

Abstract

Virtual ligand screening of a publicly available database of antimalarial hits using a pharmacophore derived from antimalarial MMV008138 identified TCMDC-140230, a tetrahydro-β-carboline amide, as worthy of exploration. All four stereoisomers of this structure were synthesized, but none potently inhibited growth of the malaria parasite Plasmodium falciparum. Interestingly, 7e, a minor byproduct of these syntheses, proved to be potent in vitro against P. falciparum and was orally efficacious (40 mg/kg) in an in vivo mouse model of malaria.
Keywords: Drug discovery, Plasmodium, synthesis, DMPK, in vivo
Worldwide, malaria is one of the deadliest diseases, causing more than 600,000 deaths in 2020.1 In our search for new and safe drugs that can kill resistant strains of Plasmodium falciparum parasites, we were drawn to the 2-C-methyl-d-erythritol-4-phosphate (MEP) pathway for synthesis of isoprenoid precursors, since this pathway is not present in humans.2 Using the isopentenyl pyrophosphate (IPP) chemical rescue approach3 to screen the 400-compound Malaria Box,4 only MMV008138 (1a, Figure 1) was found to be an inhibitor of this pathway.5 Other researchers determined that 1a inhibits IspD, the third enzyme in the MEP pathway.6,7 We subsequently synthesized and assayed dozens of analogs of 1a that differed in stereochemistry and substitution of the B-, C-, and D-rings.8−10 From these studies a clear picture emerged: (1R,3S)- stereochemistry, 2′,4′-halogen disubstitution of the D-ring (e.g., 1a–d), and a 3-carboxylic acid or methyl amide (e.g., 1a and 3a) were required for antimalarial potency. However, to date none of these potent PfIspD inhibitors has shown oral efficacy in mice models of malaria.
Figure 1.

MEP pathway inhibiting antimalarials based on the MMV008138 (1a) scaffold.8−10
Since 1a, as a member of the 400-compound Malaria Box, was selected from a much larger initial hit set (20K compounds with 3D7 strain half-maximal effective concentration (EC50) < 1 μM),4 we wondered if other structurally related compounds were present in the initial set. We thus performed virtual ligand screening (VLS) of the GlaxoSmithKline (GSK) portion of this larger collection (∼13K compounds),11 using a 3D pharmacophore derived from 1a and 92 close analogs. From this exercise the compound TCMDC-140230 emerged (Figure 2).
Figure 2.

VLS hit TCMDC-140230 and derived lead 7e.
Several features of this compound stand out; in addition to the undisclosed stereochemistry, 3′,4′-dichloro substitution and the large amide substituent were not suggestive of good antimalarial potency within the established pharmacophore of 1a (Figure 1, cf. 1e vs 1a; 4a–6a vs 3a). As described below, the stereoisomers of tetrahydro-β-carboline TCMDC-140230 proved to be dead ends, but the β-carboline analog 7e (tested as the HCl salt, Table 1) was found to have excellent antimalarial activity in vitro and in vivo.
Table 1. In VitroP. falciparum Growth Inhibition of 1a, 13e Stereoisomers, and 7e·HCl.
| Compound | EC50 (nM)a | IPP rescueb |
|---|---|---|
| 1a | 250 ± 70c | 100% @ 2.5 μM |
| (1R,3S)-(−)-13e | 1,950 ± 240 | 50% @ 5 μM |
| (1S,3S)-(−)-13e | 1,300 ± 100 | nd |
| (1S,3R)-(+)-13e | 3,690 ± 310 | nd |
| (1R,3R)-(+)-13e | 1,430 ± 70 | nd |
| 7e·HCl | 108 ± 7 | 0% @ 2.5 μM |
Growth inhibition (72 h) of asexual blood stages determined using SYBR Green I assay. Values represent average ± SEM from at least three independent biological replicates. The Dd2 strain is resistant to chloroquine, pyrimethamine, and mefloquine.
Recovery of parasite growth in the presence of 200 μM IPP, at the indicated concentration of the test compound (Supporting Information); nd designates “not determined”.
