

Abstract
Nonalcoholic steatohepatitis (NASH) is characterized by accumulation of lipids in the hepatocytes (steatosis) and chronic inflammation. Liver resident macrophages (Kupffer cells) play a pivotal role in inducing inflammation. Cross-talk between hepatocytes and Kupffer cells (KCs) regulate both steatosis and inflammation during the pathogenesis of NASH. Isolated hepatocytes and KC serve as important tools to study mechanistic events during NASH in an in vitro setting. Because mice and humans share identical genes, primary mouse hepatocytes and KC are valuable ex vivo models for NASH studies. However, isolation of mouse liver cells is challenging and requires specific technical procedure and skills. Here, we elaborate a method for effective isolation of both primary hepatocytes and KC from adult liver of the same mouse. This protocol can be used for isolation of liver cells from both wild-type (WT) and genetically-engineered mice. The principle of the method is based on a two-step collagenase perfusion technique in which the liver is washed by perfusion, liver cells are segregated by collagenase treatment, and hepatocytes and KC are then purified and cultured. We optimized this protocol in terms of reproducibility, yield of different population of liver cells, and viability.
Keywords: Hepatocytes, Kupffer cells, Nonalcoholic steatohepatitis (NASH), Primary culture, Collagenase, Two-step separation technique
1. Introduction
Obesity and metabolic syndrome have become a global epidemic in the twenty-first century. Nonalcoholic fatty liver disease (NAFLD) is the hepatic consequence of obesity and metabolic syndrome. NAFLD is estimated to affect one among three persons in many developed and developing countries [1]. A subset of patients with NAFLD develop an inflammatory condition, termed nonalcoholic steatohepatitis (NASH), that is characterized by hepatocyte ballooning, steatosis, and lobular inflammation in liver histology [2]. As 70–85% of the liver volume is occupied by parenchymal hepatocytes, isolation of hepatocytes is important to study liver diseases like NASH [3]. In addition, hepatocytes are involved in many physiological processes, such as protein synthesis, metabolism, detoxification of drugs, as well as synthesis and secretion of cholesterol, bile acids, and phospholipids. Because of this diverse array of functions in the context of liver biology, the use of freshly isolated primary hepatocytes has become an important tool to investigators studying various aspects of liver function. In addition to hepatocytes, non-parenchymal liver resident macrophages (Kupffer cells) play a key role in regulating the inflammatory events associated with NASH, and hepatocyte-Kupffer cell cross-talk drives the steato-inflammatory process [4]. Kupffer cells (KCs) constitute 20% of the non-parenchymal cells of the rodent liver [5]. Although KCs constitute less than 5% of the liver volume, they represent approximately 80–90% of the total fixed macrophage population in the body of rodents [6].
Liver cells should be used immediately after isolation to avoid changes in gene regulation, polarization, and dedifferentiation [7]. The two-step collagenase perfusion technique for the isolation of liver cells was first reported by Berry and Friend for the isolation of rat hepatocytes [8]. Different nonenzymatic isolation procedures were used to obtain liver cells before the widespread implementation of the enzymatic digestion method with collagenase [9, 10]. These procedures include liver homogenization, trituration of liver samples, and filtration of the tissues through fine sieves. However, the above-mentioned procedures have considerable drawbacks, for example, noticeable damage to hepatocytes obtained from these procedures was observed. Other digestive enzymes such as trypsin, pronase, and lysozyme have been used to obtain single-cell suspension of liver, but yield of viable hepatocytes was affected [11, 12].
The protocol described in this chapter is aimed at isolating hepatocytes and KCs from the same adult mouse liver. The two-step collagenase perfusion technique is the method of choice to date for the isolation of a large number of viable liver cells. In the first step, the inferior vena cava is cannulated after giving anesthesia and the liver is perfused to chelate calcium and wash out blood. Removal of calcium ions (by using EDTA or citrate) from epithelial cells results in rapid destruction of intercellular junctions so that the cell–cell contacts are lost [13]. In the second step, the enzyme collagenase is introduced into the liver lobes and the perfused collagenase dissociates the extracellular matrix of liver cells. The procedure is unique as the liver is perfused in situ via the inferior vena cava. Finally, the liver is separated from the mouse, and liver cells are purified by centrifugation. This protocol holds some advantages over similar protocols [7, 8, 10]. The main improvements of this protocol are higher reproducibility, reduced technical challenge, and increased yield of viable liver cells. Our protocol will be useful for experiments demanding a large number of hepatocytes and for studying hepatocytes and Kupffer cells that have been isolated from a single mouse.
2. Materials
All solutions should be freshly prepared with Milli-Q purified water.
2.1. Liver Perfusion
2.1.1. Reagents
KCl buffer: Add 0.5 g KCl to 10 ml 1 M HEPES. Vortex until dissolved.
Perfusion Buffer #1: To a 500 ml bottle of Dulbecco’s phosphate buffered saline (DPBS) without Ca2+/Mg2+, add 2.5 ml 1 M d-glucose solution, 0.25 ml 0.5 M EDTA, and 5 ml KCl buffer. Mix, adjust pH to 7.4 with NaOH, and filter sterilize using a 500 ml 0.22 μm PES filter system (see Note 1).
