

Abstract
Aberrant activation of the JAK-STAT signaling pathway has been implicated in the pathogenesis of a range of hematological malignancies and autoimmune disorders. Here we describe the design, synthesis, and characterization of JAK2/3 PROTACs utilizing a phenyl glutarimide (PG) ligand as the cereblon (CRBN) recruiter. SJ10542 displayed high selectivity over GSPT1 and other members of the JAK family and potency in patient-derived ALL cells containing both JAK2 fusions and CRLF2 rearrangements.
Keywords: Janus kinase, cereblon, GSPT1, PROTAC, phenyl glutarimide (PG), JAK-STAT, ALL
Genetic alterations resulting in constitutive JAK-STAT signaling are driver events in several subtypes of acute lymphoblastic leukemia (ALL). In particular, many cases of Philadelphia-like (Ph-like) ALL exhibit activation of the JAK-STAT signaling pathway with approximately half of all cases harboring rearrangements or sequence mutations leading to deregulation of cytokine receptor-like factor 2 (CRLF2), commonly with concomitant genetic alterations augmenting signaling, most commonly sequence mutations of Janus kinases 2 (JAK2).1−3 In these cases, CRLF2 is deregulated and overexpressed because of rearrangement of the IGH enhancer (IGH-CRLF2) or the P2R8Y promoter (P2R8Y-CRLF2).4−6 The associated JAK mutations include mutations at or near p.Arg683 in the JAK2 pseudokinase domain and less commonly in the pseudokinase and kinase domains of JAK1, JAK2, and JAK3. Ph-like ALL is associated with high-risk clinical features and poor outcome in both childhood and adult ALL.1,7 Thus, the Janus kinase family of proteins, particularly JAK2, is a potential therapeutic target in JAK-STAT activated Ph-like ALL. In engineered cell lines, CRLF2 and JAK2 mutations result in cytokine independent proliferation and activation of JAK-STAT signaling which can be abrogated by type I JAK inhibitors such as ruxolitinib.6 However, variable inhibition of signaling and proliferation has been observed in human ALL xenograft models.8 This limited efficacy is in part due to trans-phosphorylation of JAK2 inhibited in its active conformation by JAK1 and TYK2 leading to persistent signaling.9 The persistent signaling has been associated with reactivation of JAK-STAT signaling resulting from heterodimerization between activated JAK2 and JAK1/TYK2. This phenomenon of increased JAK2 heterodimerization and sustained JAK-STAT activation was observed in cell lines, mouse models, and patients treated with JAK2 inhibitors.9 Consequently, therapies that result in JAK2 degradation may retain efficacy in persistent cells and provide additional benefit to patients with JAK2-dependent malignancies treated with JAK2 inhibitors. Therefore, we hypothesized that a JAK2 PROTAC would circumvent the issues of type I inhibitors with persistent signaling.10
Cereblon (CRBN)-directing PROTACs are often the preferred approach because of favorable physicochemical properties of immunomodulatory drugs (IMiDs), such as pomalidomide and lenalidomide, that are commonly used as CRBN-directing ligands.11 However, IMiDs also act as molecular glues that recruit neosubstrates such as IKZF1 and GSPT1 (G1 to S phase transition 1).12−14 GSPT1 is a translation termination factor whose degradation was shown to mediate potent antitumor activity, including in acute leukemia, and therefore can hinder the evaluation and interpretation of target-specific PROTAC pharmacology.15 We previously reported the development and characterization of IMiD-based PROTACs targeting multiple JAK proteins, which displayed various degrees of IKZF1 and GSPT1 degradation.12 An earlier study found that PROTACs based on inhibitor of apoptosis protein (IAP) ligand induced significantly more JAK degradation over Von Hippel–Lindau (VHL) and CRBN– directing analogues but required high micromolar concentrations to induce degradation, further highlighting challenges in developing potent and selective JAK2 PROTACs.16
To address these challenges, we sought to evaluate phenyl glutarimide 1a (PG) as a CRBN-directing warhead in the design of JAK2-selective PROTACs. We, and others,17 recently reported incorporation of the PG ligand in BET-PROTACs which displayed greatly improved degradation and cellular potency compared to IMiD-based analogues.18 In addition, PG contains a phenyl group substituted for the phthalimide present in IMiDs, and we hypothesized that it may display reduced off-target recruitment of neosubstrates such as GSPT1. Furthermore, we were interested to find if PG-based PROTACs may display greater selectivity for JAK2 over other members of the JAK family and elucidate the role of JAK2 degradation in leukemia progression.
