258–7261. 10.1039/c6cc08599g. [DOI] [PubMed] [Google Scholar]
- Lin H.; McConnell C. R.; Jilus B.; Liu S.-Y.; Jäkle F. Changing up BN-Polystyrene: Effect of Substitution Pattern on the Free-Radical Polymerization and Polymer Properties. Macromolecules 2019, 52, 4500–4509. 10.1021/acs.macromol.9b00466. [DOI] [Google Scholar]
- van de Wouw H. L.; Lee J. Y.; Awuyah E. C.; Klausen R. S. ABN Aromatic Ring Strategy for Tunable Hydroxy Content in Polystyrene. Angew. Chem., Int. Ed. 2018, 57, 1673–1677. 10.1002/anie.201711650. [DOI] [PubMed] [Google Scholar]
- van de Wouw H. L.; Lee J. Y.; Klausen R. S. Gram-Scale Free Radical Polymerization of an Azaborine Vinyl Monomer. Chem. Commun. 2017, 53, 7262–7265. 10.1039/c7cc02300f. [DOI] [PubMed] [Google Scholar]
- Pan J.; Kampf J. W.; Ashe A. J. 1,2-Azaboratabenzene: A Heterocyclic π-Ligand with an Adjustable Basicity at Nitrogen. Organometallics 2004, 23, 5626–5629. 10.1021/om049399g. [DOI] [Google Scholar]
- Pan J.; Kampf J. W.; Ashe A. J. Tricarbonylchromium Complexes of 1,2-Dihydro-1,2-benzazaborines. Organometallics 2009, 28, 506–511. 10.1021/om800383v. [DOI] [Google Scholar]
- Baggett A. W.; Vasiliu M.; Li B.; Dixon D. A.; Liu S.-Y. Late-Stage Functionalization of 1,2-Dihydro-1,2-azaborines via Regioselective Iridium-Catalyzed C-H Borylation: The Development of a New N,N-Bidentate Ligand Scaffold. J. Am. Chem. Soc. 2015, 137, 5536–5541. 10.1021/jacs.5b01916. [DOI] [PubMed] [Google Scholar]
- Marwitz A. J. V.; Matus M. H.; Zakharov L. N.; Dixon D. A.; Liu S.-Y. A Hybrid Organic/Inorganic Benzene. Angew. Chem., Int. Ed. 2009, 48, 973–977. 10.1002/anie.200805554. [DOI] [PubMed] [Google Scholar]
- Braunschweig H.; Geetharani K.; Jimenez-Halla J. O. C.; Schäfer M. Direct Synthetic Route to Functionalized 1,2-Azaborinines. Angew. Chem., Int. Ed. 2014, 53, 3500–3504. 10.1002/anie.201309707. [DOI] [PubMed] [Google Scholar]
- Lu J.-S.; Ko S.-B.; Walters N. R.; Kang Y.; Sauriol F.; Wang S. Formation of Azaborines by Photoelimination of B,N-Heterocyclic Compounds. Angew. Chem., Int. Ed. 2013, 52, 4544–4548. 10.1002/anie.201300873. [DOI] [PubMed] [Google Scholar]
- Müller M.; Maichle-Mössmer C.; Bettinger H. F. BN-Phenanthryne: Cyclotetramerization of an 1,2-Azaborine Derivative. Angew. Chem., Int. Ed. 2014, 53, 9380–9383. 10.1002/anie.201403213. [DOI] [PubMed] [Google Scholar]
- Ashe A. J.; Fang X. A Synthesis of Aromatic Five- and Six-Membered B–N Heterocycles via Ring Closing Metathesis. Org. Lett. 2000, 2, 2089–2091. 10.1021/ol0001113. [DOI] [PubMed] [Google Scholar]
- Ashe A. J.; Fang X.; Fang X.; Kampf J. W. Synthesis of 1,2-Dihydro-1,2-azaborines and Their Conversion to Tricarbonyl Chromium and Molybdenum Complexes. Organometallics 2001, 20, 5413–5418. 10.1021/om0106635. [DOI] [Google Scholar]
