

SUMMARY
By combining 6 druggable genome resources, we identify 6,083 genes as potential druggable genes (PDGs). We characterize their expression, recurrent genomic alterations, cancer dependencies, and therapeutic potentials by integrating genome, functionome, and druggome profiles across cancers. 81.5% of PDGs are reliably expressed in major adult cancers, 46.9% show selective expression patterns, and 39.1% exhibit at least one recurrent genomic alteration. We annotate a total of 784 PDGs as dependent genes for cancer cell growth. We further quantify 16 cancer-related features and estimate a PDG cancer drug target score (PCDT score). PDGs with higher PCDT scores are significantly enriched for genes encoding kinases and histone modification enzymes. Importantly, we find that a considerable portion of high PCDT score PDGs are understudied genes, providing unexplored opportunities for drug development in oncology. By integrating the druggable genome and the cancer genome, our study thus generates a comprehensive blueprint of potential druggable genes across cancers.
Graphical Abstract
In brief
Jiang et al. generate a comprehensive blueprint of potential druggable genes (PDGs) across cancers by a systematic integration of the druggable genome and the cancer genome. This resource is publicly available to the cancer research community in The Cancer Druggable Gene Atlas (TCDA) through the Functional Cancer Genome data portal.
INTRODUCTION
The “druggable genome,” a term coined by Hopkins and Groom (2002), defines a group of genes in our genome-encoding proteins that may be modulated by drug-like molecules. A large proportion of successful small-molecule drugs achieve their activity by competing for a binding site on the target protein with an endogenous biological molecule in cells. Sequence/structure similarities in the conserved domains of a family of proteins are usually indicative of a general conservation of binding site architecture. This would suggest that, if one member of a protein family can be modulated by a small molecule, the other members may also be modulated by compounds with appropriate pharmacological properties (Hopkins and Groom, 2002). Therefore, by analyzing the sequences of drug-binding domains of known drug target proteins, researchers are able to predict potential druggable protein families containing the same domains (Hopkins and Groom, 2002). Using this rationale, the druggable genome has been defined by multiple approaches on a whole-genome-wide scale (Brown et al., 2018; Campbell et al., 2012; Finan et al., 2017; Hopkins and Groom, 2002; Kumar et al., 2013; Overington et al., 2006; Rask-Andersen et al., 2011, 2014; Russ and Lampel, 2005; Southan et al., 2015). Excitingly, many efforts have been launched recently to improve scientific understanding of these putative druggable genes, such as the Illuminating the Druggable Genome (IDG) project by NIH. However, current drug development in oncology is still narrowly focused on a relatively small proportion of genes, due to challenges in target identification and prioritization. Strategies to identify potential targets of small-molecule compounds for cancer therapy are mostly driven by possibilities from a medical chemistry viewpoint rather than by cancer genomic or functional profiles. During the last decade, cancer genomes have been comprehensively characterized by high-throughput profiling technologies in large sample cohorts. Characterization of recurrent genomic alterations has provided a power tool for identification and prioritization of drug targets in oncology (Bailey et al., 2018; Beroukhim et al., 2010; Garraway and Lander, 2013; Kandoth et al., 2013; Lawrence et al., 2013; Sanchez-Vega et al., 2018; Vogelstein et al., 2013; Yuan et al., 2014; Zack et al., 2013). Meanwhile, recent advances in genome-wide loss-of-function genetic screenings in large-scale cancer cell lines have also provided rich functional information for mapping cancer dependency and prioritizing potential therapeutic targets (Behan et al., 2019; Tsherniak et al., 2017). Therefore, we propose that integrated analysis of gene expression, recurrent genomic alterations, and cancer dependencies for putative druggable genes across cancers can systematically identify and prioritize potential therapeutic targets for treatment of cancer.
RESULTS
Definition of potentially druggable genes in the human genome
To define potentially druggable genes (PDGs) in the human genome, we integrated the PDG candidates generated by six independent studies in which the druggable genes were systematically annotated by different strategies at a whole-genome level (Brown et al., 2018; Finan et al., 2017; Hopkins and Groom, 2002; Kumar et al., 2013; Russ and Lampel, 2005; Southan et al., 2015). We initially identified 11,280 genes that were predicted to be PDGs by at least one of the PDG sources (Figure 1A). Among them, 714 genes were annotated by all 6 studies, while 5,445 genes were only reported by one source (Figure 1B). The most recent database on the druggable genome, Open Targets (Brown et al., 2018), contributed the largest numbers of those unique PDGs (n = 4,923) (Figure 1C), suggesting that the number of predicted PDGs was remarkably increased during the last decade. In this study, we defined genes annotated by at least two sources as PDGs. Notably, if a gene is defined as a PDG, this does not necessarily mean that its protein product has been successfully targeted in the clinic. For example, plenty of pharmacological approaches have been developed to target mutant p53 or restore wild-type p53 (Vassilev et al., 2004); thus, TP53 was defined as a PDG. However, most of these approaches have failed in early clinical development. Second, a small portion of genes that are not able to be directly targeted may also be defined as PDGs, due to pharmacological strategies that target their associated protein complexes. A typical example is represented by genes in the cyclin family, such as CCND1 and CCNE1. To be consistent with the current definition for PDGs, we included the above two classes of genes as PDGs if they were annotated by more than one source. Finally, given rapid advances in development of epigenetic drugs (i.e., small-molecule compounds directly modulate histone modification enzymes [HMEs]) in oncology, HMEs were included in the PDG list. Taken together, a total of 6,083 PDGs were analyzed in this study (Table S1). Using the information from the Target Center Resource Database (Lin et al., 2017; Nguyen et al., 2017), we analyzed the gene family categories and the target development levels (TDLs) of the above PDGs. Consistent with previous reports, a large percentage of PDGs fell into four well-known PDG families: kinases (8.9%), G-protein-coupled receptors (GPCRs) (12.9%), ion channels (ICs) (4.5%), and nuclear hormone receptors (NRs) (0.8%). PDGs belonging to these four well-known PDG families were commonly shared by all six sources (Figures 1C and 1D). Consistent with their percentage in the human genome (20.7%), enzymes make up the largest gene function category (24.8% of PDGs), representing 1,507 genes encoding enzymes. In contrast, although there are 1,396 transcription factors (TFs) in our genome, only 40 transcription factors were defined as PDGs (druggable TFs, 0.7% of PDGs), indicating that targeting TFs remains challenging in drug development. Notably, although 9.2% and 20.6% of the PDGs were targeted by approved drugs (TDL: Tclin) and small molecules that satisfy the activity thresholds (TDL: Tchem), respectively, the majority of the PDGs (53.4%) still lack chemical compounds to manipulate their functions (Figures 1D and 1E). Importantly, 15.9% of them were defined as TDL Tdark (i.e., their biological functions were still unknown). Consistently, when analyzing the related publications for each PDG, we found that a majority of PDGs (69.2%) were understudied genes (PubTator score < 150) based on their PubTator scores (Wei et al., 2019) (Figure 1E). For example, among the 4 major druggable families, 339 genes were defined as understudied genes by IDG.
Figure 1. Definition of PDGs in the human genome.

(A) Venn diagram shows the gene numbers of six resources. Size of circles: the gene numbers in each dataset.
(B) Venn diagram shows the numbers of PDGs that overlap among the six resources. From the inner to the outer circles, the diagrams represent the numbers of the PDGs shared by six (n = 714), five (n = 698), four (n = 1,030), three (n = 755), and two (n = 2,638) datasets, respectively.
(C) Heatmap shows the similarity among the six resources, which were ordered by unsupervised clustering. The core gene families contributed a considerable number of overlapping PDGs.
(D) Classification of PDGs based on gene family category (left) and target development level (TDL) (right).
(E) River plot shows the relationships among gene family category, TDL, and PubTator scores of the PDGs. The width of the bar is proportional to the number of PDGs in each category.
Expression of the PDGs across cancers
The RNA sequencing profiles were retrieved from the GTEx and TCGA (Figure S1; Tables S2 and S3). We found that 81.5% of PDGs (n = 4,957) were expressed in cancers, while 18.5% (n = 1,126) were defined as undetectable genes (Figure 2A). Among the expressed PDGs, 34.6% (n = 2,107) exhibited a ubiquitous expression pattern across cancers. Notably, 46.9% (n = 2,850) of PDGs were only detectable in a portion of tumor specimens across cancers (defined as selectively expressed). As expected, genes targeted by FDA-approved cancer therapy drugs were significantly enriched in this selective expression group (p = 0.018, OR = 1.6). We were able to further classify the selectively expressed PDGs into four categories based on their expressional distribution (Figures 2A and 2B; Table S4). Across cancers, a total of 2,099 PDGs showed a lineage-enriched expression pattern, and a median of 86 lineage-enriched PDGs were identified in each tumor type (Figure 2C). Most druggable gene families were widely expressed in cancers, although the GPCRs and ICs had significantly larger portions (67.6% and 23.1%, respectively) defined as unexpressed (Figure 2D). The HMEs and druggable TFs showed higher percentages of ubiquitous expression, while the expression patterns of the ICs, NHRs, and transporters were more selective. To unbiasedly and systematically identify the PDGs that are highly expressed in cancers (caPDGs), we generated an expressional score for each PDG by comparing its RNA expression level in a certain tumor type (TCGA cohort) to all normal tissue specimens (GTEx cohort) using a computational strategy recently developed by the Functional Cancer Genome (FCG) project (Hu et al., 2021) (Figure 2E). We identified a total of 697 caPDGs, which were relatively highly expressed in at least one cancer type (a median of 28 caPDGs for each cancer type) (Figures 2C and 2F). Based on their expressional scores, we further classified the caPDGs into three tiers (i.e., high, moderate, and low confidence). Notably, although the majority of the caPDGs were identified in a single cancer type, we found that 29.6% (206/697) of caPDGs were shared by more than one tumor type (Figure 2F), suggesting that these caPDGs may be upregulated by common tumorigenic signals. Taken together, a large portion of PDGs were expressed in cancer and many of them showed selective expression patterns or relatively higher expression in cancer, thus providing potential therapeutic windows for cancer drug development.
Figure 2. Expression of PDGs across cancers.

(A) Mosaic plots show the classification of the PDGs based on their expression patterns.
(B) Expressional distribution of typical examples of selectively expressed PDGs across cancers. Cancer type of each sample in the density plots is indicated by color code under the plots.
(C) Summary of the numbers of lineage-enriched PDGs and PDGs with relatively higher expression in cancer (caPDGs) in each cancer type. Size of circles: number of genes. Orange, lineage-enriched PDGs; red, caPDGs.
(D) Percentage of genes in different expression categories for each gene family.
(E) Workflow of identifying caPDGs. Five principally different computational strategies were applied to identify caPDGs.
(F) Expression levels of typical examples of identified caPDGs across normal and tumor specimens. Cancer types in which the caPDGs were identified are labeled by colors. Based on specificity scores, the identified potential caPDGs were classified into three tiers (high, moderately, and low confident).
Somatic copy number alterations of the PDGs across cancers
We identified cancer-associated PDGs driven by recurrent focal somatic copy number alterations (SCNAs) in each cancer type using a computational strategy recently developed by the FCG (Hu et al., 2021) (Figure 3A). After initially identifying 35,697 (8,705 amplification and 26,992 deletion) recurrent focal SCNA events harboring PDGs across 33 tumor types, a G score was estimated for each PDG located in recurrent SCNA loci. We removed the non-detectable PDGs, then analyzed the correlations between RNA expression and DNA copy number, and a positive and significant correlation was observed for 50.7% (4,539/8,949) of PDG SCNA events identified in the first 3 steps. Collectively, across the TCGA cohort, we identified 1,993 PDGs that met all 4 criteria in at least one tumor type. To estimate the SCNAs for PDGs at a pan-cancer level, we also calculated an overall G score (Table S5). Across 33 cancer types, 464 PDGs showed an overall G score above a cutoff (Figure 3B). The most well-known SCNA-driven targets with FDA-approved drugs were successfully identified and prioritized as the top rank (Figures 3C and S2). Importantly, after the PDGs were classified to eight categories based on their functions, we observed that kinases and HMEs were significantly enriched in the PDGs with recurrent SCNAs, while GPCRs and ICs were significantly enriched in the PDGs without recurrent SCNAs (Figures 3D and S3). This indicates that a large portion of kinases and HMEs may play “driver” roles in tumorigenesis, thereby serving as promising anticancer drug targets. Notably, HMEs were the most significantly altered gene class among the PDGs in both copy number gains and losses, indicating that epigenetic dysregulation may serve as one of the major vulnerabilities in cancer for treatment. Finally, we analyzed the TDL for each PDG estimated by the IDG and found that more than half of the SCNA-driven PDGs (61.0%) were classified as Tbio or Tdark (Figure 3E), providing large and unexplored opportunities for development of anticancer drugs. Consistently, when we searched in the PubTator database for the numbers of research publications for each PDG, 64.2% (298/464) of the SCNA-driven PDGs were classified as understudied genes.
