

Abstract
Purpose
To evaluate intraperitoneal (IP) nab-paclitaxel in patients with advanced malignancies that are primarily confined to the peritoneal cavity in a phase I trial.
Methods
Using a 3+3 dose escalation of IP nab-paclitaxel on days 1, 8, and 15 of a 28-day cycle we evaluated 6 dose levels (35–175mg/m2/dose). Maximum tolerated dose (MTD) and pharmacokinetics (PK) of IP nab-paclitaxel were determined.
Results
There were no dose limiting toxicities (DLTs) in cohorts 1–3. There was a DLT in one of six patients in cohort 4 (112.5mg/m2) (grade 3 neutropenia causing treatment delay >15 days) and a DLT in one of three patients in cohort 6 (175mg/m2) (grade 4 neutropenia and grade 3 abdominal pain). A second patient in cohort 6 experienced a serious adverse event (cycle 1, grade 4 ANC ≤7 days, cycle 4, grade 2 left ventricular dysfunction). This dose level was determined to be above the MTD. No DLTs were seen in seven patients treated in cohort 5 (140mg/m2). The MTD of IP nab-paclitaxel was established at 140mg/m2 on days 1, 8, and 15 of a 28-day cycle. There was a PK advantage for IP nab-paclitaxel, with an IP plasma area under the concentration-time curve (AUC) ratio of 147-fold (range 50–403) and therapeutic range systemic drug levels. Eight of 27 enrolled patients had progression free survival ≥6 months. One patient experienced complete response, and one patient experienced partial response. Six patients had stable disease.
Conclusions
Weekly IP nab-paclitaxel has a favorable toxicity profile, a significant pharmacologic advantage, and promising clinical activity.
Keywords: intraperitoneal chemotherapy, nab-paclitaxel, pharmacologic advantage, peritoneal carcinomatosis, ovarian cancer
Introduction
Peritoneal carcinomatosis is a typical presentation of advanced ovarian carcinoma and is less commonly the initial presentation for other epithelial carcinomas. Potentially curative therapy exists for ovarian carcinoma at initial presentation; however, therapeutic options for other malignancies presenting with these findings are limited. In ovarian cancer, there is a definite dose-response relationship, with higher doses of chemotherapeutic agents showing higher response rates [1]. Delivery of chemotherapeutic agents to these tumors via intraperitoneal (IP) administration allows increased dose intensity to tumors with the primary site of disease in the peritoneal cavity.
This route of administration confers a pharmacologic advantage allowing large concentrations of active agents to reach the tumor while minimizing systemic toxicity. Several chemotherapeutic agents have been utilized for IP administration including melphalan, cisplatin, carboplatin, 5-fluorouracil, methotrexate, cytosine arabinoside, paclitaxel, gemcitabine, docetaxel, and bleomycin. The pharmacologic advantage of various drugs, defined as the ratio of the peak IP drug level (the highest IP concentration of a drug) to corresponding plasma values, is agent specific and ranges from 7 to almost 8000.
Three sequential randomized Phase III GOG trials demonstrated a survival advantage for IP versus intravenous (IV) chemotherapy in patients with advanced epithelial ovarian cancer with low volume residual disease after primary debulking surgery [2–4]. The first two studies were not adopted by the oncology community, due to criticism for using an “outdated” platinum regimen which did not include paclitaxel [2] or concern for severe toxicity and inability to deliver IP chemotherapy to a large number of patients[3]. GOG 172, however, had a more significant clinical impact on the treatment of ovarian cancer compared to previous studies [4], where Armstrong et al demonstrated improved PFS (23.8 vs 18.3 months) and OS (65.6 vs 49.7 months) in patients with advanced, optimally debulked ovarian cancer who received the experimental arm of IP cisplatin and IV/IP paclitaxel compared to IV cisplatin/paclitaxel. Even with the substantial benefit in OS, toxicity of the IP arm of the GOG 172 trial was substantial, explaining why many physicians were reluctant to adopt IP chemotherapy. Nevertheless, the improvement in survival led to the 2006 NCI clinical announcement recommending the use of IP therapy in optimally debulked ovarian patients [5].
Following the publication of these landmark clinical trials and NCI acknowledgement of improved clinical outcomes associated with IP chemotherapy in ovarian cancer, investigators in several centers and cooperative groups initiated a series of IP trials with other chemotherapy agents or combination regimens, with the ultimate goal to maintain the advantage of regional administration of chemotherapy, while improving the toxicity.
Between 1998 and 2007 our institution reported several phase I studies evaluating IP administration of iododeoxyuridine, docetaxel, and gemcitabine, demonstrating a 67–850 fold peritoneal advantage depending on the agent [6–8].
By the mid-2000s, IV nab-paclitaxel (a solvent-free, albumin-bound paclitaxel) had demonstrated antitumor activity in women with metastatic breast cancer, and the IV weekly administration of this agent showed a 15% objective response in patients whose disease progressed despite conventional taxane therapy [9]. The regimen was well tolerated. Evidence of nab-paclitaxel activity in recurrent ovarian cancer after prior taxane exposure, was first demonstrated in patients with platinum sensitive disease [10].
The current study reports the results of a phase I trial in patients with peritoneal carcinomatosis and evaluates IP nab-paclitaxel on days 1, 8, and 15 of a 28-day cycle. The goal of the study was to determine the maximum tolerated dose (MTD) and to evaluate its toxicity. Secondary goals were to evaluate the pharmacokinetics (PK) of IP nab-paclitaxel and to further explore peripheral neuropathy through pre-treatment and sequential evaluation of the Neuropathic Pain Syndrome Inventory and Serial Nerve Conduction Studies.
Materials and Methods
Patient Eligibility
The following patients were eligible for this study: Adults with histologically confirmed advanced cancer primarily confined to the peritoneal cavity for which no “standard” chemotherapy regimens existed with an Eastern Cooperative Group (ECOG) performance status (PS) of 0–2, adequate hematological, hepatic (total bilirubin within normal institutional limits, liver enzymes <2.5 x upper limit of normal, ULN), and renal function (estimated creatinine clearance >60 mL/min/1.73 m2). Prior IP chemotherapy was allowed. Patients with ovarian cancer having residual disease at second-look laparotomy or following secondary debulking were also eligible and were enrolled >4 weeks after surgery. There was no limit on the number of prior lines of chemotherapy, but the protocol required a 4 week washout from previous chemotherapy or radiotherapy (6 weeks for nitrosoureas or mitomycin C). Prior taxane exposure was allowed if pre-existing sensory neuropathy was ≤grade 1.