- Appiarius Y.; Stauch T.; Lork E.; Rusch P.; Bigall N. C.; Staubitz A. From a 1,2-Azaborinine to Large BN-PAHs via Electrophilic Cyclization: Synthesis, Characterization and Promising Optical Properties. Org. Chem. Front. 2021, 8, 10–17. 10.1039/d0qo00723d. [DOI] [Google Scholar]
- Wang X.-Y.; Zhuang F.-D.; Zhou X.; Yang D.-C.; Wang J.-Y.; Pei J. Influence of Alkyl Chain Length on the Solid-State Properties and Transistor Performance of BN-Substituted Tetrathienonaphthalenes. J. Mater. Chem. C 2014, 2, 8152–8161. 10.1039/c4tc01369g. [DOI] [Google Scholar]
- Chrostowska A.; Xu S.; Lamm A. N.; Mazière A.; Weber C. D.; Dargelos A.; Baylère P.; Graciaa A.; Liu S.-Y. UV-Photoelectron Spectroscopy of 1,2- and 1,3-Azaborines: A Combined Experimental and Computational Electronic Structure Analysis. J. Am. Chem. Soc. 2012, 134, 10279–10285. 10.1021/ja303595z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wouw H. L.; Lee J. Y.; Siegler M. A.; Klausen R. S. Innocent BN Bond Substitution in Anthracene Derivatives. Org. Biomol. Chem. 2016, 14, 3256–3263. 10.1039/c5ob02309b. [DOI] [PubMed] [Google Scholar]
- Abbey E. R.; Zakharov L. N.; Liu S.-Y. Boron in Disguise: The Parent “Fused” BN Indole. J. Am. Chem. Soc. 2011, 133, 11508–11511. 10.1021/ja205779b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann J.; Jacquemin D.; Hissler M.; Staubitz A. BN-Substituted coronene diimide donor-acceptor-donor triads: photophysical, (spectro)-electrochemical studies and Lewis behavior. J. Mater. Chem. C 2021, 9, 13926–13934. 10.1039/d1tc03034e. [DOI] [Google Scholar]
- Ishibashi J. S. A.; Marshall J. L.; Mazière A.; Lovinger G. J.; Li B.; Zakharov L. N.; Dargelos A.; Graciaa A.; Chrostowska A.; Liu S.-Y. Two BN Isosteres of Anthracene: Synthesis and Characterization. J. Am. Chem. Soc. 2014, 136, 15414–15421. 10.1021/ja508813v. [DOI] [PubMed] [Google Scholar]
- Zhuang F. D.; Sun Z. H.; Yao Z. F.; Chen Q. R.; Huang Z.; Yang J. H.; Wang J. Y.; Pei J. BN-Embedded Tetrabenzopentacene: A Pentacene Derivative with Improved Stability. Angew. Chem., Int. Ed. 2019, 58, 10708–10712. 10.1002/anie.201905601. [DOI] [PubMed] [Google Scholar]
- Abengózar A.; Sucunza D.; García-García P.; Sampedro D.; Pérez-Redondo A.; Vaquero J. J. A New Member of the BN-Phenanthrene Family: Understanding the Role of the B-N Bond Position. J. Org. Chem. 2019, 84, 7113–7122. 10.1021/acs.joc.9b00800. [DOI] [PubMed] [Google Scholar]
- Wang X.-Y.; Narita A.; Feng X.; Müllen K. B2N2-Dibenzo[a,e]pentalenes: Effect of the BN Orientation Pattern on Antiaromaticity and Optoelectronic Properties. J. Am. Chem. Soc. 2015, 137, 7668–7671. 10.1021/jacs.5b05056. [DOI] [PubMed] [Google Scholar]
