

Abstract

Myostatin is a key negative regulator of skeletal muscle growth, and myostatin inhibitors are attractive tools for the treatment of muscular atrophy. Previously, we reported a series of 14–29-mer peptide myostatin inhibitors, including a potent derivative, MIPE-1686, a 16-mer N-terminal-free l-peptide with three unnatural amino acids and a propensity to form β-sheets. However, the in vivo biological stability of MIPE-1686 is a concern for its development as a drug. In the present study, to develop a more stable myostatin inhibitory d-peptide (MID), we synthesized various retro-inverso versions of a 16-mer peptide. Among these, an arginine-containing derivative, MID-35, shows a potent and equivalent in vitro myostatin inhibitory activity equivalent to that of MIPE-1686 and considerable stability against biodegradation. The in vivo potency of MID-35 to increase the tibialis anterior muscle mass in mice is significantly enhanced over that of MIPE-1686, and MID-35 can serve as a new entity for the prolonged inactivation of myostatin in skeletal muscle.
Keywords: Inhibitor, d-peptide, muscle atrophic disorder, myostatin, stability
Myostatin (growth differentiation factor-8, GDF-8), a member of the transforming growth factor-β (TGF-β) superfamily, suppresses the growth of skeletal muscle and is promising for the treatment of muscle atrophic disorders, such as muscular dystrophy, sarcopenia, and cancer-associated cachexia.1−3 Intracellularly produced myostatin precursors are dimerized and cleaved into prodomains and a mature dimer by furin-like proteases. This mature dimer, myostatin, can bind to activin type I and II receptors and activate the intracellular Smad2/3 signaling to negatively regulate muscle growth.4−7 The development of high-molecular-weight inhibitory molecules such as antibodies and soluble decoy receptors8−10 has progressed, but none of these products are currently in clinical use for muscle wasting.11,12 Therefore, it is appropriate to examine a new therapeutic strategy involving compounds that can overcome muscular dystrophy.13 Recently, the cooperative regulation of multiple TGF-β superfamily members, including myostatin, GDF-11, and activin, has received much attention in drug development laboratories.14−17 Latres et al. suggested that the inhibition of both myostatin and activin is more effective for muscle building than myostatin-specific inhibition.14 Follistatin like-3 (Fstl3), an endogenous molecule inhibitory against TGF-β family ligands, also has attracted attention for its enhancement of muscle growth.17,18 For instance, the antibody Fc-fused Fstl3 protein, which inhibits myostatin, GDF-11, and activin, was reported as a new agent that increases muscle mass.17
Since 2015, we have reported a series of 14–29-mer peptide inhibitors of myostatin.19−26 Among these, a 16-mer inhibitor, MIPE-1686 (Figure 1), is the most potent peptide encountered to date.24 In terms of the mechanism, we proposed that the inhibitory peptide binds to myostatin at the activin type I receptor binding site described in our recent study.27 MIPE-1686 has a predominant β-sheet-forming property, and this contributes to a potent myostatin inhibitory activity and a high stability in vitro against recombinant enzymes such as aminopeptidase N, chymotrypsin C, and trypsin 3.24,28 However, the in vivo stability of MIPE-1686 under biological conditions remains a concern because its N-terminus is unprotected, and it is basically composed of native l-amino acids with the exception of three unnatural amino acids, one d-Trp and two l-cyclohexylglycine residues.
Figure 1.

Sequences of previously reported 16-mer peptides MIPE-1686 and 7c(24) and newly synthesized peptides 7c-ri, MID-35, MID-36, and MID-39 using an Fmoc-based solid-phase peptide synthesis (SPPS) method. The numbers above each amino acid indicate the position relative to the N-terminus. The lowercase letter indicates the d-amino acid. The underline indicates the substituted amino acid from peptide 7c or 7c-ri. The hyphen means deletion of the corresponding amino acid.
