

SUMMARY
Powassan virus (POWV) is a tick-borne pathogen for which humans are an incidental host. POWV infection can be fatal or result in long-term neurological sequelae; however, there are no approved vaccinations for POWV. Integral to efficacious vaccine development is the identification of correlates of protection, which we accomplished in this study by utilizing a murine model of POWV infection. Using POWV lethal and sub-lethal challenge models, we show that (1) robust B and T cell responses are necessary for immune protection, (2) POWV lethality can be attributed to both viral- and host-mediated drivers of disease, and (3) knowledge of the immune correlates of protection against POWV can be applied in a virus-like particle (VLP)-based vaccination approach that provides protection from lethal POWV challenge. Identification of these immune protection factors is significant as it will aid in the rational design of POWV vaccines.
In brief
Stone et al. describe Powassan virus (POWV) adaptive immune protection in a murine model of infection. This understanding culminates in a vaccination approach that elicits protective adaptive immune responses against POWV morbidity and mortality. These findings will aid in fulfilling the unmet need for rational design of POWV vaccinations.
Graphical Abstract
INTRODUCTION
Powassan virus (POWV) is a flavivirus and emerging arthropod-borne virus of public health concern. Predictions that the range of ticks transmitting POWV to humans will expand as the climate warms situate POWV as a pathogen of national importance (Clow et al., 2017; Sonenshine, 2018; Gasmi et al., 2018; Ogden et al., 2008; Pierson and Diamond, 2020). Although rare, incidence of POWV infection in humans has increased substantially in the past two decades, a phenomenon that cannot wholly be explained by increased reporting and surveillance (Centers for Disease Control and Prevention, 2020; Nofchissey et al., 2013; Kemenesi and Bányai, 2019; Hinten et al., 2008). A high proportion (>50%) of reported POWV infections are accompanied by long-term neurological sequelae, which range in severity from mild to debilitating (Hermance and Thangamani, 2017; Kemenesi and Bányai, 2019; Goldfield et al., 1973; Tavakoli et al., 2009) and in 10%–15% of cases is fatal (Kemenesi and Bányai, 2019; Tavakoli et al., 2009). Although nucleic acid-based vaccination approaches can prevent severe disease and neurological sequelae in murine models of POWV infection (Choi et al., 2020, Vanblargan et al., 2018), there are no approved vaccines or therapeutics for individuals infected with POWV. Currently, there are six approved vaccinations for the flavivirus tick-borne encephalitis virus (TBEV) that can prevent the development of severe disease and neurological sequelae (Chernokhaeva et al., 2016; Chumakov et al., 1963; Kubinski et al., 2020), but these vaccines are not effective in preventing POWV infection (Chernokhaeva et al., 2016; Shamanin et al., 1991, Mcauley et al., 2017). Taken together, the success of TBEV vaccination strategies in humans, and the advent of successful POWV vaccinations in murine models, suggests that a protective vaccine against POWV is achievable.
Despite its increasing incidence, the immune correlates of protection for POWV are unknown. This is largely because POWV is a rare and relatively recently described virus (Mclean and Donohue, 1959). The enzootic cycle of POWV is thought to be dependent upon transmission from the bite of an infected tick on an uninfected host during a bloodmeal, although other modes of transmission have been described as important (Costero and Grayson, 1996; Nonaka et al., 2010). There are two lineages of POWV, known as lineage I (POWV-LB) and lineage II (POWV-Sponer [POWV-Spo], or deer tick virus). Of these two lineages, POWV-LB is predominantly transmitted by Ixodes cookei ticks, which primarily feed on small woodland mammals such as groundhogs (Marmota monax) (Ebel et al., 2001). As Ixodes cookei ticks have less promiscuous feeding patterns relative to other Ixodid ticks, human infection with POWV-LB is less frequent. POWV-Spo, on the other hand, is transmitted by black-legged deer ticks (Ixodes scapularis). As these ticks have a broader range of host species, most incidence of POWV infection in humans is attributable to POWV-Spo infection. POWV-LB infection in humans is more common in far eastern portions of Russia, while in North America, POWV-Spo is the predominant human-infecting lineage. Despite these distinct geographic ranges and host species, the two lineages are serologically indistinguishable, and there is a large degree of nucleic acid (~86%) and amino acid (aa) (~96%) similarity between the two viruses (Ebel et al., 2001). How these differences inform the viral life cycle, tissue tropism, and interactions with host immune systems are all active areas of research (Robertson et al., 2009; Best Sonja et al., 2005; Mlera et al., 2017; Hermance et al., 2016).
POWV is a member of the family Flaviviridae and a single-stranded positive sense RNA virus with a genome ~11 kbp in length. The genome encodes a single polyprotein cleaved by viral and host proteases into three structural proteins (pre-membrane or prM, capsid or C, and envelope or E) and seven nonstructural (NS) proteins: NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5. The E protein is the main antigenic determinant for flaviviruses, such as TBEV (Kuivanen et al., 2014), and is 500 aa in length with three functional domains: DI, DII, and DIII. It has been well documented that antibodies directed against the E protein can be either neutralizing or non-neutralizing (Ershova et al., 2016; Matveev et al., 2019; Kuivanen et al., 2014). Epitopes recognized by T cells have also been documented in the NS (Lampen et al., 2018; Aberle et al., 2015) and structural proteins of TBEV (Varnaite et al., 2020; Schwaiger et al., 2014), including the E protein, although the contribution of E-specific T cells in flavivirus infection is an active area of research (Hassert et al., 2018, 2019, 2021).
Among flaviviruses, POWV is a member of the tick-borne encephalitis (TBE) serocomplex of viruses, examples of which include TBEV and Langat virus (LGTV) (Pesko et al., 2010; Ebel et al., 2001). In this serocomplex of viruses,TBEV is the most common infection in humans and therefore the most well studied. It is the only TBE serocomplex member for which there are licensed vaccines available for human use. The six available vaccinations cover three different TBEV subtypes and are immunogenic inactivated, whole virus-based vaccines (Kubinski et al., 2020). While it is widely appreciated that cellular and humoral components of adaptive immunity are important for protective responses following TBEV vaccination (Aberle et al., 2015; Schwaiger et al., 2014; Blom et al., 2015, 2018; Best Sonja et al., 2005; Robertson et al., 2009), the correlates of protection for POWV remain unknown. While others have characterized the contribution of monoclonal antibodies to POWV immunity, the polyclonal response is less well characterized (Errico et al., 2020, Vanblargan et al., 2021). Furthermore, multiple studies have demonstrated that approved TBEV vaccines do not provide protection against POWV in humans (Shamanin et al., 1991; Chernokhaeva et al., 2016), although it is unclear why. Identifying the correlates of immune protection is critical to aid development of therapeutics and vaccines, a yet unmet need for POWV infection.
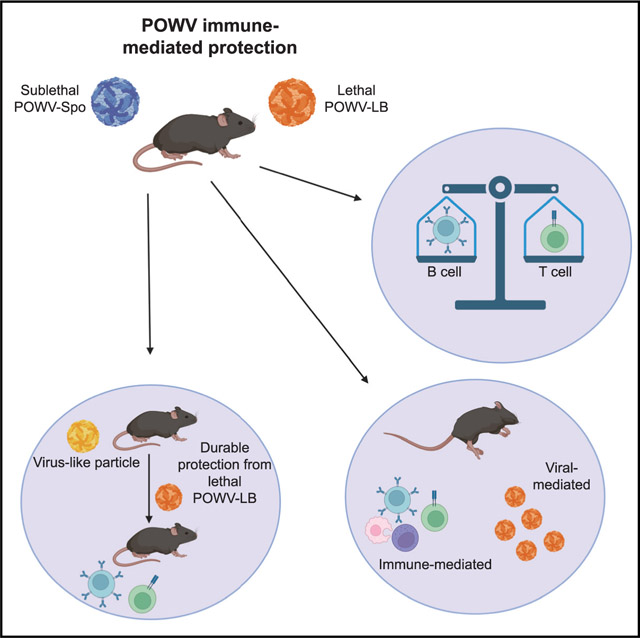
Integral to the development of future vaccinations is establishing the immune correlates of protection for POWV. In these studies, we describe a murine model of POWV infection, which we used to determine the correlates of protection and demonstrate that both the B and T cell compartments of adaptive immunity contribute to durable protection against POWV. In addition, we identify CD8+ and CD4+ T cell epitopes in the structural proteins of POWV and show that CD8+ T cell responses against POWV can drive immune-mediated pathology. We also demonstrate that a virus-like particle (VLP)-based vaccination strategy can be effective against POWV-LB, eliciting robust and durable neutralizing antibodies and detectable CD8+ T cell responses. The adaptive immune response elicited by POWV-LB VLPs resulted in uniform protection from lethal POWV-LB challenge. Because these studies will allow for a more targeted design of POWV vaccines–which has previously been limited to empirical approaches–the findings presented here will have important implications for future POWV vaccine development.
RESULTS
POWV-LB and POWV-Spo cause morbidity and mortality in C57BL/6 mice
With the goal of studying POWV-specific adaptive immune responses and identifying factors associated with protection, we established a susceptible mouse model of POWV infection using the POWV lineage 1 strain LB (POWV-LB). We infected C57BL/6 (B6) mice subcutaneously (s.c.) with 103 or 102 infectious units (focus-forming units [FFU]) of POWV-LB and monitored mice for 15 days post infection (dpi). As observed by others following challenge of B6 mice, POWV-LB infection with 103 FFU resulted in high mortality in B6 mice (Santos et al., 2016; Mlera et al., 2017; Hermance et al., 2015). In our studies, challenge with POWV-LB s.c. resulted in all mice succumbing to infection 7–13 days dpi with a median survival time (MST) of 7 days when challenged with 103 FFU (Figure 1A). At a lower infectious dose, 102 FFU, POWV-LB challenge resulted in uniform mortality, but with a slightly longer MST of 8.5 days.
Figure 1. POWV-LB and POWV-Spooner cause morbidity and mortality in C57BL/6 mice.

(A) Mortality of C57BL/6 (B6) mice challenged subcutaneously (s.c.) with 103(n = 6) or 102(n = 12) focus-forming units (FFU) of POWV-LB and monitored for 14 days post infection (dpi).
(B) Weight loss for B6 mice challenged s.c. with POWV-LB and monitored for 14 dpi.
(C) Clinical scoring results for B6 mice challenged s.c. with 103 FFU (middle, n = 5), and 102 FFU (right, n=8) of POWV-LB and monitored for 16 dpi.
(D) Mortality of B6 mice challenged s.c. with 103 (n = 12) or 102(n = 12) FFU of POWV-Spo and monitored for 30 dpi.
(E) Weight loss for B6 mice challenged s.c. with POWV-Spo and monitored for weight loss for 16 dpi.
(F) Clinical scoring results for mice B6 mice challenged s.c. with 103 FFU(left, n = 9) or 102 FFU(right, n = 10) of POWV-Spo and monitored for 16 dpi. Prior exposure to 102 FFU of POWV-Spooner significantly (p = 0.0002, Mantel-Cox test) improved POWV-LB survival. All data are reported as mean±SEM.
