

Abstract
Background and aims
The atherosclerotic plaque microenvironment is highly complex, and selective agents that modulate plaque stability are not yet available. We sought to develop a scRNA-seq analysis workflow to investigate this environment and uncover potential therapeutic approaches. We designed a user-friendly, reproducible workflow that will be applicable to other disease-specific scRNA-seq datasets.
Methods
Here we incorporated automated cell labeling, pseudotemporal ordering, ligand-receptor evaluation, and drug-gene interaction analysis into a ready-to-deploy workflow. We applied this pipeline to further investigate a previously published human coronary single-cell dataset by Wirka et al. Notably, we developed an interactive web application to enable further exploration and analysis of this and other cardiovascular single-cell datasets.
Results
We revealed distinct derivations of fibroblast-like cells from smooth muscle cells (SMCs), and showed the key changes in gene expression along their de-differentiation path. We highlighted several key ligand-receptor interactions within the atherosclerotic environment through functional expression profiling and revealed several avenues for future pharmacological development for precision medicine. Further, our interactive web application, PlaqView (www.plaqview.com), allows lay scientists to explore this and other datasets and compare scRNA-seq tools without prior coding knowledge.
Conclusions
This publicly available workflow and application will allow for more systematic and user-friendly analysis of scRNA datasets in other disease and developmental systems. Our analysis pipeline provides many hypothesis-generating tools to unravel the etiology of coronary artery disease. We also highlight potential mechanisms for several drugs in the atherosclerotic cellular environment. Future releases of PlaqView will feature more scRNA-seq and scATAC-seq atherosclerosis-related datasets to provide a critical resource for the field, and to promote data harmonization and biological interpretation.
Keywords: Atherosclerosis, scRNA, cardioinformatics, drug discovery, pipeline, web application, precision medicine
Graphical Abstract
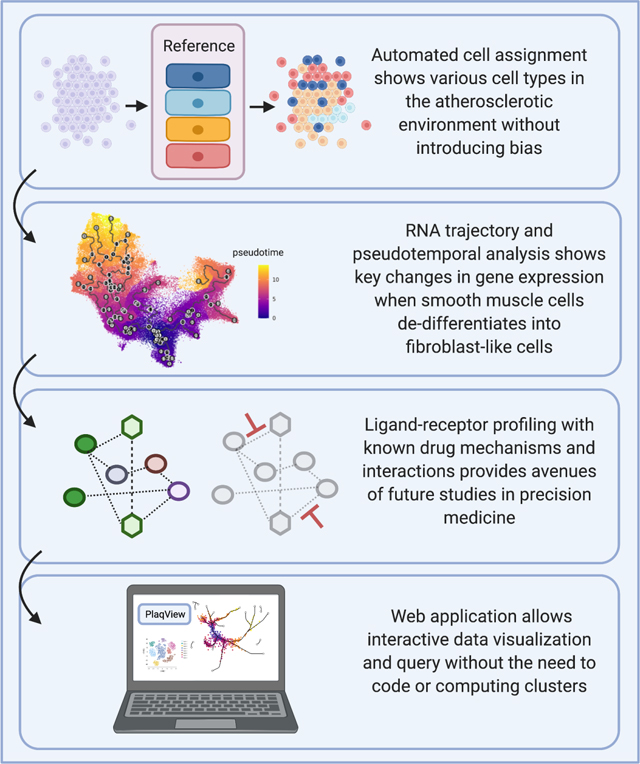
1. Introduction
Atherosclerosis, a complex process involving chronic inflammation and hardening of the vessel wall, represents one of the major causes of coronary artery disease (CAD), peripheral artery disease, and stroke [1]. Rupture of an unstable atherosclerotic lesion can lead to the formation of a thrombus, causing complete or partial occlusion of a coronary artery [2]. The contribution of smooth muscle cells (SMCs) to both lesion stability and progression has recently been established by numerous groups. However, the exact mechanisms by which SMCs modulate the atherosclerotic microenvironment and whether pharmacological agents can be used to selectively counter SMC-related deleterious effects are still under investigation [3–5].
Recent advances in single-cell RNA-sequencing (scRNA-seq) have enabled ultra-fine gene expression profiling of many diseases at the cellular level, including atherosclerotic CAD [5]. As sequencing costs continue to decline, there has also been a consistent growth in scRNA datasets, data analysis tools and applications [6]. Currently, a major challenge with scRNA-seq analysis is the inherent bias introduced during manual cell labeling, in which cells are grouped by clusters and their identities assigned collectively based on their overall differential gene expression profiles [7]. Another drawback inherent to commonly used scRNA-seq protocols is that they destroy the samples, making time-series analyses of the same cells impossible. Instead, these studies must rely on time-points from separate libraries to monitor processes such as clonal expansion and cell differentiation [8,9].
Recently, new approaches have been developed to compensate for both shortcomings, namely automatic cell labeling, pseudotemporal analysis, and trajectory inference. Tools such as ‘SingleR’, ‘scCATCH’, ‘Seurat’, and ‘Garnett’ have been used to assign unbiased identities to individual cells using reference-based and machine learning algorithms [7,10–12]. Moreover, tools such as ‘Monocle3’ and ‘scVelo’ align and project cells onto a pseudotemporal space where each cell becomes a snapshot within the single-cell time continuum [13,14]. In essence, the single scRNA-seq dataset is effectively transformed into a time series [13,15,16]. Although the pseudotemporal scale does not reflect the actual time scale, it is a reliable approximation to characterize cell fate and differentiation events, e.g., during organogenesis, disease states, or in response to SARS-CoV-2 infections [14,15,17].
In this study, we present the application of an enhanced, scalable, and user-friendly scRNA-seq analysis workflow on four previously published human and mouse atherosclerosis scRNA-seq datasets [4,5,18]. Focusing on the only available human coronary artery dataset [5], we performed unbiased automatic cell identification at the single-cell level, pseudotemporal analysis, cell-to-cell communication profiling, and drug repurposing analysis. Our results demonstrate potential new mechanisms by which SMCs contribute to the atherosclerotic phenotype and signaling within the lesion microenvironment. More importantly, we revealed attractive candidate avenues and prospective therapeutic targets for experimental studies of pharmacological candidates. We also developed an interactive web application, PlaqView (www.plaqview.com), to allow users to explore this and other human and mouse cardiovascular single-cell datasets. This reproducible analysis pipeline and application can also be easily modified to incorporate different tissue data sources and single-cell modalities such as scATAC-seq or CITE-seq, and will serve as a template to analyze and visualize single-cell datasets in other disease models.