Figure 3. Somatic copy number alterations of PDGs across cancers.

(A) Workflow of somatic copy number alterations (SCNA) analysis.
(B) Scatterplots show distribution of overall amplification or deletion G scores of all protein-coding genes, arranged in ascending order of G scores. Heatmaps show PDGs by gene families in the same order as the scatterplots. Bar plots (right) show enrichment of amplified or deleted PDGs in the corresponding gene families. Purple, enriched; orange, depleted.
(C) Bubble plots show the SCNA G scores of the top 100 PDGs driven by SCNAs across cancers. Left, copy number gain; right, copy number loss. Size of bubbles, G score; red, gain; blue, loss. Heatmap (left) show the PubTator scores. Green, <150 (understudied genes); red, >150. Target development level of each gene is indicted by color codes.
(D) Pie diagrams show the percentage of amplified and deleted PDGs in each gene family. Yellow line indicates the overall percentage across all PDGs.
(E) Mosaic plots show the distribution of amplified and deleted PDGs in each TDL.
Somatic mutations of the PDGs across cancers
We integrated five complementary approaches to identify recurrent somatic mutations using a computational strategy recently developed by the FCG (Hu et al., 2021) (Figure 4A). A mutation score (M score) was estimated for each mutated PDG in a given tumor type. Collectively, across 33 tumor types, we identified 361 PDGs that have recurrent mutations in at least one cancer type. To estimate the recurrent mutations of PDGs at a pan-cancer level, an overall M score was also estimated (Table S6). Across 33 cancer types, 117 PDGs showed an overall M score above a cutoff (Figures 4B and S4). As in the SCNA analysis, the most well-known mutation-driven targets with FDA-approved drugs were successfully identified and prioritized as the top rank (Figure 4C). Notably, we observed 40/117 (34.2%) recurrent mutant PDGs harboring hotspots defined by the Cancer Hotspots database (Chang et al., 2016). Among them, mutations in 14 PDGs were predicted as gain-of-function mutations based on the OncoKB database (Chakravarty et al., 2017) (Figure 4D), suggesting that they may serve as oncogenes during tumorigenesis. Other mutation-driven PDGs may function as tumor suppressors given that these mutations may lead to partial loss of function of these genes. Importantly, we found that kinases and HMEs were significantly enriched in the PDGs with recurrent mutations, while GPCRs, ICs, and transporters were significantly enriched in the PDGs without recurrent mutations (Figure 4B). This strongly indicates that a large portion of kinases and HMEs play causal roles in tumorigenesis, thereby serving as potential drug targets in cancer. Notably, kinases showed significant enrichment for gain-of-function hotspot mutations, while HMEs had higher percentages of both gain- and loss-of-function mutations (Figures 4B and 4E), suggesting that different targeting strategies should be designed to drug these two groups of PDGs. Finally, only a small portion of the mutation-driven PDGs were classified as Tbio, and no mutation-driven PDGs were classified as Tdark for their TDLs (Figures 4F and S5), suggesting that a large effort has been made to understand the functions of these genes with recurrent mutations in cancer.
Figure 4. Somatic mutations of PDGs across cancers.

(A) Workflow of mutation analysis.
(B) Scatterplots show distribution of overall M scores of all protein-coding genes, arranged in ascending order of M scores. Heatmaps show PDGs with (upper) or without (lower) hotspot mutations, displayed by gene families and in the same order as the scatterplots. Bar plots (right) show enrichment of mutated PDGs in the corresponding gene families. Purple, enriched; orange, depleted.
(C) Bubble plot show the mutation frequencies and recurrent mutation indexes of the top 100 cancer-associated PDGs driven by somatic mutations across cancers. Size of bubbles, overall mutation frequency; intensity of color, recurrent mutation index. Heatmap (left) shows PubTator scores. Green, <150 (understudied genes); red, >150. Target development level of each gene is indicted by color codes.
(D) Bubble plot show frequencies of hotspot mutations in the PDGs presented in (B) (genes are arranged in the same order). Size of bubbles: hotspot mutation frequency. Hotspot mutations that were predicted as gain-of-function mutations are indicated as red.
(E) Pie diagrams show the percentage of mutated PDGs with (red) or without (orange) hotspot mutations in each gene family. Blue line indicates the overall percentage of mutation across all PDGs.
(F) Mosaic plots show the distribution of mutated PDGs for each TDL. Left, overall mutation, right, hotspot mutation.
Transcript fusions of PDGs across cancers
A total of 10,811 fusion transcripts (9,554 fusion pairs) involving 3,392 PDGs were identified across 33 cancer types. Among them, 7,319 (67.7%), 1,348 (12.5%), 1,400 (12.9%), and 743 (6.9%) events were defined as tier 1, tier 2, tier 3, and tier 4, respectively (Table S7). After applying the Elbow method to determine the cutoff for the fusion events whose numbers were significantly higher than background, we found that both overall fusion events and recurrent fusion events (which occurred at least twice in the same cancer type) were significantly enriched in the families of kinases, druggable TFs, and HMEs (Figure S6A), suggesting that these PDG families may play crucial roles in tumorigenesis. Notably, only 775 of 10,811 (7.2%) PDG fusion transcripts were recurrent events, representing 186 of 9,554 fusion pairs. TMPRSS2-ERG (n = 177), FGFR3-TACC3 (n = 36), and RPS6KB1-VMP1 (n = 29) were the most frequent fusions across 33 cancer types (Figures S6B and S7). Although both TMPRSS2 and ERG have been considered as potentially druggable, TMPRSS2-ERG fusion has been notoriously difficult to target in the clinic (Wang et al., 2017). To better assess the targetable potentials of PDGs with recurrent fusions, we further analyzed the integrity of PDG partners in each recurrent fusion transcript and found that 618 of 775 recurrent PDG fusion transcripts contained full or partial coding sequences (CDS) of PDG genes (Figure S6C). The most frequent fusion pattern was a joining of CDS regions of both 5′ and 3′ partners in frame, followed by 5′ UTR of 5′ partner joining with CDS of 3′ partner and joining of 5′ CDS with 3′ CDS out of frame. After removing the fusions without CDS of PDGs, the frequencies of PDGs contained recurrent fusion events in each cancer types were obtained (Figure S6D). Taken together, except for TMPRSS2-ERG, transcript fusions in PDGs are common but low-frequency genomic events across adult cancers.
Cancer dependency of PDGs across cancer cell lines
We retrieved genome-wide RNAi/CRISPR screening profiles from the DepMap (Dempster et al., 2019; McFarland et al., 2018; Meyers et al., 2017). Among 5,937 PDGs with dependency information, 784 (13.2%) of them were identified as genes required for cancer cell growth (“common essential” or “strongly selective” cancer-dependent PDGs) by either RNAi or CRISPR screen, including 495 strongly selective PDGs (Table S8). Druggable TFs, NRs, kinases, and HMEs were most significantly enriched in the PDGs that were defined as cancer-dependent genes (Figure 5A). This indicates that a large portion of PDGs in these four categories play crucial roles in cell growth and proliferation, thereby serving as potential anticancer (anti-proliferation) drug targets. Notably, more than half of the cancer-dependent PDGs (57.5%) were classified as Tbio or Tdark (Figure 5B). Consistently, when we searched the PubTator database for the numbers of research publications for each PDG, 60.5% of the cancer-dependent PDGs were classified as understudied genes. Importantly, the PDGs with recurrent genomic alterations identified from TGCA pan-cancer analyses were significantly enriched in the cancer-dependent PDGs (OR = 2.9, p = 2.3 × 10−24), strongly indicating cancer “driver” roles during tumorigenesis. Consistent with previous reports, cancer cell lines harboring hotspot gain-of-function mutations of PDGs were significantly sensitive to knockdown of these driver mutations (Figure 5C). A large portion of cancer-dependent PDGs (e.g., 40.7% of cancer-dependent PDGs identified from RNAi screening) showed a significant correlation between dependency and gene expression (FDR < 10%). They were able to be divided into two groups: PDGs for which high expression was correlated with increased sensitivity to RNAi knockdown, and PDGs for which low expression was correlated with increased sensitivity to RNAi knockdown (referred to as groups I and II, respectively) (Figure 5D). Similar behavior was also observed in the CRISPR screening (Figure 5E). Among the cancer-dependent PDGs, kinases, NRs, and druggable TFs were significantly enriched in group I, while group II contained more enzymes (Figure 5F). Next, we analyzed the correlation between dependence and copy number alteration for the cancer-dependent PDGs that exhibited recurrent SCNAs in TCGA pan-cancer analysis. Among the amplified cancer-dependent PDGs screened by RNAi assay, 14/68 (20.6%) showed significantly positive correlations between dependence and copy number (i.e., cells with copy number gain were more sensitive to knockdown), and 13 of them also showed positive correlation between dependence and RNA expression (SCNAgain/group I). Unexpectedly, we also identified 13 amplified cancer-dependent PDGs whose dependencies were significantly and negatively correlated with both copy numbers and RNA expression levels (SCNAgain/group II) (Figure 5G). Among the deleted cancer-dependent PDGs screened by RNAi assay, 20/47 (42.6%) showed significantly negative correlation between dependence and copy number, and 13 of them also showed negative correlation between gene dependence and RNA expression (SCNAloss/group II). Only two PDGs showed dependencies that were positively correlated with both copy number and RNA expression at a borderline level (SCNAloss/group I) (Figure 5H). These observations were further confirmed by an independent CRISPR screen dataset across 739 cancer cell lines (Figures 5I and 5J). Importantly, analyses by the GISTIC (Mermel et al., 2011) and ABSOLUTE algorithms (Carter et al., 2012) also demonstrated that the copy number losses in SCNAloss/group II genes appeared hemizygous, confirming that complete deletion of these genes may be lethal for tumor cells. These results suggest that both SCNAgain/group I and SCNAloss/group II PDGs may serve as potential therapeutic targets for anti-proliferation, although different strategies should be considered. Supporting this idea, 8/13 (61.5%) SCNAgain/group I PDGs had targeted therapy drugs that were approved by the FDA or are under development in the clinic. However, although it has been proposed for over two decades that a gene with hemizygous loss may cause vulnerabilities in cancer (Frei, 1993; Kronke et al., 2015; Nichols et al., 2020; Nijhawan et al., 2012; Paolella et al., 2017; Rendo et al., 2020), no SCNAloss/group II gene has directly targeted drugs approved in the clinic. To experimentally validate that those deletions of SCNAloss/group II genes can provide therapeutic windows, we chose CDK7, which showed hemizygous loss in 27.3% (2,991/10,950) of patients at a pan-cancer level in TCGA cohort and was recurrently deleted in four cancer types. Consistently, as a typical group SCNAloss/group II gene, CDK7 was hemizygous deleted in 29.8% of DepMap cancer cell lines, and its copy number was significantly and positively correlated with RNA expression (p = 8.3 × 10−73). Importantly, both CDK7 copy number losses and lower levels of RNA expression were significantly associated with increased sensitivity to CDK7 siRNAs (p = 1.6 × 10−6 and p = 1.5 × 10−6, respectively). Notably, CDK7-specific inhibitors have been advanced into early clinical trials (Hu et al., 2019a). We analyzed a large-scale CDK7i, THZ1 treatment response screen in cancer cell lines (n = 580, non-hematological malignant lines) (Kwiatkowski et al., 2014), and observed a significant and positive correlation between CDK7 copy number loss and increased THZ1 sensitivity (p = 0.037, adjusting for cancer lineage). This was also confirmed at the CDK7 RNA level (p = 0.024, adjusting for cancer lineage), and was further experimentally validated by colony formation assays in a series of cancer cell lines (Figure 5K). Finally, using two gRNAs that target the genomic sequences located in the 5′ and 3′ UTRs of CDK7 (Figure 5L), we completely deleted a single copy of the full-length CDK7 gene (42.5 kb) in OVCAR5 cells that harbor neutral CDK7 (Figures 5M and 5N). Notably, although >30 clones were examined and multiple hemizygous clones were identified, no homozygous knockout clone was found, indicating that CDK7 is an essential gene for cell survival. Importantly, we found that the CDK7-deleted clones were significantly more sensitive to THZ1 compared with their parental clone (Figures 5O and 5P).
