

Abstract
Noncommunicable diseases are chronic diseases that contribute to death worldwide, but these diseases can be prevented and mitigated with regular exercise. Exercise activates signaling molecules and the transcriptional network to promote physiological adaptations, such as fiber type transformation, angiogenesis, and mitochondrial biogenesis. AMP-activated protein kinase (AMPK) is a master regulator that senses the energy state, promotes metabolism for glucose and fatty acid utilization, and mediates beneficial cellular adaptations in many vital tissues and organs. This review focuses on the current, integrative understanding of the role of exercise-induced activation of AMPK in the regulation of system metabolism and promotion of health benefits.
Keywords: AMPK, exercise, metabolism, glucose uptake, fatty acid oxidation, adaptive responses
INTRODUCTION
Noncommunicable diseases (NCDs) caused 71% of all deaths (41 million) worldwide in 2018, reaching pandemic proportions (1). Regular exercise is indisputably the most powerful intervention for prevention and treatment of most NCDs. However, despite decades of research, the underlying mechanisms by which exercise training induces the health benefits are poorly understood. This is particularly true for the molecular and signaling mechanisms that modulate the adaptive processes of exercise. One such signaling modulator of exercise adaptation is AMP-activated protein kinase (AMPK), which is ubiquitously expressed in literally all cells, tissues, and organs, including the skeletal muscle, heart, liver, adipose tissue, and brain. Ever since its discovery in the 1970s during the search for a kinase that regulates 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase in control of cholesterol synthesis (2), attention has been drawn to many other regulatory functions of this kinase, including its regulatory roles in the metabolic and cellular processes induced by exercise in the aforementioned tissues and organs (3, 4). AMPK is a highly conserved serine/threonine kinase that plays a critical role in energy homeostasis. AMPK monitors energy availability and positively regulates glucose and fatty acid uptake and oxidation and catabolic pathways, such as autophagy. Simultaneously, AMPK negatively regulates fatty acid, sterol, and protein synthesis and other anabolic processes (5, 6). Previous reviews have eloquently discussed the role of AMPK in cellular metabolism and adaptation (6–8). This review focuses on an integrative understanding of exercise-induced AMPK activation in cellular and metabolic adaptations in vital tissues and organs, including skeletal muscle, heart, liver, adipose tissue, and brain. We also review the evidence of exercise-induced AMPK activation in mitigating NCDs.
AMPK STRUCTURE AND FUNCTION
AMPK is a heterotrimer composed of alpha (α1 or α2 isoform), beta (β1 or β2 isoform), and gamma (γ1, γ2, or γ3 isoform) subunits. The α subunit contains the catalytic domain, whereas the β subunit functions as scaffolding to link the α and γ subunits. The γ subunit senses relative levels of AMP or ADP to ATP to induce conformational change of the α subunit in favor of AMPK activation through phosphorylation by upstream kinases. The γ subunit possesses four cystathionine-β-synthase (CBS) domains, of which only CBS1, CBS3, and CBS4 bind AMP/ADP/ATP (9, 10). The binding of AMP or ADP to the γ subunit contributes to three mechanisms of AMPK activation: (a) allosteric activation, (b) promotion of AMPK activation by phosphorylation of the α subunit at threonine 172 (Thr172), and (c) inhibition of protein phosphatases, preventing dephosphory-lation at Thr172 (9, 11). Of particular interest is the mechanism by which AMPK is activated by upstream kinases, as these can be activated by different cellular cues, making AMPK activation more versatile. It is now known that AMPK activation by phosphorylation of Thr172 is achieved by liver kinase B1 (LKB1) and calcium/calmodulin-dependent protein kinase 2 (CaMKK2). Activated AMPK exerts its roles over metabolic regulation and cellular adaptive processes by phosphorylating downstream effectors, leading to their activation or inhibition. AMPK activation has been implicated in the regulation of glucose, lipid, and protein metabolism as well as cellular adaptive processes, such as autophagy and mitochondrial remodeling.
AMPK ACTIVATION BY EXERCISE IN SKELETAL MUSCLE METABOLISM AND ADAPTATION
In response to exercise, there is a dramatic increase in energy demand in skeletal muscle that requires increased energy turnover. Exercise-induced molecular signaling is intimately involved in the regulation of metabolism to meet the need of increased energy demand and adaptive cellular processes to prepare for future challenges. Muscle contractions during exercise lead to increases in calcium, nitric oxide, reactive oxygen species, and AMP/ADP as cues of stress, which in turn activate many kinases and signaling pathways, such as mitogen-activated protein kinases (ERK, p38, JNK), CaMKKs, and AMPK (7). Activation of these signaling pathways contributes to not only increased glucose and fatty acid uptake and oxidation as a fuel source to maintain exercise but also adaptative processes critical for enhanced muscle contractile and metabolic functions in the long run. AMPK has been strongly implicated in these regulatory processes, as activation of AMPK with increased Thr172 phosphorylation and activity occurs during and immediately after exercise (Figure 1). The first direct evidence of AMPK activation in skeletal muscle by exercise was reported in rats following as little as 5 min of treadmill running with concurrent reduction of acetyl-CoA carboxylase (Acc) activity, possibly through phosphorylation at serine 79 (Ser79) of Acc1 and Ser212 of Acc2, and malonyl-CoA concentration (12). Cycling exercise of moderate intensity (90 min) in humans resulted in increased AMPK activity and phosphorylation in muscle biopsies (13). Treadmill running in mice resulted in increased phosphorylation of AMPK at Thr172 in all recruited glycolytic and oxidative muscles (14). Because ADP increases from approximately 40 μM to between 130 and 200 μM, which is above the Kd of AMPK (80 μM) during moderate to high-intensity exercise, while AMP increased significantly but stayed below the Kd of AMPK, Oakhill et al. (15) argued that ADP is the predominant agonist of AMPK in skeletal muscle during exercise. Furthermore, pharmacological activation of AMPK in skeletal muscle by 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR), an endogenous substrate that can induce accumulation of 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranosyl monophosphate (ZMP) to mimic AMP, indicates that AMPK activation is sufficient to promote glucose and fatty acid uptake and oxidation (16–20). An elegant global phosphoproteomics analysis of human skeletal muscle biopsies after acute exercise in untrained healthy individuals revealed more than 1,000 phosphosites, of which 55 overlapped with the phosphosites found in AICAR-stimulated myotubes in cell culture (21). This is to date the most comprehensive analysis of the AMPK phos-phoproteome network in skeletal muscle in the context of exercise. These findings suggest that exercise-induced AMPK activation may cause the increased glucose and fatty acid uptake and oxidation in skeletal muscle. However, it was not until the recent advancements in molecular genetics that animal models of AMPK gene deletions allowed studies to address whether AMPK activation is required for exercise-induced glucose and fatty acid uptake and oxidation in skeletal muscle.
Figure 1.
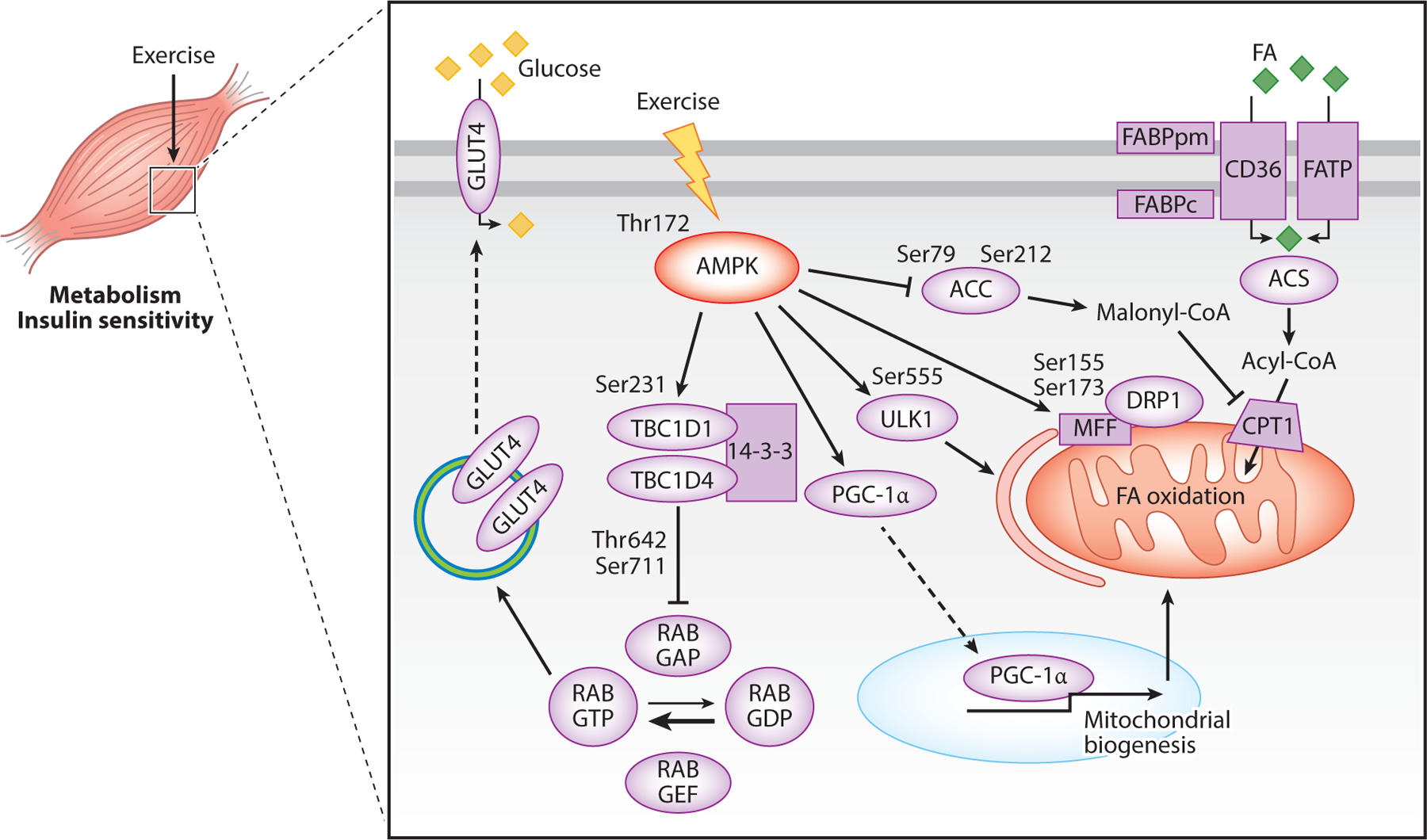
Mechanisms by which exercise-induced AMPK modifies metabolism in skeletal muscle. Phosphorylation of AMPK at Thr172 increases glucose uptake by phosphorylating TBC1D1 and TBC1D4, thus promoting their binding to 14-3-3, which inhibits RAB GAP activity. This leads to increased RAB GTP, which promotes mobilization of GLUT4 to the membrane. AMPK activation also increases FA oxidation. FAs are taken up into the cell by a group of FA transport proteins; FAs are then converted to acyl-CoA and taken into the mitochondria by CPT1 for oxidation. AMPK activation promotes FA uptake by phosphorylating and inhibiting ACC. This inhibits ACC synthesis of malonyl-CoA and prevents malonyl-CoA from inhibiting CTP1, thus promoting FA uptake into the mitochondria and FA oxidation. Lastly, AMPK activation promotes PGC-1α activity, resulting in translocation of PGC-1α to the nucleus, where it functions to promote transcription of mitochondrial genes. Abbreviations: ACC, acetyl-CoA carboxylase; ACS, acyl-CoA synthetase; AMPK, AMP-activated protein kinase; CD36, fatty acid translocase; CPT1, carnitine palmitoyl transferase 1; DRP1, dynamin-related protein 1; FA, fatty acid; FABPc, fatty acid–binding protein cytosolic; FABPpm, fatty acid–binding protein plasma membrane; FATP, fatty acid transport protein; GLUT4, glucose transporter type 4; MFF, mitochondrial fission factor; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1 alpha; RAB GAP, GTPase-activating protein–bound form of RAB; RAB GDP, guanosine diphosphate–bound form of RAB; RAB GEF, guanine nucleotide exchange factor–bound form of RAB; RAB GTP, guanosine-5′-triphosphate-bound form of RAB; Ser, serine; TBC1D1, TBC1 domain family member 1; TBC1D4, TBC1 domain family member 4; Thr, threonine; ULK1, Unc-51-like autophagy activating kinase 1.