- Baggett A. W.; Guo F.; Li B.; Liu S.-Y.; Jäkle F. Regioregular Synthesis of Azaborine Oligomers and a Polymer with asyn Conformation Stabilized by N–H···π Interactions. Angew. Chem., Int. Ed. 2015, 54, 11191–11195. 10.1002/anie.201504822. [DOI] [PubMed] [Google Scholar]
- Dou C.; Ding Z.; Zhang Z.; Xie Z.; Liu J.; Wang L. Developing Conjugated Polymers with High Electron Affinity by Replacing a C–C Unit with a B←N Unit. Angew. Chem., Int. Ed. 2015, 54, 3648–3652. 10.1002/anie.201411973. [DOI] [PubMed] [Google Scholar]
- Wang X.-Y.; Zhuang F.-D.; Wang R.-B.; Wang X.-C.; Cao X.-Y.; Wang J.-Y.; Pei J. A Straightforward Strategy toward Large BN-Embedded π-Systems: Synthesis, Structure, and Optoelectronic Properties of Extended BN Heterosuperbenzenes. J. Am. Chem. Soc. 2014, 136, 3764–3767. 10.1021/ja500117z. [DOI] [PubMed] [Google Scholar]
- Hatakeyama T.; Hashimoto S.; Seki S.; Nakamura M. Synthesis of BN-Fused Polycyclic Aromatics via Tandem Intramolecular Electrophilic Arene Borylation. J. Am. Chem. Soc. 2011, 133, 18614–18617. 10.1021/ja208950c. [DOI] [PubMed] [Google Scholar]
- Saint-Louis C. J.; Magill L. L.; Wilson J. A.; Schroeder A. R.; Harrell S. E.; Jackson N. S.; Trindell J. A.; Kim S.; Fisch A. R.; Munro L.; et al. The Synthesis and Characterization of Highly Fluorescent Polycyclic Azaborine Chromophores. J. Org. Chem. 2016, 81, 10955–10963. 10.1021/acs.joc.6b01998. [DOI] [PubMed] [Google Scholar]
- Bosdet M. J. D.; Jaska C. A.; Piers W. E.; Sorensen T. S.; Parvez M. Blue Fluorescent 4a-Aza-4b-boraphenanthrenes. Org. Lett. 2007, 9, 1395–1398. 10.1021/ol070328y. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Zhang L.; Sun C.; Sun W.; Liu X. BN-Phenanthrenes: Synthesis, Reactivity, and Optical Properties. Org. Lett. 2019, 21, 3476–3480. 10.1021/acs.orglett.9b00530. [DOI] [PubMed] [Google Scholar]
- Birks J. B.Photophysics of Aromatic Molecules; Wiley-Interscience: London, 1970. [Google Scholar]
- Berlman I. B.Handbook of Fluorescence Spectra of Aromatic Molecules; Academic Press: New York, 1971. [Google Scholar]
- Vollmann H.; Becker H.; Corell M.; Streeck H. Beiträge zur Kenntnis des Pyrens und seiner Derivate. Liebigs Ann. Chem. 1937, 531, 1–159. 10.1002/jlac.19375310102. [DOI] [Google Scholar]
- Figueira-Duarte T. M.; Müllen K. Pyrene-Based Materials for Organic Electronics. Chem. Rev. 2011, 111, 7260–7314. 10.1021/cr100428a. [DOI] [PubMed] [Google Scholar]
- Jin P.; Song T.; Xiao J.; Zhang Q. Recent Progress in Using Pyrene-4,5-diketones and Pyrene-4,5,9,10-tetraketones as Building Blocks to Construct Large Acenes and Heteroacenes. Asian J. Org. Chem. 2018, 7, 2130–2146. 10.1002/ajoc.201800039. [DOI] [Google Scholar]
