

Abstract
NMDA receptors (NMDARs) are essential components in glutamatergic synaptic signaling. The NMDAR antagonist MK-801 has been a valuable pharmacological tool in evaluating NMDAR function because it binds with high affinity to the NMDAR ion channel pore and is non-competitive with ligand binding. MK-801 has also been used to selectively inhibit NMDAR current in only the cell being recorded by including the drug in the intracellular recording solution. Here, we report that intracellular MK-801 (iMK-801) only partially inhibits synaptic NMDAR currents at both cortical layer 4 to layer 2/3 and hippocampal Schaffer collateral to CA1 synapses. Furthermore, iMK-801 incompletely inhibits heterologously expressed NMDAR currents consistent with a model of iMK-801 having a very slow binding rate and consequently ~30,000 times lower affinity than MK-801 applied to the extracellular side of the receptor. While iMK-801 can be used as a qualitative tool to study reduced postsynaptic NMDAR function, it cannot be assumed to completely block NMDARs at concentrations typically used in experiments.
1. Introduction
NMDARs are ion channels that open in response to binding of the agonist glutamate and co-agonist glycine (or D-serine; Johnson and Ascher, 1987; Kleckner and Dingledine, 1988) in addition to coincident depolarization that relieves Mg2+ block of the ion channel pore (Mayer et al., 1984; Nowak et al., 1984). NMDAR current contributes to synaptic depolarization and has a large Ca2+ conductance (MacDermott et al., 1986; Jahr and Stevens, 1993; Burnashev et al., 1995) which is involved in initiating intracellular signaling events, including synaptic plasticity (Lynch et al. 1983; Luscher and Malenka, 2012). Many pharmacological tools have been developed to manipulate NMDAR function (Traynelis et al., 2010).
The NMDAR antagonist MK-801 has been particularly useful in studying fundamental properties of glutamatergic synapses. MK-801 binds with high affinity when applied extracellularly (~5–30 nM dissociation constant at −70 mV; Dravid et al. 2007) to the NMDAR ion channel pore only when the receptor is active and prevents ionic current through the channel (Huettner and Bean, 1988). The combination of state-dependence and high-affinity binding has made possible the measurement of several facets of glutamatergic synaptic signaling. For example: MK-801 has been used to measure the biophysical property of ion channel open probability in response to glutamate (Jahr, 1992), the probability of vesicle release from the presynaptic terminal (Rosenmund et al., 1993; Murphy et al., 2003), and the motility of NMDARs in the postsynaptic membrane (Tovar and Westbrook, 2002). MK-801 has also been used to selectively inhibit NMDARs in single cells by introducing the drug into the intracellular environment through a recording pipette (Berretta and Jones, 1996). Because intracellular MK-801 (iMK-801) can only access NMDAR in the selected cell, it has been used to investigate the NMDAR contribution to synaptic signaling and integration independent of possible network effects of blocking NMDARs globally (e.g. Lavzin et al., 2012; Smith et al., 2013). This manipulation has also led to the suggestion that, at some synapses, NMDARs may be functioning presynaptically (Berretta and Jones, 1996; Humeau et al., 2003; Sjőstrom et al., 2003; Bender et al., 2006; Nevian and Sakmann, 2006; Corlew et al., 2007; Rodriguez-Moreno et al., 2011; Bouvier et al., 2015), or postsynaptically in a manner independent of ion flux through the NMDAR (Carter and Jahr, 2016). However, many of these interpretations of iMK-801 effects rely on the assumption that iMK-801 blocks NMDARs completely.
In this study, we tested whether iMK-801 was effective in inhibiting NMDAR currents. At the synapse between cortical layer 4 (L4) to layer 2/3 (L2/3) in rat somatosensory cortex, the commonly used concentration of iMK-801, 1 mM, reduced, but did not eliminate NMDAR currents. Similarly, at the hippocampal Schaffer collateral to CA1 neuron synapse, iMK-801 reduced, but did not eliminate NMDAR currents. We then tested iMK-801 in heterologously expressed NMDARs and found a similar reduction of NMDAR current, but the inhibition was incomplete. These results were recapitulated in a NMDAR kinetic model in which the rate of iMK-801 binding was ~30,000 times slower than extracellularly applied MK-801. These results show that iMK-801 can be used to qualitatively reduce NMDAR currents, but complete inhibition cannot be assumed.
2. Materials and Methods
2.1. Cortical slice preparation and electrophysiology
Young (postnatal day 13–21) Sprague-Dawley rats (Charles River) of either sex were anesthetized with isoflurane and decapitated. The brain was removed into warm ACSF consisting of (in mM): 119 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 10 Glucose, 1.3 sodium Ascorbate, 3 sodium pyruvate, equilibrated with 95% O2 / 5% CO2 (chemicals from Sigma). The brain was blocked at 35° from the coronal plane (Agmon and Connors, 1991), and 300 μm slices containing Barrel Cortex were cut with a vibratome (Leica VT1200S) in warm 37 °C ACSF. Slices were recovered in 37 °C ACSF for 30 minutes and maintained at room temperature (~22 °C) until use. Animal handling and procedures followed OHSU IACUC approved protocols.