In this POWV mouse model, we observed substantial morbidity among infected mice, with weight loss beginning at 5 dpi and continuing until mice succumb to infection (Figure 1B). Time to exhibit signs of disease for mice infected with OWV-LB did not appear to be dose dependent, and typically occurred at 6 dpi (Figure 1C). POWV-LB-infected mice displayed a limp tail and ruffled appearance, progressing rapidly to moribund posture and shallow breathing with few disease indicators in the days preceding death (Figure 1C). In the case of a lower infectious dose, more mice adopted a ruffled posture, limp tail, and hindlimb weakness, but displayed few other signs of disease before succumbing (Figure 1C). As others have noted, we observed hindlimb paralysis usually corresponding to the site of injection, albeit in a smaller number (<1%) of POWV-LB infections (Mlera et al., 2017). We did, however, note substantial weakness and reduced usage of the hindlimbs as disease progressed and, in the case of the higher infectious doses, we noted that disease progression may have been too rapid for hindlimb paralysis to be observed.
Because of the constraints posed by uniform mortality in this model of POWV-LB infection, we wanted to establish a model of infection with POWV lineage 2 strain, Spooner (POWV-Spo). Interestingly, at the same infectious doses used for POWV-LB (103 or 102 FFU), we observed a lineage-specific survival rate phenomenon, as infection by the same s.c. route resulted in 58% and 42% survival, respectively, but uniform mortality at 105 FFU (Figure 1D). This contrasts sharply with the uniform mortality seen in POWV-LB challenge. Like POWV-LB, weight loss following infection with POWV-Spo began at 5 dpi. But, unlike POWV-LB, surviving POWV-Spo-infected mice recovered lost weight at 12–16 dpi (Figure 1E). In general, mice challenged with POWV-LB exhibited no or few signs of disease before succumbing. In contrast, POWV-Spo-challenged mice displayed signs of disease around the same 6 dpi window, but worsened disease progression between days 9 and 14 was apparent (Figure 1F). Hindlimb weakness tended to be exacerbated in these mice. Of note, we did not observe significant differences in morbidity or mortality on the basis of sex in either POWV-LB or POWV-Spo mice (Figure S1). The similar but delayed onset of morbidity associated with POWV-Spo compared with POWV-LB supported the use of these two highly similar viruses to study the correlates of protection for POWV.
Observing distinct disease phenotypes between these two similar POWV lineages, we next evaluated the kinetics of viral replication of POWV-LB and POWV-Spo in our murine model, and found that POWV-LB tended to replicate more quickly and to higher titers than POWV-Spo, consistent with previous reports (Figure S2) (Grabowski et al., 2019). We also wanted to examine the structural envelope protein of the two POWVs in conjunction with other neuroinvasive arthropod-borne flaviviruses for which the correlates of protection are established or licensed vaccines available. To this end, we performed an aa alignment of the envelope (E) proteins of both lineages of POWV, as well as TBFVs LGTV, and TBEV, and the mosquito-borne virus West Nile virus (WNV). There is a high degree of homology in the envelope (E) protein between POWV-LB and POWV-Spo lineages, but POWV is notably distinct from LGTV, TBEV, and WNV in terms of envelope aa identity (Figures S3A and S3B). Importantly for our study, the sequence similarity between the two lineages, combined with the survival differences between POWV-LB and POWV-Spo, enabled our group to use the two viruses to interrogate different elements of POWV immune-mediated protection. In this work, we examined antigen-specific adaptive immune responses to POWV using the sub-lethal POWV-Spo challenge model and determined factors critical for immune protection using the lethal POWV-LB challenge model.
Having demonstrated that POWV-Spo infection resulted in distinct disease outcomes despite striking aa identity with the more lethal POWV-LB lineage, we considered whether mice recovered from the serologically indistinguishable POWV-Spo lineage would generate a memory response providing protection from lethal, but biologically relevant POWV-LB challenge (Ebel and Kramer, 2004). To this end, we challenged mice recovered from POWV-Spo with a lethal dose of POWV-LB. We observed that POWV-Spo convalescent mice were protected from lethal POWV-LB infection (p = 0.002, Figure 1G). These mice demonstrated no appreciable indicators of disease nor signs of weight loss following challenge (Figure 1H). These results indicated that POWV-Spo immune memory was protective against POWV-LB and thereby suitable for evaluating immune protection for POWV.
POWV-specific antibodies are present at 7 dpi
Prior POWV-Spo infection provided protection against lethal POWV-LB infection, suggesting that adaptive immune responses can mitigate POWV disease. With the goal of determining whether POWV-specific antibodies are present when mice succumb to infection, we examined the kinetics of the antibody response to POWV-LB, as we had previously done with Zika virus (ZIKV) (Hassert et al., 2018, 2019). However, unlike in the case of ZIKV, POWV-LB-infected B6 mice all succumb to infection within 2 weeks, therefore determination of the functionality and kinetics of the adaptive immune response was severely limited. We began to characterize the humoral response by terminally bleeding mice infected with 102 FFU POWV-LB at 7 dpi to examine serum binding and neutralization capacity. We were able to detect POWV-specific IgG responses by 7 dpi using whole virion antigen in an indirect enzyme-linked immunosorbent assay (ELISA), and confirmed that the two POWV lineages are serologically indistinguishable (Figures 2A and 2B). We did not detect binding to either lineage by IgG at 3 dpi. IgG raised against POWV-LB bound to virions of both lineages, with a slight but significant preference for POWV-LB (p = 0.0272, Figure 2C). We were able to detect neutralization of POWV-LB in a focus reduction neutralization test (FRNT), with an average reciprocal serum dilution of 1.73×105 for 50% neutralization (Figure 2D). Analysis of IgM levels at 3 and 7 dpi with POWV-LB showed detectable binding at 3 dpi, which was increased at 7 dpi (Figures 2E and 2F). Our analysis of the kinetics of the POWV-Spo antibody binding yielded similar results, with detectable IgG levels by 8 dpi and increasing at 15 and 30+ dpi (Figures S4A–S4C). Levels of IgM for POWV-Spo-infected mice were increased from 4 to 8 dpi and decreased at 15 and 30+ dpi as IgG binding increased (Figures S4D and S4E). The results of this kinetics study suggest that, while POWV-specific B cells are present and producing IgG and IgM antibodies during the acute infection, they are incapable of protecting mice from a lethal POWV-LB infection as these mice uniformly succumb to infection by 13 dpi (Figure 1A).
Figure 2. POWV-specific antibodies are present at 7 dpi.

(A) Whole virion POWV-LB IgG ELISA using sera of B6 mice challenged s.c. with 102 FFU POWV-LB and harvested at 3 dpi (black, n = 6) and 7 dpi (blue, n = 8).
(B) Whole virion POWV-Spooner (POWV-Spo) IgG ELISA using sera of B6 mice (n = 8) challenged s.c. with 102 FFU POWV-LB and harvested at 3 dpi (black, n = 6) or 7 dpi (blue, n = 8).
(C) Area under the curve for IgG ELISAs as shown in (A and B) (p = 0.0272, Student’s unpaired t test).
(D) FRNTs performed on baby hamster kidney cells (BHK-21 clone 13 ATCC) examining the neutralization capacity against 102 FFU POWV-LB of sera from mice challenged s.c. with 102 FFU POWV-LB and harvested 7 dpi (n = 9).
(E) Whole virion POWV-LB IgM ELISA using sera of B6 mice challenged s.c. with 102 FFU POWV-LB and harvested 3 dpi (n = 6) or 7 dpi (n = 3).
(F) Area under the curve for IgM ELISAs as shown in (E). Data are reported as mean ± SEM.
POWV-specific T cells are present during infection
We next sought to describe the kinetics of the T cell response to POWV. Previous studies with TBEV have described both protective and pathogenic roles for T cells during infection (Hassert et al., 2018, 2019; Grifoni et al., 2019, 2020; Brien et al., 2008; Růžek et al., 2009). To describe the contribution of the T cell compartment to POWV immunity, we used our H2b restricted B6 model of infection to map the CD4+ and CD8+ T cell epitopes within the structural proteins of POWV-LB. Using approaches previously adapted for ZIKV epitope mapping in our lab, we designed a peptide library of 157 peptides, each 15 aa in length and 10 with aa of overlap (Hassert et al., 2018, 2019). This library spanned the structural proteins capsid (C), pre-membrane (prM), and envelope (E) (Figure 3A).
Figure 3. POWV-specific T cells are present during infection.

(A) Schematic representation of the POWV-LB polyprotein (not to scale). Structural proteins capsid (red) (C) pre-membrane (pink, prM), and envelope (purple) (E) are located near the N terminus. Also depicted is the coverage of the POWV peptide library, comprised of a 15-mer peptide library designed with a 10-amino-acid (aa) overlap.
(B) Potential CD8+ T cell epitope hits as determined by interferon-gamma (IFN-γ) production by CD8+ T cells (CD19− CD8+) in response to the peptide library illustrated in (A). Candidate epitopes were determined in two independent experiments by stimulating splenocytes ex vivo from POWV-LB infected B6 mice s.c. infected with 103 FFU (n = 2) 7 dpi in the presence of brefeldin A for 6 h. Positive hits were considered those that elicited a T cell response 2-fold above background (dashed line).
(C) Potential CD4+ T cell epitope hits determined in the same manner as (B), at 11 dpi (n = 2).
(D) Representative flow plots depicting CD8+ T cell responses to POWV-LB infection described in (B). The cytokines tumor necrosis factor alpha (TNF-α) and IFN-γ are shown on the x and y axes, respectively. Reponses to no stimulation (top left), α-CD3 (top right), POWV56/57 peptide (bottom left) and POWV70/71 peptide (bottom right) are shown.
(E) Representative flow plots depicting CD4+ T cell responses to POWV-LB infection described in (B). Reponses to no stimulation (left), α-CD3 (middle), POWV104/105 peptide (top right), and POWV125/126 peptide (bottom right) are shown.
(F) Representative image of tetramer staining for POWV-E282-specific CD8+ T cells using H2-b restricted Db tetramer from naive B6 mice (left) or mice challenged s.c. with 103 FFU POWV-LB and harvested 7 dpi. Cells are gated on CD19− CD8+.
(G) Frequency of POWV-E282-specific CD8+ T cells by tetramer staining relative to total CD8+ T cells from B6 mice challenged s.c. with 103 FFU POWV-LB and bled longitudinally over a period of 7 days. All data are reported as mean ± SEM.