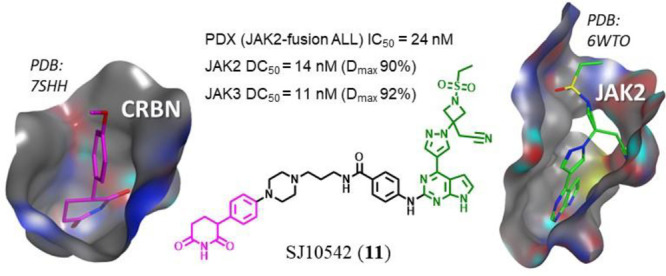
We previously designed a series of JAK PROTACs based on thalidomide as a CRBN-directing warhead and the FDA-approved JAK inhibitors ruxolitinib and baricitinib (Figure 1). The design was guided by our crystal structures of ruxolitinib and baricitinib bound to JAK2 (PDB: 6WTN and 6WTO respectively).12 A 4-amino-benzamide attached to the solvent exposed C2-carbon of the pyrimidine was used as a handle for PROTAC linkers. This structural modification was found to be well tolerated in the JAK binding pocket (PDB: 6WTP and 6WTQ)12,19 and lead to the synthesis of our initial set of IMiD-based PROTACs that degraded the JAK family of proteins (JAK1–3 and TYK2) to varying degrees in the CRLF2-rearranged, JAK2 I682F mutated MHH-CALL-4 ALL cell line (Table 1).5,6,12
Figure 1.

Chemical structures of the following: (A) Phenyl glutarimide (PG) and its PG-OMe and PG-NH2 analogues. (B) The FDA-approved JAK inhibitors baricitinib and ruxolitinib, both containing the pyrimidine core where the C2-carbon is solvent-exposed providing a suitable linker attachment site.
Table 1. PROTACs Protein Degradation and CRBN Affinity.


Protein levels remained compared to a DMSO control in MHH-CALL-4 cells determined by immunoblotting (Supp. Figures S1 and S2), after incubation with 100 nM compound for 24 h (average of at least two biological replicates).
CRBN Fluorescent Polarization assay. Experiments were performed in biological triplicate and the average reported with SD. ND: Not determined.
However, most of the PROTACs also displayed potent degradation of GSPT1, a known off-target neosubstrate of IMiD-based PROTACs,14,20,15 which limited their use to study the biological effects of specific to JAK protein degradation (Table 1).12
These findings inspired us to investigate if the phenyl glutarimide (PG), an alternative CRBN-binding ligand for PROTAC design,18 could help address the undesirable degradation of GSPT1. To evaluate findings from our earlier in silico modeling studies, we generated the crystal structure of the CRBN thalidomide binding domain (TBD) from M. gryphiswaldense (M.g.),21,22 in the presence of PG-OMe (1b) to a resolution of 1.9 Å (Figure 2A and Supp. Figure S3). As shown in Figure 2B, the PG-methoxy compound is buried inside the pocket and accommodated by an aromatic cage composed of three conserved tryptophan residues (Trp79, Trp85, and Trp99).22 On the opposite side, the glutarimide moiety forms three hydrogen-bonds: one with the hydroxyl group of Tyr101, and one each with backbone atoms of Trp79 and Phe77. This positions the glutarimide moiety of 1b in a similar position to the glutarimide of thalidomide in a previously published M.g. TBD structure (Figure 2C and Supp. Figure S4).22 Protruding outside the pocket, the methoxy phenyl moiety makes hydrophobic interactions with Asn50, Pro51, and Trp85. As our initial in silico modeling studies suggested, the PG phenyl group was found to be deeper in the pocket when compared with thalidomide (Figure 2C). Hence, we hypothesized that it may be less accessible for interactions with the Gly-containing β-hairpin loop degron present in neosubstrates such as GSPT1 and therefore may display improved overall degradation selectivity.
Figure 2.

(A) Crystal structure of (R)-PG-OMe 1b (purple) bound to CRBN (M.g. TBD; PDB: 7SHH). (B) Annotated LigPlot+23 diagram with hydrogen bonds shown as dashed green lines and distances in Å and hydrophobic contacts represented as spoked arcs. (C) Superimposed structures of PG-OMe (PDB: 7SHH) and thalidomide (yellow, PDB: 4V2Y) bound to CRBN (M.g. TBD); atoms forming the protein surface are shown in gray (C), blue (N), and red (O).
