

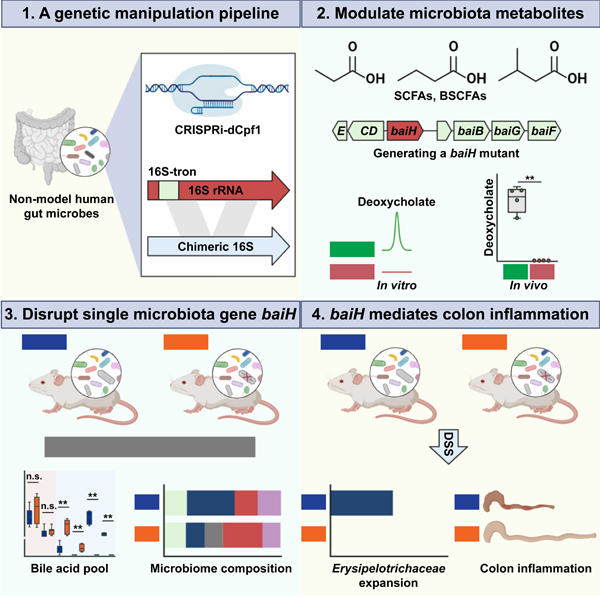
Summary
Hundreds of microbiota genes are associated with host biology/disease. Unraveling the causal contribution of a microbiota gene to host biology remains difficult because many are encoded by non-model gut commensals and not genetically targetable. A general approach to identify their gene transfer methodology and build their gene manipulation tools would enable mechanistic dissections of their impact on host physiology. We developed a pipeline that identifies the gene transfer methods for multiple non-model microbes spanning 5 phyla, and we demonstrated the utility of their genetic tools by modulating microbiome-derived short-chain fatty acids and bile acids in vitro and in the host. In a proof-of-principle study, by deleting a commensal gene for bile acid synthesis in a complex microbiome, we discover an intriguing role of this gene in regulating colon inflammation. This technology will enable genetically engineering the non-model gut microbiome and facilitate mechanistic dissection of microbiota-host interactions.
Graphical Abstract
In Brief:
A pipeline for the genetic manipulation of non-model gut microbes enables single- gene, functional interrogation of bacteria in a complex microbial system
INTRODUCTION
Multi-omics studies uncover many microbiota genes that are associated with host biology. However, it remains challenging to unravel the causal mechanisms underlying microbiota gene-host biology interactions, mainly because many are encoded by non-model gut microbes like Firmicutes/Clostridia. While genetic toolsets are readily available for model bacteria like E. coli or B. thetaiotaomicron, many non-model gut bacteria (e.g., Lachnospiraceae, Prevotella) are not genome sequenced, and it is unknown how to introduce exogenous DNA or which gene manipulation tool to select (Waller et al., 2017). Until now, there is no efficient, standardized, and in vitro pipeline to identify their gene transfer methods and build their genetic manipulation systems without prior knowledge of their genome information.
Such a pipeline should be prioritized for three reasons: 1) Multi-omics studies have uncovered significant associations between microbiota genes and diseases. Many of these genes are exclusively expressed in non-model microbes such as Firmicutes/Clostridia (Lloyd-Price et al., 2019; Thomas et al., 2019; Wang et al., 2012; Wirbel et al., 2019; Yachida et al., 2019; Zhou et al., 2019). A pipeline addressing this need would be a first step to manipulating these genes in vivo and causally connecting them with host diseases. 2) The gut microbiota plays an essential role in regulating host biology, but little is known about which bacteria and genes are responsible. This pipeline would enable gene toggling in previously non-targetable microbes and boost in-depth mechanistic studies of microbiota-host physiology interactions. 3) The microbiota impacts multiple therapies such as fecal microbiota transplantation and cancer immunotherapy (Helmink et al., 2019; Roy and Trinchieri, 2017), but the molecular mechanisms behind them largely remain elusive. The pipeline would not only facilitate dissection of the effect of microbiota on the associated treatments but would also enable genetic engineering of the gut microbiome, as a whole, for improved therapeutics.
Here, we establish a genetic manipulation (GM) pipeline to identify gene transfer methodology and build a genetic tool for non-model human gut commensals on a large scale (200 gut isolates from >140 species in five phyla) (Fig. 1). This pipeline efficiently identified the gene transfer methods for 88 non-model gut bacterial isolates and built their tools for targeted gene manipulation (Fig. 1). Of note, gut Firmicutes/Clostridia comprises one of the most abundant bacterial groups in healthy human guts, yet its genetic manipulation is largely unexplored (Waller et al., 2017). Via a multifactorial optimization of their conjugation/transformation conditions, we identified the gene transfer methods for 38 non-model gut Clostridia, and set up CRISPRi or Group II intron-based genetic tools in 27 of them. We demonstrated the utility of these toolsets by modulating short-chain fatty acids (SCFAs) and secondary bile acids in vitro and in the context of host colonization. As a proof of principle, we selected one Clostridia specific pathway-bile acid 7α dehydroxylation-for further functional investigation. By genetically tagging the Clostridia commensal, we were able to manipulate its bai gene in a complex microbiome. We provide evidence that the bai gene significantly impacts host gut microbiome and bile acid composition, and mediates colon inflammation in a complex microbiome.
Figure 1. Overview of the genetic manipulation (GM) pipeline for non-model gut commensals.
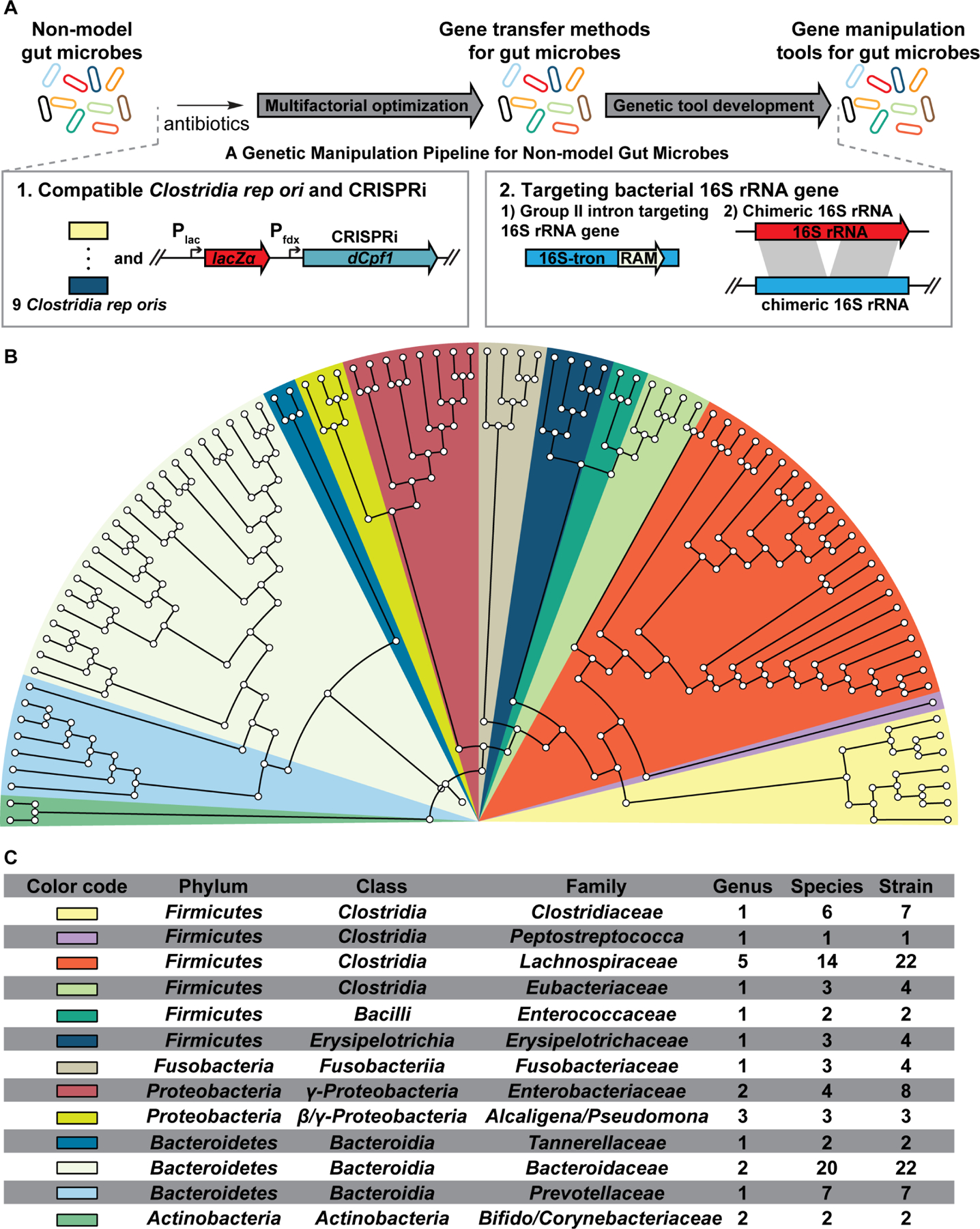
(A) A total of 200 human gut isolates from >140 species and 5 phyla were subject to the GM pipeline. The pipeline identifies gene transfer methods for 88 non-model gut microbes and build gene manipulation tools for 71 of them (Table S1). For Gram-negative gut microbes, identifying their gene transfer methods and building their gene insertion tools are achieved in one step via the chimeric-16S rRNA strategy.
(B) Phylogenetic tree (colored by Family) of the 16S rRNA sequences from the genetically targetable microbes identified via the GM pipeline.
(C) Detailed phylogenetic information of the genetically targetable microbes identified in this study.
The pipeline described here and the related findings represent a large-scale identification of gene transfer methodology for non-model Firmicutes/Clostridial isolates that greatly expands the manipulatable genes/pathways coded by the gut microbiota. For instance, microbiota pathways encoded by the gut bacteria that previously had no tractable genetic tools, like that for butyrate or bile acid 7α-dehydroxylation, were identified in our library of genetically targetable commensals and manipulated. This library of targetable gut isolates and their genetic tools will serve as a starting point for precisely controlling microbiome molecular output and interrogating their effects on host biology. The GM pipeline efficiently identifies gene transfer methods for gut Clostridial isolates and develops their gene manipulation tools without prior knowledge of their genome sequences. Both features suggest its application as a useful technology to delineate the genetics for non-model gut Firmicutes/Clostridia commensals.
RESULTS
An overview of the GM pipeline
The overall GM workflow is summarized in Fig. 1A. There are three challenges toward building such a pipeline. First, there is no previously reported antibiotic marker that universally functions in most of the non-model microbes. By assessing the antibiotic resistance and testing different conjugation donors to introduce antibiotic markers into these gut isolates, we found that one chloramphenicol resistant marker operates in many of the transformed microbes (Fig. 1B). Second, Firmicutes/Clostridia microbes are highly abundant in healthy human guts, yet the genetic manipulation of this physiologically important, host-associated bacterial group remains largely unexplored (Waller et al., 2017). Lacking their genetic tools greatly limits mechanistic dissection of the effects of Firmicutes/Clostridia genes on host biology. By optimizing multiple factors, we managed to identify gene transfer methods for multiple non-model gut Clostridia microbes (Fig. 1). Third, when we started building the pipeline, the genomes of many isolates had not been sequenced, posing a considerable roadblock in establishing their targeted gene manipulation tools on a large scale. To overcome this hurdle, we incorporated CRISPRi and a lacZα transcription reporter or developed strategies to genetically target the bacterial 16S rRNA gene (Fig. 1A). For consistency, we consider a gut microbe as genetically targetable if exogenous DNA (shuttle or suicide plasmids) can be repeatedly introduced into the microbe in vitro. A genetic manipulation tool is established if targeted manipulation of its gene/gene expression is achieved in the microbes of interest.
Selection of gut microbes and screening their culture conditions and antibiotic resistance
We prioritized Firmicutes and Bacteroidetes microbes that dominate healthy human guts (Cho and Blaser, 2012), but many (like Clostridia and Prevotella, e.g.) do not have gene transfer methods and tractable genetic tools. We diversified the screened pool by selecting commensals from a variety of genera/species (Table S1). We first identified the culture conditions supporting the growth of these gut microbes (Fig. 1A, Tables S1 and S3). Next, we screened these microbes against a collection of antibiotics to identify the following (Fig. 1A, Table S1): 1) the MIC of an antibiotic to which they are susceptible, allowing its resistance gene to be used as a selective marker, and 2) an antibiotic which they are resistant to but is active against E. coli, enabling suppression of E. coli growth after conjugation. For 1), we determined the MIC of thiamphenicol that inhibits the growth of almost all the tested microbes (Table S1), and for 2), almost all the Clostridia are resistant to D-cycloserine or kanamycin, and all the Prevotella to gentamycin or D-cycloserine (Table S1).
A multifactorial optimization to identify gene transfer methodology for non-model Clostridia
Multiple reasons, including incompatible origins of replication (rep oris) and antibiotic markers, host endogenous defense systems, and very inefficient homologous recombination (HR), cause the genetics of gut Firmicutes/Clostridia commensals to be poorly investigated compared to its counterpart Bacteroides. (Waller et al., 2017). Therefore, herein we have performed a multifactorial optimization of the transformation/conjugation parameters to identify gene transfer conditions for previously untransformed gut Firmicutes/Clostridia commensals (Fig. 2A, STAR Methods):
Figure 2. Developing a genetic manipulation pipeline for non-model gut commensals.
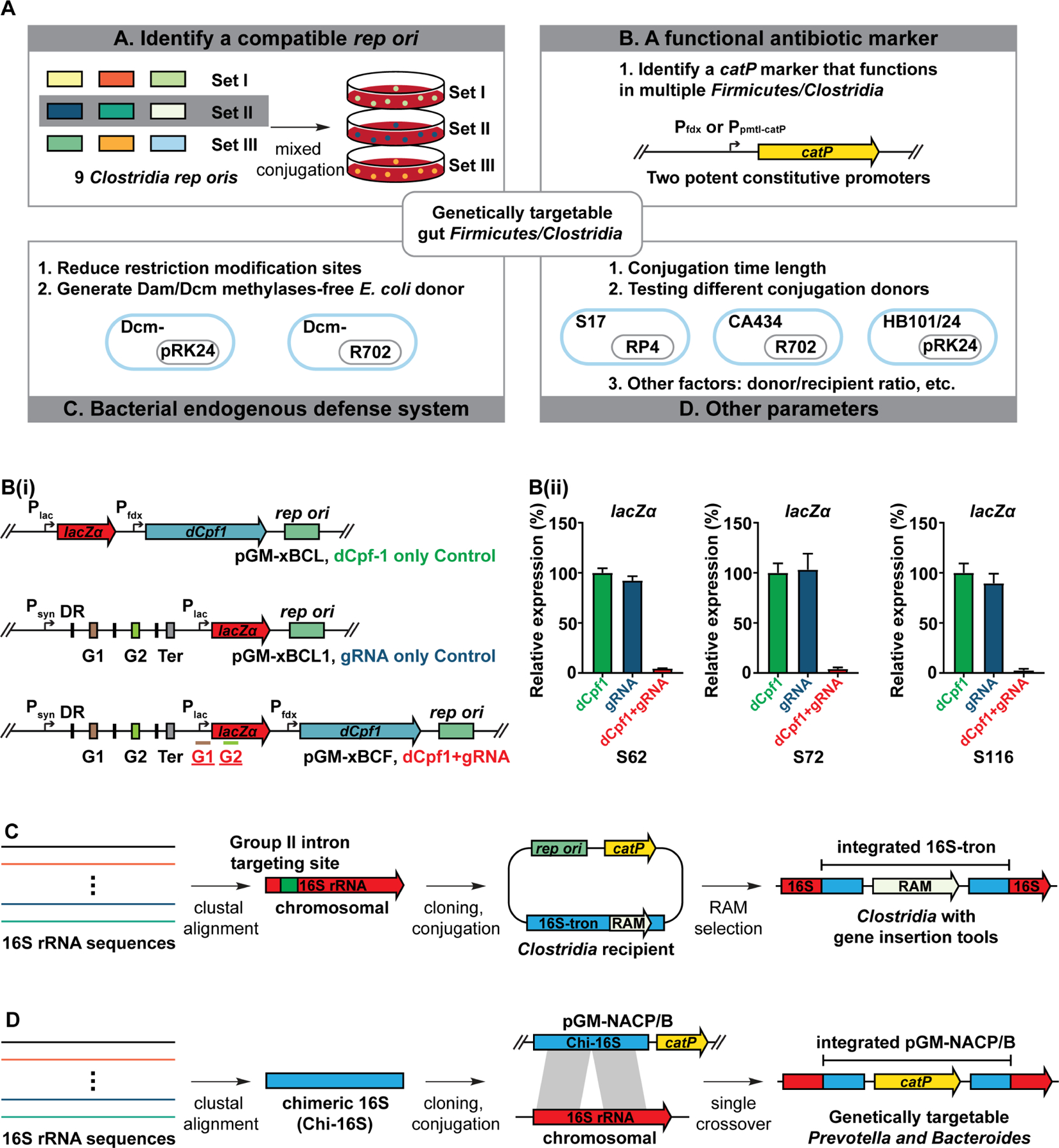
(A) Schematic view of a multifactorial optimization of the conjugation/transformation parameters to identify gene transfer conditions for 38 non-model gut Firmicutes/Clostridia that are mostly untransformed. Please see STAR Methods for more details.
(B) Establishment of a dCpf1-lacZα platform for non-model gut Firmicutes/Clostridia. (i) Schematic view of a dCpf1-lacZα system. The promoter and CDS region of lacZα are targeted by a duplex gRNA G1 and G2. (ii) The dCpf1-lacZα system efficiently suppresses lacZα expression in 25 Clostridia microbes. The panel shows the mean gene expression of three biological replicates as determined by qPCR. The dCpf-1-only and gRNA-only vectors are used as negative controls. Three qPCR results are shown. The numbering of the strains corresponds to the strain information shown in Table S1. Error bar: S.E.M. DR: direct repeat, G1: guide RNA-coding sequence 1, G2: guide RNA-coding sequence 2, Ter: terminator.
(C) Schematic view of the 16S-tron strategy for non-model Clostridia. The Clostridia 16S rRNA sequences were aligned to identify a conserved target site of Group II intron. The 16S targeting Group II intron (16S-tron) was introduced into the targetable Clostridia commensals due to RAM (retrotransposition-activated marker) availability. We identified 15 Clostridia whose chromosomes have been integrated by the 16S-tron.
(D) Schematic view of a Bacteroidia/Prevotella GM pipeline. The Prevotella 16S rRNA sequences were aligned to generate a ~1kb chimeric 16S (Chi-16S) fragment. The Chi-16S was assembled to get a suicide vector pGM-NAC2P. The pGM-NAC2P (NAC2B for Bacteroides) was conjugated to multiple Prevotella, Bacteroides, and Parabacteroides commensals targeting their chromosomal 16S rRNA genes (Table S1). We identified 31 targetable Bacteroidia whose 16S rRNA genes have been integrated by pGM-NAC2P (or NAC2B) (Table S1).
See also Tables S1 and S2, and Figures S1 and S2.
First, because our initial attempt to introduce the four most-used Clostridium rep oris (Heap et al., 2009) into several gut Clostridia like C. bolteae were unsuccessful, we expanded the repertoire of Clostridia rep oris and developed a mixed-conjugation strategy to introduce compatible rep ori into non-model gut Clostridia (Figs. 2A, S1, and STAR Methods). Second, we utilized a universal catP marker regulated by a potent constitutive promoter, Ppmtl-catP or Pfdx-cs (identified via a promoter library screen in multiple non-model Clostridia, Fig. S1), to confer antibiotic resistance during conjugation/transformation. This effort significantly reduced the workload of marker-switching when applying the pipeline to a large number of previously non-targetable Clostridial microbes (Fig. 2A). Third, we attempted different approaches, including utilization of an E. coli methylase-free ‘sExpress’ conjugation donor (Woods et al., 2019), decreasing restriction-modification (RM) recognition sites (Mermelstein et al., 1992; Purdy et al., 2002; Yang et al., 2016), and/or pre-methylate transforming DNA (Jennert et al., 2000; Pyne et al., 2014), to reduce the effect of Clostridia host defense systems during conjugation/transformation (Fig. 2A, STAR Methods). Last, several other parameters are optimized in this study, including conjugation time length, conjugation donor/recipient ratio, different conjugative plasmids, etc. (Fig. 2A, Table S1). These optimized parameters are summarized in Table S1, and detailed protocols for conjugation/electroporation are reported in STAR Methods.