In the present study, we sought to develop a myostatin inhibitory d-peptide (MID) to avoid the proteolytic degradation entirely. The preparation of such a retro-inverso peptide is one of several challenging strategies to produce stable alternatives to biologically active l-peptides.29 Snyder et al., for example, successfully developed the retro-inverso version of an anti-cell proliferative peptide derived from the C-terminal region of p53, which sustainably suppresses the cancer growth.30 This appears, however, to be an exceptional case, and it is generally recognized that retro-inverso isomerization is less effective in reproduction of the biological activity of the original l-peptide.31 In this case, we focused on the l-peptide (7c) (Figure 1), which has been reported as a potent inhibitor prior to the d-Trp incorporation step in the production of MIPE-1686.24 Peptide 7c-ri and its derivatives, retro-inverso versions of peptide 7c (Figure 1), were synthesized with >98% purity, and their myostatin inhibitory activities were evaluated. As reported previously,19,24 these peptides were synthesized manually using an Fmoc-based solid-phase peptide synthesis (SPPS) method. A Smad2/3-responsive luciferase reporter assay using human embryonic kidney 293 (HEK293) cells was used to evaluate the myostatin inhibitory activity of the synthesized peptides. We found that peptide 7c-ri possessed potent myostatin inhibitory activity similar to that of peptide 7c (Figure 2A). The physicochemical properties of peptide 7c-ri, however, required attention because as in 7c and MIPE-1686,24 some insoluble aggregates were observed during its purification, and the yield was only 10%. (For details, see the Supporting Information (SI)). The nonspecific binding (NSB) of MIPE-1686 to the experimental apparatus was observed in our previous study.28 Therefore, to improve the physicochemical property of peptide inhibitors, we designed the novel basic arginine-containing derivative MID-35, in which both neutral d-Ser and d-Gln residues at positions 5 and 8 of 7c-ri were replaced by d-Arg. These two hydrophilic amino acid residues were chosen because hydrophobic amino acids are known to be important for effective myostatin inhibition.22,32 Regarding the incorporation of d-Arg, the advantageous effect on the inhibitory activity in comparison with Lys, as previously reported,24 was considered. No problematic aggregates were observed during the purification step in the MID-35 synthesis, and the yield increased to 21%. (See the SI.) In the reporter assay, the myostatin inhibitory activity observed in 7c-ri was well maintained in MID-35 (Figure 2B).
Figure 2.

Results of a luciferase reporter assay of the activities of myostatin inhibitory d-peptides. (A) 7c-ri, (B) MID-35, and (C) MID-36 and MID-39. Cells, HEK293-cell-transfected Smad-responsive reporter (pGL4.48[luc2P/SBE/Hygro]) and control (pGL4.74[hRluc/TK]) plasmids; peptide concentration, 0.3 μM; positive control (prodomain, mouse recombinant) concentration, 10 nM; myostatin concentration, 8 ng/mL (0.32 nM); starvation with serum-free DMEM, 8 h; incubation with or without peptides, 4 h. Values represent means ± SD (N = 3).
To investigate the possibility of chain shortening, we synthesized MID-36 and MID-39, deleted forms of N- and C-terminal amino acids of MID-35, respectively (Figure 1). As shown in Figure 2C, the myostatin inhibitory activities of both of these 15-mer d-peptides (MID-36 and MID-39) were similar to or weaker than that of MID-35 at a concentration of 0.3 μM. This result suggests that the C-terminal d-Trp residue is more important than the N-terminal d-Leu residue for effective inhibition. Nevertheless, 15-mer peptides such as MID-36 maintain potent activity similar to that of our previously reported 22-mer derivatives 3d(22) and 11,23 with IC50 values of ∼0.3 μM. However, the both termini-deleted 14-mer peptide (rxkrwirxkiwriy-amide) lost the inhibitory activity at a concentration of 0.3 μM (data not shown). Recently, we also reported a structure–activity relationship (SAR) study of follistatin-derived myostatin inhibitory 14-mer peptides.25,26 The most potent derivative among them, DF-100,26 has an IC50 value of 4.0 μM. These findings imply that a 15-mer or larger is required to obtain the potent IC50 value of ∼0.2 to 0.3 μM. The proposed binding site of the inhibitory peptides27 may impose a molecular size limitation because the activin type I receptor binding site on myostatin, for example, has a broad concavity.