We next synthesized several PG-based PROTACs that were direct IMiD analogues for comparison (Table 1). All PG-based PROTACs displayed high CRBN binding affinity, with low nanomolar IC50 values in CRBN fluorescence polarization assay (Table 1 and Supp. Figure S5).15,24 We also found that PG–PROTACs 7 and 11 retained affinity similar to baricitinib12 for the JAK family kinases, including JAK2 with Kd values of 0.14 and 0.022 nM, respectively (Supp. Table S2).
Next, we compared the degradation profiles of PG–PROTACs to their direct IMiD analogues in MHH-CALL4 cells. We found that PG–PROTAC 6 with the NH attachment group showed reduced GSPT1 degradation compared with the IMiD analogue 2, with 73% and 10% JAK2 protein remaining, respectively (Table 1). A similar trend was observed with analogues containing an oxygen-attachment, where the treatment of MHH-CALL4 cells with PG–PROTACs 7 and 8 resulted in 79% and 88% of GSPT1 protein remaining, respectively, compared with IMiD PROTAC 3 with only 18% GSPT1 remaining. Similarly, incubation with piperidine analogues resulted in 87% GSPT1 remaining for PG PROTAC 9, compared with 65% for IMiD analogue 4. A piperazine attachment proved the best for both PG and IMiD PROTACs, with 90% GSPT1 remaining for 5 and 99% for 10. However, 10 displayed a hook effect in the degradation of JAK2 at relatively low concentrations compared with the other PG–PROTACs, with less than 20% of JAK2 degraded at 1 μM (Supp. Figure S2E). An analogue with a single carbon longer linker, 11, did not display the same hook effect and was also found to be selective over GSPT1 (Table 1). Interestingly, further modifications around the piperazine moiety, such as in 12 and 13, were less tolerated, resulting in loss of JAK2 degradation efficacy, despite of the retained CRBN affinity (Table 1). In addition to JAK2 all PG–PROTACs displayed a weaker degradation of JAK3, while not affecting the levels of JAK1 and TYK2 proteins (Figure 3A, and Supp. Table 1). In all cases, the PG analogue had decreased GSPT1 degradation compared with the IMiDs.
Figure 3.

(A) Immunoblots for JAK1–3, TYK2, and GSPT1 degradation dose–response in MHH-CALL-4 cells when treated with 11 for 24 h. (B) Graphical representation of degradation dose–response of JAK2 by 11 in MHH-CALL-4 and KOPN49 cell lines. (C) Immunoblots for JAK2 and GSPT1 degradation time course in MHH-CALL-4 cells when treated with 11 at 100 nM. (D) Immunoblots for JAK2 and GSPT1 degradation dose–response in KOPN49 cells when treated with 11 for 24 h. (E) Cytotoxicity dose–response curve for PROTAC 11, PG-NH2, ruxolitinib, and baricitinib in MHH-CALL-4 cells. All experiments were repeated in biological triplicate and the average reported with SD and representative data shown. Immunoblotting bands were normalized to beta-actin, and degradation of target proteins was represented as a ratio compared with the DMSO control.
Following the SAR exploration, we selected 11 (SJ10542) as a lead compound on the basis of its overall degradation profile. The JAK2 degradation dose–response of 11 in MHH-CALL-4 cells gave a DC50 value of 24 nM and Dmax 82%, while a Dmax value of 50% prevented reliable DC50 determination for JAK3. The other two members of the JAK family were not significantly degraded (Figure 3A,B). The time course experiment performed over 32 h at 100 nM concentration established that the maximum degradation of JAK2 was achieved within 16 h (Figure 3C, Supp. Figure S6).
Treatment of the CRBN knocked down MHH-CALL-4 (CRBN KD)12 cell line with degrader 11 did not result in degradation of JAK2 by immunoblotting, indicating that degradation of JAK2 was CRBN-dependent (Supp. Figure 9B). The overall cytotoxicity profile of 11 in the MHH-CALL-4 CRBN knocked down12 cell line was decreased compared to the MHH-CALL-4 wild type cells, while the parent inhibitors ruxolitinib and baricitinib had similar cytotoxicities in both cell lines (Supp. Figure S9C). PROTAC 11 was also found to be a potent and selective JAK2 degrader in another CRLF2-rearranged, JAK2 R683G cell line KOPN49 (Figure 3D).25 The degradation potency was somewhat lower than MHH-CALL-4, where the DC50 for 11 was 74 nM compared with 24 nM in MHH-CALL-4 (Figure 3B), while JAK1 and TYK2 protein levels were not affected (Supp. Figure S7).