Perfusion Buffer #2: To a 500 ml bottle of DPBS without Ca2+/Mg2+, add 2.5 ml 1 M d-glucose solution, 10 ml 1 M HEPES, 1 ml 0.5 M CaCl2, and 5 ml KCl buffer. Mix, adjust pH to 7.4 with NaOH, and filter sterilize using a 500 ml 0.22 μm PES filter system (see Note 1).
Collagenase solution: Add 45 mg Collagenase Type I (Worthington) to 100 ml pre-warmed (37 °C) Perfusion Buffer #2. Filter sterilize using a 150 ml 0.22 μm PES filter system (see Note 2).
Povidone-Iodine solution (Ricca).
Isoflurane.
70% ethanol.
2.1.2. Equipment and Consumables
100 ml sterile glass or plastic bottles.
150 ml and 500 ml 0.22 μm PES filter systems.
Sterile drapes and 4″ × 4″ gauze.
Sterile dissecting instruments (forceps and scissors).
Shaking water bath set to 37 °C.
Flask holders for water bath.
Peristaltic pump set to 7–8 ml/min.
Silicone double pump segment manifold tubing.
Sterile pyrex Erlenmeyer flasks (125, 250 ml).
Tubing filters (Swinnex).
Metal luer-lock adapters.
20G 1.25″ IV catheters.
Anesthesia vaporizer (VetEquip IMPAC).
95%O2/5%CO2 mix.
Mouse dissection tray.
Spray bottle.
50 ml conical tubes.
Swinging bucket refrigerated centrifuge with 50 ml tube adapters.
70 or 100 μm cell strainers.
Precision or analytical balance.
pH meter and NaOH.
2.2. Hepatocyte Culture
2.2.1. Reagents
0.4% Trypan blue solution.
Williams’ E Media with l-glutamine and Penicillin–Streptomycin.
Insulin solution, human.
10 mM dexamethasone.
2.2.2. Equipment and Consumables
Rat-tail collagen coated tissue culture dishes.
CO2 incubator, 37 °C.
Hemocytometer or automated cell counter.
Large orifice filter pipet tips.
2.3. Kupffer Cell Isolation and Culture
2.3.1. Reagents
1000× Tricine solution: 3.7 mg EDTA, 269 mg Tricine, 425 mg NaCl in 50 ml DPBS, filter sterilized.
OptiPrep density gradient medium (diluted to 40%, 17.6%, and 8.2% in Tricine solution, kept on ice) (Sigma).
RPMI 1640, with l-Glutamine and Penicillin–Streptomycin.
Fetal Bovine Serum.
DPBS without Ca2+/Mg2+.
0.05% Trypsin-EDTA.
2.3.2. Equipment and Consumables
10 cm petri dishes.
Tissue culture plates.
Hemocytometer or automated cell counter.
CO2 incubator, 37 °C.
3. Methods
Pre-warm one 100 ml bottle each of Perfusion Buffer #1 and Perfusion Buffer #2. Ideally, the buffers should be maintained in a water bath set to 39 °C directly beside the surgical area during the entire perfusion process, but if this is not possible, the buffers may be kept warm in a nearby bath and brought out of the bath just before use.
Prepare the surgical area by placing a sterile drape over the dissection tray, and laying out sterilized dissection instruments on another sterile drape. Connect the perfusion tubing to the pump and begin running Perfusion Buffer #1 through the tubing. Ensure that no bubbles are present in the tubing (see Note 3).
Anesthetize the mouse in an induction chamber. Conduct a toe pinch to ensure the animal is fully anesthetized and restrain limbs.
Sterilize the abdominal area by spraying with 70% ethanol and wiping off excess with sterile gauze. Apply povidone-iodine, wiping off excess, followed by two washes with 70% ethanol. Place pieces of sterile gauze on either side of the abdomen as well as over the hind legs.
Using sterile scissors and forceps, remove the skin from the abdomen. With a second pair of scissors, remove the muscle layer from the abdominal cavity. Gently shift the intestines to the side and locate the inferior vena cava and hepatic portal vein (Fig. 1).
As parallel to the vein as possible, insert the catheter (with the needle bevel facing up) into the inferior vena cava approximately 5–6 mm below its entrance into the liver (see Note 4). Once the needle has been inserted, carefully push the catheter forward while simultaneously pulling the needle out. Once the needle has been removed, blood flowing from the end of the catheter will indicate a successful cannulation of the vein (Fig. 2).
Carefully adjust the cannula so that it is inserted approximately 3–4 mm inside the vein. It should move freely (see Note 5).
Double-check tubing for the presence of bubbles and connect the perfusion tubing to the cannula. The liver should immediately begin to swell. Once this is observed, cut the hepatic portal vein with a fresh pair of scissors. The liver should fully blanch within a few seconds, indicating blood being flushed from the liver (see Note 6). Ensure that the buffer is not leaking from the junction of catheter and tubing. Place trays or absorbent padding along sides of the dissection tray to collect the buffer as it flows from the liver.