Glucose Uptake and Insulin Sensitivity
During exercise, an immediate increase in glucose uptake by the muscle cells is needed as fuel to maintain exercise. Muscle contractions stimulate glucose transporter type 4 (GLUT4) translocation from storage vesicles to the surface of the sarcolemma, the plasma membrane of muscle, which permits facilitated diffusion of glucose down its concentration gradient into the muscle cells (Figure 1). Two important proteins that regulate GLUT4 mobilization are TBC1 domain family member 1 (TBC1D1) and TBC1D4 (AS160). TBC1D1 and TBC1D4 promote GLUT4 translocation to the sarcolemma by inhibiting Rab-GTPase-activating protein (Rab-GAP). Phosphorylation of TBC1D1 and TBC1D4 can occur at many sites on each protein, some of which are phosphorylated by AMPK. AMPK has long been considered a critical kinase in promoting exercise-induced glucose uptake. This notion has been supported by evidence of the pharmacological activation of AMPK by AICAR, which has been shown to be sufficient to induce translocation of GLUT4 to the sarcolemma (22–24), resulting in increased glucose uptake (25). However, the evidence for a definitive role of AMPK activation in exercise-induced glucose uptake is somewhat conflicting and has led to a dispute over the causal role of AMPK activation in the increased glucose uptake during exercise (26). An early study reported that muscle-specific AMPKβ1β2 knockout (AMPKβ1β2 MKO) mice have reduced clearance of glucose during exercise, suggesting that AMPK activation is required for increased glucose uptake by working skeletal muscle (27). Conversely, more recent studies in muscle-specific AMPKα1α2 KO (AMPKα1α2 MKO) and in tamoxifen-inducible, muscle-specific AMPKα1α2 KO mice (AMPKα1α2 iMKO) suggested that AMPK is not required for exercise-induced glucose uptake (28, 29). In addition to exercise, muscle contraction by electric stimulation has also been used to induce glucose uptake and assess the role of AMPK activation. Some researchers found that AMPK activation is not required in contraction-induced glucose uptake in a variety of AMPK gene depletion models (16, 30–32), but others found contraction-induced glucose uptake is impaired in muscle-specific AMPKα2 kinase-dead (AMPKα2 KD) transgenic mice, suggesting that contraction-induced glucose uptake is dependent on AMPK activation (17, 33). Tbc1d1 and Tbc1d4 can be phosphorylated by AMPK, but these events are not required for glucose uptake (34). Phosphorylation of Tbc1d1 at Ser231 and Tbc1d4 at Ser711 is elevated during in situ contraction, which is abated in AMPKα1α2 MKO mice, suggesting that these phosphorylation sites are caused by AMPK activation (Figure 1). However, glucose uptake is normal in these KO animals during contraction, suggesting that these phosphorylation events are not important in glucose uptake during exercise (34). As pointed out by previous publications (26, 29), glucose uptake was measured after muscle contraction. The remaining possibility is that AMPK-mediated phosphorylation of these substrates regulates glucose uptake during recovery from exercise.
In support of a role for AMPK activation in promoting glucose uptake during recovery from exercise, a recent study of AMPKα1α2 MKO and AMPKα1α2 iMKO mice confirmed that AMPK is not required for glucose uptake during exercise but is required for this process during recovery (29). The authors proposed that AMPK activation delays Glut4 endocytosis (through internalization of Glut4 back to storage vesicles in the cytosol) while insulin and muscle contraction increase Glut4 exocytosis. Both mechanisms ultimately act to elevate Glut4 abundance on the sarcolemma. Studies in global Tbc1d1 KO mice confirmed the role of Tbc111 in glucose uptake during recovery from exercise (29). Together, these data suggest that the AMPK-TBC1D1 axis regulates muscle glucose during recovery from exercise. It is of note that all of the genetic interventions were either global or muscle-specific deletions of the AMPK genes or the muscle-specific overexpression of a dominant negative kinase-dead AMPK isoform, which inevitably disrupt stoichiometry and/or elicit compensatory changes that may confound some of the analyses. Thus, there is an urgent need to develop more sophisticated molecular genetic animal models with targeted mutations of AMPK activation phosphorylation sites, such as Thr172, and of the AMPK phosphorylation sites of the effector proteins to definitively prove the role of AMPK activation to control glucose uptake during and after exercise.
A single bout of endurance exercise or repeated muscle contractions promote muscle insulin sensitivity several hours later. Because AMPK is activated by exercise or muscle contraction, the question is whether AMPK activation is causal to the enhanced insulin sensitivity in skeletal muscle. Kjøbsted et al. (34) showed that in situ contraction and in vivo treadmill running increase muscle and whole-body insulin sensitivity in wild-type (WT) mice, respectively, but not in AMPKα1α2 MKO mice. The data strongly support that AMPK activation is necessary for improved insulin-stimulated glucose uptake following an acute bout of exercise. This is likely mediated by the AMPK-TBC1D4 signaling axis, as enhanced insulin-stimulated Tbc1d4 Thr649 (equivalent to Thr642 in human TBC1D4) and Ser711 phosphorylation by prior in situ contraction is abolished in AMPKα1α2 MKO mice (34). Similarly, prior activation of AMPK with AICAR increases insulin sensitivity and insulin-induced glucose uptake, which are absent in AMPKα1α2 MKO mice. Importantly, prior treatment with AICAR causes greater Tbc1d4 phosphorylation at Thr649 and Ser711 under the condition of insulin stimulation (35). These findings support the role of AMPK activation during exercise or muscle contraction in promoting insulin sensitivity in skeletal muscle converging on signaling molecules that control GLUT4 trafficking as a mechanism of the benefit of exercise (Figure 1). Targeting this pathway may prove to be an effective intervention against insulin resistance and type 2 diabetes.
Fatty Acid Uptake and Oxidation
Fatty acid uptake by muscle cells requires fatty acid translocase (CD36), fatty acid–binding protein (FABP), and fatty acid transport protein (FATP) (36). Once inside the cell, fatty acids are converted to fatty acyl-CoA by acyl-CoA synthetase for oxidation or further storage. Acyl-CoA can be shuttled into the mitochondria through carnitine palmitoyl transferase 1 (CPT1) for β oxidation. Acyl-CoA transport into the mitochondria is, under basal condition, limited due to the inhibitory function of the ACC product, malonyl-CoA, toward CPT1. Malonyl-CoA concentrations are reduced, and fatty acid oxidation is increased during exercise (37). When AMPK is activated, ACC is directly phosphorylated by AMPK, resulting in reduced ACC activity, and this AMPK activation-mediated ACC inhibition is therefore considered responsible for enhanced fatty acid oxidation during exercise (12, 22, 38) (Figure 1).
In support of evidence of a causal role of AMPK activation in increased fatty acid oxidation in skeletal muscle during exercise, AMPKα1α2 MKO mice show a greater respiratory exchange ratio, a physiological parameter of carbohydrate oxidation over fatty acid oxidation measured by calorimetry, during treadmill exercise compared with WT mice (39). Findings from muscle contractions of isolated muscles in the same study further support this. However, these animals also have reduced levels of Cd36 and Fabp (39); these unwanted changes in proteins important for fatty acid transportation could result in decreased fatty acid uptake capacity, confounding interpretation of the role of AMPK activation in controlling fatty acid oxidation during exercise. Indeed, there are conflicting findings suggesting that AMPK is not required for fatty acid uptake and oxidation during exercise. In several AMPK loss-of-function genetic models, the respiratory exchange ratio during exercise is decreased or unchanged, suggesting that fatty acid utilization is increased or unchanged in the absence of AMPK activation (27, 40). Additionally, fatty acid utilization during muscle contraction is not impaired in AMPK loss-of-function genetic models (20, 27, 28, 31, 40). Therefore, further studies are needed to ascertain the definitive role of AMPK activation in controlling fatty acid oxidation during exercise, as the current genetic models are inherently disruptive of the stoichiometry of the metabolic network in skeletal muscle that regulates fatty acid metabolism.