- Stępień M.; Gońka E.; Żyła M.; Sprutta N. Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications. Chem. Rev. 2017, 117, 3479–3716. 10.1021/acs.chemrev.6b00076. [DOI] [PubMed] [Google Scholar]
- Wang C.-Z.; Pang Z.-J.; Yu Z.-D.; Zeng Z.-X.; Zhao W.-X.; Zhou Z.-Y.; Redshaw C.; Yamato T. Short Axially Asymmetrically 1,3-Disubstituted Pyrene-Based Color-Tunable Emitters: Synthesis, Characterization and Optical Properties. Tetrahedron 2021, 78, 131828. 10.1016/j.tet.2020.131828. [DOI] [Google Scholar]
- Crawford A. G.; Dwyer A. D.; Liu Z.; Steffen A.; Beeby A.; Pålsson L.-O.; Tozer D. J.; Marder T. B. Experimental and Theoretical Studies of the Photophysical Properties of 2- and 2,7-Functionalized Pyrene Derivatives. J. Am. Chem. Soc. 2011, 133, 13349–13362. 10.1021/ja2006862. [DOI] [PubMed] [Google Scholar]
- Hu J.-Y.; Pu Y.-J.; Nakata G.; Kawata S.; Sasabe H.; Kido J. A Single-Molecule Excimer-Emitting Compound for Highly Efficient Fluorescent Organic Light-Emitting Devices. Chem. Commun. 2012, 48, 8434–8436. 10.1039/c2cc33463a. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Chen S.; Lam J. W. Y.; Wang Z.; Lu P.; Mahtab F.; Sung H. H. Y.; Williams I. D.; Ma Y.; Kwok H. S.; et al. Pyrene-Substituted Ethenes: Aggregation-Enhanced Excimer Emission and Highly Efficient Electroluminescence. J. Mater. Chem. 2011, 21, 7210–7216. 10.1039/c0jm04449k. [DOI] [Google Scholar]
- Fujii A.; Sekiguchi Y.; Matsumura H.; Inoue T.; Chung W.-S.; Hirota S.; Matsuo T. Excimer Emission Properties on Pyrene-Labeled Protein Surface: Correlation between Emission Spectra, Ring Stacking Modes, and Flexibilities of Pyrene Probes. Bioconjugate Chem. 2015, 26, 537–548. 10.1021/acs.bioconjchem.5b00026. [DOI] [PubMed] [Google Scholar]
- Cho H.; Lee S.; Cho N. S.; Jabbour G. E.; Kwak J.; Hwang D.-H.; Lee C. High-Mobility Pyrene-Based Semiconductor for Organic Thin-Film Transistors. ACS Appl. Mater. Interfaces 2013, 5, 3855–3860. 10.1021/am4005368. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wang H.; Liu Y.; Di C.-a.; Sun Y.; Wu W.; Yu G.; Zhang D.; Zhu D. 1-Imino Nitroxide Pyrene for High Performance Organic Field-Effect Transistors with Low Operating Voltage. J. Am. Chem. Soc. 2006, 128, 13058–13059. 10.1021/ja064580x. [DOI] [PubMed] [Google Scholar]
- Birks J. B.; Dyson D. J.; Munro I. H. “Excimer” Fluorescence. II. Lifetime Studies of Pyrene Solutions. Proc. R. Soc. A 1963, 275, 575–588. 10.1098/rspa.1963.0187. [DOI] [Google Scholar]
- Bains G.; Patel A. B.; Narayanaswami V. Pyrene: A Probe to Study Protein Conformation and Conformational Changes. Molecules 2011, 16, 7909–7935. 10.3390/molecules16097909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub P.Molecular Dynamics Study of Pyrene Excimer Formation and Oxidation in Lipid Bilayer Models, PhD Thesis, Université de Strasbourg, Strasbourg, France, 2015. [Google Scholar]
