

Abstract
Few tools are available for noninvasive imaging of synapses in the living mammalian brain. Current paradigms require the use of genetically modified mice or viral delivery of genetic material to the brain. To develop an alternative chemical approach, utilizing the recognition of synaptic components by organic small molecules, we designed an imaging-based, high-content screen in cultured cortical neurons to identify molecules based on their colocalization with fluorescently tagged synaptic proteins. We used this approach to screen a library of ~7000 novel fluorescent dyes, and identified a series of compounds in the xanthone family that exhibited consistent synaptic labeling. Follow-up studies with one of these compounds, CX-G3, demonstrated its ability to label acidic organelles and in particular synaptic vesicles at glutamatergic synapses in cultured neurons and murine brain tissue, indicating the potential of this screening approach to identify promising lead compounds for use as synaptic markers, sensors, and targeting devices.
Keywords: Synapse, synaptic vesicle, high-content screening, xanthone, fluorescence, imaging probe
Graphical Abstract

Synapses are the fundamental building blocks of neural circuits, and synapse loss and dysfunction are underlying causes of many neuropsychiatric and neurodegenerative diseases, including autism, schizophrenia, and Alzheimer’s disease.1–5 Indeed, the ability to monitor synapse density and function would be extremely useful for the diagnosis and treatment of brain disorders; however, no tools are currently available to image specific synapse types in the living human brain (e.g., glutamatergic versus GABAergic synapses). Current efforts to image synapses and synaptic function in animal models have focused on genetically encoded markers and sensors (e.g., fluorescent proteins, genetically encoded calcium indicators, and channelrhodopsins).6–10 Although these are powerful and widely used approaches, they also require genetically altered organisms or viral delivery of genetic material to the brain. We therefore seek to provide an alternative “chemical” approach. Not only could small molecule chemical probes potentially cross the blood-brain barrier and reach synaptic targets in a noninvasive manner, but such markers could themselves function as optical sensors of synaptic activity or function. Furthermore, small molecule synaptic markers could serve as targeting elements for the delivery of other sensors (e.g., chemicals, proteins, and nanoparticles) specifically to synapses, unlocking exciting opportunities to monitor or modulate synaptic function in multiple imaging modalities.
Only a few small molecule probes have been developed to label synapses in living neurons or brain tissue. These include FM dyes, fluorescent false neurotransmitters (FFNs), and fluorescent analogues of the neurotoxin MPP+, such as APP +.11–14 FM dyes label recycling synaptic vesicles, allowing for visualization of active presynaptic terminals irrespective of synapse type,11 but they cannot be used in vivo due to their high level of background staining. FFNs function as fluorescent tracers of native neurotransmitters, acting as both presynaptic markers and functional reporters, and enabling imaging of synaptic vesicle fusion at individual presynaptic release sites in a number of species and experimental systems, including acute mammalian brain tissue.13,15–19 However, FFN probes are largely limited to monoaminergic synapses. APP+ also labels synaptic vesicles in dopaminergic terminals, but its utility as a synaptic marker in vivo is limited by the fact that it also localizes to organelles such as mitochondria, and is likely to have neurotoxic side effects.14 Therefore, the discovery of chemical probes for other synapse types, particularly glutamatergic synapses, which comprise the majority of synapses in the brain, would provide the neuroscience community with novel imaging tools, and the medical community with potential probes for diagnosing and treating human neurological diseases.
To date, synaptic small molecule probes, such as FFNs, have been created using a rational design approach.18 Although high-throughput screening has been amply validated as a powerful approach for drug discovery, this method has not yet been harnessed for synaptic probe discovery due to the lack of structurally diverse collections of organic dyes. However, such a library of novel fluorescent dyes has recently been generated based on diversification of common fluorophore structural cores spanning a wide spectroscopic range (~7000 dyes),20 and successfully utilized for the discovery of a number of cell labeling reagents (e.g., neuronal and neural stem cell probes).21–24 We have therefore developed an image-based, high-throughput screening platform to detect synaptic labeling, which was then used to screen this dye library for potential synaptic markers. We find that a group of compounds in the xanthone family exhibit high colocalization with known presynaptic proteins. Follow-up studies with the leading candidate, compound CX-G3, demonstrates its ability to label glutamatergic terminals in culture, brain slice, and in vivo, indicating the potential of xanthone compounds as synaptic markers.
RESULTS AND DISCUSSION
Our major objectives in this study were to establish a robust platform for screening fluorescent dyes in cultured neurons, and to utilize this platform to identify promising compounds for use as synaptic markers (hit compounds). For our screening assay, we used cultured rat cortical neurons plated in 96-well format and lentivirally transduced at 3 days in vitro (DIV) with one of two fluorescently tagged, synaptic vesicle-associated proteins: GFP-Synapsinla (GFP-Syn) or mCherry-Rab3 (mCh-Rab3).25–27 Initial optimization experiments, performed in 14–16 DIV cultures following synapse maturation, indicated that >60% of neurons expressed these proteins, and that synaptic GFP-Syn and mCh-Rab3 puncta were readily detectable in images acquired on the HTS imaging system (Figure 1a,b). Colocalization via immunostaining of GFP-Syn or mCh-Rab3-expressing cultures with antibodies against known synaptic (SV2, VGLUT1, Synaptophysin, and Synapsin) and nonsynaptic (MAP2 and LAMP1) proteins (see Methods for details) reliably distinguished synaptic markers (fractional colocalization values between 0.6 and 0.8; fraction of GFP-Syn/mCh-Rab3 puncta containing the immunostaining/compound signal; see Methods) from those that were not (colocalization values between 0.11 and 0.19; Table S1 and Figure 1c,d). The colocalization algorithm was further tested in live GFP-Syn-expressing neurons using FM4–64, a lipophilic dye taken up by recycling synaptic vesicles and an established marker for active presynaptic boutons.11 FM4–64 exhibited a lower signal-to-noise ratio than immunostained synaptic proteins due to its nonspecific labeling of cellular membranes, and thus served as a good test case (and the only available chemical tool-based positive control) for the colocalization algorithm in live neurons. Although the fractional colocalization values for FM4–64 were lower than those for immunostained synaptic proteins, the average value (0.50 ± 0.15, mean ± SD; Figure 1E) was still significantly higher than those seen for nonsynaptic proteins (Table S1; it should be noted that FM4–64 requires stimulation of cortical neuronal culture with 90 mM KCl, followed by washing to remove excess dye).
Figure 1.