Figure 5. Cancer dependency of PDGs across cancer cell lines.

(A) Bar plot shows enrichment of cancer-dependent PDGs in the corresponding gene families. Cancer-dependent PDGs were defined as common essential or strongly selective in the DepMap project. Purple, enriched; orange, depleted.
(B) Mosaic plots show the distribution of TDL classes among cancer-dependent PDGs.
(C) Volcano plot summarizes correlations between dependency and gene mutation for cancer-dependent PDGs. Each dot represents one cancer-dependent PDG with recurrent mutations. Of the genes whose mutations were significantly correlated with either increased or decreased sensitivity to RNAi knockdown (purple or green, respectively; FDR < 10%), genes with hotspot gain-of-function mutations were highlighted with red circles.
(D) Volcano plot summarizes correlations between dependency and gene expression for cancer-dependent PDGs. At the FDR 10% level, the genes whose higher expression levels were significantly correlated with either increased or decreased sensitivity to RNAi knockdown were categorized as group I (purple) or group II (green), respectively.
(E) Correlation of gene dependency (x axis, RNAi; y axis, CRISPR) with RNA expression for cancer-dependent PDGs. Purple or green, significant in either RNAi or CRISPR; borders, significant in both analyses; gray, not significant. Coordinates: “signed log q values” by linear regression; negative/positive sign: higher gene expression associated with increased/decreased sensitivity.
(F) Percentage of group I (purple) and group II (green) genes in each gene family.
(G and H) Correlation of gene dependency (RNAi) with copy number (x axis) and RNA expression (y axis) for amplified PDGs (G) and deleted PDGs (H). Points in pink/green or orange/blue indicate significance in either copy number or expression analysis; points within borders indicate significance in both analyses; points in gray indicate non-significance. Coordinates: “signed log q values” by linear regression; negative sign: high gene expression or copy number associated with increased sensitivity; positive sign: high gene expression or copy number associated with decreased sensitivity; distance from 0: q value; FDR: false discovery rate.
(I and J) Correlation of gene dependency (x axis, RNAi; y axis, CRISPR) with copy number for cancer-dependent amplified PDGs (I) and deleted PDGs (J). Each dot represents one cancer-dependent PDG with recurrent copy number alterations (G score for amplification >0.61 or G score for deletion >0.66). Pink/green or orange/blue, significant in either RNAi or CRISPR analysis; borders, significant in both analyses; gray, not significant. Coordinates: “signed log q values” by linear regression; negative/positive sign: higher copy number associated with increased/decreased sensitivity.
(K) Cancer cell lines with hemizygous losses of CDK7 were sensitive to CDK7i. Representative colony formation assay of a panel of cancer cell lines treated with a series of dosages of THZ1 for 6 days. CDK7 copy number status of each line was assessed by GISTIC.
(L) Manipulation of CDK7 copy number by CRISPR-Cas9.
(M) PCR results of wild-type OVCAR5 and two CDK7 hemizygously deleted clones. Bands of 1.7 and 1.1 kb indicate CDK7-deleted and wild-type alleles, respectively.
(N) qRT-PCR analysis (top) and western blot (bottom) show CDK7 RNA and protein expression among the indicated cells, respectively.
(O and P) Representative colony formation assay (O) and survival fraction (P) of wild-type OVCAR5 and two CDK7 hemizygously deleted clones treated with a series of dosages of THZ1 for 6 days. All experiments were performed in triplicate. Statistical analysis by Student’s t test, *p < 0.05; n = 3. Error bars represent means ± SD.
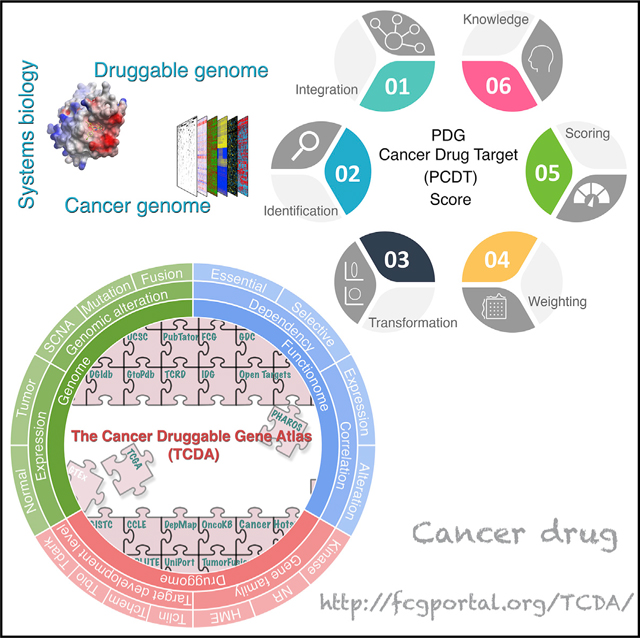
Systematic integration of multidimensional profiles of PDGs across cancers
Our above multi-omics analysis suggests remarkable unexplored opportunities for identification of drug targets in oncology; however, the key challenge is how to prioritize these potential druggable candidates at a genome-wide scale. We hypothesize that integration of expressional, genomic, functionomic, and pharmacological profiles of PDGs across cancers can comprehensively identify and prioritize potential therapeutic targets for treatment of cancer. In this regard, a PDG cancer drug target score (PCDT score) for each PDG was estimated by a systems biology approach (Figures 6A, 6B, and S8A–S8C). First, at both individual cancer type and pan-cancer levels, we integrated the multi-omics profiles from healthy individuals (GTEx, n = 7,429), primary cancer specimens (TCGA, n = 11,160), and cancer cell lines (DepMap, n = 1,775). Then, we comprehensively collected a total of 16 cancer-related features for a given PDG and generated a quantitative measurement for each feature (including 12 continuous and 4 discrete variables). Based on intrinsic characteristics of these features in oncology, 3 cancer drug target prediction modules were built, including an expression module, a genomics module and a dependency module. After transformation of the raw data, all features were scaled to have values ranging from 0 to 1 to facilitate downstream analysis. Finally, to optimize the performance of our PCDT score, we applied a grid search procedure to determine the weight of each feature within a module and the weight of each module for the PCDT score. Using known targets of FDA-approved small-molecule drugs in oncology as positive controls, the grid search procedure iteratively assessed the ability of the PCDT score to prioritize known cancer drug targets over a range of plausible weight values. Based on the optimized weights, a core PCDT score was estimated for each PDG at a pan-cancer level (Figures 6C; Table S9). Compared with other target identification score systems that were recently estimated for cancer treatment (Behan et al., 2019), the PCDT score not only specifically focuses on druggable genes (more practical for drug development), but also comprehensively considers multiple features that may contribute to prioritizing target candidates. For example, as expected, the dependency module of the PCDT score shows significant and positive correlation with the target priority scores, which were based on cancer dependency (Behan et al., 2019), however, the expression and genomic modules of the PCDT score provide additional information that was not be covered by other score systems (Figure S8D).
Figure 6. Systematic integration of multidimensional profiles of PDGs across cancers.

(A) Illustration of generation of a PCDT score for each PDG in cancer.
(B) Workflow of estimation of the PCDT score.
(C) A four-module score system provides comprehensive information for identification and prioritization of potential candidates for drug targets in oncology.
Consistent with our analysis on individual profiling platforms, among the PDGs with high core PCDT scores (i.e., top 10% of all PDGs, referred as to high PCDT score group), kinases, druggable TFs, HMEs, and NRs were significantly enriched, whereas GPCRs, ICs, and transporters contributed to smaller fractions to this group (Figures 7A and 7B). Notably, even in the high PCDT score group, only 49.8% of PDGs were defined as Tclin and Tchem for TDLs, suggesting large opportunities for further drug development in oncology (Figure 7C). As expected, kinases were the most highly represented PDG family in the high PCDT score group and had a considerably higher percentage of genes (81.5%) with chemical compounds in both clinical and preclinical stages. Although promising, PCDT scores were observed for many HMEs; drug development efforts for these genes are still unmet, and only 41.1% of HMEs in the high PCDT score group have existing compounds targeted against them (Figure 7D). Finally, we collected additional information about our current knowledge for each PDG, such as numbers of publications (Wei et al., 2019), approved drugs, and drugs in clinical development (Nguyen et al., 2017), as well as predicted tractability (Brown et al., 2018). After adding these factors to the core PCDT score, an extension PDG cancer drug target score (extension PCDT score) was estimated to further assist prioritization of cancer drug targets (Figure 6C). Collectively, by systematically integrating expressional, genomic, dependency, and pharmacological profiles, we computationally prioritized PDGs for potential application in oncology at a genome-wide scale, which may facilitate the development of therapeutics as well as the selection of patients for precision cancer treatment. A publicly accessible database, The Cancer Druggable Gene Atlas (TCDA), has also been developed (Figure 7E).
Figure 7. Large and unexplored opportunities for development of anticancer drugs.

(A) Density plots show distribution of core PCDT scores among PDGs stratified by gene families.
(B) Bar plot shows enrichment of PDGs with high core PCDT scores in the corresponding gene families.
(C) Mosaic plots show distribution of TDL classes within PDGs with high core PCDT scores.
(D) Word clouds of the high core PCDT score PDGs in three gene families. Size of fonts: core PCDT score. Color of fonts: target tractability defined by the Open Targets database; red, clinical precedence; pink, discovery precedence; gray, others.
(E) Overview of the TCDA data portal.
DISCUSSION
After the concept of the druggable genome was proposed in 2002, more than 6,000 genes have been estimated to be part of PDGs whose activities may be modulated by pharmaceuticals (Brown et al., 2018; Campbell et al., 2012; Finan et al., 2017; Hopkins and Groom, 2002; Kumar et al., 2013; Overington et al., 2006; Rask-Andersen et al., 2011, 2014; Russ and Lampel, 2005; Southan et al., 2015). However, <10% of these druggable genes are currently targeted by drugs approved by the FDA (Oprea et al., 2018; Santos et al., 2017), and a small portion of them have been applied in oncology (Rubio-Perez et al., 2015; Yap and Workman, 2012), reflecting opportunities for the next generation of drug development for cancer treatment. By systematically reviewing six comprehensive PDG resources, we observed that the PDG lists have remarkably increased during the past decade, indicating that more and more proteins can be modulated by small molecules.
Although the majority of PDGs are reliably detectable in cancer, only 46.9% of them show selective expression patterns, including 2,099 lineage-enriched PDGs. Selective expression of a PDG not only indicates its potential roles during tumorigenesis, but also provides a better therapeutic window for drug development. Supporting this idea, we found that the targets of approved cancer drugs were indeed significantly enriched in selectively expressed PDGs. In addition, by comparing expression of PDGs in cancer with their expression in a large-scale normal tissue cohort (not only corresponding adjacent specimens of a give cancer type), we identified 697 caPDGs that are highly expressed in at least one cancer type. For example, many DNA damage repair-related PDGs are highly expressed in cancers, indicating that tumor cells may rely on their functions for survival. Collectively, a large portion of PDGs are expressed in cancers, and their expression patterns, provide rich information for target selection and prioritization. The recurrence of genomic alterations of a PDG is another strong indicator of its therapeutic potential (Garraway and Lander, 2013; Vogelstein et al., 2013). We comprehensively characterized genomic alterations across cancers, and estimated quantitative scores for recurrent SCNAs, mutations, and transcript fusions of each PDG at both individual and pan-cancer levels. PDGs with gain-of-function hotspot mutations have been the most widely identified as targets for cancer drugs, although these candidates have been largely exhausted during the first wave of development of targeted therapy (Huang et al., 2020). Focally recurrent copy number gains serve as the second most important resource for target identification. However, a focally amplified genomic locus usually contains multiple genes, including both cancer “driver(s)” and co-altered “passengers.” Identification of functional drivers is still a challenging step in prioritizing SCNA-driven PDGs in cancer. A combination of genomic profiling and genetic screening may assist in reducing noise from passenger alterations (Beroukhim et al., 2010; Zack et al., 2013). Finally, despite relatively low frequencies of fusions of PDGs in adult cancers, recurrent transcript fusion events serve as promising and actionable targets for considerable numbers of patients.