- Hoche J.; Schmitt H.-C.; Humeniuk A.; Fischer I.; Mitrić R.; Röhr M. I. S. The Mechanism of Excimer Formation: An Experimental and Theoretical Study on the Pyrene Dimer. Phys. Chem. Chem. Phys. 2017, 19, 25002–25015. 10.1039/c7cp03990e. [DOI] [PubMed] [Google Scholar]
- Sahoo D.; Narayanaswami V.; Kay C. M.; Ryan R. O. Pyrene Excimer Fluorescence: A Spatially Sensitive Probe To Monitor Lipid-Induced Helical Rearrangement of Apolipophorin III. Biochemistry 2000, 39, 6594–6601. 10.1021/bi992609m. [DOI] [PubMed] [Google Scholar]
- Pokhrel M. R.; Bossmann S. H. Synthesis, Characterization, and First Application of High Molecular Weight Polyacrylic Acid Derivatives Possessing Perfluorinated Side Chains and Chemically Linked Pyrene Labels. J. Phys. Chem. B 2000, 104, 2215–2223. 10.1021/jp9917190. [DOI] [Google Scholar]
- Daems D.; Van den Zegel M.; Boens N.; De Schryver F. C. Fluorescence decay of pyrene in small and large unilamellar L,α-Dipalmitoylphosphatidylcholine vesicles above and below the phase transition temperature. Eur. Biophys. J. 1985, 12, 97–105. 10.1007/bf00260432. [DOI] [PubMed] [Google Scholar]
- Yang J.-S.; Lin C.-S.; Hwang C.-Y. Cu2+-Induced Blue Shift of the Pyrene Excimer Emission: A New Signal Transduction Mode of Pyrene Probes. Org. Lett. 2001, 3, 889–892. 10.1021/ol015524y. [DOI] [PubMed] [Google Scholar]
- Lee Y. H.; Lee M. H.; Zhang J. F.; Kim J. S. Pyrene Excimer-Based Calix[4]arene FRET Chemosensor for Mercury(II). J. Org. Chem. 2010, 75, 7159–7165. 10.1021/jo101113f. [DOI] [PubMed] [Google Scholar]
- Haedler A. T.; Misslitz H.; Buehlmeyer C.; Albuquerque R. Q.; Köhler A.; Schmidt H.-W. Controlling the π-Stacking Behavior of Pyrene Derivatives: Influence of H-Bonding and Steric Effects in Different States of Aggregation. ChemPhysChem 2013, 14, 1818–1829. 10.1002/cphc.201300242. [DOI] [PubMed] [Google Scholar]
- Fink R. F.; Seibt J.; Engel V.; Renz M.; Kaupp M.; Lochbrunner S.; Zhao H.-M.; Pfister J.; Würthner F.; Engels B. Exciton Trapping in π-Conjugated Materials: A Quantum-Chemistry-Based Protocol Applied to Perylene Bisimide Dye Aggregates. J. Am. Chem. Soc. 2008, 130, 12858–12859. 10.1021/ja804331b. [DOI] [PubMed] [Google Scholar]
- This conclusion was made based on a patent search at espacenet.com. Using the general keywords “oled”, “2000” and “emitting”, the molecule keyword “pyrene” was com-pared to the group of following keywords: “rubrene”,” perylene” and “alq3”. The ratio between pyrene patents to the average amount of patents was determined to be 1:1.6.