The following patients were not eligible: patients with ongoing abdominal infections, bowel obstruction, and peritoneal adhesions that precluded the placement of an IP catheter; patients with known brain metastases; pregnant or breastfeeding women; patients with a history of allergic reactions to compounds similar to nab-paclitaxel; and patients with serious or uncontrolled intercurrent illness. Patients with “massive ascites” requiring therapeutic paracentesis were evaluated on an individual basis prior to enrollment. The Institutional Review board (IRB) of the two participating centers, City of Hope, CA and Swedish Cancer Institute, WA approved the conduct of the trial (ClinicalTrials.gov Identifier NCT00825201), which was in accordance with the Declaration of Helsinki and Good Clinical Practice. Written informed consent was obtained from all patients prior to inclusion onto the study according to institutional guidelines.
Study treatment
The study was designed as a 3+3 dose escalation clinical trial. Treatment was administered on an outpatient basis. Nab-paclitaxel was administered by IP infusion weekly on days 1, 8, and 15 of a 28-day cycle in successive cohorts of patients with no intra-patient dose escalation. The starting dose was determined by decreasing the usual IV dose by 60% and escalating according to a modified Fibonacci scheme. Doses explored were 35, 70, 90, 112.5, 140, and 175 mg/m2. All patients required surgical placement of an IP catheter by standard surgical procedures. Sequential assessment of the sensory neurotoxicity was conducted during the course of treatment and included optional quantitative sensory and nerve conduction tests, peripheral neuropathy composite score, and neuropathic pain syndrome inventory. The assessment was done at baseline, prior to cycle three, and at the end of study. Additional testing was offered to patients who developed grade 3 neuropathy.
Toxicity evaluation, dose escalation rules, and response assessment
Dose limiting toxicities (DLTs) and adverse events (AE) were graded by the NCI Common Terminology Criteria of Adverse Events (CTC-AE) version 3.0. Patients were enrolled per dose level in cohorts of three. The MTD was defined as <2 patients out of six with a DLT and required at least six patients to be treated at that dose. To be evaluable for toxicity, a patient must have completed two out of the three weekly administrations of nab-paclitaxel during the first cycle of treatment or have experienced a DLT. Patients who were not evaluable for toxicity were replaced. All patients who did not experience a DLT were observed for a minimum of four weeks after the start of the first course, before the dose level was escalated.
DLTs were defined as any of the following events during the first course of treatment and attributable to nab-paclitaxel: grade 4 neutropenia lasting more than 7 days or associated with fever or infection; grade 4 thrombocytopenia, or any grade 3 or 4 non hematological toxicity with the following exceptions: grade 3 or 4 nausea or vomiting that occurred without maximal antiemetic therapy; grade 3 or 4 diarrhea due to patient noncompliance with loperamide; grade 3 alopecia; and grade 3 fatigue. Failure to complete at least 66% of planned dose during cycle 1 due to toxicity and any AE that resulted in a delay of treatment for >15 days were also considered DLTs. All patients underwent baseline radiologic evaluation (CT or PET/CT). Restaging scans were obtained every 8 weeks until progressive toxicity, intolerable toxicity, or patient’s request to discontinue treatment. RECIST 1.0 criteria were used for response assessment.
Plasma and peritoneal samples for PK studies were obtained on cycle 1, day 1 and cycle 1, day 15 pre-instillation, hour 0, 1, 2, 4, 6, 8, 12, 24, and 48 following completion of nab -paclitaxel administration. Total and free paclitaxel in plasma were measured using a modification of the LC-MS/MS method of Gardner et al [11]. Briefly, after the addition of paclitaxel-d5 (Cambridge Isotope Laboratories, Tewksbury, MA, USA) as an internal standard, total paclitaxel was extracted from plasma by protein precipitation, and free paclitaxel was extracted by ultracentrifugation using a Centrifree micropartition device (EMD Millipore, Billerica, MA, USA). Following extraction, paclitaxel concentrations were determined by reversed-phase liquid chromatography and tandem mass spectrometry. The lower limit of quantitation for paclitaxel was 4 ng/ml, and the intra- and inter-day accuracy and precision of the assay were within ± 10% of target values.
PK data analyses were performed using non-compartmental methods according to the rule of linear trapezoids. Individual PK parameter estimates (Cmax and AUC0-t) for total plasma and peritoneal paclitaxel were determined and tabulated using summary statistics (medians and ranges). The pharmacologic advantage of IP nab-paclitaxel was defined as the AUC peritoneal/AUC plasma.
Results
Patient characteristics
Twenty seven patients with peritoneal carcinomatosis secondary to gynecologic (n=14) and gastrointestinal (n=12) malignancies and peritoneal mesothelioma (n=1) were enrolled on this study. The starting dose level was 35 mg/m2 and escalated to 175 mg/m2.
Between April 2009 and November 2014 treatment was initiated in 27 patients. Two patients were not evaluable for cycle 1 toxicity; one patient in the 35 mg/m2 cohort had port failure, and a second patient in the 70 mg/m2 cohort dose was held due to leukopenia that did not qualify as a DLT. The clinico-pathologic characteristics of the 27 treated patients are shown in Table 1.