While the buffer is perfusing through the liver, prepare the collagenase solution as described in Subheading 2.1.1. After sterile filtration, mix 5 ml of the collagenase solution and 15 ml Perfusion Buffer #2 (25% strength) in a 50 ml conical tube. Maintain both solutions at 37 °C until needed.
When <5 ml of Perfusion Buffer #1 is remaining in the bottle, press stop on the pump, quickly transfer the tubing end into the bottle of filtered collagenase solution, and press start on the pump (see Note 7).
Once all of the buffer has perfused through the liver (Fig. 3), stop the pump. With a fresh pair of scissors, carefully remove the liver (it will be soft) and place into a sterile flask. In order to prevent bacterial contamination, special care should be taken to avoid nicking the intestines.
With a fresh pair of scissors, mince the liver in the flask. Add the 25% strength collagenase solution and swirl. Place the flask in a flask holder and shake the suspension in the water bath for 3 min at 125 rpm.
In a laminar flow hood, filter the liver suspension through sterile gauze into a 50 ml conical tube to remove any remaining pieces of undigested liver. Centrifuge for 3 min at 50 × g, 4 °C. Remove the supernatant and continue wash steps with the pellet (step 14). If purifying KCs, collect the supernatant containing the non-parenchymal cells and continue at step 19 (see Note 8).
(For hepatocyte culture) Carefully remove supernatant and add 20 ml of Perfusion Buffer #2 to the cell pellet with a 25 ml pipette. Gently pipette up and down once or twice. Further mixing can be accomplished by gently inverting the tube. Centrifuge for 3 min at 50 × g, 4 °C.
(For hepatocyte culture) Repeat step 14 two times, for a total of three washes (Fig. 4) (see Note 9).
(For hepatocyte culture) Resuspend the pellet in 10 ml hepatocyte culture media using a 25 ml pipette and filter through a cell strainer to separate out any cell clumps that have developed (see Note 10).
(For hepatocyte culture) Using wide-bore pipette tips for ali-quoting, calculate cell concentration and viability with a 1:1 dilution of trypan blue. Typically, yield will range from 20 to 45 million cells per liver.
(For hepatocyte culture) Plate cells on collagen coated tissue culture dishes according to the numbers shown in Table 1 and maintain in a humidified 37 °C CO2 incubator. The cells should be plated to confluency, as they will not divide. Gently change media after 3–4 h, or when all cells have adhered. Media should be changed each day that cells are in culture (see Note 11). Experiments with primary hepatocytes should be started no later than the day after plating. A representative image of mouse hepatocytes is shown in Fig. 5.
(For KC culture) Centrifuge supernatant from step 13 at 650 × g for 10 min, 4 °C. Remove supernatant and resuspend the cell pellet in 20 ml cold 17.6% Optiprep. As slowly as possible, pipette 20 ml of cold 8.2% Optiprep over the cell suspension so that two distinct layers are created (see Note 12).
(For KC culture) Centrifuge 1400 × g for 30 min, 4 °C, with low or no brake. After this centrifugation, non-parenchymal cells (NPCs) will be visible in a distinct band at the interface between the two layers of gradient medium (Fig. 6). There will be a small pellet at the bottom of the tube containing any remaining hepatocytes.
(For KC culture) Remove the top 15 ml solution from the tube and discard. Proceed to collect the next 10–15 ml making sure to collect the entire band of cells. Transfer this to a fresh tube and add 35–40 ml of cold complete RPMI media.
(For KC culture) Centrifuge at 650 × g for 10 min, 4 °C. Aspirate supernatant and resuspend in 10 ml prewarmed complete media. Plate cells in a 10 cm petri dish (not tissue culture treated) and maintain in a humidified 37 °C CO2 incubator for 30 min (see Note 13).
(For KC culture) Aspirate media from dish and wash plate 3× with DPBS. Add 1.5 ml Trypsin-EDTA and keep in a 37 °C CO2 incubator for 5 min.
(For KC culture) Neutralize the trypsin by adding 4 ml complete media and count cells.
(For KC culture) Plate 5–6 × 105 cells/well of a 24-well plate or 4-chamber slide for further analysis. Yield will range from 1 to 3 million cells per liver. A representative image of KC stained with KC-specific F4/80 antibody is shown in Fig. 7.
Fig. 1.