Reported previously.8
Since stereochemistry proved so crucial to the antimalarial activity of 1a, we undertook the enantioselective synthesis of the four stereoisomers of TCMDC-140230. 7-Methylindole 8 was converted to 9, which then underwent Negishi coupling with the organozinc reagent derived from N-Boc-iodoalanine methyl ester (R)-10,12 affording the protected 7-methyltryptophan ester (S)-11. Deprotection, followed by Pictet–Spengler reaction8 with 3,4-dichlorobenzaldhyde afforded (1R,3S)- and (1S,3S)-12e (Scheme 1). The relative configuration of the 12e diastereomers was assigned on the basis of the 1H–1H coupling constants.13 Conversion to the amides (1R,3S)- and (1S,3S)-13e was achieved with N-methylethylene diamine in MeOH at reflux under nitrogen. The remaining enantiomeric pair of diastereomers of 13e was then prepared from (S)-10 (Supporting Information). Interestingly, in the amidation of (1S,3S)- and (1R,3R)-12e, oxidized byproduct 7e was isolated in 14–17% yield (Scheme 1 and Supporting Information). To confirm its structure, 7e was independently prepared by oxidation of the mixture of the trans- and cis-esters 12e to β-carboline 14e, using PhI(OAc)214 (Scheme 2). Amidation of 14e in neat amine and HCl treatment yielded 7e·HCl in 75% yield. With these compounds in hand, we examined their activity against asexual blood stage P. falciparum (Table 1).
Scheme 1. Synthesis of the Stereoisomers of VLS Hit TCMDC-140230 and Serendipitous Isolation of 7e.

Scheme 2. Independent Synthesis of 7e·HCl.

None of the stereoisomers of tetrahydro-β-carboline 13e potently inhibited the growth of P. falciparum. Since (1R,3S)-13e has the same configuration of 1a, the ability of IPP supplementation (200 μM) to reverse growth inhibition was assessed; the 50% rescue seen at 5 μM drug suggested this stereoisomer is a weak MEP pathway inhibitor. Fortuitously, the β-carboline amide 7e, isolated initially as a byproduct, did potently inhibit parasite growth with EC50 = 108 ± 7 nM. Interestingly, supplementation with IPP did not rescue parasite growth. Thus, the antimalarial activity of 7e·HCl is not related to inhibition of the MEP pathway.
We then prepared a range of analogs of 7e that differed in the C3 carbonyl substituent (Scheme 3).
Scheme 3. Synthesis of Acid and Amide Variants of 7e.

Hydrolysis of 14e afforded carboxylic acid 15e; amidation of 14e yielded neutral amides 16e–18e and amides 19e–23e, which resemble 7e in having a pendant basic amine. These were tested for growth inhibition of P. falciparum, and a clear pattern emerged (Table 2). As can be seen, ester 14e and acid 15e did not inhibit growth at the highest concentration tested, and only amides with a pendant basic amine (7e and 19e–23e) had submicromolar potency. Within the limited set of compounds explored, it appears that the N-(2-methylamino)ethyl amide (i.e., 7e) confers the best antimalarial potency.
Table 2. In VitroP. falciparum Growth Inhibition of Acid and Amide Variants of 7e.
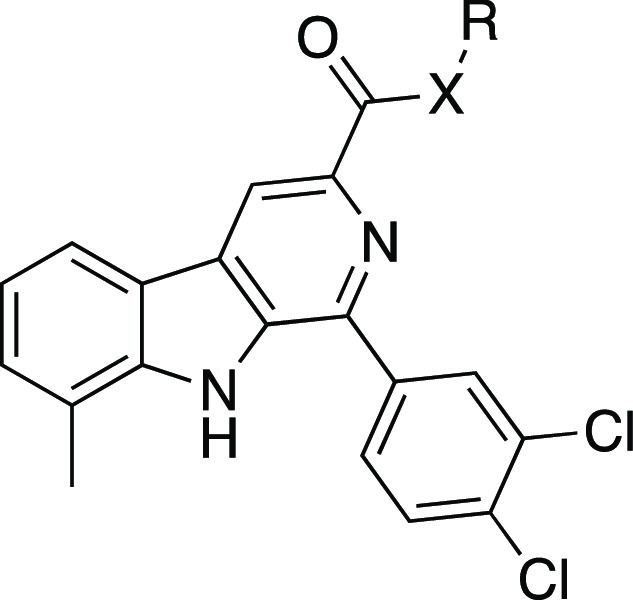
| Compound | X | R | EC50 (nM)a |
|---|---|---|---|
| 7e·HCl | NH | (CH2)2NHMe·HCl | 108 ± 7 |
| 14e | O | Me | >10,000 |
| 15e | O | H | >10,000 |
| 16e | NH | Me | >10,000 |
| 17e | NH | (CH2)2OH | 3,270 ± 40 |
| 18e | NH | (CH2)3CH3 | 1,020 ± 20 |
| 19e·HCl | NH | (CH2)2NH2·HCl | 263 ± 25 |
| 20e·HCl | NH | (CH2)2NMe2·HCl | 264 ± 26 |
| 21e·HCl | NH | (CH2)3NH2·HCl | 726 ± 57 |
| 22e·HCl | NH | (CH2)3NHMe·HCl | 217 ± 28 |
| 23e·HCl | NH | (CH2)3NMe2·HCl | 240 ± 23 |
Growth inhibition (72 h) of asexual blood stages determined using SYBR Green I assay. Values represent average ± SEM from at least three independent biological replicates. The Dd2 strain is resistant to chloroquine, pyrimethamine, and mefloquine.