Slices were transferred to a recording chamber perfused with 35 °C ACSF at a rate of ~2 mL/min. Barrel cortex was visually identified and two Θ glass stimulation pipettes filled with ACSF were placed in layer 4 and used to stimulate two independent synaptic pathways. L2/3 pyramidal neurons in the same column were then visually identified and patched with borosilicate glass pipettes (2–4 MΩ) filled with an internal solution consisting of (in mM): 108 cesium methanesulfonate or cesium gluconate, 2.8 NaCl, 20 HEPES, 0.4 EGTA, 5 tetraethylammonium chloride, pH adjusted to 7.3 with CsOH. (+)-MK-801 (Tocris) was added to the internal solution from a 100 mM (in water) stock solution. After break-in, the L2/3 neuron was voltage-clamped at −70 mV and L4 was stimulated at 0.1 Hz and inward currents were measured. Picrotoxin (50 μM, Sigma) and NBQX (5 μM, Tocris) were then applied to block GABAA and AMPA receptors, respectively. The cell was then held at +40 mV to relieve Mg2+ block and measure outward NMDAR currents, followed by bath application of 10 μM R-CPP (Tocris) to block NMDAR currents. Peak inward and outward currents were measured from averaged traces in each condition. Data were acquired using a Multiclamp 700B (Molecular devices), controlled by Scanimage software (Pologruto et al., 2003), sampled at 10 kHz, and analyzed using Igorpro (Wavemetrics).
2.2. Hippocampal slice preparation and electrophysiology
All mice were of C57BL/6J background and housed according to IACUC guidelines at the University of California Davis. P18–24 mice were anesthetized in isoflurane, and decapitated. Brains were rapidly removed and placed in ice-cold sucrose cutting buffer, containing (in mM) 210 sucrose, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 7 glucose, 7 MgCl2, 0.5 CaCl2. Transverse 300μm hippocampal slices were cut on a Leica VT1200 vibratome (Buffalo Grove, IL) in ice-cold cutting buffer. Slices were recovered in 32°C artificial cerebrospinal fluid (ACSF) solution, containing (in mM) 119 NaCl, 26.2 NaHCO3, 11 glucose, 2.5 KCl, 1 NaH2PO4, 2.5 CaCl2 and 1.3 MgSO4, for 1 hour before recording. Slices were transferred to a submersion chamber on an upright Olympus microscope, perfused in room temperature normal ACSF containing picrotoxin (0.1 mM) and saturated with 95%O2/5%CO2. CA1 neurons were visualized by infrared differential interference contrast microscopy. Cells were patched with 3–5MΩ borosilicate pipettes filled with intracellular solution, containing (in mM) 135 cesium methanesulfonate, 8 NaCl, 10 HEPES, 0.3 Na-GTP, 4 Mg-ATP, 0.3 EGTA, and 5 QX-314 (Sigma, St Louis, MO). (+)-MK-801 (Abcam) was added to the internal solution from a 100 mM (in DMSO) stock solution to a final concentration of 1 mM and 1% DMSO. Control experiments with simultaneous whole cell recordings with internal solutions containing 1% DMSO and 0% DMSO showed no difference in NMDAR-EPSCs (data not shown). Excitatory postsynaptic currents (EPSCs) were evoked by electrical stimulation of Schaffer collaterals with a bipolar electrode (MicroProbes, Gaithersburg, MD). AMPAR-EPSCs were measured at a holding potential of −70 mV, and NMDAR-EPSCs were measured at +40 mV in the presence of 10 μM NBQX. Series resistance was monitored and not compensated, and cells were discarded if series resistance varied more than 25%. All recordings were obtained with a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA), filtered at 2 kHz, digitized at 10 kHz. Analysis was performed with the Clampex software suite (Molecular Devices, Sunnyvale, CA) and Prism 7 software (GraphPad).
2.3. Recombinant expression and recording
HEK293 were transfected with plasmid cDNAs encoding GluN1and GluN2 subunits at a ratio of 1:2 using the calcium phosphate precipitation method as previously described (Hansen et al., 2014). Whole-cell voltage-clamp recordings were performed using an Axopatch 1D amplifier (Molecular Devices, Union City, CA) at room temperature. The holding potential was −60 mV. The electrodes were filled with internal solution containing (in mM) 110 D-gluconate, 110 CsOH, 30 CsCl, 5 HEPES, 4 NaCl, 0.5 CaCl2, 2 MgCl2, 5 BAPTA, 2 NaATP, and 0.3 NaGTP (pH 7.35 with CsOH), and the extracellular recording solution was composed of (in mM) 150 NaCl, 10 HEPES, 3 KCl, 0.5 CaCl2, 0.01 EDTA, 20 mM D-mannitol (pH 7.4 with NaOH). Rapid solution exchange (open tip solution exchange had 10–90% rise times below 1 ms) was achieved using a two-barrel theta-glass pipette controlled by a piezobimorph. Data were acquired at 20 kHz, filtered at 5–10 kHz, and analyzed with Axograph software (axographx.com) and IgorPro (Wavemetrics).
2.4. Statistical analysis
Values are reported as the mean ± SEM. At least two animals were used per group. Comparisons were made using paired or unpaired t-test where appropriate. Statistical significance is reported p < 0.05. Experiments were not performed blind to the condition of the experiments. Sample sizes are similar to those generally used in the field, and no statistical methods were used to predetermine the sample size. Randomization was not used to determine experimental conditions.