- Sasaki S.; Suzuki S.; Igawa K.; Morokuma K.; Konishi G.-i. The K-Region in Pyrenes as a Key Position to Activate Aggregation-Induced Emission: Effects of Introducing Highly Twisted N,N-Dimethylamines. J. Org. Chem. 2017, 82, 6865–6873. 10.1021/acs.joc.7b00996. [DOI] [PubMed] [Google Scholar]
- Ge Y.; Wen Y.; Liu H.; Lu T.; Yu Y.; Zhang X.; Li B.; Zhang S.-T.; Li W.; Yang B. A key stacking factor for the effective formation of pyrene excimer in crystals: degree of π-π overlap. J. Mater. Chem. C 2020, 8, 11830–11838. 10.1039/d0tc02562c. [DOI] [Google Scholar]
- Zhang Y.; Tan L.; Shi J.; Ji L. Iridium-catalysed borylation of pyrene - a powerful route to novel optoelectronic materials. New J. Chem. 2021, 45, 14869–14878. 10.1039/d1nj00538c. [DOI] [Google Scholar]
- Bosdet M. J. D.; Piers W. E.; Sorensen T. S.; Parvez M. 10a-Aza-10b-borapyrenes: Heterocyclic Analogues of Pyrene with Internalized BN Moieties. Angew. Chem., Int. Ed. 2007, 46, 4940–4943. 10.1002/anie.200700591. [DOI] [PubMed] [Google Scholar]
- Wang S.; Yang D.-T.; Lu J.; Shimogawa H.; Gong S.; Wang X.; Mellerup S. K.; Wakamiya A.; Chang Y.-L.; Yang C.; et al. In Situ Solid-State Generation of (BN)2-Pyrenes and Electroluminescent Devices. Angew. Chem., Int. Ed. 2015, 54, 15074–15078. 10.1002/anie.201507770. [DOI] [PubMed] [Google Scholar]
- Dewar M. J. S.; Poesche W. H. New Heteroaromatic Compounds. XXI.1 Some Tetracyclic Systems2. J. Org. Chem. 1964, 29, 1757–1762. 10.1021/jo01030a023. [DOI] [Google Scholar]
- Chissick S. S.; Dewar M. J. S.; Maitlis P. M. New Heteraromatic Compounds Containing two Boron Atoms. Tetrahedron Lett. 1960, 1, 8–10. 10.1016/s0040-4039(00)70270-x. [DOI] [Google Scholar]
- Ashton P. R.; Harris K. D. M.; Kariuki B. M.; Philp D.; Robinson J. M. A.; Spencer N. A Borazaaromatic Analogue of Isophthalic Acid. J. Chem. Soc., Perkin Trans. 2 2001, 2166–2173. 10.1039/b104794a. [DOI] [Google Scholar]
- Jaska C. A.; Piers W. E.; McDonald R.; Parvez M. Synthesis, Characterization, and Fluorescence Behavior of Twisted and Planar B2N2-Quaterphenyl Analogues. J. Org. Chem. 2007, 72, 5234–5243. 10.1021/jo0706574. [DOI] [PubMed] [Google Scholar]
- Desiraju G. R.; Gavezzotti A. From Molecular to Crystal Structure; Polynuclear Aromatic Hydrocarbons. J. Chem. Soc., Chem. Commun. 1989, 621–623. 10.1039/c39890000621. [DOI] [Google Scholar]
- Mas-Torrent M.; Hadley P.; Bromley S. T.; Ribas X.; Tarrés J.; Mas M.; Molins E.; Veciana J.; Rovira C. Correlation between Crystal Structure and Mobility in Organic Field-Effect Transistors Based on Single Crystals of Tetrathiafulvalene Derivatives. J. Am. Chem. Soc. 2004, 126, 8546–8553. 10.1021/ja048342i. [DOI] [PubMed] [Google Scholar]