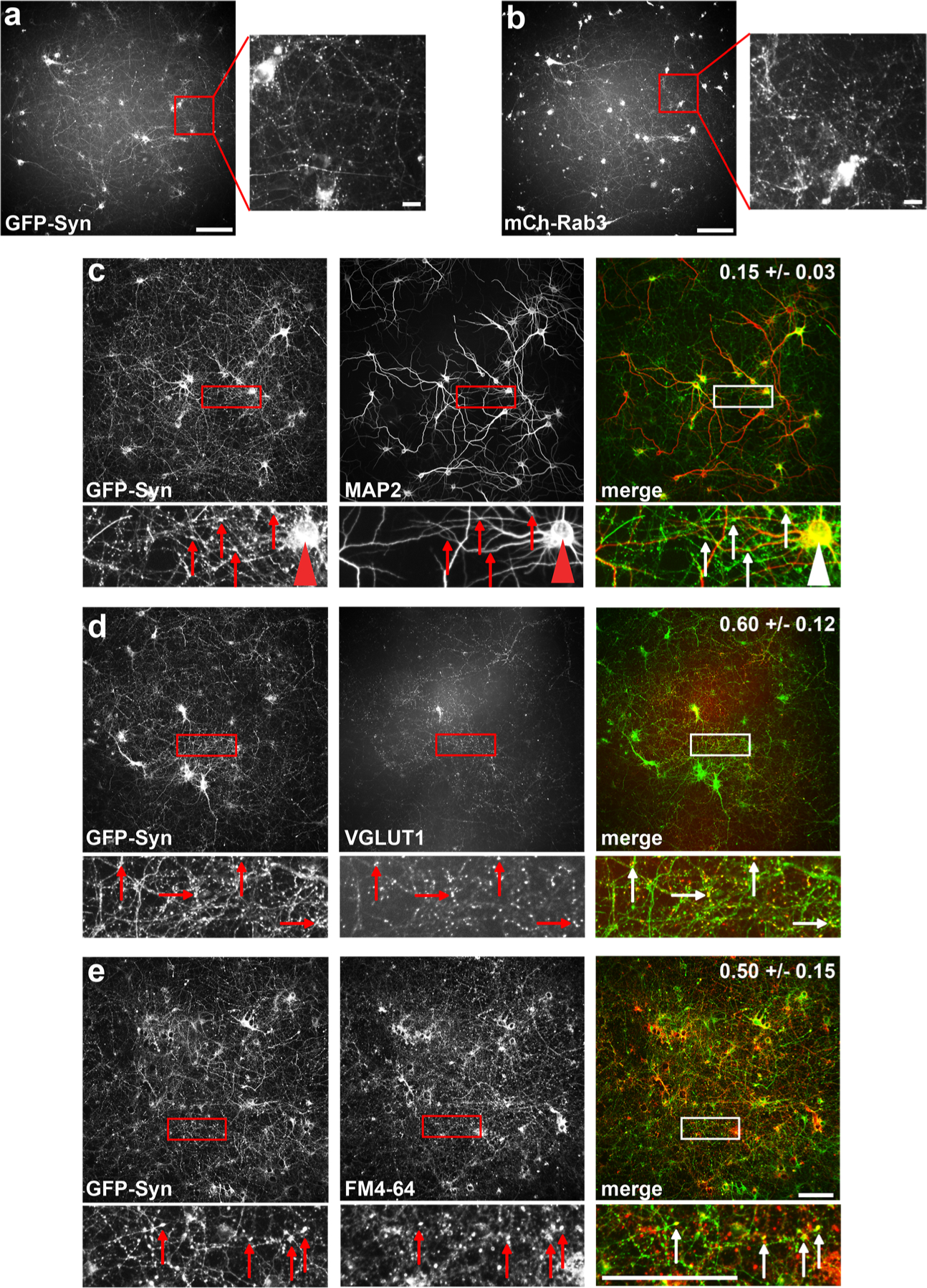
Development of high-throughput screening platform to detect synaptic labeling. (a) Original and zoomed images of GFP-Synapsin-expressing cortical neurons in 96-well plate format, acquired via high-throughput screening microscope. Size bar is 100 μm for original image, 10 μm for zoomed image. (b) Same as (a), but neurons are expressing mCh-Rab3. (c–e) Original and zoomed images showing colocalization of GFP-Syn with MAP2, VGLUT1, and FM4–64. The fraction of GFP-Syn puncta that colocalize with each marker is indicated on the merged image. Arrows indicate GFP-Syn puncta and corresponding immunostaining of the marker; arrowhead in (c) indicates a neuronal cell body. Size bars are 100 μm.
With an optimized platform for detecting synaptic labeling, we next screened the fluorescent dye library (6,992 compounds) in 14–16 DIV neurons. Prior to screening, compounds were divided into groups based on their excitation/emission spectra, and each group was screened together with neurons expressing the spectrally compatible synaptic marker (GFP-Syn or mCh-Rab3; Table S2; see Methods for details). For each compound group, average colocalization with the synaptic marker was calculated, and hits were defined as compounds with colocalization values > 2.5 standard deviations above the group mean. This selection method led to the identification of 53 hits across 11 of the compound groups (Table S3). Images of each hit were manually inspected to evaluate synaptic labeling. Sixteen were excluded due to bleed-through into the marker channel, as some of the compound groups exhibited broader excitation and/or emission spectra (indicated by ** in Table S3). Of the remaining 37 hits, the 22 most promising (based on highly punctate labeling that colocalized with GFP-Syn; Figure 2b,c) were from a single plate (Q4) of the compound group designated “DAPI:DAPI” due to its excitation/emission profile in the 4’,6-diamidino-2-phenylindole (DAPI) range (~360 nm excitation, 460 nm emission).
Figure 2.
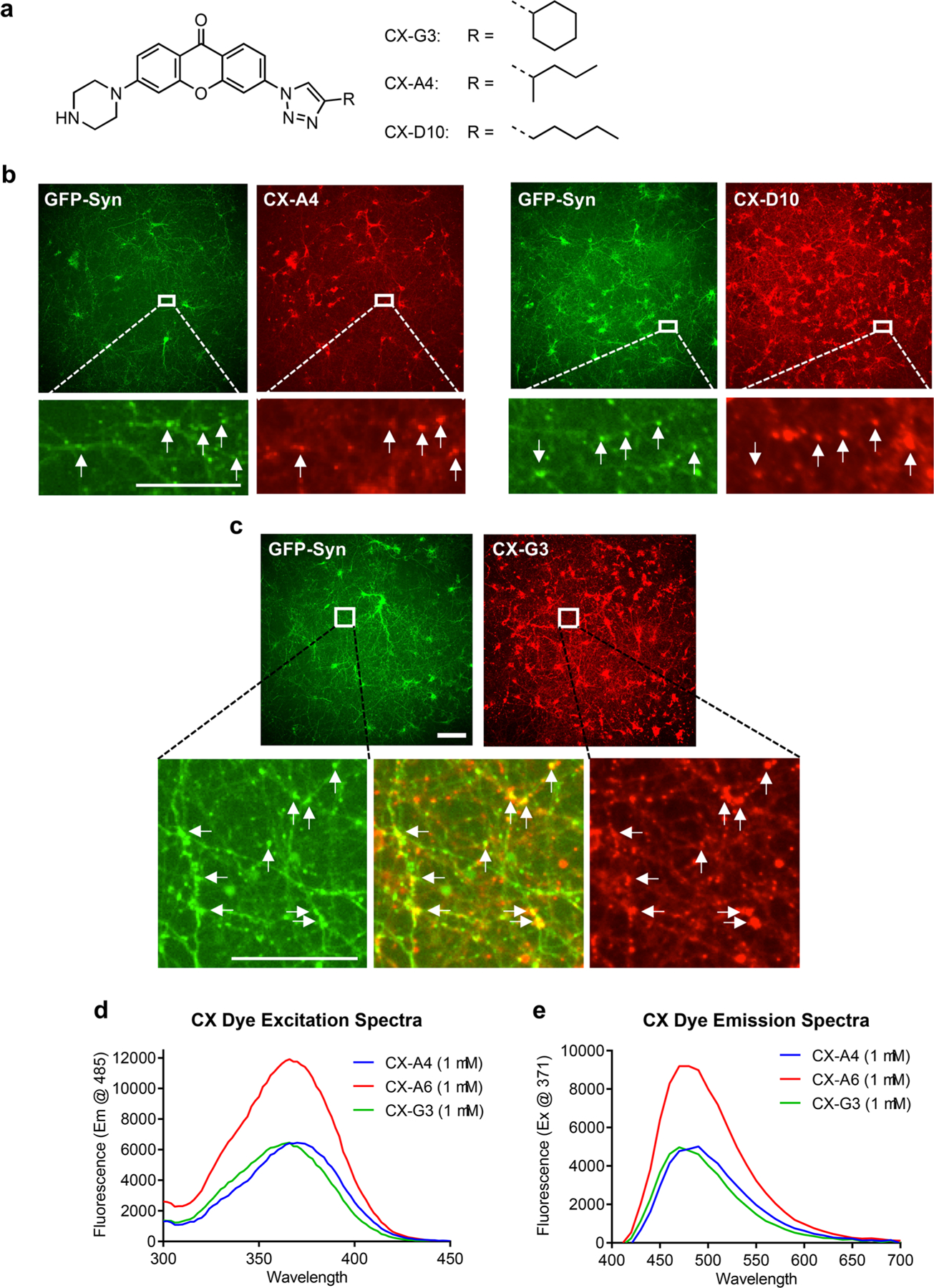
CX xanthone compounds exhibit colocalization with GFP-Syn synaptic marker. (a) Structure ofxanthone core and the R groups from CX-G3, A4, and D10 compounds. (b) Original and zoomed screening images of CX-A4 and CX-D10 with GFP-Syn in cortical neurons. Arrows indicate sites of colocalization. Size bar is 100 μm for original image, 25 μm for zoomed image. (c) Original and zoomed images of CX-G3, which has the highest GFP-Syn colocalization of the CX compounds. Arrows indicate sites of colocalization. Size bar is 100 μm for original image, 50 μm for zoomed image. (d,e) Excitation and emission spectra of select CX compounds in DMSO. Excitation spectra were measured with emission at 485 nm; emission spectra with excitation at 371 nm.