These concerted efforts allowed us to identify their gene transfer conditions with an overall 41% success rate (Figs. 2A and S1), suggesting the possibility of developing associated gene manipulation systems. As may be anticipated, multiple factors need to be optimized simultaneously to successfully introduce plasmids into previously untransformed Clostridia (Table S1). For instance, introducing plasmids into S71 C. bartlettii (that harbors a putative Type-IV RM system) requires a compatible Clostridia rep ori, a functional catP marker driven by a strong promoter (and plating on plates supplemented with thiamphenicol at MIC), an E. coli ‘sExpress’ donor that does not methylate plasmid DNA and harbors R702 conjugative plasmid, and combination with other optimized parameters such as conjugation time and conjugation antibiotics detailed in Table S1. Interestingly, some Clostridia accept different rep oris even if they are closely related (e.g., C. bolteae isolates, see Table S1), demonstrating the necessity of expanding the collection of Clostridia rep oris.
Testing CRISPRi-dCpf1 system in multiple Clostridia commensals
The following critical step toward developing a Clostridia GM pipeline is identifying a genetic manipulation tool that enables targeted gene manipulation in most Clostridia. As with Cas9-initiated cutting and dCas9-mediated interference, CRISPR-based systems have been recently applied to C. sporogenes (Cañadas et al., 2019; Guo et al., 2019) and C. difficile (McAllister et al., 2017). We prioritized the CRISPRi-dCpf1 (deactivated Cpf1) system (Hong et al., 2018; Hur et al., 2016; Kim et al., 2017; Peters et al., 2019; Tang et al., 2017; Zetsche et al., 2015; Zhang et al., 2017) mainly because the dCpf1 does not initiate a DNA double-strand break, and the dCpf1 plasmids showed less toxicity and higher conjugation efficiency than Cas9 or Cpf1. In comparison, our preliminary test found that the double-stranded cut by Cas9/Cpf1 is lethal to many Clostridia because of their very inefficient HR. The CRISPRi-dCpf1 system incorporates a catalytically dead dCpf1 and a guide RNA (gRNA) repurposed for gene regulation in bacteria. During regulation, the dCpf1/crRNA complex binds to the template strand of a target gene and blocks the transcription elongation, thus suppressing gene expression (Kim et al., 2017; Zhang et al., 2017).
To test CRISPRi-dCpf1 in the genetically targetable Clostridia, we assembled the dCpf1 and lacZα (as a transcription reporter) with the pGM plasmids harboring the nine rep origins (Fig. 2Bi, STAR Methods, and Data S1B). LacZα was selected because of its small size (~300 bp) and robust expression in multiple Clostridia. We designed a duplex gRNA targeting both the promoter and the template strand of lacZα (Fig. 2Bii). We found that dCpf1 leads to efficient knockdowns (~3 to over 100 fold) of lacZα transcription in multiple Clostridia (Figs. 2B and S2, Table S1). Several tested Clostridia could not take in this set of vectors, probably because the conjugation efficiency is greatly diminished due to this vector’s large size (>10kb) (Guo et al., 2019; Zhang et al., 2018). Altogether our data suggest that the CRISPRi-dCpf1 system regulates gene transcription in many Clostridia that uptake extracellular plasmid DNA.
A strategy targeting bacterial 16S rRNA genes to generate targeted gene insertion tools in non-model gut commensals
Besides CRISPRi, a targeted gene insertion tool will also facilitate studying the molecular functions of Clostridia genes. Over half of the targetable Clostridia are not genome sequenced. We wondered whether targeting their universally conserved DNA sequences (as ‘an archery target’) could enable selective genetic insertion of a Clostridia gene without prior knowledge of its genome sequence. However, highly conserved genes are generally functionally essential (Isenbarger et al., 2008), and a genetic mutation to these genes could be lethal. To find such a target, we interrogated the 16S rRNA gene that has been used to assess microbiome diversity and construct bacterial phylogeny. We believe that the 16S rRNA gene is an optimal target for two reasons: 1) a microbe usually has multiple copies, such that the disruption of one will not be lethal; 2) it is highly conserved among bacteria (Isenbarger et al., 2008). The same set of 16S-targeting vectors can be applied to different bacteria, thus significantly saving time and effort in sequencing and cloning.
Group II intron-directed mutagenesis systems, such as Targetron (Zhong et al., 2003) or Clostron (Heap et al., 2007), utilize base-pairing (between the excised intron lariat RNA and the target site DNA) for DNA target recognition to direct the site-specific insertion of a retrotransposition-activated selectable marker (RAM) into the targeted DNA loci. The RAM itself is interrupted by a self-splicing group I intron and only confers the corresponding antibiotic resistance after splicing out the group I intron and successful insertion into the Clostridial chromosome (Heap et al., 2007; Zhong et al., 2003). We proposed that a Group II intron targeting the 16S gene will likely integrate into the 16S loci of multiple Clostridia. To test this assumption, we aligned their 16S rRNA genes (from the HMP reference genomes (Turnbaugh et al., 2007)) and identified several potential, highly conserved target sites of Group II intron (Fig. 2C). We then introduced the 16S-targeting Group II intron (16S-tron), along with their compatible rep origins and antibiotic RAM, into 18 targetable Clostridia (Fig. 2C, and Data S1D). The RAM provides antibiotic resistance only upon integration into the Clostridia chromosome. We found 15 Clostridia whose chromosomes were inserted by the 16S-tron, indicating that the Group II intron-based gene insertion machinery functions in the corresponding microbe (Fig. 2C, and Data S1D, Table S1). These data suggest that this strategy can be applied to develop gene insertion tools for non-model gut Clostridia.
We then tested whether a similar strategy can be applied to non-model Gram-negative gut commensals. We prioritized Prevotella microbes because there are limited genetic tools available for this genus (Li et al., 2021; Naito et al., 2021). Gram-negative bacteria, in general, have more efficient HR. We synthesized a chimeric 16S (Chi-16S) sequence with high homology to the Prevotella 16S rRNA genes (Fig. 2D). We introduced the Chi-16S with a suicide vector into 21 human-associated Prevotella isolates (Kraal et al., 2014) (Table S1). We found 7 Prevotella whose 16S loci were inserted by pGM-NAC2P (Fig. 2D, and Table S1). The Chi-16S strategy was also applied to 45 Bacteroides/Parabacteroides microbes and 33 gut-associated Gram-negative microbes from other phyla (some with reported genetic tools (Bencivenga-Barry et al., 2020; Fung et al., 2016; García-Bayona and Comstock, 2019; Hmelo et al., 2015; Keller et al., 2011; Peluso et al., 2020; Salyers et al., 1999; Taketani et al., 2020)), leading us to identify the gene transfer methods for 36 of them (Fig. 1C, and Table S1). These data demonstrate that the HR-based Chi-16S strategy efficiently identifies their gene transfer methods and generates gene insertion tools in non-model Prevotella, and gut-associated microbes from other phyla that are amenable to genetic manipulations.
Constructing mutants to modulate Clostridia gene transcription and microbiome metabolites production
To demonstrate the utility of genetic tools developed in this study, we selected a widely distributed gene bcat and modulated its expression in 11 Clostridia and 1 Bifidobacterium (Fig. 3Ai). The BCAT protein deaminates branched-chain amino acids into their keto acid form (Hur et al., 2016). (Figs. 3Aii, S3, and Table S1). A duplex bcat-targeting gRNA along with dCpf1 was introduced, and bcat transcription was repressed in all the mutants with the dCpf1+gRNAs, compared to control with only dCpf1 (Fig. 3Aii).
Figure 3. Modulating Clostridia gene expression and microbiome-derived metabolites using gene manipulation tools developed via the GM pipeline.
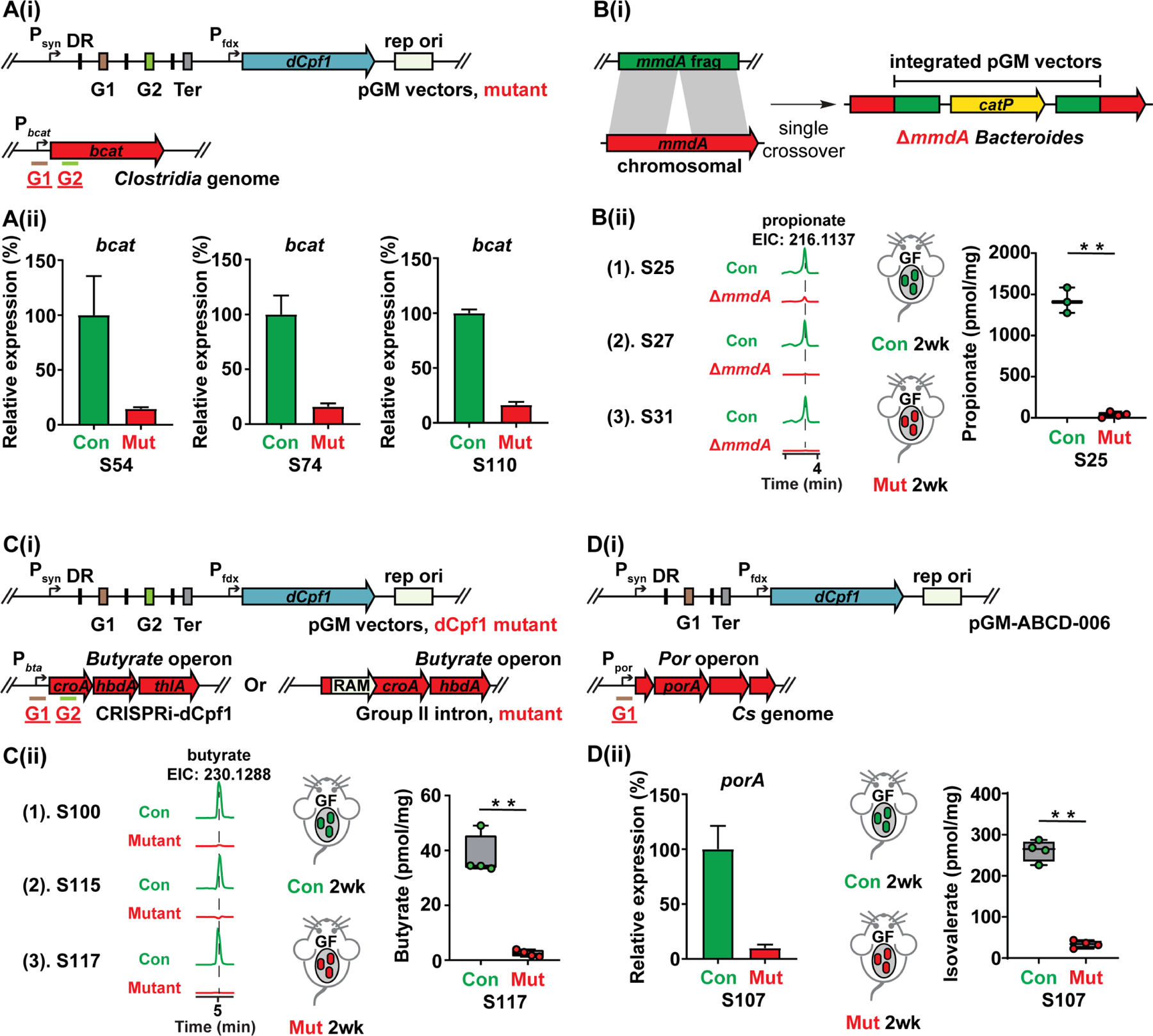
(A) (i) Schematic view of a duplex gRNA targeting the branched-chain amino acid aminotransferase bcat in the Clostridia commensals. (ii) The bcat gene of 11 Clostridia microbes was efficiently repressed using dCpf1. The panel shows the mean gene expression of three biological replicates as determined by qPCR. Only three representative results (S54, S74, and S110, Table S1) are shown.
(B) (i) Schematic view of knocking out the Bacteroides mmdA genes using pGM vectors. The pMG vector was assembled with ~1kb fragment of the mmdA genes, and the mmdA genes of three Bacteroides microbes were knocked out via single crossover integration. (ii) Three Bacteroides ΔmmdA mutants (S25, S27, and S31, Table S1) deplete propionate in vitro. The bacterial culture supernatant was derivatized and propionate production was examined using LC-MS (EIC: 216.1137). The Bacteroides ΔmmdA mutant depletes propionate in vivo. Germ-free Swiss Webster mice (n = 3 or 4 per group) were mono-colonized with the S25 control strain (Con, 16S integrated by pGM-NAC2B) and ΔmmdA mutant (Mut). Propionate was depleted in the host by mmdA deletion. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**). Error bar: standard deviation.
(C) (i) Schematic view of modulating butyrate production in the Clostridia commensals using dCpf1 or Group II intron. (ii) The butyrate production (quantified by LC-MS) was significantly reduced in three Clostridia microbes S100 (by dCpf1), S115 (by Group II intron), and S117 (by dCpf1) (Table S1). The fecal butyrate (quantified by LC-MS) in the germ-free Swiss Webster mice mono-colonized with S117 mutant (Mut, dCpf1+gRNAs) is significantly lower compared to the control (Con, dCpf1 only). Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**). Error bar: standard deviation.
(D) In vitro and in vivo depletion of branched short-chain fatty acids (BSCFAs) by S107 using CRISPR-dCpf1. (i) Schematic view of targeting the BSCFAs gene porA using CRISPR-dCpf1. The dCpf1 gRNA (G1) targets the porA promoter region. (ii) The porA expression (by qPCR) is significantly reduced in the mutant (Mut, dCpf1 with gRNA) compared to the control (Con, dCpf1 only) in vitro. Germ-free Swiss Webster mice (n = 4 per group) mono-associated with porA repression mutant have much less isovalerate (quantified by LC-MS) in their feces than the control (dCpf1 only). Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**). Error bar: S.E.M. For (A), (C), and (D), DR: direct repeat, G1: guide RNA-coding sequence 1, G2: guide RNA-coding sequence 2, Ter: terminator. The numbering of the strains corresponds to the strain information shown in Table S1.
We next sought to utilize these gene manipulation tools to modulate the production of microbiome-derived metabolites in vitro and in the context of host colonization. We selected short-chain fatty acids (SCFAs) propionate and butyrate, as well as branched-SCFAs (BSCFAs), because of their vital role in maintaining host immune homeostasis and metabolic health (Blander et al., 2017; Cani et al., 2019; Rooks and Garrett, 2016). We first identified several gut commensals as abundant producers of the corresponding metabolites by analyzing the SCFA profiles of our targetable commensals. Next, we generated a series of mutants that reduce their production in vitro by targeting the corresponding metabolic genes. For propionate, we deleted the methylmalonate mutase (mmdA) genes of three Bacteroides microbes that convert methylmalonate to propionate (Figs. 3B and S3, and Table S1) (Fischbach and Sonnenburg, 2011) (Reichardt et al., 2014). For butyrate, we targeted the crotonase gene (croA) (Vital et al., 2014) and used either dCpf1 to downregulate its expression or group II intron to knock out the gene (Figs. 3C and S3, and Table S1). For BSCFAs, we applied the dCpf1 tool to suppress porA expression in C. sporogenes (Guo et al., 2019). For all the mutants we generated, we found that the in vitro production of the corresponding metabolites is significantly reduced compared to the control, and their levels in the mono-colonized mice are also much less than that of the control (Figs. 3D and S3, and Table S1). Taken together, these data show that we can utilize the genetic tools developed via the GM pipeline to modulate microbiome-derived metabolites in vitro and in the context of host colonization, suggesting their potential in systematically linking microbiota genes with their responsible metabolites and associated host biology.
A case study of Clostridia-specific bile acid 7α-dehydroxylation
We sought to use these genetic tools to study the effect of one microbiota gene on host biology. We selected the bai operon for 7α-dehydroxylating of CA (cholate)/CDCA (chenodeoxycholate) to DCA (deoxycholate)/LCA (lithocholate) for follow-up studies. Three reasons motivated us to choose this pathway (Fig. 4, and Data S1): 1) Interestingly, we found one commensal Faecalicatena contorta S122 (S122) that efficiently converts CA(1)/CDCA to DCA(3)/LCA (Figs. 4A and 4B). Previous works have stepwise elucidated the chemistry and enzymology of 7α-dehydroxylation (Funabashi et al., 2020; Ridlon et al., 2006, 2016). However, a key impediment to investigating bai operon biology is that all the identified bai-coding Clostridia have no published gene transfer methods and tractable genetic tools (Ridlon et al., 2016). 2) DCA/LCA and their derivatives dominate the host secondary bile acid pool (Arab et al., 2017). 3) Amphipathic bile acids have intriguing biological activities: they inhibit the growth of enteric pathogens (Buffie et al., 2015), regulate mucosal immunity (Chen et al., 2019; Fiorucci et al., 2018), and promote liver cancer (Yoshimoto et al., 2013).
Figure 4. Knocking out baiH in gnotobiotic mice.
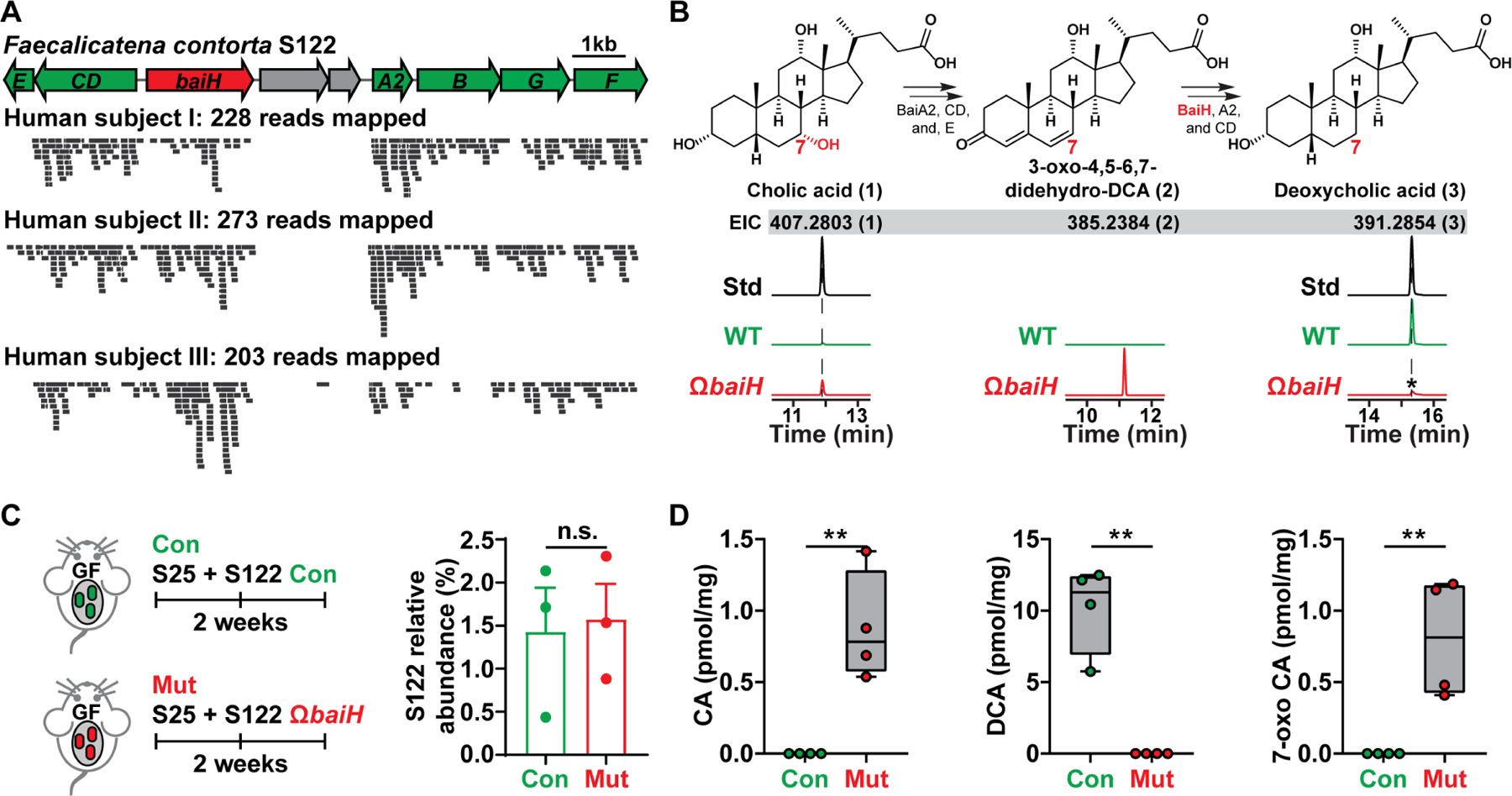
(A) The orientation of the S122 bai operon for bile acid 7α-dehydroxylation. The mutated gene baiH (by Group II intron) is highlighted in red. The S122 bai operon is actively transcribed under host colonization, and three representative results of metatranscriptomic analyses of the S122 bai operon are shown.