1. Methods
2.1. Data retrieval and pre-processing
Human coronary artery scRNA data read count matrix was retrieved from the Gene Expression Omnibus (GEO) using #GSE131780 and loaded into R 4.1.1, and was preprocessed using standard parameters of the R packages ‘Seurat’ v.4, and ‘Monocle3’ as required [11,12,15,56]: read count matrix was read into R and converted to a ‘Seurat object’ using the CreateSeuratObject() function. Then, the object underwent removal of mitochondrial and low-quality reads, followed by normalization and variable feature selection using the NormalizeData() and FindVariableFeature() functions, respectively. Uniform manifold approximation projections (UMAPs) were then calculated using the runUMAP() function using the first 30 dimensions. Custom scripting was created to export UMAP of the clusters from ‘Seurat’ into ‘Monocle3’ before pseudotemporal analysis.
2.2. Automatic cell Identification
scRNA read matrices were read into ‘SingleR’ as previously described for cell labeling [7]. Here, we exported the processed scRNA matrix from Seurat into a ‘SingleCellExperiment’ object using the GetAssay() function, and fed into ‘SingleR’ along with the human primary cell atlas as the reference via the ‘celldex’ package [7]. ‘SingleR’ compares each cell’s gene expression profile with known human primary cell atlas data and gives the most likely cell identity independently. ‘SingleR’ first corrects for batch effects, then calculates the expression correlation scores for each test cell to each cell type in the reference, and the cell identity is called based on reference cell type exhibiting the highest correlation. Annotations provided by ‘SingleR’ were then abbreviated for clarity.
To validate the labeling provided by singleR, we tested two additional labeling tools: ‘scCATCH’ [11] and ‘Seurat’ [11,19] using the Tabula sapiens data (abbreviated as Seurat/TS). Tabula sapiens is the latest and largest human single-cell reference available to-date [20]. For ‘scCATCH’, the findmarkergenes() function was first used to identify marker genes in the query dataset using the minimum percentage of cell expression cut off of 0.25, minimum log-fold change of 0.25, and p-value of 0.05. We then ran the main labeling function scCATCH() and specified the tissue type as ‘Blood Vessel’, ‘Heart’, ‘Myocardium’, and ‘Serum’.
The CZ Biohub recently released the first comprehensive human single-cell blueprint consisting of high-quality sequencing from 25 organs and eight normal human subjects, and a large portion is dedicated to vasculature tissue [20]. This atlas is extremely helpful in that many reference-based labeling tools lack the necessary reference for correct tissue types. We obtained this dataset and converted it into a Seurat-ready file using the Convert() function in the ‘SeuratDisk’ package. We then used the FindTransferAnchors() function in ‘Seurat V4’ to find the low-dimensional representations of our dataset, then used the TransferData() function to label the test dataset. The annotation provided by ‘Seurat’ and the ‘Tabula sapiens’ dataset were abbreviated for clarity.
2.3. Pseudotemporal ordering and trajectory inference
Pseudotemporal analyses were performed as previously described in the analysis of embryo organogenesis [15]. The processed count matrix was imported into a ‘cds’ object for ‘Monocle3’ using the new_cell_data_set() function, and preprocessed again using the preprocess_cds() function. To have consistent UMAPs across these tools, we used custom scripts to transfer the UMAP calculated in ‘Seurat’ into ‘Monocle3’ before running the main functions ‘learn_graph()’ and ‘order_cells()’ to get trajectory and pseudotime. The starting nodes, or root nodes, were selected in the SMC cluster using an automated function that picks the node most heavily surrounded by ‘early cells. We picked SMCs as the starting cell type based on prior evidence suggesting that SMCs can ‘dedifferentiate’ [5,18]. The SMCs and related clusters were then subsetted for detailed sub-clustering and analysis. For each cluster, Moran’s I statistics were calculated, which identify genes that are differentially expressed along their trajectories. The Moran’s I statistic helped inform the selection of genes to display in Figure 1D. We reviewed the most significant candidates of the Moran’s I calculation and selected 12 to present for clarity. Additionally, we explored 60+ other trajectory inference methods using the ‘Dynverse’ package [25]. We first used guidelines_shiny() function to filter out methods that are not suitable for disconnected topography, as in the case of human tissues where cells are developed from multiple progenitors. We then exported the ‘Seurat’ object into a ‘dyno’ object using the wrap_expression() function. infer_trajectory() function was used to calculate the trajectory based on the chosen algorithm, including ‘SlingShot’ [23], ‘SCROPIUS’ [26], and ‘PAGA’ [24]. Detailed codes to reproduce the figures and custom scripts aforementioned can be found at the Miller Lab Github (see availability of data and materials).
Figure 1.

Unbiased automatic cell labeling of human coronary scRNA data using ‘Seurat v4’ had high concordance with manual labels and pseudotemporal and ultra-fine clustering reveals FB-like cells derivation from SMCs.
(A) Top panel: UMAP clustering of 9798 cells derived from human coronary artery explants with labels based on Seurat v4 using the Tabula sapiens reference; middle panel: automatic labels provided by singleR using Human Primary Cell Atlas; bottom: cluster-based manual annotation provided by original authors Wirka et al. (B) Population breakdown by percentage based on Seurat/TS labels. (C) Left panel: RNA trajectory (line) shows direct paths from the SMCs toward FBs; middle: ultra-fine clustering shows the logical transition stages (microclusters) from SMCs to FBs, and each cluster is numbered for clarity; right: pseudotemporal analysis confirms that the cell and clusters existing along a logical single-cell continuum. (D) Selected genes that were shown to vary over pseudotime by Moran’s I test were visualized. Abbreviations: SMC: smooth muscle cells, EC: endothelial cells, FB: fibroblasts, Mø: macrophages.
2.4. Ligand-receptor cell communication analysis
We analyzed candidate ligand-receptor interactions to infer cell communication using the R package ‘scTalk’, as previously described in the analysis of glial cells [31] and CellChat [32]. For scTalk, we exported statistically significant differentially expressed genes from ‘Seurat’ using the ‘FindMarkers()’ function and imported the preprocessed data. Then, overall edges of the cellular communication network were calculated using the ‘GenerateNetworkPaths()’ function, which reflects the overall ligand-receptor interaction strength between each cell type. Then, the cell types of interest were specified and treeplots were generated using the ‘NetworkTreePlot()’ function. The final figures were re-rendered in BioRender manually for clarity. For CellChat, the processed Seurat object was fed into the ‘createcellchat()’ function and processed using its standard pipeline. Briefly, the Seurat count matrices were extracted along with the Seurat/TS annotation generated previously. CellChatDB.human was loaded and differentially expressed genes and interactions were identified in the CellChat object via identifyOverExpressedGenes() and identifyOverExpressedInteractions(), respectively. The CellChat algorithm was then run to calculate the probable interactions and pathways via computeCommunProb() and computeCommunProbPathway(). We also ran filterCommunication() to filter out interactions with less than 10 cells in each cell type.