Altogether, the data described above suggest that AMPK may be dispensable for increased fatty acid oxidation during exercise, but there are data to support a role of AMPK in fatty acid oxidation during recovery from exercise. AMPKα2 KO mice have lower fatty acid oxidation concurrent with higher glucose oxidation compared with WT mice during recovery from a bout of treadmill running, suggesting that AMPK, at least the dominant α2 isoform in skeletal muscle, is required for enhanced fatty acid oxidation during recovery from exercise (41). The same study showed that AMPK activation is necessary for induced protein expression of pyruvate dehydrogenase kinase 4 (Pdk4) during recovery. Because PDK4 can directly phosphorylate pyruvate dehydrogenase at its inhibitory phosphorylation site Ser293, and pyruvate dehydrogenase is responsible for the conversion of pyruvate to acetyl-CoA for oxidation, AMPK-dependent PDK4 activation may be responsible for the inhibition of glucose oxidation and promotion of fatty acid oxidation during recovery from an acute bout of exercise (41). Collectively, these data point to AMPK as a regulator of energy homeostasis and substrate partitioning.
Exercise Adaptations
Exercise-induced skeletal muscle adaptations ensure an improved ability to deal with future metabolic or contractile challenges. It is well known that endurance exercise training leads to numerous phenotypic changes, including improved mitochondrial quality, increased glucose uptake, and enhanced insulin sensitivity. Because we know that AMPK activation occurs in response to muscle contraction and exercise, a fundamentally important question is whether AMPK activation in skeletal muscle underlies the adaptive responses and processes induced by exercise. In an early study, Rodnick et al. (42) showed that voluntary wheel running induces an increase in Glut4 expression in rat plantaris muscle, a muscle that is heavily recruited during this habitual exercise. Later, many animal studies showed that endurance exercise training promotes Glut4 mRNA and protein expression in recruited skeletal muscles, which is considered one of the major metabolic adaptations to endurance exercise (7). However, studies using molecular genetic models suggest that AMPK is dispensable for the increase in Glut4 abundance following exercise training. For example, four weeks of treadmill training or wheel running in global AMPKα2 KO mice led to similar degrees of increased Glut4 protein and transcript compared to WT mice, suggesting that AMPK is not required for these adaptations (43, 44). A recent study of two-week endurance training in rat triceps transfected with dominant negative AMPK showed reduced basal Glut4 but a similar percentage increase of Glut4 after exercise training. This suggests that AMPK plays a role in controlling basal GLUT4 expression in skeletal muscle but is not required for the exercise training-induced GLUT4 upregulation (45).
Enhanced mitochondrial function in skeletal muscles is one of the most important cellular adaptations induced by endurance exercise, where mitochondria remodeling occurs to achieve optimal function through a collection of processes of mitochondrial biogenesis, mitochondrial dynamics, and mitophagy. AMPK has been implicated in promoting mitochondrial remodeling in skeletal muscle in response to exercise training. Regarding the regulation of mitochondrial biogenesis, it has been shown that both endurance exercise and pharmacological activation of AMPK in skeletal muscle stimulate gene expression of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α), a master regulator of mitochondrial biogenesis (46). Using primary muscle cells and Pgc-1α KO mice, Jäger et al. (47) showed that AMPK phosphorylates Pgc-1α protein directly at Thr177 and Ser538 both in vivo and in vitro, and these phosphorylation events are required for the Pgc-1α-dependent induction of the Pgc-1α promoter (Figure 1). Importantly, both AMPKβ1β2 and AMPKα1α2 MKO mice have reduced mitochondrial biogenesis, mitochondrial content, and impaired mitochondrial function (27, 30). Increases of mitochondrial content in response to 6.5 weeks of exercise training are blunted in dominant negative AMPKα2 KD transgenic mice, suggesting that exercise training–induced mitochondrial biogenesis is AMPK dependent (48). In contrast, AMPKα2 KO mice have been shown to have normal increases of mitochondrial protein abundance after two weeks of exercise training, suggesting that AMPK is not essential for mitochondrial adaptation in skeletal muscle in response to exercise (44). In summary, the role of AMPK in promoting skeletal muscle mitochondrial biogenesis in response to exercise training is not yet fully elucidated.
AMPK activation has also been implicated in controlling mitochondrial dynamics in response to exercise. In a rat model of endurance exercise, Ding et al. (49) showed reduced mitofusin 1/2 (Mfn1/2) mRNA and protein as well as increased mitochondrial fission 1 (Fis1) mRNA and protein during a relatively long duration of treadmill running (150 min). This suggests changes in mitochondrial dynamics that favor mitochondrial fission. In a recent study, Laker et al. (50) showed that exercise-induced Drp1 phosphorylation at Ser616 and Ser637 remains intact in muscle-specific dominant negative AMPKα1 KD transgenic mice, whereas muscle-specific AMPKγ1 constitutively active transgenic mice have normal Drp1 phosphorylation at both sites. These findings suggest that Drp1 is phosphorylated by another kinase during exercise. Recent proteomic and bioinformatic screens have identified two phosphorylation sites, Ser155 and Ser173, of the mitochondrial fission factor (MFF) (51), the primary receptor for DRP1 on the mitochondrial outer membrane for mitochondrial fission (Figure 1). Indeed, activation of AMPK causes the localization of DRP1 to mitochondria that are dependent on AMPK-mediated MFF phosphorylation (51). Thus, AMPK activation may promote mitochondrial fission by phosphorylating MFF, which in turn controls the localization of DRP1. Finally, an AMPK activation compound, 991, stimulates mitochondrial fission via phosphorylation of Mff at Ser146 (equivalent to human Ser172) and induces mitophagy in cultured muscle cells (52). It is unknown whether exercise-induced AMPK activation is required for MFF phosphorylation and the consequential changes in mitochondrial dynamics.
Finally, exercise may also activate mitophagy through AMPK activation in skeletal muscle. Laker et al. (50) recently showed that a single bout of treadmill running is sufficient to induce mitophagy in skeletal muscle. Because mitophagy activation in response to acute exercise requires the simultaneous activation of several processes (e.g., autophagosome biogenesis, mitochondrial fission, and autophagosome-lysosome fusion), it makes biological sense to have an energy sensor kinase as a critical node for coordinated activation of these converging processes. Using muscle-specific dominant negative AMPKα1 KD and constitutively active AMPKγ1 transgenic mice, Laker et al. (50) confirmed that immediately following an acute bout of endurance exercise in mice, AMPK is necessary and sufficient for increased phosphorylation of Unc-51-like autophagy activating kinase 1 (Ulk1) at Ser555 in skeletal muscle; Ulk1 is a kinase required for mitophagy (53) (Figure 1). Importantly, both muscle-specific dominant negative AMPKα1 KD transgenic mice and Ulk1 MKO mice showed impaired mitophagy following a single bout of treadmill running, suggesting that activation of the AMPK-ULK1 regulatory axis is required for exercise-induced mitophagy.
Taken together, activation of AMPK and the resulting activation of downstream effectors may together stimulate mitochondrial biogenesis, mitochondrial fission, and mitophagy to promote mitochondrial quality and function in skeletal muscle, which are fundamentally important for skeletal muscle metabolic and contractile functions. While this review was written, Drake et al. (54) reported that specific isoforms of AMPKα1/α2/β2/γ1 are localized on the outer mitochondrial membrane, referred to as mitoAMPK, in various tissues in mice and humans. mitoAMPK is activated by mitochondrial energetic stress, and inhibition of mitoAMPK activity attenuates exercise-induced mitophagy in skeletal muscle in vivo. These findings support that mitoAMPK-mediated mitochondrial quality control underscores the complexity of sensing cellular energetics and controlling mitochondrial adaptation in vivo.
AMPK ACTIVATION BY EXERCISE IN CARDIAC METABOLISM AND FUNCTION
During exercise, the body needs to increase cardiac output three- to fourfold to meet the demand for oxygen in working muscles. Animal studies indicate that acute exercise and exercise training increase cardiac AMPK phosphorylation and activity (55, 56) (Figure 2). Exercise and in vitro cardiomyocyte contraction increase fatty acid uptake and glucose uptake along with Glut4 translocation to the sarcolemma of cardiac myocytes (56, 57). AMPK activation by AICAR in rat ventricular papillary muscles increases Glut4 translocation and glucose uptake (58). Furthermore, pharmacological activation of AMPK results in increased fatty acid uptake and oxidation in isolated cardiomyocytes concurrent with increased mRNA and protein expression of fatty acid uptake proteins, Fat/Cd36 and Got2 (equivalent to human FABPpm), which are abolished by the AMPK inhibitor adenine 9-β-d-arabinofuranoside (59). Lastly, AICAR treatment increases sar-colemmal levels of Fat/Cd36 and Glut4, suggesting that AMPK activation increases both fatty acid and glucose uptake in cardiomyocytes (57). Collectively, these studies suggest that AMPK activation is sufficient to induce glucose and fatty acid metabolism (Figure 2), but a causal relationship between AMPK activation in control of cardiac metabolism has not been ascertained or elucidated.
Figure 2.