Table 1:
Patient Characteristics (N=27)
| Age: median (range) | 59 (38–77) |
|
| |
| Gender | |
| male | 7 |
| female | 20 |
|
| |
| ECOG score at screening | |
| 0 | 3 |
| 1 | 20 |
| 2 | 4 |
|
| |
| Number of Prior Systemic Chemotherapy Treatments | |
| 1 | 7 |
| 2 | 12 |
| > 2 | 8 |
|
| |
| Tumor Types | |
| GI | |
| colon/appendix/rectosigmoid/rectum | 8 |
| stomach | 2 |
| pancreas | 1 |
| bile duct | 1 |
| GYN | |
| Ovary/fallopian | 9 |
| Uterus | 2 |
| Cervix | 3 |
| Other (mesothelioma) | 1 |
DLT and the maximum tolerated dose
No DLTs were observed in the first three cohorts. One patient treated in cohort 4 (112.5 mg/m2) experienced grade 3 neutropenia resulting in a treatment delay >15 days, which qualified as a DLT. This cohort was expanded to a total of 6 patients with no additional DLTs. One DLT was noted on the first patient treated in cohort 6 (175 mg/m2) and combined with a second serious AE (SAE); this dose level was closed and determined to be above the MTD. The patient with the DLT had both grade 4 neutropenia and grade 3 abdominal pain possibly related to the IP administration of nab-paclitaxel. The second patient treated in cohort 6 experienced asymptomatic grade 2 left ventricular systolic dysfunction during cycle 4, possibly related to nab-paclitaxel, detected on a routine 2D-echocardiogram. This patient had been exposed to doxorubicin (total dose 180 mg/m2) and one cycle of pegylated liposomal doxorubicin 40 mg/ m2 prior to nab-paclitaxel, but a baseline 2D- echocardiogram prior to enrollment onto this clinical trial had demonstrated a normal ejection fraction. The patient had no prior history of coronary disease and no cardiac complaints at the time the systolic dysfunction was detected. This event did not qualify for a DLT but was considered a SAE. Although a third patient treated in cohort 6 tolerated the treatment well with no major toxicities, this dose level was considered intolerable, and a decision was made to stop accrual to cohort 6 (175 mg/m2). The previous cohort, cohort 5, (140 mg/m2) had initially enrolled three patients with no DLTs, and this cohort was expanded and enrolled four additional patients. This dose level was well tolerated, and no DLTs were observed in the seven patients treated in this cohort. The MTD of IP nab-paclitaxel was established at 140 mg/m2 on days 1, 8, and 15 of a 28-day cycle. Table 2 shows dose level summary and Table 3 shows detailed toxicities.
Table 2:
Treatment Summary
| Nab-paclitaxel (mg/m2) |
Patients treated |
Patients excluded from course one tox. eval. | Patients excluded from response evaluation | Completed cycles median (range) (excluding ineligible patients for response) |
Patients with DLTs | DLT description |
Best responses during therapy (all eligible patients for response) |
|---|---|---|---|---|---|---|---|
| Cohort 1 – 35 | 4 | 1* | 1* | 2 (2–2) | 0 | - | PD – 3 NA – 1 |
| Cohort 2 – 70 | 4 | 1** | 0 | 15 (6–18) | 0 | - | CR - 1 SD – 3 |
| Cohort 3 – 90 | 3 | 0 | 0 | 2 (2–10) | 0 | - | SD – 1 PD – 2 |
| Cohort 4 – 112.5 | 6 | 0 | 2*** | 2 (1–3) | 1 | Gr3 Leukocyte Cnt Decr and Gr3 Neutrophil Cnt Decr | SD – 2 PD – 2 NA – 2 |
| Cohort 5 – 140 | 7 | 0 | 0 | 2 (2–12) | 0 | - | PR – 1 SD – 3 PD – 3 |
| Cohort 6 – 175 | 3 | 0 | 1# | 2 (1–4) | 1## | Gr4 Neutrophil Cnt Decr and Gr3 Abdominal Pain | SD – 1 PD – 1 NA – 1 |
Second patient did not receive full course of course 1 treatment due to port complications; terminated treatment prior to completion of course 2.
Second patient held treatment on day 8 of course 1 due to low wbc (not a DLT).
Second patient terminated treatment due to toxicity and the third patient died (sepsis) prior to the first disease evaluation.
Third patient off treatment for toxicity prior to first disease evaluation.
Second patient on arm 6 had grade 4 ANC, and decr ejection fraction cycle 4.
Table 3:
Treatment-related toxicity, arm by grade, n (%)
| Adverse Event | TREATMENT ARM BY GRADE, n(%) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Abraxane 35 mg/m2 (n=4) | Abraxane 70 mg/m2 (n=4) | Abraxane 90 mg/m2 (n=3) | Abraxane 112.5 mg/m2 (n=6) | Abraxane 140 mg/m2 (n=7) | Abraxane 175 mg/m2 (n=3) | |||||||||||||
| 2 | 3 | 4 | 2 | 3 | 4 | 2 | 3 | 4 | 2 | 3 | 4 | 2 | 3 | 4 | 2 | 3 | 4 | |
| AST, SGOT | 0 | 0 | 0 | 0 | 0 | 0 | 2(67%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Albumin, serum-low | 0 | 0 | 0 | 1(25%) | 0 | 0 | 0 | 0 | 0 | 1(17%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Alkaline phosphatase | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1(33%) | 0 | 1(17%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Amylase | 0 | 0 | 0 | 0 | 0 | 1(25%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Anorexia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1(17%) | 0 | 1(14%) | 0 | 0 | 0 | 0 | 0 |
| Diarrhea | 0 | 0 | 0 | 1(25%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1(14%) | 0 | 0 | 0 | 0 | 0 |
| Distension/bloating, abdominal | 0 | 0 | 0 | 1(25%) | 0 | 0 | 0 | 0 | 0 | 2(33%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Fatigue | 0 | 0 | 0 | 2(50%) | 0 | 0 | 1(33%) | 0 | 0 | 1(17%) | 1(17%) | 0 | 3(43%) | 2(29%) | 0 | 1(33%) | 1(33%) | 0 |
| Glucose, serum-high | 0 | 0 | 0 | 1(25%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hair loss/alopecia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1(17%) | 0 | 0 | 1(14%) | 0 | 0 | 0 | 0 | 0 |
| Hemoglobin | 1(25%) | 0 | 0 | 2(50%) | 0 | 0 | 2(67%) | 0 | 0 | 1(17%) | 1(17%) | 0 | 1(14%) | 2(29%) | 0 | 2(67%) | 0 | 0 |
| Infection | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1(33%) | 0 |
| Left ventricular systolic dysfunction | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1(33%) | 0 | 0 |
| Leukocytes | 0 | 0 | 0 | 1(25%) | 1(25%) | 0 | 0 | 0 | 0 | 2(33%) | 1(17%) | 1(17%) | 3(43%) | 1(14%) | 0 | 1(33%) | 1(33%) | 0 |
| Lipase | 0 | 0 | 0 | 0 | 0 | 1(25%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Lymphopenia | 0 | 0 | 0 | 0 | 0 | 2(50%) | 1(33%) | 0 | 0 | 0 | 2(33%) | 0 | 0 | 2(29%) | 0 | 0 | 0 | 0 |
| Mucositis/stomatitis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1(33%) | 0 | 0 |
| Neuropathy | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1(14%) | 0 | 0 | 0 | 0 | 0 |
| Neutrophils/granulocytes | 0 | 0 | 0 | 1(25%) | 1(25%) | 0 | 0 | 0 | 0 | 2(33%) | 1(17%) | 1(17%) | 0 | 2(29%) | 0 | 1(33%) | 0 | 2(67%) |
| Pain | 2(50%) | 0 | 0 | 1(25%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1(14%) | 0 | 0 | 1(33%) | 1(33%) | 0 |
| Pancreatitis | 0 | 0 | 0 | 0 | 1(25%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Phosphate, serum-low | 0 | 0 | 0 | 1(25%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Platelets | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1(17%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Proteinuria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1(14%) | 0 | 0 | 0 | 0 | 0 |
| Rash/desquamation | 0 | 0 | 0 | 0 | 1(25%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Sodium, serum-low | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1(17%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ulcer, GI | 0 | 0 | 0 | 0 | 0 | 0 | 1(33%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Vomiting | 1(25%) | 0 | 0 | 2(50%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Response
Clinical response was not an end point on this study due to the potential heterogeneity of the patients. However, eight of 27 enrolled patients had progression-free survival (PFS) ≥6 months. One patient with relapsed ovarian cancer (biochemical recurrence) achieved a complete biochemical response and decided to discontinue the clinical trial after cycle 6 due to abdominal pain and intense treatment schedule interfering with the quality of life. A patient with recurrent carcinosarcoma of the uterus had a partial response and received 12 cycles of IP nab-paclitaxel before progressing on treatment. Six patients had stable disease (Table 4).