2.5. Gene-drug interaction analysis
The above identified ligand and receptor interaction pairs were fed into the Drug-Gene Interaction database (DGIdb 3.0) to reveal candidate drug-gene interactions [37]. Ligands and receptors that were deemed significant from ‘scTalk’ and genes identified in Monocle3 were evaluated using the ‘queryDGIdb()’ function of the ‘rDGIdb’ R package [37,57]. Additionally, we queried CTD2, OMIM, ClinVar, Pharos, GnomAD, and the ExAC databases using the docker-based tool ‘DrugThatGene’ [58]. We included all top FDA-approved drugs and experimental or investigational drugs with verified inhibitory or antagonistic activities, as well as drugs that may influence or be influenced by changes in the receptor. Figures 3A-D were modified using BioRender for clarity.
Figure 3.

Integrative analysis with drug-gene interaction database DGIdb 3.0 reveals potential pharmacological inhibition of SMC and FB signaling and dedifferentiation.
(A) ‘scTalk’ shows that FBs signals to T-cells, pericytes, and SMCs through pro-inflammatory molecules like C3, which can be targeted by the experimental drug compstatin. (B) SMCs interact with FBs through at least four pairs of experimentally verified interactions. Of these, complement and EFEMP1-EGFR signaling is the most druggable as revealed by DGIdb 3.0. (C) Druggable genome analysis revealed agents that can target genes along the SMC-FB de-differentiation trajectory, such as C3, C-XC Motif Chemokine Ligand 12 (CXCL12), and matrix metalloproteinase (MMP2).
2.6. Development of PlaqView
PlaqView is written in R and Shiny, and is hosted on the web at www.plaqview.com using dedicated DC/OS servers at the University of Virginia, but can be run locally through RStudio. The raw data was first processed as previously described, but packaged into .rds objects. The data were placed into a housing directory within the PlaqView application. Custom scripts were written to recall selected datasets on the user-interface and loaded on-demand. Users-inputs were made interactive using Shiny’s reactive scripting practices, and calculations were done ad hoc. Packages such as Seurat, CellChat, and singleR are wrapped and rebuilt during each update in the web server. This application is open-sourced and its code and data is available at github.com/MillerLab-CPHG/PlaqView. Additional datasets are actively being recruited and will be made available in future updates.
3. Results and Discussion
3.1. Unbiased automatic cell labeling is comparable to manual annotation, and reveals abundant cells with chondrocyte and fibroblast characteristics.
Recently, automatic cell identification tools have been implemented to overcome the subjective nature of manual cell-cluster labeling [7]. Here, we compared two popular reference-based cell type annotation methods, ‘SingleR’ [7] and ‘Seurat’ [19], by applying these tools to a previously published human coronary artery scRNA-seq dataset [5], and found that ‘Seurat’ in combination with the Tabula sapiens (TS) reference [20] had the highest concordance with author-supplied manual labels when compared to labels produced by SingleR (Figure 1). For example, smooth muscle cells (SMC), pericytes, endothelial cells, and blood cells such as T-cells, B-cells, and mast cells are labeled in similar clusters. From Seurat/TS, we found that the majority of cell types were fibroblasts (FB, 27%), macrophages (Mø, 26%), endothelial cells (14%), and SMCs (11.5%, Figure 1B). In contrast, more cells were labeled as endothelial cells by both singleR (16.21%, Supplemental Figure 1A) and Wirka et al. (16.3%, Supplemental Figure 1B). Furthermore, Seurat/TS identified 11.5% cells as SMCs, whereas singleR and Wirka et al. found 13.8% and 6.2%, respectively. Although the true population percentage is likely somewhere in-between, this highlights the need to use multiple tools when analyzing single-cell datasets. We also applied an additional reference-based cell annotation tool ‘scCATCH,’ and found that it underperforms relative to Seurat and SingleR, and fails to provide consistent cell type assignment when provided with similar tissue priors (Supplemental Table 1). These and additional labeling tools have been extensively benchmarked elsewhere [21].
Although Seurat/TS did not identify the novel cell state that Wirka et al., refer to as ‘fibromyocytes’, a clear transition from SMCs to fibroblasts (FB) within the overall cluster was noted (Figure 1, top). However, Seurat/TS underperformed in some clusters. For example, SingleR identified neurons/astrocytes and PreB_CD34- cells in concordance with Wirka et al., whereas Seurat/TS labels neurons/astrocytes as Erythrocytes and did not identify the cluster PreB_CD34-. In their original publication, Wirka et al., did not identify clusters 10, 11, 12, 14, and 18 [5]. However, using reference-based annotation, more cells were confidently identified, presumably due to the larger and newer references used and that individual cell expression is not masked by grouping of cells. SingleR identified cells that have osteoblast and chondrocyte (CH)-like cells, whose roles in atherosclerotic plaque stability are under active investigation [18,22]. However, the exact identity and behaviors of these cells in this dataset will require further validation by comparing with other tissues with advanced calcification. In the UMAP clusters reflecting single-cell identities provided by SingleR (Figure 1, middle), there was an infiltration of osteoblast (OS), stem cell (SC), and chondrocyte (CH)-like cells within and next to the SMC cluster. Similar but more transitional cell-mixing is also observed using Seurat/TS (Figure 1). Although UMAP representations often reflect global gene expression similarity, subtle changes in key phenotypic genes that cause cells to appear more similar to CHs or OSs are often difficult to observe in human single-cell datasets. Although the exact consequences of these phenotypic modulations are still under investigation, data from lineage-tracing mouse models suggest that SMCs transition to a panoply of phenotypes such as stem-like and osteogenic phenotypes, and that the osteogenic transcription factor Klf4 contributes to plaque destabilization [18].
3.2. Pseudotemporal ordering identifies distinct fibroblast-like cells originating from SMCs.
To evaluate putative cell fate decisions or differentiation events (e.g., SMC phenotypic transition states), we performed pseudotemporal analysis and ultra-fine clustering using ‘Monocle3’, a method previously applied to normal and diseased states, e.g., embryo organogenesis and response to coronavirus infection, respectively [14,15,17]. We compared over 60 additional trajectory inference (TI) methods including Slingshot [23], PAGA [24], and SCORPIUS using the ‘Dynverse’ package [25,26]. However, we found that most TI algorithms were unsuitable for complex tissue environments such as atherosclerotic plaques due to their inability to distinguish disconnected topologies (Supplemental Table 2). Consistent with Wirka et al., we found that SMCs directly give rise to FB-like cells as revealed by Monocle3 (Figure 1C, left). This corroborates earlier findings showing that SMCs may transition or de-differentiate into ‘fibromyocytes’—SMCs that have undergone a phenotypic modulation to an extracellular matrix producing cell type within the atherosclerotic lesions [5,8]. Although Monocle3 cannot distinguish a clear boundary where SMCs become so-called ‘fibromyocytes’, it infers a possible course by which SMCs transition to FB or FB-like cells through a series of smaller ‘transition states’ or fine-clusters (Figure 1C, middle). Additionally, pseudotemporal analysis using SMCs as a starting point shows clear alignment in the transition pathway, which further strengthens the idea that many of these FB are indeed closely related to SMCs (Figure 1C, right). These findings are consistent with mouse data using lineage tracing to establish that multiple cell types including ECM-producing and fibroblast-like cells are derived from SMCs [18].