The cytotoxic activity in the MHH-CALL-4 cell line of selective JAK2 degrader 11 was found to be considerably higher than the parent inhibitors ruxolitinib and baricitinib, whereas, as expected, the CRBN ligand 1c on its own was inactive (Figure 3E). However, none of the PROTACs achieved full efficacy (100% killing), which is in part due to JAK2 redundancy in the context of JAK-STAT signaling pathway activation. Prior studies have shown that while coexpression of CRLF2 and JAK2 play a critical role in leukemia induction,6 JAK2 may be partly dispensable for leukemia cell maintenance, at least in the MHH-CALL-4 cell line, as JAK2 knockdown had an antiproliferative rather than cytotoxic effect (Supp. Figures S10 and S11).26
We next examined the activity of 11, baricitinib and 1c in a range of JAK-STAT driven patient-derived ALL cells ex vivo. While 1c was inactive, PROTAC 11 was found active in all PDX models, with IC50 values <120 nM (Supp. Table S4, and Supp. Figure S13). Only PDX SJBALL020589 showed modest sensitivity to baricitinib (Figure 4A). PROTAC 11 was also most potent in this PDX, displaying over 20-times higher cytotoxicity than baricitinib, with IC50 of 24 and 536 nM, respectively. PDX SJBALL020589 was also the most sensitive to our previously reported IMiD-based selective PROTACs, consistent with these results.12 In this PDX, PG–PROTAC 11 achieved a DC50 of 14 nM for JAK2 (Figure 4B,C), with similar degradation potency observed in the other tested PDXs (Supp. Figure S12). This PDX harbors an ATF7IP-JAK2 fusion which along with other JAK2 chimeric fusions were previously reported to show sensitivity to JAK2 inhibition (Supp. Table S5).3 Interestingly, whereas JAK1, TYK2, and GSPT1 protein levels were largely unaffected in the PDX, the extent of JAK3 degradation induced by 11 was similar to JAK2 (Figure 4C). This marked difference in the effect of 11 on JAK3 levels when compared with MHH-CALL-4 and KOPN49 cells (Figure 3B) could be a consequence of differential protein expression or post-translational modification pattern in the PDX. The other tested PDX models harbored CRLF2-rearrangements such as IGH-CRLF2 and P2RY8-CRLF2 which typically are less sensitive to JAK2 inhibitors and have shown transactivation of JAKs and parallel signaling pathway activation, which could account for lower cytotoxicity of JAK2-selective PROTACs.6,8
Figure 4.

(A) Cytotoxicity dose–response curves for PROTAC 11, baricitinib, and PG-NH2 in PDX SJBALL020589 with genetic alterations: ATF7IP-JAK2, KMT2C R3382W, PIK3CD F146I. (B) Immunoblots for JAK1–3, TYK2, and GSPT1/2 degradation dose–response in PDX cells SJBALL020589 when treated with 11 for 24 h. Bands were normalized to beta-actin and degradation of target proteins represented as a ratio compared with the DMSO control. (C) Graphical representation of 11 degradation dose–response for JAK2 and JAK3 in PDX cells SJBALL020589. Experiments were performed in biological duplicate, and the average was reported, with representative blots shown.
Here we described the design, synthesis, and in vitro characterization of JAK2/3-selective PROTACS in JAK-STAT driven cell lines and in vitro PDX models. To overcome common off-target degradation by IMiD-based PROTACs, we utilized PG as a CRBN-directing warhead. In all instances, the PG analogues displayed reduced GSPT1 degradation (70–99% GSPT1 remaining) compared with their IMiD analogues (10–90% GSPT1 remaining), which further demonstrated the value of PG as an alternative to IMIDs for PROTAC design. JAK2/3 PG PROTAC 11, SJ10542, was found to be variably effective in CRLF2-rearranged cell lines. In MHH-CALL-4 and KOPN49 cell lines, 11 showed greater efficacy compared with the parental JAK2 inhibitors. However, it did not achieve full efficacy, which is in part due to JAK2 redundancy in the context of JAK-STAT signaling pathway activation.26 However, PROTAC 11 was found to be potent in several PDX models containing both JAK2 fusions and CRLF2 rearrangements with IC50 values of <120 nM, and it was most potent in the JAK2 fusion PDX SJBALL020589 with an IC50 of 24 nM. This result may indicate that sensitivity to selective JAK2 degradation is in part genotype-dependent. Indeed, genotype-dependent sensitivity to JAK2 degradation was corroborated by publicly available synthetic lethality screening data from Project DRIVE, showing an ATARiS Sensitivity Score for JAK2 ranging from 0.784 to −1.924 (Supp. Table S6). Other studies have assessed combinatorial treatments with JAK2 inhibitors which have shown synergism and could be a viable option for further improving the effectiveness of JAK2 PROTACs as a therapy for JAK-STAT driven ALL.3,27 In summary, JAK2/3-selective PG–PROTACs proved valuable tools for studying the effects of JAK2/3 degradation in JAK-STAT driven ALL.