(B) The biosynthetic scheme of bile acid 7α-dehydroxylation. The baiH encodes an oxidoreductase that reduces the 6,7-olefinic bond of the intermediate 3-oxo-4,5–6,7-didehydro-DCA (2, EIC: 385.2384). The S122 ΩbaiH mutant accumulates the predicted intermediate (2, EIC: 385.2384) and no longer converts CA (1, EIC: 407.2803) to DCA (3, EIC: 391.2854) in vitro. The structure of the intermediate (2) was determined by comparing its retention time and exact mass to the published literature. The asterisk indicates a residual amount of DCA that is a contaminant from the CA chemical standard. EIC: extracted ion chromatogram.
(C) Germ-free C57BL/6J mice (n = 3 or 4 per group) were co-colonized with S25 plus the S122 control (Con) or ΩbaiH mutant (Mut) (by Group II intron) strain. The relative abundances of S122 in the control and mutant group were assessed by 16S rRNA sequencing and were comparable.
(D) Depleting baiH using Group II intron abolishes gut 7α-dehydroxylating activity and modifies gut bile acid pool in gnotobiotic mice. CA, DCA, and 7-oxo CA (see Data S1E for their structures) were quantified using LCMS.
Data in (C) and (D) were analyzed using unpaired two-tailed Student’s T-test. The asterisk indicates p-value < 0.05 (*) or < 0.01 (**). The numbering of the strains corresponds to the strain information shown in Table S1.
We sequenced S122 and identified its bai operon (Fig. 4A). Our bioinformatic analyses revealed three intriguing features of S122: 1) The strain is widely distributed among the healthy human population in two independent cohorts (41.30% (Lloyd-Price et al., 2019), 85.98% (Yatsunenko et al., 2012)). 2) Like other 7α-dehydroxylating Clostridia, S122 has a low intestinal abundance (~0.016%), but its bai operon is actively transcribed under conditions of host colonization (Fig. 4A). 3) S122 and its close relatives are more prevalent and abundant than C. hiranonis or C. scindens (Figs. S4A and S4B), indicating they play a significant role in regulating gut 7α-dehydroxylating activity.
To manipulate the bai pathway in vivo, we generated a baiH insertion mutant (S122 ΩbaiH) (Figs. 4B and S4C, Data S1). The baiH gene encodes an oxidoreductase that reduces 3-oxo-4,5–6,7-didehydro-DCA (2) to 3-oxo-4,5-didehydro-DCA (Fig. 4B) (Funabashi et al., 2020; Kang et al., 2008). The ΩbaiH mutant depleted DCA and accumulated the intermediate (2) and 7-oxo CA in vitro (Fig. 4B). Unexpectedly, our attempt to efficiently mono-associate GF mice with S122 to induce in vivo DCA production proved unsuccessful. Instead, we found S122 can stably co-colonize the GF mice with S25, and knocking out baiH eliminates gut 7α-dehydroxylating activity: The control accumulated ~12 pmol/mg DCA (Figs. 4C and 4D), while the mutant abolished DCA but accumulated 7-oxo-CA (Figs. 4D and S4D). Moreover, S122 can stably co-colonize GF mice with 55 other genetically targetable microbes identified in this study (Fig. S4E). The relative abundance of S122 is low in both cases (Figs. 4C and S4E), but robust CA to DCA conversion can be detected, suggesting that S122 is a highly active 7α-dehydroxylating bacterium in the host.
The baiH gene has significant effects on the host bile acid pool and microbiota composition
This finding motivates us to knock out baiH in a complex microbiota, like that of Specific Pathogen Free (SPF) mice. Manipulating microbiota genes in a complex microbiome provides a direct readout of their effects on microbial composition, which can be critical to explaining its impact on host biology. Unlike GF mice, the GI tract of SPF mice already harbors a complex microbiome with robust 7α-dehydroxylating activity, leaving a limited niche for S122 to occupy. To overcome this challenge, we genetically tagged the control and ΩbaiH mutant with a thiamphenicol-resistant marker. We supplemented their drinking water with thiamphenicol (15 μg/ml) and erythromycin (10 μg/ml) at very low concentrations for two reasons: 1) to facilitate the colonization of the tagged S122 strains that are resistant to these two antibiotics, and 2) to eliminate the background 7α-dehydroxylating activity conferred by the existing bai-coding Clostridia. This strategy led us to stably colonize the SPF mice with S122 control and the ΩbaiH mutant at about the same level with comparable total bacterial load (Figs. 5A, and 5B) for about 4 weeks. Because supplemented antibiotics minimally accumulate in the feces (~5 pmol/mg for thiamphenicol and not detectable for erythromycin), they do not reduce the total bacterial load compared to the SPF mice (Fig. S5H). Their effect on the gut microbiome is also controlled under this experimental setting.
Figure 5. Knocking out baiH in the context of a complex microbiota impacts the host bile acid pool and the gut microbiome.
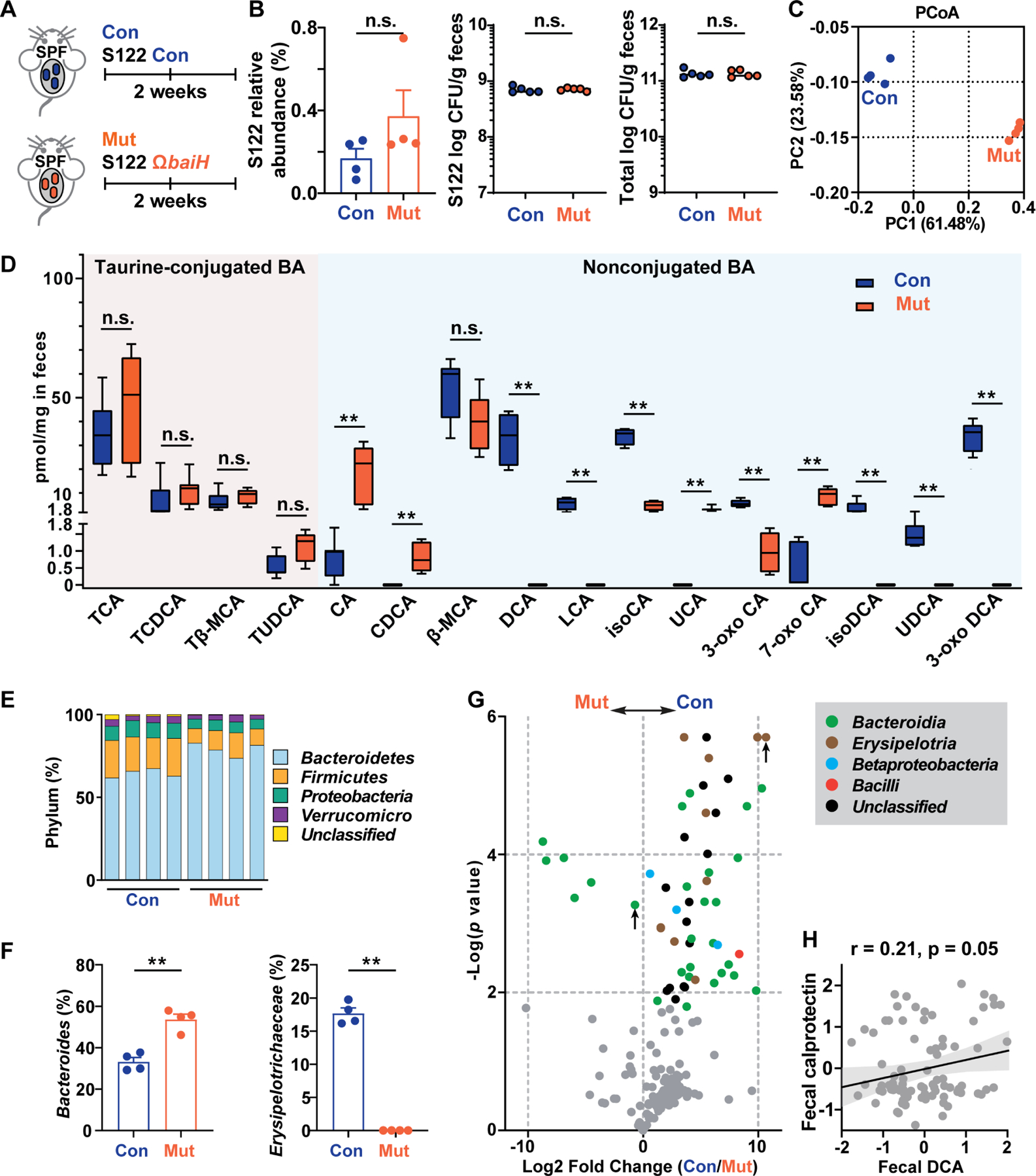
(A) SPF C57BL/6J mice (n = 4 or 5 per group) given low dose antibiotic water (15 μg/ml thiamphenicol and 10 μg/ml erythromycin) were colonized with genetically tagged S122 control (Con) or ΩbaiH mutant (Mut) (by Group II intron) strain.
(B) The relative abundances of S122 in the control and mutant group were assessed by 16S rRNA sequencing and were comparable. The SPF mice are stably colonized with S122 control (Con) and the ΩbaiH mutant (Mut) at about the same level with a comparable total bacterial load.
(C) Principal coordinates analysis (PCoA) of the fecal microbiome of the control and ΩbaiH mutant mice.
(D) Targeted metabolomics analyses (quantified by LC-MS) of the stool bile acid (BA) compositions of the control (Con) and ΩbaiH mutant (Mut) colonized SPF mice.
(E) The relative abundance of taxonomic phyla in the gut microbiota of the control and ΩbaiH mutant mice.
(F) Relative abundances of inflammation-associated gut microbial taxa in the stool microbiome of the control and ΩbaiH mutant mice.
(G) Volcano plot of differential bacterial OTU abundances calculated from 16S rRNA gene sequencing. Significantly different OTUs (n= 56, FDR < 0.05) are colored and plotted. The Bacteroidia OTU and Erysipelotrichaceae OTU with high relative abundances (>10%) are marked with an upward pointing arrow.
(H) Gut 7α-dehydroxylating activity is weakly associated with fecal calprotectin level in nonIBD people. In (B), (D), and (F), data were analyzed using unpaired two-tailed Student’s T-test, and the asterisk indicates p-value < 0.01 (**). The data in (C), (E), (F), and (G) are representative of two independent experiments with n =4 or 5 per group, and only the changes in taxonomic groups that are consistent between the two experiments are shown. Data are shown as mean ± SEM. The numbering of the strains corresponds to the strain information shown in Table S1.
To examine whether baiH deletion affects gut bile acid composition and the microbiome, we performed metabolomics and 16S rRNA sequencing analyses on mouse stool samples (Figs. 5 and S5). Principal coordinate analysis (PCoA) demonstrated that stool samples are clustered by genotype (Fig. 5C). We drew two observations from these data: First, the control and ΩbaiH colonized mice have different intestinal bile acid pools. Both groups have similar levels of conjugated bile acids like TCA and TCDCA (Fig. 5D), indicating baiH depletion does not significantly modify microbiome bile salt hydrolyzing activity. DCA and its derivatives (such as isoDCA and 3-oxo DCA) are accumulated in the control group at levels comparable to host physiological levels. In contrast, the mutant group has higher levels of CA and its derivatives, including 7-oxo CA and UCA (Fig. 5D).
Second, knocking out baiH modifies host gut microbiome composition. Both the control and mutant groups harbor a highly complex stool microbiota, and their overall phylum composition was maintained (Fig. 5E). The control group has a lower abundance of Bacteroidetes, higher Proteobacteria, and a significantly elevated Erysipelotrichaceae (Figs. 5F, S5C, and S5D). This compositional shift has been associated with worsened intestinal inflammation (Kaakoush, 2015; Kaser et al., 2010; Palm et al., 2014). A total of 56 operational taxonomic units (OTUs) were differentially abundant between groups, and they belong predominantly to the Bacteroidia, Betaproteobacteria, and Erysipelotria (Fig. 5G). Of note, the control has significantly more Erysipelotrichaceae that have high IgA coating and are associated with exacerbated colon inflammation (Figs. 5F and 5G) (Kaakoush, 2015; Palm et al., 2014). Aligned with our findings in the SPF mice, a higher stool DCA is positively associated with Erysipelotrichaceae abundance (Fig. S5F) and fecal calprotectin (a marker for the level of intestinal inflammation) (Fig. 5H) in nonIBD human stools. However, this correlation is not observed in patients with ulcerative colitis or Crohn’s disease whose gut microbiota are usually structurally altered and whose gut 7α-dehydroxylation activity is disrupted because of an exaggerated immune response (Lloyd-Price et al., 2019). These data indicate a potential modulatory role of bai operon in human gut microbiota and the onset of intestinal inflammation.
Assessing the effect of baiH on intestinal inflammation
Because knocking out baiH shifts the gut microbiome to a less proinflammatory state (Kaser et al., 2010), we assessed whether baiH regulates intestinal inflammatory responses in a dextran sodium sulfate (DSS)-induced murine colitis model. The control and ΩbaiH mutant colonized SPF mice were given drinking water with DSS (Figs. 6A and 6F). We found that both the S122 control and ΩbaiH mutant strains stably colonized the mice (Figs. S6F and S6I), and the control group still has significantly higher Erysipelotrichaceae during DSS treatment (Fig. S6G). As colonic inflammation progressed, baiH indeed played a modulatory role in intestinal inflammation: The control lost more weight and experienced more severe inflammation as shown by enhanced colonic pathology, shorter colon lengths, increased fecal lipocalin-2 levels, higher hematochezia score, and upregulation of inflammatory genes (Figs. 6B–E, S6A, S6C, and S6E). Interestingly, the same DSS treatment successfully triggered an inflammation response in the GF C57BL/6 mice co-colonized with the S122 control or ΩbaiH mutant and S25, but knocking out baiH has no notable effect on intestinal inflammation (Figs. 6G–J, S6B, and S6D). Taken together, these data indicate that baiH-mediated inflammatory responses are microbiota dependent, and baiH depletion in a complex microbiota reshapes host bile acid profiles and presets gut microbiome composition to a more protective state against DSS-induced colitis. More importantly, using a combination of microbiome genetics, metabolomics, and colitis mouse models, we demonstrate how a single commensal gene of a low intestinal abundance may significantly impact host biology by reshaping bile acid metabolism and the gut microbiota ecosystem.
Figure 6. baiH modulates intestinal inflammation in the context of complex gut microbiota.
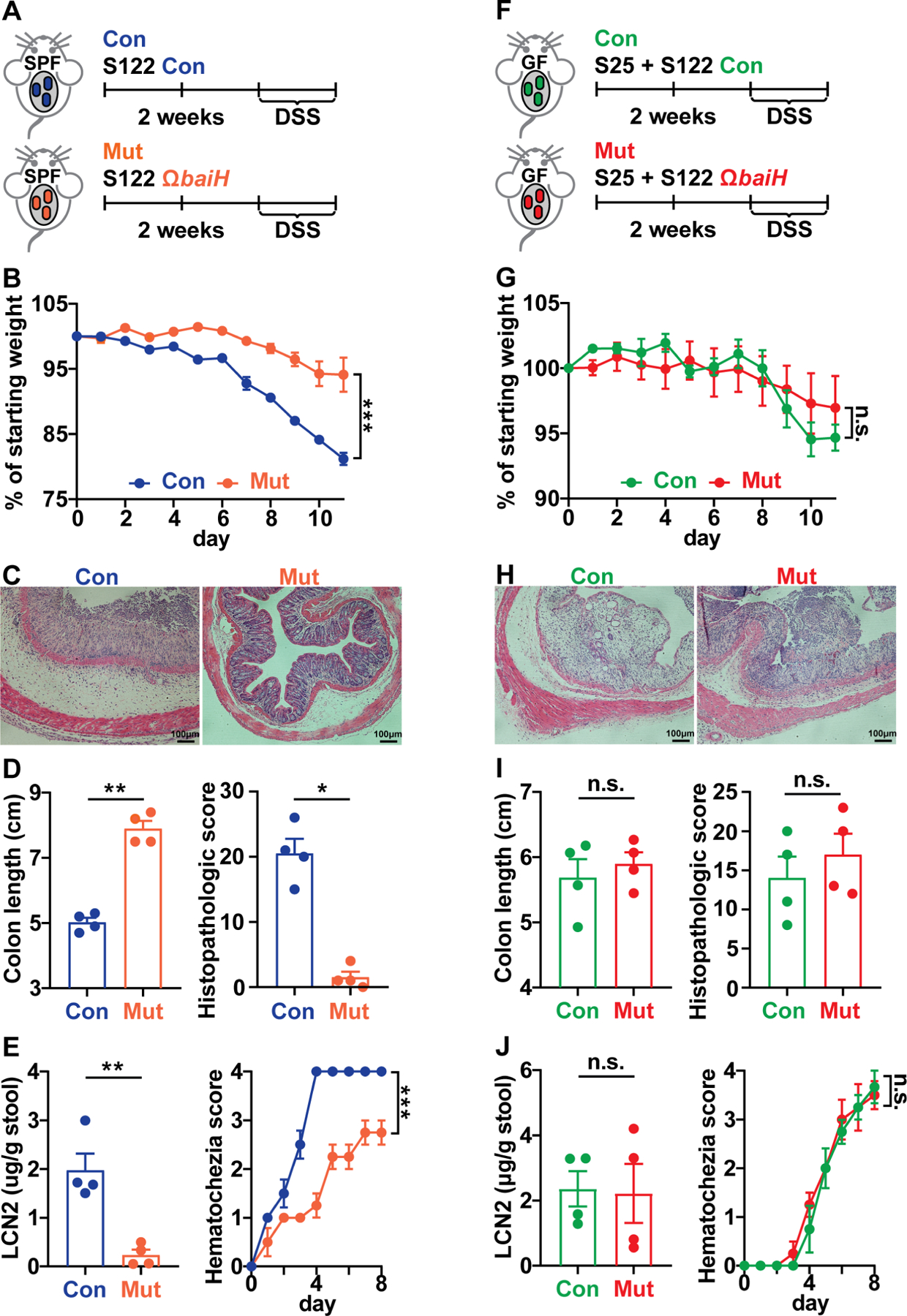
(A, F) DSS-induced murine colitis model was applied to the SPF or gnotobiotic mice colonized with the genetically tagged S122 control (Con) and ΩbaiH mutant (Mut). Mice were colonized with the control or mutant strain for at least two weeks before giving DSS, SPF mice were given 2.5% DSS (in water supplemented with 15 μg/ml thiamphenicol and 10 μg/ml erythromycin) for 8 days, and gnotobiotic mice were given 2.0% DSS (in water supplemented with 15 μg/ml thiamphenicol) for 7 days. The disease state was monitored by weight loss (B, G), hematoxylin and eosin (H&E) staining of the distal colon (C, H), colon shortening, and histopathologic score (D, I), and fecal lipocalin-2 and daily hematochezia score (E, J).
Data shown in (B-E, G-J) are representations of n = 4 to 5 mice per group replicated in two or more independent experiments. In (B, G), % of starting weight was calculated by normalizing weights at sacrifice to starting weight. In (D, I) and (E, J), colon length and LCN2 data were analyzed using unpaired two-tailed Student’s T-test. In (B, G) and (E, J), % of starting weight and hematochezia score data were analyzed using Two-way ANOVA followed by the Bonferroni post hoc test (n=4). In (D, I), histopathologic score data were analyzed using the Mann-Whitney test. Data are shown as mean ± SEM. The asterisk indicates p-value < 0.05 (*), < 0.01 (**) or < 0.001 (***). The numbering of the strains corresponds to the strain information shown in Table S1.