Further analysis of the pseudotemporal graph using Moran’s I statistic is performed to identify specific changes associated in this transition pathway. Genes associated with healthy SMC phenotypes, such as MYH11 (a canonical marker of SMC), IGFBP2 (associated with decreased visceral fat), and PPP1R14A (which enhances smooth muscle contraction), are decreased by approximately 50–75% along the SMC trajectory as these cells become more FB-like (Supplemental Table 3, Figure 1D, p < 0.1E-297) [3,27]. Similar results were found by another group using mouse lineage-traced models where MYH11 expression was decreased in SMC-derived modulated “intermediate cell states” [4].
More importantly, specific inflammatory markers and proteins associated with thrombotic events during CAD, including complement proteins C7 and C3, FBLN1, and CXCL12, are increased along the same trajectory (Figure 1D) [2,28]. Recent evidence suggests that CAD-associated CXCL12 secreted from endothelial cells may promote atherosclerosis [29]. Our results point to a potentially new source of CXCL12 that could be targeted to inhibit SMC-to-FB dedifferentiation. Together, and in corroboration of recent studies, our pseudotemporal analysis demonstrates that SMC and modulated, FB-like cells exist within a continuum, with phenotypes toward the FB phenotype associated with worse clinical observations [4,5]. This is further supported by a recent study, in which blocking of SMC-derived intermediate cells coincides with less severe atherosclerotic lesions [4]. Precisely how these modulated cells might influence the overall stability of atherosclerotic lesions and clinical outcomes requires additional longitudinal studies using genetic models and deep phenotyping of human tissues [3,4,8].
3.3. Ligand-receptor analysis shows complex intercellular communications in the human coronary micro-environment
To examine the potential cross-talk between different cell types using scRNA-seq data, we performed cell-to-cell communication analysis using ‘scTalk’, a network-based modeling method that uses confirmed interactions from StringDB [30,31]. The resulting networks are highly dependent on the prior labeling methods (Figure 2A). While FB cells labeled from Seurat/TS were shown to have weak outgoing signaling, FB from manual and singleR labeling have stronger outgoing signals. The strength of the interaction, or path weight, is calculated based on the summed weight of a four-layer node network as described in [31]. Cells that are labeled as osteoblasts from SingleR had significant autocrine signaling as well as outgoing signals, but corresponding cells in the same UMAP regions (e.g., ‘pericyte’ in the manual labels) do not exhibit the same signaling pattern. This further highlights the need to explore various labeling priors as well as develop label-agnostic inference tools.
Figure 2.

Comprehensive ligand-receptor analysis reveals the major inflammatory and immune signaling pathways in FBs.
(A) Circle plots representation of the inferred intercellular communications within the coronary artery environment generated using scTalk when provided with different prior labeling methods. (B) CellChat identifies major signaling pathways involving FBs and major contributions to the activation of these pathways. (C and D) Hierarchy plots showing MHC-I and II signaling among the cell types. Hierarchy plots are useful in examining autocrine signaling or when circle plots are complicated.
Recently, ‘CellChat’ and its companion database were introduced by Jin et al. CellChat infers the probabilities of cell-cell communications using the law of mass action, and is particularly useful in visualizing pathway-specific interactions [32]. We provided CellChat with the labels and produced from Seurat/TS and scRNA-seq count matrices, and discovered several significant pathways: collagen, laminin, fibronectin (FN1), and complement signaling (Figure 2B). In particular, SMCs exert a strong signal in FN1 and collagen signaling, and FB have significant contributions in laminin and complement pathways. Like collagen type IV, laminin isoforms form the basement membrane and have been studied in the context of atherosclerosis, particularly in endothelial cells [33]. However, the exact mechanisms of how FB and FB-like cells contribute to plaque formation and/or stability requires future mechanistic studies.
In addition to complement activation, other immune system involvement is of particular interest in the atherosclerosis field [34]. We examined the overall MHC-I and MHC-II signaling activation within the atherosclerotic environment (Figure 2C-D). We found that almost all cell types exert putative signaling toward NK and T-cells via MHC-I (Figure 2C), principally through HLA-A (Supplemental Figure 2A), whereas high MHC-II class interactions are focused on only macrophages (Figure 2D). While macrophages under basal conditions express low levels of MHC-II molecules [35], our result suggest that a majority of the cells within the lesion may contribute to macrophage activation through interactions such as HLA-DPA1:CD4, HLA-DMB:CD4, and HLA-DRA:CD4 interactions (Supplemental Figure 2B). In fact, many of these MHC-II class molecules, such as HLA-DRA and HLA-DMB, have been shown to have possible prognostic value in identifying atherosclerotic plaque rupture [36].
To identify putative actionable signaling targets, we first evaluated the overall expression and signaling patterns between SMCs, FB, pericytes, and T-cells using singleR, then cross-referenced these interactions against known druggable databases using DGIdb 3.0 [37]. As more cell types are included, the interaction network becomes increasingly complex (Supplemental Figure 2), thus more simplified schematics depicting the overall results are presented (Figure 3A-B). Unlike scCATCH, singleR does not delineate the interactions by specific signaling pathways but rather by cell type-specific expression, which is particularly useful in providing a global overview of signaling for each cell type. We first highlighted the signaling from fibroblast (FB) or FB-like cells to T-cells, pericytes, and SMCs using labels provided by Seurat/TS. These signaling pairs involved various known inflammatory and repair mechanisms like C3 complement, fibulin-1 (FBLN1), and matrix metalloprotease 2 (MMP2, Figure 3A, Supplemental Figure 2). We then examined how SMCs may signal to the other aforementioned cell types (Figure 3B), and we found that COL1A2 and C3 are common ligands used by SMCs and FBs.