3. Results
3.1. Synaptic NMDAR currents are reduced, but not eliminated by 1 mM iMK-801
To test the effect of iMK-801 on synaptic NMDARs, we recorded synaptic currents from L2/3 neurons in rat somatosensory cortical slices in response to L4 stimulation. Figure 1A shows an example recording using control internal solution (without added MK-801). After breaking into the L2/3 neuron, voltage-clamp was established and the neuron was held at −70 mV while L4 was stimulated at 0.1 Hz. NBQX (5 μM) and Picrotoxin (50 μM) were then added to block AMPA and GABAA receptors, respectively. The neuron was then held at +40 mV to relieve Mg2+ block, outward currents were measured, and the NMDAR antagonist R-CPP (10 μM) was added. AMPAR and NMDAR currents were then isolated by subtraction of traces obtained in the presence of the specific antagonists (Figure 1A, inset). In this example, the peak AMPAR current was −198 pA and the peak NMDAR current was 276 pA, giving a ratio of NMDAR/AMPAR current of 1.39. Figure 1B shows a similar experiment except the internal recording solution contained 1 mM MK-801. Inward currents measured at −70 mV were blocked by NBQX, and outward currents measured at +40 mV were blocked by R-CPP. In this example recording, the peak AMPAR current was −542 pA and the peak NMDAR current was 111 pA, giving a ratio of 0.20. NBQX decreased inward currents using either control internal solution (Figure 1C, −277.5 ± 30.4 pA to −28.1 ± 3.5 pA, N=25 synaptic pathways from 13 neurons, t(24)=9.07, p=3.2e-9, paired t test), or internal solution containing 1 mM MK-801 (from −357.0 ± 54.1 pA to −28.4 ± 3.5 pA, N=24 synaptic pathways from 13 neurons, t(23)=6.32, p=1.9e-6, paired t test). R-CPP reduced outward currents in experiments using control internal solution (Figure 1D, from 246.0 ± 40.2 pA to 13.5 ± 3.4 pA, N=25, t(24)=5.82, p=5.4e-6, paired t test), as well as in experiments using 1 mM iMK-801 containing internal solution (Figure 1D, from 61.3 ± 10.8 pA to 8.5 ± 1.4 pA, N=24, t(23)=5.07, p=3.9e-5, paired t test). Figure 1E shows a summary of the AMPAR and NMDAR currents isolated by antagonist subtraction. There was no significant difference between the AMPAR current level measured using either control or 1 mM iMK-801 internal solution (control internal: −256.9 ± 27.9 pA, N=25, iMK-801: −335.8 ± 51.8 pA, N=24, t(47)=1.34, p=0.19, t test). The NMDAR current level was significantly reduced to ~25% of control by 1 mM iMK-801 (control: 241.4 ± 39.7 pA, N=25, iMK-801: 61.5 ± 10.6 pA, N=24, t(47)=4.38, p=6.7e-5, t test). In addition, the synaptic NMDAR/AMPAR ratio was significantly reduced in the iMK-801 condition relative to control (Figure 1F, control NMDAR/AMPAR ratio: 0.93 ± 0.09, N=25, iMK-801 ratio: 0.20 ± 0.03, N=24, t(47)=7.27, p=3.2e-9).
Figure 1. Intracellular MK-801 inhibits synaptic NMDAR current, but not completely.

(A) example recording of synaptic AMPAR and NMDAR currents from a L2/3 neuron while stimulating L4 at 0.1 Hz. Left: time course of peak synaptic currents, measuring inward currents while holding the postsynaptic neuron at −70 mV, and outward currents at +40 mV. 5 μM NBQX was added to block AMPAR currents, and 10 μM CPP was added to block NMDAR currents. Right: AMPAR (downward current) and NMDAR current (upward currents ) isolated by subtraction of traces before and after antagonist addition. Scale bars are 100 ms, 100 pA.
(B) example time course (left) and traces (right) from a recording with 1 mM MK-801 included in the internal solution. Scale bars are 100 ms, 200 pA.
(C) summary of inward currents measured at −70 mV before (ACSF) and after 5 μM NBQX addition using control internal solution (left, N = 25 synaptic pathways from 13 neurons, t(24)=9.07, p=3.2e-9, paired t test ), and with 1 mM iMK-801 (right, N = 24 synaptic pathways from 13 neurons, t(23)=6.32, p=1.9e-6, paired t test).
(D) summary of outward currents measured at +40 mV before and after 10 μM R-CPP addition using control internal solution (left, N = 25 synaptic pathways from 13 neurons, t(24)=5.82, p=5.4e-6, paired t test), and with 1 mM iMK-801 (right, N = 24 pathways from 13 neurons, t(23)=5.07, p=3.9e-5, paired t test).
(E) summary of AMPAR currents isolated by NBQX subtraction with or without MK-801 in the pipette, (ns, not significant, t(47)=1.34, p=0.19, t test), and NMDAR currents isolated by CPP subtraction, t(47)=4.38, p=6.7e-5, t-test).
(F) the ratio of NMDAR current to AMPA receptor currents was reduced in the iMK-801 condition (right) compared to control internal solution (left, t(47)=7.27, p=3.2e-9, t test). Red symbols in (C),(D), and (F) are data from the examples shown in (A) and (B).