The baiH-mediated microbiota composition shift exacerbates DSS-Induced colitis in gnotobiotic mice
Motivated by our findings that baiH mediates colon inflammation in a complex microbiome, we proceeded to examine if microbiota composition shift induced by baiH deletion (Figs. 5F and S6G) could be related to the different intestinal inflammatory responses in the SPF mice under DSS treatment. First, we determined whether Erysipelotrichaceae expansion is baiH-dependent. Indeed, Erysipelotrichaceae isolates are more resistant to high concentrations of DCA and 3-oxo DCA compared to Bacteroides (Fig. 7A). In a 10-member synthetic consortium we prepared in vitro, Erysipelotrichaceae also expands in the presence of baiH and its product DCA (Figs. 7B and 7C).
Figure 7. The baiH-mediated microbiota composition shift exacerbates DSS-induced colitis in gnotobiotic mice.
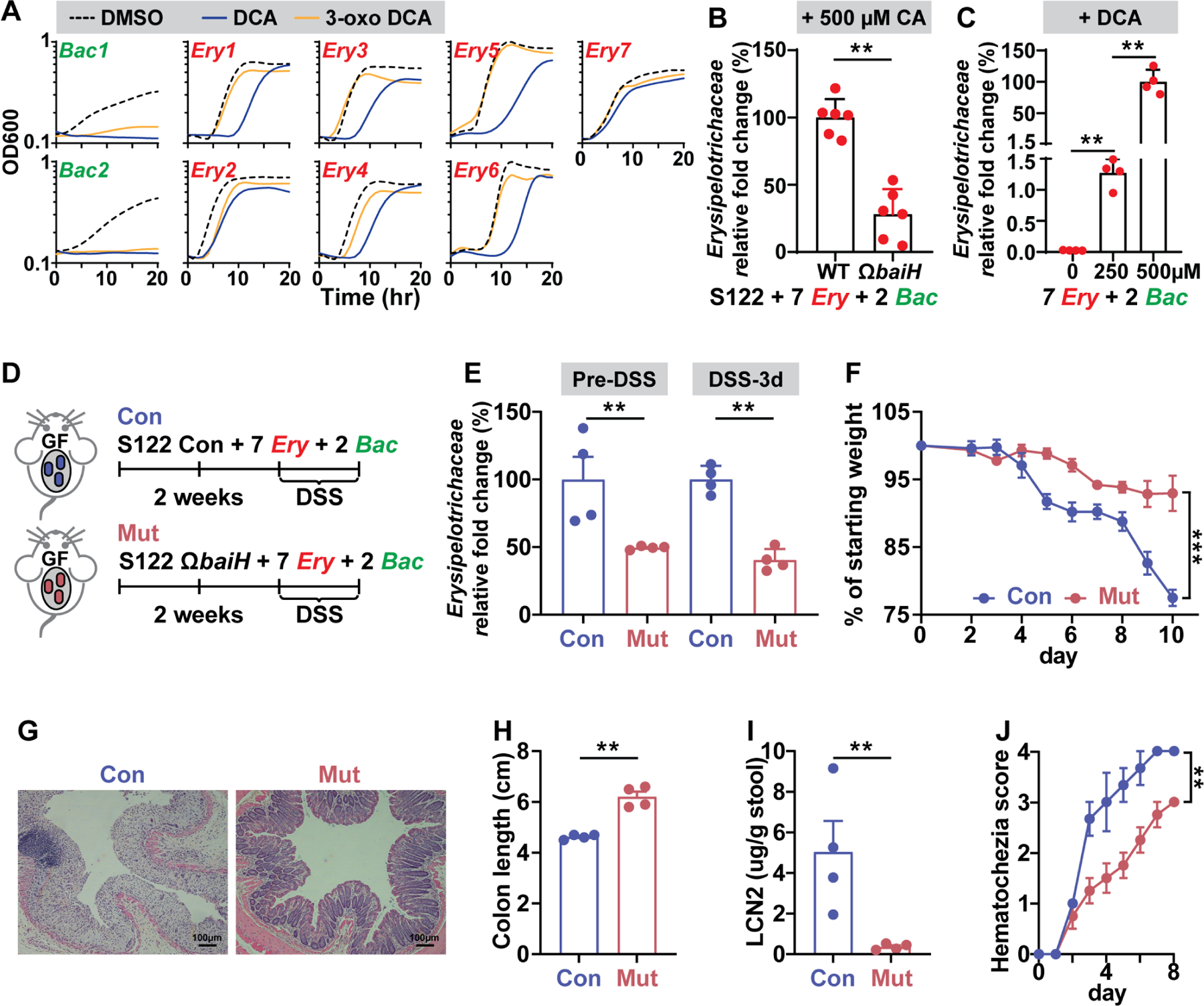
(A) The growth curve of two Bacteroides (bac) microbes and seven Erysipelotrichaceae (Ery) microbes in the presence of 500 μM DCA, 500 μM 3-oxo DCA, or DMSO control. The Erysipelotrichaceae microbes are more resistant to DCA and 3-oxo DCA than the Bacteroides microbes.
(B) The baiH gene drives expansion of Erysipelotrichaceae microbes in an in vitro consortium consisting of 2 Bacteroides (Bac) and 7 Erysipelotrichaceae microbes (Ery) with either the S122 control or ΩbaiH strain. 500 µM CA was supplemented as the substrate for the bai pathway. The relative fold change of Erysipelotrichaceae was assessed by qPCR.
(C) DCA drives expansion of Erysipelotrichaceae microbes in an in vitro consortium consisting of 2 Bacteroides (Bac) microbes and 7 Erysipelotrichaceae (Ery) microbes. DCA was supplemented at 0, 250, and 500 µM, respectively. The relative fold change of Erysipelotrichaceae was assessed by qPCR.
(D) DSS-induced murine colitis model was applied to the gnotobiotic mice colonized with a synthetic consortium consisting of the genetically tagged S122 control (Con) or ΩbaiH mutant (Mut) (by Group II intron) along with 2 Bacteroides (Bac) microbes and 7 Erysipelotrichaceae (Ery) microbes tested in (A), (B), and (C). Mice were colonized with the control or mutant strain for at least two weeks followed by 2.5% DSS for 8 days.
(E) The baiH gene drives expansion of Erysipelotrichaceae microbes in the context of host colonization before and during DSS treatment assessed by qPCR. The disease state was monitored by weight loss (F), hematoxylin and eosin (H&E) staining of the distal colon (G), colon shortening (H), fecal lipocalin-2 (I), and daily hematochezia score (J).
The data in (A to C) are from a representative experiment with three technical replicates (A), or with six or four biological replicates (B, C). Data shown in (F, H, I, J) are representations of n = 4 mice per group replicated in two or more independent experiments. In (F), % of starting weight was calculated by normalizing weights at sacrifice to starting weight. In (H) and (I), colon length and LCN2 data were analyzed using unpaired two-tailed Student’s T-test. In (F) and (J), % of starting weight and hematochezia score data were analyzed using Two-way ANOVA followed by the Bonferroni post hoc test (n=4). Data are shown as mean ± SEM. The asterisk indicates p-value < 0.05 (*), < 0.01 (**) or < 0.001 (***). The numbering of the strains corresponds to the strain information shown in Table S1.
Next, we asked whether baiH drives Erysipelotrichaceae expansion in vivo, and whether this microbiota composition shift affects colon inflammatory responses in the DSS colitis model. We colonized two groups of germ-free C57BL6/N mice with the same 10-member synthetic consortium (S122 control or ΩbaiH mutant with 7 Erysipelotrichaceae and 2 Bacteroides, Figs. 7B and 7D) and applied the DSS treatment two weeks post colonization (Fig. 7D). As expected, baiH also drives the expansion of Erysipelotrichaeceae in the context of host colonization (Fig. 7E). The control group has exacerbated colon inflammation in this gnotobiotic setting as evaluated by severe weight loss (Fig. 7F), enhanced colonic pathology (Fig. 7G), shorter colon lengths (Fig. 7H), increased fecal lipocalin-2 levels (Fig. 7I), and higher hematochezia score (Fig. 7J). The same DSS treatment also induced a robust inflammation response in the GF C57BL/6 mice co-colonized with the S122 control or ΩbaiH mutant with only the two Bacteroides (three-member, Fig. S7G).However, depleting baiH in this gnotobiotic setting has no notable effect on intestinal inflammation (Figs. S7H to S7L). The S122 control and ΩbaiH mutant colonized the mice at comparable levels under both gnotobiotic settings (10-member vs. 3 member) (Figs. S7A and S7B), and their fecal bile acid profiles are comparable (Figs. S7C to S7F), suggesting that the different intestinal inflammatory response observed in the 10-member consortium colonized mice is more likely due to the expansion of Erysipelotrichaeceae driven by baiH. Altogether, these data indicate that a baiH-mediated microbiota composition shift could exacerbate DSS-Induced colitis in the gnotobiotic mice, and the similar shift observed in the SPF mice (Figs. 5F and S6G) could be potentially related to the different intestinal inflammatory responses induced by baiH depletion in a complex microbiota. Of note, members of the synthetic consortium were selected based on the information we obtained by depleting baiH in a highly diverse microbiome, demonstrating the usefulness and necessity of studying the function of a microbiota gene in the background of a complex microbiota.
DISCUSSION
Our GM pipeline, for identifying genetically targetable gut microbes and building their gene manipulation tools, can synergize with other microbiome-based genetic toolsets (Lim et al., 2017; Mimee et al., 2015; Shiver et al., 2021; Taketani et al., 2020; Whitaker et al., 2017) to add additional approaches to engineering the gut microbiome and facilitating a better understanding of microbe-host interaction. The GM-screen is performed in an in vitro, efficient, high-throughput manner and significantly expands the manipulatable territory of the gut microbiome (Table S1). Moreover, by incorporating CRISPR machinery and targeting bacterial 16S rRNA genes (e.g., 16S-tron, Chi-16S), we established gene manipulation tools in multiple phylogenetically diverse microbes (Table S1) in a ‘screened’ approach without prior knowledge of their genome sequence. We provide the gene transfer methods for these non-model gut commensals and their genetic tools as a resource for the scientific community to further study the molecular functions of their genes or dissect the molecular mechanisms behind their interactions with the host.
The most precise way to interrogate a microbiome gene is to manipulate it in the background of a complex microbiota and compare gut microbiome composition and host phenotypes that differ only in this gene. This method is analogous to a conditional gene knockout in a host. While such technology has been widely applied to eliminate a host gene in a specific tissue, no comparable toolset is currently available for the gut microbiome study. Herein by selectively deleting the baiH gene in a complex gut microbiome, we found that baiH impacts intestinal bile acid metabolism and modulates host inflammation in a microbiota-dependent manner. More interestingly, guided by the microbiota composition shift we observed in the SPF mice, we further established a synthetic consortium in the germ-free mice, which allows us to demonstrate that baiH-driven microbiota composition change could affect host colon inflammatory responses. This technology and approach could potentially be applied to study the biological functions of other microbiota genes and associated metabolic pathways and uncover host biology regulated by the shift of gut microbiome composition.
Our GM pipeline will greatly facilitate functional studies that causally connect microbiota genes to diseases or dissect the molecular mechanisms underlying microbe-host interactions. The human microbiome is a vital component of our “pan-genome” whose biological functions are potentially regulatable by genetic manipulation. While no such technology currently allows genetic manipulation of the gut microbiome as a whole, we believe that the GM pipeline, an efficient and generalizable approach of identifying genetically targetable non-model gut commensal and building their genetic tools, will be a first step toward establishing such a tool in the human gut microbiome.
LIMITATION OF THE STUDY
Despite these advances, our pipeline and approach have several limitations. First, despite our consideration of multiple factors and significant efforts to optimize the GM pipeline, there are still over 55% of non-model gut microbes for which we could not identify their gene transfer conditions/methodology. This barrier could be due to multiple reasons, such as incompatible conjugation donor/rep oris/antibiotic markers, bacterial host defense systems (Johnston et al., 2019), and other unknown factors. Further optimizations might be needed for specific bacteria to identify their gene transfer methodology. Second, the genetic tools we developed have limitations, and additional optimizations will be required. For instance, CRISPRi and Group II introns have off-target effects (ectopic integrations), or a single crossover mutation (in Prevotella) is less stable than a double crossover and subject to other limitations (Li et al., 2021). And complementation analysis is needed to restore the function of a mutated gene. These limitations can be overcome by introducing a new set of gene editing systems. For example, a recent major advance in the microbial genetics field is the identification of new CRISPR-based gene editing systems including that of CRISPR RNA-guided integrase (Klompe et al., 2019; Strecker et al., 2019; Vo et al., 2021). Such an integration system directs precise gene insertion independent of HR machinery, suggesting its potential to improve gene editing in gut microbes such as Firmicutes/Clostridia, which have inefficient HR. Third, our findings in the mouse studies should be cautiously interpreted. Mice have different bile salts composite and gut microbiome from that of humans, and thus the contribution of baiH to human intestinal inflammation warrants further investigation. Our findings reveal that baiH strongly affects gut inflammation in a complex microbiota, and baiH-driven expansion of Erysipelotrichaceae could worsen DSS-induced colitis in the gnotobiotic mice. Meanwhile, as we observed global changes in their bile acid pool and gut microbiota in the SPF background, it is possible that other factors, including some immunoregulatory bile acids (Campbell et al., 2020; Hang et al., 2019; Sinha et al., 2020; Song et al., 2020; Zhao et al., 2018), or other colitis-related gut microbes, could contribute to this regulation. Therefore, more work is needed to deconvolute the contribution of single bile acids (or other microbiome-derived metabolites) or a gut microbial species/strain to this modulation.
STAR★METHODS
RESOURCES AVAILABLITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Chun-Jun Guo (cj@guo-group.org).
Materials availability
Plasmids and other resources are available upon request to the lead contact.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and culture conditions
All strains and their culture conditions, plasmids, and primers used are listed in Tables S1 and S2. TYGB, BHIB, Mega, RCM, CMM, or LB liquid broth and/or agar plates were used for majority of the gut microbes. See Table S3 for detailed medium recipes. The culture plates were pre-reduced in the anaerobic chamber for at least 10 hrs (and 48 hrs for liquid broth with a loosened cap). All the anaerobic bacterial strains were grown anaerobically in an anaerobic chamber at 37 °C under an atmosphere of 5% CO2, 7.5% H2, 87.5% N2. Strains were first restreaked from a glycerol stock onto the pre-reduced agar plates (Table S1). A single colony was then sub-cultured into 1 mL pre-reduced liquid broth (Table S1). All the aerobic strains, including the E. coli conjugation donors, were cultivated aerobically at 37 °C shaking at 220 rpm.
Plasmid assembly and cloning
Plasmids pGM-ABCM to pGM-IBCM were used for identifying the gene transfer method for non-model Clostridia. Five extra Clostridia origins of replication (rep oris) (Table S2) were PCR amplified and assembled with the pGM-ABCM backbone digested by AscI and FseI. All this set of shuttle plasmids and the plasmids described in the latter sections are verified by restriction digestion followed by Sanger sequencing of their core functional components.
Plasmid series pGM-ABCL to pGM-IBCL and pGM-ABCF to pGM-IBCF were used for testing the CRISPRi-dCpf1 lacZα system in multiple non-model gut Clostridia. To construct these vectors, plasmid pMTL83153 (Heap et al., 2009) was double-digested with XbaI and XhoI, and ligated with a fusion PCR product of dCpf-1 CDS to give plasmid pGM-BBCD. Next, the nine Clostridia rep oris were PCR amplified using primers pMTL_rep_origin_F and pMTL_rep_origin_R (Table S2) and then assembled with the pGM-BBCD backbone via Gibson assembly, yielding a set of plasmids pGM-ABCD to pGM-IBCD. Similarly, the lacZα CDS (and the gRNA targeting the lacZα CDS) was introduced into this set of plasmids to yield plasmids pGM-ABCL to pGM-IBCL (and plasmids pGM-ABCF to pGM-IBCF).
Plasmid series pGM-ACAQ to pGM-ICAQ and pGM-ACBQ to pGM-ICBQ were used for testing the 16S-tron strategy for non-model gut Clostridia. The 16S-targeting intron (targeting the conserved motif in the 16S rRNAs of Clostridia microbes) was amplified using primers EBS universal primer + WBJ_16S_tgt_685_IBSN + WBJ_16S_tgt_685_EBS1d + WBJ_16S_tgt_685_EBS2 and Gibson assembled with the backbone amplified from plasmid pGM-BCAR-001 (laboratory stock) to give pGM-BCAQ. Next, the Clostridia rep oris were PCR amplified and assembled with the pGM-BCAQ backbone via Gibson assembly, yielding plasmids pGM-ACAQ to pGM-ICAQ. Last, if needed, the retrotransposition-activated marker (RAM, ermB) was replaced by aad9 to give plasmids pGM-ACBQ to pGM-ICBQ.
Plasmid series pGM-NAC2B, NAC2P, NACO were used for identifying the gene transfer methods for Bacteroidia and microbes of other phyla. The synthetic Bacteroides chimeric 16S rRNA (Chi-16S) was amplified using primers CJG_syn16S_F and CJG_syn16S_R and Gibson assembled with the pExchange backbone to get pGM-NAEB, and the CDS of the antibiotic marker ermB was then replaced with catP to give pGM-NAC2B. The Chi-16S of Bacteroides was replaced by Chi-16S of Prevotella to give pGM-NAC2P. The promoter Ppex and the antibiotic marker ermB in the vector pExchange was replaced with the promoter Ppmtl-catP and the marker catP, followed by Gibson assembling with the Chi-16S or the PCR amplified 16S rRNA fragment of microbes from other phyla to give plasmid series pGM-NACO1–7.
Mice
All animals were housed and maintained in a certified animal facility at the Weill Cornell Medicine Research Animal Resource Center (RARC). All animal care and experimental procedures were approved by the Weill Cornell Medicine Animal Care and Use Committee (IACUC). The experiments of modulating microbiome-derived metabolites in gnotobiotic mice were performed on germ-free Swiss Webster or C57BL/6 mice of 7-to-10-week-old sex-matched mice (male or female, n = 3 or 4 per group), which were bred within sterile vinyl isolators and maintained at the gnotobiotic mouse facility at Weill Cornell Medicine. Specific-pathogen-free (SPF) C57BL/6 mice were purchased from the Jackson Laboratory and were bred and maintained in the SPF mouse facility at Weill Cornell Medicine. 6-to-8-week-old male SPF or germ-free C57BL/6 mice (n = 4 or 5 per group, bred in the facility) were used for the experiments of modulating the baiH gene in a complex microbiome and the DSS treatments.
METHOD DETAILS
Antibiotics sensitivity test
All the detailed information about antibiotics used during conjugation/electroporation can be found in Table S1. To find the antibiotic and its optimal concentration that suppresses the growth of donor E. coli in conjugation, the recipient strains (Clostridia, Bacteroidia, Desulfovibrio, and Proteus microbes) were restreaked on plates supplemented with 250 µg/mL D-cycloserine or 200 µg/mL gentamicin (to suppress the growth of conjugation donor E. coli CA434 or S17), or with 200 µg/mL kanamycin (to suppress the growth of conjugation donor E. coli HB101/pRK24). Next, these resistant recipient strains were screened against agar plates supplemented with different concentrations of thiamphenicol (for Clostridia, Bacteroidia, and Fusobacterium), chloramphenicol (for Desulfovibrio and Proteus), carbenicillin, or kanamycin (for Klebsiella) to determine their minimal inhibitory concentrations (MICs) (Table S1). In most cases, the thiamphenicol resistant gene can be exploited as a universal marker to select transconjugants or transformants that can uptake and stably maintain extracellular plasmid DNA.
Identify gene transfer methods for Clostridia gut commensals
All the E. coli conjugation donors used in this study were verified by miniprepping the plasmid harbored by the E. coli donor followed by restriction digestion and Sanger sequencing of their core functional components if necessary. The ‘sExpress’ E. coli donor was generated following the published protocol (Woods et al., 2019). Please refer to Table S1 for detailed conjugation/transformation parameters. The same gene transfer conditions were applied when introducing dCpf-1-based or Clostron-based plasmids.