3.4. Integrative analysis of cell-cell communication reveals cell-specific druggable targets
To investigate potential pharmacological interventions that may disrupt deleterious intercellular communications, we performed an integrative analysis of cell-cell communication with known druggable genome databases. Key mediators C3, MMP2, and integrins (ITGA1, ITGB1, ITGB5, all expressed by FBs) interact with T-cells, pericytes, and SMCs, and can be disrupted by drugs such as compstatin, tanomastat, SAN-300, volciximab, and cilengitide, respectively (Figure 3A). Similar components, such as collagen (COL1A2) and C3, also appear to be significantly expressed in SMCs that could signal to FBs, pericytes, and T-cells (Figure 3B). Interestingly, multiple studies have linked C3 and the complement system to atherosclerotic lesion maturation in mouse models [38], and a recent case study showed that compstatin-derived C3 inhibitor (AMY-101) may prevent cardiovascular complications in patients with severe COVID-19 pneumonia [39,40]. Further, a recent study demonstrated that microRNA-9 repression of Syndecan-2 (SDC2) impedes atherosclerosis formation [41], while MMP2 alteration also contributes to atherosclerosis in mouse models [42]. Here, we show that SMCs may communicate with FBs within the atherosclerotic environment via SDC2-MMP2, and reveal additional potential upstream candidate drug therapies that may influence atherosclerosis progression (Figure 3A). In general, these results provide a potential mechanistic explanation by which FBs and SMCs can modulate the inflammatory environment and plaque formation.
Surprisingly, anti-EGFR (epidermal growth factor receptor)-based cancer treatments such as erlotinib, cetuximab, and gefitinib were identified as potential key mediators of signaling pathways between SMCs and FBs via EFEMP1 (EGF Containing Fibulin Extracellular Matrix Protein 1) and EGFR (Figure 3B). EFEMP1 has been suggested as a prognostic marker for atherosclerotic plaque rupture [36]. Although the overlap between CAD and cancer etiology has been previously noted, the long-term efficacy and cardiovascular impact of chemotherapy drugs, such as erlotinib, requires further translational studies to investigate their potential use in cancer patients to treat CAD [43–45].
While the exact clinical outcome of SMC de-differentiation is not fully resolved [5,18], such events observed in different single-cell studies present opportunities for pre-clinical pharmacological intervention and testing. Here, we performed integrative analysis of gene expression variation along the SMC-to-FB RNA trajectory with DGIdb. We found that the expression of complement genes such as C3 and C7, and chemokine CXCL12 are increased as SMCs become more FB-like. Although CXCL12 derived from endothelial cells has been recognized to promote atherosclerosis in mouse models [29], here we provide a potentially new source of CXCL12 using human data and found several pharmacological agents such as tinzaparin, an FDA-approved anticoagulant, to investigate in future interventional studies. Together, this combination of cell-cell communication and trajectory analyses reaffirm the druggable potential of these target genes.
3.5. PlaqView is a user-friendly web application to share and explore atherosclerosis-related single-cell datasets
To enable other researchers to explore the transcriptomic landscape of the atherosclerotic environment easily and rapidly, we developed a web interface called PlaqView (www.plaqview.com, Figure 4A). This interactive R- and Shiny-based tool allows for multiple gene queries and comparisons of gene expression, cell-labeling methods such as SingleR, Seurat/TS, and scCATCH, RNA-trajectory tools such as Monocle3 and Slingshot, integrative drug-gene analysis using DGIdb 3.0, and outputs high quality graphs and detailed tables. Calculations are done ad hoc according to users’ input and the application has been optimized for rapid exploration of these datasets by academic researchers, clinicians, and lay scientists. To our knowledge, there are no publicly available tools to visualize atherosclerosis-related single-cell datasets without prior coding knowledge. Further, PlaqView is under active development and will be releasing new datasets coincidently with future atherosclerosis-related publications. At the time of writing, three human and four mouse datasets from four independent studies are available in PlaqView (Supplemental Table 4). PlaqView is also open-source and is fully maintained at https://github.com/MillerLab-CPHG/PlaqView. We have written PlaqView to be easily repurposed for other single-cell studies; all description files are written in basic R markdown, and the data preprocessing pipeline can be adapted to any tissue from mouse or human. As the database in PlaqView expands along with the growing number of single-cell datasets, we anticipate that it will become an essential tool for the atherosclerosis research field. Furthermore, PlaqView will serve as a template for other fields of single-cell biology to rapidly disseminate relevant and cutting-edge data with minimal web-development skills required.
Figure 4.

PlaqView and its analysis pipelines are reproducible, user-friendly tools for single-cell data analysis and presentation.
(A) Screenshot of the PlaqView web application. (B) A roadmap of enhanced scRNA-seq analysis steps. Instead of cluster-based grouping, our pipeline uses automatic cell labeling, coupled with pseudotime trajectory, cellular network interaction and drug targeting, and provides a reproducible process for scRNA datasets. From this roadmap, it is easy to add additional analysis tools and modify workflow as more tools and datasets become available.
3.6. Limitations
Despite the advances presented in this workflow, we are still working to improve several limitations. For instance, the default reference in ‘SingleR’ and Tabula sapiens [20] do not contain more recently discovered cellular phenotypes such as ‘fibromyocytes’ [5,7,29,46], which may require a combination of manual and automated labeling methods for accurate identification. Factors such as the intrinsic heterogeneity of the tissue sample, disease stage, and tissue processing artifacts are difficult to isolate computationally and could influence automated labeling methods. Our analysis also demonstrates that upstream analysis such as cell type annotation greatly affects the outcome of downstream results, such as cell-cell communication inference. Nonetheless, interactive viewers and tools such as PlaqView, which incorporate multiple methods and visualizations in one location, could help users separate out the technical and biological variation from various single-cell datasets. Additionally, the modular nature of PlaqView will also allow for future improvement of labeling methods as more precise reference datasets are made available.
While we cannot verify the directionality of the RNA trajectories presented, future releases of PlaqView will feature RNA ‘velocity’ as described in [13] where directionality will be calculated using splice/unspliced RNA ratios from raw data. Lastly, the true efficacy of proposed drugs cannot be verified without extensive pre-clinical testing and clinical trials. Still, these findings may catalyze future investigative efforts to develop more targeted therapies.
3.7. Conclusions
Our findings show that an enhanced, reproducible pipeline for scRNA-seq analysis has the potential to improve upon current standard scRNA-seq bioinformatics protocols. For instance, we provide new insights into intricate vascular cell differentiation and communication pathways while providing actionable and testable targets for future experimental studies (Figure 4B). Our combined analysis suggests that SMCs give rise to a population of FBs that express genetic signatures associated with inflammation and extracellular matrix degradation. For example, our cell-cell communication analysis suggests that SMCs signal to FBs via inflammatory molecules like C3 complement and MMP2, whose expression increases along the SMC-to-FB trajectory. We revealed possible therapeutic avenues that may disrupt these cell-to-cell communications and alter the atherosclerotic pathology. Furthermore, several FDA-approved drugs (e.g., erlotinib, cetuximab, and gefitinib) were shown as potential effectors of SMC signaling to FB, and merit molecular studies to determine whether they may be used to treat CAD in cancer patients to simplify or augment current drug regimens [43]. This is consistent with recent reports showing beneficial effects of the acute promyelocytic drug all-trans-retinoic acid (ATRA) in atherosclerosis mouse models [4]. Further investment in scRNA-seq may also help resolve the balance of anti-tumor efficacy and atheroprotection for immune checkpoint inhibitors as well as immunomodulators at the interface of cardio-immuno-oncology [47].