Directly targeting loss-of-function genomic alterations with small-molecule drugs remains a challenge (Huang et al., 2020). Recurrent hemizygous copy number loss of an essential PDG represents a promising but largely understudied resource for cancer drug development, although it has been proposed for two decades that CYCLOPS (copy number alterations yielding cancer liabilities owing to partial loss) may cause vulnerabilities that can be exploited for treatment (Frei, 1993; Kronke et al., 2015; Nichols et al., 2020; Nijhawan et al., 2012; Paolella et al., 2017; Rendo et al., 2020). Recently, a few CYCLOPS genes have successfully been evaluated in preclinical models (Nichols et al., 2020; Nijhawan et al., 2012; Paolella et al., 2017; Rendo et al., 2020). More excitingly, the example of targeting a CYCLOPS gene, CK1α, by lenalidomide (Revlimid) has been applied in the clinic to treat myelodysplastic syndrome with loss of chromosome 5q (Kronke et al., 2015). Thus, identification of druggable CYCLOPS genes may provide an avenue for precision patient selection for existing cancer drugs. Although loss-of-function mutations in a considerable number of PDGs have been observed, they may not be able to serve as direct drug targets. Instead, synthetic lethality may be an efficient approach to target the vulnerabilities induced by these loss-of-function alterations (Huang et al., 2020). Finally, genome-scale assessments of the effects of each PDG on tumor cell growth (cancer dependency) provide a strong functional indicator for target identification and prioritization (Behan et al., 2019; Tsherniak et al., 2017), especially when combined with genomic profiles of primary tumors.
A large percentage of the PDGs (69.3%) were defined as Tbio and Tdark for their TDLs, and indeed the majority of the PDGs (n = 4,222) were classified as understudied genes based on the numbers of related publications. More importantly, many of these less characterized PDGs showed dysregulated expression, recurrent alterations, and/or functional dependencies in cancers. For example, among the high PCDT score PDGs, 50.2% of them fell into Tbio and Tdark categories without existing chemical compounds. This strongly indicates large and unexplored opportunities for future drug development in oncology. Notably, potential causal events were significantly enriched in a few druggable families, such as kinases, NRs, and HMEs, whereas most GPCRs and ICs may play relatively limited functions during tumorigenesis. This result is supported by the fact that most currently approved targeted therapy drugs in oncology target kinases, NRs or HMEs (Rubio-Perez et al., 2015; Yap and Workman, 2012). However, the drug development levels among these different families are still unbalanced. Although considerable numbers of kinase inhibitors have been developed and many of them are advancing into early clinical trials, the need for potent and selective HME modulators is still unmet. Most importantly, unlike kinases, HMEs predominantly show ubiquitous expression patterns in normal healthy tissues and loss-of-function alterations in cancers. Thus, which patient population should be selected and how a therapeutic window can be achieved are key clinical challenges for future drug development in oncology. Finally, although many TFs show promising PCDT scores, the numbers of predicted druggable TFs have been very limited to date.
Strategies to identify and prioritize druggable targets for cancer treatment would represent a significant advance in therapeutic development in oncology (Rubio-Perez et al., 2015; Yap and Workman, 2012). However, most current approaches to identify potential targets for small-molecule compounds for cancer therapy are largely driven by possibilities from a medical chemistry viewpoint rather than by cancer genomic profiles. By an integration of “the druggable genome” and the “cancer genome,” our present study provided a comprehensive “blueprint” of PDGs across cancers. Based on this informative blueprint, we quantified 16 cancer-related features of PDGs and estimated a core PCDT score to prioritize their therapeutic potentials in oncology. In addition, our current knowledge on clinical applications of PDGs and their predicted tractability were also integrated into this score system as an extension PCDT score. A publicly accessible database, TCDA, was also developed through the FCG data portal (http://fcgportal.org/TCDA/).
Limitations of the study
There are some limitations to our study. The list of PDGs may dynamically change in advances of medical chemistry. For example, KRAS was previously considered a typically undruggable gene, despite its dominant cancer-driver function in tumorigenesis (Moore et al., 2020). Recent advances in KRAS (G12C) inhibitors have shifted this paradigm, and several KRAS inhibitors are advanced in early clinical trials (Moore et al., 2020). Meanwhile, not every PDG is able to be successfully translated to the clinic. Examples include TP53 and TMPRSS2-ERG, which have historically been considered attractive PDGs in cancer therapy (Vassilev et al., 2004; Wang et al., 2017). The list of PDGs has thus been dynamically changing and may continuously increase. For example, proteolysis targeting chimera technology may remarkably change the current definition of druggable genes in the following years. In addition, as drug development progresses, the definition of “druggable” in oncology has expanded from genes targeted by small molecules to genes targeted by biotherapeutic drugs such as antibodies and cellular therapies (Brown et al., 2018). More than 2,000 genes on the current PDG list encode cell membrane surface proteins (Brown et al., 2018), which are potentially targetable by antibody-based drugs. However, the actual number of cell surface protein may be far larger than that (Bausch-Fluck et al., 2018; Hu et al., 2021). In this study, we integrated the six most comprehensive PDG resources with a uniform approach to best reflect our current knowledge on the druggable genome. Finally, most current large-scale genetic screens are based on in vitro proliferation assays (Behan et al., 2019; Tsherniak et al., 2017). Beyond cell growth, cancer-driven PDGs play functions in many distinct cancer-related pathways, such as angiogenesis, metastasis, and immune response, leading to additional challenges in selecting and prioritizing drug targets.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Xiaowen Hu (xiaowenh@pennmedicine.upenn.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Information of gene family category and target development level were generated by the IDG project, which are publicly available through the PHAROS database (https://pharos.nih.gov/) and the Target Center Resource Database (TCRD; http://juniper.health.unm.edu/tcrd/). The genomic profiles of human cancers were generated by the TCGA project, which are publicly available through the Genomic Data Commons portal (GDC, https://gdc-portal.nci.nih.gov). The RNA expression profiles of human normal healthy tissues were by the GTEx project, which are publicly available through the GTEx portal (https://gtexportal.org/home/). Genetic screening profiles in human cancer cell lines were generated by the DepMap and the Score projects, which are publicly available through the DepMap portal (https://depmap.org/portal/), and the Score projects (https://score.depmap.sanger.ac.uk/). The genomic data were retrieved, processed and analyzed through a master computational protocol developed by the Functional Cancer Genome project (FCG, http://fcgportal.org/home/) as described by our previous publications (Hu et al., 2019b, 2021; Shan et al., 2020; Yuan et al., 2018) as well as the STAR Method section. The data generated by this study are public available through the Functional Cancer Genome data portal (http://fcgportal.org/home) and the Cancer Druggable Gene Atlas (TCDA) website (http://fcgportal.org/TCDA/). This paper does not report original code. Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Cancer cell lines were purchased from the ATCC or NCI Development Therapeutics Program. SKOV3, OVCAR5, OVCAR3, OVCAR4, MCF7, HCC1937, CAOV3, MDA-MB-468, HCC38 and SKBR3 were cultured in RPMI1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (VWR). UWB1.289 was cultured in 50% RPMI1640 medium and 50% MEGM (Lonza, CC-3150) supplemented with 3% fetal bovine serum. All cell lines were maintained at 37°C and 5% CO2.
METHOD DETAILS
Definition of the potentially druggable genes
To define the potentially druggable genes (PDGs), candidates from 6 comprehensive druggable gene resources (Carvalho-Silva et al., 2019; Finan et al., 2017; Hopkins and Groom, 2002; Kumar et al., 2013; Russ and Lampel, 2005; Southan et al., 2015) were integrated. The PDG protein/gene names of each resource were retrieved from the Drug Gene Interaction Database (DGIdb; https://www.dgidb.org/) (Cotto et al., 2018), Open Targets Platform (https://www.opentargets.org/) (Carvalho-Silva et al., 2019) or original publications (Carvalho-Silva et al., 2019; Finan et al., 2017; Hopkins and Groom, 2002; Kumar et al., 2013; Russ and Lampel, 2005; Southan et al., 2015). After converting the protein/gene names to the ENSEMBL gene annotation (Version 80), genes annotated by at least 2 sources were defined as PDGs in current study. Given rapid advances in epigenetic drug development for cancer treatment, histone modification enzymes that were systematically annotated by the Structural Genomics Consortium (https://www.thesgc.org/) were included in the PDG list. Information of gene family category, target development level and Pubtator score of PDGs were retrieved from the PHAROS database (https://pharos.nih.gov/) and the Target Center Resource Database (TCRD v5.2.0; http://juniper.health.unm.edu/tcrd/) from the IDG project of NIH (Nguyen et al., 2017).
RNA-seq data processing and gene expression analysis
The poly(A)+ RNA-seq data for primary tumors and their adjacent tissues were generated by the University of North Carolina and the British Columbia Cancer Agency Genome Sciences Centre as part of the TCGA project. The poly(A)+ RNA-seq data for normal healthy tissues were generated by the Broad Institute of Harvard and MIT as part of the GTEx project. The poly(A)+ RNA-seq data for hematopoietic cells were download from Sequence Read Archive (SRA, accession number SRP125125), and the poly(A)+ RNA-seq data for lymphatic tissues were download from the Human Protein Atlas (HPA), Illumina’s Human BodyMap 2.0 project, and Encyclopedia of DNA Elements (ENCODE). All RNA-seq data were processed through a pipeline developed by the UCSC Toil RNAseq Recompute Compendium, which allowed us to consistently process large-scale RNA-seq data without computational batch effects (Vivian et al., 2017). For TCGA RNA-seq data, if more than one sample existed for a participant, one single tumor sample (and matched adjacent sample, if applicable) was selected based on the following rules: (1) tumor sample type: primary (01) > recurrent (02) > metastatic (06); (2) order of sample portions: higher portion numbers were selected; and (3) order of plate: higher plate numbers were selected. Expression of a PDG in a given tissue or cancer type was defined as positive if its mRNA expression was reliably detected in at least 50% of specimens (i.e., the 50th percentile of fragments per kilobase of transcript per million mapped reads [FPKM] value ≥ 1).
Classification of expressional distribution
Genes were classified into 6 categories based on their expression levels across the TCGA samples: (I) undetectable genes: genes that showed undetectable RNA expression for all 33 TCGA cancer types (FPKM < 1 for more than 50% tumor samples of each cancer type); (II) ubiquitously expressed genes: genes that were expressed (FPKM > 1) for the majority of tumor samples (95%); (III) lineage-enriched genes: genes with elevated (five-fold) RNA expression levels in an individual cancer type or a group of cancer types (a maximum of seven cancer types) compared to all other cancer types; (IV) right-skewed genes: genes whose expression had skewness above 0.5 and were at least 125 times more likely to have been sampled from a right-skewed distribution than a normal distribution (i.e. skewed-LRT value > 125); (V) bimodal-like genes: genes whose expression had bimodal index (BI) >1.2 and were at least 125 times more likely to have been sampled from a bimodal distribution than a normal distribution (i.e. bimodal-LRT value > 125); (VI) unclassified: genes that were not assigned to any of the above five groups. The hierarchy of groups used to classify genes: undetectable > ubiquitously expressed > lineage-enriched > right-skewed > bimodal-like > unclassified. Genes from the “lineage-enriched”, “right-skewed”, “bimodal-like”, and “unclassified” groups were considered as selectively expressed genes.
Identification of PDGs specifically expressed in cancers (caPDGs)
caPDGs were identified independently for each individual cancer type by comparing mRNA expression levels of PDGs between a given cancer type (TCGA) and normal tissues from 29 organs (GTEx). Because cancer-testis genes often encode immunogenic antigens for cancer immunotherapy (Hofmann et al., 2008; Zhang et al., 2016), normal testis tissues were excluded from the normal tissue pools (except for analysis on testicular germ cell cancer [TGCT]). To reduce false positives, we applied five independent computational algorithms to identify cancer-specific genes: specificity measure (SPM) (Xiao et al., 2010), TissueEnrich (Jain and Tuteja, 2019; Uhlen et al., 2015), specificity index probability (pSI) (Dougherty et al., 2010), sample set enrichment analysis (SSEA) (Subramanian et al., 2005), and differential expression analysis by Mann-Whitney-Wilcoxon test (MWW test). These algorithms were categorized into two groups based on their principles: Group I, including TissueEnrich and SPM, which calculated a metric to assess the specificity of each gene independently; Group II, including pSI, SSEA and MWW test, which required an additional step to calculate a rank for each gene across all genes based on the specificity metrics. Notably, distinct input data matrices were used by these algorithms: for pSI, SPM and TissueEnrich, median FPKM values of a given gene in each tissue or cancer type were used to represent the expression levels; for SSEA and MWW test, FPKM values of a given gene in each individual sample were used for analysis. For each method, both stringent and less stringent criteria were applied to define caPDGs with high and moderate confidence, respectively.