Table 4:
Patients who had progression free survival ≥6 months
| Patient number |
Dose Level | Prior Regimens* | Response | Disease site | Cycles |
|---|---|---|---|---|---|
| 5 | 2 | 4 | CR | Ovary | 6 (off for abdominal pain) |
| 6 | 2 | 1 | SD | Pylorus (signet ring cell) | 12 (off for PD) |
| 7 | 2 | 1 | SD | Pancreas | 18 (off for PD) |
| 8 | 2 | 2 | SD | Appendix (mucin) | 18 (off for PD) |
| 10 | 3 | 4 | SD | Appendix (mucin producing) | 10 (off for PD) |
| 13 | 4 | 2 | SD | Cervix uteri | 1 (off for DLT) |
| 18 | 5 | 1 | PR | Uterus | 12 (off for PD) |
| 24 | 5 | 2 | SD | Cervix uteri | 6 (chemical peritonitis and pleural effusion) |
count includes adjuvant chemotherapy and subsequent regimens for recurrent disease
Pharmacokinetics
Plasma and peritoneal nab-paclitaxel PK data were available from 16 and 13 patients, respectively. The data are summarized in Table 5 and time series are shown in Figure 1 where the separation between plasma and peritoneal concentrations are shown. Total paclitaxel exposure in both the peritoneum and plasma increased in a dose-dependent manner. Furthermore, the intrapatient variability in plasma and peritoneal PK between days 1 and 15 of the first cycle was low (data not shown). The median peritoneal total paclitaxel Cmax and AUC0-t at the defined MTD of 140 mg/m2 on days 1 and 15 were 73.6 mg/L (range 53.2–97.0) and 259.1 mg/L x hr (range 244.0–339.6). The median plasma Cmax and AUC0-t at the MTD days 1 and 15 were 0.5 mg/L (range 0.2–1.0) and 4.4 mg/L x hr (range 2.8–11.8). Across all dose levels, the median pharmacologic advantage of IP administration of nab-paclitaxel was 147-fold (range 50–403).
Table 5:
Pharmacokinetics nab-paclitaxel
| Dose Level (mg/m2) |
Study Day | N | Cmax (mg/L)* | AUC 0-t (mg/L x hr) | |||
|---|---|---|---|---|---|---|---|
| Plasma | Peritoneal | Plasma | Peritoneal | Ratio | |||
| 35 | Cycle 1, Day 1 | 1 | 0.06 | 25.1 | 0.6 | 89.3 | 148 |
| 35 | Cycle 1, Day 15 | 1 | 0.06 | 31.1 | 0.5 | 93.2 | 186 |
| 70 | Cycle 1, Day 1 | 1 | 0.1 | 24.9 | 1.5 | 299.2 | 199 |
| 70 | Cycle 1, Day 15 | 1 | 0.1 | 30.3 | 1.6 | 321.4 | 201 |
| 90 | Cycle 1, Day 1 | 2 | 0.2 (0.1–0.2) | 39.8 | 1.6 (1.1–2.2) | 206.8 | 94 |
| 90 | Cycle 1, Day 15 | 2 | 0.2 (0.1–0.2) | 46.1 (33.9–58.4) | 1.9 (1.1–2.7) | 193.9 | 72 |
| 112.5 | Cycle 1, Day 1 | 4 | 0.3 (0.04–0.4) | 65.8 (29.3–1473.1) | 3.0 (0.8–4.2) | 304.9 (210.5–427.2) | 134 (50–389) |
| 112.5 | Cycle 1, Day 15 | 4 | 0.2 (0.03—0.3) | 34.5 (28.0–79.4) | 2.5 (0.6–3.7) | 289.6 (229.9–358.0) | 118 (88–403) |
| 140 | Cycle 1, Day 1 | 5 | 0.5 (0.2–0.5) | 65.8 | 4.3 (3.6–6.3) | 339.6 | 116 |
| 140 | Cycle 1, Day 15 | 5 | 0.5 (0.3–1.0) | 73.6 (53.2–97.0) | 4.5 (2.8–11.8) | 251.5 (244.0–259.1) | 239 (87–392) |
| 170 | Cycle 1, Day 1 | 3 | 0.4 (0.3–0.5) | 243.3 | 6.0 (4.8–7.2) | 622.7 | 260 |
| 170 | Cycle 1, Day 15 | 3 | 0.7 (0.4–1.0) | 146.8 | 3.2 | 978.0 | 306 |
Median (range)
Figure 1.