We thus prepared a set of analogs of 7e·HCl that differed in the 8-substituent of the A-ring and in the substitution of the D-ring (Scheme 4).
Scheme 4. Synthesis of A- and D-Ring Variants of 7e·HCl.

Racemic 7-methyl tryptophan methyl ester (±)-24 was prepared from 8 by adapting a literature method;15 it and (S)-25 were subjected to Pictet–Spengler reaction with the requisite benzaldehydes; oxidation with PhI(OAc)2 yielded the corresponding β-carboline methyl esters 14a,f–h and 26e–h. Amidation and treatment with methanolic HCl afforded the desired A- and D-ring variants of 7e·HCl. The antimalarial potencies of these compounds and 7e·HCl are shown in Table 3.
Table 3. Antimalarial Potencies of 7e·HCl and Its A-/D-Ring Variants.

| Compound | R | X | EC50 (nM)a |
|---|---|---|---|
| 7a·HCl | CH3 | 2′,4′-Cl2 | 583 ± 91 |
| 7e·HCl | CH3 | 3′,4′-Cl2 | 108 ± 7 |
| 7f·HCl | CH3 | H | 4,000 ± 900 |
| 7g·HCl | CH3 | 4′-Cl | 274 ± 12 |
| 7h·HCl | CH3 | 3′-Cl | 342 ± 7 |
| 27e·HCl | H | 3′,4′-Cl2 | 227 ± 33 |
| 27f·HCl | H | H | 2,500–5,000 |
| 27g·HCl | H | 4′-Cl | 496 ± 60 |
| 27h·HCl | H | 3′-Cl | 641 ± 11 |
Growth inhibition (72 h) of asexual blood stages determined using SYBR Green I assay. Values represent average ± SEM from at least three independent biological replicates. The Dd2 strain is resistant to chloroquine, pyrimethamine, and mefloquine.
As can be seen, 3′,4′-Cl2 substitution of the D-ring (i.e., 7e·HCl and 27e·HCl) affords the best potency in both the R = CH3 and R= H series. Unsubstituted analogs 7f·HCl and 27f·HCl are approximately 10-fold less potent than 7e·HCl and 27e·HCl. Single Cl-substitutions in the 4′- or 3′-positions (e.g., 7g,h·HCl /27g,h·HCl) recover some potency relative to the unsubstituted analogs 7f·HCl/27f·HCl but are less potent than 3′,4′-Cl2 analogs 7e·HCl/27e·HCl. Compound 7a·HCl provides one example of 2′,4′-Cl2 disubstitution (cf. PfIspD inhibitor 1a), but it is less potent than 7e·HCl. Lastly, in this small series it appears that 8-methyl substitution may confer some advantage for in vitro potency (cf. 7e·HCl and 27e·HCl).
Since 7e·HCl emerged as the best compound, we evaluated it for potency against a number of different strains of P. falciparum (Table 4).