Acknowledgments
We are grateful for the support of the ALSAC and St. Jude Children’s Research Hospital, and would like to thank the patients, their families, and the staff at our institution. We thank the Compound Management team at the Department of Chemical Biology and Therapeutics for performing general QC and compound plate reformatting for screening and the Analytical and Technology Center in the Department of Chemical Biology and Therapeutics for performing HRMS of target compounds, the X-ray Center, and the Biorepositry of St. Jude for continuous support. Crystallographic data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. SER-CAT is supported by its member institutions. We thank the Pharmacotyping Resource in the Department of Pharmacy and Pharmaceutical Sciences at St. Jude for supporting this project.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00650.
Supplementary figures, crystallographic, biophysical and biological assays data, as well as full chemical synthetic methods and 1H and 13C NMR of target compounds (PDF)
Author Contributions
C.G.M. and Z.R. conceived the study. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by a Translational Research Program award from the Leukemia Lymphoma Society and the SnowDome Foundation, an NCI Outstanding Investigator Award R35 CA197695 and a St. Baldrick’s Foundation Robert J. Arceci Innovation Award (to C.G.M.); St Jude Cancer Center Support Grant CA021765, (to C.G.M.); R35 GM142772 (to M.F.), R01CA264837 (to J.J.Y.) and the American Syrian Associated Charities of St. Jude Children’s Research Hospital.
The authors declare the following competing financial interest(s): J.J.Y. receives research funding from Takeda Pharmaceutical Company. C.G.M. has received research funding from AbbVie and Pfizer; consulting fees from Illumina and speaking fees and holds equity in Amgen. Z.R. receives consulting fees from Revolution Medicines, Orum Therapeutics, and Nyrada, Inc.
Special Issue
Published as part of the ACS Medicinal Chemistry Letters virtual special issue “New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science”.
Supplementary Material
References
- Roberts K. G.; Li Y.; Payne-Turner D.; Harvey R. C.; Yang Y.-L.; Pei D.; McCastlain K.; Ding L.; Lu C.; Song G.; Ma J.; Becksfort J.; Rusch M.; Chen S.-C.; Easton J.; Cheng J.; Boggs K.; Santiago-Morales N.; Iacobucci I.; Fulton R. S.; Wen J.; Valentine M.; Cheng C.; Paugh S. W.; Devidas M.; Chen I.-M.; Reshmi S.; Smith A.; Hedlund E.; Gupta P.; Nagahawatte P.; Wu G.; Chen X.; Yergeau D.; Vadodaria B.; Mulder H.; Winick N. J.; Larsen E. C.; Carroll W. L.; Heerema N. A.; Carroll A. J.; Grayson G.; Tasian S. K.; Moore A. S.; Keller F.; Frei-Jones M.; Whitlock J. A.; Raetz E. A.; White D. L.; Hughes T. P.; Guidry Auvil J. M.; Smith M. A.; Marcucci G.; Bloomfield C. D.; Mrózek K.; Kohlschmidt J.; Stock W.; Kornblau S. M.; Konopleva M.; Paietta E.; Pui C.-H.; Jeha S.; Relling M. V.; Evans W. E.; Gerhard D. S.; Gastier-Foster J. M.; Mardis E.; Wilson R. K.; Loh M. L.; Downing J. R.; Hunger S. P.; Willman C. L.; Zhang J.; Mullighan C. G. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N Engl J. Med. 2014, 371 (11), 1005–1015. 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts K. G.; Morin R. D.; Zhang J.; Hirst M.; Zhao Y.; Su X.; Chen S.-C.; Payne-Turner D.; Churchman M. L.; Harvey R. C.; Chen X.; Kasap C.; Yan C.; Becksfort J.; Finney R. P.; Teachey D. T.; Maude S. L.; Tse K.; Moore R.; Jones S.; Mungall K.; Birol I.; Edmonson M. N.; Hu Y.; Buetow K. E.; Chen I.