Total splenocytes from POWV-LB infected B6 mice were harvested 8 or 10 dpi, corresponding to the peak of the CD8+ and CD4+ T cell responses, respectively. T cells were stimulated ex vivo in the presence of brefeldin A and individual peptides in the library for 6 h. As a positive control, anti-CD3 was used to stimulate antigen-experienced CD4+ and CD8+ T cells and a well of unstimulated cells served as a negative control. Following stimulation, cells were fixed, permeabilized, and stained for flow cytometric analysis to identify the production of cytokines IFN-γ and TNF-α. Putative epitope candidates were identified as peptides that elicited a response greater than 2-fold over background determined from the unstimulated wells. We were unable to detect cytokine production following peptide stimulation in uninfected animals (Figure S5A). Using this approach, we identified two CD8+ and two CD4+ candidate epitopes (Figures 3B–3E). The POWV peptide CD8+ and CD4+ T cell epitopes are named with the same nomenclature used previously for both WNV and ZIKV, with the flavivirus followed by the abbreviated name of the viral protein and number of the aa, based on the POWV open reading frame (ORF) (Hassert et al., 2018, 2019; Brien et al., 2008) (e.g., POWV-E282 epitope is located in the E protein at the 282nd aa in the ORF). H2b restricted CD4+T cell epitopes are listed as 15-mer peptides (Table S1). Because our study examined structural POWV proteins, there are likely CD4 T cell epitopes within the NS proteins that were not screened in this study. The possibility that there are multiple unidentified POWV-specific CD4+ T cell epitopes within NS proteins is supported by studies with other flaviviruses in both humans and mice (Blom et al., 2015; Hassert et al., 2018, 2019; Elong Ngono et al., 2019; Brien et al., 2007, 2008; Graham et al., 2020; Grifoni et al., 2019, 2020).
For putative CD8+ T cell candidates, we sought to determine the optimal 8-mer or 9-mer H2b restricted POWV CD8+ T cell peptides. To do this, we generated a panel of possible POWV CD8+T epitopes based on previously identified Kb and Db conserved peptide anchor residues (Hoof et al., 2009) and repeated the experiments in the presence of predicted 8- or 9-mer peptides (Figures 3D; Table S1). Notably, the E351–361 CD8+T cell epitope–although it consistently yielded strong responses as a 15-mer peptide (POWV70/71)–could not be optimized to determine the ideal 8- or 9-mer peptide, nor could the Kb and Db restriction be determined. The difficulty determining the optimal epitope was attributable to the stronger response to the E282–291 CD8+T cell epitope. This epitope–designated POWV-E282–elicited the strongest CD8+ T cell response and appears to be a prominent target for CD8+ T cells during POWV-LB and POWV-Spo challenge. We determined via RMA-S stabilization assay that this epitope is H2b-Db restricted (Figures S5B and S5C) and can be quantified by tetramer staining (Figure 3F).
To describe the kinetics of the POWV-specific CD4+ and CD8+ T cell response, we challenged mice with 103 FFU of POWV-LB and isolated blood over the course of infection for tetramer staining (Figure 3G). We found that the POWV-E282 CD8+T cell response is detectable 5 dpi with 1% of CD8+ T cells responding to POWV-E282 and increases until 7 dpi with 3% of CD8+ T cells responding to POWV-E282. Of these, a low but detectable number of POWV-E282-specific T cells produce granzyme B. We were limited to blood collected at or before 7 dpi, as B6 mice succumb to POWV-LB infection within this time frame. The CD4+ T cell response was not strong enough to be detected with the identified peptides at the 1–7 dpi time points, and cytokines produced in response to anti-CD3 were difficult to detect before 10 dpi. However, a detectable CD4+ T cell response was present from 10 dpi until mice succumb to infection, suggesting that the CD4+ T cell compartment was not sufficient for control of POWV replication, as was observed with CD8+ T cells. Examinations of POWV-E282 in POWV-LB- or POWV-Spo-infected mice showed no significant differences in the number of antigen-specific CD8+ T cells in splenocytes, nor did we see differences in granzyme B production (Figures S5D–S5F). We did see a significantly (p = 0.0025) higher number of antigen-specific T cells in the brains of POWV-LB-infected mice at 8 dpi relative to POWV-Spo-infected mice (Figure S5G). The results of the kinetics study suggest that, while POWV-specific CD4+ and CD8+ T cells are primed, producing cytokines, and infiltrating infected tissues during the acute infection, they are not sufficient for protecting mice from lethal POWV-LB infection.
POWV-Spo infection elicits partially protective antibody responses against lethal POWV-LB
Our studies of POWV-LB infection of B6 mice revealed detectable antibody binding and neutralization responses as early as 7 dpi, as well as polyfunctional CD8+ T cell responses peaking at 7 dpi and CD4+ T cell responses at 10 dpi. These responses were not sufficient to protect mice from POWV-LB challenge, and infection resulted in uniform mortality.
We wanted to examine the protective capacity of convalescent POWV-Spo sera in immune competent mice. To do this, we passively transferred sera from recovered POWV-Spo or an irrelevant alphavirus-negative control (Rift Valley fever virus vaccine strain, MP-12) mice into naive B6 mice. B6 mice were challenged s.c. with 103 FFU POWV-Spo or 102 FFU MP-12. We saw no mortality from the mice infected with the control MP12 and a survival rate of 60% of mice challenged with POWV-Spo as observed previously (Figure 4A). POWV-Spo- and MP12-infected mice that survived >30 dpi, recovered weight loss, and had no apparent indicators of disease, were considered recovered from infection and terminally bled for collection of sera. Sera was diluted 1:10 in saline and passively transferred into naive B6 mice. These mice were challenged s.c. with 102 FFU POWV-LB within 24 h of serum transfer and monitored daily (Figure 4B).
Figure 4. POWV-Spooner infection elicits partially protective antibody responses against lethal POWV-LB.

Mortality (A) and weight loss (B) of alphavirus-negative control (Rift Valley fever virus [RVFV] vaccine strain MP-12) (n = 4) or POWV-Spooner (n = 7) sera-recipient B6 mice following lethal challenge with 102 FFU POWV-LB s.c.. One day prior to challenge, mice were treated with sera from convalescent MP-12 or POWV-Spo mice diluted 1:10 in saline and administered via intraperitoneal (i.p.) injection. Mice receiving POWV-For generation of convalescent sera, B6 mice were phase 30 dpi. All data are reported as mean ± SEM.
We found that B6 mice which received sera from POWV-Spo immune mice were somewhat protected from lethal POWV-LB challenge, with a significantly improved survival rate of 60% relative to no protection for MP-12 recipient mice (p = 0.0005, Figure 4A). POWV-Spo recipient mice experienced mild weight loss beginning at around 13 dpi, later than MP-12 recipient mice, which began losing weight at around 6 dpi (Figure 4B). These mice displayed few indicators of disease compared with control mice that received sera from MP-12 immune mice (not shown). For POWV-Spo recipient mice that recovered from POWV-LB challenge, weight loss was recovered by 18 dpi. We did not observe long-term neurological sequelae in surviving mice following the resolution of disease indicators and weight loss. Our results using passive transfer of convalescent POWV-Spo sera into B6 mice suggest that–while humoral responses raised against POWV-Spo can delay disease onset in isolation–only the combination of cellular and humoral adaptive immune responses is sufficient to provide protection from lethal POWV-LB challenge in this model.
POWV-LB neuroinvasive disease and death occurs independent of host immune response
As we had previously observed that 7–8 dpi corresponded to the peak of the CD8+ T cell response in our mouse model of both ZIKV and POWV, we wanted to further investigate the role of the T cell compartment in POWV-LB challenge. To determine whether mortality following POWV-LB infection was attributable to the T cell compartment, we infected recombination-activating gene 1-deficient (Rag1−/−) mice and NOD SCID gamma (NSG) mice with 102 FFU of POWV-LB. Rag1−/− mice are T and B cell deficient and NSG mice are also deficient in T and B cells, as well as NK cells and hemolytic complement, and have defects in macrophage and dendritic cell functions (Mombaerts et al., 1992; Shultz et al., 2005; Coughlan et al., 2016). By challenging with a low dose of POWV-LB, we anticipated that we would gain insight into whether POWV-LB could cause morbidity and mortality independent of the T and B cell compartments.
Following infection of B6, Rag1−/−, and NSG mice with 102 FFU POWV-LB, mice were monitored for 21 days (Figure 5). We observed that Rag1−/− and NSG mice had significantly longer survival times relative to B6 mice (p = 0.0493, p = 0.0443) (Figure 5A). However, both groups uniformly succumb to infection, with Rag1−/− mice succumbing to disease with an MST of 12.8 dpi and NSG mice succumbing to disease with an MST of 13.2 dpi, compared with an MST of 8.5 dpi for B6 mice. Weight loss for Rag1−/− mice began at 6 dpi and persisted until mice succumbed (Figure 5B). Similarly, for NSG mice, weight loss began at 6 dpi and persisted until mice succumbed to infection, with no significant differences between immunocompromised mice (p = 0.65) (Figure 5B). Interestingly, we observed distinct disease pathology in Rag1−/− and NSG mice relative to infected B6 mice. Whereas B6 mice began to show signs of infection as early as 5 dpi, and by 8 dpi all showed evidence of infection, Rag1−/− and NSG mice had delayed onset of disease. In immunocompromised mice, the first indicators of disease did not appear until 8 dpi, with all mice showing evidence of disease by 10 dpi. We also noted distinct signs of disease among Rag1−/− and NSG mice. POWV-LB infection in B6 mice typically results in ruffled fur and hunched posture, limp tail, hindlimb and forelimb weakness, and eventually moribund posture, before succumbing to infection (Figure 1C). In Rag1−/− and NSG mice, however, we observed that, after 10 dpi, mice developed a shaking or twitching phenotype, and often displayed a reduction in or loss of coordination. Occasionally, mice had muscle spasms, and one Rag1−/− mouse developed right hindlimb paralysis. Because these mice exhibited different disease progression, disease scoring by the same system established for B6 mice was not possible and is therefore not represented in this dataset. In summary, infection of mice lacking functional B and T cell compartments (Rag1−/− and NSG) with POWV-LB resulted in prolonged survival and distinct disease progression. These findings suggest that the adaptive immune response could drive morbidity and mortality. Importantly, this finding also suggests that, in this murine model, POWV-LB lethality involves two components: one independent of the adaptive immune response and one that is immune mediated.
Figure 5. POWV-LB neuroinvasive disease and death occurs independent of host immune response.

(A and B) Mortality (A) and weight loss (B) of NOD SCID gamma (NSG) (n = 5), recombination-activating gene 1-deficient (Rag1−/−, n = 5), and C57BL/6 (B6, n = 6) mice challenged s.c. with 102 FFU POWV-LB and monitored for 14 dpi. Immunocompromised mice survived significantly longer than B6 counterparts (pNSG = 0.0443, pRag1−/− = 0.0493, Mantel-Cox test). There was no difference between NSG and Rag1−/−mice (p = 0.65, Mantel-Coxtest).
(C and D) Mortality (C) and weight loss (D) for saline Rag1−/− (n = 8) mice, as well as recipient Rag1−/− mice that received POWV-Spo convalescent sera diluted 1:10 (green, n = 5) or 1:1 (blue, n = 5) prior to s.c. challenge with 102 FFU POWV-LB. Rag1−/− mice that received POWV-Spo sera diluted 1:10 (**p = 0.031) or 1:1 (**p = 0.013) had significantly prolonged survival times relative to Rag1−/− mice receiving saline (Mantel-Cox test). All data are reported as mean ± SEM.