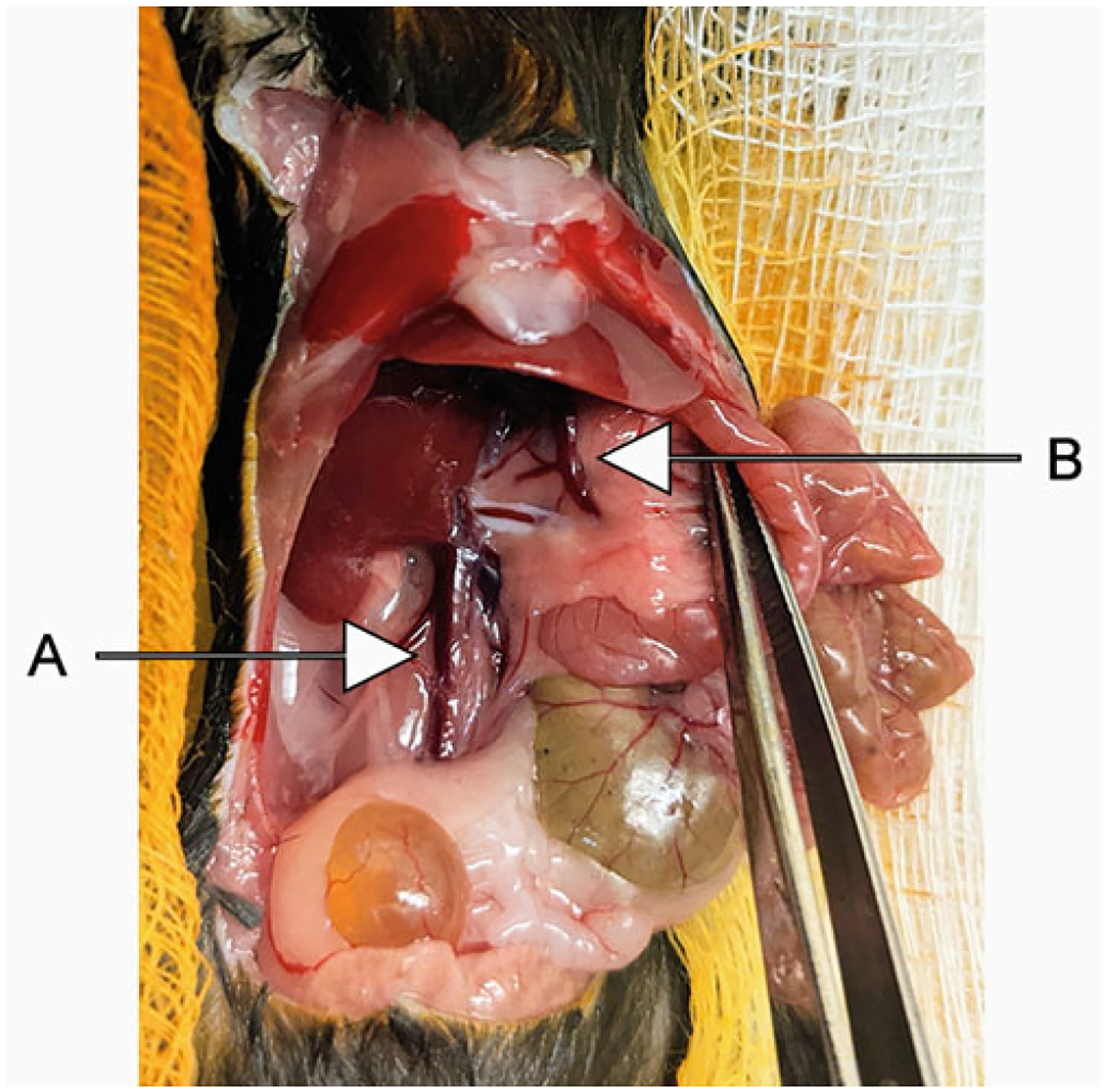
Once the skin and muscle layers are removed, intestines and other organs are pushed to the side and the inferior vena cava (A) and hepatic portal vein (B) are identified
Fig. 2.
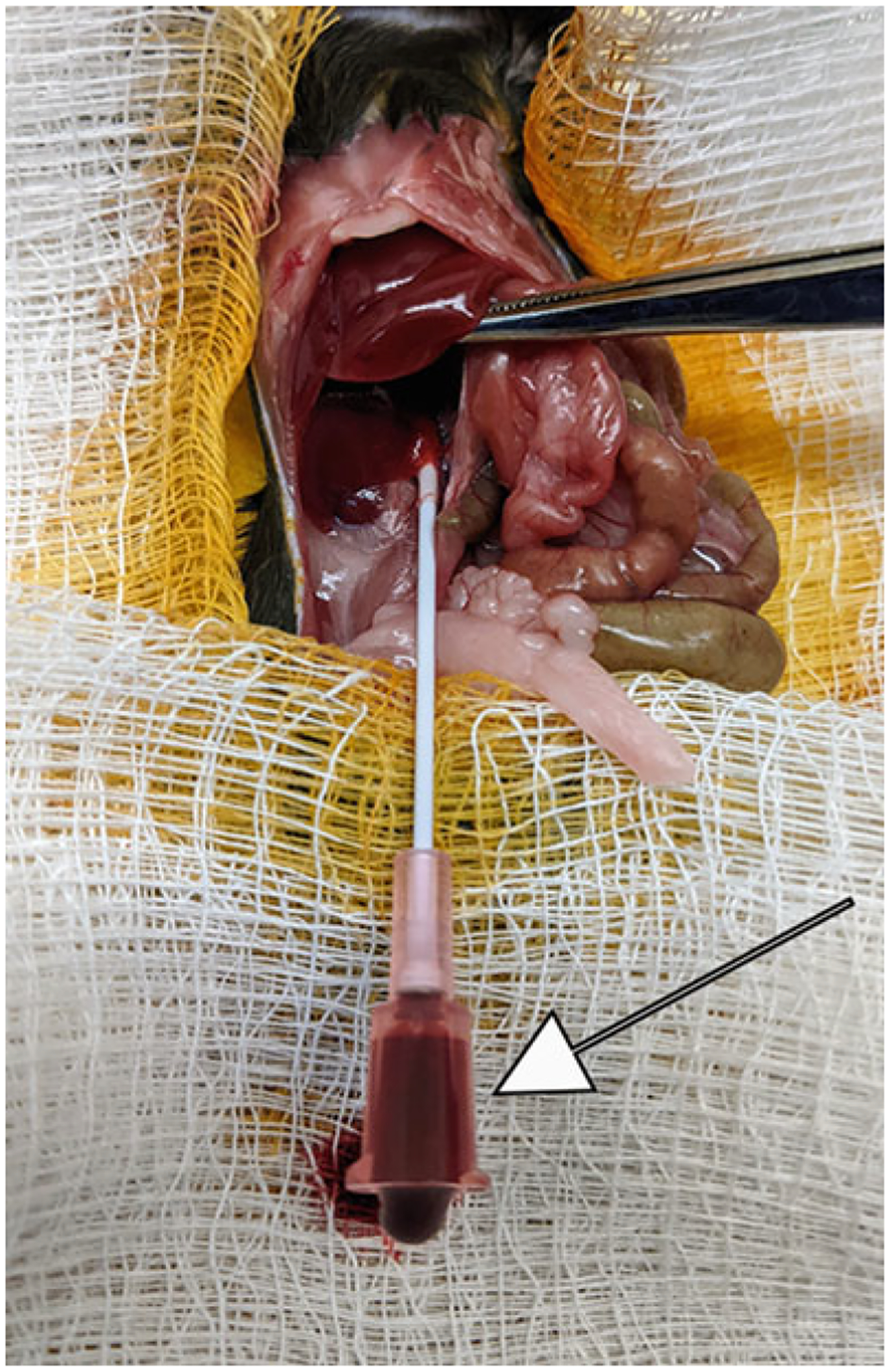
Successful cannulation of the inferior vena cava indicated by the flow of blood (arrow)