-M.; Carroll W. L.; Wei L.; Ma J.; Kleppe M.; Levine R. L.; Garcia-Manero G.; Larsen E.; Shah N. P.; Devidas M.; Reaman G.; Smith M.; Paugh S. W.; Evans W. E.; Grupp S. A.; Jeha S.; Pui C.-H.; Gerhard D. S.; Downing J. R.; Willman C. L.; Loh M.; Hunger S. P.; Marra M. A.; Mullighan C. G. Genetic Alterations Activating Kinase and Cytokine Receptor Signaling in High-Risk Acute Lymphoblastic Leukemia. Cancer Cell 2012, 22 (2), 153–166. 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts K. G.; Yang Y.-L.; Payne-Turner D.; Lin W.; Files J. K.; Dickerson K.; Gu Z.; Taunton J.; Janke L. J.; Chen T.; Loh M. L.; Hunger S. P.; Mullighan C. G. Oncogenic Role and Therapeutic Targeting of ABL-Class and JAK-STAT Activating Kinase Alterations in Ph-like ALL. Blood Adv. 2017, 1 (20), 1657–1671. 10.1182/bloodadvances.2017011296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoda A.; Yoda Y.; Chiaretti S.; Bar-Natan M.; Mani K.; Rodig S. J.; West N.; Xiao Y.; Brown J. R.; Mitsiades C.; Sattler M.; Kutok J. L.; DeAngelo D. J.; Wadleigh M.; Piciocchi A.; Dal Cin P.; Bradner J. E.; Griffin J. D.; Anderson K. C.; Stone R. M.; Ritz J.; Foà R.; Aster J. C.; Frank D. A.; Weinstock D. M. Functional Screening Identifies CRLF2 in Precursor B-Cell Acute Lymphoblastic Leukemia. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (1), 252–257. 10.1073/pnas.0911726107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell L. J.; Capasso M.; Vater I.; Akasaka T.; Bernard O. A.; Calasanz M. J.; Chandrasekaran T.; Chapiro E.; Gesk S.; Griffiths M.; Guttery D. S.; Haferlach C.; Harder L.; Heidenreich O.; Irving J.; Kearney L.; Nguyen-Khac F.; Machado L.; Minto L.; Majid A.; Moorman A. V.; Morrison H.; Rand V.; Strefford J. C.; Schwab C.; Tönnies H.; Dyer M. J. S.; Siebert R.; Harrison C. J. Deregulated Expression of Cytokine Receptor Gene, CRLF2, Is Involved in Lymphoid Transformation in B-Cell Precursor Acute Lymphoblastic Leukemia. Blood 2009, 114 (13), 2688–2698. 10.1182/blood-2009-03-208397. [DOI] [PubMed] [Google Scholar]
- Mullighan C. G.; Collins-Underwood J. R.; Phillips L. A. A.; Loudin M. G.; Liu W.; Zhang J.; Ma J.; Coustan-Smith E.; Harvey R. C.; Willman C. L.; Mikhail F. M.; Meyer J.; Carroll A. J.; Williams R. T.; Cheng J.; Heerema N. A.; Basso G.; Pession A.; Pui C.-H.; Raimondi S. C.; Hunger S. P.; Downing J. R.; Carroll W. L.; Rabin K. R. Rearrangement of CRLF2 in B-Progenitor- and Down Syndrome-Associated Acute Lymphoblastic Leukemia. Nat. Genet. 2009, 41 (11), 1243–1246. 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts K. G.; Gu Z.; Payne-Turner D.; McCastlain K.; Harvey R. C.; Chen I.-M.; Pei D.; Iacobucci I.; Valentine M.; Pounds S. B.; Shi L.; Li Y.; Zhang J.; Cheng C.; Rambaldi A.; Tosi M.; Spinelli O.; Radich J. P.; Minden M. D.; Rowe J. M.; Luger S.; Litzow M. R.; Tallman M. S.; Wiernik P. H.; Bhatia R.; Aldoss I.; Kohlschmidt J.; Mrózek K.; Marcucci G.; Bloomfield C. D.; Stock W.; Kornblau S.; Kantarjian H. M.; Konopleva M.; Paietta E.; Willman C. L.; Mullighan C. G. High Frequency and Poor Outcome of Philadelphia Chromosome-Like Acute Lymphoblastic Leukemia in Adults. J. Clin Oncol 2017, 35 (4), 394–401. 10.1200/JCO.2016.69.0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasian S. K.; Doral M. Y.; Borowitz M. J.; Wood B. L.; Chen I.-M.; Harvey R. C.; Gastier-Foster J. M.; Willman C. L.; Hunger S. P.; Mullighan C. G.; Loh M. L. Aberrant STAT5 and PI3K/MTOR Pathway Signaling Occurs in Human CRLF2-Rearranged B-Precursor Acute Lymphoblastic Leukemia. Blood 2012, 120 (4), 833–842. 