As we had previously seen that POWV-Spo infection could generate an antibody response with binding and neutralization capabilities that provided partial protection in B6 mice, we next sought to understand the protective capacity of POWV-specific antibodies against lethal POWV-LB challenge independent of the T cell compartment. To this end, we harvested sera from recovered POWV-Spo mice, which was diluted in saline and passively transferred via intraperitoneal (i.p.) injection into Rag1−/− mice 24 h before lethal POWV-LB challenge. Mice were then monitored daily for weight loss and signs of disease.
We found that POWV-Spo sera diluted 10-fold and passively transferred into Rag1−/− mice significantly prolonged survival time (p = 0.0031) but was insufficient to provide protection against lethal POWV-LB challenge (Figure 5C). Instead, Rag1−/− mice that received POWV-Spo sera displayed delayed onset of weight loss and indicators of disease. Mice that received more concentrated POWV-Spo sera (diluted only 2-fold) showed significantly prolonged survival (p = 0.0013) relative to Rag1−/− mice receiving no sera (Figure 5C). Notably, these mice still succumb to disease by 22 dpi. Whereas Rag1−/− mice that received saline alone displayed signs of weight loss at 6 dpi, weight loss for Rag1−/− mice receiving POWV-Spo sera diluted 10-fold showed signs of weight loss around 11 dpi, which persisted until mice succumbed between 15 and 21 dpi (Figure 5D). Rag1−/− mice that received more concentrated POWV-Spo sera (diluted only 2-fold) displayed signs of weight loss at around 15 dpi but succumbed at around 21 dpi (Figure 5D). Signs of disease exhibited by passive transfer recipients were effectually identical to the saline-treated mice, but occurred later, at around 13–16 dpi. Altogether, the inability of convalescent POWV-Spo sera to provide protection from lethal POWV-LB challenge in Rag1−/− mice suggests that humoral responses alone are insufficient for long-term control of POWV-LB infection as polyclonal antibodies in sera, and that the T cell compartment is an important factor in sustained protection from POWV-LB.
T cell-mediated immunity alone is insufficient for protection against POWV-LB
We have observed a detectable T cell response specific to POWV-LB that arises in wild-type B6 mice and observed that Rag1−/− and NSG mice lacking adaptive immune cell compartments have a longer MST compared with immune competent mice. However, passive transfer of sera alone is insufficient for durable protection in Rag1−/− mice. Furthermore, CD4+ and CD8+ T cells have been shown to be important for immune protection from tick-borne flaviviruses (Blom et al., 2015, 2018). Taken together with our data, this suggested to us that the T cell compartment does contribute to durable protection against POWV. We therefore undertook T cell adoptive transfer studies. As before, donor B6 mice for adoptive T cell transfers were challenged s.c. with 103 FFU POWV-Spo or 102 FFU MP-12. Upon recovery, total CD4+ and CD8+ T cells were harvested from splenocytes and purified by negative selection before 1 × 106 total T cells were administered intravenously (i.v.) to B6 mice. Within 24 h, mice were s.c. challenged with 102 FFU POWV-LB. Unlike most previous studies with flaviviruses (Brien et al., 2008; Hassert et al., 2018, 2019; Elong Ngono et al., 2019; Watson et al., 2016; Bassi et al., 2015; Regla-Nava et al., 2018; Wen et al., 2017; Yauch et al., 2009), we saw no significant differences between survival time of naive recipient mice and mice that received T cells from POWV-Spo immune donors (p = 0.97, Figure 6A). We also saw no differences in weight loss between the naive and POWV-Spo T cell recipient groups (Figure 6B).
Figure 6. T cell-mediated immunity alone is insufficient for protection against POWV-LB.

(A and B) Mortality (A) and weight loss (B) for mice receiving adoptively transferred T cells (both CD4+ and CD8+) from POWV-Spooner (red, POWV-Spo, n = 8) experienced, alphavirus-negative control (RVFV vaccine strain MP-12) experienced T cells (blue, MP12, n = 2) or naive T cells (black, n = 8) into naive B6 mice. B6 mice were challenged s.c. with 102 FFU POWV-Spo or saline and harvested in memory phase 30 dpi. CD4+ and CD8+ T cells were enriched via negative selection and 1×3 106 cells were administered i.v. to B6 mice. Within 24 h, recipient mice were challenged s.c. with 102 FFU POWV-LB and monitored for 21 dpi. No significant differences in mortality were observed between naive and POWV-Spo recipient mice (p = 0.3592, Mantel-Cox test).
(C and D) Mortality (C) and weight loss (D) for B6 mice s.c. challenged with 102 FFU POWV-LB in control (black, n = 3) or CD4+ T cell-deplete (green, n = 3) conditions and monitored for 14 dpi. No significant difference in mortality (p = 0.6877) as determined by Mantel-Cox test.
(E and F) Mortality (E) and weight loss (F) for B6 mice s.c. challenged with 102 FFU POWV-LB in control (black, n = 6) or CD8+ T cell deplete (red, n = 7) conditions and monitored for 14 dpi (p = 0.0003, Mantel-Cox test). All data are reported as mean ± SEM.
To further assess the contribution of the T cell compartment in POWV-LB infection, we performed CD4+ and CD8+ T cell depletions by administering CD4+ or CD8+ T cell-depleting antibody before challenge with 102 FFU POWV-LB. Confirmation of T cell depletion was performed by flow cytometric analysis of peripheral blood collected 3 dpi (Figure S6). We saw no significant differences in survival or weight loss between control and CD4+ T cell-depleted mice (Figures 6C and 6D). We also saw no significant differences in signs of disease between control and CD4+ T cell-deplete groups (not shown). Depletion of CD8+ T cells, however, was able to significantly prolong survival relative to control mice (p = 0.0003, Figure 6E). Despite prolonged survival times of CD8+ T cell-deplete mice, we saw no significant weight loss differences between depleted and non-depleted groups (Figure 6F). However, in CD8+ T cell-deplete animals we observed slightly worsened signs of disease (not shown). Taken together, our T cell adoptive transfer and depletion studies show that memory T cells primed in POWV-Spo infection are not capable of providing protection against lethal POWV-LB challenge alone. In addition, the prolonged survival observed following CD8+ T cell depletion in the lethal POWV-LB challenge model suggests that CD8+ T cells may be partly responsible for immunopathogenesis in B6 mice.
VLP-based vaccination confers protection against lethal POWV-LB challenge
Applying what we learned about the requirement for humoral and cell-mediated immunity for control of POWV-LB infection, we sought to utilize a VLP-based vaccination approach to protect against lethal POWV-LB challenge. Expression of flavivirus prM and E proteins in cultured cells has previously been shown to result in secretion of VLPs into culture media (Garg et al., 2017; Espinosa et al., 2018; Salvo et al., 2018). We constructed a cell line that inducibly expresses POWV-prME upon addition of doxycycline resulting in VLP secretion (Figures S7A–S7C). VLPs were purified from culture supernatant and used as the antigen in vaccination experiments. We adopted a prime-boost strategy for POWV-VLP vaccination in B6 mice. In our approach, mice were administered 2×107 plaque-forming units equivalents of POWV-LB VLP intramuscularly (i.m.) in aluminum hydroxide gel (Alum) adjuvant for a primary vaccination. At 21 days post vaccination, mice were boosted in a manner identical to the primary vaccination and bled for B and T cell analysis (Figure 7A).
Figure 7. VLP-based vaccination confers protection against lethal POWV-LB challenge.

(A) Vaccination strategy and relevant time points for 10- to 14-week-old B6 mice. Mice were vaccinated with 300 μg per mouse of 2% Alhydrogel (Alum only) ± 2 μg POWV-LB VLP i.m. and allowed to develop a memory response for 21 days post vaccination (dpv). At 21 dpv, mice were boosted with identical formulations of Alum ± 2 mg POWV-LB VLP i.m. Three days after boosting, submandibular bleeds were performed to examine the binding and neutralization activity via ELISA and FRNT. Thirty-one days following initial vaccination, mice were challenged s.c. with 102 FFU POWV-LB and monitored for morbidity and mortality. At 3 dpi, submandibular bleeds were performed to examine CD4+ and CD8+ T cell responses.
(B) Representative graph from two independent experiments with ELISAs examining binding to whole virions using sera collected from mice 24 dpv with Alum ± 2 μg POWV-LB VLP (n = 8 and n = 9, respectively). Binding indicated by absorbance (A450) to POWV-LB.
(C and D) POWV-Spooner (C) and West Nile virus (WNV) (D) is shown.
(E) Representative graph from two independent experiments with FRNTs performed on BHK-21 cells examining neutralization activity against 102 FFU POWV-LB for sera collected 24 dpv (n = 2 and n = 4, respectively).
(F) Representative flow plots showing T cell responses in blood of mice VLP vaccinated (n = 4) or Alum adjuvant only (n = 3)-treated mice 3 days after 102 FFU POWV-LB challenge. T cells were treated ex vivo with either no stimulus (left), 10 μM peptide (middle), or α-CD3 (right) in the presence of BFA for 6 h and stained and analyzed as in Figure 3.
(G and H) Mortality (G) and weight loss (H) of B6 mice vaccinated and boosted with Alum ± 2 μg POWV-LB VLP(n = 8 and n = 9, respectively) and challenged s.c. with 102 FFU POWV-LB and monitored for 30 dpi. Mice vaccinated with POWV-VLP were protected from lethal POWV-LB challenge (p < 0.0001, Mantel-Cox test). All data are reported as mean ± SEM.
We performed an indirect ELISA to examine the capacity of POWV-VLP sera to bind whole POWV-LB, POWV-Spo, and WNV virions. We found that POWV-VLP vaccination induces antibodies that bind both POWV lineages (Figures 7B and 7C). We also see a lower degree of cross-binding to WNV, suggesting the presence of antibodies recognizing conserved portions of structural proteins (Figure 7D).
In addition to binding capacity, we wanted to examine the neutralization capacity of antibodies raised during VLP vaccination. To this end, we performed FRNTs as described previously (Brien et al., 2013; Smith and Hirsch, 2020). We found that POWV-LB neutralization was low but detectable following primary VLP vaccination (Figure 7E). We observed with a limited subset of samples that neutralization increased following boosting (not shown). We also wanted to assess the durability of antibody responses using this approach. To this end, we vaccinated mice with a single dose of adjuvanted VLP or adjuvant only control in the same manner as Figure 7A. Blood from vaccinated (n = 3) or Alum control mice (n = 2) was collected via submandibular cheek bleed 15 weeks following vaccination and neutralization determined by FRNT against POWV-LB. We were able to detect neutralizing antibody titers for the duration of the experiment, up to 15 weeks post vaccination (Figure S7D). This suggested that POWV-VLP-based vaccination could induce durable neutralizing antibody responses.
To determine whether VLP-based vaccination-induced T cell responses, we stimulated T cells ex vivo using the epitopes identified in Figure 4 and Table S1, followed by flow cytometric analysis, to examine cytokine production. Responses were not detectable in peripheral blood following the boost (Figure S7E), but we did observe polyfunctional POWV-E282-specific CD8+ T cells in the blood 3 days after POWV-LB challenge (Figure 7F). This response was similar in magnitude to anamnestic responses observed in POWV-Spo convalescent mice 4 days after challenge with POWV-LB (Figure S7F). This response was absent in Alum-only-vaccinated mice, indicating that POWV-VLP-vaccinated mice mounted a POWV-specific T cell response. However, we were unable to detect CD8+ T cell responses to the POWV351–361 epitope and the POWV-specific CD4+ T cell epitopes at the indicated time points.