Next, we examined the dose dependency of MID-35 against myostatin, GDF-11, activin A, and TGF-β1 (Figure 3), all of which activate Smad2/3 signaling. The IC50 values of MID-35 and MIPE-1686 are shown in Table 1. MID-35 has slightly better inhibitory IC50 values (0.19 and 0.89 μM, respectively) against myostatin and activin A than against MIPE-1686 (0.26 and 1.4 μM, respectively). In the inhibition of GDF-11 and TGF-β1, MID-35 (0.63 and 1.6 μM) has two times and four times more potent activity than MIPE-1686 (1.4 and 6.7 μM), respectively. These results suggest that MID-35 shows the redundant inhibition of TGF-β superfamily members compared with MIPE-1686, although the IC50 value for myostatin is at least three times more potent than that for GDF-11. The observed potent IC50 values (<1 μM) of MID-35 for GDF-11 and activin A were the first observed among our peptidic inhibitors. GDF-11, reported to be a negative regulator of muscle growth,33 is closely related to myostatin, with which it has 90% homology. The observed redundant inhibition is therefore reasonable. Because the importance of regulating multiple TGF-β superfamily members, including GDF-11 and activin, has been suggested by several groups, as previously mentioned,14−17 the observed multi-inhibitory capability of MID-35 could be a suitable property to support effective muscle growth.
Figure 3.

Luciferase reporter assays to evaluate the dose-dependent inhibition by MIPE-1686 and MID-35 against myostatin (A), GDF-11 (B), activin A (C), and TGF-β1 (D). Cells, HEK293-cell-transfected Smad-responsive reporter (pGL4.48[luc2P/SBE/Hygro]) and control (pGL4.74[hRluc/TK]) plasmids; peptide concentration, 0.025–2 μM (A), 0.08–6 μM (B), 0.025–18 μM (C), and 0.025–18 μM for MID-35 or 0.074–54 μM for MIPE-1686 (D); positive control (SB431542) concentration, 5 μM; myostatin, GDF-11, and activin A concentrations, 8 ng/mL (0.32 nM); TGF-β1 concentration, 2.5 ng/mL (0.1 nM); starvation with serum-free DMEM, 8 h; incubation with or without peptides, 4 h. Values represent means ± SD (N = 3). Curve fitting was performed using KaleidaGraph 4.5. The sigmoidal curve is normalized with the minimum values in each assay. The vertical dotted line (gray) represents a concentration of 1 μM.
Table 1. IC50 Values (μM) of MIPE-1686 and MID-35a.
| inhibited protein | MIPE-1686 | MID-35 |
|---|---|---|
| myostatin | 0.26 ± 0.04 | 0.19 ± 0.05 |
| GDF-11 | 1.4 ± 0.2 | 0.63 ± 0.05 |
| activin A | 1.4 ± 0.5 | 0.89 ± 0.09 |
| TGF-β1 | 6.7 ± 0.6 | 1.6 ± 0.2 |
Prior to the in vivo study, we tested the in vitro proteolytic stability of MID-35 and MIPE-1686 in a solution of isolated digestive enzymes derived from the bovine pancreas. As previously described, MIPE-1686 is highly stable in the human-derived recombinant enzyme solutions containing aminopeptidase N, chymotrypsin C, and trypsin 3, notwithstanding its being an N-terminal-free l-amino-acid-based peptide.28 It was also confirmed through reverse-phase high-performance liquid chromatography (RP-HPLC) analysis that MIPE-1686 is stable in solutions of trypsin (Figure 4A and Figure S2A) or α-chymotrypsin (Figures S1A and S2B), although the peak area of the intact form after 400 min of incubation was significantly decreased to 77 and 72%, respectively. To validate the activity of bovine enzymes used, we treated the previously reported myostatin inhibitory l-peptide (SRIEWIKIQIWSKLRL-amide; 4b in ref (24), 2 in ref (28)).24,28 This peptide was easily degraded to <1% of intact forms by trypsin and α-chymotrypsin within 45 and 90 min, respectively (Figure S3). The reduced recovery of MIPE-1686 seems to be partially due to the loss of its NSB to the experimental apparatus, as previously reported.28 However, some new small peaks from the enzyme treatments appeared, as depicted in both HPLC charts at 400 min (Figure 4A and Figure S1A). We successfully determined by mass spectrometric analysis that the peak detected with a retention time of 18.7 min in Figure 4A is the metabolite tm1 (Figure 4C). This is a 15-mer peptide formed from the hydrolysis by trypsin of the amide bond of the C-terminal Arg15–Leu16 sequence of MIPE-1686. These results suggests that MIPE-1686 is highly but not entirely stable against proteolytic degradation. However, the incubation of MID-35 for 400 min in trypsin (Figure 4B) or α-chymotrypsin (Figure S1B) solution produced no new peak, as expected, and 99 and 97% of the intact form survived, respectively. This means that no obvious NSB occurred and suggests that MID-35 composed of all d-amino acids possesses not only complete proteolytic stability but also better physicochemical properties than MIPE-1686.