Table 4. In Vitro Antimalarial and ADME-Tox Data, Physicochemical Parameters, and In Vivo PK Data for 7e·HCl.
| Entry | Assay | Value |
|---|---|---|
| 1 | Dd2 EC5072h (nM)a,b | 108 ± 7 |
| 2 | 3D7 EC5072h (nM)a,c | 53 ± 7 |
| 3 | Dd2-KAE609R EC5072h (nM)a,d | 101 ± 5 |
| 4 | W2 EC5072h (nM)a,e | 100 ± 8 |
| 5 | 4G EC5072h (nM)a,f | 110 ± 17 |
| 6 | 3D7 3H–H IC5048h (nM)g | 70 ± 10 |
| 7 | 3D7 LDH IC5072h (nM)h | 117 ± 11 |
| 8 | HEK293 CC50 (nM) | 32,000 ± 4,000 |
| 9 | hPHep CC5096h (nM)i | 8,500 ± 1,900 |
| 10 | E. coli MIC (nM) | >250,000 |
| 11 | hERG IC50 (nM) | 510 |
| 12 | Mini-Ames Panel | negative |
| 13 | MW (g/mol, free base) | 427 |
| 14 | tPSA (Å2) | 65.5 |
| 15 | Log D (pH 7.4)j | 3.3 |
| 16 | PBS Solubility (μM, pH 7.4) | 9.5 |
| 17 | PPB (mouse) | 99.49% |
| 18 | Mouse plasma stability | 100% at 1 h |
| 19 | Mouse microsomal t1/2 (min) | 110 |
| 20 | Clint (mouse μg/mL/min) | 12.6 |
| 21 | %F (rat)k | 40% |
| 22 | Cmax (nM, oral 40 mg/kg, rat)k | 800 ± 25 |
| 23 | Rat plasma t1/2 (h)k | 8 |
| 24 | Vdss (L/kg, rat)k | 21.8 |
Growth inhibition determined using the SYBR Green I assay for all EC50 determinations. Values represent average ± SEM from at least three independent biological replicates.
Resistant to chloroquine (CQ), pyrimethamine (PY), and mefloquine (MQ).
Sensitive to CQ, PY, and MQ.
KAE609-resistant strain, courtesy of E. Winzeler.
Resistant to CQ, quinine, PY, cycloguanil, and sulfadoxine.
Dihydroartemisinin- and CQ-resistant field strain, courtesy of D. E. Kyle.
[3H]hypoxanthine incorporation assay.
Lactate dehydrogenase activity.
Primary human hepatocytes.
Experimentally determined.
See Supporting Information for details.
Compound 7e·HCl had similar EC50 values against the susceptible 3D7 strain and a number of resistant strains of P. falciparum (Table 4, entries 1–5). Thus, 7e·HCl is apparently not subject to the resistance mechanisms of a number of antimalarials such as chloroquine, mefloquine, quinine, pyrimethamine, cycloguanil, sulfadoxazine, KAE609, and dihydroartemisinin. Furthermore, the antimalarial potency against the 3D7 strain was similar when assessed for DNA replication, purine biosynthesis, and metabolic activity (Table 4, entries 2, 6, and 7). 7e·HCl was also not generally cytotoxic, as demonstrated by 78- to 320-fold higher half-maximal cytotoxic concentration (CC50) values against mammalian cells and no effect at 250 μM against human microbiome constituent E. coli (Table 4, entries 8–10). The compound showed a hERG liability, but it was not mutagenic in a mini-Ames panel (Table 4, entries 11–12). The physicochemical properties of 7e·HCl are suggestive of acceptable oral bioavailability (Table 4, entries 13–15). The compound is highly plasma-protein-bound but has excellent plasma and microsomal stability and, consequently, low Clint (Table 4, entries 17–20). In rat, 7e·HCl has moderate oral bioavailability (40%) and a Cmax of 800 ± 25 nM (at 40 mg/kg po); the long plasma half-life (8 h) and relatively high volume of distribution (21.8 L/kg) are also positive features for an antimalarial candidate (Table 4, entries 21–24).
With this data in hand, the in vivo antimalarial activity of 7e·HCl was assessed in a P. berghei model of murine malaria (Figure 3).
Figure 3.