SPM: SPM was adopted from TiSGeD (Xiao et al., 2010), by which the specificity measure for each gene in a given cancer type was calculated as the cosine value of the intersection angle between the gene’s observed expression pattern and a pre-defined artificial expression pattern. The observed expression pattern was represented as a vector of expression values of the gene corresponding to the given cancer type and each normal tissue type. An artificial expression pattern was pre-defined, representing the extreme case in which the gene was expressed in the given cancer type while its expression level was zero in all normal tissue types. Genes with SPM values greater than 0.99 and 0.9 were considered as highly confident (stringent criteria) and moderately confident (less stringent criteria), respectively.
TissueEnrich: The function teGeneRetrieval of TissueEnrich R package (Jain and Tuteja, 2019) was used to classify genes into six different groups according to pairwise expression fold change among tissue types. Genes classified as “Tissue-Enriched” in a given cancer type (i.e., its expression level in a given cancer type was at least five-fold higher than all normal tissue types) were considered as highly confident (stringent criteria). Genes classified as “Tissue-Enhanced” in a given cancer type (i.e., its expression level in a given cancer type was at least five-fold higher than the average of all normal tissue types) were considered as moderately confident (less stringent criteria).
pSI statistic: The R package pSI, developed by Dougherty et al. (Dougherty et al., 2010), was applied to calculate a pSI value for each gene in a given cancer type. Genes with pSI values less than 0.001 and 0.01 in a given cancer type were considered as highly confident (stringent criteria) and moderately confident (less stringent criteria), respectively.
SSEA: SSEA was adopted from the Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005): the R package fgsea was applied for testing differential expression between a given cancer type and each normal tissue type. For each pairwise comparison (a given cancer type vs. a given normal tissue type), all samples were ranked according to expression level of a specific gene. Querying the sample set of cancer against the ranked sample list yielded a normalized enrichment score (higher score means stronger enrichment of expression in cancer). We ranked genes within each pairwise comparison by NES and assigned percentile ranks (e.g., a percentile rank of 0.95 implies the gene ranked in the top 5th percentile of all genes analyzed). Each of the percentile ranks obtained from comparisons against different normal tissue types were then combined. The genes with an average percentile rank above 0.99 were considered as highly confident (stringent criteria); the genes with a minimum percentile rank above 0.9 were considered moderately confident (less stringent criteria).
MWW test: Differential expression of a gene between a given cancer type and each normal tissue type was estimated by the function Wilcox_test of R package coin (Torsten et al., 2006). For each pairwise comparison (a given cancer type vs. a given normal tissue type), the difference in rank position of expression levels of the two groups was estimated (higher positive value means stronger enrichment of expression in cancer). We ranked genes within each pairwise comparison by difference in rank position and assigned percentile ranks (e.g., a percentile rank of 0.95 implies the gene ranked in the top 5th percentile of all genes analyzed). Each percentile rank obtained from comparisons against different normal tissue types were then combined. The genes with an average percentile rank above 0.99 were considered as highly confident (stringent criteria); the genes with a minimum percentile rank above 0.9 were considered moderately confident (less stringent criteria).
To integrate the results generated by different methods, we summed the potential caPDG lists from all five algorithms based on the confidence levels, then estimated a specificity score for each potential caPDG. For each algorithm, 2 = positive by stringent criteria; 1 = positive by less stringent criteria; and 0 = negative.
where
After a cut-off (specificity score ≥ 3) was estimated to define the caPDGs in a given cancer type, the caPDGs were further divided into three tiers. Tier 1 (high confident caPDGs): the caPDGs were identified by at least two algorithms with stringent criteria; Tier 2 (moderately confident caPDGs): the caPDGs were identified by at least one algorithm with stringent criteria and one algorithm with less stringent criteria; Tier 3 (low confident caPDGs): the caPDGs were identified by at least three algorithms with less stringent criteria. Finally, to reduce the expressional interference from tumor-infiltrating immune cells in tumor specimens, PDGs that are highly expressed in immune cells were excluded (except for analysis on hematopoietic malignancies) based on the RNA-seq profiles from 30 distinct types of hematopoietic cells and six lymphatic tissues.
SNP array data collection and processing
Single-nucleotide polymorphism (SNP) array data (Affymetrix Genome-Wide Human SNP Array 6.0) in CEL format across 33 cancer types were retrieved from the TCGA Data Portal (https://tcga-data.nci.nih.gov/tcga/). Segmentation files of TCGA tumor samples processed by circular binary segmentation (CBS) algorithm (Olshen et al., 2004) were retrieved from the TCGA GDAC Firehose of the Broad Institute (http://gdac.broadinstitute.org/; retrieval date: Jan, 3, 2018). If multiple samples existed for a participant, one pair of tumor and matched control was selected for ABSOLUTE analysis and one tumor sample was kept for focal SCNA analysis. Sample selection was based on the following rules: (1) sample type: for tumor tissues, primary (01) > recurrent (02) > metastatic (06); for normal control tissues, blood (10) > solid (11); (2) molecular type of analyte for analysis: preference for D analytes (native DNA) over G, W, or X (whole-genome amplified); (3) order of sample portions: higher portion numbers were selected; and (4) order of plate: higher plate numbers were selected.
Recurrent focal SCNA estimation
The Genomic Identification of Significant Targets in Cancer (GISTIC 2.0) algorithm (Mermel et al., 2011) (https://www.broadinstitute.org/cancer/cga/gistic) was used to identify significantly recurrent focal genomic regions that were gained or lost in a given tumor type. Segmentation files retrieved from the TCGA GDAC Firehose of the Broad Institute were used as input. GISTIC deconstructed copy number alterations into broad and focal events and applied a probabilistic framework to identify location and significance levels of SCNAs. For recurrent focal SCNA estimation, the significance levels (q values) were calculated by comparing the observed gains/losses at each locus to those obtained by randomly permuting the events along the genome. Tumors which had more than 2,000 segments were excluded from our analysis. Default parameters of GISTIC were used with the confidence level set to 0.99 (by -conf). Focal events with q-value below 0.25 were considered as significantly recurrent. Significant focal events in individual samples were then classified into four categories according to the amplitude threshold of GISTIC: GISTIC status=0, below threshold; GISTIC status=1, amplified (gain); GISTIC status=2, highly amplified (amplification); GISTIC status =−1, deleted (loss); GISTIC status=−2, highly deleted (deletion). In each cancer type, a GISTIC score (G-score), which accounts for both frequency and amplitude of a given SCNA event (Mermel et al., 2011), was generated by GISTIC for each gene and separately for gain or loss. Genes with a G-score < 0.1 were excluded from downstream analysis due to low frequency and/or amplitude. For a given gene, an overall G-score across all cancer types was calculated by an unweighted sum of G-scores in every cancer type.
Correlation analysis between copy number and RNA expression
To identify genes that had positive correlations between their RNA expression levels and copy number alterations, the putative gene-level copy number of a given gene was estimated by the GISTIC algorithm. Genes that were detectable in at least 10% of tumor specimens (90th percentile of FPKM value ≥1) in a given cancer type were subjected to correlation analysis. Pearson correlation analysis was performed by R software and the threshold of significant correlation between the estimated copy number and RNA expression level for each gene was set to p<0.001 (Pearson’s correlation).
Identification of the putative cancer-associated PDGs driven by SCNAs
At the individual cancer type level, we identified putative cancer-associated PDGs driven by SCNAs using four criteria: 1) location in a peak region of a significantly recurrent focal SNCA locus estimated by GISTIC (q≤0.25); 2) alteration with high frequency and large amplitude (G-score ≥0.1); 3) mRNA expression reliably detected in at least 10% of tumor specimens in a given cancer type (the 90th percentile of FPKM value ≥1); and 4) expression level of mRNA significantly and positively correlated with the estimated copy numbers (p-value of Pearson’s correlation coefficient between log[FKPM+0.001] and log ratio < 0.001). To estimate SCNAs for these putative cancer-associated GESP genes at a pan-cancer level, we calculated an overall G-score by an unweighted numeric sum of G-scores that met all four criteria in each individual cancer type.
Whole-exome sequencing data collection and processing
Mutation Annotation Format (MAF) profiles for 33 cancer type were downloaded from the TCGA Multi-Center Mutation Calling in Multiple Cancers (MC3) project (https://doi.org/10.7303/syn7214402), a variant calling project of TCGA (Ellrott et al., 2018). The MC3 data was generated through seven independent mutation calling algorithms, including Pindel (INDEL), MuSE (SNV), Radia (SNV) (Radenbaugh et al., 2014), VarScan2 (SNV/INDEL), MuTect (SNV), Indelocator (INDEL) and SomaticSniper (SNV). Variants from each caller were merged, QC filtered and stored in MAF file (Ellrott et al., 2018). If multiple samples existed for a participant in the MAF, one single pair of tumor/matched control sample was kept following these rules: (1) sample type: for tumor tissues, primary (01) > recurrent (02) > metastatic (06); for normal tissues, blood (10) > solid (11); (2) molecular type of analyte for analysis: preference for D analytes (native DNA) over G, W, or X (whole-genome amplified); (3) order of sample portions: higher portion numbers were selected; and (4) order of plate: higher plate numbers were selected. We excluded all mutations that were not tagged with PASS or WGA alone in all cancer types.
Recurrent mutation gene estimation
To predict the putative cancer-associated genes driven by mutation, five independent methods were integrated and applied to identify recurrent mutations: (1) MutSigCV (http://software.broadinstitute.org/cancer/software/genepattern/modules/docs/MutSigCV), which identifies genes that are significantly mutated in cancer genomes using a model with mutational covariates. It analyzes the mutations of each gene to identify genes that were mutated more often than expected by chance, given the background model; (2) Oncodrivefm (http://bg.upf.edu/group/projects/oncodrive-fm.php), which computes a metric of functional impact using three well-known methods (SIFT, PolyPhen2 and MutationAssessor) and assesses how the functional impact of variants found in a gene across several tumor samples deviates from a null distribution to detect candidate driver genes; (3) OncodriveCLUST (http://bg.upf.edu/group/projects/oncodrive-clust.php), which is designed to exploit the feature that mutations in cancer genes, especially oncogenes, often cluster in particular positions of the protein and change their functions; thus, this feature can be used to nominate candidate driver genes; (4) ActiveDriver (http://reimandlab.org/software/activedriver/), which identifies post-translational modification (PTM) sites in proteins (i.e., active sites such as signaling sites, protein domains, regulatory motifs) that are significantly mutated in cancer genomes; and (5) HotSpot3D (https://github.com/ding-lab/hotspot3d), which identifies mutation hotspots from linear protein sequence and correlates the hotspots with known or potentially interacting domains and mutations. MC3 MAF files excluding hypermutated samples were used as input for the above programs, and default parameters were used for all five programs. A mutation index x (ranging from 0 to 5) was assigned to genes which passed the threshold of x out of five programs for a given cancer type. In addition, a mutation score (M-score) was calculated for each mutated gene in a given cancer type, which takes into account both the mutation index and frequency of mutation across samples (i.e., M score = mutation index × mutation frequency). Genes with mutation index ≥ 2 (identified as positive by at least two programs) were considered to be recurrently mutated. An overall M-score was generated to measure the recurrent mutation level of a given gene across all cancers, by unweighted sum of M-scores estimated for each individual cancer type.