Colocalization values of Q4 plate compounds were consistent across three independent experiments (~0.30–0.40 for the top 10 hits; Table S4), and similar to that of FM dye with GFP-Syn (0.50 ± 0.15). Dyes from the Q4 plate are based on the xanthone scaffold (Figure 2a),28 with peak excitation and emission values between 360 and 380 nm and 480–520 nm, respectively (Figure 2d,e). Acetyl derivatives of this click xanthone (CX) library were previously reported to selectively label mouse embryonic stem cells and to exhibit minimal cellular toxicity over multiple days of incubation,28 indicating the suitability of this family for use as probes in living cells.
To understand which features of CX compounds enable synaptic labeling, we took advantage of the fact that Q4 plate compounds, which differ only in their side chains (Figure S1a), exhibited variable degrees of colocalization with GFP-Syn (from ~5% to 40%; Table S4). Analysis of side chain lipophilicity revealed that there was a bell-shaped correlation (R2 = 0.37) between calculated log P values of the side chain and number of puncta that colocalized with GFP-Syn (Figure S1b). Compounds containing aliphatic groups within the LogP range of 2–4 exhibited the most punctate labeling, while those with polar aromatic and hydroxy groups or long aliphatic side chains exhibited less punctate labeling (Figure S1). Clearly, compounds with moderately lipophilic side chains were better able to penetrate neuronal membranes and label intracellular compartments such as synaptic vesicles, while those with nonlipophilic or highly lipophilic side chains could not, presumably due to either their inability to cross the plasma membrane or their strong associations with lipid bilayers.
We next performed follow-up studies with CX-G3, the compound with the highest average colocalization with GFP-Syn (Table S4). CX-G3 showed strong absorption in the near-UV extending into the visible spectral region, with a maximum at 360 nm (molar absorptivity of 21,000 M−1cm−1) and emission maximum at 484 nm (Φf = 0.50, 4.0 ns lifetime) (Figure 3a–c). Moreover, the excitation and emission peaks of CX-G3 were strongly pH-dependent, with 3-fold higher fluorescence intensity at acidic pH, such as that found within synaptic vesicles (pH ~ 5.0), compared to the neutral pH found in the cellular cytosol or extracellular space (pH ~ 7.4, Figure 3d). This pH-dependence is a result of protonation of the outer piperazine amino group (pKa ~ 6.5), which eliminates the photoinduced electron transfer quenching of the xanthone by the amine lone pair.29,30 Based on these properties, we hypothesized that the moderately high lipophilicity of CX-G3 allows the compound to cross membranes and enter intracellular compartments, while its pH-dependent fluorescence selectively enhances its signal within acidic organelles. Moreover, increasing the degree of protonation of CX-G3 under acidic conditions likely traps the compound within low pH compartments. Before testing these concepts in cultured hippocampal neurons, we first performed serial dilutions to determine the optimal concentration of CX-G3 based on its signal-to-noise ratio and colocalization with GFP-Syn (Figure 3e–g; see Methods). As both parameters were maximized around 0.5–1 μM, we used 0.5 μM for subsequent in vitro experiments.
Figure 3.

Photophysical properties and optimal concentration of CX-G3. (a) Absorption spectrum of CX-G3 (10 μM) in aqueous solution at room temperature. (b) Fluorescence excitation spectrum (blue; λem = 480 nm) and emission spectrum (red; λex = 360 nm) of CX-G3 (10 μM) in aqueous solution at room temperature. (c) Fluorescence decay trace of CX-G3 in aqueous solution monitored at 480 nm with pulsed excitation at 335 nm (red). Instrument’s response function is in black, (d) Graph depicting pH-dependence of CX-G3 photon excitation and emission. (e) Sample images from serial dilution of CX-G3 in cultured cortical neurons in 96-well plate format. Concentration is indicated on the left. Size bar is 50 μm. (f) Graph of CX-G3 concentration vs intensity (expressed as fraction of G3 intensity at GFP-Syn puncta to average intensity over the entire plate). Higher values correspond to higher signal-to-noise ratio (values calculated from 3 wells/condition, confirmed by two independent dilution experiments). (g) Graph of CX-G3 concentration vs colocalization with GFP-Syn (values calculated from 3 wells/condition, confirmed by two independent dilution experiments).
To test whether CX-G3 labels acidic intracellular compartments, we examined its colocalization with LysoTracker Red (Thermofisher), a dye that labels acidic organelles in live cells, in 14–16 DIV hippocampal neurons. The two dyes exhibited relatively high colocalization (0.45 ± 0.06; Figure 4a,b), indicating that CX-G3 indeed labels acidic compartments. In contrast, CX-G3 exhibited lower colocalization with the mitochondrion-selective dye MitoTracker (0.29 ± 0.08, Figure 4a,b), indicating its preference for acidic organelles such as SVs or lysosomes. Confirming this concept, we found that the fluorescence of CX-G3 puncta decreased by nearly 85% following intracellular deacidification with 50 mM NH4Cl (Figure 4c).
Figure 4.