Although the utilization of this workflow can compensate for many of the shortcomings of current scRNA-seq analyses, we are still unable to perform cell-lineage tracing that reflects actual timescales without additional gene engineering experiments in vivo [48]. However, leveraging mitochondrial DNA variants in snATAC-seq data has enabled lineage tracing analysis in human cells [49,50]. Likewise, these analyses can ultimately be extended to integrate spatial omics and other multi-modal data [19]. As spatial transcriptomics, scATAC-seq, and/or CITE-seq data become more widely available, this workflow can be easily modified to discover signaling pathways or differentiation events at specific tissue locations and timepoints, allowing for more disease-relevant drug-gene interaction analyses (Figure 4B). Nonetheless, this pipeline can be applied immediately to datasets from other tissues or diseases to generate informative directions for follow-up studies, and is more user-friendly and reproducible compared to standard scRNA analyses. PlaqView serves as a central repository for interactive analysis and exploration for atherosclerosis-related single-cell datasets. As PlaqView incorporates additional relevant single-cell datasets, we anticipate that this application will become an indispensable resource for the community and for the growing field of ‘cardioinformatics’ [51–53]. Most importantly, web applications such as PlaqView democratize the access and analysis of single-cell data, which will promote collaboration, reproducibility, and innovation across disciplines [54,55].
Supplementary Material
Supplemental Figure 1. (A) Population percentage when cells are labeled using singleR. (B) Population when using author-provided labels.
Supplemental Figure 2. Comprehensive ligand-receptor pairs as revealed by ‘scTalk’ when provided with Seurat/TS or SingleR labels. Top: signaling from fibroblasts to macrophages, pericytes, smooth muscle cells, T-cells, or natural killer cells. Middle: signaling from smooth muscle cells. Bottom: signaling from smooth muscle cells but with labels from SingleR. Simplified figures of A and B were presented in Figure 3 in the main text.
Supplemental Figure 3. Detailed contributions of MHC-I and MHC-II pairs from Figure 3C-D calculated using CellChat.
Automated cell annotation and pseudotemporal analyses show key changes in gene expression during smooth muscle cell (SMC) de-differentiation.
Ligand-receptor profiling reveals potential drug targets that modulate SMC and fibroblast signaling and gene expression responses during atherosclerosis.
Reproducible analysis template allows for detailed examination of scRNA-seq datasets with minimal coding and eliminates some user-induced bias.
PlaqView web application allows for interactive analysis of human and mouse scRNA-seq datasets without prior coding knowledge and facilitates sharing and reanalysis of valuable data.
Acknowledgements
The authors acknowledge Research Computing at The University of Virginia for providing computational resources and technical support that have contributed to the results reported within this publication. URL: https://rc.virginia.edu. The authors also acknowledge Robert Wirka and Thomas Quertermous at Stanford University for providing more details on the coronary artery dataset.
Fuinancial support
Funding support was provided by grants from the National Institutes of Health (NIH): R00HL125912 (CLM), R01HL148239 (CLM), K12HL143959 (BBK) and T32HL007284 (CJH and DW); American Heart Association (AHA): POST35120545 (AWT) and Leducq Foundation Transatlantic Network of Excellence (‘PlaqOmics’) (CLM, AWT, GKO, GFA, SWvL, GP, MM).
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Declaration of competing interests
BBK is the founder of Dock Therapeutics, Inc. The remaining authors have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z, Vascular smooth muscle cells in atherosclerosis, Nat Rev Cardiol. 16 (2019) 727–744. 10.1038/s41569-019-0227-9. [DOI] [PubMed] [Google Scholar]
- [2].Argraves WS, Tanaka A, Smith EP, Twal WO, Argraves KM, Fan D, Haudenschild CC, Fibulin-1 and fibrinogen in human atherosclerotic lesions, Histochem Cell Biol. 132 (2009) 559–565. 10.1007/s00418-009-0628-7. [DOI] [PubMed] [Google Scholar]
- [3].Bennett MR, Sinha S, Owens GK, Vascular Smooth Muscle Cells in Atherosclerosis., Circ Res. 118 (2016) 692––702. 10.1161/circresaha.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pan H, Xue C, Auerbach BJ, Fan J, Bashore AC, Cui J, Yang DY, Trignano SB, Liu W, Shi J, Ihuegbu CO, Bush EC, Worley J, Vlahos L, Laise P, Solomon RA, Connolly ES, Califano A, Sims PA, Zhang H, Li M, Reilly MP, Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human, Circulation. (2020). 10.1161/circulationaha.120.048378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, Kundu R, Nagao M, Coller J, Koyano TK, Fong R, Woo YJ, Liu B, Montgomery SB, Wu JC, Zhu K, Chang R, Alamprese M, Tallquist MD, Kim JB, Quertermous T, Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis, Nat Med. 25 (2019) 1280––1289. 10.1038/s41591-019-0512-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yifan C, Fan Y, Jun P, Visualization of cardiovascular development, physiology and disease at the single-cell level: Opportunities and future challenges, J Mol Cell Cardiol. 142 (2020) 80–92. 10.1016/j.yjmcc.2020.03.005. [DOI] [PubMed] [Google Scholar]
- [7].Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ, Bhattacharya M, Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage, Nat Immunol. 20 (2019) 163–172. 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang Y, Nanda V, Direnzo D, Ye J, Xiao S, Kojima Y, Howe KL, Jarr K-U, Flores AM, Tsantilas P, Tsao N, Rao A, Newman AAC, Eberhard AV, Priest JR, Ruusalepp A, Pasterkamp G, Maegdefessel L, Miller CL, Lind L, Koplev S, Björkegren JLM, Owens GK, Ingelsson E, Weissman IL, Leeper NJ, Clonally expanding smooth muscle cells promote atherosclerosis by escaping efferocytosis and activating the complement cascade., P Natl Acad Sci Usa. 117 (2020) 15818–15826. 10.1073/pnas.2006348117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].DiRenzo D, Owens GK, Leeper NJ, “Attack of the Clones,” Circ Res. 120 (2017) 624––626. 10.1161/circresaha.116.310091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pliner HA, Shendure J, Trapnell C, Supervised classification enables rapid annotation of cell atlases, Nat Methods. 16 (2019) 983–986. 10.1038/s41592-019-0535-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shao X, Liao J, Lu X, Xue R, Ai N, Fan X, scCATCH: automatic annotation on cell-types of clusters from single-cell RNA-seq data, Iscience. 23 (2020) 100882. 10.1016/j.isci.2020.100882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zagar M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LB, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R, Integrated analysis of multimodal single-cell data, Biorxiv. (2020) 2020.10.12.335331. 10.1101/2020.10.12.335331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ, Generalizing RNA velocity to transient cell states through dynamical modeling, Biorxiv. (2019) 820936. 10.1101/820936. [DOI] [PubMed] [Google Scholar]