With the understanding that B and T cell responses are critical for POWV-LB immune-mediated protection, we challenged mice vaccinated with POWV-VLP + Alum or Alum only with 102 FFU POWV-LB. The POWV-VLP-vaccinated mice uniformly survived lethal POWV-LB challenge (Figure 7G). POWV-VLP-vaccinated mice experienced no weight loss or apparent indicators of disease (Figure 7H). Together, these results suggest that the combination of humoral and cellular responses were necessary for protection against lethal POWV-LB challenge, and that a prime-boost strategy for POWV-LB-based VLPs can provide durable protection in a lethal POWV-LB challenge model.
DISCUSSION
Using B6 mice, we have gained insights into immune protection against POWV disease. We have demonstrated that POWV-Spo infection in our model can drive a protective immune response against POWV-LB. We also described detectable binding and neutralizing antibody profiles in POWV-LB-infected mice. However, we showed via passive transfer studies in B6 and Rag1−/−mice that POWV-Spo sera alone appears to delay, but not entirely mitigate, POWV-LB morbidity and mortality. Furthermore, we identified two CD8+ and 2 CD4+ T cell epitopes within the structural proteins of POWV-LB, including one H2-Db-restricted CD8+ T cell epitope, POWV-E282. Finally, we demonstrate, via T cell adoptive transfer of POWV-Spooner primed cells and depletion studies, that POWV-specific T cells are insufficient for complete protection from lethal POWV-LB challenge. Ultimately, our findings reveal that B and T cell-mediated protection is critical for protection, as exemplified by the successful implementation of a POWV-LB VLP-based vaccination strategy. In these studies, we utilized a mouse model of POWV-LB infection to establish factors associated with immune protection against lethal POWV-LB challenge. Our main findings include: (1) the requirement of B and T cell responses for protection against lethal POWV-LB challenge, (2) viral- and host-mediated components of POWV-LB mortality, and (3) application of this knowledge to design a successful VLP vaccination approach.
In addition to establishing a mouse model of POWV-LB infection, our work using immunocompromised mouse models indicate that the POWV-LB neurotropism can drive mortality independent of the host adaptive immune system. This was best illustrated by uniform mortality in B and T cell-deficient NSG and Rag1−/− mice challenged with POWV-LB. These mice displayed a distinct disease progression, implying that POWV neurological involvement in mice is possibly independent of adaptive immunity. Future studies should consider whether cellular targets of infection or replication kinetics are altered in NSG and Rag1−/− mice, which may impact POWV disease.
The implication that adaptive immunity may contribute to immune-mediated pathogenesis is noteworthy. The disease phenotype observed in B and T cell-deficient (NSG, Rag1−/−) mice was similar to depletion of CD8L+ T cells in B6 mice prior to POWV-LB challenge, indicating that CD8+ T cells are important drivers of immune-mediated pathogenesis in our model. This finding may be relevant to POWV morbidity and mortality, and the development of neurological symptoms during POWV disease. CD8+ T cells contributing to immune-mediated pathogenesis in POWV infection is consistent with reports for other flaviviruses (Hassert et al., 2020, 2021) and flaviviruses causing TBE (Růžek et al., 2009; Blom et al., 2018). Strikingly,immuno-compromised SCID mice challenged with TBEV have longer survival times than BALB/c or B6 mice. Furthermore, studies of mice lacking CD8+ T cells during TBEV challenge found survival time was extended (Ruzek et al., 2009) and adoptive transfer of TBEV-specific CD8+ T cells into SCID reduced survival times (Růžek et al., 2009). This suggests that the presence of CD8+ T cells is sufficient for immune-mediated pathogenesis of these neurotropic tick-borne flaviviruses. This phenomenon has relevance for efforts to develop vaccinations against tick-borne flaviviruses and should be the subject of further study.
Interestingly, the POWV351–359 region containing a CD8+ T cell epitope also contains an POWV-specific B cell epitope identified by Choi et al. (2020) in their synthetic enhanced DNA-based POWV vaccination approach (Choi et al., 2020). These concomitant findings, as well as other studies (Cimica et al., 2021), underscore the benefit of a VLP-based vaccination strategy that elicits responses to B and T cell epitopes. This finding also highlights the need to better understand immune responses elicited by different vaccine formulations (i.e., different adjuvants driving T-helper profiles, TH1 versus TH2) to provide optimal immunity against POWV-LB. Future studies should explore the role of CD8+ T cell responses in VLP-based vaccination strategies against POWV-LB to elucidate the role of CD8+ T cells in protection versus pathogenesis during POWV infection. Notably, in our studies of CD8+ T cell responses within the POWV model, mice mount a response lower in magnitude relative to other flaviviruses (ZIKV, WNV) utilized in our lab. This is true for both lineages, although the mechanisms underlying this reduced response remain unclear to us. It is also notable that antibody responses detected against POWV-LB were similar in magnitude to responses against POWV-Spo, suggesting that the ability to generate an antibody response to POWV does not solely dictate survival outcome among B6 mice infected with the two lineages.
The contribution of CD4+ T cells in the context of POWV immunity is unclear. Although our studies did not examine the impact of adoptively transferring POWV-experienced CD4+ T cells alone, we observed no significant differences in morbidity or mortality of POWV-LB challenged mice when pan T cells were transferred to B6 mice. We also saw no significant differences in mice depleted ofCD4+ T cells prior to POWV-LB challenge. It has been shown for other flaviviruses that CD4+ T cell responses are important for protection from disease (Brien et al., 2008; Elong Ngono et al., 2019; Hassert et al., 2018; Watson et al., 2016). In particular, the role of CD4+ T cells in POWV infection and CNS disease (Ciurkiewicz et al., 2020) should be a subject of further inquiry.
From our studies, it appears that no single component of POWV immunity in isolation is sufficient for durable protection against POWV, as only sera transferred to B6 mice with a functional T cell compartment resulted in limited protection from death and severe disease.
Limitations of the study
One limitation of our studies of the POWV-LB-adaptive immune response is the necessity of using POWV-Spo for the generation of donor sera and T cells. The discrepancies in disease progression between the two lineages underscore the need to better each POWV lineage, but also to dissect the molecular mechanisms that drive the distinct disease outcomes observed among two lineages that differ very little in terms of aa identity. Overall, our studies suggest that the balance of T and B cell-mediated immunity is critical for prevention of severe POWV disease, and that POWV neurological sequelae are the result of viral-and host-mediated pathologies. Our findings support the careful consideration of a balanced B and T cell response elicited by future POWV vaccines to provide protection from disease. The finding that balanced T and B cell responses are critical for immune-mediated protection from POWV disease will aid the rational design of future POWV vaccines.
STAR*METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Amelia K. Pinto (amelia.pinto@health.slu.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact upon request.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Viruses and cells
POWV-LB (lineage I) (Mandl et al., 1993) and POWV-Spooner (lineage II) (Ebel et al., 1999) were obtained courtesy of G. Ebel and passaged once on BHK-21 clone 13 cells (Mesocricetus auratus, Syrian golden hamster kidney fibroblasts) purchased from American Type Culture Collection (ATCC CCL-10). Rift Valley Fever Virus vaccine (strain MP-12, GenBank accession numbers: DQ375404 (L), DQ380208 (M), and DQ380154 (S)) (Ermler et al., 2013) was gifted from A. Hise and M. Buller and passaged twice in African green monkey kidney epithelial cells (Vero-WHO) purchased from American Type Culture Collection (ATCC CCL-81). All supernatants of these cultures were clarified of cellular debris by centrifugation at 3,500 RPM prior to being aliquoted and frozen at 80 °C. Infectious virus titer was measured using a standard focus forming assay (FFA) on Vero cells for MP-12 or BHK-21 (clone-13) cells for POWV as described previously (Brien et al., 2013; Smith and Hirsch, 2020).
Ethics statement
All animal studies were conducted in accordance with the Guide for Care and Use of Laboratory Animals of the National Institutes of Health and approved by the Saint Louis University Animal Care and Use Committee (IACUC protocol # 2777).
Mice and infections
Wild type C57BL/6J (B6, 000664), Nod SCID gamma (NSG, 005557) and recombination activating genes 1 (Rag –/-, 002216) mice were purchased commercially from Jackson Laboratories and housed in a pathogen-free mouse facility at the Saint Louis University School of Medicine. Female Swiss Webster mice (SW-F) were purchased from Taconic Biosciences and housed in the same manner. Unless otherwise noted, approximately equal ratios of male and female mice were utilized for these studies. For CD8+ and CD4+ T cell depletion studies, 10- to 12-week-old B6 mice were infected subcutaneously (s.c.) via footpad injection with 102 focus forming units (FFU) of POWV-LB. For epitope identification, wild type B6 mice were injected with heat-inactivated serum from POWV-immune mice at −1 days post infection (DPI). Within 24 hours, serum-treated mice were infected s.c. with 102 FFU of POWV-LB virus. For Pan T-cell adoptive transfer studies, 10–12-week-old B6 mice were infected with 103 FFU of POWV-LB one day after adoptive transfer of cells. During the course of infection mice were assessed for weight loss, signs of neurological disease, and mortality daily. Signs of disease range and in the most severe cases accelerate in the following manner from no apparent disease, limp tail, hind limb weakness, hind limb paralysis, complete paralysis and death. Clinical scoring system was as follows: 0=clinically normal, 1 = limp tail, slight ruffled appearance, 2 = highly ruffled appearance, hunched posture, 3 = hindlimb weakness, conjunctivitis, 4 = hindlimb paralysis, severe conjunctivitis, urinary incontinence, 5 = moribund, animal is not moving, 6 = deceased. Mice displaying multiple signs of disease at once, such as limp tail accompanied by hindlimb weakness, are scored as the more severe sign of disease (e.g. hindlimb weakness).