Figure 4.

(A,B) Representative analytical RP-HPLC chromatograms of (A) MIPE-1686 and (B) MID-35 incubated in a solution of trypsin from bovine pancreas (1 μg/mL) on 50 mM Tris buffer (pH 7.5) containing 150 mM NaCl, 10 mM CaCl2, and 0.05% Brij-35 at 37 °C for 400 min. Column, COSMOSIL 5C18-AR-II (4.6 × 150 mm); binary solvent system, a linear gradient of CH3CN (25–40%, 30 min) in 0.1% aqueous TFA; flow, 1.0 mL/min; detection, UV 220 nm. Asterisks identify unknown peaks. (C) Sequence and mass spectrometric data of metabolite tm1 derived from MIPE-1686.
Finally, we completed an in vivo study using wild-type C57BL/6J 8-week old male mice. At day 0, the peptide solution (30 nmol, 40 μL) and saline were injected into the left and right tibialis anterior (TA) muscles, respectively. At day 28, the TA muscles were collected and weighed. In our previous study using Duchenne muscular dystrophy (DMD) model mdx mice, the muscle-building effect of MIPE-1686 on the TA muscle (increase to 114% compared with saline) was obtained at day 42 after two intramuscular injections (30 nmol/injection), on days 0 and 14.24 We were able to confirm that MIPE-1686 significantly induces the increase in TA muscle mass in C57BL/6J mice (Figure 5A) in a single dose, and the weight of the MID-35-injected TA muscle became significantly heavier than that of the saline-injected muscle (Figure 5B). As shown in Figure 5C, the % increase of the TA weight following the injection of MID-35 (saline-injected TA weight = 100%) was 133 ± 10%, which was significantly higher than that following MIPE-1686 injection (109 ± 2.3%). This result suggests that MID-35 functions as a more potent myostatin inhibitor than MIPE-1686 in skeletal muscle growth. The higher enzymatic stability of MID-35 shown in Figure 4 is probably related to the effective muscle building that was observed. This result might be an increase in the residence time of MID-35 in the muscle, resulting in the long-lasting inhibition of myostatin and leading to effective muscle hypertrophy and regeneration. MIPE-1686 has similar good stability in the solution of isolated enzymes from the bovine pancreas in the present study in addition to the previous human-derived recombinant enzymes.28 Therefore, the complete enzymatic stability of MID-35 would be highly expected to be maintained in other species. Moreover, the better inhibitory activity of MID-35 against multiple TGF-β superfamily members might enhance the effectiveness of the muscle growth.14−17 Further biological assessments including an appropriate dosing regimen would be required to clarify these possibilities and more detailed pharmacological functions of all d-peptidic inhibitors represented by MID-35 in skeletal muscle tissue. Also, to enhance the drug development, it will be necessary to pursue the applicability of MID-35 by using muscle atrophic models such as the DMD model mdx and tumor-bearing cachexia mice. Myostatin is completely conserved between humans and mice, and hence the data obtained in various mouse models would be extremely valuable for extrapolation to the effect on humans.
Figure 5.