Fig. 3.
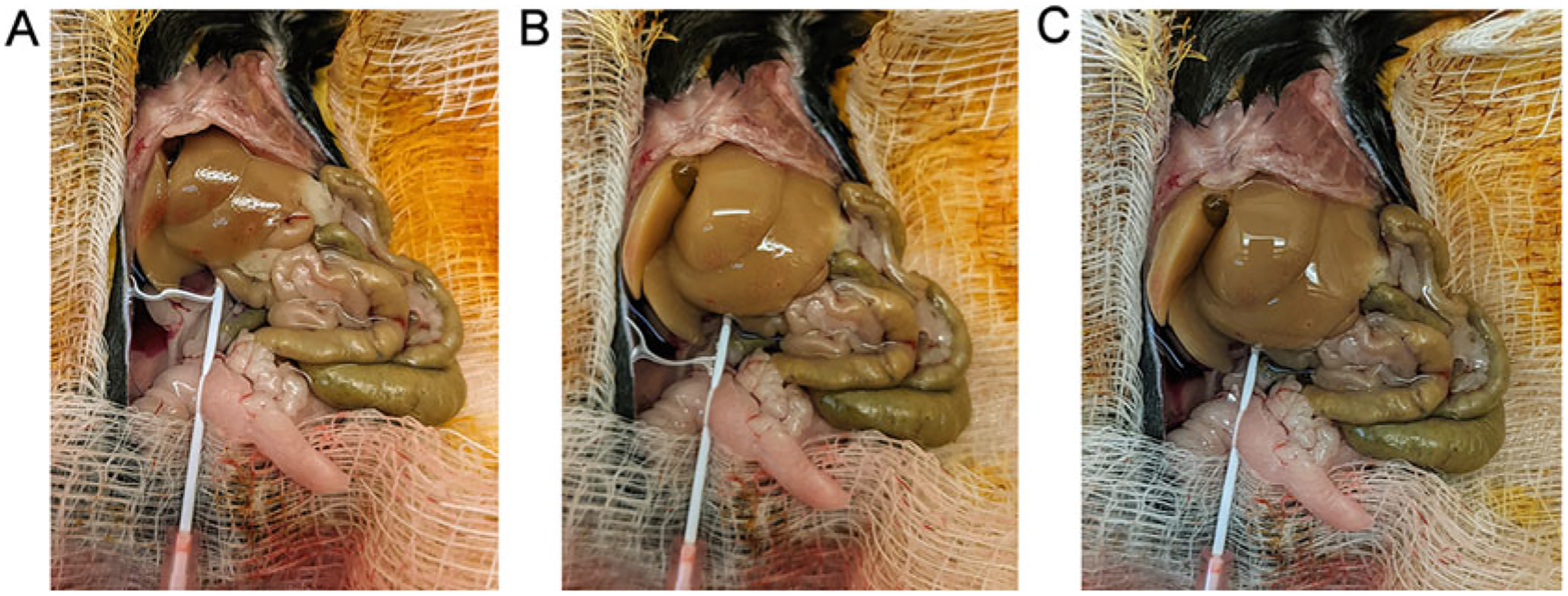
Perfusion progression. (A) 10 min after cannulation nearing the end of Perfusion Buffer 1. (B) 20 min after cannulation, collagenase digestion of the liver evident. (C) 25 min after cannulation, digestion nearly complete
Fig. 4.