3.2. Incomplete block of NMDAR currents by intracellular MK-801 in hippocampal CA1 neurons
In addition to L4-to-L2/3 synapses, the efficacy of iMK-801 at blocking synaptic NMDAR currents was also examined at the prototypical CA3-to-CA1 synapse. As a control, extracellular MK-801 (50 μM) was shown to effectively block synaptic NMDAR currents within 15 minutes of repeated stimulation of the Schaffer collateral axons (Figure 2A). To increase the probability of synaptic vesicle release at individual synapses without inducing postsynaptic plasticity, a paired-pulse stimulation protocol (two pulses 50 ms apart repeated at 0.1 Hz) was utilized. To examine the efficacy of iMK-801, we performed simultaneous whole cell recordings from neighboring CA1 pyramidal neurons with either control internal solution or internal solution containing 1 mM iMK-801 (Figure 2B). Simultaneous paired whole-cell recordings at CA3-to-CA1 synapses provide a rigorous, quantitative, and internally controlled comparison of the effects of iMK-801. Importantly, at the commonly used concentration of 1 mM, iMK-801 may have off-target effects, though no effects were observed on AMPAR-EPSCs (Figure 2C; control: 61.3 ± 5.8 pA, iMK-801: 71.6 ± 11.0 pA; n=9, t(8)=0.8023, p=0.446, paired t test). NMDAR-EPSCs were then examined at +40 mV in the presence of 10 μM NBQX to block AMPARs. After 20 minutes of simulating Schaffer collaterals at a neutral rate of 0.1 Hz, iMK-801 only blocked ~50% of synaptic NMDAR currents (Figure 2D; control: 185.7 ± 33.2 pA, iMK-801: 96.4 ± 17.4 pA; n=8, t(7)=4.782, p=0.0020, paired t test). Increase synaptic release probability using the paired-pulse protocol only increased NMDAR block to ~60% (Figure 2E; control: 74.0 ± 21.2 pA, iMK-801: 26.7 ± 5.2 pA; n=6, t(5)=2.904, p=0.0336, paired t test). Thus, while increase stimulation led to a modest increase in synaptic NMDAR block (Figure 2F; 0.1 Hz for 20 min: 53.6 ± 3.8% block, n=8; paired pulse: 40.9 ± 4.2% block, n=6; t(12)=2.223, p=0.046, t test), the block was still incomplete.
Figure 2. Incomplete block of synaptic NMDAR currents by intracellular MK-801 at the Schaffer collateral to CA1 synapse.

(A) Complete block of evoked NMDAR-EPSC amplitudes in 50 μM extracellular MK-801. Baseline NMDAR-EPSC amplitudes were obtained for 2 min then a paired-pulse stimulation protocol (two pulses 50 msec apart repeated at 0.1 Hz) was applied. Data represent the mean ± SEM of the NMDAR-EPSC amplitude normalized to the average baseline amplitude (n=7). Inset, sample traces of NMDAR-EPSCs at baseline (black) and after paired-pulse stimulation (gray) in 50 μM MK-801; scale bars represent 100 pA, 200 msec.
(B) Schematic of simultaneous whole-cell recordings from neighboring CA1 neurons with internal containing either 1 mM iMK-801 or control solution.
(C) AMPAR-EPSCs are unchanged by 1 mM iMK-801. Scatterplot of AMPAR-EPSC amplitudes from individual neuron pairs (open circles) and averaged pair ± SEM (solid circle) (control: 61.3 ± 5.8 pA, iMK-801: 71.6 ± 11.0 pA; n=9, t(8)=0.8023, p=0.446, paired t test). Dashed lines represent linear regression and 95% confidence interval. Inset, sample traces of AMPAR-EPSCs (black, control; gray iMK-801); scale bars represent 20 pA, 20 msec.
(D-F) Incomplete block of NMDAR-EPSCs by iMK-801. Amplitudes were measured after 20 min of either 0.1 Hz stimulation (D) or 20 min of a paired-pulse stimulation protocol (two pulses at a 50 msec interval repeated at 0.1 Hz)
(E) Scatterplots represent NMDAR-EPSC amplitudes from individual neuron pairs (open circles) and mean ± SEM (solid circle). Insets, sample traces of NMDAR-EPSCs (black, control; gray iMK-801); scale bars represent 100 pA, 200 msec.
(F) Paired-pulse stimulation lead to significantly more, yet still incomplete, NMDAR blockade (0.1Hz for 20 min: 53.6 ± 3.8% of control cell, n=8; paired pulse: 40.9 ± 4.2% of control, n=6; t(12)=2.223, p=0.046, t test).
3.3. Recombinant NMDARs are inhibited, but not completely blocked by 1 mM intracellular MK-801
The recordings of synaptic currents shown in Figures 1 and 2 were obtained from intact neurons in brain slices that have processes extending for hundreds of micrometers, and the exact location of the stimulated synapses was not known. While the inhibition of NMDAR currents by iMK-801 indicates that the drug reached and interacted with the synaptic receptors, the concentration of MK-801 at the synapse was not known with certainty. Therefore, we tested the effects of 1 mM iMK-801 on NMDAR currents in a heterologous expression system (Figure 3). GluN1 and GluN2A subunits were expressed in HEK293 cells and whole cell voltage clamp recordings were then made from isolated cells. L-glutamate (1 mM) was delivered by a fast-flow exchange system to activate the NMDARs. Figure 3A shows a recording using the control internal solution while voltage-clamping the cell at −60 mV. In this example recording, the NMDAR current reached a peak of −839 pA which was followed by a desensitization to a steady-state level 63% of the peak current level. In 7 cells using control internal solution, the peak NMDAR current induced by 1 mM glutamate was −1.92 ± 0.77 nA. The steady-state NMDAR current level was 55 ± 6 % of the peak current and developed over a time course that could be fit with a single exponential with a time constant of 1.06 ± 0.23 seconds. Figure 3B shows an example recording of NMDAR currents with 1 mM MK-801 in the recording pipette. In this example, after reaching a peak inward current of −3.45 nA, the current decayed to a steady-state level 13% of the peak level. In 4 cells recorded with 1 mM intracellular MK-801, the mean peak inward current was −2.59 ± 0.53 nA, not significantly different from the control recordings (t(9)=0.71, p=0.50, t test). The steady-state current level with 1 mM intracellular MK-801 was on average 17 ± 4 % of the peak current, ~31% of the control steady-state current level (Figure 3C, t(9)=4.56, p=1.9e-3, t test). The time course of decay of current in the presence of 1 mM intracellular MK-801 could be fit with a double exponential function in which the fast component was set to the average time constant of desensitization measured in the control condition (1.06 seconds), and the second, slower component was 4.95 ± 1.87 seconds.