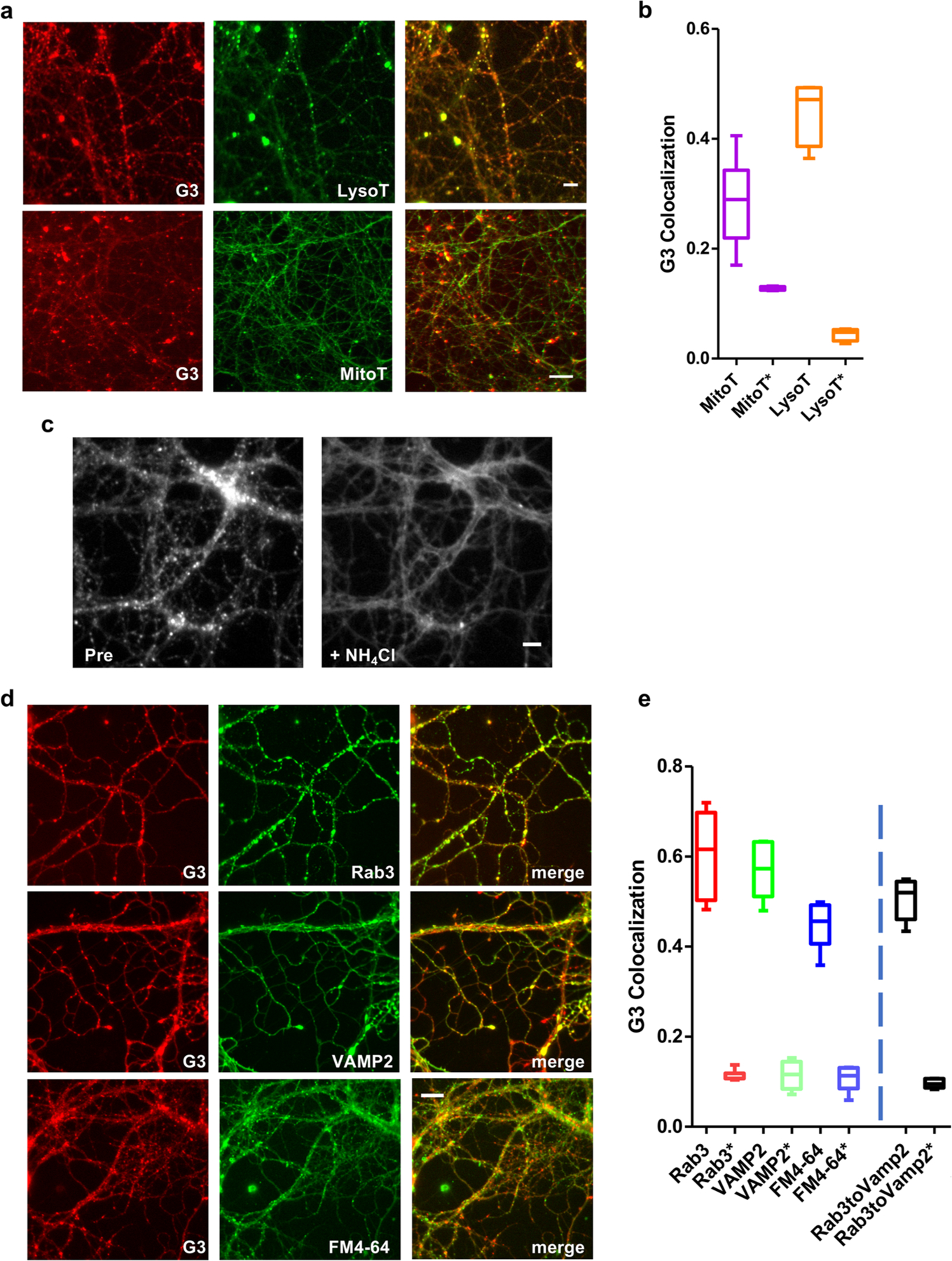
CX-G3 is a pH-sensitive dye that labels synaptic vesicles in cultured neurons. (a) Images showing colocalization of CX-G3 with LysoTracker Red and MitoTracker Red in hippocampal neurons. Size bar is 10 μm. Note that MitoTracker images were acquired using a different imaging system (see Methods). (b) Graph of CX-G3 colocalization with LysoTracker and MitoTracker, defined as fraction of CX-G3 puncta which contain fluorescent marker. Colocalization of G3 with these dyes in randomized images (depicted with *) are included for comparison (n = 2–4 images per batch, acquired from two independent batches of neurons). (c) Images of CX-G3-labeled neurons before (pre) and after deacidification with 50 mM NH4CL (d) Images showing colocalization of CX-G3 with mCh-tagged SV markers (Rab3 or VAMP2) or FM4–64 to label active terminals. Size bar is 10 μm. (e) Graph of CX-G3 colocalization with SV markers in cultured hippocampal neurons, defined as fraction of CX-G3 puncta which contain fluorescent marker. Colocalization of G3 with Rab3, VAMP2, and FM in randomized images (depicted with *), and colocalization of mCh-Rab3 with endogenous VAMP2 via immunostaining are included for comparison (n = 2–4 images per batch, acquired from two independent batches of neurons).
To evaluate the ability of CX-G3 to label SVs, we examined its colocalization with the SV-associated proteins VAMP2 and Rab3 (Figure 4d,e). Since the dye is not fixable, we also performed these experiments by live imaging in 14–16 DIV neurons lentivirally transduced on 3 DIV with mCh-tagged VAMP2 or Rab3. Again, we found that CX-G3 exhibited a high degree of colocalization with both VAMP2-mCh (0.57 ± 0.06) and mCh-Rab3 (0.61 ± 0.09; Figure 4d,e), comparable to colocalization values measured for mCh-Rab3 and endogenous VAMP2 (0.51 ± 0.05; Figure 4d,e). By comparison, Lysotracker Red, previously used as a marker of SV pools,31 exhibited much lower colocalization with SV markers (0.26 ± 0.05 with GFP-Syn; Figure S1c) than CX-G3, indicating that CX-G3 labels a wider range of acidic vesicles. CX-G3 also strongly colocalized with FM4–64 in hippocampal cultures (0.45 ± 0.05; Figure 4d,e). However, unlike FM4–64, CX-G3 did not exhibit dramatic destaining in response to 10 Hz electrical stimulation for 1 min (Figure S2a–d), indicating a more stable association with SV-associated lipids. Supporting this concept, we found that mild extraction of neurons with 0.01% Triton X-100 completely eliminated CX-G3 signal (Figure S2e). However, additional experiments are needed to fully characterize the mechanism of how CX-G3 labels SVs.
While a chemical marker capable of rapid, in vitro synaptic labeling has useful applications, such a marker would have even greater value if applicable in more complex experimental systems like acute brain slices or in vivo preparations. We therefore evaluated the ability of CX-G3 to label glutamatergic presynaptic terminals in hippocampal brain slices. For these experiments, slices were prepared from mice injected in the dorsal hippocampus with AAV8 to express mCh-Synaptophysin in glutamatergic neurons of the hippocampus. Following a 30 min incubation with 2 μM CX-G3, images were acquired by two-photon microscopy, where peak excitation of CX-G3 was found to be 720–740 nm (Figure S2f), and colocalization with mCh-Synaptophysin was assessed (Figure 5B–E). Not only did CX-G3 exhibit punctate labeling in the stratum radiatum near the CA1 layer that was similar to its labeling pattern in neuronal cultures, but it also exhibited a high degree of colocalization with mCh-Synaptophysin (0.66 ± 0.08; Figure 5E). As a final test, we assessed the ability of CX-G3 to label synapses in vivo. Here, a cranial window was prepared above the somatosensory cortex, CX-G3 was perfused onto the surface of the tissue for 30 min at 5 μM, and the cortical layer 1 (0–100 μm depth; Figure 5A) was imaged via two-photon microscopy. As in hippocampal slice, CX-G3 exhibited a punctate labeling pattern throughout Layer 1, an area of dense synaptic terminals (Figure 5F). Together, these experiments demonstrate the ability of CX-G3 to rapidly and stably label presynaptic terminals in a variety of preparations (hippocampal cultures, brain slices, and in vivo). Given the similarity of other compounds on the Q4 plate to CX-G3, and their high colocalization with GFP-Syn in screening experiments (Table S4), it is likely that multiple members of the CX xanthone library share this property.