- [14].Cao J, O’Day DR, Pliner HA, Kingsley PD, Deng M, Daza RM, Zager MA, Aldinger KA, Blecher-Gonen R, Zhang F, Spielmann M, Palis J, Doherty D, Steemers FJ, Glass IA, Trapnell C, Shendure J, A human cell atlas of fetal gene expression, Science. 370 (2020) eaba7721. 10.1126/science.aba7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, Trapnell C, Shendure J, The single-cell transcriptional landscape of mammalian organogenesis, Nature. 566 (2019) 496–502. 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Manno GL, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, Fan J, Borm LE, Liu Z, van Bruggen D, Guo J, He X, Barker R, Sundström E, Castelo-Branco G, Cramer P, Adameyko I, Linnarsson S, Kharchenko PV, RNA velocity of single cells, Nature. 560 (2018) 494––498. 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F, Debnath O, Thürmann L, Kurth F, Völker MT, Kazmierski J, Timmermann B, Twardziok S, Schneider S, Machleidt F, Müller-Redetzky H, Maier M, Krannich A, Schmidt S, Balzer F, Liebig J, Loske J, Suttorp N, Eils J, Ishaque N, Liebert UG, von Kalle C, Hocke A, Witzenrath M, Goffinet C, Drosten C, Laudi S, Lehmann I, Conrad C, Sander L-E, Eils R, COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis, Nat Biotechnol. (2020) 1–10. 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- [18].Alencar GF, Owsiany KM, Sukhavasi SK,K, Mocci G, Nguyen A, Williams CM, Shamsuzzaman S, Mokry M, Henderson CA, Haskins R, Baylis RA, Finn AV, McNamara CA, Zunder ER, Venkata V, Pasterkamp G, Björkegren J, Bekiranov S, Owens GK, The Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis, Circulation. (2020). 10.1161/circulationaha.120.046672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data, Cell. 177 (2019) 1888–1902.e21. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Consortium TTS, Quake SR, The Tabula Sapiens: a single cell transcriptomic atlas of multiple organs from individual human donors, Biorxiv. (2021) 2021.07.19.452956. 10.1101/2021.07.19.452956. [DOI] [Google Scholar]
- [21].Huang Q, Liu Y, Du Y, Garmire LX, Evaluation of Cell Type Annotation R Packages on Single-cell RNA-seq Data, Genom Proteom Bioinform. (2020). 10.1016/j.gpb.2020.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Perisic L, Aldi S, Sun Y, Folkersen L, Razuvaev A, Roy J, Lengquist M, Åkesson S, Wheelock CE, Maegdefessel L, Gabrielsen A, Odeberg J, Hansson GK, Paulsson-Berne G, Hedin U, Gene expression signatures, pathways and networks in carotid atherosclerosis., J Intern Med. 279 (2016) 293––308. 10.1111/joim.12448. [DOI] [PubMed] [Google Scholar]
- [23].Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, Dudoit S, Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics, Bmc Genomics. 19 (2018) 477. 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wolf FA, Hamey FK, Plass M, Solana J, Dahlin JS, Göttgens B, Rajewsky N, Simon L, Theis FJ, PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells, Genome Biol. 20 (2019) 59. 10.1186/s13059-019-1663-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Saelens W, Cannoodt R, Todorov H, Saeys Y, A comparison of single-cell trajectory inference methods, Nat Biotechnol. 37 (2019) 547–554. 10.1038/s41587-019-0071-9. [DOI] [PubMed] [Google Scholar]
- [26].Cannoodt R, Saelens W, Sichien D, Tavernier S, Janssens S, Guilliams M, Lambrecht B, Preter KD, Saeys Y, SCORPIUS improves trajectory inference and identifies novel modules in dendritic cell development, Biorxiv. (2016) 079509. 10.1101/079509. [DOI] [Google Scholar]
- [27].Carter S, Lemieux I, Li Z, Alméras N, Tremblay A, Bergeron J, Poirier P, Després J-P, Picard F, Changes in IGFBP-2 levels following a one-year lifestyle modification program are independently related to improvements in plasma apo B and LDL apo B levels, Atherosclerosis. 281 (2018) 89–97. 10.1016/j.atherosclerosis.2018.12.016. [DOI] [PubMed] [Google Scholar]
- [28].Carter AM, Complement Activation: An Emerging Player in the Pathogenesis of Cardiovascular Disease, Sci. 2012 (2012) 1–14. 10.6064/2012/402783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Döring Y, van der Vorst EPC, Duchene J, Jansen Y, Gencer S, Bidzhekov K, Atzler D, Santovito D, Rader DJ, Saleheen D, Weber C, CXCL12 Derived From Endothelial Cells Promotes Atherosclerosis to Drive Coronary Artery Disease, Circulation. 139 (2019) 1338–1340. 10.1161/circulationaha.118.037953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, von Mering C, The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible, Nucleic Acids Res. 45 (2017) D362–D368. 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, Ho JW, Nordon RE, Harvey RP, Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury, Elife. 8 (2019) e43882. 10.7554/elife.43882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan C-H, Myung P, Plikus MV, Nie Q, Inference and analysis of cell-cell communication using CellChat, Nat Commun. 12 (2021) 1088. 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yang T-L, Lee P-L, Lee D-Y, Wang W-L, Wei S-Y, Lee C-I, Chiu J-J, Differential regulations of fibronectin and laminin in Smad2 activation in vascular endothelial cells in response to disturbed flow, J Biomed Sci. 25 (2018) 1. 10.1186/s12929-017-0402-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wolf D, Ley K, Immunity and Inflammation in Atherosclerosis, Circ Res. 124 (2019) 315–327. 10.1161/circresaha.118.313591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Xaus J, Comalada M, Barrachina M, Herrero C, Goñalons E, Soler C, Lloberas J, Celada A, The Expression of MHC Class II Genes in Macrophages Is Cell Cycle Dependent, J Immunol. 165 (2000) 6364–6371. 10.4049/jimmunol.165.11.6364. [DOI] [PubMed] [Google Scholar]