(A) Cell suspension after filtering through gauze. Cell pellet after first centrifugation (B), after first wash (C), after second wash (D), and after third wash (E)
Table 1.
Number of hepatocytes to be plated according to the vessel size
| Culture vessel | Number of plated cells |
|---|---|
| 96-well plate | 5 × 103 to 1×104 cells/well |
| 48-well plate | 1 × 104 cells/well |
| 24-well plate, 4-chamber slide | 1–1.5 × 105 cells/well |
| 6-well plate, 35 mm dish | 5 × 105 cells/well |
| 60 mm dish | 1–1.2 × 106 cells/well |
| 100 mm dish | 3–3.5 × 106 cells/well |
Fig. 5.
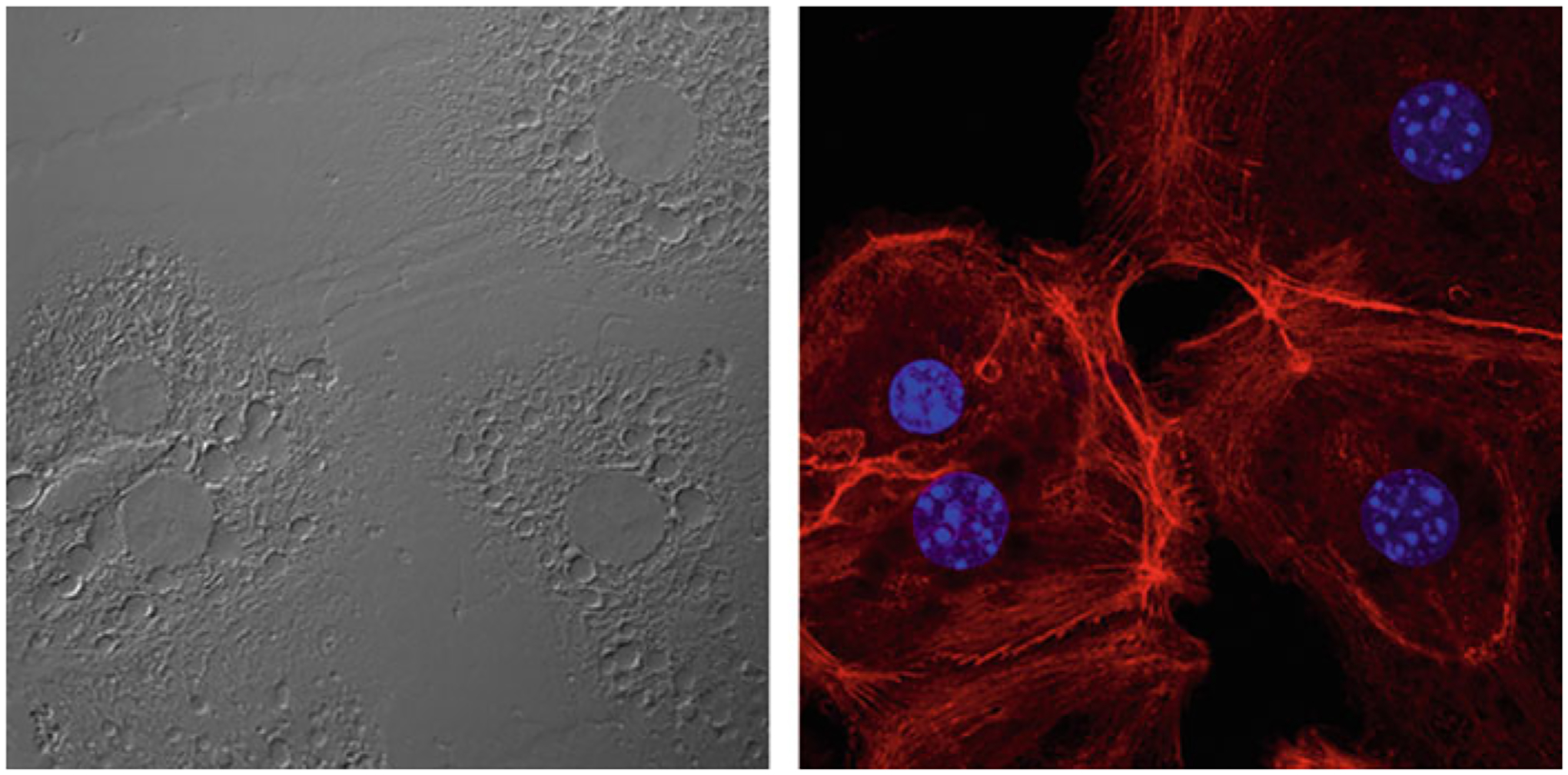
Representative image of the isolated hepatocytes. (Left panel) bright-field image. (Right panel) Stained with Rhodamine phalloidin to visualize the actin cytoskeleton (red) and DAPI (blue)
Fig. 6.