1). Mixed-conjugation:
To identify gene transfer methods for non-model Clostridia, E. coli donors harboring shuttle plasmids were separated into three groups, including group I: pGM-ABCM, BBCM, and CBCM; group II: pGM-DBCM, EBCM, and FBCM; and group III: pGM-GBCM, HBCM, IBCM, and a negative rep ori-less control. A 1.0 mL culture of each E. coli (cultivated overnight in LB supplemented with tetracycline (15 µg/mL) and chloramphenicol (25 µg/mL)) within the same group were mixed and centrifuged at 1500 x g for 2 min. The culture supernatant was discarded, and the cell pellet was gently washed with 500 µL PBS buffer (Gibco, pH = 7.4), followed by centrifugation at 1500 x g for 2 min and removal of the PBS supernatant. The three cell pellets were transferred on ice into the anaerobic chamber, and each were mixed gently with 300 μL overnight culture of the recipient Clostridia microbe. A 35 μL cell mixture was dotted on pre-reduced agar plates. After 48 hrs to 72 hrs, the cell dots were scraped using a sterile pre-reduced inoculation loop and resuspended in 300 μL pre-reduced PBS (pH 7.4) buffer. About 100 µL of the cell suspension was plated on plates supplemented with 15 µg/mL thiamphenicol (or MIC, see Table S1), along with 250 µg/mL D-cycloserine or 200 µg/mL gentamicin (inhibiting E. coli CA434), or 200 µg/mL kanamycin (inhibiting E. coli HB101/pRK24). Colonies typically appeared after 36–48 hrs.
2). Single strain E. coli conjugation verification:
The same procedure (as mixed-conjugation) was applied to each recipient Clostridia except that only one E. coli donor (1.5 ml) harboring a compatible rep ori was used in each conjugation. The same protocol and parameters were applied when introducing plasmid-based gene manipulation tools like CRISPRi-dCpf1 and Group II intron (Clostron) into Clostridia commensals.
3). Electroporation of Clostridia microbes:
All the experimental procedures described below are carried out in an anaerobic chamber. A single colony was inoculated in 1 mL of pre-reduced liquid broth and incubated anaerobically at 37 °C for overnight. 1 mL of the seed culture was inoculated in 45 mL of pre-reduced liquid broth (see Table S1). Culture with an OD600 of 0.6–0.8 was chilled on ice for at least 10 min (from this time point, all manipulations were performed at 4°C using an ice-bath and pre-chilled buffer). Cells were harvested by centrifugation at 4000 × g and 4°C for 10 min. The pellet was washed twice with 10 mL pre-reduced, filter-sterilized SMP buffer (270 mM sucrose, 1 mM MgCl2, and 5 mM sodium phosphate, pH 6.9), and resuspended in 1.8 mL SMP buffer. Plasmid was pre-methylated using CpG (M. SssI) and GpC (M.CviPI) methyltransferases following the manufacturer’s protocol (by NEB). 2 μg of plasmid was added into 600 µL competent cell and mixed gently by flicking. The DNA-cell mixture was transferred to a pre-chilled electroporation cuvette (4mm, Fisher Scientific). After incubating on ice for 10 min, a single exponential decay pulse was applied using an ECM 630 Electroporation System (BTX) set at 2.0 kV, 25 µF, and 400 Ω. Immediately following pulse delivery, 900 µL of liquid broth containing 0.2 M sucrose was added into the electroporation cuvette, and the entire suspension was transferred to 400 µL of the same medium. The DNA-cell suspension, along with the same amount of competent cell without adding plasmid DNA (as a negative control), was recovered at 37 °C overnight, then 200 µL of the recovered culture was plated onto TSAB or BHIB agar plates with 15 µg/mL thiamphenicol (or MIC, see Table S1).
Six to twelve transconjugants/transformants were restreaked onto plates with the same set of antibiotics to purity. The single colonies were verified by aerotolerance test (Sheridan et al., 2019), 16S rRNA sanger sequencing, and PCR using primers specific for the plasmids (rep oris, etc) followed by sanger sequencing of the PCR products.
Identify gene transfer methods for other gut-associated microbes
Please refer to Table S1 for detailed conjugation/transformation parameters. The same gene transfer conditions were applied when introducing propionate deletion plasmids.
1). E. coli filter mating conjugation:
The E. coli S17 harboring the chimeric-16S vector was inoculated in the LB broth supplemented with carbenicillin (100 µg/mL) grown at 37 °C with aerobic shaking at 220 rpm. After ~12–16 hrs, 6mL of E. coli S17 culture (OD600: 0.8 to 1.0) was centrifuged at 1500 x g for 2 min. The cell pellet was washed twice with 3 mL PBS buffer (Gibco, pH = 7.4). The washed E. coli S17 cell pellet was resuspended in 3 mL overnight culture of the recipient strain and gently mixed by pipetting. The mixture was filtered through a 50 mL Tube Top Vacuum Filter System (0.22 μm) from Corning Life Sciences (Cat. #430320). The filter with the cell mixture was placed (top surface facing the agar) onto a pre-reduced TSAB plate. The plate was incubated aerobically at 37 °C incubator for 24 hrs. The filter was then soaked in 2 mL of pre-reduced TYGB medium followed by gentle vortexing. The mixture was then transferred into an anaerobic chamber, and 100 µL was plate onto a pre-reduced TSAB plate + 200 µg/mL gentamycin + 15 µg/mL thiamphenicol (or MICs, see Table S1). Colonies of the target strain typically appeared after 36–48 hrs. The protocol for doting mating is similar as described for Clostridia commensals, except that some conjugations were performed aerobically if the recipient microbe is not sensitive to oxygen.
2). Electroporation:
The electroporation protocol is similar as described for Clostridia commensals, except that some electroporation were performed aerobically and different parameters were used (see Table S1). For Fusobacterium, the resulting cell pellet collected from 45 ml culture (OD600 ~1.2) was washed twice with 25 mL of pre-reduced, filter-sterilized ddH2O, and the competent cell was resuspended in 1 mL of 10% (v/v) cold glycerol. 100 µL competent cell was mixed with 2µg plasmid and transferred to a pre-chilled electroporation cuvette (1mm, Fisher Scientific). Immediately following pulse delivery (2.5 kV, 25 µF, and 200 Ω), 1 mL liquid broth was added into the electroporation cuvette, and the entire suspension was recovered anaerobically at 37 °C for 3 hrs. A 200 µL portion of the recovery culture (along with no-plasmid control) was plated onto CBAB agar plates containing thiamphenicol at their corresponding MICs. Colonies typically appeared after 48–72 hrs.
For Klebsiella and Proteus, the resulting cell pellet collected from 45 ml culture (OD600 ~0.6) was washed once with 25 mL of pre-reduced, filter-sterilized ddH2O, followed by two wash using 2 mL 10% (v/v) cold glycerol. 100 µL competent cell was mixed with 2µg plasmid and transferred to a pre-chilled electroporation cuvette (1mm, Fisher Scientific). Immediately following pulse delivery (2.5 kV, 25 µF, and 200 Ω), 500 μL of LB was added into the cuvette, and the entire suspension (along with no plasmid control) was recovered at 37 °C for 1 hr, then 100 µL of the recovery culture was plated onto LB agar containing the corresponding selective antibiotics (Table S1). Colonies typically appeared after 24–48 hrs.
Six to twelve transconjugants/transformants were restreaked onto plates with the same set of antibiotics to purity. The single colonies were verified by aerotolerance test if necessary, 16S rRNA sanger sequencing, and diagnostic PCR (Data S1D) followed by sanger sequencing of the PCR products.
Selecting the Group II intron (Clostron)-integrated Clostridia mutants
This method section applies to the test of 16S-tron in various non-model gut Clostridia (Fig. 2) and generating the S122 baiH mutant (Fig. 4). Please refer to Data S1 for more details. The Clostridia transconjugants harboring the Group II intron-based plasmids were restreaked to purity onto agar plates supplemented with the same set of antibiotics used during conjugation. Then, three single colonies were cultivated into 1 mL liquid broth with the same antibiotics. After 24–36 hrs, 50 µL to 200 µL culture were spread onto corresponding agar plates supplemented with antibiotics for RAM selection (Table S1). The putative integration mutants typically appeared after 36–48 hrs. At least eight integration mutants were restreaked to isolate single colony, and each colony was inoculated into 3 mL liquid broth supplemented with antibiotics for RAM selection (Table S1). After 24–36 hrs, the genomic DNA was extracted and diagnostic PCR was performed (Data S1D), and the PCR product with the expected size was purified and sent for Sanger sequencing.
Diagnostic PCR and sanger sequencing of the PCR products
Please see Fig. S1 and Data S1D for detailed diagnostic PCR strategy. The isolated single colony (via re-streak) was cultivated in 3 mL liquid culture supplemented with the same set of antibiotics used during plating (see Table S1). The genomic DNA was isolated, then multiplex diagnostic PCR was performed to assess which plasmid was uptaken by the conjugation recipient Clostridia microbe (Fig. S1). Diagnostic PCR following the scheme in Data S1D was performed to verify the single crossover integration of the suicide vector or the genome insertion of Group II intron. The genome insertion of Group II intron was further verified by PCR monitoring the intron splicing and motility (Heap et al., 2007). All the amplified PCR fragments were verified by Sanger sequencing to avoid non-specific PCR amplifications.
Identifying 7α-dehydroxylating Clostridia microbes
To identify if there are any 7α-dehydroxylating bacteria in the group of genetically targetable Clostridia commensals, two single colonies of one Clostridia was cultivated in 1mL liquid medium supplemented with 100 µM CA and 100 µM CDCA. After 48 hrs, 1ml of the culture was centrifuged at 15000 g for 20 min, and the supernatant was subject to LC-MS analysis to examine if CA and CDCA were 7α-dehydroxylated to DCA and LCA.
Whole-genome sequencing of Faecalicatena contorta S122
A single colony of S122 was cultivated in 3 mL Mega liquid broth for 24 hrs and the genomic DNA was extracted and sent for whole genome sequencing (BGI). The raw sequencing reads were filtered (for quality control), and de novo assembled (Geneious). The assembled contig in fasta format was further annotated using Prokka (v1.12) (Seemann, 2014). To locate the bai operon in the S122 genome, a tblastn search was performed for each bai gene annotated in the genome of C. scindens ATCC 35704 and a cluster of nine genes as a candidate bai operon was identified in the S122 genome (Fig. 4A).
Quantification of SCFAs, BSCFAs, and bile acids using LC-MS
In all the LCMS analyses, peaks were assigned by comparison with authentic standards and relative analyte concentrations were quantified by comparing their peak areas with those of internal standards.
For isovalerate, propionate, and butyrate, a 10 µL aliquot of the bacterial culture or cell suspension was mixed with 190 µL of short-chain fatty acids (SCFAs) derivatization solution (1 mM 2,2’-dipyridyl disulfide, 1 mM triphenylphosphine, and 1 mM 2-hydrazinoquinoline dissolved in acetonitrile) (Lu et al., 2013). The resulting mixture was incubated at 60 °C with shaking at 950 rpm for 1 hr. The mixture was centrifuged at 21000 x g for 20 min, and the supernatant was analyzed using an Agilent 1290 LC system coupled to an Agilent 6530 quadrupole time-of-flight (QTOF) mass spectrometer with a 130Å, 1.7 μm, 2.1 mm × 100 mm ACQUITY UPLC BEH C18 column (Waters). The following solvent system was used: A: H2O with 0.1% formic acid; B: Methanol with 0.1% formic acid. 1 µL of each sample was injected, and the flow rate was 0.35 mL/min with a column temperature of 40 °C. The gradient for HPLC-MS analysis was: 0–6.0 min, 99.5%−70.0% A; 6.0–9.0 min, 70.0%−2.0% A; 9.0–9.4 min, 2.0% A; 9.4–9.6min, 2.0%−99.5% A.
For bile acids detection and quantification, 100 µL of culture was centrifuged at 21000 x g for 20 min, and the supernatant was analyzed using an Agilent 1290 LC system coupled to an Agilent 6530 quadrupole time-of-flight (QTOF) mass spectrometer with a 1.7 μm, 2.1 mm × 100 mm Kinetex C18 column (Phenomenex). The following solvent system was used: A: H2O with 0.05% formic acid; B: Acetone with 0.05% formic acid. 1 µL of each sample was injected, and the flow rate was 0.35 mL/min with a column temperature of 40 °C. 0–1 min, 75% A; 1–25 min, 75%−25% A; 25–26 min, 25%−0% A; 26–30 min, 0% A; 30–32 min 0%−75% A.
To quantify SCFAs, BSCFAs, and bile acids in mouse biological samples, a ~ 10mg fecal samples (or cecal samples) were resuspended in 50 µL of 50% MeOH (in H2O) and vortexed for 10 min (some beads were added to disperse the fecal/cecal material). Then the mixture was spun down, and the following treatment is the same as that of culture supernatant. The calculated concentrations were normalized to the fecal/cecal weight.
Growth curve of bacteria
Bacteroides (S14 (Bac1) and S33 (Bac2)) and Erysipelotrichaceae (S94 (Ery1), S95 (Ery2), S97 (Ery3), S96 (Ery4), S87 (Ery5), S88 (Ery6) and Holdemania filiformis DSM 12042 (Ery7)) (Fig. 7, Table S1) were restreaked and inoculated into 1 ml of Mega broth and cultured anaerobically for overnight at 37 °C. Cells were diluted 1,000-fold into Mega broth to reach late-log phase. Then 5 μL of the culture was resuspended in 145 μL broth, loaded into a 96-well plated, and incubated anaerobically at 37 °C in Multiskan Sky Microplate Spectrophotometer (Thermo Fisher Scientific). Four bile acids (CA, 7-oxoCA, DCA, and 3-oxoDCA, 500 µM each). were tested with their solvent DMSO as control. OD600 was recorded every 30 min until the cultures reached the stationary phase. Growth curves were performed in triplicate, with each biological replicate derived from a single colony.
Quantitative PCR (qPCR)
For the qPCR of dCpf1 targeting genes, three isolated transconjugants of control or mutant strains were restreaked onto the agar plates with the same set of antibiotics used during conjugation. A single colony was subcultured and then inoculated into 5 mL pre-reduced Mega medium with antibiotics. When cells reach late-exponential phase, total RNA was extracted using Quick RNA fungal/bacterial kit (Zymo Research). Reverse transcription of extracted RNA into cDNA was performed using PrimeScript™ RT Reagent Kit (TaKaRa). Real-time quantitative PCR (qPCR) was performed on diluted cDNA using SYBR green chemistry (Applied Biosystems). Reactions were run on a real-time quantitative PCR system (ABI 7500; Applied Biosystems). Expressions of target genes were normalized to the 16S rRNA of each strain. Three biological replicates and two technical replicates were used for each control or mutant strains.
To assess the relative fold change of Erysipelotrichaceae in liquid culture, 5 µL of overnight culture of two Bacteroides and seven Erysipelotrichaceae in Mega (~1 × 107 CFU) were inoculated into 1ml fresh Mega broth with different concentrations of DCA (0 µM, 250 µM, 500 µM), or co-inoculated together with S122 control or S122 ΩbaiH mutant into Mega with 500 µM CA. After incubating anaerobically for 24h, gDNA was extracted using Quick RNA fungal/bacterial kit (Zymo Research). To assess the relative fold change of Erysipelotrichaceae in fecal samples, gDNA in fecal samples was extracted using QIAamp Fast DNA Stool Mini Kit (Cat. # 51604). qPCR was performed using primers Bac_Erysi_16S_qPCR_F-2 + Bac_Erysi_16S_qPCR_R-2 that amplify total 16S of Bacteroides and Erysipelotrichaceae as reference, and primers Erysi_16S_qPCR_F + Erysi_16S_qPCR_R to amplify Erysipelotrichaceae-specific 16S.
To assess the expression of inflammatory genes in the colon, colonic samples were homogenized, and then RNA was extracted from the resulting homogenate using Quick RNA fungal/bacterial kit (Zymo Research) following the manufacturer’s protocol. Extracted RNA was reverse transcribed into cDNA for qPCR. Expressions were normalized to Hprt1.
Colonize germ-free and SPF mice with the control and mutant bacteria
Three isolates of each genetically manipulated bacterial strain were restreaked and mixed before colonization. For mono-associating germ-free mice with a bacterial control or mutant (Fig. 3), a 200 µL portion of bacterial overnight culture (~1 × 107 CFU) were given to germ-free mice (n = 3 or 4 per group) via oral gavage. Successful colonization of the bacterium was determined by plating fecal samples and colony-forming unit (CFU) counting. For co-colonizing germ-free mice with S122 and other gut microbes (S25 in Fig. 4, or Bacteroides S14 (Bac1) and S33 (Bac2) and Erysipelotrichaceae S94 (Ery1), S95 (Ery2), S97 (Ery3), S96 (Ery4), S87 (Ery5), S88 (Ery6) and Holdemania filiformis DSM 12042 (Ery7) in Figs. 7, and S7), 1 ml of their overnight culture (~1 × 107 CFU) were mixed and co-colonized with germ-free mice (n = 4 per group) via oral gavage (300 µL per mouse). Successful colonization of S122 and S25 (Fig. 4) was determined by PCR and Sanger sequencing of their 16S rRNA genes, and 16S rRNA sequencing. Successful colonization of S122 and other microbes (Figs. 7 and S7) was determined by CFU (for S122 and total bacterial load), and 16S rRNA sequencing. For colonizing SPF mice with S122 control or its baiH mutant, a 300 µL portion of their overnight culture (~1 × 107 CFU) were given to SPF mice (n = 4 per group) via oral gavage, twice per day for 3 days in a row. Successful colonization of the SPF mice was monitored weekly and determined by CFU plating and PCR amplification of S122 specific gene.
Colony-forming unit (CFU) quantification of mouse fecal samples
A ~5 mg fecal material was resuspended in 200 µL pre-reduced Gibco™ PBS buffer, pH 7.4, followed by a 10-fold serial dilution (to 10−4) in the same buffer on a 96-well plate. 50 µL from each well was plated on pre-reduced TSAB agar and was incubated anaerobically at 37 °C. After 24 to 48 hrs, colonies will appear, and the CFUs of mouse fecal samples is calculated after normalizing to fecal weight. For Fig. 5, CFU quantification was performed weekly and TSAB agar was supplemented with 250 µg/mL D-cycloserine + 15 µg/mL thiamphenicol + 10 µg/mL erythromycin. Diagnostic PCR was performed to verify the colonies on the plates were indeed S122 by amplifying its specific gene and Sanger sequencing.
DSS treatment of gnotobiotic and SPF mice
All DSS experiments used sex-matched mice at age 8–10 weeks. Dextran sulfate sodium salt (DSS) of colitis-grade with an average MW of 36,000–50,000 Da (MP Biomedicals) was added to drinking water at day 0. DSS was administered until substantial inflammation was induced as evidenced by significant weight loss. Titration experiments were performed to determine the DSS concentration required to induce similar weight loss and inflammation across manufacturer lots (if needed). In Fig. 6F, 2% DSS in water supplemented with 15 μg/ml thiamphenicol was given for 7 days. In Fig. 6A, 2.5% DSS in water with 15 μg/ml thiamphenicol and 10 μg/ml erythromycin was given for 8 days. In Figs 7D and S7G, 2.5% DSS was given for 9 days. After DSS treatment, the same water without DSS was used during recovery.
Assess colon inflammation in DSS-treated mice
Throughout DSS treatment and recovery, mice were weighed daily at the same time of day at indicated time points, and feces were collected daily at the same time points. Mice were then euthanized by CO2 asphyxiation. Colon length was measured, proximal colon/distal colon/ileum tissue samples were collected for histology, and colon/ileum tissue samples were collected for qPCR.
For fecal lipocalin-2 quantification, fecal samples were collected and suspended in PBS with 1% Bovine Serum Albumin (1g/100mL) to a final concentration of 100 mg/mL and vortexed for 20 min to get a homogenous fecal suspension. Samples were then centrifuged for 10 min at 14000 g and 4°C to remove aggregates, and the resulting supernatant was collected. A sandwich ELISA was performed following appropriate dilution using mouse lipocalin-2/NGAL DuoSet ELISA (R & R&D Systems). Fecal hematochezia score was assessed using Hemoccult II SENSA Dispensapak Plus kit (Backman Coulter) following the manufacturer’s instructions.