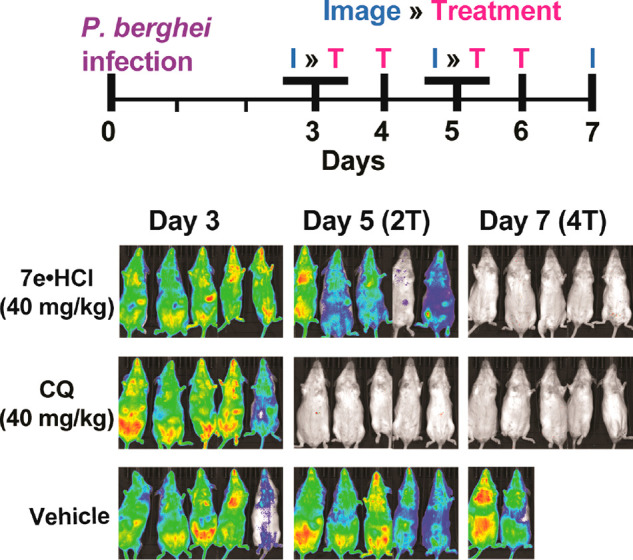
7e·HCl (40 mg/kg/day) has oral efficacy in P. berghei-infected mice. A transgenic P. berghei ANKA strain (l676m1cl1 line, PbGFP-Luccon) was used, that expresses a fusion GFP (mutant 3) and firefly luciferase (LucIAV). CQ is chloroquine. Note: three of the five vehicle-treated mice died before Day 7.
Mice were infected on Day 0 with 103P. berghei-infected erythrocytes and treated for 4 days starting on Day 3 after inoculation with P. berghei. Mice were imaged (luminescence) before dosing on Days 3 and 5; final images were taken on Day 7. As can be seen in Figure 3, at 40 mg/kg (po), 7e·HCl substantially reduced parasitemia in 3–4 doses. At the same dose, chloroquine cleared infection in 1–2 doses. No signs of toxicity (lethargy, gait, weight loss, ruffled fur, and differences in behavior) were observed in any of the groups. Results were reproduced in two independent trials with independently synthesized samples of 7e·HCl at 40 mg/kg. Note that no reduction in parasitemia was seen at 20 mg/kg, using the same dosing/imaging protocol shown in Figure 3. Thus, further increases in in vitro protency will be needed. Nevertheless, to the best of our knowledge, this is the first example of oral antimalarial efficacy within the β-carboline (as opposed to the tetrahydro-β-carboline) scaffold. In addition to its good druglike properties and low general toxicity, 7e·HCl has a number of other promising characteristics, which we will disclose in due course.
Acknowledgments
P.R.C., M.B.C., and M.T. thank the National Institutes of Health (AI128362 and AI157445) for financial support. We gratefully acknowledge E. Winzeler and D. A Kyle for providing the Dd2-KAE609R and 4G strains of P. falciparum, respectively. Part of the data presented in this manuscript are included in patent WO2021/195603 A1 (PCT/US2021/024542), filed on behalf of Virginia Polytechnic Institute and State University, Virginia Tech Intellectual Properties, Inc., and the University of Georgia Research Foundation Inc.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00663.
Synthetic procedures, tabulations of analytical data, and NMR spectra of all tested compounds; in vitro growth inhibition procedures; details on the ADME-Tox assays, pharmacokinetics, and in vivo efficacy studies (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- World Malaria Report 2021. The World Health Organization, available at https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2021 (accessed 2021-12-13).