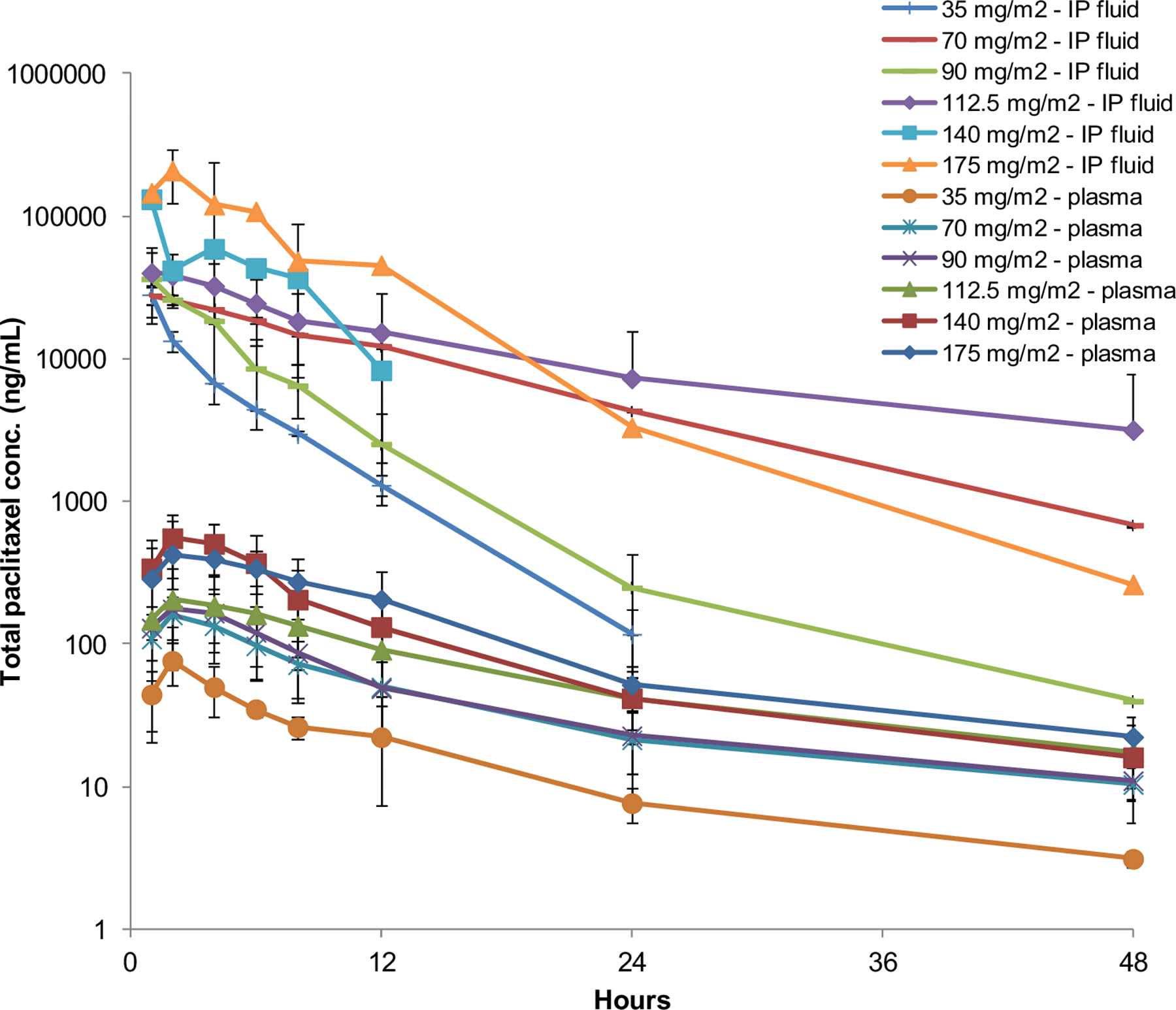
Total paclitaxel concentrations in intraperitoneal fluid and plasma. Symbols represent the means and the error bars are standard deviations.
Discussion
Nab-paclitaxel is a cremophor-free 130 nm nanoparticle of albumin-stabilized paclitaxel. This formulation can increase the intratumoral concentration of paclitaxel by a receptor-mediated transport process across the endothelial cell wall [12]. Additional advantages include the avoidance of the cremophor EL medium and ease of administration. The major toxicity of this agent is hematologic. Nab-paclitaxel has demonstrated a high degree of activity in metastatic breast, lung, and pancreatic cancer [9,13,14] and also in ovarian and gastric malignancies [10,15,16]. Teneriello et al. demonstrated an objective response rate of 64% in platinum sensitive ovarian patients treated with nab-paclitaxel 260 mg/m2 [10]. The weekly regimen of nab-paclitaxel was also evaluated in platinum and taxane refractory ovarian patients with evidence of a 23% partial response rate and 36% stable disease [15].
Nab paclitaxel appears an attractive chemotherapy option in taxane-sensitive malignancies due to similar or improved efficacy compared to conventional solvent-based paclitaxel (sb-paclitaxel) and also due to a favorable toxicity profile.
Several clinical studies have demonstrated the favorable PK of paclitaxel administered IP [17–21], with cytotoxic drug levels maintained in the peritoneal cavity for several days [18,22]. A preclinical study using a mouse peritoneal model with subcutaneous xenografts evaluated the antitumor activity of nab-paclitaxel administered IV or IP compared to conventional IP paclitaxel administered at equitoxic doses [23]. Treatment with either IV or IP nab-paclitaxel achieved greater survival benefit compared to conventional IP paclitaxel.
Our study represents the first trial of IP nab-paclitaxel in patients with peritoneal carcinomatosis. We enrolled 14 patients with gynecologic malignancies (nine of which had ovarian cancer) 12 patients with gastrointestinal tumors (eight of which had colon cancer) and 1 patient with peritoneal mesothelioma. The MTD of IP nab-paclitaxel was established at 140 mg/m2 on day 1, 8, and 15 of a 28-day cycle. We demonstrated that this regimen was feasible and had a favorable toxicity profile. IP catheter complications in our study were minimal and led to discontinuation of treatment in only two patients. Only three patients experienced abdominal pain related to the IP administration of nab-paclitaxel. Eight out of 27 enrolled patients had PFS of more than 6 months.