For histology scores, distal colon sections were obtained and fixed in 10% neutral buffered formalin overnight at room temperature and then were transferred to 70% ethanol. Then sections were paraffin-embedded, sectioned, and stained with hematoxylin and eosin by IDEXX BioAnalytics company. Blinded histological evaluation was conducted on a scale of 1–3 or 4 for the following histologic parameters: area involved (0–4), erosion/ulceration (0–4), follicles (0–3), edema (0–3), fibrosis (0–3), crypt loss (0–4), granulocytes (0–3), mononuclear cells (0–3), and crypt damage/apoptosis (0–4). Scores were accumulated to give a total score of inflammation.
16S rRNA gene sequencing of fecal samples
For the 16S rRNA gene sequencing, gDNA was extracted from mouse fecal samples using QIAamp Fast DNA Stool Mini Kit (Cat. # 51604), and the concentration of double-stranded gDNA in the extracted gDNA was measured using Quant-iT™ dsDNA Assay Kit (Cat. # Q33120). Then gDNA was normalized to 20 ng/µL and sent for 16S rRNA gene sequencing by Genewiz or WCM Microbiome Core Lab.
Bioinformatic analyses
1). Phylogenetic analyses:
16S rRNA sequences were extracted from their sequenced genomes, Silva database (Quast et al., 2013), or from the Sanger sequencing result. The 16S rRNA sequences were aligned using Clustal Omega, and the aligned sequences were used to construct a phylogenetic tree (neighbor-joining) with a bootstrap test of 5000 using Mega11 (Kumar et al., 2016).
2). Metatranscriptomic analyses of the S122 bai operon:
To determine if the S122 bai operon is actively transcribed under the condition of host colonization, we built a local DNA sequence database consisting of all the Clostridia bai operons identified so far. We mapped metatranscriptomic reads from the stool of healthy human subjects (David et al., 2014) to this database using Bowtie 2 (local, high sensitivity); representative mapping results are shown in Fig. 4A.
3). Correlation analyses:
The metabolomics data, fecal calprotectin data, and relative abundances of specific taxonomic groups of gut microbes whose relative abundances are affected by baiH depletion in our experiment were downloaded and extracted from the iHMP-IBD website (https://ibdmdb.org/). Longitudinal data of the same participant were Z-transformed (with a mean of 0 and an SD of 1). The correlation between 1) fecal DCA and fecal calprotectin level, and 2) fecal DCA and specific gut microbial taxonomic groups, were assessed using Pearson correlation, with a pre-specific alpha level of 0.05 to assess statistical significance. A correlation of 0.2 or higher or of −0.2 or lower was considered moderate. In addition, a linear mixed model with random intercept was used to assess the association between fecal DCA and fecal calprotectin/gut microbe relative abundances, accounting for the longitudinal measurements obtained from the same individual. Z-transformed values (dots) and the fitted values based on the linear mixed model (line) were presented in Figs. 5 and S5.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were obtained from three or more biological replicates (n) and were processed with software GraphPad Prism 8 for statistical purposes. Mean, SD, SEM, and statistical significance were used in the figure preparation as indicated in the figure legends.
Supplementary Material
(A) Schematic view of a mixed-conjugation strategy to identify the Clostridia that stably maintains exogenous DNA. Ten pGM vectors, each harboring a single rep ori (9 Clostridia specific and 1 rep ori-less), were separated into three sets and mixed-conjugated to a Clostridia recipient.
(B) Multiplex PCR strategy was used to identify which rep ori-contained plasmid was introduced into which Gram+ Clostridia strain in mixed-conjugation. For the mixed-conjugation with set I, primers pMTL_laz_diag_F (universal forward primer) + pGM-ABCM_rep_R_1500bp + pGM-BBCM_rep_R_1000bp + pGM-CBCM_rep_R_2000bp were used for diagnostic PCR. A 1.5 kb (or 1.0 kb, or 2.0 kb) PCR band would be seen if pGM-ABCM (or BBCM, or CBCM) is uptaken by the Clostridia microbe. The primers for the set II and set III mixed-conjugation are shown in Table S2.
(C) Distribution of Clostridial rep oris tested in this study based on phylogeny. The phylogenetic tree was constructed using the 16S rRNA sequences of the 42 gut microbes (38 Firmicutes/Clostridia, 2 Enterococcus, and 2 Actinobacteria) with a compatible rep ori identified in this study. The sequences were aligned using Clustal Omega, and a neighbor-joining tree was constructed with a bootstrap test of 5000.
(D) Multiple Clostridia and promoters are screened, and the red arrow indicates the original promoter Ppmtl-catP used in the Clostridia conjugation vectors (one representative promoter screening result is shown). Error bar: standard deviation.
The numbering of the strains corresponds to the strain information shown in Table S1.
(A) qPCR results showing that CRISPRi-dCpf1 precisely and efficiently suppressed lacZα expression in Gram-positive Clostridia and Bifidobacterium microbes, using both dCpf-1-only and gRNA-only as controls.
(B) qPCR results showing that CRISPRi-dCpf1 precisely and efficiently suppressed lacZα expression in other Gram-positive Clostridia microbes and Gram-negative microbes from other phyla, using dCpf-1-only as control.
For each strain shown in (A), conjugation was conducted with E. coli harboring plasmids with dCpf1-lacZα (dCpf1), gRNA-lacZα (gRNA), or gRNA-dCpf1-lacZα (dCpf1+gRNA). For each strain shown in (B), conjugation was conducted with E. coli harboring plasmids with dCpf1-lacZα (dCpf1, Con), or gRNA-dCpf1-lacZα (dCpf1+gRNA, Mut). Then transconjugants were cultured, and RNA was extracted and reverse transcribed to cDNA. Quantitative PCR (qPCR) was used to assess the expression of lacZα after normalizing to the expression of the 16S rRNA gene of each strain. Error bar: standard deviation. The numbering of the strains corresponds to the strain information shown in Table S1.
(A) CRISPRi-dCpf1 precisely and efficiently suppressed bcat expression in 11 Clostridia and 1 Bifidobacterium microbes with sequenced genomes. Quantitative PCR (qPCR) was used to assess bcat expression after normalizing to the 16S rRNA gene expression of each strain.
(B) The S25 ΔmmdA mutant and control strains, (C) the S117 croA knockdown mutant and the control strains, (D) and the S107 porA knockdown mutant and the control strains colonized the germ-free mice (n = 3 or 4 per group) at comparable levels. Colonized germ-free mice were maintained on a standard diet and supplied with water containing 15 µg/mL thiamphenicol and 2 mg/mL Sweet N Low sweetener. We calculated colony-forming units (CFU) in fecal pellets to estimate the density of intestinal colonization. n.s.: not statistically significant.
Error bar: standard deviation. The numbering of the strains corresponds to the strain information shown in Table S1.
The prevalence (A) and percent relative abundance (B) of gut Clostridia commensals harboring the bai operon were examined using a large publicly available 16S rRNA dataset (Yatsunenko et al., 2012), including stool samples from 528 individuals. S122 and its closely related relatives are more prevalent than C. hylemonae, C. scindens, and D. sp. D27_3, but less than that of C. hiranonis (A). S122 and its closely related strains are the most abundant 7α-dehydroxylating commensal in this cohort (B). For (A), the prevalence data were analyzed using Fisher’s exact test, and mean ± SEM was plotted. For (B), the relative abundance data were first analyzed using the D’Agostino & Pearson test for normality. The relative abundance of S122 was compared to other commensals using the Mann-Whitney test. A Median with a 95% confidence interval (CI) was plotted. The asterisk in (A) and (B) indicates p-value < 0.0001 (****).
(C) Successful insertion of baiH was determined by amplifying DNA using primers flanking the target gene baiH. The expected size for the control is ~2kb, and ~4kb for the S122 ΩbaiH mutant.
(D) Metabolomics analyses of bile acids in S25 + S122 ΩbaiH mutant (Mut) and S25 + S122 control (Con) co-colonized germ-free mice. Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**), n.s.: not statistically significant, n.d.: not detected.
(E) Co-colonization of germ-free mice with S122 and 55 other genetically targetable microbes identified in this study. Successful colonization of S122 was determined by CFU and the detection of DCA in LCMS, and colonization of other strains was confirmed by 16S rRNA sequencing.
The numbering of the strains corresponds to the strain information shown in Table S1.
(A-B) The Chao 1 index (A) and Shannon index (B) of the fecal microbiota of the SPF mice colonized with the control (Con) and the ΩbaiH mutant (Mut). Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**).
(C-D) Relative abundances of Bacteroidetes and Proteobacteria in the stool microbiome of the control and ΩbaiH mutant colonized SPF mice. ΩbaiH mutant (Mut) colonized mice harbor significantly higher abundances of Bacteroidetes and lower abundances of Proteobacteria compared to the control group (Con). Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**).
(E) Relative abundances of gut microbial taxa (at the family level) in the stool microbiome of the control and ΩbaiH mutant colonized SPF mice. Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**).
(F-G) In nonIBD human stools, the fecal DCA level is weakly associated with the relative abundance of microbes in the Erysipelotrichaceae family and with that of the B. eggerthii species.
(H) The density of total intestinal bacteria of SPF control mice compared with SPF mice colonized with S122 control and ΩbaiH mutant. SPF control mice (SPF) were maintained with a standard diet and water, and S122 colonized SPF mice with standard diet and water with 15 µg/mL thiamphenicol and 10 µg/mL erythromycin. We calculated colony-forming units in fecal pellets on TSAB agar plates to estimate the density of intestinal bacteria. Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**), n.s.: not statistically significant.
(A) Macroscopic observations of the colon length of SPF mice colonized with S122 control (Con) and ΩbaiH mutant (Mut) post-DSS treatment.
(B) Macroscopic observations of colon length of germ-free mice colonized with S122 control (Con) and ΩbaiH mutant (Mut) post-DSS treatment.
(C) Quantification of fecal lipocalin-2 (LCN-2) in SPF mice colonized with S122 control (Con) and ΩbaiH mutant (Mut) post-DSS treatment on the day-5, 6, 7, and day-sacrifice, the amount of lipocalin-2 was significantly higher in the control group. Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**).
(D) Quantification of fecal lipocalin-2 (LCN-2) in germ-free mice colonized with S122 control (Con) and ΩbaiH mutant (Mut) post-DSS treatment at the day-sacrifice, no significant difference was found. Data are shown as mean ± SEM. Student’s T-test was performed, n.s.: not statistically significant.
(E) Comparison of colonic expression of inflammatory genes in SPF mice colonized with S122 control (Con) and ΩbaiH mutant (Mut) post-DSS treatment. Expression of inflammatory genes was normalized to Hprt1 and shown as the fold change relative to the mutant group. Colonic expression of inflammatory genes was significantly higher in the control group. Data are shown as mean ± SEM. Student’s T-test was performed, and asterisk indicates p-value < 0.05 (*) or < 0.01 (**).
(F) Density of intestinal colonization of SPF mice supplemented with low dose antibiotics (15 µg/mL thiamphenicol and 10 µg/mL erythromycin in drinking water) (n = 4 per group) by the S122 control (Con) and ΩbaiH mutant (Mut) (Fig. 6A). The colony-forming units (CFU) were calculated before DSS treatment and on day 3 and day 6 of DSS treatment. Student’s T-test was performed. n.s.: not statistically significant.
(G) Relative abundances (by 16S rRNA sequencing) of Erysipelotrichaeceae in the stool microbiome of the S122 control (Con) and ΩbaiH mutant (Mut) colonized SPF mice (n =4 per group) at day 3 and day 6 of DSS treatment (Fig. 6A). Student’s T-test was performed. The asterisk indicates p-value < 0.01 (**) n.s.: not statistically significant.
(H) Fecal DCA levels of the SPF mice (grey bar), and the SPF mice supplemented with low dose antibiotics (n = 4 per group) colonized with the S122 control (Con) strain (before and at day 3 of DSS treatment) (Fig. 6A). Student’s T-test was performed. n.s.: not statistically significant.
(I) The relative abundances of S122 in the control and mutant group (n = 4 per group) of the DSS germ-free experiment (Fig. 6B) were assessed by 16S rRNA sequencing and were comparable. Student’s T-test was performed. n.s.: not statistically significant.
(J) Targeted metabolomics analyses of the stool bile acid (BA) compositions of the S122 control (Con) and ΩbaiH mutant (Mut) colonized gnotobiotic mice (n = 4 per group) at day 3 after the DSS treatment (Fig. 6F). Student’s T-test was performed. The asterisk indicates p-value < 0.01 (**) n.s.: not statistically significant, n.d.: not detected.
(A) Density of intestinal colonization (assessed by CFU) of gnotobiotic mice colonized with S122 control (Con) and ΩbaiH mutant (Mut), and total intestinal bacterial load in the 10-member community as shown in Fig. 7D. We calculated colony-forming units in fecal pellets to estimate the density of intestinal colonization. n.s.: not statistically significant.
(B) Density of intestinal colonization (assessed by CFU) of gnotobiotic mice colonized with S122 control (Con) and ΩbaiH mutant (Mut), and total intestinal bacterial load in the 3-member community as shown in Fig. S7G. We calculated colony-forming units in fecal pellets to estimate the density of intestinal colonization. n.s.: not statistically significant.
(C-D) Metabolomics analyses of fecal bile acids in feces of two Bacteroides (bac) microbes and seven Erysipelotrichaceae (Ery) microbes + S122 control (Con) / S122 ΩbaiH mutant (Mut) co-colonized germ-free mice before DSS treatment (C) and on day 3 of DSS treatment (D). Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**), n.s.: not statistically significant, n.d.: not detected.
(E-F) Metabolomics analyses of bile acids in feces of two Bacteroides (bac) microbes + S122 control (Con) / S122 ΩbaiH mutant (Mut) co-colonized germ-free mice before DSS treatment (E) and on day 3 of DSS treatment (F). Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**), n.s.: not statistically significant, n.d.: not detected.
(G-L) DSS-induced murine colitis model was applied to the gnotobiotic mice colonized with a synthetic consortium consisting of the genetically tagged S122 control (Con) or ΩbaiH mutant (Mut) along with 2 Bacteroides (Bac) microbes only. Mice were colonized with the consortium for at least two weeks, followed by 2.5% DSS for 9 days. The disease state was monitored by weight loss (H), hematoxylin and eosin (H&E) staining of the distal colon (I), colon shortening (J), fecal lipocalin-2 (K), and daily hematochezia score (L).
Data shown in (H, I, J, K, L) are representations of n = 4 mice per group replicated in two independent experiments. In (H), % of starting weight was calculated by normalizing weights at sacrifice to starting weight. In (J) and (K), colon length and LCN2 data were analyzed using unpaired two-tailed Student’s T-test. In (H) and (L), % of starting weight and hematochezia score data were analyzed using Two-way ANOVA followed by the Bonferroni post hoc test (n=4). Data are shown as mean ± SEM. The asterisk indicates p-value < 0.05 (*), < 0.01 (**) or < 0.001 (***). The numbering of the strains corresponds to the strain information shown in Table S1.
Table S1. Bacterial strains, their culture conditions, and their gene transfer methods, related to Figures 1, 2, 3, and STAR Methods.
Table S2. Primers and plasmids used in this study, related to Figures 2, 3, 4, and STAR Methods.
Table S3. Recipes for all the culture media used in this study and their ingredient details, related to Figure 1 and STAR Methods.