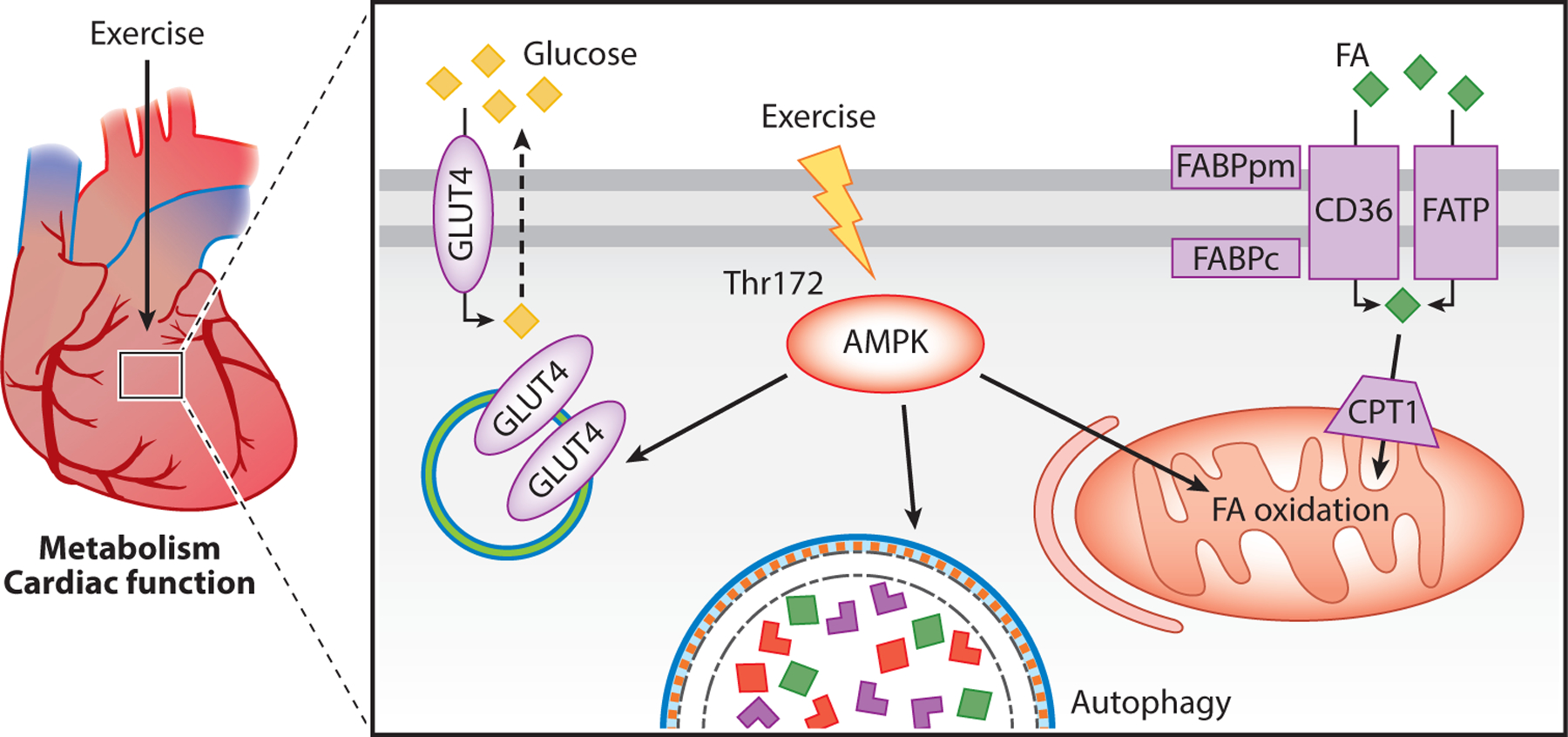
Exercise-induced AMPK activation in cardiac metabolism and function. Phosphorylation of AMPK leads to increased GLUT4 mobilizing to the sarcolemma to increase glucose uptake. AMPK activation also increases autophagy and oxidation of FA. Abbreviations: AMPK, AMP-activated protein kinase; CD36, fatty acid translocase; CPT1, carnitine palmitoyl transferase 1; FA, fatty acid; FABPc, fatty acid–binding protein cytosolic; FABPpm, fatty acid–binding protein plasma membrane; FATP, fatty acid transport protein; GLUT4, glucose transporter type 4; Thr, threonine.
It is well known that exercise training promotes metabolic and structural adaptations in the heart. Depending on the type, intensity, and duration of exercise, the adaptations lead to improved energetic and contractile machineries that better sustain cardiac contractility. In fact, regular exercise has emerged as a powerful intervention for many cardiovascular disease conditions, such as chronic heart failure. Using a mouse model of pathological cardiac hypertrophy with fibrosis, Ma et al. (55) showed that four weeks of swim training alleviate isoproterenol-induced cardiac fibrosis and oxidative stress in WT but not AMPKα2 KO mice. In another model of heart failure, exercise training (four weeks of treadmill running) in rats attenuates transverse aortic constriction–induced cardiac dysfunction, fibrosis, and endoplasmic reticulum stress-related apoptosis. It also significantly improves autophagy and increases AMPK phosphorylation, both of which are abolished by AMPK inhibitor compound C or autophagy inhibitor 3-methyladenine (60). These findings suggest that exercise training can alleviate pressure overload–induced left ventricle dysfunction and remodeling via AMPK-dependent autophagy (Figure 2). Importantly, voluntary wheel running in WT mice results in a 53% increase of protein expression of sarco/endoplasmic reticulum Ca2+-ATPase (Serca2a) in the heart, a cornerstone molecule for restoring cytosolic concentration of Ca2+ during the diastole of the cardiac cycle (61). In contrast, dominant negative AMPKα2 KD transgenic mice display a reduction in basal Serca2a expression and an absence of induction by exercise (61). These findings strongly support the importance of AMPK activation by exercise in the heart in mediating adaptive responses against cardiac dysfunction under various disease conditions. It is worth noting that treatment with a pan-AMPK activator, MK-8722, improves whole-body glucose homeostasis but promotes cardiac hypertrophy and glycogen accumulation in rodents and rhesus monkeys (23). Another pan-AMPK activator, O304, increases cardiac glucose uptake, reduces cardiac glycogen levels, and improves cardiac function in mice with no complication of cardiac hypertrophy (62). Thus, pharmacological activators of AMPK may have therapeutic potential for cardiometabolic disorders.
AMPK ACTIVATION BY EXERCISE IN HEPATIC GLUCONEOGENESIS AND LIPID METABOLISM
The liver is critical in maintaining whole-body glucose homeostasis within a narrow range under energy-demanding conditions such as exercise (63). Exercise typically causes an increase in glucose production through glycogenolysis and gluconeogenesis (64). These processes result in glucose release from the liver during short-term, moderate exercise concurrent with an elevated glucose disposal primarily in the working muscle and heart to maintain the circulating glucose level. Hepatic glucose production is controlled by many hormones released during exercise (65). Specifically, glucagon released from pancreatic α cells promotes glycogenolysis and gluconeogenesis in the liver (66). Importantly, enhanced glucose synthesis requires extensive consumption of ATP (67), which results in energetic stress and an elevated AMP/ATP ratio, a strong cue for AMPK activation. It has been hypothesized that glucose homeostasis is regulated by AMPK because endurance exercise, both acute and long term, increases phosphorylation of AMPK in the liver (12, 68–72) (Figure 3). However, evidence from molecular genetic models is somewhat counterintuitive to physiological conditions. For example, liver-specific Lkb1 or AMPKα2 KO mice exhibit hyperglycemia (73, 74), whereas liver-specific overexpression of AMPKα2 has the opposite change in blood glucose (75). Importantly, these findings were obtained at rest and therefore may not reflect the metabolic responses influenced by AMPK activation under the condition of exercise. More relevant evidence of the role of AMPK activation in controlling hepatic glucose homeostasis comes from a study in which liver-specific AMPKα1α2 KO (AMPKα1α2 LKO) mice fail to maintain euglycemia during exercise due to reduced glycogenolysis, whereas gluconeogenesis remains at a normal level (64). However, AMPKα1α2 LKO mice have reduced glycogen deposition in the liver, which can impede glycogenolysis. Nevertheless, these findings show that AMPK activation is not required for exercise-induced gluconeogenesis in the liver.
Figure 3.
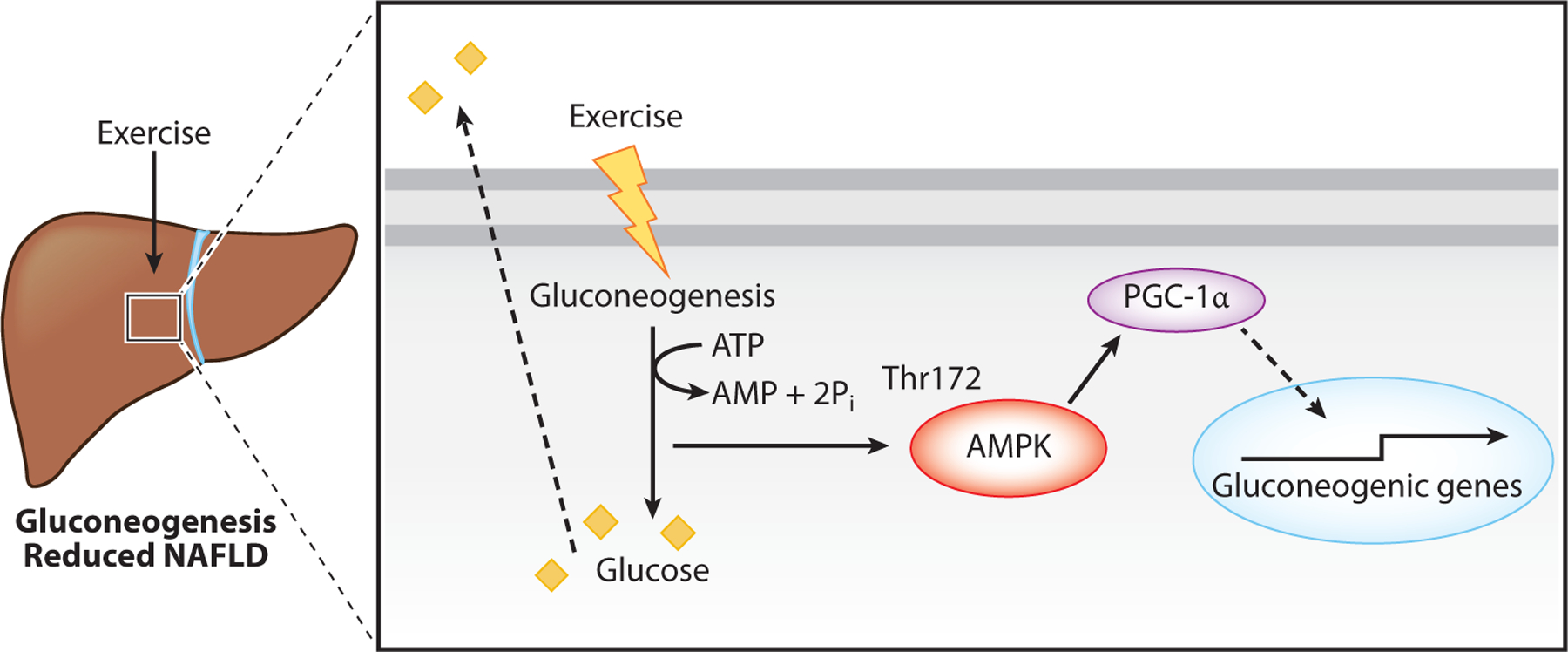
Exercise and AMPK activation in hepatic gluconeogenesis. Exercise increases gluconeogenesis, leading to increased glucose for redistribution to the heart and muscle. Gluconeogenesis is an ATP-consuming process resulting in an increased AMP/ATP ratio that activates AMPK. This leads to PGC-1α activation and increased transcription of gluconeogenic genes. Abbreviations: AMPK, AMP-activated protein kinase; NAFLD, nonalcoholic fatty liver disease; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1 alpha; Pi, inorganic phosphate; Thr, threonine.