Figure 3. Incomplete inhibition of GluN1/GluN2A recombinant receptor currents by intracellular MK-801.
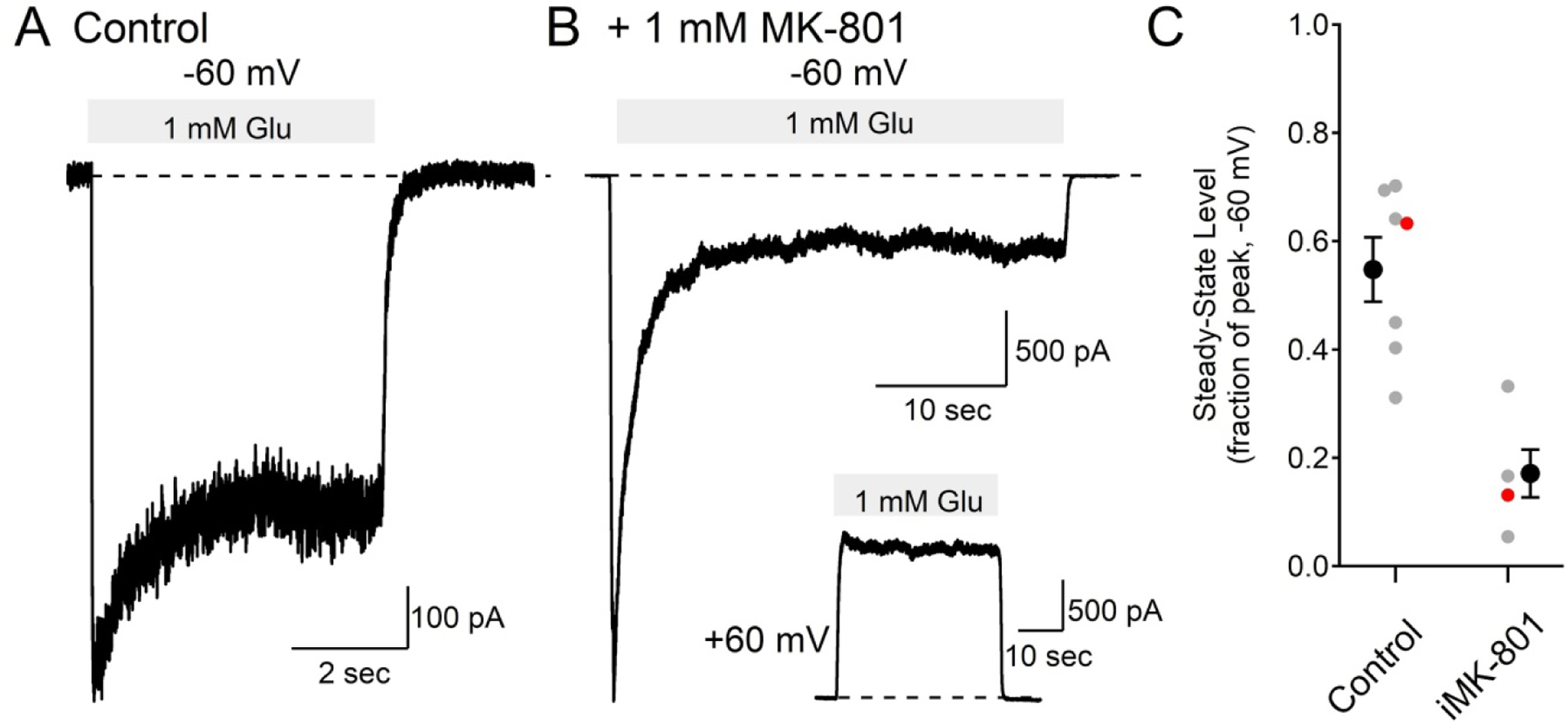
(A) using control internal solution and holding at −60 mV, 1 mM L-Glutamate induced an inward current that desensitized to 63% of the peak level.
(B) with 1 mM MK-801 in the intracellular solution, glutamate induced current fell to 13% of the peak value (note the different time scale from panel (A)). The inset shows a recording from the same cell held at +60 mV.
(C) summary of steady-state current as a fraction of the peak current at −60 mV in 7 cells using control internal solution (0.55 ± 0.06, mean ± SEM) and in 4 cells with 1 mM intracellular MK-801 (0.17 ± 0.04, mean ± SEM, t(9)=4.56, p=1.4e-3, t test). The red markers are from the example traces in (A) and (B).