Table S4. 16S rRNA gene sequencing of fecal samples from SPF mice colonized with Faecalicatena contorta S122 (S122) control (Con) and ΩbaiH mutant (Mut), related to Figure 5.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| 12-keto Lithocholic Acid (12-oxo DCA) | Cayman Chemical | Cat# 29547 |
| 12-oxo-CA | Avanti | Cat# 700236P |
| 2,2′-dipyridyl disulfide | Alfa Aesar | Cat# A11118 |
| 2-hydrazinoquinol | TCI | Cat# H0178 |
| 3-oxo DCA (3-OH oxidized) | ChemCruz | Cat# sc-477977 |
| 3-oxo-CDCA | Avanti | Cat# 700255P |
| 3-Oxocholic Acid (3-oxo CA) | Cayman Chemical | Cat# 28403 |
| 3β-Cholic Acid-2,2,4,4-d4 (iso CA−-2,2,4,4-d4) | Cayman Chemical | Cat# 27376 |
| 7-Ketodeoxycholic acid (7-oxo CA) | Sigma | Cat# SMB00806 |
| Acetic acid | Fisher Chemical | Cat# A38-500 |
| Acetone | Fisher Chemical | Cat# A18-4 |
| Acetonitrile | Fisher Chemical | Cat# A21-4 |
| Ascorbic acid | Fisher Chemical | Cat# BP351500 |
| BBL™ Bacto Tryptone | BD | Cat# 211921 |
| Butyric acid | ACROS ORGANICS | Cat# 108111000 |
| CaCl2 | ACROS ORGANICS | Cat# 349615000 |
| Carbenicillin | GOLDBIO | Cat# C-103-25 |
| Chenodeoxycholic acid (CDCA) | Alfa Aesar | Cat# J60364 |
| Chloramphenicol | VWR | Cat# 0230 |
| Cholic acid (CA) | Alfa Aesar | Cat# A11257 |
| D-(−)-Fructose | VWR | Cat# 0226-1KG |
| D-(+)-Cellobiose | Alfa Aesar | Cat# A14553 |
| D-(+)-Glucose | VWR | Cat# BDH9230 |
| D-(+)-Maltose monohydrate | VWR | Cat# 1B1184 |
| D-cycloserine | TCI | Cat# C1189 |
| Deoxycholic acid (DCA) | Alfa Aesar | Cat# B20061 |
| Dextran sulfate sodium salt (DSS) | MP Biomedicals | Cat# 160110 |
| Difco™ Brain Heart Infusion Agar | BD | Cat# 241830 |
| Difco™ Columbia agar | BD | Cat# 279240 |
| Difco™ Columbia Broth | BD | Cat# 294420 |
| Difco™ Luria-Bertani (LB) broth | BD | Cat# BP1426-2 |
| Difco™ Tryptic Soy Agar | BD | Cat# 236950 |
| Difco™ Trypticase Peptone | BD | Cat# 211921 |
| Difco™ Yeast Extract | Fisher Bioreagents | Cat# BP1422-2 |
| DMSO | ACROS ORGANICS | Cat# 61097-1000 |
| Erythromycin | VWR | Cat# 0219 |
| Ethanol | Fisher Bioreagents | Cat# BP2818-4 |
| FeSO4 | ACROS ORGANICS | Cat# 423731000 |
| Formic acid | Fisher Chemical | Cat# A117-50 |
| Gentamicin | GOLDBIO | Cat# G-400-25 |
| Glycerol | Fisher Bioreagents | Cat# BP229-4 |
| Glycine | Alfa Aesar | Cat# J62407 |
| Ground beef (fat-free) | RESEARCH DIETS | Cat# D12451i |
| Horse blood | Hemostat Laboratories | Cat# 637291-1 |
| Isovalerate | ACROS ORGANICS | Cat# 156691000 |
| Isovaleric acid | ACROS ORGANICS | Cat# 156691000 |
| K2HPO4 | ACROS ORGANICS | Cat# 424195000 |
| Kanamycin | GOLDBIO | Cat# K-120-25 |
| KH2PO4 | ACROS ORGANICS | Cat# 42420-5000 |
| L-Cysteine hydrochloride | Fisher Bioreagents | Cat# BP376-100 |
| Lithocholic acid (LCA) | ICC | Cat# 123530 |
| Meat extract | Sigma | Cat# 70164 |
| Methanol | Fisher Chemical | Cat# A456-4 |
| MgSO4·7H2O | Fisher Chemical | Cat# M65-500 |
| Na2HPO4 | Alfa Aesar | Cat# 13437 |
| Na2SO4 | Fisher Chemical | Cat# S421-1 |
| NaCl | Fisher Chemical | Cat# S271-1 |
| Na-DL-lactate | Sigma | Cat# 71720 |
| NaH2PO4 | Fisher Bioreagents | Cat# BP329-500g |
| NaHCO3 | ACROS ORGANICS | Cat# 424270010 |
| Na-thioglycolate | TCI | Cat# M0053 |
| NH4Cl | Fisher Chemical | Cat# 12125-02-9 |
| PBS | Gibco | Cat# 10010-031 |
| Propionic acid | ACROS ORGANICS | Cat# 14930-0010 |
| Reinforced Clostridial Medium | BD | Cat# 218081 |
| Resazurin | ACROS ORGANICS | Cat# 418900250 |
| Soluble starch | ACROS ORGANICS | Cat# 177132500 |
| Spectinomycin | ENZO | Cat# BML-A281-0010 |
| Sucrose | Fisher Bioreagents | Cat# BP220-1 |
| Taurochenodeoxycholic Acid (sodium salt) (TCDCA) | Cayman Chemical | Cat# 20275 |
| Taurocholic Acid (sodium salt) (TCA) | Cayman Chemical | Cat# 16215 |
| Taurodeoxycholic acid (TDCA) | Cayman Chemical | Cat# 15935 |
| Tauroursodeoxycholic acid (TUDCA) | Cayman Chemical | Cat# 20277 |
| Tetracycline | GOLDBIO | Cat# T-101-25 |
| Thiamphenicol | ACROS ORGANICS | Cat# 455450250 |
| Triphenylphosphine | Alfa Aesar | Cat# L02502 |
| Tween 80 | VWR | Cat# 0442-1L |
| Ursocholic acid (UCA) (7-OH isomer) | Avanti | Cat# 700229 |
| Ursodeoxycholic acid (UDCA) (7-OH isomer) | Avanti | Cat# 700199 |
| Vitamin K1 | Alfa Aesar | Cat# L10575 |
| Vitamin K3 | MP | Cat# 102259 |
| Critical commercial assays | ||
| AscI restriction enzyme | NEB | Cat# R0558S |
| Blue sapphire DNA polymerase | TAKARA | Cat# RR350 |
| CpG (M. SssI) methyltransferases | NEB | Cat# M0226S |
| DNA Clean and Concentrator | Zymo Research | Cat# D4003 |
| Esp3I restriction enzyme | NEB | Cat# R0734S |
| FseI restriction enzyme | NEB | Cat# R0588S |
| Gibson Assembly Cloning Kit | NEB | Cat# E5510S |
| GpC (M.CviPI) methyltransferases | NEB | Cat# M0227S |
| Hemoccult II SENSA Dispensapak Plus kit | Backman Coulter | Cat# 61130 |
| Instant Sticky-end Ligase | NEB | Cat# M0370S |
| Mouse lipocalin-2/NGAL DuoSet ELISA | R&D Systems | Cat# DY1857-05 |
| NotI restriction enzyme | NEB | Cat# R3189S |
| Plasmid Midiprep Kit | Zymo Research | Cat# D4201 |
| Primer star DNA polymerase | TAKARA | Cat# R045 |
| PrimeScript™ RT Reagent Kit | Takara | Cat# RR047A |
| QIAamp Fast DNA Stool Mini Kit | Qiagen | Cat# 51604 |
| Qiagen Miniprep Kits | Qiagen | Cat# 27104 |
| Quant-iT™ dsDNA Assay Kit, high sensitivity | Invitrogen | Cat# Q33120 |
| Quick DNA fungal/bacterial kit | Zymo Research | Cat# D6005 |
| Quick RNA fungal/bacterial kit | Zymo Research | Cat# R2010 |
| SbfI restriction enzyme | NEB | Cat# R3642S |
| SYBR green chemistry | Applied Biosystems | Cat# 4368706 |
| T4 DNA Ligase (HC) | Promega | Cat# M1794 |
| XbaI restriction enzyme | NEB | Cat# R0145S |
| XhoI restriction enzyme | NEB | Cat# R0146S |
| Experimental models: Organisms/strains | ||
| Strains used in this study | Lab stock | Table S1 |
| Oligonucleotides | ||
| Primers used in this study | This study | Table S2 |
| Recombinant DNA | ||
| pDEST-hisMBP-AsCpf1-EC | Addgene | Cat# 79007 |
| pExchange | Lab stock | N/A |
| pGM-ABC1M_seq-opt | This study | N/A |
| pGM-ABCD | This study | N/A |
| pGM-ABCD-006 | This study | Table S2 |
| pGM-ABCD-008 | This study | Table S2 |
| pGM-ABCD-009 | This study | Table S2 |
| pGM-ABCD-013 | This study | Table S2 |
| pGM-ABCD-015 | This study | Table S2 |
| pGM-ABCD-019 | This study | Table S2 |
| pGM-ABCF | This study | N/A |
| pGM-ABCG | This study | N/A |
| pGM-ABCL | This study | N/A |
| pGM-ABCL1 | This study | N/A |
| pGM-ABCM | This study | N/A |
| pGM-BBCD | This study | N/A |
| pGM-BBCF | This study | N/A |
| pGM-BBCL | This study | N/A |
| pGM-BBCM | This study | N/A |
| pGM-CBC1M_seq-opt | This study | N/A |
| pGM-CBCD | This study | N/A |
| pGM-CBCD-017 | This study | Table S2 |
| pGM-CBCF | This study | N/A |
| pGM-CBCL | This study | N/A |
| pGM-CBCL1 | This study | N/A |
| pGM-CBCM | This study | N/A |
| pGM-DBC1M_seq-opt | This study | N/A |
| pGM-DBCD | This study | N/A |
| pGM-DBCD-012 | This study | Table S2 |
| pGM-DBCF | This study | N/A |
| pGM-DBCL | This study | N/A |
| pGM-DBCM | This study | N/A |
| pGM-EBC1M_seq-opt | This study | N/A |
| pGM-EBCD | This study | N/A |
| pGM-EBCD-014 | This study | Table S2 |
| pGM-EBCF | This study | N/A |
| pGM-EBCL | This study | N/A |
| pGM-EBCL1 | This study | N/A |
| pGM-EBCM | This study | N/A |
| pGM-FBC1M_seq-opt | This study | N/A |
| pGM-FBCD | This study | N/A |
| pGM-FBCD-007 | This study | Table S2 |
| pGM-FBCD-010 | This study | Table S2 |
| pGM-FBCD-011 | This study | Table S2 |
| pGM-FBCD-016 | This study | Table S2 |
| pGM-FBCD-020 | This study | Table S2 |
| pGM-FBCF | This study | N/A |
| pGM-FBCL | This study | N/A |
| pGM-FBCL1 | This study | N/A |
| pGM-FBCM | This study | N/A |
| pGM-FCAR-002 | This study | Table S2 |
| pGM-FCAR-003 | This study | Table S2 |
| pGM-GBC1M_seq-opt | This study | N/A |
| pGM-GBCD | This study | N/A |
| pGM-GBCF | This study | N/A |
| pGM-GBCL | This study | N/A |
| pGM-GBCM | This study | N/A |
| pGM-HBC1M_seq-opt | This study | N/A |
| pGM-HBCD | This study | N/A |
| pGM-HBCD-018 | This study | Table S2 |
| pGM-HBCF | This study | N/A |
| pGM-HBCL | This study | N/A |
| pGM-HBCL1 | This study | N/A |
| pGM-HBCM | This study | N/A |
| pGM-IBCD | This study | N/A |
| pGM-IBCF | This study | N/A |
| pGM-IBCL | This study | N/A |
| pGM-IBCM | This study | N/A |
| pGM-NAC2B | This study | N/A |
| pGM-NAC2P | This study | N/A |
| pGM-NACM-003 | This study | Table S2 |
| pGM-NACM-004 | This study | Table S2 |
| pGM-NACM-005 | This study | Table S2 |
| pGM-NACO1 | This study | N/A |
| pGM-NACO2 | This study | N/A |
| pGM-NACO4 | This study | N/A |
| pGM-NACO5 | This study | N/A |
| pGM-NACO6 | This study | N/A |
| pGM-NACO7 | This study | N/A |
| pGM-NAKO3 | This study | N/A |
| pGM-NBC2B | This study | N/A |
| pGM-NBCC | This study | N/A |
| pGM-NAEM-001 | This study | Table S2 |
| pGM-NAEM-002 | This study | Table S2 |
| synthetic Bacteroides Chi-16S rRNA | This study | Synthesized by Twist |
| synthetic Desulfovibrio Chi-16S rRNA | This study | Synthesized by Twist |
| synthetic fragment (gBlocks) | This study | Synthesized by IDT |
| synthetic Prevotella Chi-16S rRNA | This study | Synthesized by Twist |
| synthetic Proteus Chi-16S rRNA | This study | Synthesized by Twist |
| Software and algorithms | ||
| Adobe Illustrator | Adobe | N/A |
| Agilent Masshunter Workstation | Agilent | N/A |
| Agilent Qualitative analysis | Agilent | N/A |
| Blast | NCBI | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| Bowtie 2 (local, high sensitivity) | Johns Hopkins University | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| ChembioDraw Ultra 14.0 | PerkinElmer | N/A |
| ClosTron | ClosTron | http://www.clostron.com/pMTL80000.php |
| Clustal Omega | The European Bioinformatics Institute | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| Geneious | Geneious | http://www.geneious.com/ |
| GraphPad Prism version 8.0 | GraphPad | www.graphpad.com |
| Human Microbiome Project (HMP) | Human Microbiome Project | https://hmpdacc.org/ |
| iHMP-IBD | Human Microbiome Project | https://ibdmdb.org/ |
| Mega 7 | Mega | http://www.megasoftware.net/ |
| Microsoft excel | Microsoft | N/A |
| Microsoft word | Microsoft | N/A |
| MultiGeneBlast | Medema et al., 2013 | http://multigeneblast.sourceforge.net/ |
| NEB Tm Calculator | NEB | https://tmcalculator.neb.com/#!/main |
| NEBaseChanger | NEB | http://nebasechanger.neb.com/ |
| Prokka (v1.12) | Victorian Bioinformatics Consortium | https://vicbioinformatics.com/software.prokka.shtml |
| REBASE | The Restriction Enzyme Database | http://rebase.neb.com/rebase/rebase.html |
| Scifinder | The American Chemical Society | https://scifinder.cas.org/ |
| Silva database | Max Planck Institute | https://www.arb-silva.de/ |
| SnapGene Viewer | SnapGene | http://www.snapgene.com/products/snapgene_viewer/ |
| XCMS online | The Scripps Research Institute | https://xcmsonline.scripps.edu/landing_page.php?pgcontent=mainPage |
| Other | ||
| ACQUITY UPLC BEH C18 column, 130Å, 1.7 μm, 2.1 mm × 100 mm | Waters | N/A |
| Agilent 6530 Q-TOF LC/MS equipment | Agilent | N/A |
| Anaerobic chamber | Coy Laboratory Products | N/A |
| Electroporation cuvette | USA Scientific | Cat. #9104-1050 |
| Electroporation System | BTX | N/A |
| Emulsiflex homogenizer | Avestin | N/A |
| 50 mL Tube Top Vacuum Filter System (0.22 μm) | Corning Life Sciences | Cat. #430320 |
| Kinetex C18 column, 1.7 μm, 2.1 mm × 100 mm | Phenomenex | N/A |
| Multiskan Sky Microplate Spectrophotometer | Thermo Fisher Scientific | N/A |
| Real-time quantitative PCR system (ABI 7500) | Applied Biosystems | N/A |
| Thermo Scientific Nanodrop 2000 | Thermo Scientific | N/A |
Identify gene transfer methods for non-model gut Firmicutes/Clostridia
Develop gene manipulation tools for non-model gut Firmicutes/Clostridia
Functional analysis of a microbiota gene baiH in the host with a complex microbiome
baiH mediates host bile acid pool, gut microbiome composition, and colon inflammation
Acknowledgments
We thank Dr. Michael Fischbach, and an anonymous healthy human fecal donor for their contribution of some bacterial strains we have screened in this study. We also thank Dr. Carl Nathan, Dr. Gregory Sonnenberg, members of the Guo Group, and an anonymous scientist for helpful suggestions and comments on the manuscript. Microbiome sequencing was performed by Genewiz and the Weill Cornell Microbiome Core Lab. The graphical abstract was created using BioRender. This work was supported by a pilot award from the American Gastroenterological Association, NIH grants 1DP2HD101401-01, the W.M. Keck Foundation, and the RAPP funding from the Weill Cornell Medicine (all to C.J.G.), NIH grants R01DK128257 (to R.L. and C.J.G) and R01DK114252 (to R.L.), and the National Institutes of Health (DK126871, AI151599, AI095466, AI095608, AI142213 to D.A.), LEO Foundation, Cure for IBD, Jill Roberts Institute, Sanders and Rosanne H. Silbermann Foundation (all to D.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
D.A. has contributed to scientific advisory boards at Genentech, Pfizer, Takeda, FARE, and the KRF. The other authors declare no competing interests. A provisional patent application has been filed by the Weill Cornell Center for Technology Licensing based on this work.
References
- Arab JP, Karpen SJ, Dawson PA, Arrese M, and Trauner M (2017). Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 65, 350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bencivenga-Barry NA, Lim B, Herrera CM, Stephen Trent M, and Goodman AL (2020). Genetic manipulation of wild human gut Bacteroides. Journal of Bacteriology 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander JM, Longman RS, Iliev ID, Sonnenberg GF, and Artis D (2017). Regulation of inflammation by microbiota interactions with the host. Nature Immunology 18, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al. (2015). Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, Mai C, Jin WB, Guo CJ, Violante S, et al. (2020). Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581, 475–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cañadas IC, Groothuis D, Zygouropoulou M, Rodrigues R, and Minton NP (2019). RiboCas: A Universal CRISPR-Based Editing Tool for Clostridium. ACS Synthetic Biology 8, 1379–1390. [DOI] [PubMed] [Google Scholar]
- Cani PD, Van Hul M, Lefort C, Depommier C, Rastelli M, and Everard A (2019). Microbial regulation of organismal energy homeostasis. Nature Metabolism 1, 34–46. [DOI] [PubMed] [Google Scholar]
- Chen ML, Takeda K, and Sundrud MS (2019). Emerging roles of bile acids in mucosal immunity and inflammation. Mucosal Immunology 12, 851–861. [DOI] [PubMed] [Google Scholar]
- Cho I, and Blaser MJ (2012). The human microbiome: at the interface of health and disease. Nature Reviews Genetics 13, 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorucci S, Biagioli M, Zampella A, and Distrutti E (2018). Bile acids activated receptors regulate innate immunity. Frontiers in Immunology 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach MA, and Sonnenburg JL (2011). Eating for two: How metabolism establishes interspecies interactions in the gut. Cell Host and Microbe 10, 336–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funabashi M, Grove TL, Wang M, Varma Y, McFadden ME, Brown LC, Guo C, Higginbottom S, Almo SC, and Fischbach MA (2020). A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 582, 566–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung TC, Bessman NJ, Hepworth MR, Kumar N, Shibata N, Kobuley D, Wang K, Ziegler CGK, Goc J, Shima T, et al. (2016). Lymphoid-Tissue-Resident commensal bacteria promote members of the IL-10 cytokine family to establish mutualism. Immunity 44, 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Bayona L, and Comstock LE (2019). Streamlined genetic manipulation of diverse Bacteroides and Parabacteroides isolates from the human gut microbiota. MBio 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C-J, Allen BM, Hiam KJ, Dodd D, Van Treuren W, Higginbottom S, Nagashima K, Fischer CR, Sonnenburg JL, Spitzer MH, et al. (2019). Depletion of microbiome-derived molecules in the host using Clostridium genetics. Science 366, eaav1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang S, Paik D, Yao L, Kim E, Jamma T, Lu J, Ha S, Nelson BN, Kelly SP, Wu L, et al. (2019). Bile acid metabolites control TH17 and Treg cell differentiation. Nature 576, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heap JT, Pennington OJ, Cartman ST, Carter GP, and Minton NP (2007). The ClosTron: A universal gene knock-out system for the genus Clostridium. Journal of Microbiological Methods 70, 452–464. [DOI] [PubMed] [Google Scholar]
- Heap JT, Pennington OJ, Cartman ST, and Minton NP (2009). A modular system for Clostridium shuttle plasmids. Journal of Microbiological Methods 78, 79–85. [DOI] [PubMed] [Google Scholar]
- Helmink BA, Khan MAW, Hermann A, Gopalakrishnan V, and Wargo JA (2019). The microbiome, cancer, and cancer therapy. Nature Medicine 25, 377–388. [DOI] [PubMed] [Google Scholar]
- Hmelo LR, Borlee BR, Almblad H, Love ME, Randall TE, Tseng BS, Lin C, Irie Y, Storek KM, Yang JJ, et al. (2015). Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nature Protocols 10, 1820–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong W, Zhang J, Cui G, Wang L, and Wang Y (2018). Multiplexed CRISPR-Cpf1-mediated genome editing in Clostridium difficile toward the understanding of pathogenesis of C. difficile Infection. ACS Synthetic Biology 7, 1588–1600. [DOI] [PubMed] [Google Scholar]
- Hur JK, Kim K, Been KW, Baek G, Ye S, Hur JW, Ryu SM, Lee YS, and Kim JS (2016). Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nature Biotechnology 34, 807–808. [DOI] [PubMed] [Google Scholar]
- Isenbarger TA, Carr CE, Johnson SS, Finney M, Church GM, Gilbert W, Zuber MT, and Ruvkun G (2008). The most conserved genome segments for life detection on earth and other planets. Origins of Life and Evolution of Biospheres 38, 517–533. [DOI] [PubMed] [Google Scholar]
- Jennert KCB, Tardif C, Young DI, and Young M (2000). Gene transfer to Clostridium cellulolyticum ATCC 35319. Microbiology 146, 3071–3080. [DOI] [PubMed] [Google Scholar]
- Johnston CD, Cotton SL, Rittling SR, Starr JR, Borisy GG, Dewhirst FE, and Lemon KP (2019). Systematic evasion of the restriction-modification barrier in bacteria. Proceedings of the National Academy of Sciences 116, 11454–11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaakoush NO (2015). Insights into the role of Erysipelotrichaceae in the human host. Frontiers in Cellular and Infection Microbiology 5, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D-J, Ridlon JM, Moore DR 2nd, Barnes S, and Hylemon PB (2008). Clostridium scindens baiCD and baiH genes encode stereo-specific 7alpha/7beta-hydroxy-3-oxo-delta4-cholenoic acid oxidoreductases. Biochimica et Biophysica Acta 1781, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A, Zeissig S, and Blumberg RS (2010). Inflammatory bowel disease. Annual Review of Immunology 28, 573–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller KL, Wall JD, and Chhabra S (2011). Methods for engineering sulfate reducing bacteria of the genus Desulfovibrio. Methods in Enzymology, 497, 503–517. [DOI] [PubMed] [Google Scholar]
- Kim SK, Kim H, Ahn WC, Park KH, Woo EJ, Lee DH, and Lee SG (2017). Efficient transcriptional gene repression by Type V-A CRISPR-Cpf1 from Eubacterium eligens. ACS Synthetic Biology 6, 1273–1282. [DOI] [PubMed] [Google Scholar]
- Klompe SE, Vo PLH, Halpin-Healy TS, and Sternberg SH (2019). Transposon-encoded CRISPR–Cas systems direct RNA-guided DNA integration. Nature 571, 219–225. [DOI] [PubMed] [Google Scholar]
- Kraal L, Abubucker S, Kota K, Fischbach MA, and Mitreva M (2014). The prevalence of species and strains in the human microbiome: a resource for experimental efforts. PLoS ONE 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, and Tamura K (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Molecular Biology and Evolution 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Gálvez EJC, Amend L, Almási É, Iljazovic A, Lesker TR, Bielecka AA, Schorr E, and Strowig T (2021). A versatile genetic toolbox for Prevotella copri enables studying polysaccharide utilization systems. The EMBO Journal 40:e108287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim B, Zimmermann M, Barry NA, and Goodman AL (2017). Engineered regulatory systems modulate gene expression of human commensals in the gut. Cell 169, 547–558.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, Andrews E, Ajami NJ, Bonham KS, Brislawn CJ, et al. (2019). Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Yao D, and Chen C (2013). 2-Hydrazinoquinoline as a derivatization agent for LC-MS-based metabolomic investigation of diabetic ketoacidosis. Metabolites 3, 993–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister KN, Bouillaut L, Kahn JN, Self WT, and Sorg JA (2017). Using CRISPR-Cas9-mediated genome editing to generate C. difficile mutants defective in selenoproteins synthesis. Scientific Reports 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema MH, Takano E, & Breitling R (2013). Detecting sequence homology at the gene cluster level with multigeneblast. Molecular Biology and Evolution, 30(5), 1218–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermelstein LD, Welker NE, Bennett GN, and Papoutsakis ET (1992). Expression of cloned homologous fermentative genes in Clostridium acetobutylicum ATCC 824. Bio/Technology 10, 190. [DOI] [PubMed] [Google Scholar]