10.1182/blood-2011-12-389932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppikar P.; Bhagwat N.; Kilpivaara O.; Manshouri T.; Adli M.; Hricik T.; Liu F.; Saunders L. M.; Mullally A.; Abdel-Wahab O.; Leung L.; Weinstein A.; Marubayashi S.; Goel A.; Gönen M.; Estrov Z.; Ebert B. L.; Chiosis G.; Nimer S. D.; Bernstein B. E.; Verstovsek S.; Levine R. L. Heterodimeric JAK-STAT Activation as a Mechanism of Persistence to JAK2 Inhibitor Therapy. Nature 2012, 489 (7414), 155–159. 10.1038/nature11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco M.-J.; Gardinier K. M. New Chemical Modalities and Strategic Thinking in Early Drug Discovery. ACS Med. Chem. Lett. 2020, 11 (3), 228–231. 10.1021/acsmedchemlett.9b00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T.; Ciulli A. E3 Ligase Ligands for PROTACs: How They Were Found and How to Discover New Ones. SLAS Discov 2021, 26 (4), 484–502. 10.1177/2472555220965528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.; Min J.; Jarusiewicz J.; Actis M.; Bradford S. Y.-C.; Mayasundari A.; Yang L.; Chepyala D.; Alcock L. J.; Roberts K. G.; Nithianantham S.; Maxwell D.; Rowland L.; Larsen R.; Seth A.; Goto H.; Imamura T.; Akahane K.; Hansen B.; Pruett-Miller S. M.; Paietta E. M.; Litzow M. R.; Qu C.; Yang J. J.; Fischer M.; Rankovic Z.; Mullighan C. G. Degradation of Janus Kinases in CRLF2- Rearranged Acute Lymphoblastic Leukemia. Blood 2021, 138 (23), 2313–2326. 10.1182/blood.2020006846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela M. E.; Lu G.; Ito T.; Pagarigan B.; Lu C.-C.; Miller K.; Fang W.; Wang N.-Y.; Nguyen D.; Houston J.; Carmel G.; Tran T.; Riley M.; Nosaka L.; Lander G. C.; Gaidarova S.; Xu S.; Ruchelman A. L.; Handa H.; Carmichael J.; Daniel T. O.; Cathers B. E.; Lopez-Girona A.; Chamberlain P. P. A Novel Cereblon Modulator Recruits GSPT1 to the CRL4(CRBN) Ubiquitin Ligase. Nature 2016, 535 (7611), 252–257. 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- Ishoey M.; Chorn S.; Singh N.; Jaeger M. G.; Brand M.; Paulk J.; Bauer S.; Erb M. A.; Parapatics K.; Müller A. C.; Bennett K. L.; Ecker G. F.; Bradner J. E.; Winter G. E. Translation Termination Factor GSPT1 Is a Phenotypically Relevant Off-Target of Heterobifunctional Phthalimide Degraders. ACS Chem. Biol. 2018, 13 (3), 553–560. 10.1021/acschembio.7b00969. [DOI] [PubMed] [Google Scholar]
- Nishiguchi G.; Keramatnia F.; Min J.; Chang Y.; Jonchere B.; Das S.; Actis M.; Price J.; Chepyala D.; Young B.; McGowan K.; Slavish P. J.; Mayasundari A.; Jarusiewicz J. A.; Yang L.; Li Y.; Fu X.; Garrett S. H.; Papizan J. B.; Kodali K.; Peng J.; Pruett Miller S. M.; Roussel M. F.; Mullighan C.; Fischer M.; Rankovic Z. Identification of Potent, Selective, and Orally Bioavailable Small-Molecule GSPT1/2 Degraders from a Focused Library of Cereblon Modulators. J. Med. Chem. 2021, 64 (11), 7296–7311. 10.1021/acs.jmedchem.0c01313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah R. R.; Redmond J. M.; Mihut A.; Menon M.; Evans J. P.; Murphy J. A.; Bartholomew M. A.; Coe D. M. Hi-JAK-Ing the Ubiquitin System: The Design and Physicochemical Optimisation of JAK PROTACs. Bioorg. Med. Chem. 2020, 28 (5), 115326. 10.1016/j.bmc.2020.115326. [DOI] [PubMed] [Google Scholar]
- Nasveschuk C.; Zeid R.; Yin N.; Jackson K. L.; Veits G. K.; Moustakim M.; Yap J. L.. Compounds for Targeted Degradation. Patent WO2017197046A1, 2017.