At positive voltages, extracellular MK-801 dissociates from the NMDAR more readily than at negative voltages (Huettner and Bean, 1988). Similarly, inhibition of NMDAR current by intracellular MK-801 showed voltage dependence (Figure 3B). At +60 mV, NMDAR currents in cells filled with 1 mM intracellular MK-801 reached a steady-state level 85 ± 5 % of the peak current level (mean ± SEM, N = 4), significantly larger than the steady-state current from the same cells measured at −60 mV (t(3)=8.9, p=1.2e-4, paired t test).
3.4. An NMDAR model can recapitulate the intracellular MK-801 block
Figure 4A shows a Markov model structure for NMDAR glutamate binding, and gating based on the model of Lester and Jahr, 1992. Following the binding of two glutamate molecules to the receptor, the channel can either open or enter a desensitized state. If MK-801 is present, the drug has access only to the open channel state, and when MK-801 is bound, the receptor can gate normally, but does not conduct current. The rate constants of glutamate binding and unbinding were initially set similar to recombinant GluN1/2A receptors (Maki and Popescu, 2014). Peak open probability in the model is ~0.46, similar to that measured from GluN1/2A receptors (Erregar et al., 2005), and, in the absence of MK-801, the conductance desensitizes to a steady-state level that is ~60% of the peak, similar to the value measured in the recombinant receptors (Figure 3C). The rate constant of MK-801 unbinding (0.06 s−1, at −60 mV) was set to match the rate of MK-801 unbinding measured in dissociated neurons (Huettner and Bean, 1988). The on-rate of MK-801 was then adjusted to empirically match the time course of block measured in recombinant receptors (Figure 3B). This led to an estimated on-rate of 8e-4 μM−1s−1, which captures well the time course and degree of inhibition by 1mM intracellular MK-801 on the NMDAR conductance (Figure 4B). In the absence of MK-801, the modeled conductance desensitizes to a level similar to the measured levels (Figure 4B, dashed trace). When 1 mM MK-801 is included in the model, the conductance (Figure 4B, solid red trace) matches very closely the measured NMDAR conductance time course (Figure 4B, black trace, same trace as in Figure 3B). These modeled rates correspond to an affinity of MK-801 to the NMDAR that is ~30,000 times lower when applied to the intracellular side of the receptor than when applied extracellularly.
Figure 4. A NMDAR model can recapitulate inhibition by iMK-801.

(A) Structure of the NMDAR model consisting of the unliganded receptor, R, two binding sites for glutamate, RG and RGG, a desensitized state D, open state O, and corresponding states with MK-801 bound (gray states). After two glutamate molecules bind to the receptor, the channel can open or desensitize (black states). MK-801 has access to the channel only during the open state, and, once bound the channel does not conduct, but can gate normally (gray states).
(B) Normalized modeled NMDAR conductance (dashed red trace) matches well the recorded NMDAR conductance (black trace, same recording as Figure 3A).
(C) Modeling NMDAR conductance with 1 mM iMK-801 (solid red trace) matches well the time course of recorded NMDAR conductance (black trace, same recording as Figure 3B). The red dashed trace shows the modeled current in the absence of iMK-801. The model rates used were: kon = 17 μM−1s−1, koff = 60 s−1, kd = 6.5 s−1, kr = 0.5 s−1, α = 250 s−1, β = 2000 s−1, kMk, on = 8e-4 μM−1s−1, kMk, off = 0.06 s−1.
4. Discussion
4.1. The binding site of MK-801
The binding site of iMK-801 was presumed to be the same site as extracellularly applied MK-801. Consistent with this interpretation, iMK-801 required opening of the channel to inhibit NMDAR current: the current reaches a peak before being inhibited to a lower steady-state level, and the peak current was similar in amplitude in both control and iMK-801 conditions. The inhibition by iMK-801 shows voltage dependence consistent with the expulsion of positively charged MK-801 through the channel. A single binding site for iMK-801 is also consistent with recent cryo-EM structural data that show a single MK-801 binding site in the NMDAR channel (Lu et al., 2017).
The voltage-dependent off-rate of MK-801 in the model (kMK, off = 0.64 * exp(Vm / 25.4) s−1) leads to an estimated rate of recovery from MK-801 block that matches the measured rates of recovery in isolated neuronal cells (measured as 92 ± 40 minutes at −70 mV and 1.8 ± 0.3 minutes at +30 mV, Huettner and Bean, 1988), assuming popen of 0.007, close to the estimated popen in those recordings (Huettner and Bean, 1988). This similarity in measured and modeled MK-801 off-rate is consistent with MK-801 binding at the same site whether it entered from the extra- or intracellular side of the receptor. The primary difference between intra- and extracellularly applied MK-801 was found to be the on-rate of the drug. The modeled on-rate, 8×10−4 μM−1s−1, ~30,000 times slower than the measured on-rate of extracellularly applied MK-801, 23.7 μM−1s−1 (Jahr, 1992), led to an estimated KD of ~75 μM for iMK-801 at −60 mV. The slow on-rate of iMK-801 reflects the reduced access to the binding site that is on the extracellular side of the pore-loop in the NMDAR ion channel (Lu et al., 2017).