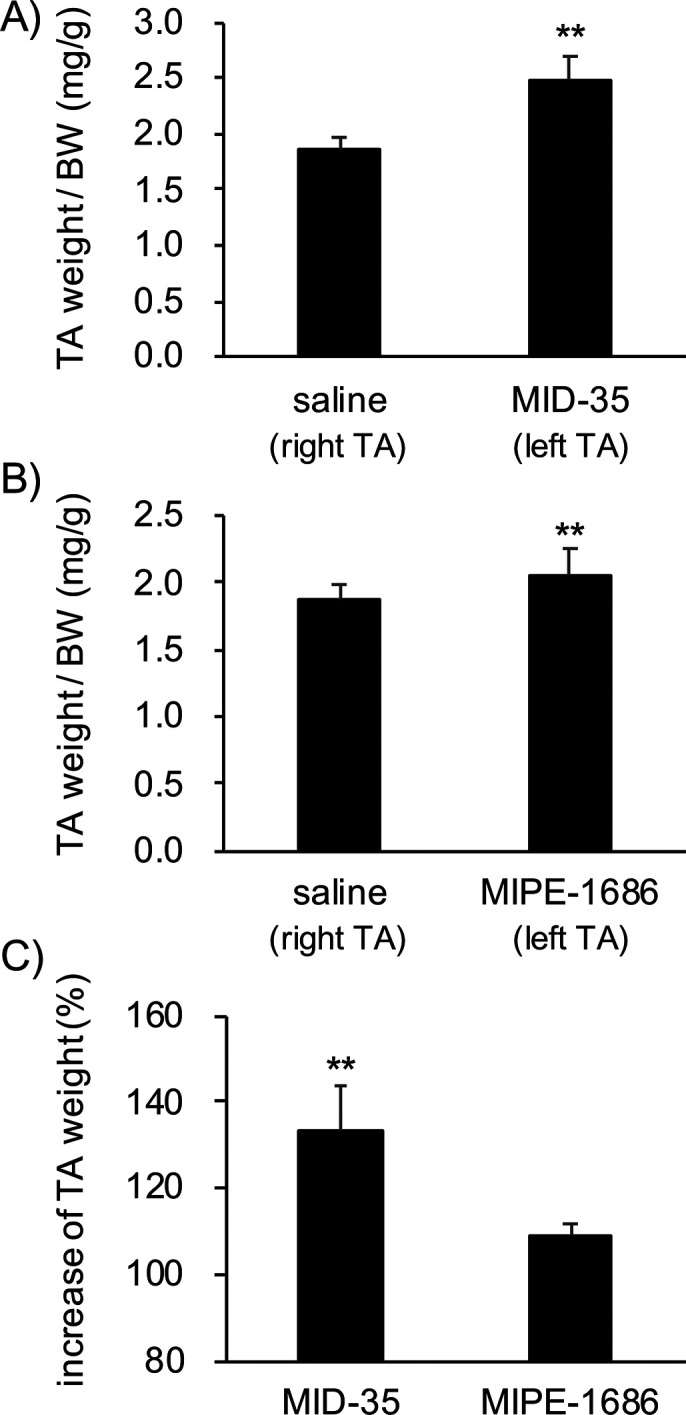
(A) Weight of tibialis anterior (TA) muscles in 8-week-old male C57BL/6J mice treated with MIPE-1686 (left TA; 0.75 mM in saline, 40 μL) or saline (right TA; 40 μL). The TA weight is normalized with the body weight (BW). Results are presented as mean values ± SD (N = 4). (B) Weight of TA muscles in 8-week-old male C57BL/6J mice treated with MID-35 (left TA; 0.75 mM in saline, 40 μL) or saline (right TA; 40 μL). The TA weight is normalized with the body weight (BW). Results are presented as mean values ± SD (N = 5). (C) Increase (%) in TA weight upon treatment with peptides. The saline-injected TA weight in each mouse is 100%. The values are presented as mean values ± SD (MID-35, N = 5; MIPE-1686, N = 4). **p < 0.01.
In conclusion, we report the discovery of a series of first-in-class myostatin inhibitors consisting entirely of d-amino acids and the synthesis of the retro-inverso peptide 7c-ri. This is one of the few successful examples of the retro-inverso strategy. An arginine-incorporated derivative of 7c-ri, MID-35, showed improved physicochemical properties and enzymatic stability compared with the previously reported l-amino-acid-based MIPE-1686. Although the in vitro myostatin inhibitory activity of MID-35 is almost equivalent to that of MIPE-1686, the in vivo efficacy and the stability of the former d-inhibitor are superior to those of the latter l-based inhibitor. MID-35 is a promising platform for expressing the long-lasting muscle-building effect in skeletal muscles. The present study also suggests that the size of 15-mer (MID-36) or 16-mer (MID-35) peptides may represent the minimum structure to exhibit IC50 values of 0.2 to 0.3 μM against myostatin. Elucidation of the binding mode to myostatin based on these peptides might be useful in the design of more potent myostatin inhibitors, leading to drug development with superior pharmacokinetic and safety properties in the future.
Glossary
Abbreviations
- BW
body weight
- DMD
Duchenne muscular dystrophy
- DMEM
Dulbecco’s modified Eagle’s medium
- Fmoc
9-fluorenylmethyloxycarbonyl
- HEK293
human embryonic kidney 293
- RP-HPLC
reverse-phase high-performance liquid chromatography
- SAR
structure–activity relationship
- SBE
Smad binding element
- SPPS
solid-phase peptide synthesis
- TA
tibialis anterior
- TGF-β
transforming growth factor-β
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00705.