At each dose level, we found a large PK advantage of IP administration of nab-paclitaxel. Across all dose levels of nab-paclitaxel, the median IP versus IV AUC0-t was 147-fold (range 50–403), resulting in increased peritoneal drug exposure. The inter- and intra-patient variability appears to be low. At the defined MTD of weekly IP nab-paclitaxel of 140 mg/m2, the median peritoneal total paclitaxel Cmax and AUC0-t were 73.6 mg/L (range 53.2–97.0) and 259.1 mg/L x hr (range 244.0–339.6), with median plasma Cmax and AUC0-t of 0.5 mg/L (range 0.2–1.0) and 4.4 mg/L x hr (2.8–11.8). These results are identical to plasma AUC0-t obtained after the weekly IV administration of nab-paclitaxel at 100–130 mg/m2. This indicates that in addition to achieving high local drug levels in the peritoneum, patients are exposed to therapeutic systemic drug levels as well. The pharmacologic advantage for IP nab-paclitaxel appears lower compared to the Cmax and AUC0-t ratios of IP conventional paclitaxel, which are approximately 800–1000 and 550–2000, respectively [17–21]. Possible explanations include differences in methods of drug concentration measurement, volume of carrier solution, treatment frequency, and patient population. Compared to other drugs that are frequently used in ovarian cancer such as cisplatin and carboplatin, which have IP to IV ratios of approximately 5–20 [24], our study demonstrated a significant peritoneal advantage for nab-paclitaxel. This chemotherapy agent had a low frequency of abdominal pain, which allowed a dose escalation at higher doses than anticipated. This compares favorably to the IP administration of conventional paclitaxel, for which abdominal pain was the DLT [17]. Fourteen patients underwent voluntary neurological exams, nerve conduction studies, and quantitative sensory nerve testing at baseline and at pre-cycle three. Although there was a slight trend towards worsening neuropathy, no significant differences were found for any of the measured parameters. Only one patient, with stage IV carcinosarcoma of the uterus, with base line grade 1 peripheral neuropathy, developed grade 2 motor neuropathy related to IP nab-paclitaxel after cycle 12 of treatment. The neuropathy was confirmed by a nerve conduction study that compared to two prior nerve conduction studies demonstrated new bilateral axonal neuropathy. The patient discontinued treatment after cycle 12 due to progressive disease and toxicity. Her grade 2 neuropathy improved to grade 1, 4 months after discontinuing nab-paclitaxel.
This study took 68 months to determine the MTD. This is more than double the national average for Phase I cancer studies and represents a significant barrier for IP investigations. Several causes were apparent: 1) starting far below the IV dose added multiple dose levels that increased the duration and may not be necessary in future IP studies; 2) the screening period was very long (average of 1 month and as long as 3 months) and involved normal screening procedures plus the additional port placement and possible additional debulking at time of port placement; this necessitated subsequent additional recovery time for up to 6 weeks; 3) the number of screen failures, due both to screening tests, port failures and other intercurrent illnesses that can occur in the screening period was high at 40% (18/45 patients consented), which due to the limited number of slots in a 3+3 provides additional delays, and 4) the patient interarrival times were longer than standard Phase I patients due to the specific candidacy requirements. Based on the assumption of a 30 day mean interarrival time, a 40% screen failure and other parameters to estimate the above logistical considerations, simulations show an expected duration of 68 months if dose level 5 was selected as the MTD, consistent with our observation. This consistent observation has led us to make several suggestions to improve the feasibility of future IP studies. First, adding an additional site could reduce the mean interarrival time to 14 days and reduce the study duration to 47 months, which is still considerable. Second, starting at 70% of the IV dose rather than 1/3 the IV dose reduces the duration to 38.9 months. Lastly, applying queue based modifications of the 3+ 3 design (manuscript in submission), results in an expected duration of 29.0 months, which is a duration that would allow IP studies to be completed with an acceptable duration.
The study completed accrual in 2014 and was reported at ASCO 2015 [25]. Around the same time, Wright et al. reported on the use and effectiveness of IP/IV chemotherapy at six National Comprehensive Cancer Network (NCCN) centers, including our institution [26]. The authors concluded that the use of IP/IV chemotherapy increased significantly at NCCN centers between 2003 and 2012, especially after the publication of GOG 172, and this approach was associated with significantly improved overall survival (3-year overall survival, 81% vs 71%; hazard ratio, 0.68; 95% CI, 0.47 to 0.99).
Our initial intent was to develop larger studies utilizing IP nab-paclitaxel either as a single agent or in combination with platinum agents in a more homogenous patient population to further demonstrate the efficacy of peritoneal administration of nab-paclitaxel. The extended duration of this IP Phase 1 trial has lead us to consider several modifications to our approach for future similar studies, including adding an additional center and adopting a queue-based Phase I design that combined can reduce the expected study duration by more than half.
However, the preliminary results of the phase III randomized GOG 252 trial added more questions than answers on the role of IP chemotherapy [27]. This study evaluated 3 arms: a standard IV arm of weekly carboplatin and weekly paclitaxel based on the encouraging results of JGOG 3016 [28], an IP dose reduced cisplatin arm, and an additional arm substituting IP carboplatin for cisplatin. All arms included IV bevacizumab. The study failed to demonstrate a PFS advantage from IP chemotherapy over IV dose-dense chemotherapy in patients who have undergone optimal primary debulking surgery. Survival data is not yet mature.
This negative trial is challenging the benefit of IP chemotherapy compared to dose-dense IV chemotherapy. Dose reductions of paclitaxel and cisplatin may have compromised the efficacy of IP chemotherapy. The addition of bevacizumab to all arms of study may have been a confounding factor that equalized the efficacy of IP treatment to that of IV dose dense treatment.
Another IP study, the Japanese iPocc trial (ClinicalTrials.gov identifier: NCT01506856) is still ongoing, but is unlikely to have an international impact because trials showing a benefit for Japanese patients have not always been confirmed in Western populations.
It is unlikely that large, conventional IP chemotherapy studies will be developed by cooperative groups. New techniques of IP chemotherapy, such as pressurized intraperitoneal aerosol chemotherapy (PIPAC) [29,30], may stimulate the interest in regional delivery of chemotherapy agents including nab-paclitaxel. In addition, novel IP trials incorporating targeted agents or immunotherapy may prove beneficial for patient with carcinomatosis due to gynecologic or gastrointestinal malignancies.
Acknowledgements
This study was approved and funded by the National Comprehensive Cancer Network (NCCN) Oncology Research Program (ORP) from general research support from Celgene Corporation.
Research reported in this publication was also supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.”
The authors thank Chris Gandhi, PhD and Nicola M. Solomon, PhD, for editorial assistance and critical review of the manuscript.
Funding
This study was approved and funded by the National Comprehensive Cancer Network (NCCN) Oncology Research Program (NCCNMIRV0016) and general research support from Celgene Corporation. Additional support for the study was provided by Cancer Center Support Grant P30CA033572.
Footnotes
Clinical trial registration
Compliance with ethical standards
Conflict of interest
MC receives personal fees from Astra Zeneca and VC is a member of the Celgene Speaker’s Bureau.