The low affinity of iMK-801 to NMDARs can explain the use of high concentrations (usually 1 mM) of iMK-801. However, this may lead to off-target effects on other ion channels or proteins inside the cell. For example, extracellular MK-801 at μM levels can affect acetylcholine receptor signaling (Galligan and North, 1990), and, at mM levels, extracellular MK-801 can affect voltage-gated K+ channels (Rothman, 1988). Because it is applied intracellularly, off-target effects will be difficult to test experimentally.
4.2. Synaptic NMDAR inhibition
Because block by MK-801 occurs in a 1:1 manner with a single drug molecule binding in the ion channel pore, the number of NMDARs in a given synapse that are blocked should follow a binomial distribution. Since glutamatergic synapses are typically thought to contain a small number of NMDARs, estimated in hippocampal CA1 neurons to be ~5 functional NMDARs on average per synapse (Nimchinsky et al., 2004), synaptic recordings would be expected to show variability consistent with binomial statistics. For example, if there are 5 NMDARs in a synapse and iMK-801 blocks NMDAR current by 70% (as in Figure 3), there would be a ~17% (i.e. 0.75) chance that all the receptors will be blocked in a given trial. This could explain some of the variability of iMK-801 block of the synaptically recorded NMDAR currents (Figure 1). This intrinsic variability of inhibition highlights the fact that iMK-801 cannot be assumed to eliminate the contribution of postsynaptic NMDAR current during synaptic stimulation.
At positive voltages, the dissociation of MK-801 from NMDARs is much faster than at negative voltages (Huettner and Bean, 1988). Using the model rates, the estimated KD of iMK-801 binding at +60 mV is ~8.5 mM. This is apparent in the recordings from the recombinant NMDARs (Figure 3) where, after blocking NMDAR current by ~70% at −60 mV, glutamate application at +60 mV led to unblock of the channels (Figure 3B, inset). This raises the question of why the synaptic NMDAR currents are blocked by iMK-801 by ~75% relative to the control synaptic current at +40 mV (Figure 1). One possible explanation is that synaptically released glutamate is only present at high concentration very briefly (~1 ms) (Clements et al., 1992), and, at least in the synaptic recordings, there may not be sufficient time for MK-801 dissociation from open receptors to reach a steady-state level. This would be in contrast to the experiments with recombinant receptors where sustained glutamate application over many seconds allows enough time for MK-801 dissociation to occur. NMDAR subunit identity is a crucial determinant of receptor kinetics (Cull-Candy and Leszkiewicz, 2004) and could also affect the binding of MK-801 and, while the synaptic NMDAR subunit composition is not known for certain, it likely contains both GluN2A and GluN2B subunits either as diheteromeric GluN1/GluN2A, GluN1/2B, or triheteromeric GluN1/2A/2B receptors (Monyer et al., 1994; Gray et al., 2011; Tovar et al. 2013). The presence of the GluN2B subunit confers a lower open probability (Erregar et al., 2005; Gray et al., 2011), which would decrease the rate of MK-801 dissociation.
5. Conclusions
MK-801 has been an extremely useful tool in probing NMDAR channel function, and intracellular MK-801 has been useful in distinguishing NMDAR function in single cells from network effects of NMDAR inhibition. However, we show here 1 mM iMK-801, a commonly used concentration, does not completely inhibit NMDAR currents. This incomplete inhibition can be explained by an extremely slow binding rate of MK-801 when applied to the intracellular side of the membrane. These results are consistent with prior studies that show iMK-801 significantly reduces NMDAR currents, but there is often incomplete block of the current (Humeau et al., 2003; Samson and Pare, 2005; Bender et al., 2006; Corlew et al., 2007; Rodriguez-Moreno et al., 2011; Lavzin et al., 2012; Smith et al., 2013; Larsen et al., 2014). Therefore, while this manipulation can be a useful qualitative tool to selectively reduce NMDAR current in a single recorded cell, it cannot be assumed to eliminate the contribution of NMDAR current to postsynaptic signaling.
Acknowledgements
We thank Craig Jahr and Delia Chiu for helpful discussions.
JAG was supported by the Whitehall Foundation.