METHOD DETAILS
CD8+ and CD4+ T cell depletions
B6 mice were treated with 100 μg of either CD8-depleting antibody (Anti-mouse CD8b.2, 53–5.8, Leinco, C2832) or CD4-depleting antibody (GK1.5, InVivoMab, BE0003–1) two times via intraperitoneal (i.p.) injection; once on day –3 and again on day 0. To confirm T cell depletions, blood was collected by submandibular bleed on day 3 post-infection and analyzed by flow cytometry using antibodies denoted in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-Mouse IgG (whole molecule)–Peroxidase antibody produced in goat | Sigma-Aldrich | Cat# A8924, RRID:AB_258426 |
| Alexa Fluor® 488 anti-mouse CD19 Antibody, Isotype Rat IgG2a, κ, clone 6D5 | BioLegend | Cat# MCA1439A488T, RRID:AB_1101006 |
| PerCP/cyanine5.5 anti-mouse CD8α Antibody, Isotype Rat IgG2a, κ, clone 53–6.7 | BioLegend | Cat# 100734, RRID:AB_2075238 |
| Brilliant Violet 605™ anti-mouse CD4 antibody, Rat IgG2a, κ, clone RM4-5 | BioLegend | Cat# 100548, RRID:AB_2563054 |
| APC anti-mouse IFN-gamma antibody, Rat IgG1, κ, clone XMG1.2 | BioLegend | Cat# 505810, RRID:AB_315404 |
| PE anti-mouse TNF-alpha antibody, Rat IgG1, κ, clone MP6-XT22 | BioLegend | Cat# 506306, RRID:AB_315427 |
| PE mouse anti-human Granzyme B antibody, clone GB11 | BD Biosciences | Cat# 561142, RRID:AB_10561690 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| POWV strain LB | Ebel laboratory | Mandl et al., 1993 |
| POWV-DTV strain Spooner | Ebel laboratory | Ebel et al., 1999 |
| Rift Valley Fever Virus vaccine strain (MP-12) | A. Hise & M. Buller | Ermler et al., 2013 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| POWV peptide library, accession #: NP_620099.1 | 21st Century Biochemicals | N/A |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| BHK-21 clone 13 | ATCC | ATCC Cat# CCL-10, RRID:CVCL_1915 |
| Vero CCL-81™ | ATCC | Cat# CCL-81, RRID:CVCL_0059 |
| RMA-S Cells | ATCC | RRID:CVCL_2180 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6J mice | The Jackson Laboratory | 000664 |
| B6.129S7-Rag1tm1Mom/J mice | The Jackson Laboratory | 002216 |
| NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ mice | The Jackson Laboratory | 005557 |
|
| ||
| Oligonucleotides | ||
|
| ||
| POWV LB qPCR primer 5’ - GGCTGCAAATGAGACCAATTC −3′ | IDT | N/A |
| POWV LB qPCR primer 5′’-CAGCGACACATCTCCATAGTC −3′ | IDT | N/A |
| POWV LB qPCR probe 5’-/56-FAM/TGGCATCCG/Zen/AGAAAGTGATCCTGC/3IABkFQ/-3’ | IDT | N/A |
| POWV Spooner qPCR primer 5′- GCAGCACCATAGGTAGAATGT-3′ | IDT | N/A |
| POWV Spooner qPCR primer 5′-CCACCCACTGAACCAAAGT-3′ | IDT | N/A |
| POWV Spooner qPCR probe 5’ -/56-FAM/TCTCAGTGG/Zen/TTGGAGAACACGCAT/3IABkFQ-3′ | IDT | N/A |
|
| ||
| Other | ||
|
| ||
| POWV-E282 H2-Db Tetramer, Alexa Fluor® 647 | NIH Tetramer Core | N/A |
Peptide library
A POWV peptide library was constructed based on amino acid (a.a.) sequences from POWV-LB polyprotein (Accession #: NP_620099.1). The library consists of 157 15-mer peptides, overlapping by 10 a.a. Together, it spans the nucleocapsid (Anc), pre-membrane (prM), and envelope (E) portion of the polyprotein. Lyophilized peptides were reconstituted to 10 mg/mL in 90% DMSO and stored at –80°C. until use. Within this library, all peptides appeared soluble. For epitope identification, the peptides in the library were diluted such that the final concentration of each peptide was 2μM.
Peptide stimulation
Splenocytes were harvested from mice 7 DPI in the case of CD8+ epitope identification or 10 DPI in the case of CD4+ epitope identification for acute experiments or >30 days post infection for assessment of memory and vaccination responses. Spleens were processed to a single-cell suspension over a 40 μm cell strainer and suspended in RPMI supplemented with 5% FBS and 1% HEPES. 1×106 cells were plated per well in a round-bottom 96-well plate and stimulated for 6 hours at 37°C, 5% CO2 in the presence of 10 μg/mL brefeldin A and either α-CD3 (clone 2C11) as a positive control or 10 μg of peptide.
Flow cytometry
Following peptide stimulation, cells were washed with PBS and stained for surface markers using antibodies denoted in the key resources table. The cells were analyzed by flow cytometry using an Attune NxT.
Passive transfer of sera
Ten to 12-week-old B6 mice were infected via s.c. footpad injection with 102 FFU of POWV-Spooner or Rift Valley Fever virus vaccine strain MP-12 and allowed to develop a memory response. Mice were considered convalescent when they displayed: 1) survived >30 days post infection, 2) completely recovered weight loss, and 3) showed no apparent indicators of disease. >30 DPI, mice were terminally bled via cardiac puncture and whole blood was collected and subjected to centrifugation for 15 min at 12,000 × g. Sera was stored at –80°C until further use. Sera was diluted in sterile tissue-culture grade PBS and injected i.p. 24 hours before infection with POWV.
Adoptive transfer of T cells
Ten to 12-week-old B6 mice were infected via s.c. footpad injection with 102 FFU of POWV-Spooner, Rift Valley Fever Virus vaccine strain MP-12, or PBS as a negative control. >30 DPI, splenocytes were harvested and prepared for use in a Milteyni enrichment kit. T cells were purified to >90% purity using a Miltenyi negative selection kit (130–095-130) according to manufacturer’s specifications. ~1×106 cells were administered to B6 mice i.v. 24 hours prior to infection. Successful adoptive transfer of T cells into recipient mice was confirmed by submandibular bleed at 3 DPI and flow cytometric analysis using antibodies denoted in the key resources table. When CD45.1 congenic donors were used, α-CD45.1-FITC (clone A20) was also included in the panel.
Virus-like particle (VLP) preparation and vaccination
POWV genomic RNA was isolated from a purified virus stock using Trizol reagent (Invitrogen) according to manufacturer’s protocol. POWV-LB nucleotides 361–2415 (Accession: NC_003687), encoding the transmembrane anchor and signal sequence of the C protein, prM, and E were reverse transcribed and PCR amplified using primers 50-GATCGCGGCCGCACCATGCAGAGCCTTCACATGA GAGG-3' and 5'-GATCGGAATTCCTATGCCCCAACTCCCATTGTCATC-3' (restriction sites, NotI and EcoRI respectively in bold, POWV genomic sequence underlined). The amplified product was restriction digested and cloned into vector pLVX tight-puro (Takara) to create plasmid pLVX-POWVprME. To create a cell line for inducible expression of POWV VLPs, HEK293 cells (Microbix) were transfected with pLVX-Tet-On Advanced (Takara), which expresses the tetracycline-controlled transactivator. Stable transformants were selected with G418 and cloned by limiting dilution. POWV prME-expressing pseudotyped lentiviruses were then produced by co-transfection of pLVX-POWVprME with the lentiviral packaging vector pSPAX2 (psPAX2 was a gift from Didier Trono (Addgene plasmid # 12,260; RRID: Addgene_12260) and vesicular stomatitis virus G protein expression vector pMD2.G (also from Didier Trono (Addgene plasmid # 12259; RRID: Addgene_12259). The resulting pseudotyped lentiviruses were used to transduce the tet-transactivator expressing HEK293 cells. Transductants were selected with1 μg/mL puromycin. For VLP production, cells were cultured in DMEM (Corning) supplemented with 2% fetal bovine serum, penicillin and streptomycin and 1 μg/mL doxycycline. VLPs were purified and concentrated by collecting supernatants at 7 d post doxycycline addition, filtration through a 0.45 μm filter, and centrifugation at 150,000 × g for 2 hours through a 20% sorbitol cushion. Pellets containing VLP were resuspended in PBS at 1/100th original volume and analyzed by SDS-PAGE, followed by immunoblot and detection with a POWV E-specific antibody (Anti-Langat virus E, clone 10F6, BEI Resources). Preparations of VLP were quantified by comparing dilutions of VLP to known quantities of POWV expressed at FFU equivalents/μL, and total protein quantified by BCA assay.
Ten to 12-week-old B6 mice were vaccinated i.m. in the right quadricep with 2 μg of POWV-VLP adjuvanted with 300μg of 2% aluminum hydroxide gel, Alhydrogel (InvivoGen, 21645–51-2) or 300 mg per mouse Alhydrogel alone, (denoted ‘Alum’ or ‘Alum only’). Twenty-one days later, mice were boosted with an identical formulation of POWV-VLP or Alum.
Enzyme-linked immunosorbent assays (ELISAs)
For POWV-LB or POWV-Spooner ELISAs measuring binding to whole virions, Nuc MaxiSorp Plates (ThermoFisher, 460984) were coated with 75μL/well of virus stock diluted 1:30 in carbonate coating buffer (0.1 M Na2CO3, 0.1 M NaHCO3, pH 9.3) and stored at –20°C until further use. Following coating, plates were washed with four times with 200 μL/well of ELISA wash buffer (1XPBS, 0.5% Tween) and blocked with 250 μL/well of blocking buffer (PBS + 5%BSA + 0.5% Tween) at room temperature for 1 hour. Sera from POWV-challenged mice was diluted in blocking buffer starting at 1:50 with 4-fold serial dilutions. Plates were washed with ELISA wash buffer and 50 μL/well of sera was allowed to incubate for 1 hour at 37°C. Following incubation, plates were washed with ELISA wash buffer and 50μL/well of a 1:5000 dilution of secondary antibody was allowed to incubate for 2 hours at room temperature. Plates were washed with ELISA wash buffer and 100 μL/well of enhanced TBD substrate (Neogen 308176) was allowed to incubate for 10–15 minutes during development of color change. Finally, 50μL/well of 1 N HCl was used to quench the reaction and absorbance at 450 nm was read immediately using a BioTek Instruments Epoch plate reader.
Focus forming assay (FFA)
One day prior to the assay, tissue-culture treated 96-well plates (MidSci, TP92696) were seeded with 2 × 3 104 cells/well and allowed to grow at 37°C, 5% CO2 to a target confluency of ~90%. Samples were serially diluted and added to BHK monolayers in 96-well plates for 1 h at 37°C to allow virus adsorption. Cells were overlaid with 2% methylcellulose mixed with DMEM containing 5% FBS, 1% HEPES and incubated for 24 hours at 37°C. Media was removed and cell monolayers were fixed with 5% paraformaldehyde in 1X PBS for 15 min at room temperature, rinsed, and permeabilized in Perm Wash (PBS, 0.05% Triton-X). Infected cell foci were stained by incubating cells with sera from Swiss-Webster mice challenged with 103 FFU-equivalents of β-propiolactone (BPL, Sigma-Aldrich, P5648–5ML)-inactivated POWV-LB. Sera was diluted 1:5,000 in Perm Wash and 50 μL/well was allowed to incubate for overnight at 4°C and then washed three times with Perm Wash. Foci were detected with 50 μL/well of 1:5,000 dilution of horseradish peroxidase-conjugated goat anti-mouse IgG for 2 hours. After three washes with Perm Wash, staining was visualized by addition of 50 μL/well TrueBlue detection reagent (KPL). Infected foci were enumerated by CTL Elispot.