Ethical approval
All procedures performed in the study involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
References
- 1.Levin L, Hryniuk WM (1987) Dose intensity analysis of chemotherapy regimens in ovarian carcinoma. J Clin Oncol 5 (5):756–767. doi: 10.1200/jco.1987.5.5.756 [DOI] [PubMed] [Google Scholar]
- 2.Alberts DS, Liu PY, Hannigan EV, O’Toole R, Williams SD, Young JA, Franklin EW, Clarke-Pearson DL, Malviya VK, DuBeshter B (1996) Intraperitoneal cisplatin plus intravenous cyclophosphamide versus intravenous cisplatin plus intravenous cyclophosphamide for stage III ovarian cancer. N Engl J Med 335 (26):1950–1955. doi: 10.1056/nejm199612263352603 [DOI] [PubMed] [Google Scholar]
- 3.Markman M, Bundy BN, Alberts DS, Fowler JM, Clark-Pearson DL, Carson LF, Wadler S, Sickel J (2001) Phase III trial of standard-dose intravenous cisplatin plus paclitaxel versus moderately high-dose carboplatin followed by intravenous paclitaxel and intraperitoneal cisplatin in small-volume stage III ovarian carcinoma: an intergroup study of the Gynecologic Oncology Group, Southwestern Oncology Group, and Eastern Cooperative Oncology Group. J Clin Oncol 19 (4):1001–1007. doi: 10.1200/jco.2001.19.4.1001 [DOI] [PubMed] [Google Scholar]
- 4.Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, Copeland LJ, Walker JL, Burger RA (2006) Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med 354 (1):34–43. doi: 10.1056/NEJMoa052985 [DOI] [PubMed] [Google Scholar]
- 5.National Cancer Institute Division of Cancer Treatment & Diagnosis (2006) NCI Clinical Announcement: Intraperitoneal chemotherapy for ovarian cancer. http://ctep.cancer.gov/highlights/docs/clin_annc_010506.pdf. Accessed 6/25/2018
- 6.Morgan RJ Jr., Newman EM, Doroshow JH, McGonigle K, Margolin K, Raschko J, Chow W, Somlo G, Leong L, Tetef M, Shibata S, Hamasaki V, Carroll M, Vasilev S, Akman S, Coluzzi P, Wagman L, Longmate J, Paz B, Yen Y, Klevecz R (1998) Phase I trial of intraperitoneal iododeoxyuridine with and without intravenous high-dose folinic acid in the treatment of advanced malignancies primarily confined to the peritoneal cavity: flow cytometric and pharmacokinetic analysis. Cancer research 58 (13):2793–2800 [PubMed] [Google Scholar]
- 7.Morgan RJ Jr., Doroshow JH, Synold T, Lim D, Shibata S, Margolin K, Schwarz R, Leong L, Somlo G, Twardowski P, Yen Y, Chow W, Lin P, Paz B, Chu D, Frankel P, Stalter S (2003) Phase I trial of intraperitoneal docetaxel in the treatment of advanced malignancies primarily confined to the peritoneal cavity: dose-limiting toxicity and pharmacokinetics. Clin Cancer Res 9 (16 Pt 1):5896–5901 [PubMed] [Google Scholar]
- 8.Morgan RJ Jr., Synold TW, Xi B, Lim D, Shibata S, Margolin K, Schwarz RE, Leong L, Somlo G, Twardowski P, Yen Y, Chow W, Tetef M, Lin P, Paz B, Koczywas M, Wagman L, Chu D, Frankel P, Stalter S, Doroshow JH (2007) Phase I trial of intraperitoneal gemcitabine in the treatment of advanced malignancies primarily confined to the peritoneal cavity. Clin Cancer Res 13 (4):1232–1237. doi: 10.1158/1078-0432.ccr-06-1735 [DOI] [PubMed] [Google Scholar]
- 9.Blum JL, Savin MA, Edelman G, Pippen JE, Robert NJ, Geister BV, Kirby RL, Clawson A, O’Shaughnessy JA (2007) Phase II study of weekly albumin-bound paclitaxel for patients with metastatic breast cancer heavily pretreated with taxanes. Clinical breast cancer 7 (11):850–856. doi: 10.3816/CBC.2007.n.049 [DOI] [PubMed] [Google Scholar]
- 10.Teneriello MG, Tseng PC, Crozier M, Encarnacion C, Hancock K, Messing MJ, Boehm KA, Williams A, Asmar L (2009) Phase II evaluation of nanoparticle albumin-bound paclitaxel in platinum-sensitive patients with recurrent ovarian, peritoneal, or fallopian tube cancer. J Clin Oncol 27 (9):1426–1431. doi: 10.1200/jco.2008.18.9548 [DOI] [PubMed] [Google Scholar]
- 11.Gardner ER, Dahut W, Figg WD (2008) Quantitative determination of total and unbound paclitaxel in human plasma following Abraxane treatment. Journal of chromatography B, Analytical technologies in the biomedical and life sciences 862 (1–2):213–218. doi: 10.1016/j.jchromb.2007.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desai N, Trieu V, Yao Z, Louie L, Ci S, Yang A, Tao C, De T, Beals B, Dykes D, Noker P, Yao R, Labao E, Hawkins M, Soon-Shiong P (2006) Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin Cancer Res 12 (4):1317–1324. doi: 10.1158/1078-0432.ccr-05-1634 [DOI] [PubMed] [Google Scholar]
- 13.Socinski MA, Bondarenko I, Karaseva NA, Makhson AM, Vynnychenko I, Okamoto I, Hon JK, Hirsh V, Bhar P, Zhang H, Iglesias JL, Renschler MF (2012) Weekly nab-paclitaxel in combination with carboplatin versus solvent-based paclitaxel plus carboplatin as first-line therapy in patients with advanced non-small-cell lung cancer: final results of a phase III trial. J Clin Oncol 30 (17):2055–2062. doi: 10.1200/jco.2011.39.5848 [DOI] [PubMed] [Google Scholar]