Figure 5.
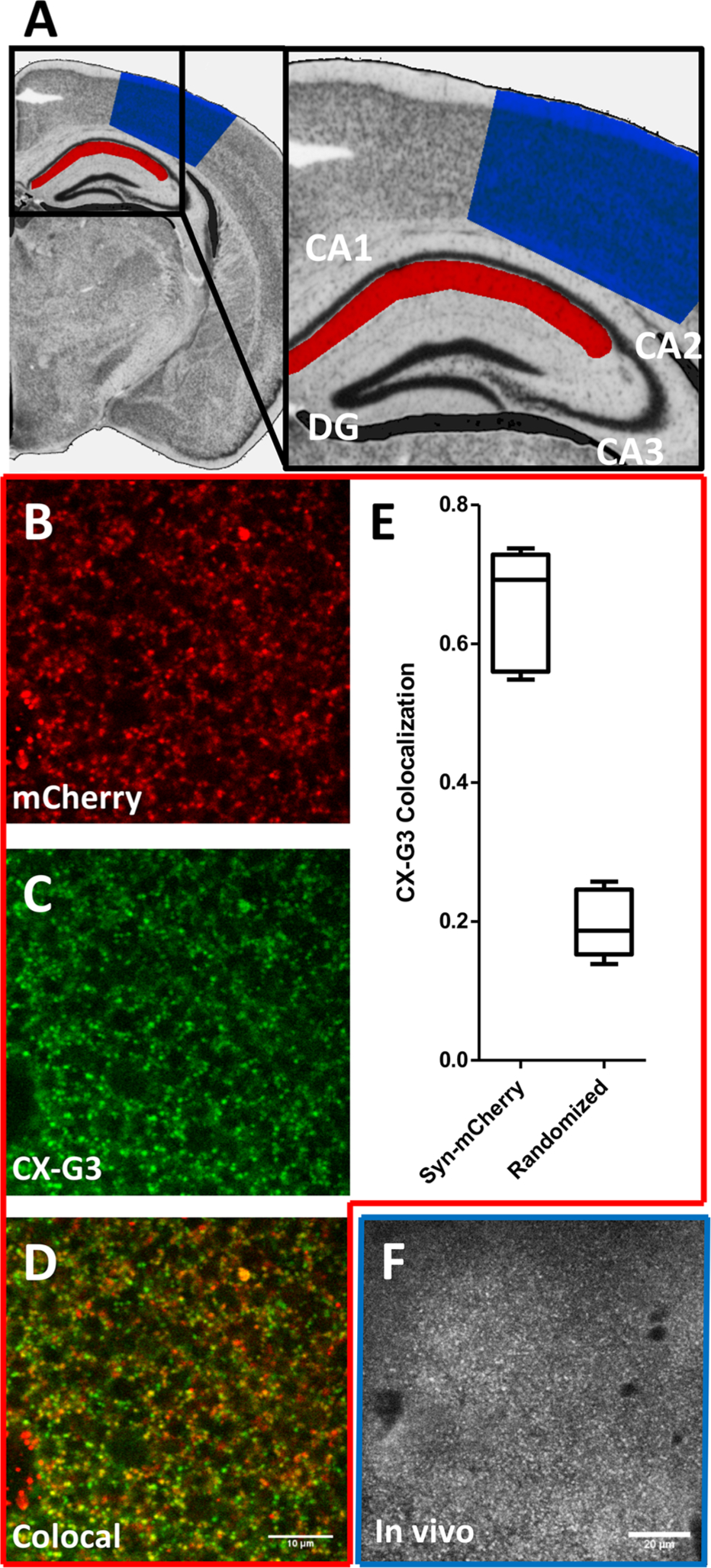
CX-G3 labels presynaptic terminals in hippocampal slice and somatosensory cortex. (A) Coronal section of the mouse brain (from The Mouse Brain Library, www.mbl.org 35) showing the sites of mCh-Synaptophysin expression in the hippocampus (red) used for acute brain slice experiments, and of CX-G3 in vivo imaging in the somatosensory cortex (blue). (B–D) Images showing CX-G3 and mCh-Synaptophysin colocalization in hippocampal slice (stratum radiatum near area CA1). Size bar is 10 μm. (E) Quantification of mCh-Syph colocalization with CX-G3. Colocalization in randomized images is shown for comparison. (F) Images of CX-G3 in somatosensory cortex of a living rodent. Size bar is 20 μm.
One caveat of CX xanthone labeling is that these compounds do not exclusively label neurons or synapses. Indeed, many of these dyes also exhibit punctate, perinuclear labeling in glia (Figure S2g). However, in the case of CX-G3, the intensity of perinuclear glial labeling was decreased to near-background levels by dilution to 0.5 μM (data not shown), and glial labeling did not interfere with the visualization of synapses in any of our preparations. We also found that most CX dyes labeled neuronal cell bodies in addition to synapses. The high colocalization of CX-G3 with LysoTracker, and its decreased fluorescence upon NH4Cl deacidification, indicates that this labeling reflects dye uptake into acidic organelles such as late endosomes and lysosomes. However, the majority of this “off-target” labeling will likely occur in cell bodies, which are usually in anatomically distinct layers/areas of the brain in acute slice and in vivo preparations.
One unresolved question is whether xanthone dyes such as CX-G3 could cross the blood-brain barrier, which would boost their utility as noninvasive synaptic markers. Our experiments in acute brain slice and in vivo suggest that the CX-G3 compound penetrates brain tissue well, and its physicochemical properties suggest that it may indeed penetrate the blood-brain barrier. Another question remains whether additional synaptic markers are present in the dye library. We plan to rescreen compounds that we were forced to disregard due to their fluorescence overlap with our FP-tagged synaptic markers, following replacement of the GFP/mCh tag with fluorescent proteins in the blue and far-red excitation/emission spectra (i.e., BFP, TagRFP657). Given the diversity of the library, we expect that additional synaptic probes could emerge.
In summary, we have used image-based, high-content screening of a novel fluorescent dye library to identify a family of small molecules, xanthone-piperidine CX compounds, that label presynaptic terminals. These compounds are lipophilic, pH-sensitive dyes, making them well-suited to serve as acidic compartment fluorescent markers. Moreover, they exhibit consistently higher rates of colocalization with SVs than Lysotracker, the dye most frequently used for labeling acidic organelles in living cells. In the absence of further development, these compounds can be used as crude synaptic markers in a variety of preparations (cultured neurons, brain slices, in vivo).
METHODS
DNA Constructs and AAV Production.
Lentiviral constructs to express EGFP-Synapsin1a, mCh-Rab3, and VAMP2-EGFP/mCh have been previously described.25,32 For AAV experiments, Rattus norvegicus synaptophysin (GenBank accession number NM_012664.3) tagged at its C-terminus with mCherry was synthesized (Genewiz) and subcloned into the pAAV-CaMKIIa-EGFP expression vector (gift from Ed Boyden; Addgene plasmid #64545) at the BamHI and EcoRI sites, replacing the EGFP cassette. This construct was sent to the University of North Carolina viral vector core for custom AAV8 production.
Neuronal Culture and Lentiviral Transduction.