Transcript fusion data collection and analysis
The gene fusion data of TCGA were retrieved from TumorFusions data portal (http://tumorfusions.org/), which analyzed transcript fusions across 33 cancer types from TCGA (Hu et al., 2018). Transcript fusion events were called by Pipeline for RNAseq Data Analysis (PRADA) (Torres-Garcia et al., 2014), and fusions detected in normal samples were excluded. Six filters controlling for sequence similarity of the partner genes, transcriptional allelic fraction, dubious junctions, germline events and presence in non-neoplastic tissue were applied (Hu et al., 2018). If more than one sample existed for a participant, one single sample was kept following these rules: (1) sample type: for tumor tissues, primary (01) > recurrent (02) > metastatic (06); (2) order of sample portions: higher portion numbers were selected; and (3) order of plate: higher plate numbers were selected.
Definition of genes associated with significant genomic alterations at a pan-cancer level
Waterfall method described by a recent publication from Cancer Cell Line Encyclopedia Consortium (Cancer Cell Line Encyclopedia Consortium and Genomics of Drug Sensitivity in Cancer Consortium, 2015) was used to define significant genomic alterations at a pan-cancer level. Specifically, the genomic alteration metrics were extracted for each protein coding gene (G-score was used for somatic copy number alterations, M-score was used for somatic mutations, and number of occurrences was used for transcript fusions). Increasing scores were then sorted to generate a waterfall distribution of all protein coding genes. The inflection point of the waterfall curve was estimated as the point on the curve with the maximal distance to a line drawn between the start and end points of the distribution. Genes with genomic alteration metric values above this inflection point were classified as significantly associated with the corresponding genomic alteration type. In somatic copy number analysis, amplification and deletion were analyzed separately. In mutation analysis, hypermutated tumor specimens were excluded for estimation of M-score. Given that TP53 showed a remarkably larger M-score value (which was considered as an outlier), TP53 was set aside when determining the inflection point. In transcript fusion analysis, genes with no fusion events were excluded. Within the PDGs, enrichment of each gene family for the genes associated with genomic alterations was calculated by a Fisher’s exact test.
Characterization of dependencies of the PDGs in cancer cell growth
Genome-wide CRISPR/Cas9 and RNAi screening profiles in a large-scale cancer cell line panel were retrieved from the Dependency Map (DepMap) portal (https://depmap.org/portal/). Criteria for definition of the “common essential” and “strongly selective” genes were described previously by the DepMap team (Dempster et al., 2019; Meyers et al., 2017). Briefly, a common essential gene (i.e., a gene universally required for viability of cancer cells in a large pan-cancer screen) was defined as a gene which ranks in the top X most depleting genes in at least 90% of cell lines. Here, X is chosen empirically using the minimum of the distribution of gene ranks in their 90th percentile least depleting lines. A strongly selective gene (i.e., a gene whose dependency is observed in a subset of cancer cells in a large pan-cancer screen) was defined as a gene whose dependency is at least 100 times more likely to have been sampled from a skewed distribution than a normal distribution (i.e., skewed-LRT value > 100). Both common essential and strongly selective genes defined by either CRISPR/Cas9 or RNAi screening were considered as essential genes for cancer cell viability. Assessment of enrichment for essential genes for cancer cell viability was performed by Fisher’s exact test across different gene classes. For the PDGs which were defined as either “common essential” or “strongly selective” by DepMap, we used the Bioconductor Limma package (Ritchie et al., 2015) to estimate the correlation between their dependencies (dependency scores) and mRNA expression or DNA copy number levels. The processed mRNA expression (RNA-seq) and DNA copy number (whole-exon seq or SNP array) profiles of the cancer cell lines were retrieved from the DepMap portal. Cohen’s effect size was scaled so that it measured the change in dependency across the interquartile range of mRNA expression or DNA copy number. For DNA copy number, log2(relative to ploidy + 1) was used. For mRNA expression, log2 transformed TPM values using a pseudo-count of 1 were used. P-values were adjusted with the Benjamini and Hochberg (BH) method (Benjamini and Hochberg, 1995).
Generation of cancer drug target score
To prioritize anti-cancer drug targets among PDGs, multidimensional omics profiles were integrated to estimate a PDG cancer drug target score (PCDT-Score) for each PDG. Three modules were established to integrate features at the expressional level, genomic level, and functional level, respectively. All features were transformed and scaled to have values ranging from 0 to 1 to facilitate integration. The expression module was based on the RNA-seq profiles from the GTEx and TCGA as well as the clinical annotation information of TCGA. Detailed methods for the raw data processing and quantification were described in the method sections of RNA-seq and expression analyses. The features in expression module included: 1) Tau tissue-specificity index (Yanai et al., 2005) assessing cancer type/tissue specificity across TCGA cancer types. 2) Distribution of expression across TCGA tumors. The expressional categories were transformed into discrete values: 0 for undetectable genes, 0.5 for ubiquitously expressed genes, and 1 for selectively expressed genes. 3) Cancer specific expression (i.e., caPDG, detailed method was described in the estimation of caPDG section). Cancer specificity score was scaled to range [0, 1]. 4) Prognostic value of gene expression. For each gene in a given cancer type, we fit a Cox proportional hazards model and obtained a z-statistic to assess whether high expression was associated with favorable or unfavorable overall survival. Then, we combined z-statistics for individual cancer types to yield a meta-z-score to assess the prognostic value at pan-cancer levels. P-values converted from meta-z-scores were log transformed and scaled to range [0, 1], with 1 representing the genes with the most significant p-values and 0 representing the genes with p-values of 1. 5) Differential expression in tumors. For each cancer type which had normal tumor-adjacent tissues, differential gene expression analysis between tumor tissues and normal tumor-adjacent tissues was performed using the DESeq2 package (Love et al., 2014) with the raw count matrix as input. A z-statistic was returned for each gene in the specific cancer type to assess whether it was up-regulated or down-regulated in tumor tissues. Then, we combined z-statistics for individual cancer types to yield a meta-z-score to assess the degree of dysregulation at the pan-cancer level. P-values converted from meta-z-scores were log transformed and scaled to range [0, 1], with 1 representing the genes with most significant p-values and 0 representing the genes with p-values of 1. The genomic module was based on the SNP array and WES profiles from the TCGA as well as the clinical annotation information of TCGA. The RNA-seq profiles of TCGA were also used to estimate this module. Detailed methods for the raw data processing and quantification (e.g., estimation of G-score, M-score and recurrent fusion) were described in the method sections of genomic alteration analyses. The features in genomic module included: 1) Overall G-score for amplification, scaled to range [0, 1]. 2) Overall G-score for deletion, scaled to range [0, 1]. 3) Prognostic value of copy number. For each gene in a given cancer type, we fit a Cox proportional hazards model and obtained a z-statistic to assess whether high copy number was associated with favorable or unfavorable overall survival. Then, we combined z-statistics for individual cancer types to yield a meta-z-score to assess the prognostic value at the pan-cancer level. P-values converted from meta-z-scores were log transformed and scaled to range [0, 1], with 1 representing the genes with most significant p-values and 0 representing the genes with p-values of 1. 4) Overall M-score, scaled to range [0, 1]. 5) Prognostic value of mutations. For each gene in a given cancer type, we fit a Cox proportional hazards model and obtained a z-statistic to assess whether presence of non-silent mutations was associated with favorable or unfavorable overall survival. Then, we combined z-statistics for individual cancer types to yield a meta-z-score to assess the prognostic value at the pan-cancer level. P-values converted from meta-z-scores were log transformed and scaled to range [0, 1], with 1 representing the genes with most significant p-values and 0 representing the genes with p-values of 1. 6) Occurrence of recurrent fusion involved events, scaled to range [0, 1]. The dependency module was based on the RNAi and CRISPR screening profiles from the DepMap as well as the cancer cell line annotation information of DepMap. The RNA-seq, WES and SNP profiles of DepMap as well as recurrent genomic alteration information from the TCGA were also used to estimate this module. Detailed methods for the raw data processing and quantification were described in the method sections of dependency analyses. The features in dependency module included: 1) Combined essentiality index. Annotation of essential genes (“common essential” or “strongly selective”) based on genome-wide RNAi/CRISPR screening was retrieved from the Dependency Map (DepMap) portal (https://depmap.org/portal/) (retrieved date: Apr 17, 2020). An essentiality index was derived as the weighted sum of binary calls of “common essential” (weight=1) and “strongly selective” (weight=2), for each of the RNAi and CRISPR datasets separately. A combined essentiality index was then summarized and scaled to range [0, 1]. 2) Predictive power of copy number for cancer dependencies. We used linear regression models (limma) (Ritchie et al., 2015) to reveal the associations between cancer dependencies and copy number across all screened cancer cell lines for each of the RNAi and CRISPR datasets. A meta p-value was combined from p-values generated on individual datasets for each gene. Meta p-values were log transformed and scaled to range [0, 1], with 1 representing the genes with most significant p-values and 0 representing the genes with p-values of 1. 3) Predictive power of expression for cancer dependencies. We used linear regression models (limma) (Ritchie et al., 2015) to reveal the associations between cancer dependencies and gene expression across all screened cancer cell lines for each of the RNAi and CRISPR datasets. A meta p-value was combined from p-values generated on individual datasets for each gene. Meta p-values were log transformed and scaled to range [0, 1], with 1 representing the genes with most significant p-values and 0 representing the genes with p-values of 1. 4) Predictive power of mutation for cancer dependencies. We used linear regression models (limma) (Ritchie et al., 2015) to reveal the associations between cancer dependencies and gene mutation across all screened cancer cell lines for each of the RNAi and CRISPR datasets. Silent mutations were not considered. A meta p-value was combined from p-values generated on individual datasets for each gene. Meta p-values were log transformed and scaled to range [0, 1], with 1 representing the genes with most significant p-values and 0 representing the genes with p-values of 1. 5) Essentiality for fusion involved events. Fusion essentiality score and significance calculation were reported by Picco et al. (Picco et al., 2019). If multiple fusion transcripts were detected for a pair of fusion genes in a specific cancer cell line, the one with most significant essentiality score was kept. Fusion pairs present in at least three cell lines were considered for downstream analysis. For each gene with assessed fusion transcripts, the significance calculation (p-values) for all fusion transcripts across cancer cell lines was combined to yield a meta-p-value. Meta-p-values were then log transformed and scaled to range [0, 1], with 1 representing the genes with most significant p-values and 0 representing the genes with p-values of 1. A weighted average was calculated to summarize all features for each gene within each module. A core cancer target score (core PCDT-score) was then calculated as the weighted average of all three modules. To optimize the performance of the core PCDT-score in terms of highlighting promising cancer targets, we applied a grid search procedure to determine the weights of features within each module as well as the weights of the three modules. Using the genes that are currently targeted by FDA-approved small molecule drugs in oncology as positive controls, the grid search procedure iteratively assessed the ability of the score to prioritize cancer drug targets over a range of plausible weight values. A final combination of weights was chosen to maximize the discriminative power of PCDT-score.
Colony formation assay
Cells were seeded in 24-well plates one day before a series of dosages of THZ1 were added to culture medium. Cells were treated for 6 days with medium change every 3 days. After treatment, cells were fixed with methanol and stained with 0.5% crystal violet (Sigma, HT901). To quantify the results, crystal violet was dissolved in 10% SDS and the colored solution was transferred to 96-well plate for recording absorbance at 570 nm by using BioTek EL800 microplate reader.
RNA isolation and qRT-PCR
Total RNA was extracted by using TRIzol Reagent (Invitrogen) and reverse-transcribed by using a High Capacity RNA-to-cDNA Kit (Applied Biosystems) under conditions provided by the supplier. Real-time quantitative polymerase chain reaction (qRT-PCR) was performed by using SYBR Green reagents (Applied Biosystems) and ABI ViiA 7 System (Applied Biosystems) according to the manufacturer’s instructions. GAPDH (glyceraldehyde3-phosphate dehydrogenase) was used as an internal control. Delta delta Ct method was used for quantification. Primers used for qRT-RCR are as follows: CDK7 forward: GCCCCCGAGTTACTATTTGG, CDK7 reverse: GGTCTGAATCTCCTGGCAAA; GAPDH forward: ACACCATGGGGAAGGTGAAG, GAPDH reverse: AAGGGGTCATTGATGGCAAC.
Protein isolation and Western blot
Whole cell extracts were prepared by directly boiling harvested cells in 6X loading buffer. Proteins were resolved by SDS-PAGE and transferred to PVDF-membrane (Millipore, IPVH00010). The membrane was blocked in 5% blotting-grade blocker (Bio-Rad, 170-6404) and incubated with primary and secondary antibodies successively. Immunoreactive proteins were visualized by using the Western HRP substrate (Millipore, WBLUF0500). Antibodies used are as follows: anti-CDK7 (Santa Cruz, sc-7344), anti-β-tubulin (Cell signaling, 2128), anti-mouse IgG HRP linked (Cell signaling, 7076), anti-rabbit IgG HRP linked (Cell signaling, 7074).