Non-parenchymal cell (NPC) purification. (A) Density gradient. An 8.2% solution of Optiprep is carefully pipetted over cells resuspended in a 17.6% solution of Optiprep. (B) NPCs are concentrated in the interface between the two concentrations of Optiprep (arrow). Any remaining parenchymal cells are pelleted at the bottom of the tube. (C). Final NPC pellet
Fig. 7.
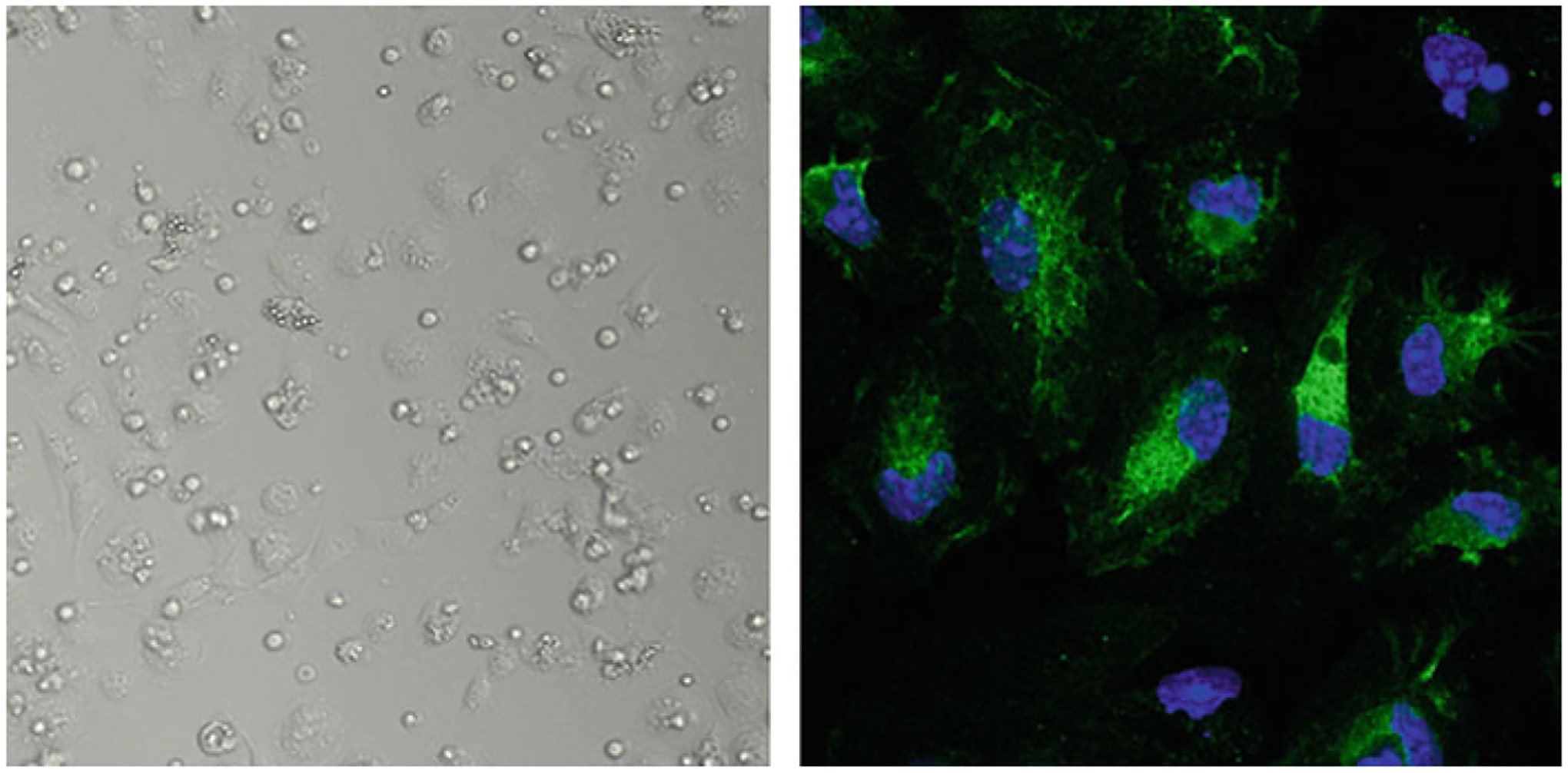
Representative image of the Kupffer cells. (Left) Bright-field image of the Kupffer cells in culture the day after plating. (Right) Kupffer cells stained with F4/80 antibody (green) and DAPI (blue)
4. Notes
While it is preferable to mix and measure pH of buffers the day of perfusion, if needed, they may be mixed the day before and stored at 4 °C.