- Jomaa H.; Wiesner J.; Sanderbrand S.; Altincicek B.; Weidemeyer C.; Hintz M.; Türbachova I.; Eberl M.; Zeidler J.; Lichtenthaler H. K.; Soldati D.; Beck E. Inhibitors of the Nonmevalonate Pathway of Isoprenoid Biosynthesis as Antimalarial Drugs. Science 1999, 285, 1573–1576. 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- Yeh E.; DeRisi J. L. Chemical Rescue of Malaria Parasites Lacking an Apicoplast Defines Organelle Function in Blood-Stage Plasmodium falciparum. PLoS. Biol. 2011, 9, e1001138. 10.1371/journal.pbio.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg T.; Burrows J. N.; Kowalczyk P.; McDonald S.; Wells T. N. C.; Willis P. The Open Access Malaria Box: A Drug Discovery Catalyst for Neglected Diseases. PLoS One 2013, 8, e62906. 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman J. D.; Merino E. F.; Brooks C. F.; Striepen B.; Carlier P. R.; Cassera M. B. Antiapicoplast and Gametocytocidal Screening To Identify the Mechanisms of Action of Compounds within the Malaria Box. Antimicrob. Agents Chemother. 2014, 58, 811–819. 10.1128/AAC.01500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.; Herrera Z.; Ebert D.; Baska K.; Cho S. H.; DeRisi J. L.; Yeh E. A chemical rescue screen identifies a Plasmodium falciparum apicoplast inhibitor targeting MEP isoprenoid precursor biosynthesis. Antimicrob. Agents Chemother. 2015, 59, 356–364. 10.1128/AAC.03342-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay L. S.; Armstrong C. M.; Masters M. C.; Li T.; Price K. E.; Edwards R. L.; Mann K. M.; Li L. X.; Stallings C. L.; Berry N. G.; O’Neill P. M.; Odom A. R. Plasmodium IspD (2-C-methyl-D-erythritol 4-phosphate cytidyltransferase), an essential and druggable antimalarial target. ACS Infect. Dis. 2015, 1, 157–167. 10.1021/id500047s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z.-K.; Krai P. M.; Merino E. F.; Simpson M. E.; Slebodnick C.; Cassera M. B.; Carlier P. R. Determination of the active stereoisomer of the MEP pathway-targeting antimalarial agent MMV008138, and initial structure-activity studies. Bioorg. Med. Chem. Lett. 2015, 25, 1515–1519. 10.1016/j.bmcl.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavami M.; Merino E. F.; Yao Z.-K.; Elahi R.; Simpson M. E.; Fernández-Murga M. L.; Butler J. H.; Casasanta M. A.; Krai P. M.; Totrov M. M.; Slade D. J.; Carlier P. R.; Cassera M. B. Biological Studies and Target Engagement of the 2-C-Methyl-D-Erythritol 4-Phosphate Cytidylyltransferase (IspD)-Targeting Antimalarial Agent (1R,3S)-MMV008138 and Analogs. ACS Infect. Dis. 2018, 4, 549–559. 10.1021/acsinfecdis.7b00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S.; Ghavami M.; Butler J. H.; Merino E. F.; Slebodnick C.; Cassera M. B.; Carlier P. R. Probing the B- & C-rings of the antimalarial tetrahydro-β-carboline MMV008138 for steric and conformational constraints. Bioorg. Med. Chem. Lett. 2020, 30, 127520. 10.1016/j.bmcl.2020.127520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo F.-J.; Sanz L. M.; Vidal J.; de Cozar C.; Alvarez E.; Lavandera J.-L.; Vanderwall D. E.; Green D. V. S.; Kumar V.; Hasan S.; Brown J. R.; Peishoff C. E.; Cardon L. R.; Garcia-Bustos J. F. Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465, 305–310. 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- Ross A. J.; Lang H. L.; Jackson R. F. W. Much Improved Conditions for the Negishi Cross-Coupling of Iodoalanine Derived Zinc Reagents with Aryl Halides. J. Org. Chem. 2010, 75, 245–248. 10.1021/jo902238n. [DOI] [PubMed] [Google Scholar]
- Cagašová K.; Ghavami M.; Yao Z.-K.; Carlier P. R. Questioning the γ-gauche effect: stereoassignment of 1,3-disubstituted-tetrahydro-β-carbolines using 1H–1H coupling constants. Org. Biomol. Chem. 2019, 17, 6687–6698. 10.1039/C9OB01139K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A.; Tangella Y.; Manasa K. L.; Sathish M.; Srinivasulu V.; Chetna J.; Alarifi A. PhI(OAc)2-mediated one-pot oxidative decarboxylation and aromatization of tetrahydro-β-carbolines: synthesis of norharmane, harmane, eudistomin U and eudistomin I. Org. Biomol. Chem. 2015, 13, 8652–8662. 10.1039/C5OB00871A. [DOI] [PubMed] [Google Scholar]
- Konda-Yamada Y.; Okada C.; Yoshida K.; Umeda Y.; Arima S.; Sato N.; Kai T.; Takayanagi H.; Harigaya Y. Convenient synthesis of 7′ and 6′-bromo-D-tryptophan and their derivatives by enzymatic optical resolution using D-aminoacylase. Tetrahedron 2002, 58, 7851–7861. 10.1016/S0040-4020(02)00909-2. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.