Exercise appears to also promote insulin sensitivity in the liver. A single bout of treadmill running (60 min) in mice results in increased insulin sensitivity in the liver and stimulates transcription of genes important for hepatic gluconeogenesis, such as Pdk4, Pgc-1α, glucose 6-phosphatase (G6pc), and phosphoenolpyruvate carboxykinase (Pck1), concurrent with activation of AMPK (Thr172 phosphorylation) (71). Similar findings were observed in gene transcript levels with delayed increases of Pck1, G6pc, and Pdk4 proteins in mouse liver following a similar treadmill running regime (76). Pgc-1α functions as a transcriptional coactivator to increase Pdk4 gene expression along with increased transcription of the other gluconeogenic genes, G6pc and Pck1 (77, 78). Pdk4 inhibits glucose oxidation and promotes gluconeogenesis by phosphorylating and inactivating the pyruvate dehydrogenase complex (79). To date, there is no experimental evidence of the causal role of AMPK activation by exercise in the liver in controlling hepatic gluconeogenic gene expression.
Recently, liver AMPK activation has emerged as a potential target for treatment of nonalcoholic fatty liver disease (NAFLD), which is becoming one of the most common diseases worldwide. NAFLD in 25% of patients progresses into nonalcoholic steatohepatitis (NASH). A hall-mark of this disease is increased triglycerides content in the liver (80). Activation of liver AMPK leads to the inhibition of ACC and, consequently, increased fatty acid oxidation, which may underlie the mechanism by which AMPK activation reduces NAFLD (81). Broadly, both aerobic and resistance exercise training are effective in treating NAFLD and reducing liver fat mass and liver triglycerides in mouse models (82–84). Exercise training also decreases the progression of fibrosis, enhances regenerative capacity, and reduces the occurrence of hepatocellular carcinoma, which develops in late stage NAFLD (84–86). AMPK activator AICAR is sufficient to recapitulate these beneficial effects of exercise training (86). Similarly, increased phosphorylation of AMPK is associated with improvements in NAFLD (80). These findings suggest that AMPK activation counteracts NAFLD by increasing fatty acid oxidation (81). However, studies of genetically engi-neered knock-in (KI) mice with mutations of AMPK phosphorylation sites in both Acc1 and Acc2 indicated that six weeks of high-intensity interval training have similar impacts on levels of ala-nine aminotransferase and aspartate transaminase, the markers of liver injury, in obese KI and WT mice. Training also suppresses hepatic glucose production induced by insulin to the same degree in these mice. Finally, endurance exercise training (eight weeks) in high-fat diet–induced obese mice results in reduced lipid droplet formation, decreased hepatic triglyceride, and enhanced lipophagy by activating the AMPK-Ulk1 regulatory axis (87). Additional research is clearly needed to better understand the functional role of AMPK activation and downstream events in response to exercise training in treating NAFLD/NASH.
AMPK ACTIVATION BY EXERCISE IN LIPOLYSIS AND BROWNING OF WHITE ADIPOSE TISSUE
Adipose tissue functions to liberate fatty acids from triacylglycerides, referred to as lipolysis, and release them into the circulation to be oxidized for energy production by other issues, including the skeletal muscle and the heart. Increased energy demand from exercise leads to increased lipolysis and reesterification in adipose tissue (88). Exercise induces AMPK activation in the adipose tissue during and after exercise, as well as after exercise training (68, 89, 90). AMPK activation in adipose tissue in response to endurance exercise appears to be dependent on β-adrenergic receptor activation induced by adrenaline (90) and is mediated by the process of lipolysis (91). As lipolysis and free fatty acid reesterification increase, the energy charge is depleted, resulting in increased AMPK activity (92) (Figure 4). AMPK activation may play a feed-forward role in lipolysis because adrenaline-induced lipolysis in isolated adipocytes can be blocked by compound C, an AMPK inhibitor (90). Interestingly, adipose tissue–specific AMPKα1α2 KO mice (AMPKα1α2 AKO) display normal fasting blood glucose, glucose tolerance, and insulin tolerance compared with WT mice (93). A definitive role of adipose tissue AMPK activation in controlling lipolysis remains to be elucidated.
Figure 4.
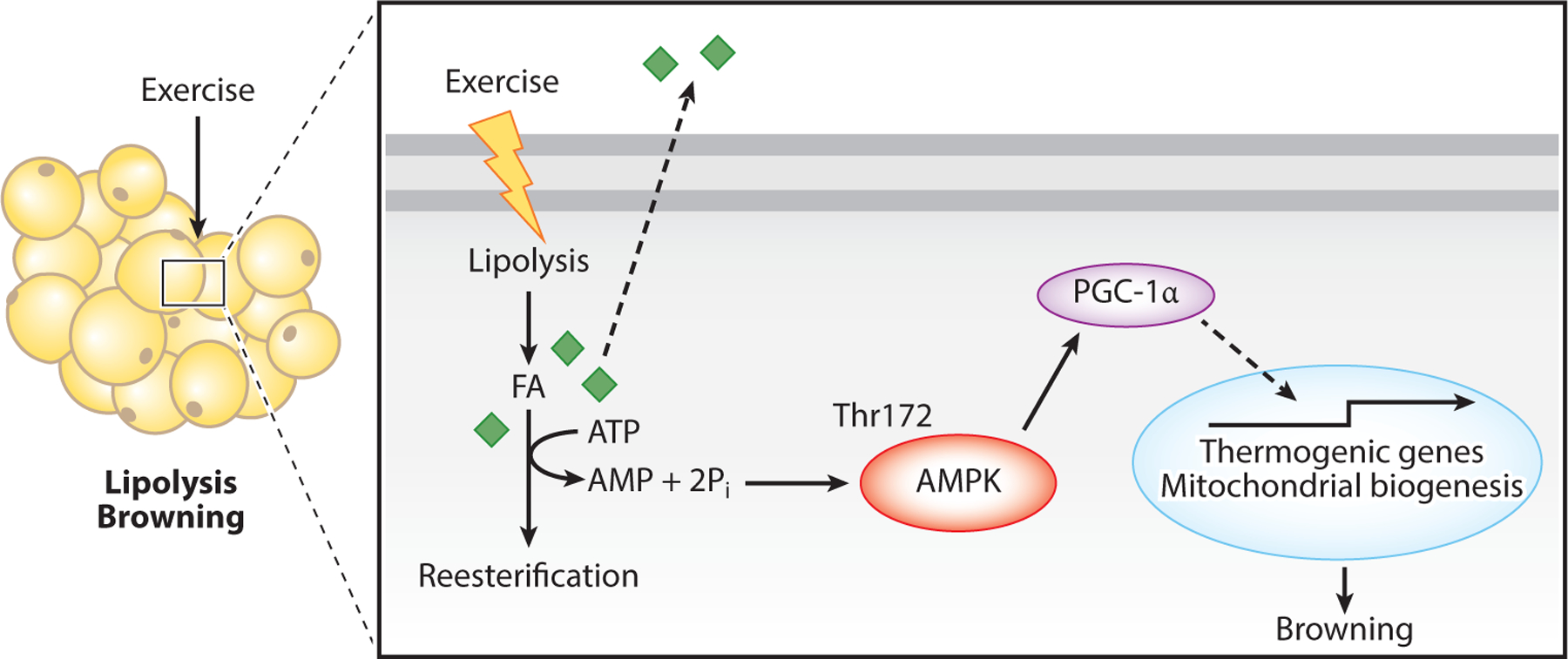
Exercise and AMPK activation in lipolysis and browning of white adipose tissue. Exercise induces lipolysis, resulting in increased FA release as fuel for other tissues/organs as well as reesterification of FA into triacylglyceride. Reesterification is an ATP-consuming process resulting in increased AMPK activation and increased PGC-1α-promoted transcription of thermogenic genes and mitochondrial biogenesis. These metabolic changes lead to adipose browning. Abbreviations: AMPK, AMP-activated protein kinase; FA, fatty acid; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1 alpha; Pi, inorganic phosphate; Thr, threonine.
Exercise training, particularly endurance exercise, is a potent lifestyle intervention against obesity. A single bout of swimming (120 min) in mice leads to reduced peroxisome proliferator-activated receptor γ2 (Pparg2) and CCAAT enhancer binding protein α (Cebpa) mRNA expression, with a concurrent increased expression of Cebpb, Cebpd, Pgc-1α, and Ucp1 in white adipose tissue (WAT) (94), suggesting that acute exercise suppresses adipogenic genes and promotes thermogenic genes. Exercise training also increases Pgc-1α expression and mitochondrial enzymes in adipose tissue, supportive of mitochondrial adaptation to exercise (95–97). AMPK activation by AICAR increases Pgc-1α mRNA expression in WAT (98). AMPKβ1 KO mice also have attenuated adipose tissue Pgc-1α mRNA expression under the condition of norepinephrine stimulation, suggesting that AMPK regulates PGC-1α and increases expression of mitochondrial proteins in adipose tissue (99) (Figure 4). Importantly, adipose-specific, inducible AMPKβ1β2 KO (AMPKβ1β2 iAKO) mice are cold intolerant and resistant to β-adrenergic activation of brown adipose tissue (BAT) and beiging of WAT, a conversion of WAT to BAT. These mice have impaired mitochondria structure and function and reduced markers of mitophagy in BAT with impaired thermogenic programs induced by chronic β-adrenergic activation (100). Overall, the role of adipose tissue AMPK activation in conversion of WAT to BAT remains unknown.
AMPK ACTIVATION BY EXERCISE IN BRAIN ADAPTATION AND COGNITIVE FUNCTION
Increasing evidence shows that regular exercise promotes mental health and cognitive function. In particular, exercise training has been shown to promote adult hippocampal neurogenesis and neuronal plasticity along with increased memory and cognitive function (101, 102). Evidence from three animal exercise models shows enhanced AMPK activity by exercise training in the hippocampus of the brain. In an accelerated aging model of d-galactose-treated rats, reduced protein expression of the AMPK/Sirt1/Pgc-1α signaling pathway is restored after 12 weeks of swimming (103). Similarly, treadmill running (10 weeks) in old rats results in increased expression and phosphorylation of Camk2a and AMPKα1 (104). Finally, voluntary wheel running restores AMPK activity and its phosphorylation in the hippocampus of senescence-accelerated mice P8 (SAMP8), with enhanced expression of mitochondrial proteins (105) (Figure 5). However, no studies show direct evidence of exercise-induced AMPK activation in the hippocampus in response to an acute bout of exercise. The aforementioned restoration of AMPK expression and phosphorylation could be a consequence of the neuronal impact of exercise training.