- [36].Xu B-F, Liu R, Huang C-X, He B-S, Li G-Y, Sun H-S, Feng Z-P, Bao M-H, Identification of key genes in ruptured atherosclerotic plaques by weighted gene correlation network analysis, Sci Rep-Uk. 10 (2020) 10847. 10.1038/s41598-020-67114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cotto KC, Wagner AH, Feng Y-Y, Kiwala S, Coffman AC, Spies G, Wollam A, Spies NC, Griffith OL, Griffith M, DGIdb 3.0: a redesign and expansion of the drug–gene interaction database, Nucleic Acids Res. 46 (2017) D1068–D1073. 10.1093/nar/gkx1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Buono C, Come CE, Witztum JL, Maguire GF, Connelly PW, Carroll M, Lichtman AH, Influence of C3 Deficiency on Atherosclerosis, Circulation. 105 (2002) 3025–3031. 10.1161/01.cir.0000019584.04929.83. [DOI] [PubMed] [Google Scholar]
- [39].Mastaglio S, Ruggeri A, Risitano AM, Angelillo P, Yancopoulou D, Mastellos DC, Huber-Lang M, Piemontese S, Assanelli A, Garlanda C, Lambris JD, Ciceri F, The first case of COVID-19 treated with the complement C3 inhibitor AMY-101, Clin Immunol. 215 (2020) 108450. 10.1016/j.clim.2020.108450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Java A, Apicelli AJ, Liszewski MK, Coler-Reilly A, Atkinson JP, Kim AHJ, Kulkarni HS, The complement system in COVID-19: friend and foe?, Jci Insight. 5 (2020). 10.1172/jci.insight.140711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang R, Song B, Hong X, Shen Z, Sui L, Wang S, microRNA-9 Inhibits Vulnerable Plaque Formation and Vascular Remodeling via Suppression of the SDC2-Dependent FAK/ERK Signaling Pathway in Mice With Atherosclerosis, Front Physiol. 11 (2020) 804. 10.3389/fphys.2020.00804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kuzuya M, Nakamura K, Sasaki T, Cheng XW, Itohara S, Iguchi A, Effect of MMP-2 Deficiency on Atherosclerotic Lesion Formation in ApoE-Deficient Mice, Arteriosclerosis Thrombosis Vasc Biology. 26 (2006) 1120–1125. 10.1161/01.atv.0000218496.60097.e0. [DOI] [PubMed] [Google Scholar]
- [43].Camaré C, Pucelle M, Nègre-Salvayre A, Salvayre R, Angiogenesis in the atherosclerotic plaque., Redox Biol 12 (2017) 18––34. 10.1016/j.redox.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tapia-Vieyra JV, Delgado-Coello B, Mas-Oliva J, Atherosclerosis and Cancer; A Resemblance with Far-reaching Implications, Arch Med Res. 48 (2017) 12–26. 10.1016/j.arcmed.2017.03.005. [DOI] [PubMed] [Google Scholar]
- [45].Libby P, Hansson GK, Taming Immune and Inflammatory Responses to Treat Atherosclerosis *, J Am Coll Cardiol. 71 (2018) 173–176. 10.1016/j.jacc.2017.10.081. [DOI] [PubMed] [Google Scholar]
- [46].Mabbott NA, Baillie JK, Brown H, Freeman TC, Hume DA, An expression atlas of human primary cells: inference of gene function from coexpression networks, Bmc Genomics. 14 (2013) 632. 10.1186/1471-2164-14-632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zaha VG, Meijers WC, Moslehi J, Cardio-Immuno-Oncology, Circulation. 141 (2020) 87–89. 10.1161/circulationaha.119.042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liu M, Gomez D, Smooth Muscle Cell Phenotypic Diversity: At the Crossroads of Lineage Tracing and Single-Cell Transcriptomics, Arteriosclerosis Thrombosis Vasc Biology. 39 (2019) 1715–1723. 10.1161/atvbaha.119.312131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lareau CA, Ludwig LS, Muus C, Gohil SH, Zhao T, Chiang Z, Pelka K, Verboon JM, Luo W, Christian E, Rosebrock D, Getz G, Boland GM, Chen F, Buenrostro JD, Hacohen N, Wu CJ, Aryee MJ, Regev A, Sankaran VG, Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling, Nat Biotechnol. (2020) 1–11. 10.1038/s41587-020-0645-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Xu J, Nuno K, Litzenburger UM, Qi Y, Corces MR, Majeti R, Chang HY, Single-cell lineage tracing by endogenous mutations enriched in transposase accessible mitochondrial DNA, Elife. 8 (2019) e45105. 10.7554/elife.45105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Khomtchouk BB, Tran D-T, Vand KA, Might M, Gozani O, Assimes TL, Cardioinformatics: the nexus of bioinformatics and precision cardiology, Brief Bioinform. 21 (2019) 2031–2051. 10.1093/bib/bbz119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Khomtchouk BB, Nelson CS, Vand KA, Palmisano S, Grossman RL, HeartBioPortal2.0: new developments and updates for genetic ancestry and cardiometabolic quantitative traits in diverse human populations, Database. 2020 (2020) baaa115-. 10.1093/database/baaa115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Khomtchouk BB, Vand KA, Koehler WC, Tran D-T, Middlebrook K, Sudhakaran S, Nelson CS, Gozani O, Assimes TL, HeartBioPortal: An Internet-of-Omics for Human Cardiovascular Disease Data, Circulation Genom Precis Medicine. 12 (2019) e002426. 10.1161/circgen.118.002426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N, Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal, Sci Signal. 6 (2013) pl1–pl1. 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Skinnider MA, Squair JW, Courtine G, Enabling reproducible re-analysis of single-cell data, Genome Biol. 22 (2021) 215. 10.1186/s13059-021-02422-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species, Nat Biotechnol. 36 (2018) 411––420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Griffith M, Griffith OL, Coffman AC, Weible JV, McMichael JF, Spies NC, Koval J, Das I, Callaway MB, Eldred JM, Miller CA, Subramanian J, Govindan R, Kumar RD, Bose R, Ding L, Walker JR, Larson DE, Dooling DJ, Smith SM, Ley TJ, Mardis ER, Wilson RK, DGIdb: mining the druggable genome, Nat Methods. 10 (2013) 1209–1210. 10.1038/nmeth.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Canver MC, Bauer DE, Maeda T, Pinello L, DrugThatGene: integrative analysis to streamline the identification of druggable genes, pathways and protein complexes from CRISPR screens, Bioinformatics. 35 (2018) 1981–1984. 10.1093/bioinformatics/bty913. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. (A) Population percentage when cells are labeled using singleR. (B) Population when using author-provided labels.
Supplemental Figure 2. Comprehensive ligand-receptor pairs as revealed by ‘scTalk’ when provided with Seurat/TS or SingleR labels. Top: signaling from fibroblasts to macrophages, pericytes, smooth muscle cells, T-cells, or natural killer cells. Middle: signaling from smooth muscle cells. Bottom: signaling from smooth muscle cells but with labels from SingleR. Simplified figures of A and B were presented in Figure 3 in the main text.
Supplemental Figure 3. Detailed contributions of MHC-I and MHC-II pairs from Figure 3C-D calculated using CellChat.