Materials, experimental procedure, and associated content. Analytical data for all peptide derivatives and analytical HPLC chromatograms (PDF)
Author Contributions
All authors were involved in the preparation of this manuscript, including the conception and design of the study (K. Takayama, Y.H.), peptide synthesis (M.O., R.O., T.S.), cell-based assay (H.O., M.S.), enzymatic stability analysis (K. Takayama, M.O.), animal research (K.H., K. Tsuchida), data analysis (K. Takayama, K.H., H.O, M.O., K. Tsuchida, Y.H.), and writing and editing of the manuscript (K. Takayama, K.H., A. Taguchi, A. Taniguchi, K. Tsuchida, Y.H.). All authors have given approval to the final version of the manuscript.
This research was supported by the Japan Society for the Promotion of Sciences (JSPS) KAKENHI, including Grants-in-Aid for Scientific Research (B) 15H04658 (Y.H.), MEXT-supported Program for the Strategic Research Foundation at Private Universities, and Intramural Research Grant (29-4 and 2-5) for Neurological and Psychiatric Disorders on NCNP (Y.H. to K. Tsuchida.).
The authors declare no competing financial interest.
Supplementary Material
References
- McPherron A. C.; Lawler A. M.; Lee S.-J. Regulation of skeletal muscle in mice by a new TGF-β superfamily member. Nature 1997, 387, 83–90. 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- Zimmers T. A.; Davies M. V.; Koniaris L. G.; Haynes P.; Esquela A. F.; Tomkinson K. N.; McPherron A. C.; Wolfman N. M.; Lee S.-J. Induction of cachexia in mice by systemically administrated myostatin. Science 2002, 296, 1486–1488. 10.1126/science.1069525. [DOI] [PubMed] [Google Scholar]
- a Ojima C.; Noguchi Y.; Miyamoto T.; Saito Y.; Orihashi H.; Yoshimatsu Y.; Watabe T.; Takayama K.; Hayashi Y.; Itoh F. Peptide-2 from mouse myostatin precursor protein alleviates muscle wasting in cancer-associated cachexia. Cancer Sci. 2020, 111, 2954–2964. 10.1111/cas.14520. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhou X.; Wang J. L.; Lu J.; Song Y.; Kwak K. S.; Jiao Q.; Rosenfeld R.; Chen Q.; Boone T.; Simonet W. S.; Lacey D. L.; Goldberg A. L.; Han H.Q. Reversal of Cancer Cachexia and Muscle Wasting by ActRIIB Antagonism Leads to Prolonged Survival. Cell 2010, 142 (4), 531–543. 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]
- Lee S.-J.; McPherron A. C. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 9306–9311. 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana J. L.; Attisano L.; Wieser R.; Ventura F.; Massagué J. Mechanism of activation of the TGF-β receptor. Nature 1994, 370, 341–347. 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- Wolfman N. M.; McPherron A. C.; Pappano W. M.; Davies M. V.; Song K.; Tomkinson K. N.; Wright J. F.; Zhao L.; Sebald S. M.; Greenspan D. S.; Lee S.-J. Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 15842–15846. 10.1073/pnas.2534946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinck A. P. Structural studies of the TGF-βs and their receptors - insights into evolution of the TGF-β superfamily. FEBS Lett. 2012, 586, 1860–1870. 10.1016/j.febslet.2012.05.028. [DOI] [PubMed] [Google Scholar]
- Bogdanovich S.; Krag T. O. B.; Barton E. R.; Morris L. D.; Whittemore L.-A.; Ahima R. S.; Khurana T. S. Functional improvement of dystrophic muscle by myostatin blockade. Nature 2002, 420, 418–421. 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- St. Andre M.; Johnson M.; Bansal P. N.; Wellen J.; Robertson A.; Opsahl A.; Burch P. M.; Bialek P.; Morris C.; Owens J. A mouse anti-myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of Duchenne muscular dystrophy and its humanized equivalent, domagrozumab (PF-06252616), increases muscle volume in cynomolgus monkeys. Skelet. Muscle 2017, 7, 25. 10.1186/s13395-017-0141-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Wang J. L.; Lu J.; Song Y.; Kwak K. S.; Jiao Q.; Rosenfeld R.; Chen Q.; Boone T.; Simonet W. S.; Lacey D. L.; Goldberg A. L.; Han H.Q. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]