- 14.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF (2013) Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369 (18):1691–1703. doi: 10.1056/NEJMoa1304369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coleman RL, Brady WE, McMeekin DS, Rose PG, Soper JT, Lentz SS, Hoffman JS, Shahin MS (2011) A phase II evaluation of nanoparticle, albumin-bound (nab) paclitaxel in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer: a Gynecologic Oncology Group study. Gynecol Oncol 122 (1):111–115. doi: 10.1016/j.ygyno.2011.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egawa T, Kemmochi T, Nishiya S, Mihara K, Ito Y, Nagashima A (2014) [Nanoparticle albumin-bound Paclitaxel for unresectable or recurrent gastric cancer]. Gan to kagaku ryoho Cancer & chemotherapy 41 (12):2251–2253 [PubMed] [Google Scholar]
- 17.Markman M, Rowinsky E, Hakes T, Reichman B, Jones W, Lewis JL Jr., Rubin S, Curtin J, Barakat R, Phillips M, et al. (1992) Phase I trial of intraperitoneal taxol: a Gynecoloic Oncology Group study. J Clin Oncol 10 (9):1485–1491. doi: 10.1200/jco.1992.10.9.1485 [DOI] [PubMed] [Google Scholar]
- 18.Francis P, Rowinsky E, Schneider J, Hakes T, Hoskins W, Markman M (1995) Phase I feasibility and pharmacologic study of weekly intraperitoneal paclitaxel: a Gynecologic Oncology Group pilot Study. J Clin Oncol 13 (12):2961–2967 [DOI] [PubMed] [Google Scholar]
- 19.Hofstra LS, Bos AM, de Vries EG, van der Zee AG, Willemsen AT, Rosing H, Beijnen JH, Mulder NH, Aalders JG, Willemse PH (2002) Kinetic modeling and efficacy of intraperitoneal paclitaxel combined with intravenous cyclophosphamide and carboplatin as first-line treatment in ovarian cancer. Gynecol Oncol 85 (3):517–523 [DOI] [PubMed] [Google Scholar]
- 20.Fushida S, Furui N, Kinami S, Ninomiya I, Fujimura T, Nishimura G, Ohta T, Yokogawa K, Miyamoto K, Miwa K (2002) [Pharmacologic study of intraperitoneal paclitaxel in gastric cancer patients with peritoneal dissemination]. Gan to kagaku ryoho Cancer & chemotherapy 29 (12):2164–2167 [PubMed] [Google Scholar]
- 21.Mohamed F, Marchettini P, Stuart OA, Yoo D, Sugarbaker PH (2003) A comparison of hetastarch and peritoneal dialysis solution for intraperitoneal chemotherapy delivery. Eur J Surg Oncol 29 (3):261–265 [DOI] [PubMed] [Google Scholar]
- 22.Markman M (1996) Intraperitoneal taxol. Cancer treatment and research 81:1–5 [DOI] [PubMed] [Google Scholar]
- 23.Kinoshita J, Fushida S, Tsukada T, Oyama K, Watanabe T, Shoji M, Okamoto K, Nakanuma S, Sakai S, Makino I, Furukawa H, Hayashi H, Nakamura K, Inokuchi M, Nakagawara H, Miyashita T, Tajima H, Takamura H, Ninomiya I, Fujimura T, Masakazu Y, Hirakawa K, Ohta T (2014) Comparative study of the antitumor activity of Nab-paclitaxel and intraperitoneal solvent-based paclitaxel regarding peritoneal metastasis in gastric cancer. Oncology reports 32 (1):89–96. doi: 10.3892/or.2014.3210 [DOI] [PubMed] [Google Scholar]
- 24.de Bree E, Tsiftsis DD (2007) Experimental and pharmacokinetic studies in intraperitoneal chemotherapy: from laboratory bench to bedside. Recent results in cancer research Fortschritte der Krebsforschung Progres dans les recherches sur le cancer 169:53–73 [DOI] [PubMed] [Google Scholar]
- 25.Cristea MC, Synold TW, Frankel PH, Rivkin SE, Lim D, Chung VM, Chao J, Wakabayashi MT, Paz IB, Han ES, Lin P, Leong LA, Hakim A, Carroll MI, Openshaw H, Prakash N, Dellinger TH, Park MS, Morgan R (2015) Pharmacologic advantage (PA) of intraperitoneal (IP) nab-paclitaxel in patients with advanced malignancies primarily confined to the peritoneal cavity. (Abstract 2553). Paper presented at the 2015 ASCO Annual Meeting, Chicago, IL, [Google Scholar]
- 26.Wright AA, Cronin A, Milne DE, Bookman MA, Burger RA, Cohn DE, Cristea MC, Griggs JJ, Keating NL, Levenback CF, Mantia-Smaldone G, Matulonis UA, Meyer LA, Niland JC, Weeks JC, O’Malley DM (2015) Use and Effectiveness of Intraperitoneal Chemotherapy for Treatment of Ovarian Cancer. J Clin Oncol 33 (26):2841–2847. doi: 10.1200/jco.2015.61.4776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker J, Brady M, DiSilvestro P, Fujiwara K, Alberts D, Zheng W, Tewari K, Cohn D, Powell M, Van Le L (2016) A phase III trial of bevacizumab with IV versus IP chemotherapy for ovarian, fallopian tube, and peritoneal carcinoma: An NRG Oncology Study. Gynecologic Oncology 141 (Supplement 1):208. doi: 10.1016/j.ygyno.2016.04.535 [DOI] [Google Scholar]
- 28.Katsumata N, Yasuda M, Isonishi S, Takahashi F, Michimae H, Kimura E, Aoki D, Jobo T, Kodama S, Terauchi F, Sugiyama T, Ochiai K, Japanese Gynecologic Oncology G (2013) Long-term results of dose-dense paclitaxel and carboplatin versus conventional paclitaxel and carboplatin for treatment of advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer (JGOG 3016): a randomised, controlled, open-label trial (The Lancet. Oncology). Lancet Oncol 14 (10):1020–1026. doi: 10.1016/S1470-2045(13)70363-2 [DOI] [PubMed] [Google Scholar]
- 29.Grass F, Vuagniaux A, Teixeira-Farinha H, Lehmann K, Demartines N, Hubner M (2017) Systematic review of pressurized intraperitoneal aerosol chemotherapy for the treatment of advanced peritoneal carcinomatosis. The British journal of surgery 104 (6):669–678. doi: 10.1002/bjs.10521 [DOI] [PubMed] [Google Scholar]
- 30.Tempfer C, Giger-Pabst U, Hilal Z, Dogan A, Rezniczek GA (2018) Pressurized intraperitoneal aerosol chemotherapy (PIPAC) for peritoneal carcinomatosis: systematic review of clinical and experimental evidence with special emphasis on ovarian cancer. Archives of gynecology and obstetrics 298 (2):243–257. doi: 10.1007/s00404-018-4784-7 [DOI] [PubMed] [Google Scholar]