- Ashe A. J.; Chan W.-T.; Smith T. W.; Taba K. M. Electrophilic aromatic substitution reactions of arsabenzene. J. Org. Chem. 1981, 46, 881–885. 10.1021/jo00318a012. [DOI] [Google Scholar]
- Hernán A. G.; Horton P. N.; Hursthouse M. B.; Kilburn J. D. New and Efficient Synthesis of Solid-Supported Organotin Reagents and Their Use in Organic Synthesis. J. Organomet. Chem. 2006, 691, 1466–1475. 10.1016/j.jorganchem.2005.11.031. [DOI] [Google Scholar]
- Hoic D. A.; Wolf J. R.; Davis W. M.; Fu G. C. Chemistry of Borabenzene: Efficient and General Synthesis of New Neutral Borabenzene–Ligand Complexes. Organometallics 1996, 15, 1315–1318. 10.1021/om9505569. [DOI] [Google Scholar]
- Schreiner P.; Machuy M. M.; Würtele C. 2,6-Bis(phenylethynyl)biphenyls and Their Cyclization to Pyrenes. Synthesis 2012, 44, 1405–1409. 10.1055/s-0031-1290754. [DOI] [Google Scholar]
- Leroux F. R.; Berthelot A.; Bonnafoux L.; Panossian A.; Colobert F. Transition-Metal-Free Atropo-Selective Synthesis of Biaryl Compounds Based on Arynes. Chem. Eur J. 2012, 18, 14232–14236. 10.1002/chem.201202739. [DOI] [PubMed] [Google Scholar]
- Spackman M. A.; Jayatilaka D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. 10.1039/b818330a. [DOI] [Google Scholar]
- Gong Y.-B.; Zhang P.; Gu Y.-r.; Wang J.-Q.; Han M.-M.; Chen C.; Zhan X.-J.; Xie Z.-L.; Zou B.; Peng Q.; et al. The Influence of Molecular Packing on the Emissive Behavior of Pyrene Derivatives: Mechanoluminescence and Mechanochromism. Adv. Opt. Mater. 2018, 6, 1800198. 10.1002/adom.201800198. [DOI] [Google Scholar]
- do Casal M. T.; Cardozo T. M. Impact of low-cost methods in the description of excimer and exciplex formation: pyrene-pyrene and pyrene-naphthalene case studies. Theor. Chem. Acc. 2020, 139, 144. 10.1007/s00214-020-02658-0. [DOI] [Google Scholar]
- Bradac C.; Xu Z.-Q.; Aharonovich I. Quantum Energy and Charge Transfer at Two-Dimensional Interfaces. Nano Lett. 2021, 21, 1193–1204. 10.1021/acs.nanolett.0c04152. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Hariharan P. C.; Pople J. A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. 10.1007/bf00533485. [DOI] [Google Scholar]
- Turner M. J.; Grabowsky S.; Jayatilaka D.; Spackman M. A. Accurate and Efficient Model Energies for Exploring Intermolecular Interactions in Molecular Crystals. J. Phys. Chem. Lett. 2014, 5, 4249–4255. 10.1021/jz502271c. [DOI] [PubMed] [Google Scholar]
- Non-time-dependent PL spectra were measured in front-face geometry (cuvette oriented at 45° in relation to the excitation beam) to suppress inner-filtering effects. Time-dependent PL spectra were measured in right-angle orientation.