Focus reduction neutralization tests (FRNTs)
One day prior to the assay, tissue-culture treated 96-well plates (MidSci, TP92696) were seeded with 2×3 104 cells/well and allowed to grow at 37°C, 5% CO2 to a target confluency of ~90%. Four-fold serial dilutions of sera of POWV-infected mice were mixed with 102 FFU of infectious virus, incubated at 37°C for 1 h, and added to BHK monolayers in 96-well plates for 1 h at 37 °C to allow virus adsorption. Cells were overlaid with 2% methylcellulose mixed with DMEM containing 5% FBS, 1% HEPES and incubated for 24 hours at 37°C. Media was removed and cell monolayers were fixed with 5% paraformaldehyde in 1X PBS for 15 min at room temperature, rinsed, and permeabilized in Perm Wash (PBS, 0.05% Triton-X). Infected cell foci were stained by incubating cells with sera from Swiss-Webster mice challenged with 103 FFU-equivalents of β-propiolactone (BPL, Sigma-Aldrich, P5648–5ML)-inactivated POWV-LB. Sera was diluted 1:5,000 in Perm Wash and 50 μL/well was allowed to incubate for overnight at 4°C and then washed three times with Perm Wash. Foci were detected with 50 μL/well of 1:5,000 dilution of horseradish peroxidase-conjugated goat anti-mouse IgG for 2 hours. After three washes with Perm Wash, staining was visualized by addition of 50 μL/well TrueBlue detection reagent (KPL). Infected foci were enumerated by CTL Elispot. FRNT curves were generated by log-transformation of the x axis followed by non-linear curve fit regression analysis using GraphPad Prism 9.
RMA-S stabilization assay
RMA-S cells were cultured in complete RPMI medium at 37 degrees Celsius, 5% CO2 until the night before the assay, when the cells were shifted to 29 degrees Celsius, 5% CO2. The cells were then incubated for 4 hours with decreasing concentrations of each peptide at 29 degrees Celsius, and then shifted back to 37 degrees Celsius, 5% CO2 for 1 hour. The cells were then washed with cold PBS and stained for Db (clone 28–14-8) MHC molecule at 4°C. Cells were washed with ice cold PBS and run on an Attune focusing flow cytometer. Fluorescence index was calculated by dividing the geometric mean fluorescence intensity (gMFI) of the peptide pulsed cells by non-peptide pulsed cells. Data is reported a percentage of the maximum fluorescence index of each peptide serial dilution.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses for were performed using GraphPad Prism 9 (version 9.1.2). Statistical differences in survival were determined using a Mantel-Cox test. Statistical significance in area under the curve for ELISAs was determined by Student’s unpaired t-test. Data for epitope candidates is represented as mean ± SEM for two individual experiments. Statistical significance has been indicated within the figures with asterisks (*p = 0.05, **p = 0.01, ***p = 0.001, ****p < 0.0001).
Supplementary Material
Highlights.
Robust B and T cell responses are necessary for protection against POWV
POWV lethality is comprised of both viral- and host-mediated mechanisms
A VLP-based vaccine protects against lethal POWV challenge
ACKNOWLEDGMENTS
This research was supported in part by the National Institutes of Health, National Institute of Allergy and Infectious Diseases (NIAID): 5R01AI152192-02, 5R01AI137424-03, and F31AI152460-01, and in part by the Department of Defense (award no. PR192269). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110388.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
REFERENCES
- Aberle JH, Schwaiger J, Aberle SW, Stiasny K, Scheinost O, Kundi M, Chmelik V, and Heinz FX (2015). Human CD4+ T helper cell responses after tick-borne encephalitis vaccination and infection. PLoS One 10, e0140545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for disease control and prevention (2020). Powassan Virus Statistics & Maps.https://www.cdc.gov/powassan/statistics.html.
- Bassi MR, Kongsgaard M, Steffensen MA, Fenger C, Rasmussen M, Skjødt K, Finsen B, Stryhn A, Buus S, Christensen JP, and Thomsen AR (2015). CD8+ T cells complement antibodies in protecting against yellow fever virus. J. Immunol. 194, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best Sonja M, Morris Keely L, Shannon Jeffrey G, Robertson Shelly J, Mitzel Dana N, Park Gregory S, Boer E, Wolfinbarger James B, and Bloom Marshall E (2005). Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne Flavivirus and identification of NS5 as an interferon antagonist. J. Virol. 79, 12828–12839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom K, Braun M, Pakalniene J, Dailidyte L, Béziat V, Lampen MH, Klingström J, Lagerqvist N, Kjerstadius T, Michaëlsson J, et al. (2015). Specificity and dynamics of effector and memory CD8 T cell responses in human tick-borne encephalitis virus infection. PLOS Pathog. 11, e1004622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom K, Cuapio A, Sandberg JT, Varnaite R, Michaëlsson J, Björkström NK, Sandberg JK, Klingström J, Lindquist L, Gredmark Russ S, and Ljunggren H-G (2018). Cell-mediated immune responses and immunopathogenesis of human tick-borne encephalitis virus-infection. Front. Immunol. 9, 2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien JD, Lazear HM, and Diamond MS (2013). Propagation, quantification, detection, and storage of West Nile virus. Curr. Protoc. Microbiol. 31, 15D.3.1–15D.3.18. [DOI] [PubMed] [Google Scholar]
- Brien JD, Uhrlaub JL, and Nikolich-Zugich J (2007). Protective capacity and epitope specificity of CD8(+) T cells responding to lethal West Nile virus infection. Eur. J. Immunol. 37, 1855–1863. [DOI] [PubMed] [Google Scholar]
- Brien JD, Uhrlaub JL, and Nikolich-Zugich J (2008). West Nile virus-specific CD4 T cells exhibit direct antiviral cytokine secretion and cytotoxicity and are sufficient for antiviral protection. J. Immunol. 181, 8568–8575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernokhaeva LL, Rogova YV, Vorovitch MF, Romanova L, Kozlovskaya LI, Maikova GB, Kholodilov IS, and Karganova GG (2016). Protective immunity spectrum induced by immunization with a vaccine from the TBEV strain Sofjin. Vaccine 34, 2354–2361. [DOI] [PubMed] [Google Scholar]
- Choi H, Kudchodkar SB, Ho M, Reuschel EL, Reynolds E, Xu Z, Bordoloi D, Ugen KE, Tebas P, Kim J, Abdel-Mohsen M, Thangamani S, Weiner DB, and Muthumani K (2020). A novel synthetic DNA vaccine elicits protective immune responses against Powassan virus. PLOS Negl. Trop. Dis. 14, e0008788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumakov MP, Gagarina AV, Vilner LM, Khanina MK, Rodin IM, Vasenovich MI, Lakina VI, and Finogenova EV (1963). [Experience in the experimental production and control of tissue culture vaccine against tick encephalitis]. Vopr Virusol 29, 415–420. [PubMed] [Google Scholar]
- Cimica V, Saleem S, Matuczinski E, Adams-Fish D, Mcmahon C, Rashid S, and Stedman TT (2021). A virus-like particle-based vaccine candidate against the tick-borne powassan virus induces neutralizing antibodies in a mouse model. Pathogens 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciurkiewicz M, Herder V, and Beineke A (2020). Beneficial and detrimental effects of regulatory T cells in neurotropic virus infections. Int. J. Mol. Sci. 21, 1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clow KM, Leighton PA, Ogden NH, Lindsay LR, Michel P, Pearl DL, and Jardine CM (2017). Northward range expansion of Ixodes scapularis evident over a short timescale in Ontario, Canada. PLoS One 12, e0189393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costero A, and Grayson MA (1996). Experimental transmission of powassan virus (Flaviviridae) by Ixodes scapularis ticks (Acari:ixodidae). Am. J. Trop. Med. Hyg. 55, 536–546. [DOI] [PubMed] [Google Scholar]
- Coughlan AM, Harmon C, Whelan S, O’brien EC, O’reilly VP, Crotty P, Kelly P, Ryan M, Hickey FB, O’farrelly C, and Little MA (2016). Myeloid engraftment in humanized mice: impact of granulocyte-colony stimulating factor treatment and transgenic mouse strain. Stem Cells Dev. 25, 530–541. [DOI] [PubMed] [Google Scholar]
- Ebel GD, Foppa I, Spielman A, and Telford SR (1999). A focus of deer tick virus transmission in the northcentral United States. Emerg. Infect. Dis. 5, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebel GD, and Kramer LD (2004). Short report: duration of tick attachment required for transmission of powassan virus by deer ticks. Am. J. Trop. Med. Hyg. 71, 268–271. [PubMed] [Google Scholar]
- Ebel GD, Spielman A, and Telford SR (2001). Phylogeny of North American powassan virus. J. Gen. Virol. 82, 1657–1665. [DOI] [PubMed] [Google Scholar]
- Elong Ngono A, Young MP, Bunz M, Xu Z, Hattakam S, Vizcarra E, Regla-Nava JA, Tang WW, Yamabhai M, Wen J, and Shresta S (2019). CD4+ T cells promote humoral immunity and viral control during Zika virus infection. PLOS Pathog. 15, e1007474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermler ME, Yerukhim E, Schriewer J, Schattgen S, Traylor Z, Wespiser AR, Caffrey DR, Chen ZJ, King CH, Gale M Jr., et al. (2013). RNA helicase signaling is critical for type i interferon production and protection against Rift Valley fever virus during mucosal challenge. J.Virol. 87,4846–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errico JM, Vanblargan LA, Nelson CA, Diamond MS, and Fremont DH (2020). Structural basis of neutralization of Powassan virus by monoclonal antibodies.J. Immunol.204,247.9. [Google Scholar]
- Ershova AS, Gra OA, Lyaschuk AM, Grunina TM, Tkachuk AP, Bartov MS, Savina DM, Sergienko OV, Galushkina ZM, Gudov VP, et al. (2016). Recombinant domains III of Tick-Borne Encephalitis Virus envelope protein in combination with dextran and CpGs induce immune response and partial protectiveness against TBE virus infection in mice. BMC Infect. Dis. 16, 544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa D, Mendy J, Manayani D, Vang L, Wang C, Richard T, Guenther B, Aruri J, Avanzini J, Garduno F, Farness P, Gurwith M, Smith J, Harris E, and Alexander J (2018). Passive transfer of immune sera induced by a Zika virus-like particle vaccine protects AG129 mice against lethal Zika virus challenge. EBioMedicine 27, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg H, Sedano M, Plata G, Punke EB, and Joshi A (2017). Development of virus-like-particle vaccine and reporter assay for Zika virus. J. Virol. 91, e00834–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasmi S, Bouchard C, Ogden NH, Adam-Poupart A, Pelcat Y, Rees EE, Milord F, Leighton PA, Lindsay RL, Koffi JK, and Thivierge K (2018). Evidence for increasing densities and geographic ranges of tick species of public health significance other than Ixodes scapularis in Québec, Canada. PLoS One 13, e0201924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfield M, Austin SM, Black HC, Taylor BF, and Altman R (1973). A non-fatal human case of powassan virus encephalitis. The American Journal of Tropical Medicine and Hygiene. Am. J. Trop. Med. Hyg. 22, 78–81. [DOI] [PubMed] [Google Scholar]