- Rankovic Z.; Min J.; Mayasundari A.; Keramatnia F.; Jonchere B.; Yang S. W.; Jarusiewicz J. A.; Actis M.; Das S.; Young B. M.; Slavish P. J.; Yang L.; Li Y.; Fu X.; Garrett S. H.; Yun M.-K.; Li Z.; Nithianantham S.; Chai S. C.; Chen T.; Shelat A. A.; Lee R. E.; Nishiguchi G.; White S. W.; Roussel M. F.; Potts P. R.; Fischer M. Phenyl-Glutarimides: Alternative Cereblon Binders for the Design of PROTACs. Angew. Chem., Int. Ed. 2021, 60, 26663. 10.1002/anie.202108848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R. R.; Li B.; Yun S. Y.; Chan A.; Nareddy P.; Gunawan S.; Ayaz M.; Lawrence H. R.; Reuther G. W.; Lawrence N. J.; Schönbrunn E. Structural Insights into JAK2 Inhibition by Ruxolitinib, Fedratinib, and Derivatives Thereof. J. Med. Chem. 2021, 64 (4), 2228–2241. 10.1021/acs.jmedchem.0c01952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Li Y.; Aguilar A.; Liu Z.; Yang C.-Y.; Wang S. Simple Structural Modifications Converting a Bona Fide MDM2 PROTAC Degrader into a Molecular Glue Molecule: A Cautionary Tale in the Design of PROTAC Degraders. J. Med. Chem. 2019, 62 (21), 9471–9487. 10.1021/acs.jmedchem.9b00846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim C.; Pliatsika D.; Mousavizadeh F.; Bär K.; Hernandez Alvarez B.; Giannis A.; Hartmann M. D. De-Novo Design of Cereblon (CRBN) Effectors Guided by Natural Hydrolysis Products of Thalidomide Derivatives. J. Med. Chem. 2019, 62 (14), 6615–6629. 10.1021/acs.jmedchem.9b00454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann M. D.; Boichenko I.; Coles M.; Zanini F.; Lupas A. N.; Hernandez Alvarez B. Thalidomide Mimics Uridine Binding to an Aromatic Cage in Cereblon. J. Struct Biol. 2014, 188 (3), 225–232. 10.1016/j.jsb.2014.10.010. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; Swindells M. B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf Model 2011, 51 (10), 2778–2786. 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Matyskiela M. E.; Zhang W.; Man H.-W.; Muller G.; Khambatta G.; Baculi F.; Hickman M.; LeBrun L.; Pagarigan B.; Carmel G.; Lu C.-C.; Lu G.; Riley M.; Satoh Y.; Schafer P.; Daniel T. O.; Carmichael J.; Cathers B. E.; Chamberlain P. P. A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J. Med. Chem. 2018, 61 (2), 535–542. 10.1021/acs.jmedchem.6b01921. [DOI] [PubMed] [Google Scholar]
- Tomoyasu C.; Imamura T.; Tomii T.; Yano M.; Asai D.; Goto H.; Shimada A.; Sanada M.; Iwamoto S.; Takita J.; Minegishi M.; Inukai T.; Sugita K.; Hosoi H. Copy Number Abnormality of Acute Lymphoblastic Leukemia Cell Lines Based on Their Genetic Subtypes. Int. J. Hematol 2018, 108 (3), 312–318. 10.1007/s12185-018-2474-7. [DOI] [PubMed] [Google Scholar]
- Kim S.-K.; Knight D. A.; Jones L. R.; Vervoort S.; Ng A. P.; Seymour J. F.; Bradner J. E.; Waibel M.; Kats L.; Johnstone R. W. JAK2 Is Dispensable for Maintenance of JAK2Mutant B-Cell Acute Lymphoblastic Leukemias. Genes Dev. 2018, 32 (11–12), 849–864. 10.1101/gad.307504.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm J. W.; Sia K. C. S.; Jones C.; Evans K.; Mariana A.; Pang I.; Failes T.; Zhong L.; Mayoh C.; Landman R.; Collins R.; Erickson S. W.; Arndt G.; Raftery M. J.; Wilkins M. R.; Norris M. D.; Haber M.; Marshall G. M.; Lock R. B. Combination Efficacy of Ruxolitinib with Standard-of-Care Drugs in CRLF2-Rearranged Ph-like Acute Lymphoblastic Leukemia. Leukemia 2021, 35, 3101. 10.1038/s41375-021-01248-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.