- Idzik K. R.; Cywiński P. J.; Kuznik W.; Frydel J.; Licha T.; Ratajczyk T. The Optical Properties and Quantum Chemical Calculations of Thienyl and Furyl Derivatives of Pyrene. Phys. Chem. Chem. Phys. 2015, 17, 22758–22769. 10.1039/c5cp03013g. [DOI] [PubMed] [Google Scholar]
- Liu M.; Gong X.; Zheng C.; Gao D. Development of Pyrene Derivatives as Promising n-Type Semiconductors: Synthesis, Structural and Spectral Properties. Asian J. Org. Chem. 2017, 6, 1903–1913. 10.1002/ajoc.201700441. [DOI] [Google Scholar]
- Weigel W.; Rettig W.; Dekhtyar M.; Modrakowski C.; Beinhoff M.; Schlüter A. D. Dual Fluorescence of Phenyl and Biphenyl Substituted Pyrene Derivatives. J. Phys. Chem. A 2003, 107, 5941–5947. 10.1021/jp026116u. [DOI] [Google Scholar]
- Idzik K. R.; Cywiński P. J.; Kuznik W.; Frydel J.; Licha T.; Ratajczyk T. The Optical Properties and Quantum Chemical Calculations of Thienyl and Furyl Derivatives of Pyrene. Phys. Chem. Chem. Phys. 2015, 17, 22758–22769. 10.1039/c5cp03013g. [DOI] [PubMed] [Google Scholar]
- Perepichka I. F.; Perepichka D. F.; Meng H.; Wudl F. Light-Emitting Polythiophenes. Adv. Mater. 2005, 17, 2281–2305. 10.1002/adma.200500461. [DOI] [Google Scholar]
- Grimsdale A. C.; Leok Chan K.; Martin R. E.; Jokisz P. G.; Holmes A. B. Synthesis of Light-Emitting Conjugated Polymers for Applications in Electroluminescent Devices. Chem. Rev. 2009, 109, 897–1091. 10.1021/cr000013v. [DOI] [PubMed] [Google Scholar]
- Moustafa R. M.; Degheili J. A.; Patra D.; Kaafarani B. R. Synthesis and Detailed Photophysical Studies of Pyrene-Based Molecules Substituted with Extended Chains. J. Phys. Chem. A 2009, 113, 1235–1243. 10.1021/jp809830x. [DOI] [PubMed] [Google Scholar]
- Geiger M. W.; Turro N. J. Pyrene Fluorescence Lifetime as a Probe for Oxygen Penetration of Micelles. Photochem. Photobiol. 1975, 22, 273–276. 10.1111/j.1751-1097.1975.tb06749.x. [DOI] [PubMed] [Google Scholar]
- Mei J.; Leung N. L. C.; Kwok R. T. K.; Lam J. W. Y.; Tang B. Z. Aggregation-Induced Emission: Together We Shine, United We Soar!. Chem. Rev. 2015, 115, 11718–11940. 10.1021/acs.chemrev.5b00263. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Xing J.; Gong Q.; Chen L.-C.; Liu G.; Yao C.; Wang Z.; Zhang H.-L.; Chen Z.; Zhang Q. Reducing Aggregation Caused Quenching Effect Through Co-Assembly of PAH Chromophores and Molecular Barriers. Nat. Commun. 2019, 10, 169. 10.1038/s41467-018-08092-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R. L. Natural Transition Orbitals. J. Chem. Phys. 2003, 118, 4775–4777. 10.1063/1.1558471. [DOI] [Google Scholar]
- Neese F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]
- Bauernschmitt R.; Ahlrichs R. Treatment of Electronic Excitations within the Adiabatic Approximation of Time Dependent Density Functional Theory. Chem. Phys. Lett. 1996, 256, 454–464. 10.1016/0009-2614(96)00440-x. [DOI] [Google Scholar]
- Hohenberg P.; Kohn W. Inhomogeneous Electron Gas. Phys. Rev. [Sect.] B 1964, 136, B864–B871. 10.1103/physrev.136.b864. [DOI] [Google Scholar]
- Kohn W.; Sham L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. [Sect.] A 1965, 140, A1133–A1138. 10.1103/physrev.140.a1133. [DOI] [Google Scholar]
- Becke A. D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. 10.1063/1.464304. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098–3100. 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- Dunning T. H. Jr Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron Through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- Schleyer P. V. R.; Maerker C.; Dransfeld A.; Jiao H.; van Eikema Hommes N. J. R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. 10.1021/ja960582d. [DOI] [PubMed] [Google Scholar]
- Møller C.; Plesset M. S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. 10.1103/physrev.46.618. [DOI] [Google Scholar]
- Hoffmann J.; Geffroy B.; Jaques E.; Hissler M.; Staubitz A. Tuning the aggregation behaviour of BN-coronene diimides with imide substituents and their performance in devices (OLEDs and OFETs). J. Mater. Chem. C 2021, 9, 14720–14729. 10.1039/d1tc02991f. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.