- Grabowski JM, Nilsson OR, Fischer ER, Long D, Offerdahl DK, Park Y, Scott DP, and Bloom ME (2019). Dissecting flavivirus biology in salivary gland cultures from fed and unfed Ixodes scapularis (Black-Legged tick). mBio 10, e02628–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham N, Eisenhauer P, Diehl SA, Pierce KK, Whitehead SS, Durbin AP, Kirkpatrick BD, Sette A, Weiskopf D, Boyson JE, and Botten JW (2020). Rapid induction and maintenance of virus-specific CD8(+) T(EMRA) and CD4(+) T(EM) cells following protective vaccination against dengue virus challenge in humans. Front. Immunol. 11, 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A, Voic H, Dhanda SK, Kidd CK, Brien JD, Buus S, Stryhn A, Durbin AP, Whitehead S, Diehl SA, De Silva AD, Balmaseda A, Harris E, Weiskopf D, and Sette A (2020). T cell responses induced by attenuated flavivirus vaccination are specific and show limited cross-reactivity with other flavivirus species. J. Virol. 94, e00089–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A, Voic H, Sidney J, De Silva AD, Durbin A, Diehl SA, Harris E, Sette A, and Weiskopf D (2019). Crosseactivity of flaviviruses specific CD8+T cell responses across different viral species. J. Immunol. 202, 76.12.. [Google Scholar]
- Hassert M, Brien JD, and Pinto AK (2020). CD8+ T cell cross-reactivity during heterologous flavivirus infection results in cross-reactive immunodomination and enhanced cytolytic capacity at the expense of virus-specific responses. J. Immunol. 204, 95.9. [Google Scholar]
- Hassert M, Harris MG, Brien JD, and Pinto AK (2019). Identification of protective CD8 T cell responses in a mouse model of Zika virus infection. Front. Immunol. 10, 1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassert M, Steffen TL, Scroggins S, Coleman AK, Shacham E, Brien JD, and Pinto AK (2021). Prior heterologous flavivirus exposure results in reduced pathogenesis in a mouse model of Zika virus infection. J. Virol. 95, Jvi0057321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassert M, Wolf KJ, Schwetye KE, Dipaolo RJ, Brien JD, and Pinto AK (2018). CD4+T cells mediate protection against Zika associated severe disease in a mouse model of infection. PLoS Pathog. 14, e1007237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermance ME, Santos RI, Kelly BC, Valbuena G, and Thangamani S (2016). Immune cell targets of infection at the tick-skin interface during powassan virus transmission. PLoS ONE 11, e0155889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermance ME, and Thangamani S (2017). Powassan virus: an emerging arbovirus of public health concern in North America. Vector Borne Zoonotic Dis 17, 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermance ME, Thangamani S, and Diamond MS (2015). Tick saliva enhances powassan virus transmission to the host, influencing its dissemination and the course of disease. J. Virol. 89, 7852–7860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinten SR, Beckett GA, Gensheimer KF, Pritchard E, Courtney TM, Sears SD, Woytowicz JM, Preston DG, Smith RP Jr., and Rand PW (2008). Increased recognition of Powassan encephalitis in the United States, 1999–2005. Vector Borne Zoonotic Dis. 8, 733–740. [DOI] [PubMed] [Google Scholar]
- Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, and Nielsen M (2009). NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemenesi G, and Bányai K (2019). Tick-borne flaviviruses, with a focus on Powassan virus. Clin. Microbiol. Rev. 32, e00106–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubinski M, Beicht J, Gerlach T, Volz A, Sutter G, and Rimmelzwaan GF (2020). Tick-borne encephalitis virus: a quest for better vaccines against a virus on the rise. Vaccines 8, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuivanen S, Hepojoki J, Vene S, Vaheri A, and Vapalahti O (2014). Identification of linear human B-cell epitopes of tick-borne encephalitis virus. Virol. J. 11, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampen MH, Uchtenhagen H, Blom K, Varnaite R, Pakalniene J, Dailidyte L, Walchli S, Lindquist L, Mickiene A, Michaelsson J, and et al. (2018). Breadth and dynamics of HLA-A2- and HLA-B7-restricted CD8(+) T cell responses against nonstructural viral proteins in acute human tick-borne encephalitis virus infection. Immunohorizons 2, 172–184. [DOI] [PubMed] [Google Scholar]
- Mandl CW, Holzmann H, Kunz C, and Heinz FX (1993). Complete genomic sequence of Powassan virus: evaluation of genetic elements in tick-borne versus mosquito-borne flaviviruses. Virology 194, 173–184. [DOI] [PubMed] [Google Scholar]
- Matveev AL, Kozlova IV, Stronin OV, Khlusevich YA, Doroshchenko EK, Baykov IK, Lisak OV, Emelyanova LA, Suntsova OV, Matveeva VA, et al. (2019). Post-exposure administration of chimeric antibody protects mice against European, Siberian, and Far-Eastern subtypes of tick-borne encephalitis virus. PLoS One 14, e0215075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcauley AJ, Sawatsky B, Ksiazek T, Torres M, Korva M, Lotrič-Furlan S, Avšič-županc T, Von Messling V, Holbrook MR, Freiberg AN, et al. (2017). Cross-neutralisation of viruses of the tick-borne encephalitis complex following tick-borne encephalitis vaccination and/or infection. npj Vaccin. 2,5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mclean DM, and Donohue WL (1959). Powassan virus: isolation of virus from a fatal case of encephalitis. Can. Med. Assoc. J. 80, 708–711. [PMC free article] [PubMed] [Google Scholar]
- Mlera L, Meade-White K, Saturday G, Scott D, and Bloom ME (2017). Modeling Powassan virus infection in Peromyscus leucopus, a natural host. PLOS Negl. Trop. Dis. 11, e0005346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, and Papaioannou VE (1992). RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877. [DOI] [PubMed] [Google Scholar]
- Nofchissey RA, Deardorff ER, Blevins TM, Anishchenko M, BoscoLauth A, Berl E, Lubelczyk C, Mutebi J-P, Brault AC, Ebel GD, and Magnarelli LA (2013). Seroprevalence of powassan virus in New England deer, 1979-2010. Am. Soc. Trop. Med. Hyg. 88, 1159–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka E, Ebel GD, and Wearing HJ (2010). Persistence of pathogens with short infectious periods in seasonal tick populations: the relative importance of three transmission routes. PLoS One 5, e11745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden NH, St-Onge L, Barker IK, Brazeau S, Bigras-Poulin M, Charron DF, Francis CM, Heagy A, Lindsay L, Maarouf A, et al. (2008). Risk maps for range expansion of the Lyme disease vector, Ixodes scapularis, in Canada now and with climate change. Int. J. Health Geographics 7,24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesko KN, Torres-Perez F, Hjelle BL, and Ebel GD (2010). Molecular epidemiology of powassan virus in North America. J. Gen. Virol. 91, 2698–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson TC, and Diamond MS (2020). The continued threat of emerging flaviviruses. Nat. Microbiol. 5, 796–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regla-Nava JA, Elong Ngono A, Viramontes KM, Huynh A-T, Wang YT, Nguyen A-VT, Salgado R, Mamidi A, Kim K, Diamond MS, and Shresta S (2018). Cross-reactive Dengue virus-specific CD8(+) T cells protect against Zika virus during pregnancy. Nat. Commun. 9, 3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson SJ, Mitzel DN, Taylor RT, Best SM, and Bloom ME (2009). Tick-borne flaviviruses: dissecting host immune responses and virus countermeasures. Immunologic Res. 43, 172–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Růžek D,Salát J, Palus M, Gritsun TS, Gould EA, Dyková I, Skallová A, Jelínek J, Kopecký J, and Grubhoffer L (2009). CD8+ T-cells mediate immunopathology in tick-borne encephalitis. Virology 384, 1–6. [DOI] [PubMed] [Google Scholar]
- Salvo MA, Kingstad-Bakke B, Salas-Quinchucua C, Camacho E, and Osorio JE (2018). Zika virus like particles elicit protective antibodies in mice. PLoS Negl. Trop. Dis. 12, e0006210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos RI, Hermance ME, Gelman BB, and Thangamani S (2016). Spinal cord ventral horns and lymphoid organ involvement in powassan virus infection in a mouse model. Viruses 8, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaiger J, Aberle JH, Stiasny K, Knapp B, Schreiner W, Fae I, Fischer G, Scheinost O, Chmelik V, and Heinz FX (2014). Specificities of human CD4+ T cell responses to an inactivated flavivirus vaccine and infection: correlation with structure and epitope prediction. J. Virol. 88, 7828–7842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamanin VA, Pletnev AG, Rubin SG, and Zlobin VI (1991). [The differentiation of viruses of the tick-borne encephalitis complex by means of RNA-DNA hybridization]. Vopr Virusol 36, 27–31. [PubMed] [Google Scholar]
- Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, et al. (2005). Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 174, 6477–6489. [DOI] [PubMed] [Google Scholar]
- Smith JL, and Hirsch AJ (2020). Analysis of serum anti-Zika virus antibodies by focus reduction neutralization test. Methods Mol. Biol. 2142, 73–80. [DOI] [PubMed] [Google Scholar]
- Sonenshine DE (2018). Range expansion of tick disease vectors in North America: implications for spread of tick-borne disease. Int. J. Environ. Res. Public Health 15, 478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavakoli NP, Wang H, Dupuis M, Hull R, Ebel GD, Gilmore EJ, and Faust PL (2009). Fatal case of deer tick virus encephalitis. New Engl. J. Med. 360, 2099–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanblargan LA, Errico JM, Kafai NM, Burgomaster KE, Jethva PN, Broeckel RM, Meade-White K, Nelson CA, Himansu S, Wang D, et al. (2021). Broadly neutralizing monoclonal antibodies protect against multiple tick-borne flaviviruses. J. Exp. Med. 218, e20210174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanblargan LA, Himansu S, Foreman BM, Ebel GD, Pierson TC, and Diamond MS (2018). An mRNA vaccine protects mice against multiple tick-transmitted flavivirus infections. Cell Rep. 25, 3382–3392.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnaite R, Blom K, Lampen MH, Vene S, Thunberg S, Lindquist L, Ljunggren HG, Rombo L, Askling HH, and Gredmark-Russ S (2020). Magnitude and functional profile of the human CD4(+) T cell response throughout primary immunization with tick-borne encephalitis virus vaccine. J. Immunol. 204, 914–922. [DOI] [PubMed] [Google Scholar]
- Watson AM, Lam LKM, Klimstra WB, and Ryman KD (2016). The 17d-204 vaccine strain-induced protection against virulent yellow fever virus is mediated by humoral immunity and CD4+ but not CD8+ T cells. PLOS Pathog 12, e1005786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Elong Ngono A, Regla-Nava JA, Kim K, Gorman MJ, Diamond MS, and Shresta S (2017). Dengue virus-reactive CD8(+) T cells mediate cross-protection against subsequent Zika virus challenge. Nat. Commun. 8, 1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauch LE, Zellweger RM, Kotturi MF, Qutubuddin A, Sidney J, Peters B, Prestwood TR, Sette A, and Shresta S (2009). A protective role for dengue virus-specific CD8+ T cells. J. Immunol. 182, 4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.