Figure 5.
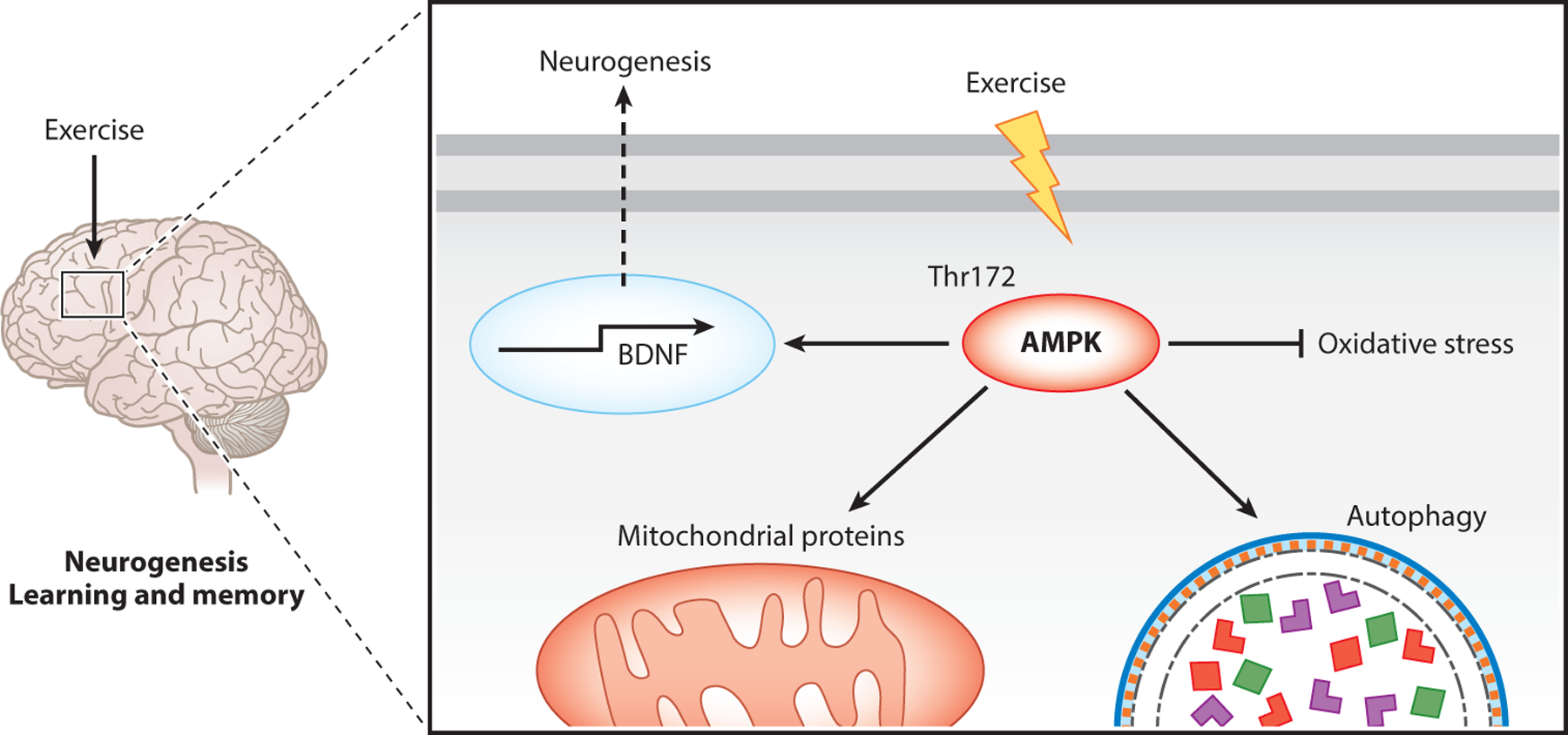
Exercise-induced AMPK activation in brain adaptation and neurogenesis. Exercise increases AMPK activity, leading to increased transcription of BDNF that promotes neurogenesis. AMPK also increases mitochondria content while promoting autophagy and inhibiting oxidative stress. Abbreviations: AMPK, AMP-activated protein kinase; BDNF, brain-derived neurotrophic factor; Thr, threonine.
Evidence of the benefits of exercise training for the brain continues to increase. Treadmill running (10 weeks) in old rats restores intersessional habituation and hippocampus morphology with reduced oxidative stress and enhanced autophagy markers (104) (Figure 5). A similar but slightly shorter-term exercise regimen (3 weeks) rescues the learning and memory deficits in restraint-stressed mice. It also restores the expression of brain-derived neurotrophic factor (Bdnf), a key molecule in plastic changes related to learning and memory; synaptophysin, a glycoprotein that is an integral part of the neuroendocrine secretory granule membrane; and postsynaptic density protein 95, a pivotal postsynaptic scaffolding protein in excitatory neurons. These changes are mimicked by 7 days of AICAR treatment (106). Treadmill running (4 weeks) rescues spatial learning and memory in mice with amyloid β injection into the cornu ammonis 1 (CA1) area of the hippocampus and restores the phosphorylation of AMPK and expression of Pgc-1α, fibronectin type III domain containing 5 (Fndc5), and Bdnf (107) (Figure 5). Finally, voluntary wheel running attenuates corticosterone-induced depression-like behavior in mice and improves neurogenesis and dendritic plasticity in the hippocampus (108). Together, these findings illustrate the many benefits of exercise training in deterring pathological changes and rescuing functional deficits in various neurodegenerative disease conditions. However, the role of AMPK activation in mediating these exercise benefits remains unexplained.
CONCLUSIONS
Exercise-induced adaptations in various tissues and organs promote health and prevent disease, and AMPK activation may play a critical role in conferring these benefits. Exercise and treatment with AMPK activators promote similar metabolic and cellular adaptations leading to improved energy homeostasis. The existing findings show that AMPK is an attractive target for prevention and treatment of NCDs. Advances in sophisticated molecular genetic models in animals will allow future experiments to improve our mechanistic understanding of the causal effects of AMPK activation in various tissues and organs during acute exercise and recovery as well as in response to exercise training.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.World Health Organ. 2018. Noncommunicable Diseases Country Profiles 2018 Geneva: World Health Organ. https://apps.who.int/iris/handle/10665/274512 [Google Scholar]
- 2.Beg ZH, Allmann DW, Gibson DM. 1973. Modulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity with cAMP and with protein fractions of rat liver cytosol. Biochem. Biophys. Res. Commun 54:1362–69 [DOI] [PubMed] [Google Scholar]
- 3.Carling D, Clarke PR, Zammit VA, Hardie DG. 1989. Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur. J. Biochem 186:129–36 [DOI] [PubMed] [Google Scholar]
- 4.Towler MC, Hardie DG. 2007. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res 100:328–41 [DOI] [PubMed] [Google Scholar]
- 5.Hardie DG, Ross FA, Hawley SA. 2012. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol 13:251–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herzig S, Shaw RJ. 2018. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol 19:121–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richter EA, Hargreaves M. 2013. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev 93:993–1017 [DOI] [PubMed] [Google Scholar]
- 8.Lundsgaard AM, Fritzen AM, Kiens B. 2020. The importance of fatty acids as nutrients during post-exercise recovery. Nutrients 12:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ross FA, MacKintosh C, Hardie DG. 2016. AMP-activated protein kinase: a cellular energy sensor that comes in 12 flavours. FEBS J. 283:2987–3001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao B, Heath R, Saiu P, Leiper FC, Leone P, et al. 2007. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449:496–500 [DOI] [PubMed] [Google Scholar]
- 11.Gowans GJ, Hawley SA, Ross FA, Hardie DG. 2013. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 18:556–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Winder WW, Hardie DG. 1996. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am. J. Physiol 270:E299–304 [DOI] [PubMed] [Google Scholar]
- 13.Clark SA, Chen ZP, Murphy KT, Aughey RJ, McKenna MJ, et al. 2004. Intensified exercise training does not alter AMPK signaling in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab 286:E737–43 [DOI] [PubMed] [Google Scholar]
- 14.Hamada T, Arias EB, Cartee GD. 2006. Increased submaximal insulin-stimulated glucose uptake in mouse skeletal muscle after treadmill exercise. J. Appl. Physiol 101:1368–76 [DOI] [PubMed] [Google Scholar]
- 15.Oakhill JS, Scott JW, Kemp BE. 2012. AMPK functions as an adenylate charge-regulated protein kinase. Trends Endocrinol. Metab 23:125–32 [DOI] [PubMed] [Google Scholar]
- 16.Jørgensen SB, Viollet B, Andreelli F, Frøsig C, Birk JB, et al. 2004. Knockout of the α2 but not α1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-β−4-ribofuranoside but not contraction-induced glucose uptake in skeletal muscle. J. Biol. Chem 279:1070–79 [DOI] [PubMed] [Google Scholar]
- 17.Lefort N, St.-Amand E, Morasse S, Côté CH, Marette A. 2008. The α-subunit of AMPK is essential for submaximal contraction-mediated glucose transport in skeletal muscle in vitro. Am. J. Physiol. Endocrinol. Metab 295:E1447–54 [DOI] [PubMed] [Google Scholar]
- 18.Kramer HF, Witczak CA, Fujii N, Jessen N, Taylor EB, et al. 2006. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes 55:2067–76 [DOI] [PubMed] [Google Scholar]
- 19.Barnes BR, Marklund S, Steiler TL, Walter M, Hjälm G, et al. 2004. The 5′-AMP-activated protein kinase γ3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J. Biol. Chem 279:38441–47 [DOI] [PubMed] [Google Scholar]
- 20.Jeppesen J, Albers PH, Rose AJ, Birk JB, Schjerling P, et al. 2011. Contraction-induced skeletal muscle FAT/CD36 trafficking and FA uptake is AMPK independent. J. Lipid Res 52:699–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffman NJ, Parker BL, Chaudhuri R, Fisher-Wellman KH, Kleinert M, et al. 2015. Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise regulated kinases and AMPK substrates. Cell Metab 22:922–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merrill GG, Kurth EJ, Hardie DH, Winder WW. 1997. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am. J. Physiol 273:E1107–12 [DOI] [PubMed] [Google Scholar]
- 23.Myers RW, Guan HP, Ehrhart J, Petrov A, Prahalada S, et al. 2017. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 357:507–11 [DOI] [PubMed] [Google Scholar]
- 24.Cokorinos EC, Delmore J, Reyes AR, Albuquerque B, Kjøbsted R, et al. 2017. Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non-human primates and mice. Cell Metab 25:1147–59 [DOI] [PubMed] [Google Scholar]
- 25.Treebak JT, Taylor EB, Witczak CA, An D, Toyoda T, et al. 2010. Identification of a novel phosphorylation site on TBC1D4 regulated by AMP-activated protein kinase in skeletal muscle. Am. J. Physiol. Cell Physiol 298:C377–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McConell GK. 2020. It’s well and truly time to stop stating that AMPK regulates glucose uptake and fat oxidation during exercise. Am. J. Physiol. Endocrinol. Metab 318:E564–67 [DOI] [PubMed] [Google Scholar]
- 27.O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jørgensen SB, et al. 2011. AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. PNAS 108:16092–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hingst JR, Kjøbsted R, Birk JB, Jørgensen NO, Larsen MR, et al. 2020. Inducible deletion of skeletal muscle AMPKα reveals that AMPK is required for nucleotide balance but dispensable for muscle glucose uptake and fat oxidation during exercise. Mol. Metab 40:101028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kjøbsted R, Roll JLW, Jørgensen NO, Birk JB, Foretz M, et al. 2019. AMPK and TBC1D1 regulate muscle glucose uptake after, but not during, exercise and contraction. Diabetes 68:1427–40 [DOI] [PubMed] [Google Scholar]
- 30.Lantier L, Fentz J, Mounier R, Leclerc J, Treebak JT, et al. 2014. AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity. FASEB J 28:3211–24 [DOI] [PubMed] [Google Scholar]