- Campbell C.; McMillan H. J.; Mah J. K.; Tarnopolsky M.; Selby K.; McClure T.; Wilson D. M.; Sherman M. L.; Escolar D.; Attie K. M. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial. Muscle Nerve 2017, 55, 458–464. 10.1002/mus.25268. [DOI] [PubMed] [Google Scholar]
- Golan T.; Geva R.; Richards D.; Madhusudan S.; Lin B. K.; Wang H. T.; Walgren R. A.; Stemmer S. M. LY2495655, an antimyostatin antibody, in pancreatic cancer: a randomized, phase 2 trial. J. Cachexia Sarcopenia Muscle 2018, 9, 871–879. 10.1002/jcsm.12331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariot V.; Joubert R.; Hourde C.; Feasson L.; Hanna M.; Muntoni F.; Maisonobe T.; Servais L.; Bogni C.; Le Panse R.; Benvensite O.; Stojkovic T.; Machado P. M.; Voit T.; Buj-Bello A.; Dumonceaux J. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat. Commun. 2017, 8, 1859. 10.1038/s41467-017-01486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latres E.; Mastaitis J.; Fury W.; Miloscio L.; Trejos J.; Pangilinan J.; Okamoto H.; Cavino K.; Na E.; Papatheodorou A.; Willer T.; Bai Y.; Hae Kim J.; Rafique A.; Jaspers S.; Stitt T.; Murphy A. J.; Yancopoulos G. D.; Gromada J. Activin A more prominently regulates muscle mass in primates than does GDF8. Nat. Commun. 2017, 8, 15153. 10.1038/ncomms15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.-J.; Lehar A.; Liu Y.; Ly C. H.; Pham Q.-M.; Michaud M.; Rydzik R.; Youngstrom D. W.; Shen M. M.; Kaartinen V.; Germain-Lee E. L.; Rando T. A. Functional redundancy of type I and type II receptors in the regulation of skeletal muscle growth by myostatin and activin A. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 30907. 10.1073/pnas.2019263117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskenderian A.; Liu N.; Deng Q.; Huang Y.; Shen C.; Palmieri K.; Crooker R.; Lundberg D.; Kastrapeli N.; Pescatore B.; Romashko A.; Dumas J.; Comeau R.; Norton A.; Pan J.; Rong H.; Derakhchan K.; Ehmann D. E. Myostatin and activin blockade by engineered follistatin results in hypertrophy and improves dystrophic pathology in mdx mouse more than myostatin blockade alone. Skelet. Muscle 2018, 8, 34. 10.1186/s13395-018-0180-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T.; Morikawa M.; Morishita Y.; Ogikubo K.; Itoh F.; Koinuma D.; Nygren P.; Miyazono K. Systemic administration of monovalent follistatin-like 3-Fc-fusion protein increases muscle mass in mice. iScience 2021, 24, 102488. 10.1016/j.isci.2021.102488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidis Y.; Mukherjee A.; Keutmann H.; Delbaere A.; Sadatsuki M.; Schneyer A. Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology 2006, 147, 3586–3597. 10.1210/en.2006-0089. [DOI] [PubMed] [Google Scholar]
- Takayama K.; Noguchi Y.; Aoki S.; Takayama S.; Yoshida M.; Asari T.; Yakushiji F.; Nishimatsu S.; Ohsawa Y.; Itoh F.; Negishi Y.; Sunada Y.; Hayashi Y. Identification of the minimum peptide from mouse myostatin prodomain for human myostatin inhibition. J. Med. Chem. 2015, 58, 1544–1549. 10.1021/jm501170d. [DOI] [PubMed] [Google Scholar]
- Ohsawa Y.; Takayama K.; Nishimatsu S.; Okada T.; Fujino M.; Fukai Y.; Murakami T.; Hagiwara H.; Itoh F.; Tsuchida K.; Hayashi Y.; Sunada Y. The inhibitory core of the myostatin prodomain: its interaction with both type I and II membrane receptors, and potential to treat muscle atrophy. PLoS One. 2015, 10, e0133713. 10.1371/journal.pone.0133713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K.; Nakamura A.; Rentier C.; Mino Y.; Asari T.; Saga Y.; Taguchi A.; Yakushiji F.; Hayashi Y. Effect of N-terminal acylation on the activity of myostatin inhibitory peptides. ChemMedChem. 2016, 11, 845–849. 10.1002/cmdc.201500533. [DOI] [PubMed] [Google Scholar]