Cortical neurons were prepared from E18 Sprague–Dawley rat embryos of both sexes, dissociated in TrypLE Express (Fisher/Life Technologies) for 20 min, washed 3X in Hank’s Balanced Salt Solution (Sigma), and plated in Neurobasal medium with B27 supplement and Glutamax (all Fisher/Life Technologies) at a density of 10 000 cells/well in 96-well plates coated with poly-L-lysine (Sigma). On 3 days in vitro (DIV), neurons were transduced with lentivirus (2 μl/well), prepared as previously described (Sheehan 2016), to express either EGFP-Synapsin1a or mCh-Rab3. Hippocampal neurons were prepared in the same way, but plated onto 22 × 22 mm2 coverslips at a density of 250 000 neurons per coverslip. Coverslips were lentivirally transduced with mCh-Rab3 or VAMP2-mCh on 5–7 DIV.
Immunofluorescence on HTS Microscope.
GFP-Synapsin or mCh-Rab3-expressing cortical cultures in 96-well plates were fixed with 4% formaldehyde in PBS for 15 min, washed 3× in PBS, then incubated with primary antibodies in blocking buffer (2% glycine, 2% BSA, 0.2% gelatin, 50 mM NH4Cl in 1× PBS) for 1 h. The following antibodies were used: MAP2, LAMP1, SV2A, VGLUT1, and GFP. Following 3× PBS washes, cells were incubated with Alexa Fluor 488 or 568 conjugated secondary antibodies for 1 h, washed 3× in PBS, and imaged using the high-throughput screening microscope (IN Cell Analyzer; GE Healthcare, 20× objective) in the Columbia Genome Center High-throughput Screening Core. Colocalization of FP-tagged Synapsin or Rab3 with the synaptic and nonsynaptic markers was calculated as described below (colocalization algorithm).
Screening.
The compound library was formatted into groups of 96-well plates based on excitation/emission spectra (Table 1). Screening was performed in batches of 12–15 plates, with each compound group screened in the same batch. The compounds (prepared in DMSO as 100 μM stock solutions) were added to the medium of 14–16 DIV cortical neurons via automated liquid handling system (Cell Explorer; PerkinElmer) for a final concentration of 2 μM. Following 20 min incubation at 37 °C, plates were washed 3× at room temperature in normal Tyrodes solution (119 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 30 mM glucose, 25 mM Hepes pH 7.4), and images acquired at room temperature in four fields of view/well using the high-throughput screening microscope (IN Cell Analyzer; GE Healthcare, 20× objective) and CCD camera (2048 × 2048 pixels) in the Columbia Genome Center HTS facility. Following screening, plates were fixed with 4% formaldehyde in PBS, washed once in PBS, and stored in the dark at 4 °C.
Colocalization Algorithm and Hit Selection.
To test the ability of the image analysis software (IN Cell Developer Toolbox) to accurately assess synaptic colocalization, we first performed immunostaining of GFP-Synapsin or mCh-Rab-expressing neurons with a series of antibodies against synaptic and nonsynaptic markers (Table S1). The vesicle segmentation algorithm of IN Cell Developer Toolbox (v.1.9) was used to threshold images based on average size of GFP-Synapsin and mCh-Rab3 puncta (1–2 pixels; only objects in range of 0.5–5 μm2 selected) and intensity (average intensity 2–2.5× over background). Thresholding of images to select synaptic puncta and exclude background signals was optimized empirically, using images of neurons immunostained with the GFP or mCh antibody, which should exhibit ~100% colocalization with GFP-Syn and mCh-Rab3, respectively. For each channel, thresholded objects were counted, and colocalization between channels calculated based on number of overlapping pixels. Colocalization values were expressed as the fraction of GFP-Syn or mCh-Rab3 puncta that exhibited overlap with the immunostained proteins, and later with each compound. For each group of compounds, average colocalization with the synaptic marker was calculated, and hits were defined as compounds with colocalization values > 2.5 standard deviations above the group mean.
Serial Dilutions.
Serial dilution was performed in 96-well plates of neurons or glia for the CX Q4 plate of compounds, using the following concentrations: 2.0, 1.0, 0.5, 0.25, 0.125, 0.0625, and 0.03125 μM. Signal-to-noise ratio of the compounds was determined by comparing their average intensity at GFP-Syn puncta to their average intensity across the entire plate.
Determination of Excitation/Emission Spectra and Lip-ophilicity.
Spectral analysis of the compounds was performed on BioTek Synergy H1 Hybrid reader. Compounds were in DMSO or normal Tyrodes buffer at pH ranging from 3 to 9 to yield final concentration of 1 μM. Excitation spectra were measured by recording emission at 485 nm; emission spectra were recorded by exciting at 371 nm. Theoretic log P values of the side chains of CX Q4 library compounds were calculated using PerkinElmer ChemDraw 14.0 software.
Photophysical Characterization.
UV–vis absorption spectra were recorded on an Agilent 8453 spectrometer. Fluorescence spectra were recorded on a Fluorolog-3P fluorometer (HORIBA Jobin Yvon). Fluorescence quantum yields were determined using 9,10-diphenylan-thracene as standard (Φref = 0.95 in ethanol).33 Sample and standard solutions with matching absorbances at the excitation wavelength were used, and the quantum yields were corrected for the refractive index of the solvent. Fluorescence lifetimes were measured by time correlated single photon counting on an OB920 spectrometer (Edinburgh Analytical Instruments) in conjunction with a pulsed LED (335 nm) as excitation light source.
Live Imaging in Cultured Hippocampal and Cortical Neurons.
Imaging experiments of CX-G3 with Lysotracker Red, NH4Cl, mCh-Rab3, VAMP2-mCh, and FM4–64 were performed with 14–18 DIV hippocampal neurons using a custom chamber designed for liquid perfusion and electrical stimulation. For Lysotracker colocalization, neurons were placed in normal Tyrodes solution with 0.5 μM CX-G3 and 10nM Lysotracker Red for 10 min, washed for 2 min in Tyrodes solution, and then imaged. Tyrodes solution with 50 mM NH4Cl was subsequently perfused in to deacidify intracellular compartments, and additional images acquired in the same field of view. For mCh-Rab3 and VAMP2-mCh imaging, neurons were labeled with CX-G3 as described above and imaged. For FM4–64 loading, neurons were incubated with FM4–64 (15 μM) in high potassium Tyrodes solution (like normal Tyrodes except for 90 mM KCl, 31.5 mM NaCl) for 45 s to stimulate FM dye uptake, then washed for ~5 min in normal Tyrodes to eliminate excess dye prior to 2 min incubation with 0.5 μM CX-G3, 2 min washing in Tyrodes, and image acquisition. Images were acquired with a 40× objective (Neofluar, NA 1.3) or a 63× objective (Neofluar, NA 1.4) on an epifluorescence microscope (Axio Observer Z1, Zeiss) with Colibri LED light source, EMCCD camera (Hamamatsu) and Zen 2012 (blue edition) software. MitoTracker experiments were performed in 19–21 DIV cortical neurons after incubation with 100 nM MitoTracker (MitoTracker Deep Red FM; Thermofisher) for 30 min. Imaging was performed using a Leica DMI4000 imaging system.
Quantification of CX-G3 Colocalization with Synaptic Markers in Cultured Hippocampal Neurons.
CX-G3 images were first background subtracted (3 μm rolling ball) using ImageJ software (National Institutes of Health). Clearly visible cell bodies were then manually excluded from the image analysis. CX-G3-positive puncta were then identified using the Multiple Thresholds plug-in (created by Damon Poburko, Simon Fraser University, Burnaby, BC, Canada) for ImageJ with the selection criteria: >1 standard deviation above background, 6–50 pixels in size, and min circularity of 0.6. CX-G3 puncta were then considered positive for the respective synaptic markers if at least 6 pixels (>0.95 μm2) of the ROI colocalized with fluorescence > 1 standard deviation above background from the corresponding fluorescent marker. As a control, the analysis was repeated on randomized image pairs. For the MitoTracker images, which were collected on a separate microscope with different resolution, this analysis was not possible. Instead these images were analyzed using the Just Another Colocalization Plugin34 setting the threshold to the >1 standard deviation and recording the Manders’ overlap coefficient.