CDK7 single allele knockout by CRISPR/Cas9
LentiCRISPRv2 and lentiviral packing vectors were purchased from Addgene. Two sgRNAs that targeted the flanking regions of CDK7 coding sequence (CDS) were subcloned into LentiCRISPRv2 vector. LentiCRISPRv2 and packaging vectors were transfected into the packaging cell line 293T (ATCC) by using the FuGENE6 Transfection Reagent (Promega). The medium containing lentivirus was collected 48h after transfection. OVCAR5 cells were simultaneously infected with lentivirus encoding two sgRNAs in the presence of 8 μg/ml polybrene (Sigma). After puromycin selection, OVCAR5 cells were expanded and picked up single clones by limiting dilution. Oligos used for sgRNA constructs are as follows: 5’ CDK7 sgRNA forward: CACCGTCAGCCACTAGATACAACTA, 5’ CDK7 sgRNA reverse: AAACTAGTTGTATCTAGTGGCTGAC; 3’ CDK7 sgRNA forward: CACCGTCACAAATCTGTAGTAGCAT, 3’ CDK7 sgRNA reverse: AAACATGCTACTACAGATTTGTGAC. Genomic DNA was extracted by using phenol/chloroform from each clone. PCR was performed to verify CDK7 wild type (WT) and knockout alleles, whose products were approximately 1,100 bp and 1,700 bp, respectively. Primers used for PCR verification are as follows: CDK7 knockout allele forward: CTAAGGGCTTTGCAG GTGTG, CDK7 knockout allele reverse: TGGCCTTGTGAGACCCTAAG; CDK7 WT allele forward: TTGAGTGCGTGTTTTCCCAG, CDK7 WT allele reverse: ACCAACTCCTAATGCCTGCT.
QUANTIFICATION AND STATISTICAL ANALYSIS
Large-scale and multi-dimensional profiling data generated by the publicly accessible databases (TCGA, GTEx, CPTAC, and DepMap) were used, therefore statistical analysis was not used to predetermine sample size in this study. For TCGA analysis, if more than one profiling file existed for a patient in TCGA, only one single file will be selected and used, and detailed methods for exclusion of duplicated profiling files are described in the STAR Method section. The computational analyses were not randomized, and the investigators were not blinded during data analyses of this study. When applicable, enrichment was tested using Fisher’s exact test with FDR correction. Linear regression models with different predictor variables (expression or copy number) were applied to fit the dependency profile of each target gene across screened cancer cell lines. Benjamini-Hochberg (BH) method was used to adjust the p-value (see method details). Cell viability and gene relative expression data were shown as means with standard deviation (SD). Comparisons between groups were performed using student t-test.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CDK7 antibody | Santa Cruz Biotechnoloy | Cat#sc-7344; RRID:AB627243 |
| Anti-b-Tubulin antibody | Cell Signaling Technology | Cat#2128; RRID AB_823664 |
| Anti-mouse IgG, HRP-linked antibody | Cell Signaling Technology | Cat#7076; RRID:AB_330924 |
| Anti-rabbit IgG, HRP-linked antibody | Cell Signaling Technology | Cat#7074; RRID:AB_2099233 |
| Chemicals, peptides, and recombinant proteins | ||
| THZ1 | Selleck Chemicals | Cat#S7549 |
| FuGENE 6 | Promega | Cat#E2691 |
| Crystal violet solution | Sigma-Aldrich | Cat#HT901 |
| Polybrene infection/transfection reagent | Sigma-Aldrich | Cat#TR-1003-G |
| PowerUp SYBR green master mix | Applied Biosystems | Cat#A25742 |
| Blotting-grade blocker | Bio-Rad | Cat#1706404 |
| Immobilon forte western HRP substrate | Millipore Sigma | Cat#WBLUF0500 |
| Critical commercial assays | ||
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368813 |
| Deposited data | ||
| TCGA genomic profiling | TCGA project | http://cancergenome.nih.gov |
| TCGA Affymetrix SNP6.0 array data (CEL) | TCGA Data Portal | https://tcga-data.nci.nih.gov/tcga/ |
| TCGA Affymetrix SNP6.0 array data (segmentation) | TCGA GDAC Firehose | http://gdac.broadinstitute.org/ |
| TCGA whole exome sequencing data | TCGA MC3 project | https://doi.org/10.7303/syn7214402 |
| TCGA RNA sequencing data | Genomic Data Commons | https://portal.gdc.cancer.gov/ |
| TCGA transcript fusion data | (Hu et al., 2018) | http://tumorfusions.org/ |
| Cancer Cell Line Encyclopedia (CCLE) | The Broad Institute | https://portals.broadinstitute.org/ccle |
| Dependency Map (DepMap) portal | The Broad Institute | https://depmap.org/portal/ |
| Drug Gene Interaction Database (DGIdb) | (Cotto et al., 2018) | https://www.dgidb.org/ |
| Open Targets Platform | (Carvalho-Silva et al., 2019) | https://platform.opentargets.org/ |
| Histone modification enzymes (HMEs) annotation | Structural Genomics Consortium | https://chromohub.thesgc.org/static/ChromoHub.html |
| PHAROS database | (Nguyen et al., 2017) | https://pharos.nih.gov/ |
| Target Central Resource Database (TCRD) | (Nguyen et al., 2017) | http://juniper.health.unm.edu/tcrd/ |
| Functional Cancer Genome data portal | This paper | http://fcgportal.org/home |
| Cancer Druggable Gene Atlas (TCDA) | This paper | http://fcgportal.org/TCDA/ |
| Experimental models: Cell lines | ||
| OVCAR3 | ATCC | HTB-161 |
| MCF7 | ATCC | HTB-22 |
| HCC1937 | ATCC | CRL-2336 |
| CAOV3 | ATCC | HTB-75 |
| MDA-MB-468 | ATCC | HTB-132 |
| HCC38 | ATCC | CRL-2314 |
| SKBR3 | ATCC | HTB-30 |
| SKOV3 | ATCC | HTB-77 |
| UWB1.289 | ATCC | CRL-2945 |
| 293T | ATCC | CRL-3216 |
| OVCAR4 | NCI Development Therapeutics Program | N/A |
| OVCAR5 | NCI Development Therapeutics Program | N/A |
| Oligonucleotides | ||
| Primers used for PCR analyses | This paper | N/A |
| Oligos used for sgRNA constructs | This paper | N/A |
| Recombinant DNA | ||
| LentiCRISPRv2 | (Sanjana et al., 2014) | Addgene #52961 |
| Software and algorithms | ||
| ABSOLUTE | (Carter et al., 2012) | https://software.broadinstitute.org/cancer/cga/absolute |
| GISTIC 2.0 | (Mermel et al., 2011) | ftp://ftp.broadinstitute.org/pub/GISTIC2.0/ |
| MutSigCV | (Lawrence et al., 2013) | http://software.broadinstitute.org/cancer/software/genepattern/modules/docs/MutSigCV |
| Oncodrivefm | (Gonzalez-Perez and Lopez-Bigas, 2012) | http://bg.upf.edu/group/projects/oncodrive-fm.php |
| OncodriveCLUST | (Tamborero et al., 2013) | http://bg.upf.edu/group/projects/oncodrive-clust.php |
| ActiveDriver | (Reimand and Bader, 2013) | http://www.baderlab.org/Software/ActiveDriver |
| HotSpot3D | (Niuetal., 2016) | https://github.com/ding-lab/hotspot3d |
| fGSEA | R package | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
Highlights.
We combine 6 druggable genome resources and define 6,083 genes as PDGs
We characterize the expression, genomic alteration, and dependency of PDGs in cancers
We estimate a PDG cancer drug target score, including 16 cancer-related features
TCDA is developed and available to the public
ACKNOWLEDGMENTS
We thank the IDG, PHAROS, DGIdb, Open Targets, TCGA, GTEx, and DepMap teams for generating and providing pharmacological, genomic, and functionomic data resources. This work was supported, in whole or in part, by the Harry Fields Professorship and Abramson Cancer Center. L.Z. was supported by the Basser Center for BRCA and the US National Institutes of Health (NIH) grants (R01CA142776, R01CA190415, R01CA225929, R01CA262070, P50CA083638, and P50CA174523). R.H. was supported by NIH grants (P01CA210944 and R01CA229803). X.H. was supported by the Ovarian Cancer Research Alliance. X.H. and Y.Z. were supported by the Foundation for Women’s Cancer. Support of the core facilities was provided by a NIH Cancer Center Support Grant (P30CA016520) to Abramson Cancer Center.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110400.
DECLARATION OF INTERESTS
L.Z. and X.H. report having received research funding from AstraZeneca, Bristol-Myers Squibb/Celgene, and Prelude Therapeutics. O.T. and H.M.C. are employees of AstraZeneca. R.H.V. is an inventor on a licensed patent relating to cancer cellular immunotherapy and receives royalties from Children’s Hospital Boston for a licensed research-only monoclonal antibody.