References
- Agmon A & Connors BW Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience 41, 365–379 (1991). [DOI] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci 26, 4166–77 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta N, & Jones RSG Tonic facilitation of glutamate release by presynaptic N-methyl-D-aspartate autoreceptors in the entorhinal cortex. Neuroscience 2, 339–344 (1996). [DOI] [PubMed] [Google Scholar]
- Bouvier G, Bidoret C, Casado M, Paoletti P Presynaptic NMDA receptors: Roles and rules. Neuroscience 311, 322–40 (2015). [DOI] [PubMed] [Google Scholar]
- Burnashev N, Zhou Z, Neher E, Sakmann B Fractional calcium currents through recombinant GluR channels of the NMDA, AMPA, and kainate receptor subtypes. J Physiol 485, 403–18 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BC & Jahr CE Postsynaptic, not presynaptic NMDA receptors are required for spike-timing-dependent LTD induction. Nat Neurosci 19, 1218–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL The time course of glutamate in the synaptic cleft. Science 258, 1498–501 (1992). [DOI] [PubMed] [Google Scholar]
- Corlew R, Wang Y, Ghermazien H, Erisir A, Philpot BD Developmental switch in the contribution of presynaptic and postsynaptic NMDA receptors to long-term depression. J. Neurosci 27, 9835–9845 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy SG & Leszkiewicz DN Role of distinct NMDA receptor subtypes at central synapses. Sci STKE 255, pp. Re16 DOI: 10.1126/stke.2552004re16 [DOI] [PubMed] [Google Scholar]
- Dravid SM, Erreger K, Yuan H, Nicholson K, Le P, Lyuboslavsky P, Almonte A, Murray E, Mosley C, Barber J, French A, Balster R, Murray TF, Traynelis SF Subunit-specific mechanisms and proton sensitivity of NMDA receptor channel block. J Physiol 581, 107–128 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erregar K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol 563, 345–58 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galligan JJ & North RA MK-801 blocks nicotinic depolarizations of guinea pig myenteric neurons. Neurosci Lett 108, 105–9 (1990). [DOI] [PubMed] [Google Scholar]
- Gray JA, Shi Y, Usui H, During MJ, Sakimura K, Nicoll RA Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: analysis of single-cell GluN2 subunit deletion in vivo. Neuron 71, 1085–1101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson KB, Ogden KK, Yuan H, Traynelis SF Distinct functional and pharmacological properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron 81, 1084–1096 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner JE & Bean BP Block of N-methyl-D-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc. Natl. Acad. Sci. U.S.A 85, 1307–11 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Shaban H, Bissiere S, Luthi A Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature 426, 841–5 (2003). [DOI] [PubMed] [Google Scholar]
- Jahr CE High probability opening of NMDA receptor channels by L-glutamate. Science 255, 470–2 (1992). [DOI] [PubMed] [Google Scholar]
- Jahr CE, & Stevens CF Calcium permeability of the N-methyl-D-aspartate receptor channel in hippocampal neurons in culture. Proc Natl Acad Sci U.S.A 90, 11573–7 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JW & Ascher P Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 325, 529–31 (1987). [DOI] [PubMed] [Google Scholar]
- Kleckner NW & Dingledine R Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science 241, 835–7 (1988). [DOI] [PubMed] [Google Scholar]
- Larsen RS, Smith IT, Miriyala J, Han JE, Corlew RJ, Smith SL, Philpot BD Synapse-specific control of experience-dependent plasticity by presynaptic NMDA receptors. Neuron 83, 879–93 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavzin M, Rapoport S, Polsky A, Garion L, Schiller J Nonlinear dendritic processing determines angular tuning of barrel cortex neurons in vivo. Nature 490, 397–401 (2012). [DOI] [PubMed] [Google Scholar]
- Lester RA & Jahr CE NMDA channel behavior depends on agonist affinity. J Neurosci 12, 635–43 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Du J, Goehring A, Gouaux E Cryo-EM structures of the triheteromeric NMDA receptor and its allosteric modulation. Science 355, eaal3729 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, & Malenka RC NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol 4 (2012). DOI: 10.1101/cshperspect.a005710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Larson J, Kelso S, Barrionuevo G, Schottler F Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature 305, 719–21 (1983). [DOI] [PubMed] [Google Scholar]
- Maki BA, & Popescu GK Extracellular Ca2+ ions reduce NMDA receptor conductance and gating. J. Gen. Physiol 144, 379–392 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature 321, 519–22 (1986). [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB Voltage-dependent block by Mg2+of NMDA responses in spinal cord neurones. Nature 309, 261–3 (1984). [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540 (1994). [DOI] [PubMed] [Google Scholar]
- Murphy GJ, Glickfield LL, Balsen Z, Isaacson JS Sensory neuron signaling to the brain: properties of transmitter release from olfactory nerve terminals. J Neurosci 24 3023–30 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevian T & Sakmann B Spine Ca2+ signaling in spike-timing dependent plasticity. J Neurosci 26, 11001–13 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Yasuda R, Oertner TG, Svoboda K The number of glutamate receptors opened by synaptic stimulation in single hippocampal spines. J Neurosci 24, 2054–64 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak L Bregestovski P, Ascher P, Herbet A, Prochiantz A Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307, 462–5 (1984). [DOI] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K ScanImage: flexible software for operating laser scanning microscopes. Biomed. Eng. Online 17, 2:13 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Moreno A, Kohl MM, Reeve JE, Eaton TR, Collins HA, Anderson HL, Paulsen O Presynaptic induction and expression of timing-dependent long-term depression by compartment-specific photorelease of a use-dependent NMDA receptor antagonist. J Neurosci 31, 8564–8569 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL Nonuniform probability of glutamate release at a hippocampal synapse. Science 262, 754–7 (1993). [DOI] [PubMed] [Google Scholar]
- Rothman S Noncompetitive N-methyl-D-aspartate antagonists affect multiple ionic currents. J Pharmacol Exp Ther 246, 137–42 (1988). [PubMed] [Google Scholar]
- Samson RD and Pare D Activity-dependent synaptic plasticity in the central nucleus of the amygdala. J Neurosci 25, 1847–1855 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39, 641–654 (2003). [DOI] [PubMed] [Google Scholar]
- Smith SL, Smith IT, Branco T, Häusser M Dendritic spikes enhance stimulus selectivity in cortical neurons in vivo. Nature 503, 115–20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR & Westbrook GL Mobile NMDA receptors at hippocampal synapses. Neuron 34, 255–64 (2002). [DOI] [PubMed] [Google Scholar]
- Tovar KR, McGinley MJ, Westbrook GL Triheteromeric NMDA receptors at hippocampal synapses. J Neurosci 33, 9150–60 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62 405–96 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]