Stereotaxic Injection.
Three-month old male mice (C57BL/6J; Jackson Laboratories) were anesthetized with isoflurane (2–5%) and placed in a stereotaxic apparatus (Digital Just for Mice Stereotaxic Instrument), and buprenorphine (0.1 mg/kg) was administered subcutaneously for analgesia. The head was fixed, and the skull was exposed. Burr holes were made and a glass micropipet (Drummond Scientific) slowly lowered into the dorsal hippocampus at −2.0 mm anteroposterior, ±1.8 mm mediolateral, and −1.8 mm dorsoventral relative to bregma. Pipets were formed with 20 μm diameter tips using a P-2000 laser puller (Sutter Instrument). AAV8-CAMKII-Synapto-physin-mCherry virus (300 nL) was pressure-injected into each hemisphere. After injection, the pipet remained in place for 10 min and was then slowly retracted. The mice were placed on a heating pad (TR-200; Fine Science Tools) throughout the duration of the surgery. After injection, the scalp was sutured, and saline and analgesic meloxicam (4mg/kg) were administered subcutaneously. The mice were placed under heating lamps during recovery from anesthesia.
Preparation of Acute Murine Brain Slices.
Mice were decapitated, and acute 300 μm thick coronal slices were cut on a Leica VT1200 vibratome (Leica Microsystems) at 4 °C and allowed to recover for 1 h in oxygenated (95% O2, 5% CO2) artificial cerebrospinal fluid (ACSF) containing (in mM): 125 NaCl, 2.5 KCl, 26 NaHCO3, 0.3 KH2PO4, 2.4 CaC2, 1.3 MgSO4, 0.8 NaH2PO4 10 glucose (pH 7.2–7.4, 292–296 mOsm/L). After 30 min of recovery at 37 °C, hippocampal slices were then moved to room temperature and used at this temperature for all imaging experiments.
Application and Two-Photon Imaging of CX-G3.
CX-G3 (2 μM) was then bath incubated with the slices for 30 min and then transferred to an imaging chamber (QE-1, Warner Instruments, Hamden, CT) and held in place with a platinum wire and nylon string custom-made holder and superfused (1 mL/min) with oxygenated ACSF. Slices were washed in the perfusion chamber for 10 min prior to imaging. CX-G3-positive structures were visualized at depths of at least 25 μm from the slice surface using a Prairie Ultima Multiphoton Microscopy System (Prairie Technologies, Middleton, WI) with a titanium-sapphire Chameleon Ultra II laser (Coherent) equipped with a 60× 0.9 NA water immersion objective. CX-G3 was excited at 760 nm and 440–500 nm light was collected. For mCherry, 760 nm light also works for two-photon excitation, and 585–630 nm light was collected. Images were captured in 16-bit 112 × 112 μm2 field of view at 512 × 512 pixel2 resolution with a dwell time of 10 μs/pixel using Prairie View 5 software.
In Vivo Imaging of CX-G3.
Mice were anesthetized with isoflurane (1–4%), and depth of anesthesia was monitored by toe-pinch and observation of respiratory rate every 5 min. The animal was then placed in a stereotactic apparatus on top of a heating pad and Puralube vet ointment was applied to the eyes. Marcaine (2 mg/kg) with saline was injected subcutaneously as a local anesthetic. The skull was then exposed and a 3 mm cranial imaging window was constructed over the somatosensory cortex with a high speed drill (Midwest Stylus 360). Application of collagen foam (Avitene Ultrafoam, Bard Davol) was used to limit any bleeding. CX-G3 (5 μM) diluted in ACSF was then loaded into a glass micropipet attached to a Nanoject II (Drummond Scientific) and injected into the somatosensory cortex. Then 100 nL of CX-G3 was injected into the brain at coordinates AP: −1.0 mm and ML: 3.0 mm, at several depths (DV: 100, 50, and 20 μm) with 3 min of delay per depth. The glass pipet was then withdrawn after a 5 min delay and a plastic ring was glued around the window with Loctite 454 to hold ACSF for imaging. A metal microscope headmount was also glued to the bone surface with Loctite 454. The mouse was then injected with ketamine (100 mg/kg) and xylazine (10 mg/kg) i.p. and weaned off of isoflurane. The mouse was then headfixed under the two-photon microscope for imaging, and body temperature was maintained with a heating pad. Two-photon images were collected as described above, except that, instead of a single plane, a 100 μm z-stack (1 μm/step) was collected from the surface of the brain tissue. The image depicted is from 50 μm from the surface.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Viktoriya Zhuravleva in the Waites lab for technical help with live imaging experiments in hippocampal cultures and Veronica Birdsall for help with data analysis.
Funding
This work was supported by the NIH Grant R21MH104803 to C. L. Waites and D. Sames We also acknowledge support by a Core Facility seed grant from Columbia University Medical Center and NIH Grant R01NS080967 to C. L. Waites, NIH R01MH108186 to D. Sames and D. Sulzer, a Swedish Research Council Fellowship to A. Beskow, and Portuguese Foundation for Science & Technology PhD fellowships to J. Vaz-Silva and T. Meira (PD/BD/105938/2014; PD/BD/113700/2015, respectively).
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneur-o.7b00263.