REFERENCES
- Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. (2018). Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385.e318. 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausch-Fluck D, Goldmann U, Muller S, van Oostrum M, Muller M, Schubert OT, and Wollscheid B (2018). The in silico human surfaceome. Proc. Natl. Acad. Sci. U S A. 115, E10988–E10997. 10.1073/pnas.1808790115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behan FM, Iorio F, Picco G, Goncalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M, et al. (2019). Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 568, 511–516. 10.1038/s41586-019-1103-9. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodological) 57, 289–300. [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KK, Hann MM, Lakdawala AS, Santos R, Thomas PJ, and Todd K (2018). Approaches to target tractability assessment - a practical perspective. Medchemcomm 9, 606–613. 10.1039/c7md00633k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell SJ, Gaulton A, Marshall J, Bichko D, Martin S, Brouwer C, and Harland L (2012). Visualizing the drug target landscape. Drug Discov. Today 15, 3–15. 10.1016/j.drudis.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Cancer Cell Line Encyclopedia Consortium; Genomics of Drug Sensitivity in Cancer Consortium (2015). Pharmacogenomic agreement between two cancer cell line data sets. Nature 528, 84–87. 10.1038/nature15736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. (2012). Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421. 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho-Silva D, Pierleoni A, Pignatelli M, Ong C, Fumis L, Karamanis N, Carmona M, Faulconbridge A, Hercules A, McAuley E, et al. (2019). Open Targets Platform: new developments and updates two years on. Nucleic Acids Res. 47, D1056–D1065. 10.1093/nar/gky1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH, et al. (2017). OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017, PO.17.00011. 10.1200/PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, Gao J, Socci ND, Solit DB, Olshen AB, et al. (2016). Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 34, 155–163. 10.1038/nbt.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotto KC, Wagner AH, Feng Y-Y, Kiwala S, Coffman AC, Spies G, Wollam A, Spies NC, Griffith OL, and Griffith M (2018). DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 46, D1068–D1073. 10.1093/nar/gkx1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster JM, Rossen J, Kazachkova M, Pan J, Kugener G, Root DE, and Tsherniak A (2019). Extracting biological insights from the project achilles genome-scale CRISPR screens in cancer cell lines. bioRxiv 720243. 10.1101/720243. [DOI] [Google Scholar]
- Dougherty JD, Schmidt EF, Nakajima M, and Heintz N (2010). Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res. 38, 4218–4230. 10.1093/nar/gkq130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, Hess J, Ma S, Chiotti KE, McLellan M, et al. (2018). Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst 6, 271–281.e277. 10.1016/j.cels.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finan C, Gaulton A, Kruger FA, Lumbers RT, Shah T, Engmann J, Galver L, Kelley R, Karlsson A, Santos R, et al. (2017). The druggable genome and support for target identification and validation in drug development. Sci. Transl Med. 9, eaag1166. 10.1126/scitranslmed.aag1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei E 3rd. (1993). Gene deletion: a new target for cancer chemotherapy. Lancet 342, 662–664. 10.1016/0140-6736(93)91764-d. [DOI] [PubMed] [Google Scholar]
- Garraway LA, and Lander ES (2013). Lessons from the cancer genome. Cell 153, 17–37. 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez A, and Lopez-Bigas N (2012). Functional impact bias reveals cancer drivers. Nucleic Acids Res. 40 (21), e169. 10.1093/nar/gks743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann O, Caballero OL, Stevenson BJ, Chen YT, Cohen T, Chua R, Maher CA, Panji S, Schaefer U, Kruger A, et al. (2008). Genome-wide analysis of cancer/testis gene expression. Proc. Natl. Acad. Sci. U S A. 105, 20422–20427. 10.1073/pnas.0810777105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins AL, and Groom CR (2002). The druggable genome. Nat. Rev. Drug Discov. 1, 727–730. 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Hu S, Marineau JJ, Rajagopal N, Hamman KB, Choi YJ, Schmidt DR, Ke N, Johannessen L, Bradley MJ, Orlando DA, et al. (2019a). Discovery and characterization of SY-1365, a selective, covalent inhibitor of CDK7. Cancer Res. 79, 3479–3491. 10.1158/0008-5472.CAN-19-0119. [DOI] [PubMed] [Google Scholar]
- Hu X, Wang Q, Tang M, Barthel F, Amin S, Yoshihara K, Lang FM, Martinez-Ledesma E, Lee SH, Zheng S, and Verhaak RGW (2018). TumorFusions: an integrative resource for cancer-associated transcript fusions. Nucleic Acids Res. 46, D1144–D1149. 10.1093/nar/gkx1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Yuan J, Long M, Jiang J, Zhang Y, Zhang T, Xu M, Fan Y, Tanyi JL, Montone KT, et al. (2021). The Cancer Surfaceome Atlas integrates genomic, functional and drug response data to identify actionable targets. Nat. Cancer 2, 1406–1422. 10.1038/s43018-021-00282-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Zhou J, Jiang J, Yuan J, Zhang Y, Wei X, Loo N, Wang Y, Pan Y, Zhang T, et al. (2019b). Genomic characterization of genes encoding histone acetylation modulator proteins identifies therapeutic targets for cancer treatment. Nat. Commun. 10, 733. 10.1038/s41467-019-08554-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A, Garraway LA, Ashworth A, and Weber B (2020). Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 19, 23–38. 10.1038/s41573-019-0046-z. [DOI] [PubMed] [Google Scholar]
- Jain A, and Tuteja G (2019). TissueEnrich: tissue-specific gene enrichment analysis. Bioinformatics 35, 1966–1967. 10.1093/bioinformatics/bty890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, Chamberlain PP, Mani DR, Man HW, Gandhi AK, et al. (2015). Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 523, 183–188. 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RD, Chang LW, Ellis MJ, and Bose R (2013). Prioritizing potentially druggable mutations with dGene: an annotation tool for cancer genome sequencing data. PLoS ONE 8, e67980. 10.1371/journal.pone.0067980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. (2014). Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511, 616–620. 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Mehta S, Kucuk-McGinty H, Turner JP, Vidovic D, Forlin M, Koleti A, Nguyen DT, Jensen LJ, Guha R, et al. (2017). Drug target ontology to classify and integrate drug discovery data. J. Biomed. Semantics 8, 50. 10.1186/s13326-017-0161-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland JM, Ho ZV, Kugener G, Dempster JM, Montgomery PG, Bryan JG, Krill-Burger JM, Green TM, Vazquez F, Boehm JS, et al. (2018). Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 9, 4610. 10.1038/s41467-018-06916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41. 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784. 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AR, Rosenberg SC, McCormick F, and Malek S (2020). RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discov. 19, 533–552. 10.1038/s41573-020-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DT, Mathias S, Bologa C, Brunak S, Fernandez N, Gaulton A, Hersey A, Holmes J, Jensen LJ, Karlsson A, et al. (2017). Pharos: collating protein information to shed light on the druggable genome. Nucleic Acids Res. 45, D995–D1002. 10.1093/nar/gkw1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CA, Gibson WJ, Brown MS, Kosmicki JA, Busanovich JP, Wei H, Urbanski LM, Curimjee N, Berger AC, Gao GF, et al. (2020). Loss of heterozygosity of essential genes represents a widespread class of potential cancer vulnerabilities. Nat. Commun. 11, 2517. 10.1038/s41467-020-16399-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhawan D, Zack TI, Ren Y, Strickland MR, Lamothe R, Schumacher SE, Tsherniak A, Besche HC, Rosenbluh J, Shehata S, et al. (2012). Cancer vulnerabilities unveiled by genomic loss. Cell 150, 842–854. 10.1016/j.cell.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu B, Scott AD, Sengupta S, Bailey MH, Batra P, Ning J, Wyczalkowski MA, Liang WW, Zhang Q, McLellan MD, et al. (2016). Protein-structure-guided discovery of functional mutations across 19 cancer types. Nat. Genet. 48 (8), 827–837. 10.1038/ng.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, and Wigler M (2004). Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572. 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- Oprea TI, Bologa CG, Brunak S, Campbell A, Gan GN, Gaulton A, Gomez SM, Guha R, Hersey A, Holmes J, et al. (2018). Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discov. 17, 377. 10.1038/nrd.2018.52. [DOI] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, and Hopkins AL (2006). How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996. 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Paolella BR, Gibson WJ, Urbanski LM, Alberta JA, Zack TI, Bandopadhayay P, Nichols CA, Agarwalla PK, Brown MS, Lamothe R, et al. (2017). Copy-number and gene dependency analysis reveals partial copy loss of wild-type SF3B1 as a novel cancer vulnerability. Elife 6, e23268. 10.7554/eLife.23268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picco G, Chen ED, Alonso LG, Behan FM, Goncalves E, Bignell G, Matchan A, Fu B, Banerjee R, Anderson E, et al. (2019). Functional linkage of gene fusions to cancer cell fitness assessed by pharmacological and CRISPR-Cas9 screening. Nat. Commun. 10, 2198. 10.1038/s41467-019-09940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radenbaugh AJ, Ma S, Ewing A, Stuart JM, Collisson EA, Zhu J, and Haussler D (2014). RADIA: RNA and DNA integrated analysis for somatic mutation detection. PLoS ONE 9, e111516. 10.1371/journal.pone.0111516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rask-Andersen M, Almén MS, and Schiöth HB (2011). Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 10, 579–590. 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- Rask-Andersen M, Masuram S, and Schiöth HB (2014). The druggable genome: evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annu. Rev. Pharmacol. Toxicol. 54, 9–26. 10.1146/annurev-pharmtox-011613-135943. [DOI] [PubMed] [Google Scholar]
- Reimand J, and Bader GD (2013). Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol. Syst. Biol. 9. 10.1038/msb.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendo V, Stoimenov I, Mateus A, Sjoberg E, Svensson R, Gustavsson AL, Johansson L, Ng A, O’Brien C, Giannakis M, et al. (2020). Exploiting loss of heterozygosity for allele-selective colorectal cancer chemotherapy. Nat. Commun. 11, 1308. 10.1038/s41467-020-15111-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Perez C, Tamborero D, Schroeder MP, Antolin AA, Deu-Pons J, Perez-Llamas C, Mestres J, Gonzalez-Perez A, and Lopez-Bigas N (2015). In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell 27, 382–396. 10.1016/j.ccell.2015.02.007. [DOI] [PubMed] [Google Scholar]
- Russ AP, and Lampel S (2005). The druggable genome: an update. Drug Discov. Today 10, 1607–1610. 10.1016/S1359-6446(05)03666-4. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, et al. (2018). Oncogenic signaling pathways in the cancer genome Atlas. Cell 173, 321–337.e310. 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 8, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, and Overington JP (2017). A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34. 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan W, Yuan J, Hu Z, Jiang J, Wang Y, Loo N, Fan L, Tang Z, Zhang T, Xu M, et al. (2020). Systematic characterization of recurrent genomic alterations in cyclin-dependent kinases reveals potential therapeutic strategies for cancer treatment. Cell Rep 32, 107884. 10.1016/j.celrep.2020.107884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH, Buneman OP, Davenport AP, McGrath JC, Peters JA, et al. (2015). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res. 44, D1054–D1068. 10.1093/nar/gkv1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamborero D, Gonzalez-Perez A, and Lopez-Bigas N (2013). Oncodrive-CLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 29 (18), 2238–2244. 10.1093/bioinformatics/btt395. [DOI] [PubMed] [Google Scholar]
- Torres-Garcia W, Zheng S, Sivachenko A, Vegesna R, Wang Q, Yao R, Berger MF, Weinstein JN, Getz G, and Verhaak RG (2014). PRADA: pipeline for RNA sequencing data analysis. Bioinformatics 30, 2224–2226. 10.1093/bioinformatics/btu169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torsten H, Kurt H, Mark AVDW, and Achim Z (2006). A lego system for conditional inference. The Am. Statistician 60, 257–263. 10.1198/000313006X118430. [DOI] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a cancer dependency map. Cell 170, 564–576.e516. 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. (2004). In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848. 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, Pfeil J, Narkizian J, Deran AD, Musselman-Brown A, et al. (2017). Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 35, 314–316. 10.1038/nbt.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., and Kinzler KW (2013). Cancer genome landscapes. Science 339, 1546–1558. 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Qiao Y, Asangani IA, Ateeq B, Poliakov A, Cieslik M, Pitchiaya S, Chakravarthi B, Cao X, Jing X, et al. (2017). Development of peptidomimetic inhibitors of the ERG gene fusion product in prostate cancer. Cancer Cell 31, 532–548.e537. 10.1016/j.ccell.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CH, Allot A, Leaman R, and Lu Z (2019). PubTator central: automated concept annotation for biomedical full text articles. Nucleic Acids Res. 47, W587–W593. 10.1093/nar/gkz389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao SJ, Zhang C, Zou Q, and Ji ZL (2010). TiSGeD: a database for tissue-specific genes. Bioinformatics 26, 1273–1275. 10.1093/bioinformatics/btq109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, et al. (2005). Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21, 650–659. 10.1093/bioinformatics/bti042. [DOI] [PubMed] [Google Scholar]
- Yap TA, and Workman P (2012). Exploiting the cancer genome: strategies for the discovery and clinical development of targeted molecular therapeutics. Annu. Rev. Pharmacol. Toxicol. 52, 549–573. 10.1146/annurev-pharmtox-010611-134532. [DOI] [PubMed] [Google Scholar]
- Yuan J, Hu Z, Mahal BA, Zhao SD, Kensler KH, Pi J, Hu X, Zhang Y, Wang Y, Jiang J, et al. (2018). Integrated analysis of genetic ancestry and genomic alterations across cancers. Cancer Cell 34, 549–560.e549. 10.1016/j.ccell.2018.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Van Allen EM, Omberg L, Wagle N, Amin-Mansour A, Sokolov A, Byers LA, Xu Y, Hess KR, Diao L, et al. (2014). Assessing the clinical utility of cancer genomic and proteomic data across tumor types. Nat. Biotechnol. 32, 644–652. 10.1038/nbt.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH, et al. (2013). Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140. 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wang J, Wang Y, Zhang Y, Castro P, Shao L, Sreekumar A, Putluri N, Guha N, Deepak S, et al. (2016). MNX1 is oncogenically upregulated in African-American prostate cancer. Cancer Res. 76, 6290–6298. 10.1158/0008-5472.CAN-16-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Information of gene family category and target development level were generated by the IDG project, which are publicly available through the PHAROS database (https://pharos.nih.gov/) and the Target Center Resource Database (TCRD; http://juniper.health.unm.edu/tcrd/). The genomic profiles of human cancers were generated by the TCGA project, which are publicly available through the Genomic Data Commons portal (GDC, https://gdc-portal.nci.nih.gov). The RNA expression profiles of human normal healthy tissues were by the GTEx project, which are publicly available through the GTEx portal (https://gtexportal.org/home/). Genetic screening profiles in human cancer cell lines were generated by the DepMap and the Score projects, which are publicly available through the DepMap portal (https://depmap.org/portal/), and the Score projects (https://score.depmap.sanger.ac.uk/). The genomic data were retrieved, processed and analyzed through a master computational protocol developed by the Functional Cancer Genome project (FCG, http://fcgportal.org/home/) as described by our previous publications (Hu et al., 2019b, 2021; Shan et al., 2020; Yuan et al., 2018) as well as the STAR Method section. The data generated by this study are public available through the Functional Cancer Genome data portal (http://fcgportal.org/home) and the Cancer Druggable Gene Atlas (TCDA) website (http://fcgportal.org/TCDA/). This paper does not report original code. Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.