- Mimee M, Tucker AC, Voigt CA, and Lu TK (2015). Programming a human commensal bacterium, Bacteroides thetaiotaomicron, to sense and respond to stimuli in the murine gut microbiota. Cell Systems 1, 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito M, Ross Belvin B, Shoji M, Gui Q, and Lewis JP (2021). Insertional inactivation of Prevotella intermedia Oxyr results in reduced survival with oxidative stress and in the presence of host cells. Microorganisms 9, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm NW, De Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, et al. (2014). Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158, 1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peluso EA, Scheible M, Ton-That H, and Wu C (2020). Genetic Manipulation and Virulence Assessment of Fusobacterium nucleatum. Current Protocols in Microbiology 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Koo BM, Patino R, Heussler GE, Hearne CC, Qu J, Inclan YF, Hawkins JS, Lu CHS, Silvis MR, et al. (2019). Enabling genetic analysis of diverse bacteria with Mobile-CRISPRi. Nature Microbiology 4, 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdy D, O’Keeffe TAT, Elmore M, Herbert M, McLeod A, Bokori-Brown M, Ostrowski A, and Minton NP (2002). Conjugative transfer of Clostridial shuttle vectors from Escherichia coli to Clostridium difficile through circumvention of the restriction barrier. Molecular Microbiology 46, 439–452. [DOI] [PubMed] [Google Scholar]
- Pyne ME, Bruder M, Moo-Young M, Chung DA, and Chou CP (2014). Technical guide for genetic advancement of underdeveloped and intractable Clostridium. Biotechnology Advances 32, 623–641. [DOI] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, and Glöckner FO (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research 41, 591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt N, Duncan SH, Young P, Belenguer A, McWilliam Leitch C, Scott KP, Flint HJ, and Louis P (2014). Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME Journal 8, 1323–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon JM, Kang DJ, and Hylemon PB (2006). Bile salt biotransformations by human intestinal bacteria. Journal of Lipid Research 47, 241–259. [DOI] [PubMed] [Google Scholar]
- Ridlon JM, Harris SC, Bhowmik S, Kang DJ, and Hylemon PB (2016). Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 7, 22–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooks MG, and Garrett WS (2016). Gut microbiota, metabolites and host immunity. Nature Reviews Immunology 16, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, and Trinchieri G (2017). Microbiota: A key orchestrator of cancer therapy. Nature Reviews Cancer 17, 271–285. [DOI] [PubMed] [Google Scholar]
- Salyers AA, Shoemaker N, Cooper A, D’Elia J, and Shipman JA (1999). 8 Genetic Methods for Bacteroides Species. Methods in Microbiology 29, 229–249. [Google Scholar]
- Seemann T (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. [DOI] [PubMed] [Google Scholar]
- Sheridan PO, Martin JC, Minton NP, Flint HJ, O’Toole PW, and Scott KP (2019). Heterologous gene expression in the human gut bacteria Eubacterium rectale and Roseburia inulinivorans by means of conjugative plasmids. Anaerobe 59, 131–140. [DOI] [PubMed] [Google Scholar]
- Shiver AL, Culver R, Deutschbauer AM, and Huang KC (2021). Rapid ordering of barcoded transposon insertion libraries of anaerobic bacteria. Nature Protocols 2021 16:6 16, 3049–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha SR, Haileselassie Y, Nguyen LP, Tropini C, Wang M, Becker LS, Sim D, Jarr K, Spear ET, Singh G, et al. (2020). Dysbiosis-Induced secondary bile acid deficiency promotes intestinal inflammation. Cell Host and Microbe 27, 659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, Geva-Zatorsky N, Jupp R, Mathis D, Benoist C, et al. (2020). Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature 577, 410–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker J, Ladha A, Gardner Z, Schmid-Burgk JL, Makarova KS, Koonin E. v., and Zhang F (2019). RNA-guided DNA insertion with CRISPR-associated transposases. Science 364, 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taketani M, Zhang J, Zhang S, Triassi AJ, Huang YJ, Griffith LG, and Voigt CA (2020). Genetic circuit design automation for the gut resident species Bacteroides thetaiotaomicron. Nature Biotechnology, 38, 962–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Lowder LG, Zhang T, Malzahn AA, Zheng X, Voytas DF, Zhong Z, Chen Y, Ren Q, Li Q, et al. (2017). A CRISPR–Cpf1 system for efficient genome editing and transcriptional repression in plants. Nature Plants 2017 3:3 3, 1–5. [DOI] [PubMed] [Google Scholar]
- Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M, Beghini F, Manara S, Karcher N, Pozzi C, et al. (2019). Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nature Medicine 25, 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, and Gordon JI (2007). The Human Microbiome Project. Nature 449, 804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vital M, Howe A, and Tiedje J (2014). Revealing the Bacterial Synthesis Pathways by Analyzing (Meta) Genomic Data. MBio 5, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo PLH, Ronda C, Klompe SE, Chen EE, Acree C, Wang HH, and Sternberg SH (2021). CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nature Biotechnology 39, 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller MC, Bober JR, Nair NU, and Beisel CL (2017a). Toward a genetic tool development pipeline for host-associated bacteria. Current Opinion in Microbiology 38, 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, et al. (2012). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60. [DOI] [PubMed] [Google Scholar]
- Whitaker WR, Shepherd ES, and Sonnenburg JL (2017). Tunable expression tools enable single-cell strain distinction in the gut microbiome. Cell 169, 538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirbel J, Pyl PT, Kartal E, Zych K, Kashani A, Milanese A, Fleck JS, Voigt AY, Palleja A, Ponnudurai R, et al. (2019). Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nature Medicine 25, 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods C, Humphreys CM, Rodrigues RM, Ingle P, Rowe P, Henstra AM, Köpke M, Simpson SD, Winzer K, and Minton NP (2019). A novel conjugal donor strain for improved DNA transfer into Clostridium spp. Anaerobe 59, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, Watanabe H, Masuda K, Nishimoto Y, Kubo M, et al. (2019). Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nature Medicine 25, 968–976. [DOI] [PubMed] [Google Scholar]
- Yang X, Xu M, and Yang ST (2016). Restriction modification system analysis and development of in vivo methylation for the transformation of Clostridium cellulovorans. Applied Microbiology and Biotechnology 100, 2289–2299. [DOI] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. (2013). Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101. [DOI] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, Van Der Oost J, Regev A, et al. (2015). Cpf1 Is a single RNA-guided endonuclease of a Class 2 CRISPR-Cas system. Cell 163, 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zong W, Hong W, Zhang ZT, and Wang Y (2018). Exploiting endogenous CRISPR-Cas system for multiplex genome editing in Clostridium tyrobutyricum and engineer the strain for high-level butanol production. Metabolic Engineering 47, 49–59. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wang J, Cheng Q, Zheng X, Zhao G, and Wang J (2017). Multiplex gene regulation by CRISPR-ddCpf1. Cell Discovery 2017, 3, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Gong Z, Du X, Tian C, Wang L, Zhou J, Xu C, Chen Y, Cai W, and Wu J (2018). Deoxycholic acid-mediated sphingosine-1-phosphate receptor 2 signaling exacerbates DSS-induced colitis through promoting cathepsin b release. Journal of Immunology Research 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Karberg M, and Lambowitz AM (2003). Targeted and random bacterial gene disruption using a group II intron (targetron) vector containing a retrotransposition-activated selectable marker. Nucleic Acids Research 31, 1656–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Sailani MR, Contrepois K, Zhou Y, Ahadi S, Leopold SR, Zhang MJ, Rao V, Avina M, Mishra T, et al. (2019). Longitudinal multi-omics of host–microbe dynamics in prediabetes. Nature 569, 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Schematic view of a mixed-conjugation strategy to identify the Clostridia that stably maintains exogenous DNA. Ten pGM vectors, each harboring a single rep ori (9 Clostridia specific and 1 rep ori-less), were separated into three sets and mixed-conjugated to a Clostridia recipient.
(B) Multiplex PCR strategy was used to identify which rep ori-contained plasmid was introduced into which Gram+ Clostridia strain in mixed-conjugation. For the mixed-conjugation with set I, primers pMTL_laz_diag_F (universal forward primer) + pGM-ABCM_rep_R_1500bp + pGM-BBCM_rep_R_1000bp + pGM-CBCM_rep_R_2000bp were used for diagnostic PCR. A 1.5 kb (or 1.0 kb, or 2.0 kb) PCR band would be seen if pGM-ABCM (or BBCM, or CBCM) is uptaken by the Clostridia microbe. The primers for the set II and set III mixed-conjugation are shown in Table S2.
(C) Distribution of Clostridial rep oris tested in this study based on phylogeny. The phylogenetic tree was constructed using the 16S rRNA sequences of the 42 gut microbes (38 Firmicutes/Clostridia, 2 Enterococcus, and 2 Actinobacteria) with a compatible rep ori identified in this study. The sequences were aligned using Clustal Omega, and a neighbor-joining tree was constructed with a bootstrap test of 5000.
(D) Multiple Clostridia and promoters are screened, and the red arrow indicates the original promoter Ppmtl-catP used in the Clostridia conjugation vectors (one representative promoter screening result is shown). Error bar: standard deviation.
The numbering of the strains corresponds to the strain information shown in Table S1.
(A) qPCR results showing that CRISPRi-dCpf1 precisely and efficiently suppressed lacZα expression in Gram-positive Clostridia and Bifidobacterium microbes, using both dCpf-1-only and gRNA-only as controls.
(B) qPCR results showing that CRISPRi-dCpf1 precisely and efficiently suppressed lacZα expression in other Gram-positive Clostridia microbes and Gram-negative microbes from other phyla, using dCpf-1-only as control.
For each strain shown in (A), conjugation was conducted with E. coli harboring plasmids with dCpf1-lacZα (dCpf1), gRNA-lacZα (gRNA), or gRNA-dCpf1-lacZα (dCpf1+gRNA). For each strain shown in (B), conjugation was conducted with E. coli harboring plasmids with dCpf1-lacZα (dCpf1, Con), or gRNA-dCpf1-lacZα (dCpf1+gRNA, Mut). Then transconjugants were cultured, and RNA was extracted and reverse transcribed to cDNA. Quantitative PCR (qPCR) was used to assess the expression of lacZα after normalizing to the expression of the 16S rRNA gene of each strain. Error bar: standard deviation. The numbering of the strains corresponds to the strain information shown in Table S1.
(A) CRISPRi-dCpf1 precisely and efficiently suppressed bcat expression in 11 Clostridia and 1 Bifidobacterium microbes with sequenced genomes. Quantitative PCR (qPCR) was used to assess bcat expression after normalizing to the 16S rRNA gene expression of each strain.
(B) The S25 ΔmmdA mutant and control strains, (C) the S117 croA knockdown mutant and the control strains, (D) and the S107 porA knockdown mutant and the control strains colonized the germ-free mice (n = 3 or 4 per group) at comparable levels. Colonized germ-free mice were maintained on a standard diet and supplied with water containing 15 µg/mL thiamphenicol and 2 mg/mL Sweet N Low sweetener. We calculated colony-forming units (CFU) in fecal pellets to estimate the density of intestinal colonization. n.s.: not statistically significant.
Error bar: standard deviation. The numbering of the strains corresponds to the strain information shown in Table S1.
The prevalence (A) and percent relative abundance (B) of gut Clostridia commensals harboring the bai operon were examined using a large publicly available 16S rRNA dataset (Yatsunenko et al., 2012), including stool samples from 528 individuals. S122 and its closely related relatives are more prevalent than C. hylemonae, C. scindens, and D. sp. D27_3, but less than that of C. hiranonis (A). S122 and its closely related strains are the most abundant 7α-dehydroxylating commensal in this cohort (B). For (A), the prevalence data were analyzed using Fisher’s exact test, and mean ± SEM was plotted. For (B), the relative abundance data were first analyzed using the D’Agostino & Pearson test for normality. The relative abundance of S122 was compared to other commensals using the Mann-Whitney test. A Median with a 95% confidence interval (CI) was plotted. The asterisk in (A) and (B) indicates p-value < 0.0001 (****).
(C) Successful insertion of baiH was determined by amplifying DNA using primers flanking the target gene baiH. The expected size for the control is ~2kb, and ~4kb for the S122 ΩbaiH mutant.
(D) Metabolomics analyses of bile acids in S25 + S122 ΩbaiH mutant (Mut) and S25 + S122 control (Con) co-colonized germ-free mice. Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**), n.s.: not statistically significant, n.d.: not detected.
(E) Co-colonization of germ-free mice with S122 and 55 other genetically targetable microbes identified in this study. Successful colonization of S122 was determined by CFU and the detection of DCA in LCMS, and colonization of other strains was confirmed by 16S rRNA sequencing.
The numbering of the strains corresponds to the strain information shown in Table S1.
(A-B) The Chao 1 index (A) and Shannon index (B) of the fecal microbiota of the SPF mice colonized with the control (Con) and the ΩbaiH mutant (Mut). Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**).
(C-D) Relative abundances of Bacteroidetes and Proteobacteria in the stool microbiome of the control and ΩbaiH mutant colonized SPF mice. ΩbaiH mutant (Mut) colonized mice harbor significantly higher abundances of Bacteroidetes and lower abundances of Proteobacteria compared to the control group (Con). Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**).
(E) Relative abundances of gut microbial taxa (at the family level) in the stool microbiome of the control and ΩbaiH mutant colonized SPF mice. Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**).
(F-G) In nonIBD human stools, the fecal DCA level is weakly associated with the relative abundance of microbes in the Erysipelotrichaceae family and with that of the B. eggerthii species.
(H) The density of total intestinal bacteria of SPF control mice compared with SPF mice colonized with S122 control and ΩbaiH mutant. SPF control mice (SPF) were maintained with a standard diet and water, and S122 colonized SPF mice with standard diet and water with 15 µg/mL thiamphenicol and 10 µg/mL erythromycin. We calculated colony-forming units in fecal pellets on TSAB agar plates to estimate the density of intestinal bacteria. Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**), n.s.: not statistically significant.
(A) Macroscopic observations of the colon length of SPF mice colonized with S122 control (Con) and ΩbaiH mutant (Mut) post-DSS treatment.
(B) Macroscopic observations of colon length of germ-free mice colonized with S122 control (Con) and ΩbaiH mutant (Mut) post-DSS treatment.
(C) Quantification of fecal lipocalin-2 (LCN-2) in SPF mice colonized with S122 control (Con) and ΩbaiH mutant (Mut) post-DSS treatment on the day-5, 6, 7, and day-sacrifice, the amount of lipocalin-2 was significantly higher in the control group. Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**).
(D) Quantification of fecal lipocalin-2 (LCN-2) in germ-free mice colonized with S122 control (Con) and ΩbaiH mutant (Mut) post-DSS treatment at the day-sacrifice, no significant difference was found. Data are shown as mean ± SEM. Student’s T-test was performed, n.s.: not statistically significant.
(E) Comparison of colonic expression of inflammatory genes in SPF mice colonized with S122 control (Con) and ΩbaiH mutant (Mut) post-DSS treatment. Expression of inflammatory genes was normalized to Hprt1 and shown as the fold change relative to the mutant group. Colonic expression of inflammatory genes was significantly higher in the control group. Data are shown as mean ± SEM. Student’s T-test was performed, and asterisk indicates p-value < 0.05 (*) or < 0.01 (**).
(F) Density of intestinal colonization of SPF mice supplemented with low dose antibiotics (15 µg/mL thiamphenicol and 10 µg/mL erythromycin in drinking water) (n = 4 per group) by the S122 control (Con) and ΩbaiH mutant (Mut) (Fig. 6A). The colony-forming units (CFU) were calculated before DSS treatment and on day 3 and day 6 of DSS treatment. Student’s T-test was performed. n.s.: not statistically significant.
(G) Relative abundances (by 16S rRNA sequencing) of Erysipelotrichaeceae in the stool microbiome of the S122 control (Con) and ΩbaiH mutant (Mut) colonized SPF mice (n =4 per group) at day 3 and day 6 of DSS treatment (Fig. 6A). Student’s T-test was performed. The asterisk indicates p-value < 0.01 (**) n.s.: not statistically significant.
(H) Fecal DCA levels of the SPF mice (grey bar), and the SPF mice supplemented with low dose antibiotics (n = 4 per group) colonized with the S122 control (Con) strain (before and at day 3 of DSS treatment) (Fig. 6A). Student’s T-test was performed. n.s.: not statistically significant.
(I) The relative abundances of S122 in the control and mutant group (n = 4 per group) of the DSS germ-free experiment (Fig. 6B) were assessed by 16S rRNA sequencing and were comparable. Student’s T-test was performed. n.s.: not statistically significant.
(J) Targeted metabolomics analyses of the stool bile acid (BA) compositions of the S122 control (Con) and ΩbaiH mutant (Mut) colonized gnotobiotic mice (n = 4 per group) at day 3 after the DSS treatment (Fig. 6F). Student’s T-test was performed. The asterisk indicates p-value < 0.01 (**) n.s.: not statistically significant, n.d.: not detected.
(A) Density of intestinal colonization (assessed by CFU) of gnotobiotic mice colonized with S122 control (Con) and ΩbaiH mutant (Mut), and total intestinal bacterial load in the 10-member community as shown in Fig. 7D. We calculated colony-forming units in fecal pellets to estimate the density of intestinal colonization. n.s.: not statistically significant.
(B) Density of intestinal colonization (assessed by CFU) of gnotobiotic mice colonized with S122 control (Con) and ΩbaiH mutant (Mut), and total intestinal bacterial load in the 3-member community as shown in Fig. S7G. We calculated colony-forming units in fecal pellets to estimate the density of intestinal colonization. n.s.: not statistically significant.
(C-D) Metabolomics analyses of fecal bile acids in feces of two Bacteroides (bac) microbes and seven Erysipelotrichaceae (Ery) microbes + S122 control (Con) / S122 ΩbaiH mutant (Mut) co-colonized germ-free mice before DSS treatment (C) and on day 3 of DSS treatment (D). Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**), n.s.: not statistically significant, n.d.: not detected.
(E-F) Metabolomics analyses of bile acids in feces of two Bacteroides (bac) microbes + S122 control (Con) / S122 ΩbaiH mutant (Mut) co-colonized germ-free mice before DSS treatment (E) and on day 3 of DSS treatment (F). Data are shown as mean ± SEM. Student’s T-test was performed, and the asterisk indicates p-value < 0.05 (*) or < 0.01 (**), n.s.: not statistically significant, n.d.: not detected.
(G-L) DSS-induced murine colitis model was applied to the gnotobiotic mice colonized with a synthetic consortium consisting of the genetically tagged S122 control (Con) or ΩbaiH mutant (Mut) along with 2 Bacteroides (Bac) microbes only. Mice were colonized with the consortium for at least two weeks, followed by 2.5% DSS for 9 days. The disease state was monitored by weight loss (H), hematoxylin and eosin (H&E) staining of the distal colon (I), colon shortening (J), fecal lipocalin-2 (K), and daily hematochezia score (L).
Data shown in (H, I, J, K, L) are representations of n = 4 mice per group replicated in two independent experiments. In (H), % of starting weight was calculated by normalizing weights at sacrifice to starting weight. In (J) and (K), colon length and LCN2 data were analyzed using unpaired two-tailed Student’s T-test. In (H) and (L), % of starting weight and hematochezia score data were analyzed using Two-way ANOVA followed by the Bonferroni post hoc test (n=4). Data are shown as mean ± SEM. The asterisk indicates p-value < 0.05 (*), < 0.01 (**) or < 0.001 (***). The numbering of the strains corresponds to the strain information shown in Table S1.
Table S1. Bacterial strains, their culture conditions, and their gene transfer methods, related to Figures 1, 2, 3, and STAR Methods.
Table S2. Primers and plasmids used in this study, related to Figures 2, 3, 4, and STAR Methods.
Table S3. Recipes for all the culture media used in this study and their ingredient details, related to Figure 1 and STAR Methods.
Table S4. 16S rRNA gene sequencing of fecal samples from SPF mice colonized with Faecalicatena contorta S122 (S122) control (Con) and ΩbaiH mutant (Mut), related to Figure 5.
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.