The collagenase solution should be mixed just before use. We generally do this while Perfusion Buffer #1 is perfusing through the liver. The optimal concentration of collagenase can vary with different lots, and each new lot purchased should be optimized for ideal yield and viability.
If a bubble flows into the liver, it can block the flow of the buffer to a section of the liver. This will reduce final yield of cells.
In older mice (>20 weeks) or mice with a higher fat content, the view of the inferior vena cava can be partially or fully obscured by fat, and in this case, it will be difficult to insert the catheter/needle properly. It is possible to cannulate the vein closer to the liver if needed, but this is not always successful.
If the cannula is pushed too far into the vein, the buffers will leak from the junction of cannula and perfusion tubing. If the cannula is not inserted far enough into the vein, the force of the solution pumping can cause the cannula to slip out of the vein.
If the liver does not blanch within a few seconds, there is a problem. This could be caused by a bubble entering the vein or the needle may have nicked the vein in a second place upon entry.
Minor swelling and complete blanching of the liver should be noted after perfusion with Buffer #1. The color should resemble the liver in Fig. 3. If the shade is off (usually lighter), there is most likely a problem with the buffers. The liver should be significantly larger once perfusion with Buffer #2 has completed. If this is not the case, it is unlikely that perfusion was successful and final yield will be extremely low.
Steps from this point on should be conducted under sterile conditions.
Pellet size may decrease slightly after the first wash. Supernatant should become clearer after each wash.
Hepatocyte culture media: 500 ml Williams Media E, 500 μl insulin solution, 50 μl 10 mM dexamethasone, 5 ml l-Gluta-mine, 5 ml penicillin–streptomycin.
Larger diameter dishes and plates are preferred, as the hepatocytes have a tendency to clump in the center of the plate, and it is easier to disperse the cells in a larger area.
Prepare 1× Tricine solution in DPBS. Prepare 40% Optiprep (10 ml Optiprep + 5 ml 1× Tricine solution), 17.6% Optiprep (8.5 ml 40% Optiprep + 11.5 ml 1× Tricine solution), and 8.2% Optiprep (4 ml 40% Optiprep + 16 ml 1× Tricine solution). Keep on ice.
Kupffer cells should readily attach to petri dishes, while other NPCs will not. Using a tissue culture treated plate will make it more difficult to remove the cells without scraping and may lead to other NPCs contaminating the KCs.
Acknowledgments
This work was supported in part by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) under Grant 1R01DK107451-01A1, the National Cancer Institute (NCI) under Grants 1R01CA230561-01A1, 1R01CA240004-01, and 1R01CA244993-01, and the Department of Defense (DOD) under Grant CA170048.
References
- 1.Anstee QM, Reeves HL, Kotsiliti E et al. (2019) From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 16:411–428 [DOI] [PubMed] [Google Scholar]
- 2.Ibrahim SH, Hirsova P, Gores GJ (2018) Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut 67:963–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou Z, Xu MJ, Gao B (2016) Hepatocytes: a key cell type for innate immunity. Cell Mol Immunol 13:301–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lefere S, Tacke F (2019) Macrophages in obesity and non-alcoholic fatty liver disease: cross-talk with metabolism. JHEP Rep 1:30–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krenkel O, Tacke F (2017) Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol 17:306–321 [DOI] [PubMed] [Google Scholar]
- 6.Oses C, Aouadi M (2020) Kupffer cell and hepatocyte isolation from a single mouse liver by gradient centrifugation. Methods Mol Biol 2164:1–10 [DOI] [PubMed] [Google Scholar]
- 7.Severgnini M, Sherman J, Sehgal A et al. (2012) A rapid two-step method for isolation of functional primary mouse hepatocytes: cell characterization and asialoglycoprotein receptor based assay development. Cytotechnology 64:187–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berry MN, Friend DS (1969) High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. J Cell Biol 43:506–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schreiber G, Schreiber M (1973) The preparation of single cell suspensions from liver and their use for the study of protein synthesis. Subcell Biochem 2:307–353 [PubMed] [Google Scholar]
- 10.Li WC, Ralphs KL, Tosh D (2010) Isolation and culture of adult mouse hepatocytes. Methods Mol Biol 633:185–196 [DOI] [PubMed] [Google Scholar]
- 11.Howard RB, Christensen AK, Gibbs FA, Pesch LA (1967) The enzymatic preparation of isolated intact parenchymal cells from rat liver. J Cell Biol 35:675–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seglen PO (1976) Preparation of isolated rat liver cells. Methods Cell Biol 13:29–83 [DOI] [PubMed] [Google Scholar]
- 13.Troyanovsky S (2005) Cadherin dimers in cell-cell adhesion. Eur J Cell Biol 84:225–233 [DOI] [PubMed] [Google Scholar]