- Takayama K.; Rentier C.; Asari T.; Nakamura A.; Saga Y.; Shimada T.; Nirasawa K.; Sasaki E.; Muguruma K.; Taguchi A.; Taniguchi A.; Negishi Y.; Hayashi Y. Development of potent myostatin inhibitory peptides through hydrophobic residue-directed structural modification. ACS Med. Chem. Lett. 2017, 8, 751–756. 10.1021/acsmedchemlett.7b00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentier C.; Takayama K.; Saitoh M.; Nakamura A.; Ikeyama H.; Taguchi A.; Taniguchi A.; Hayashi Y. Design and synthesis of potent myostatin inhibitory cyclic peptides. Bioorg. Med. Chem. 2019, 27, 1437–1443. 10.1016/j.bmc.2019.02.019. [DOI] [PubMed] [Google Scholar]
- Takayama K.; Asari T.; Saitoh M.; Nirasawa K.; Sasaki E.; Roppongi Y.; Nakamura A.; Saga Y.; Shimada T.; Ikeyama H.; Taguchi A.; Taniguchi A.; Negishi Y.; Hayashi Y. Chain-shortened myostatin inhibitory peptides improve grip strength in mice. ACS Med. Chem. Lett. 2019, 10, 985–990. 10.1021/acsmedchemlett.9b00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh M.; Takayama K.; Hitachi K.; Taguchi A.; Taniguchi A.; Tsuchida K.; Hayashi Y. Discovery of a follistatin-derived myostatin inhibitory peptide. Bioorg. Med. Chem. Lett. 2020, 30, 126892. 10.1016/j.bmcl.2019.126892. [DOI] [PubMed] [Google Scholar]
- Saitoh M.; Takayama K.; Roppongi Y.; Shimada T.; Taguchi A.; Taniguchi A.; Hayashi Y. Strategic structure–activity relationship study on a follistatin-derived myostatin inhibitory peptide. Bioorg. Med. Chem. Lett. 2021, 46, 128163. 10.1016/j.bmcl.2021.128163. [DOI] [PubMed] [Google Scholar]
- Asari T.; Ikeyama H.; Taguchi A.; Taniguchi A.; Hayashi Y.; Takayama K. Proposal for the binding mode of the 23-mer inhibitory peptide to myostatin. Bioorg. Med. Chem. 2021, 40, 116181. 10.1016/j.bmc.2021.116181. [DOI] [PubMed] [Google Scholar]
- Takayama K.; Odagiri M.; Taguchi A.; Taniguchi A.; Hayashi Y. Enzymatic stability of myostatin inhibitory 16-mer peptides. Chem. Pharm. Bull. 2020, 68, 512–515. 10.1248/cpb.c20-00158. [DOI] [PubMed] [Google Scholar]
- Doti N.; Mardirossian M.; Sandomenico A.; Ruvo M.; Caporale A. Recent applications of retro-inverso peptides. Int. J. Mol. Sci. 2021, 22, 8677. 10.3390/ijms22168677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder E. L.; Meade B. R.; Saenz C. C.; Dowdy S. F. Treatment of terminal peritoneal carcinomatosis by a transducible p53-activating peptide. PLoS Biol. 2004, 2, e36. 10.1371/journal.pbio.0020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Pazgier M.; Li J.; Li C.; Liu M.; Zou G.; Li Z.; Chen J.; Tarasov S. G.; Lu W. Y.; Lu W. Limitations of peptide retro-inverso isomerization in molecular mimicry. J. Biol. Chem. 2010, 285, 19572–19581. 10.1074/jbc.M110.116814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asari T.; Takayama K.; Nakamura A.; Shimada T.; Taguchi A.; Hayashi Y. Structural basis for the effective myostatin inhibition of the mouse myostatin prodomain–derived minimum peptide. ACS Med. Chem. Lett. 2017, 8, 113–117. 10.1021/acsmedchemlett.6b00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerman M. A.; Cadena S. M.; Gilbert J. A.; Meyer A.; Nelson H. N.; Swalley S. E.; Mallozzi C.; Jacobi C.; Jennings L. L.; Clay I.; Laurent G.; Ma S.; Brachat S.; Lach-Trifilieff E.; Shavlakadze T.; Trendelenburg A. U.; Brack A. S.; Glass D. J. GDF11 increases with age and inhibits skeletal muscle regeneration. Cell. Metab. 2015, 22, 164–174. 10.1016/j.cmet.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.