- 31.Steinberg GR, O’Neill HM, Dzamko NL, Galic S, Naim T, et al. 2010. Whole body deletion of AMP-activated protein kinase β2 reduces muscle AMPK activity and exercise capacity. J. Biol. Chem 285:37198–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merry TL, Steinberg GR, Lynch GS, McConell GK. 2010. Skeletal muscle glucose uptake during contraction is regulated by nitric oxide and ROS independently of AMPK. Am. J. Physiol. Endocrinol. Metab 298:E577–85 [DOI] [PubMed] [Google Scholar]
- 33.Mu J, Brozinick JT, Valladares O, Bucan M, Birnbaum MJ. 2001. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol. Cell 7:1085–94 [DOI] [PubMed] [Google Scholar]
- 34.Kjøbsted R, Munk-Hansen N, Birk JB, Foretz M, Viollet B, et al. 2017. Enhanced muscle insulin sensitivity after contraction/exercise is mediated by AMPK. Diabetes 66:598–612 [DOI] [PubMed] [Google Scholar]
- 35.Kjøbsted R, Treebak J, Fentz J, Lantier L, Viollet B, et al. 2015. Prior AICAR stimulation increases insulin sensitivity in mouse skeletal muscle in an AMPK-dependent manner. Diabetes 64:2042–55 [DOI] [PubMed] [Google Scholar]
- 36.Fritzen AM, Lundsgaard AM, Kiens B. 2020. Tuning fatty acid oxidation in skeletal muscle with dietary fat and exercise. Nat. Rev. Endocrinol 16:683–96 [DOI] [PubMed] [Google Scholar]
- 37.Hutber CA, Rasmussen BB, Winder WW. 1997. Endurance training attenuates the decrease in skeletal muscle malonyl-CoA with exercise. J. Appl. Physiol 83:1917–22 [DOI] [PubMed] [Google Scholar]
- 38.Park H, Kaushik VK, Constant S, Prentki M, Przybytkowski E, et al. 2002. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. J. Biol. Chem 277:32571–77 [DOI] [PubMed] [Google Scholar]
- 39.Fentz J, Kjøbsted R, Birk JB, Jordy AB, Jeppesen J, et al. 2015. AMPKα is critical for enhancing skeletal muscle fatty acid utilization during in vivo exercise in mice. FASEB J 29:1725–38 [DOI] [PubMed] [Google Scholar]
- 40.Dzamko N, Schertzer JD, Ryall JG, Steel R, Macaulay SL, et al. 2008. AMPK-independent pathways regulate skeletal muscle fatty acid oxidation. J. Physiol 586:5819–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fritzen AM, Lundsgaard AM, Jeppesen J, Christiansen ML, Biensø R, et al. 2015. 5′-AMP activated protein kinase α2 controls substrate metabolism during post-exercise recovery via regulation of pyruvate dehydrogenase kinase 4. J. Physiol 593:4765–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodnick KJ, Henriksen EJ, James DE, Holloszy JO. 1992. Exercise training, glucose transporters, and glucose transport in rat skeletal muscles. Am. J. Physiol 262:C9–14 [DOI] [PubMed] [Google Scholar]
- 43.Gong H, Xie J, Zhang N, Yao L, Zhang Y. 2011. MEF2A binding to the Glut4 promoter occurs via an AMPKα2-dependent mechanism. Med. Sci. Sports Exerc 43:1441–50 [DOI] [PubMed] [Google Scholar]
- 44.Jørgensen SB, Treebak JT, Viollet B, Schjerling P, Vaulont S, et al. 2007. Role of AMPKα2 in basal, training-, and AICAR-induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle. Am. J. Physiol. Endocrinol. Metab 292:E331–39 [DOI] [PubMed] [Google Scholar]
- 45.Koh JH, Hancock CR, Han DH, Holloszy JO, Nair KS, Dasari S. 2019. AMPK and PPARβ positive feedback loop regulates endurance exercise training-mediated GLUT4 expression in skeletal muscle. Am. J. Physiol. Endocrinol. Metab 316:E931–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, et al. 2005. Exercise stimulates Pgc-1α transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem 280:19587–93 [DOI] [PubMed] [Google Scholar]
- 47.Jäger S, Handschin C, St.-Pierre J, Spiegelman BM. 2007. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. PNAS 104:12017–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brandauer J, Andersen MA, Kellezi H, Risis S, Frøsig C, et al. 2015. AMP-activated protein kinase controls exercise training- and AICAR-induced increases in SIRT3 and MnSOD. Front. Physiol 6:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ding H, Jiang N, Liu H, Liu X, Liu D, et al. 2010. Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim. Biophys. Acta 1800:250–56 [DOI] [PubMed] [Google Scholar]
- 50.Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, et al. 2017. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun 8:548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Toyama EQ, Herzig S, Courchet J, Lewis TL Jr., Losón OC, et al. 2016. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351:275–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seabright AP, Fine NHF, Barlow JP, Lord SOS, Musa I, et al. 2020. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J 34:6284–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, et al. 2008. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 112:1493–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drake JC, Wilson RJ, Laker RC, Guan Y, Spaulding HR, et al. 2021. Mitochondria-localized AMPK responds to local energetics and contributes to exercise and energetic stress-induced mitophagy. PNAS 118:e2025932118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma X, Fu Y, Xiao H, Song Y, Chen R, et al. 2015. Cardiac fibrosis alleviated by exercise training is AMPK-dependent. PLOS ONE 10:e0129971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coven DL, Hu X, Cong L, Bergeron R, Shulman GI, et al. 2003. Physiological role of AMP-activated protein kinase in the heart: graded activation during exercise. Am. J. Physiol. Endocrinol. Metab 285:E629–36 [DOI] [PubMed] [Google Scholar]
- 57.Luiken JJ, Coort SL, Willems J, Coumans WA, Bonen A, et al. 2003. Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes 52:1627–34 [DOI] [PubMed] [Google Scholar]
- 58.Russell RR 3rd, Bergeron R, Shulman GI, Young LH. 1999. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am. J. Physiol 277:H643–49 [DOI] [PubMed] [Google Scholar]
- 59.Chabowski A, Momken I, Coort SL, Calles-Escandon J, Tandon NN, et al. 2006. Prolonged AMPK activation increases the expression of fatty acid transporters in cardiac myocytes and perfused hearts. Mol. Cell. Biochem 288:201–12 [DOI] [PubMed] [Google Scholar]
- 60.Ma Z, Qi J, Gao L, Zhang J. 2020. Role of exercise on alleviating pressure overload-induced left ventricular dysfunction and remodeling via AMPK-dependent autophagy activation. Int. Heart J 61:1022–33 [DOI] [PubMed] [Google Scholar]
- 61.Morissette MP, Susser SE, Stammers AN, Moffatt TL, Wigle JT, et al. 2019. Exercise-induced increases in the expression and activity of cardiac sarcoplasmic reticulum calcium ATPase 2 is attenuated in AMPKα2 kinase-dead mice. Can. J. Physiol. Pharmacol 97:786–95 [DOI] [PubMed] [Google Scholar]
- 62.Cyganek L, Tiburcy M, Sekeres K, Gerstenberg K, Bohnenberger H, et al. 2018. Deep phenotyping of human induced pluripotent stem cell-derived atrial and ventricular cardiomyocytes. JCI Insight 3:e99941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Camacho RC, Galassetti P, Davis SN, Wasserman DH. 2005. Glucoregulation during and after exercise in health and insulin-dependent diabetes. Exerc. Sport Sci. Rev 33:17–23 [PubMed] [Google Scholar]
- 64.Hughey CC, James FD, Bracy DP, Donahue EP, Young JD, et al. 2017. Loss of hepatic AMP-activated protein kinase impedes the rate of glycogenolysis but not gluconeogenic fluxes in exercising mice. J. Biol. Chem 292:20125–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wasserman DH, Cherrington AD. 1991. Hepatic fuel metabolism during muscular work: role and regulation. Am. J. Physiol 260:E811–24 [DOI] [PubMed] [Google Scholar]
- 66.Wasserman DH, Spalding JA, Lacy DB, Colburn CA, Goldstein RE, Cherrington AD. 1989. Glucagon is a primary controller of hepatic glycogenolysis and gluconeogenesis during muscular work. Am. J. Physiol 257:E108–17 [DOI] [PubMed] [Google Scholar]
- 67.Hems R, Ross BD, Berry MN, Krebs HA. 1966. Gluconeogenesis in the perfused rat liver. Biochem. J 101:284–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Takekoshi K, Fukuhara M, Quin Z, Nissato S, Isobe K, et al. 2006. Long-term exercise stimulates adenosine monophosphate-activated protein kinase activity and subunit expression in rat visceral adipose tissue and liver. Metabolism 55:1122–28 [DOI] [PubMed] [Google Scholar]
- 69.Lezi E, Lu J, Burns JM, Swerdlow RH. 2013. Effect of exercise on mouse liver and brain bioenergetic infrastructures. Exp. Physiol 98:207–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marcinko K, Sikkema SR, Samaan MC, Kemp BE, Fullerton MD, Steinberg GR. 2015. High intensity interval training improves liver and adipose tissue insulin sensitivity. Mol. Metab 4:903–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoene M, Lehmann R, Hennige AM, Pohl AK, Häring HU, et al. 2009. Acute regulation of metabolic genes and insulin receptor substrates in the liver of mice by one single bout of treadmill exercise. J. Physiol 587:241–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Camacho RC, Donahue EP, James FD, Berglund ED, Wasserman DH. 2006. Energy state of the liver during short-term and exhaustive exercise in C57BL/6J mice. Am. J. Physiol. Endocrinol. Metab 290:E405–8 [DOI] [PubMed] [Google Scholar]