Methods; colocalization of GFP-Syn and mCh-Rab3 with synaptic and nonsynaptic markers; organization of fluorescent dyes for screening; list of hit compounds; colocalization of CX compounds with GFP-synapsin; CX compound lipophilicity vs number of puncta; properties of CX-G3 labeling in neurons (PDF)
REFERENCES
- (1).van Spronsen M, and Hoogenraad CC (2010) Synapse pathology in psychiatric and neurologic disease. Curr. Neurol. Neurosci. Rep. 10, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Waites CL, and Garner CC (2011) Presynaptic function in health and disease. Trends Neurosci. 34, 326–337. [DOI] [PubMed] [Google Scholar]
- (3).Sheng M, Sabatini BL, and Sudhof TC (2012) Synapses and Alzheimer’s disease. Cold Spring Harbor Perspect. Biol. 4, a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Takahashi RH, Nagao T, and Gouras GK (2017) Plaque formation and the intraneuronal accumulation of beta-amyloid in Alzheimer’s disease. Pathol. Int. 67, 185–193. [DOI] [PubMed] [Google Scholar]
- (5).Bourgeron T (2015) From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci. 16, 551–563. [DOI] [PubMed] [Google Scholar]
- (6).Li H, Foss SM, Dobryy YL, Park CK, Hires SA, Shaner NC, Tsien RY, Osborne LC, and Voglmaier SM (2011) Concurrent imaging of synaptic vesicle recycling and calcium dynamics. Front. Mol. Neurosci. 4, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, and Kim DS (2013) Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gross GG, Junge JA, Mora RJ, Kwon HB, Olson CA, Takahashi TT, Liman ER, Ellis-Davies GC, McGee AW, Sabatini BL, Roberts RW, and Arnold DB (2013) Recombinant probes for visualizing endogenous synaptic proteins in living neurons.Neuron 78, 971–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Saunders A, Johnson CA, and Sabatini BL (2012) Novel recombinant adeno-associated viruses for Cre activated and inactivated transgene expression in neurons. Front. Neural Circuits 6, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Dreosti E, and Lagnado L (2011) Optical reporters of synaptic activity in neural circuits. Exp Physiol 96, 4–12. [DOI] [PubMed] [Google Scholar]
- (11).Hoopmann P, Rizzoli SO, and Betz WJ (2012) Imaging synaptic vesicle recycling by staining and destaining vesicles with FM dyes. Cold Spring Harb Protoc 2012, 77–83. [DOI] [PubMed] [Google Scholar]
- (12).Sames D, Dunn M, Karpowicz RJ Jr., and Sulzer D (2013) Visualizing neurotransmitter secretion at individual synapses. ACS Chem. Neurosci. 4, 648–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Merchant P, Sulzer D, and Sames D (2015) Synaptic optical imaging platforms: Examining pharmacological modulation of neurotransmitter release at discrete synapses. Neuropharmacology 98, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Karpowicz RJ Jr., Dunn M, Sulzer D, and Sames D (2013) APP+, a fluorescent analogue of the neurotoxin MPP+, is a marker of catecholamine neurons in brain tissue, but not a fluorescent false neurotransmitter. ACS Chem. Neurosci. 4, 858–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Pereira DB, Schmitz Y, Meszaros J, Merchant P, Hu G, Li S, Henke A, Lizardi-Ortiz JE, Karpowicz RJ Jr., Morgenstern TJ, Sonders MS, Kanter E, Rodriguez PC, Mosharov EV, Sames D, and Sulzer D (2016) Fluorescent false neurotransmitter reveals fUnctionally silent dopamine vesicle clusters in the striatum. Nat. Neurosci. 19, 578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Freyberg Z, Sonders MS, Aguilar JI, Hiranita T, Karam CS, Flores J, Pizzo AB, Zhang Y, Farino ZJ, Chen A, Martin CA, Kopajtic TA, Fei H, Hu G, Lin YY, Mosharov EV, McCabe BD, Freyberg R, Wimalasena K, Hsin LW, Sames D, Krantz DE, Katz JL, Sulzer D, and Javitch JA (2016) Mechanisms of amphetamine action illuminated through optical monitoring of dopamine synaptic vesicles in Drosophila brain. Nat. Commun. 7, 10652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gubernator NG, Zhang H, Staal RG, Mosharov EV, Pereira DB, Yue M, Balsanek V, Vadola PA, Mukherjee B, Edwards RH, Sulzer D, and Sames D (2009) Fluorescent false neurotransmitters visualize dopamine release from individual presynaptic terminals. Science 324, 1441–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lee M, Gubernator NG, Sulzer D, and Sames D (2010) Development of pH-responsive fluorescent false neurotransmitters. J. Am. Chem. Soc. 132, 8828–8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Rodriguez PC, Pereira DB, Borgkvist A, Wong MY, Barnard C, Sonders MS, Zhang H, Sames D, and Sulzer D (2013) Fluorescent dopamine tracer resolves individual dopaminergic synapses and their activity in the brain. Proc. Natl. Acad. Sci. U. S. A. 110, 870–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Vendrell M, Zhai D, Er JC, and Chang YT (2012) Combinatorial strategies in fluorescent probe development. Chem. Rev. 112, 4391–4420. [DOI] [PubMed] [Google Scholar]
- (21).Er JC, Leong C, Teoh CL, Yuan Q, Merchant P, Dunn M, Sulzer D, Sames D, Bhinge A, Kim D, Kim SM, Yoon MH, Stanton LW, Je SH, Yun SW, and Chang YT (2015) NeuO: a Fluorescent Chemical Probe for Live Neuron Labeling. Angew. Chem. Int. Ed. 54, 2442–2446. [DOI] [PubMed] [Google Scholar]
- (22).Leong C, Lee SC, Ock J, Li X, See P, Park SJ, Ginhoux F, Yun SW, and Chang YT (2014) Microglia specific fluorescent probes for live cell imaging. Chem. Commun. (Cambridge, U. K.) 50, 1089–1091. [DOI] [PubMed] [Google Scholar]
- (23).Kim B, Feng S, Yun SW, Leong C, Satapathy R, Wan SY, and Chang YT (2016) A Fluorescent Probe for Neural Stem/Progenitor Cells with High Differentiation Capability into Neurons,. ChemBioChem 17, 2118–2122. [DOI] [PubMed] [Google Scholar]
- (24).Yun SW, Leong C, Zhai D, Tan YL, Lim L, Bi X, Lee JJ, Kim HJ, Kang NY, Ng SH, Stanton LW, and Chang YT (2012) Neural stem cell specific fluorescent chemical probe binding to FABP7. Proc. Natl. Acad. Sci. U. S. A. 109, 10214–10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Leal-Ortiz S, Waites CL, Terry-Lorenzo R, Zamorano P, Gundelfinger ED, and Garner CC (2008) Piccolo modulation of Synapsin1a dynamics regulates synaptic vesicle exocytosis. J. Cell Biol. 181, 831–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bragina L, Fattorini G, Giovedi S, Melone M, Bosco F, Benfenati F, and Conti F (2012) Analysis of Synaptotagmin, SV2, and Rab3 Expression in Cortical Glutamatergic and GABAergic Axon Terminals. Front. Cell. Neurosci. 5, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Pinto JG, Jones DG, and Murphy KM (2013) Comparing development of synaptic proteins in rat visual, somatosensory, and frontal cortex. Front. Neural Circuits 7, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ghosh KK, Ha HH, Kang NY, Chandran Y, and Chang YT (2011) Solid phase combinatorial synthesis of a xanthone library using click chemistry and its application to an embryonic stem cell probe. Chem. Commun. (Cambridge, U. K.) 47, 7488–7490. [DOI] [PubMed] [Google Scholar]
- (29).Bissell RA, Calle E, de Silva AP, Desilva SA, Gunaratne HQN, Habibjiwan JL, Peiris SLA, Rupasinghe RADD, Samarasinghe TKSD, Sandanayake KRAS, and Soumillion JP (1992) Luminescence and Charge-Transfer 0.2. Aminomethyl Anthracene-Derivatives as Fluorescent Pet (Photoinduced Electron-Transfer) Sensors for Protons. J. Chem. Soc., Perkin Trans. 22, 1559–1564. [Google Scholar]
- (30).Saleh N, Al-Soud YA, and Nau WM (2008) Novel fluorescent pH sensor based on coumarin with piperazine and imidazole substituents. Spectrochim. Acta, Part A 71, 818–822. [DOI] [PubMed] [Google Scholar]
- (31).Abreu BJ, Guimaraes M, Uliana LC, Vigh J, von Gersdorff H, Prado MA, and Guatimosim C (2008) Protein kinase C modulates synaptic vesicle acidification in a ribbon type nerve terminal in the retina. Neurochem. Int. 53, 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sheehan P, Zhu M, Beskow A, Vollmer C, and Waites CL (2016) Activity-Dependent Degradation of Synaptic Vesicle Proteins Requires Rab35 and the ESCRT Pathway. J. Neurosci. 36, 8668–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Morris JV, Mahaney MA, and Huber JR (1976) Fluorescence Quantum Yield Determinations - 9,10-Diphenylanthracene as a Reference-Standard in Different Solvents. J. Phys. Chem. 80, 969–974. [Google Scholar]
- (34).Bolte S, and Cordelieres FP (2006) A guided tour into subcellular colocalization analysis in light microscopy,. J. Microsc. 224, 213–232. [DOI] [PubMed] [Google Scholar]
- (35).Williams RW. (2000) Mapping genes that modulate mouse brain development: a quantitative genetic approach, Springer, New York. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.