- 73.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, et al. 2005. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310:1642–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Andreelli F, Foretz M, Knauf C, Cani PD, Perrin C, et al. 2006. Liver adenosine monophosphate-activated kinase-α2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology 147:2432–41 [DOI] [PubMed] [Google Scholar]
- 75.Foretz M, Ancellin N, Andreelli F, Saintillan Y, Grondin P, et al. 2005. Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes 54:1331–39 [DOI] [PubMed] [Google Scholar]
- 76.Knudsen JG, Biensø RS, Hassing HA, Jakobsen AH, Pilegaard H. 2015. Exercise-induced regulation of key factors in substrate choice and gluconeogenesis in mouse liver. Mol. Cell. Biochem 403:209–17 [DOI] [PubMed] [Google Scholar]
- 77.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, et al. 2001. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413:131–38 [DOI] [PubMed] [Google Scholar]
- 78.Wende AR, Huss JM, Schaeffer PJ, Giguère V, Kelly DP. 2005. PGC-1α coactivates PDK4 gene expression via the orphan nuclear receptor ERRα: a mechanism for transcriptional control of muscle glucose metabolism. Mol. Cell. Biol 25:10684–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Holness MJ, Bulmer K, Smith ND, Sugden MC. 2003. Investigation of potential mechanisms regulating protein expression of hepatic pyruvate dehydrogenase kinase isoforms 2 and 4 by fatty acids and thyroid hormone. Biochem. J 369:687–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Woods A, Williams J, Muckett P, Mayer F, Liljevald M, et al. 2017. Liver-specific activation of AMPK prevents steatosis on a high-fructose diet. Cell Rep 18:687–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smith BK, Marcinko K, Desjardins EM, Lally JS, Ford RJ, Steinberg GR. 2016. Treatment of nonalcoholic fatty liver disease: role of AMPK. Am. J. Physiol. Endocrinol. Metab 311:E730–40 [DOI] [PubMed] [Google Scholar]
- 82.Bae JY. 2020. Resistance exercise regulates hepatic lipolytic factors as effective as aerobic exercise in obese mice. Int. J. Environ. Res Public Health; 17:8307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ok D, Ko K, Bae J. 2018. Exercise without dietary changes alleviates nonalcoholic fatty liver disease without weight loss benefits. Lipids Health Dis 17:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guarino M, Kumar P, Felser A, Terracciano LM, Guixé-Muntet S, et al. 2020. Exercise attenuates the transition from fatty liver to steatohepatitis and reduces tumor formation in mice. Cancers 12:1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Piguet AC, Saran U, Simillion C, Keller I, Terracciano L, et al. 2015. Regular exercise decreases liver tumors development in hepatocyte-specific PTEN-deficient mice independently of steatosis. J. Hepatol 62:1296–303 [DOI] [PubMed] [Google Scholar]
- 86.Linecker M, Frick L, Kron P, Limani P, Kambakamba P, et al. 2020. Exercise improves outcomes of surgery on fatty liver in mice: a novel effect mediated by the AMPK pathway. Ann. Surg 271:347–55 [DOI] [PubMed] [Google Scholar]
- 87.Gao Y, Zhang W, Zeng LQ, Bai H, Li J, et al. 2020. Exercise and dietary intervention ameliorate high-fat diet-induced NAFLD and liver aging by inducing lipophagy. Redox Biol 36:101635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hargreaves M, Spriet LL. 2018. Exercise metabolism: fuels for the fire. Cold Spring Harb. Perspect. Med 8:a029744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Watt MJ, Holmes AG, Pinnamaneni SK, Garnham AP, Steinberg GR, et al. 2006. Regulation of HSL serine phosphorylation in skeletal muscle and adipose tissue. Am. J. Physiol. Endocrinol. Metab 290:E500–8 [DOI] [PubMed] [Google Scholar]
- 90.Koh HJ, Hirshman MF, He H, Li Y, Manabe Y, et al. 2007. Adrenaline is a critical mediator of acute exercise-induced AMP-activated protein kinase activation in adipocytes. Biochem. J 403:473–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gauthier MS, Miyoshi H, Souza SC, Cacicedo JM, Saha AK, et al. 2008. AMP-activated protein kinase is activated as a consequence of lipolysis in the adipocyte: potential mechanism and physiological relevance. J. Biol. Chem 283:16514–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McKie GL, Wright DC. 2020. Biochemical adaptations in white adipose tissue following aerobic exercise: from mitochondrial biogenesis to browning. Biochem. J 477:1061–81 [DOI] [PubMed] [Google Scholar]
- 93.Choi RH, McConahay A, Johnson MB, Jeong HW, Koh HJ. 2019. Adipose tissue-specific knockout of AMPKα1/α2 results in normal AICAR tolerance and glucose metabolism. Biochem. Biophys. Res. Commun 519:633–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shen Y, Zhou H, Jin W, Lee HJ. 2016. Acute exercise regulates adipogenic gene expression in white adipose tissue. Biol. Sport 33:381–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stallknecht B, Vinten J, Ploug T, Galbo H. 1991. Increased activities of mitochondrial enzymes in white adipose tissue in trained rats. Am. J. Physiol 261:E410–14 [DOI] [PubMed] [Google Scholar]
- 96.Sutherland LN, Bomhof MR, Capozzi LC, Basaraba SA, Wright DC. 2009. Exercise and adrenaline increase PGC-1α mRNA expression in rat adipose tissue. J. Physiol 587:1607–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stephenson EK, Lessard SJ, Rivas DA, Watt MJ, Yaspelkis BB, et al. 2013. Exercise training enhances white adipose tissue metabolism in rats selectively bred for low- or high-endurance running capacity. Am. J. Physiol. Endocrinol. Metab 305:E429–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gaidhu MP, Fediuc S, Anthony NM, So M, Mirpourian M, et al. 2009. Prolonged AICAR-induced AMP-kinase activation promotes energy dissipation in white adipocytes: novel mechanisms integrating HSL and ATGL. J. Lipid Res 50:704–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wan Z, Root-McCaig J, Castellani L, Kemp BE, Steinberg GR, Wright DC. 2014. Evidence for the role of AMPK in regulating PGC-1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity 22:730–38 [DOI] [PubMed] [Google Scholar]
- 100.Mottillo EP, Desjardins EM, Crane JD, Smith BK, Green AE, et al. 2016. Lack of adipocyte AMPK exacerbates insulin resistance and hepatic steatosis through brown and beige adipose tissue function. Cell Metab 24:118–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kobilo T, Liu QR, Gandhi K, Mughal M, Shaham Y, van Praag H. 2011. Running is the neurogenic and neurotrophic stimulus in environmental enrichment. Learn. Memory 18:605–9 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Christie BR, Eadie BD, Kannangara TS, Robillard JM, Shin J, Titterness AK. 2008. Exercising our brains: how physical activity impacts synaptic plasticity in the dentate gyrus. Neuromol. Med 10:47–58 [DOI] [PubMed] [Google Scholar]
- 103.Lin J, Kuo W, Baskaran R, Kuo C, Chen Y, et al. 2020. Swimming exercise stimulates IGF1/PI3K/Akt and AMPK/SIRT1/PGC1α survival signaling to suppress apoptosis and inflammation in aging hippocampus. Aging 12:6852–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu W, Xia Y, Kuang H, Wang Z, Liu S, et al. 2019. Proteomic profile of carbonylated proteins screen the regulation of calmodulin-dependent protein kinases-AMPK-beclin1 in aerobic exercise-induced autophagy in middle-aged rat hippocampus. Gerontology 65:620–33 [DOI] [PubMed] [Google Scholar]
- 105.Bayod S, Guzmán-Brambila C, Sanchez-Roige S, Lalanza JF, Kaliman P, et al. 2015. Voluntary exercise promotes beneficial anti-aging mechanisms in SAMP8 female brain. J. Mol. Neurosci 55:525–32 [DOI] [PubMed] [Google Scholar]
- 106.Kim DM, Leem YH. 2016. Chronic stress-induced memory deficits are reversed by regular exercise viaAMPK-mediated BDNF induction. Neuroscience 324:271–85 [DOI] [PubMed] [Google Scholar]
- 107.Azimi M, Gharakhanlou R, Naghdi N, Khodadadi D, Heysieattalab S. 2018. Moderate treadmill exercise ameliorates amyloid-β-induced learning and memory impairment, possibly via increasing AMPK activity and up-regulation of the PGC-1α/FNDC5/BDNF pathway. Peptides 102:78–88 [DOI] [PubMed] [Google Scholar]
- 108.Wang P, Liang Y, Chen K, Yau SY, Sun X, et al. 2020. Potential involvement of adiponectin signaling in regulating physical exercise-elicited hippocampal neurogenesis and dendritic morphology in stressed mice. Front. Cell. Neurosci 14:189. [DOI] [PMC free article] [PubMed] [Google Scholar]